
INLYTA tabletti, kalvopäällysteinen 1 mg, 5 mg
Vaikuttavat aineet ja niiden määrät
Inlyta 1 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 1 mg aksitinibia.
Inlyta 5 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 5 mg aksitinibia.
Apuaineet, joiden vaikutus tunnetaan
Inlyta 1 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 33,6 mg laktoosimonohydraattia.
Inlyta 5 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 58,8 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti).
Kliiniset tiedot
Käyttöaiheet
Inlyta on tarkoitettu edennyttä munuaissolukarsinoomaa sairastavien aikuispotilaiden hoitoon aiemman sunitinibi- tai sytokiinihoidon epäonnistuttua.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Inlyta-hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus
Aksitinibin suositeltu annos on 5 mg kaksi kertaa vuorokaudessa.
Hoitoa on jatkettava niin kauan kuin kliinistä hyötyä todetaan tai kunnes esiintyy haittaavaa toksisuutta, jota ei voida hoitaa käyttämällä muita lääkevalmisteita samanaikaisesti tai muuttamalla annosta.
Jos potilas oksentaa tai annos jää ottamatta, lisäannosta ei saa ottaa, vaan seuraava hoito-ohjelman mukainen annos otetaan tavanomaiseen aikaan.
Annoksen muuttaminen
Annosta suositellaan suurentamaan tai pienentämään potilaan yksilöllisen turvallisuuden ja sietokyvyn mukaisesti.
Jos potilas sietää aksitinibin aloitusannoksen 5 mg kaksi kertaa vuorokaudessa eikä hänellä esiinny astetta 2 vaikeampia haittavaikutuksia (eli ei esiinny the Common Terminology Criteria for Adverse Events [CTCAE] version 3.0 ‑haittavaikutuskriteerien mukaisia vaikea-asteisia haittavaikutuksia) kahden peräkkäisen viikon aikana, potilaan annos voidaan suurentaa 7 mg:aan kaksi kertaa vuorokaudessa edellyttäen, että potilaan verenpaine ei ole > 150/90 mmHg eikä potilas saa verenpainelääkitystä. Jos potilas sietää aksitinibiannoksen 7 mg kaksi kertaa vuorokaudessa, hänen annoksensa voidaan näiden samojen kriteerien mukaisesti suurentaa enintään annokseen 10 mg kaksi kertaa vuorokaudessa.
Joidenkin haittavaikutusten hoito saattaa edellyttää aksitinibihoidon keskeyttämistä tilapäisesti tai pysyvästi ja/tai aksitinibiannoksen pienentämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos annosta on tarpeen pienentää, aksitinibiannos voidaan pienentää 3 mg:aan kaksi kertaa vuorokaudessa ja edelleen 2 mg:aan kaksi kertaa vuorokaudessa.
Annosta ei tarvitse mukauttaa potilaan iän, rodun, sukupuolen tai painon perusteella.
Voimakkaiden CYP3A4/5-estäjien samanaikainen käyttö
Aksitinibin käyttö samanaikaisesti voimakkaiden CYP3A4/5-estäjien kanssa saattaa suurentaa aksitinibipitoisuutta plasmassa (ks. kohta Yhteisvaikutukset). Samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei estä CYP3A4/5-isoentsyymiä tai estää sitä mahdollisimman vähän.
Aksitinibiannoksen muuttamista ei ole tutkittu voimakkaita CYP3A4/5-estäjiä käyttävillä potilailla, mutta jos voimakkaiden CYP3A4/5-estäjien samanaikainen käyttö on välttämätöntä, aksitinibiannosta suositellaan pienentämään noin puoleen (esim. aloitusannos tulisi pienentää annoksesta 5 mg kaksi kertaa vuorokaudessa annokseen 2 mg kaksi kertaa vuorokaudessa). Joidenkin haittavaikutusten hoito saattaa edellyttää aksitinibihoidon keskeyttämistä tilapäisesti tai pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos voimakkaan estäjän samanaikainen käyttö lopetetaan, on harkittava palaamista voimakkaan CYP3A4/5-estäjän aloitusta edeltäneeseen aksitinibiannokseen (ks. kohta Yhteisvaikutukset).
Voimakkaiden CYP3A4/5-induktorien samanaikainen käyttö
Aksitinibin käyttö samanaikaisesti voimakkaiden CYP3A4/5-induktorien kanssa saattaa pienentää aksitinibipitoisuutta plasmassa (ks. kohta Yhteisvaikutukset). Samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei indusoi CYP3A4/5-isoentsyymiä tai indusoi sitä mahdollisimman vähän.
Aksitinibiannoksen muuttamista ei ole tutkittu voimakkaita CYP3A4/5-induktoreja käyttävillä potilailla, mutta jos voimakkaiden CYP3A4/5-induktorien samanaikainen käyttö on välttämätöntä, suositellaan aksitinibiannosta suurennettavan asteittain. Suurina annoksina käytettyjen voimakkaiden CYP3A4/5-induktorien indusoivan vaikutuksen on raportoitu olevan suurimmillaan yhden viikon kuluessa induktorihoidon aloittamisesta. Jos aksitinibiannosta suurennetaan, potilaan tilaa on seurattava tarkoin toksisuuden havaitsemiseksi. Joidenkin haittavaikutusten hoito saattaa edellyttää aksitinibihoidon keskeyttämistä tilapäisesti tai pysyvästi ja/tai aksitinibiannoksen pienentämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos voimakkaan induktorin samanaikainen käyttö lopetetaan, on palattava heti voimakkaan CYP3A4/5-induktorin aloitusta edeltäneeseen aksitinibiannokseen (ks. kohta Yhteisvaikutukset).
Erityispotilasryhmät
Iäkkäät (≥ 65-vuotiaat)
Annoksen muuttaminen ei ole tarpeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka). Tietoja ei ole saatavilla aksitinibihoidosta potilailla, joiden kreatiniinipuhdistuma on < 15 ml/min.
Maksan vajaatoiminta
Annosta ei ole tarpeen muuttaa, kun aksitinibia annetaan lievää maksan vajaatoimintaa sairastavalle potilaalle (Child-Pugh-luokka A). Annosta suositellaan pienentämään, kun aksitinibia annetaan kohtalaista maksan vajaatoimintaa sairastavalle potilaalle (Child-Pugh-luokka B) (esim. aloitusannos on pienennettävä annoksesta 5 mg kaksi kertaa vuorokaudessa annokseen 2 mg kaksi kertaa vuorokaudessa). Aksitinibia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh-luokka C) eikä sitä pitäisi käyttää tälle potilasryhmälle (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Inlytan turvallisuutta ja tehoa lasten ja nuorten (alle 18-vuotiaiden) hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Aksitinibi on otettava suun kautta. Tabletit on otettava kaksi kertaa vuorokaudessa noin 12 tunnin välein joko aterian yhteydessä tai tyhjään mahaan (ks. kohta Farmakokinetiikka). Ne on nieltävä kokonaisina lasillisen vettä kanssa.
Vasta-aiheet
Yliherkkyys aksitinibille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Tiettyjä turvallisuuteen liittyviä tapahtumia on arvioitava ennen aksitinibihoidon aloittamista ja seurattava säännöllisesti hoidon aikana kuten seuraavassa kuvataan.
Sydämen vajaatoiminta
Munuaissolukarsinoomaa sairastavilla potilailla tehdyissä aksitinibin kliinisissä tutkimuksissa raportoitiin sydämen vajaatoimintatapauksia (mukaan lukien sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiopulmonaalinen vajaatoiminta, vasemman kammion toiminnanvajaus, pienentynyt ejektiofraktio ja oikean kammion vajaatoiminta) (ks. kohta Haittavaikutukset).
Sydämen vajaatoiminnan merkkejä ja oireita on seurattava säännöllisesti koko aksitinibihoidon keston ajan. Sydämen vajaatoiminnan hoito saattaa edellyttää aksitinibihoidon keskeyttämistä tilapäisesti tai pysyvästi ja/tai aksitinibiannoksen pienentämistä.
Hypertensio
Munuaissolukarsinoomaa sairastavilla potilailla tehdyissä aksitinibin kliinisissä tutkimuksissa raportoitiin hypertensiota hyvin yleisesti (ks. kohta Haittavaikutukset).
Kontrolloidussa kliinisessä tutkimuksessa hypertensio (systolinen verenpaine > 150 mmHg tai diastolinen verenpaine > 100 mmHg) ilmaantui keskimäärin kuukauden kuluessa aksitinibihoidon aloittamisesta ja verenpaineen nousua on havaittu jo 4 vuorokauden kuluttua aksitinibin aloittamisesta.
Verenpaineen on oltava hyvässä hoitotasapainossa ennen aksitinibihoidon aloittamista. Potilasta on seurattava hypertension ilmaantumisen havaitsemiseksi ja hypertensio on tarvittaessa hoidettava tavanomaisilla verenpainelääkkeillä. Hypertension jatkuessa verenpainelääkkeiden käytöstä huolimatta aksitinibiannosta on pienennettävä. Jos potilaalle kehittyy vaikea-asteinen hypertensio, aksitinibihoito on keskeytettävä tilapäisesti ja aloitettava uudelleen pienemmällä annoksella sen jälkeen, kun potilaan verenpaine on korjaantunut normaaliksi. Jos aksitinibihoito keskeytetään, verenpainelääkitystä käyttävää potilasta on seurattava hypotension havaitsemiseksi (ks. kohta Annostus ja antotapa).
Jos potilaalla ilmenee vakavaa tai pitkään jatkuvaa valtimohypertoniaa ja posterioriseen korjautuvaan enkefalopatiaoireyhtymään (PRES) viittaavia oireita (ks. jäljempänä), on harkittava diagnostista aivojen magneettikuvausta.
Kilpirauhasen toimintahäiriö
Munuaissolukarsinoomaa sairastavilla potilailla tehdyissä aksitinibin kliinisissä tutkimuksissa raportoitiin hypotyreoosia ja harvemmin hypertyreoosia (ks. kohta Haittavaikutukset).
Kilpirauhasen toiminta on määritettävä ennen aksitinibihoidon aloittamista ja sitä on seurattava säännöllisesti koko hoidon keston ajan. Hypotyreoosi tai hypertyreoosi on hoidettava tavanomaisen hoitokäytännön mukaisesti normaalin kilpirauhastoiminnan ylläpitämiseksi.
Valtimoiden tromboemboliset ja tromboottiset tapahtumat
Aksitinibilla tehdyissä kliinisissä tutkimuksissa raportoitiin valtimoiden tromboembolisia ja tromboottisia tapahtumia (kuten ohimeneviä aivoverenkiertohäiriöitä, sydäninfarkteja, aivohalvauksia ja verkkokalvon valtimotukoksia) (ks. kohta Haittavaikutukset).
Aksitinibihoidossa on oltava varovainen, jos potilaalla on tällaisten tapahtumien riski tai jos hänellä on aiemmin esiintynyt tällaisia tapahtumia. Aksitinibia ei ole tutkittu potilailla, joilla on ollut valtimoiden tromboembolinen tai tromboottinen tapahtuma edeltäneiden 12 kuukauden aikana.
Laskimoiden tromboemboliset ja tromboottiset tapahtumat
Aksitinibilla tehdyissä kliinisissä tutkimuksissa raportoitiin laskimoiden tromboembolisia ja tromboottisia tapahtumia (kuten keuhkoemboliaa, syviä laskimotukoksia ja verkkokalvon laskimotukoksia/trombooseja) (ks. kohta Haittavaikutukset).
Aksitinibihoidossa on oltava varovainen, jos potilaalla on tällaisten tapahtumien riski tai jos hänellä on aiemmin esiintynyt tällaisia tapahtumia. Aksitinibia ei ole tutkittu potilailla, joilla on ollut laskimoiden tromboembolinen tai tromboottinen tapahtuma edeltäneiden 6 kuukauden aikana.
Hemoglobiini- tai hematokriittiarvojen nousu
Aksitinibihoidon aikana saattaa esiintyä hemoglobiini- tai hematokriittiarvojen nousua, mikä kuvastaa veren punasolumassan lisääntymistä (ks. kohta Haittavaikutukset polysytemia). Veren punasolumassan lisääntyminen saattaa suurentaa tromboembolisten ja tromboottisten tapahtumien riskiä.
Hemoglobiini tai hematokriitti on määritettävä ennen aksitinibihoidon aloittamista ja niitä on seurattava säännöllisesti koko hoidon keston ajan. Jos hemoglobiini- tai hematokriittiarvot nousevat normaaliarvoja korkeammiksi, potilaalle on annettava tavanomaisen hoitokäytännön mukaista hoitoa hemoglobiini- tai hematokriittiarvojen laskemiseksi hyväksyttävälle tasolle.
Verenvuoto
Aksitinibilla tehdyissä kliinisissä tutkimuksissa on raportoitu verenvuototapahtumia (ks. kohta Haittavaikutukset).
Aksitinibia ei ole tutkittu potilailla, joilla on viitteitä hoitamattomasta aivometastaasista tai joilla on äskettäin esiintynyt ruoansulatuskanavan verenvuoto, eikä sitä tulisi käyttää näille potilaille. Jos verenvuoto vaatii lääketieteellisiä toimenpiteitä, aksitinibihoito on keskeytettävä tilapäisesti.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava huolellisesti ennen Inlyta-hoidon aloittamista potilailla, jolla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Ruoansulatuskanavan perforaatio ja fistelimuodostus
Aksitinibilla tehdyissä kliinisissä tutkimuksissa on raportoitu ruoansulatuskanavan perforaatioita ja fisteleitä (ks. kohta Haittavaikutukset).
Ruoansulatuselimistön perforaatioiden ja fistelien oireita on seurattava säännöllisesti koko aksitinibihoidon ajan.
Haavojen paranemiseen liittyvät komplikaatiot
Aksitinibin vaikutuksesta haavojen paranemiseen ei ole tehty varsinaisia tutkimuksia.
Aksitinibihoito on lopetettava vähintään 24 tuntia ennen suunniteltua leikkausta. Aksitinibihoidon jatkamisesta leikkauksen jälkeen on päätettävä haavan riittävästä parantumisesta tehdyn kliinisen arvion perusteella.
Posteriorinen korjautuva enkefalopatiaoireyhtymä (posterior reversible encephalopathy syndrome, PRES)
Aksitinibilla tehdyissä kliinisissä tutkimuksissa on raportoitu posteriorista korjautuvaa enkefalopatiaoireyhtymää (ks. kohta Haittavaikutukset).
Posteriorinen korjautuva enkefalopatiaoireyhtymä on neurologinen häiriö, jonka yhteydessä voi esiintyä päänsärkyä, kouristuskohtauksia, letargiaa, sekavuutta, sokeutta sekä muita näköaistiin liittyviä ja neurologisia häiriöitä. Lievää tai vaikeaa hypertensiota saattaa esiintyä. Posteriorisen korjautuvan enkefalopatiaoireyhtymän diagnoosi on varmistettava magneettikuvauksella. Jos potilaalla on posteriorisen korjautuvan enkefalopatiaoireyhtymän oireita tai löydöksiä, aksitinibihoito on keskeytettävä tilapäisesti tai lopetettava pysyvästi. Jos potilaalla on aiemmin ollut posteriorinen korjautuva enkefalopatiaoireyhtymä, ei tiedetä, onko aksitinibihoito turvallista aloittaa uudelleen.
Proteinuria
Aksitinibilla tehdyissä kliinisissä tutkimuksissa on raportoitu proteinuriaa, myös vaikeusasteeltaan asteen 3 ja 4 proteinuriaa (ks. kohta Haittavaikutukset).
Virtsan proteiinin määritystä suositellaan ennen aksitinibihoidon aloittamista ja seuraamista säännöllisesti koko hoidon keston ajan. Jos potilaalle kehittyy keskivaikea tai vaikea proteinuria, aksitinibiannosta on pienennettävä tai aksitinibihoito on keskeytettävä tilapäisesti (ks. kohta Annostus ja antotapa). Aksitinibihoito on lopetettava, jos potilaalle kehittyy nefroottinen oireyhtymä.
Maksaan kohdistuvat haittavaikutukset
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa raportoitiin maksaan kohdistuneita haittavaikutuksia. Yleisimmin raportoituja maksaan liittyneitä haittavaikutuksia olivat alaniiniaminotransferaasiarvon (ALAT), aspartaattiaminotransferaasiarvon (ASAT) ja veren bilirubiinipitoisuuden suureneminen (ks. kohta Haittavaikutukset). ALAT-arvon suurenemista (yli kolminkertaiseksi viitearvojen ylärajaan nähden) ja bilirubiinipitoisuuden suurenemista (yli kaksinkertaiseksi viitearvojen ylärajaan nähden) ei havaittu samanaikaisesti.
Kliinisessä annoshakututkimuksessa havaittiin yhdellä aksitinibia saaneella potilaalla samanaikaisesti ALAT-arvon suureneminen (12-kertaiseksi viitearvojen ylärajaan nähden) ja bilirubiinipitoisuuden suureneminen (2,3-kertaiseksi viitearvojen ylärajaan nähden), mikä katsottiin lääkkeeseen liittyneeksi maksatoksisuudeksi. Potilas sai aloitusannoksena 20 mg kaksi kertaa vuorokaudessa (nelinkertainen annos suositeltuun aloitusannokseen verrattuna).
Maksan toiminta on määritettävä ennen aksitinibihoidon aloittamista ja sitä on seurattava säännöllisesti koko hoidon keston ajan.
Maksan vajaatoiminta
Aksitinibilla tehdyissä kliinisissä tutkimuksissa keskivaikeaa maksan vajaatoimintaa (Child-Pugh-luokka B) sairastavien tutkimuspotilaiden systeeminen altistus aksitinibille oli noin kaksinkertainen verrattuna tutkimuspotilaisiin, joiden maksan toiminta oli normaali. Annoksen pienentämistä suositellaan annettaessa aksitinibia keskivaikeaa maksan vajaatoimintaa sairastavalle potilaalle (Child-Pugh-luokka B) (ks. kohta Annostus ja antotapa).
Aksitinibia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh-luokka C) eikä sitä pidä käyttää tälle potilasryhmälle.
Iäkkäät (≥ 65-vuotiaat) ja etninen tausta
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa 34 % aksitinibihoitoa saaneista potilaista oli 65-vuotiaita tai vanhempia. Suurin osa potilaista oli valkoihoisia (77 %) tai aasialaista alkuperää (21 %). Haittavaikutusten ilmaantumista herkemmin joillekin iäkkäille tai aasialaista alkuperää oleville potilaille ei voida sulkea pois, mutta yleisesti ottaen aksitinibin turvallisuudessa ja tehossa ei havaittu merkittäviä eroja ≥ 65-vuotiaiden tai tätä nuorempien potilaiden välillä, eikä valkoihoisten tai muuta etnistä alkuperää olevien potilaiden välillä.
Annostusta ei tarvitse muuttaa potilaan iän tai etnisen taustan perusteella (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Apuaineet
Laktoosi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasin puutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kalvopäällysteinen tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
In vitro ‑tiedot osoittavat, että aksitinibi metaboloituu pääasiassa CYP3A4/5-entsyymin välityksellä ja vähäisemmässä määrin CYP1A2- ja CYP2C19-entsyymien sekä uridiinidifosfaattiglukuronosyylitransferaasi (UGT) 1A1:n välityksellä.
CYP3A4/5-estäjät
Voimakas CYP3A4/5-estäjä ketokonatsoli annoksina 400 mg kerran vuorokaudessa 7 vuorokauden ajan käytettynä suurensi aksitinibin suun kautta otetusta 5 mg:n kerta-annoksesta terveille vapaaehtoisille koehenkilöille aiheutuvan keskimääräisen AUC-arvon (pitoisuus-pinta-alakäyrän alle jäävän alueen) kaksinkertaiseksi ja Cmax-arvon 1,5-kertaiseksi. Aksitinibin samanaikainen käyttö voimakkaiden CYP3A4/5-estäjien (esim. ketokonatsolin, itrakonatsolin, klaritromysiinin, erytromysiinin, atatsanaviirin, indinaviirin, nefatsodonin, nelfinaviirin, ritonaviirin, sakinaviirin ja telitromysiinin) kanssa voi suurentaa aksitinibipitoisuutta plasmassa. Greippihedelmä saattaa myös suurentaa aksitinibipitoisuutta plasmassa. Samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei estä CYP3A4/5-isoentsyymiä tai estää sitä mahdollisimman vähän. Jos voimakkaan CYP3A4/5-estäjän samanaikainen käyttö on välttämätöntä, aksitinibiannosta suositellaan muutettavan (ks. kohta Annostus ja antotapa).
CYP1A2- ja CYP2C19-estäjät
CYP1A2 ja CYP2C19 ovat aksitinibin metabolian sivureittejä (< 10 %). Näiden isoentsyymien voimakkaiden estäjien vaikutusta aksitinibin farmakokinetiikkaan ei ole tutkittu. Näiden isoentsyymien voimakkaita estäjiä käyttävien potilaiden hoidossa on oltava varovainen plasman aksitinibipitoisuuden suurenemisriskin vuoksi.
CYP3A4/5-induktorit
Voimakas CYP3A4/5-induktori rifampisiini annoksina 600 mg kerran vuorokaudessa 9 vuorokauden ajan käytettynä pienensi aksitinibin 5 mg:n kerta-annoksesta terveille vapaaehtoisille koehenkilöille aiheutuvaa keskimääräistä AUC-arvoa 79 % ja Cmax-arvoa 71 %.
Aksitinibin samanaikainen käyttö voimakkaiden CYP3A4/5-induktorien (esim. rifampisiinin, deksametasonin, fenytoiinin, karbamatsepiinin, rifabutiinin, rifapentiinin, fenobarbitaalin ja Hypericum perforatum -rohdoksen eli mäkikuisman) kanssa saattaa pienentää aksitinibipitoisuutta plasmassa. Samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei indusoi CYP3A4/5-isoentsyymiä tai indusoi sitä mahdollisimman vähän. Jos voimakkaan CYP3A4/5-induktorin samanaikainen käyttö on välttämätöntä, aksitinibiannosta suositellaan muutettavan (ks. kohta Annostus ja antotapa).
In vitro‑tutkimukset CYP- ja UGT-entsyymien estymisestä ja induktiosta
In vitro ‑tutkimukset osoittivat, että aksitinibi ei estä terapeuttisina pitoisuuksina plasmassa CYP2A6-, CYP2C9-, CYP2C19-, CYP2D6-, CYP2E1- ja CYP3A4/5-isoentsyymejä tai UGT1A1:stä.
In vitro ‑tutkimukset osoittivat, että aksitinibi saattaa estää CYP1A2-isoentsyymiä. Aksitinibin samanaikainen käyttö CYP1A2-substraattien kanssa saattaa siksi johtaa suurentuneeseen CYP1A2-substraatin (esim. teofylliinin) pitoisuuteen plasmassa.
In vitro ‑tutkimukset osoittivat myös, että aksitinibi saattaa estää CYP2C8-isoentsyymiä. Aksitinibin ja tunnetun CYP2C8-substraatin paklitakselin samanaikainen käyttö ei kuitenkaan aiheuttanut edennyttä syöpää sairastaneille potilaille suurentuneita paklitakselipitoisuuksia plasmassa, mikä osoittaa, ettei kliinistä CYP2C8:n estymistä tapahdu.
In vitro ‑tutkimukset ihmisen maksasoluilla osoittivat myös, ettei aksitinibi indusoi isoentsyymejä CYP1A1, CYP1A2 ja CYP3A4/5. Aksitinibin ei siksi odoteta pienentävän in vivo samanaikaisesti annetun CYP1A1-, CYP1A2- tai CYP3A4/5-substraatin pitoisuutta plasmassa.
P-glykoproteiinitutkimukset in vitro
In vitro ‑tutkimukset osoittivat, että aksitinibi estää P-glykoproteiinia. Aksitinibin ei kuitenkaan odoteta terapeuttisina pitoisuuksina plasmassa estävän P-glykoproteiinia. Aksitinibin samanaikaisen käytön ei siksi odoteta suurentavan digoksiinin tai muiden P-glykoproteiinin substraattien pitoisuutta plasmassa in vivo.
Raskaus ja imetys
Raskaus
Aksitinibin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Aksitinibi saattaa farmakologisten ominaisuuksiensa perusteella aiheuttaa sikiölle haittaa, jos sitä käytetään raskauden aikana. Eläinkokeissa on havaittu lisääntymistoksisuutta, myös epämuodostumia (ks. kohta Prekliiniset tiedot turvallisuudesta). Aksitinibia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa tällä lääkevalmisteella.
Hedelmällisessä iässä olevien naisten on käytettävä tehokasta ehkäisyä hoidon aikana ja yhden viikon ajan hoidon jälkeen.
Imetys
Ei tiedetä, erittyykö aksitinibi ihmisen rintamaitoon. Rintaruokittuun lapseen kohdistuvia riskejä ei voida sulkea pois. Aksitinibia ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Aksitinibi saattaa non-kliinisten löydösten perusteella heikentää ihmisen lisääntymistoimintoja ja hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Aksitinibilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaille on kerrottava, että heillä saattaa esiintyä aksitinibihoidon aikana mm. heitehuimausta ja/tai uupumusta.
Haittavaikutukset
Turvallisuustietojen yhteenveto
Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on tarkempaa tietoa seuraavista riskeistä sekä asianmukaisista toimenpiteistä niiden yhteydessä: sydämen vajaatoimintatapaukset, hypertensio, kilpirauhasen toimintahäiriö, valtimoiden tromboemboliset tapahtumat, laskimoiden tromboemboliset tapahtumat, hemoglobiini- tai hematokriittiarvojen nousu, verenvuoto, ruoansulatuskanavan perforaatio ja fistelimuodostus, haavojen paranemiseen liittyvät komplikaatiot, PRES, proteinuria ja maksaentsyymiarvojen suureneminen.
Yleisimmät (≥ 20 %) aksitinibihoidon haittavaikutukset olivat ripuli, hypertensio, uupumus, heikentynyt ruokahalu, pahoinvointi, painonlasku, äänen käheys, käsi-jalkaoireyhtymä, verenvuoto, hypotyreoosi, oksentelu, proteinuria, yskä ja ummetus.
Haittavaikutustaulukko
Taulukossa 1 esitetään kliinisissä tutkimuksissa raportoidut haittavaikutukset, jotka on saatu yhdistämällä 672 aksitinibia saaneen munuaissolukarsinoomapotilaan tiedot (ks. kohta Farmakodynamiikka). Mukana ovat myös haittavaikutukset, jotka on tunnistettu valmisteen markkinoilletulon jälkeisissä kliinisissä tutkimuksissa.
Haittavaikutukset on lueteltu elinluokan, esiintymistiheyden ja vaikeusasteen mukaan. Esiintymistiheyksiksi on määritelty: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, <1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Tällä hetkellä aksitinibin turvallisuustietokannassa ei ole tarpeeksi tietoa, jotta harvinaiset ja hyvin harvinaiset haittavaikutukset voitaisiin tunnistaa.
Esiintymistiheysluokat on määritelty perustuen absoluuttisiin esiintymistiheyksiin yhdistetyistä kliinisistä tutkimustiedoista. Haittavaikutukset on esitetty kussakin elinluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Munuaissolukarsinoomatutkimuksissa aksitinibia saaneilla potilailla raportoidut haittavaikutukset (N = 672)
Elinjärjestelmä | Esiintymis-tiheys | Haittavaikutukseta | Kaikki vaikeus-asteetb % | Aste 3b % | Aste 4b % |
Veri ja imukudos | Yleinen | Anemia | 6,3 | 1,2 | 0,4 |
Trombosytopenia | 1,6 | 0,1 | 0 | ||
Polysytemiac | 1,5 | 0,1 | 0 | ||
Melko harvinainen | Neutropenia | 0,3 | 0,1 | 0 | |
Leukopenia | 0,4 | 0 | 0 | ||
Umpieritys | Hyvin yleinen | Hypotyreoosic | 24,6 | 0,3 | 0 |
Yleinen | Hypertyreoosic | 1,6 | 0,1 | 0,1 | |
Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Heikentynyt ruokahalu | 39,0 | 3,6 | 0,3 |
Yleinen | Elimistön kuivuminen | 6,7 | 3,1 | 0,3 | |
Hyperkalemia | 2,7 | 1,2 | 0,1 | ||
Hyperkalsemia | 2,2 | 0,1 | 0,3 | ||
Hermosto | Hyvin yleinen | Päänsärky | 16,2 | 0,7 | 0 |
Makuaistin häiriöt | 11,5 | 0 | 0 | ||
Yleinen | Heitehuimaus | 9,1 | 0,6 | 0 | |
Melko harvinainen | Posteriorinen korjautuva enkefalopatiaoireyhtymäe | 0,3 | 0,1 | 0 | |
Kuulo ja tasapainoelin | Yleinen | Tinnitus | 3,1 | 0 | 0 |
Sydän | Yleinen | Sydämen vajaatoiminta-tapauksetc,d,f | 1,8 | 0,3 | 0,7 |
Verisuonisto | Hyvin yleinen | Hypertensiog | 51,2 | 22,0 | 1,0 |
Verenvuotoc,d,h | 25,7 | 3,0 | 1,0 | ||
Yleinen | Laskimoiden tromboemboliset ja tromboottiset tapahtumatc,d,i | 2,8 | 0,9 | 1,2 | |
Valtimoiden tromboemboliset ja tromboottiset tapahtumatc,d,j | 2,8 | 1,2 | 1,3 | ||
Tuntematon | Aneurysmat ja valtimon dissekaatiotd | - | - | - | |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hengenahdistusd | 17,1 | 3,6 | 0,6 |
Yskä | 20,4 | 0,6 | 0 | ||
Dysfonia | 32,7 | 0 | 0,1 | ||
Yleinen | Suun ja nielun kipu | 7,4 | 0 | 0 | |
Ruoansulatuselimistö | Hyvin yleinen | Ripuli | 55,4 | 10,1 | 0,1 |
Oksentelu | 23,7 | 2,7 | 0,1 | ||
Pahoinvointi | 33,0 | 2,2 | 0,1 | ||
Vatsakipu | 14,7 | 2,5 | 0,3 | ||
Ummetus | 20,2 | 1,0 | 0 | ||
Stomatiitti | 15,5 | 1,8 | 0 | ||
Dyspepsia | 11,2 | 0,1 | 0 | ||
Yleinen | Ylävatsakipu | 9,4 | 0,9 | 0 | |
Ilmavaivat | 4,5 | 0 | 0 | ||
Peräpukamat | 3,3 | 0 | 0 | ||
Glossodynia | 2,8 | 0 | 0 | ||
Ruoansulatuskanavan perforaatiot ja fistelic,k | 1,9 | 0,9 | 0,3 | ||
Maksa ja sappi | Yleinen | Hyperbilirubinemia | 1,3 | 0,1 | 0,1 |
| Kolekystiittin | 1,0 | 0,6 | 0,1 | ||
Iho ja ihonalainen kudos | Hyvin yleinen | Käsi-jalkaoireyhtymä | 32,1 | 7,6 | 0 |
Ihottuma | 14,3 | 0,1 | 0 | ||
Ihon kuivuminen | 10,1 | 0,1 | 0 | ||
Yleinen | Kutina | 6,0 | 0 | 0 | |
Eryteema | 3,7 | 0 | 0 | ||
Alopesia | 5,7 | 0 | 0 | ||
Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelsärky | 17,7 | 1,9 | 0,3 |
Raajakipu | 14,1 | 1,0 | 0,3 | ||
Yleinen | Lihassärky | 8,2 | 0,6 | 0,1 | |
Munuaiset ja virtsatiet | Hyvin yleinen | Proteinurial | 21,1 | 4,8 | 0,1 |
Yleinen | Munuaisten vajaatoimintam | 1,6 | 0,9 | 0,1 | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus | 45,1 | 10,6 | 0,3 |
Asteniad | 13,8 | 2,8 | 0,3 | ||
Limakalvotulehdus | 13,7 | 1,0 | 0 | ||
Tutkimukset | Hyvin yleinen | Painonlasku | 32,7 | 4,9 | 0 |
Yleinen | Suurentunut lipaasipitoisuus | 3,7 | 0,7 | 0,7 | |
Suurentunut alaniiniamino-transferaasipitoisuus (ALAT) | 6,5 | 1,2 | 0 | ||
Suurentunut amylaasipitoisuus | 3,4 | 0,6 | 0,4 | ||
Suurentunut aspartaattiamino-transferaasipitoisuus (ASAT) | 6,1 | 1,0 | 0 | ||
Suurentunut alkalisen fosfataasin pitoisuus (AFOS) | 4,8 | 0,3 | 0 | ||
Suurentunut kreatiniinipitoisuus | 5,7 | 0,4 | 0 | ||
Suurentunut tyreotropiinipitoisuus | 7,9 | 0 | 0 |
a Haittavaikutusten luokitteluperusteena hoidon yhteydessä havaittu, syy-yhteydestä riippumaton esiintymistiheys.
b National Cancer Institute Common Terminology Criteria for Adverse Events, Version 3.0 ‑haittavaikutuskriteerit
c Ks. kohta ”Eräiden kliinisesti tärkeiden haittavaikutusten kuvaus”.
d Kuolemaan johtaneita (asteen 5) tapauksia raportoitiin.
e Mukaan lukien leukoenkefalopatia.
f Mukaan lukien sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiopulmonaalinen vajaatoiminta, pienentynyt ejektiofraktio, vasemman kammion toiminnanvajaus ja oikean kammion vajaatoiminta.
g Mukaan lukien pahanlaatuinen hypertensio, verenpaineen nousu, hypertensio ja hypertensiivinen kriisi.
h Mukaan lukien pidentynyt aktivoitu partiaalinen tromboplastiiniaika, peräaukon verenvuoto, valtimoverenvuoto, verta virtsassa, keskushermoston verenvuoto, aivoverenvuoto, pidentynyt hyytymisaika, sidekalvon verenvuoto, kontuusio, verinen ripuli, kohdun dysfunktionaalinen vuotohäiriö, nenäverenvuoto, mahalaukun verenvuoto, ruoansulatuskanavan verenvuoto, ienverenvuoto, verioksennus, veriuloste, pienentynyt hematokriittiarvo, hematooma, verivirtsaisuus, pienentynyt hemoglobiiniarvo, veriyskä, verenvuoto, sepelvaltimon verenvuoto, virtsateiden verenvuoto, peräpukamaverenvuoto, hemostaasi, mustelmataipumuksen lisääntyminen, INR-arvon (international normalized ratio) kasvu, alemman ruoansulatuskanavan verenvuoto, meleena, petekiat, nieluverenvuoto, pidentynyt protrombiiniaika, keuhkoverenvuoto, purppura, peräsuoliverenvuoto, pienentynyt punasolumäärä, munuaisverenvuoto, kovakalvon verenvuoto, kivespussin hematoseele, pernan hematooma, kynnenalainen viivamainen verenvuoto, lukinkalvonalainen verenvuoto, kieliverenvuoto, ylemmän ruoansulatuskanavan verenvuoto ja emätinverenvuoto.
i Mukaan lukien Budd-Chiarin oireyhtymä, syvä laskimotukos, kaulalaskimon tukos, lantion alueen laskimotukos, keuhkoembolia, verkkokalvon laskimotukos, verkkokalvon laskimotromboosi, solislaskimon tukos, laskimotukos ja raajan laskimotukos.
j Mukaan lukien akuutti sydäninfarkti, embolia, sydäninfarkti, verkkokalvon valtimotukos ja ohimenevä aivoverenkiertohäiriö.
k Ruoansulatuskanavan perforaatio ja fisteli sisältää seuraavat suositellut termit: vatsaontelon märkäpesäke, peräaukon märkäpesäke, anaalifisteli, fisteli, maha-suolikanavan anastomoosivuoto, ruoansulatuskanavan perforaatio, paksusuolen perforaatio, esofagobronkiaalinen fisteli ja vatsakalvontulehdus.
l Proteinuria sisältää seuraavat suositellut termit: proteiinivirtsaisuus, proteiinia virtsassa ja proteinuria.
m Mukaan lukien akuutti munuaisten vajaatoiminta.
n Kolekystiitti sisältää akuutin kolekystiitin, kolekystiitin, infektiivisen kolekystiitin.
Eräiden kliinisesti tärkeiden haittavaikutusten kuvaus
Sydämen vajaatoiminta(ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla (N = 359) tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa 1,7 %:lla aksitinibia saaneista potilaista raportoitiin sydämen vajaatoimintatapauksia, mukaan lukien sydämen vajaatoiminta (0,6 %), kardiopulmonaalinen vajaatoiminta (0,6 %), vasemman kammion toiminnanvajaus (0,3 %) ja oikean kammion vajaatoiminta (0,3 %). Asteen 4 sydämen vajaatoimintaa raportoitiin 0,6 %:lla aksitinibia saaneista potilaista. Kuolemaan johtanutta sydämen vajaatoimintaa raportoitiin 0,6 %:lla aksitinibia saaneista potilaista.
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehdyissä aksitinibin monoterapiatutkimuksissa 1,8 %:lla aksitinibia saaneista potilaista raportoitiin sydämen vajaatoimintatapauksia (mukaan lukien sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiopulmonaalinen vajaatoiminta, vasemman kammion toiminnanvajaus, pienentynyt ejektiofraktio ja oikean kammion vajaatoimintaa). Asteen 3/4 sydämen vajaatoimintaa raportoitiin 1,0 %:lla ja kuolemaan johtanutta sydämen vajaatoimintaa 0,3 %:lla aksitinibia saaneista potilaista.
Kilpirauhasen toimintahäiriö (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa hypotyreoosia raportoitiin 20,9 %:lla potilaista ja hypertyreoosia raportoitiin 1,1 %:lla potilaista. Suurentuneita tyreotropiinipitoisuuksia (TSH) raportoitiin haittavaikutuksena 5,3 %:lla aksitinibia saaneista potilaista. Rutiiniluonteisissa laboratoriokokeissa todettiin, että jos potilaan tyreotropiinipitoisuus oli ennen hoitoa < 5 μU/ml, tyreotropiinipitoisuus suureni 32,2 %:lla aksitinibia saaneista potilaista arvoon ≥ 10 μU/ml.
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa hypotyreoosia raportoitiin 24,6 %:lla aksitinibia saaneista potilaista. Hypertyreoosia raportoitiin 1,6 %:lla aksitinibia saaneista potilaista.
Laskimoiden tromboemboliset ja tromboottiset tapahtumat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa raportoitiin laskimoiden tromboembolisia ja tromboottisia haittavaikutuksia 3,9 %:lla aksitinibia saaneista potilaista, mukaan lukien keuhkoembolioita (2,2 %), verkkokalvon laskimotukoksia/-trombooseja (0,6 %) ja syviä laskimotukoksia (0,6 %). Asteen 3/4 laskimoiden tromboembolisia ja tromboottisia haittavaikutuksia raportoitiin 3,1 %:lla aksitinibia saaneista potilaista. Kuolemaan johtanut keuhkoembolia raportoitiin yhdellä aksitinibia saaneella potilaalla (0,3 %).
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa laskimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 2,8 %:lla aksitinibia saaneista potilaista. Asteen 3 laskimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 0,9 %:lla potilaista. Asteen 4 laskimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 1,2 %:lla potilaista. Kuolemaan johtaneita laskimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 0,1 %:lla aksitinibia saaneista potilaista.
Valtimoiden tromboemboliset ja tromboottiset tapahtumat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa valtimon tromboembolisia ja tromboottisia haittavaikutuksia raportoitiin 4,7 %:lla aksitinibia saaneista potilaista, mukaan lukien sydäninfarkteja (1,4 %), ohimeneviä aivoverenkiertohäiriöitä (0,8 %) ja aivoverisuonitapahtumia (0,6 %). Asteen 3/4 valtimoiden tromboembolisia ja tromboottisia haittavaikutuksia raportoitiin 3,3 %:lla aksitinibia saaneista potilaista. Kuolemaan johtanut akuutti sydäninfarkti ja aivohalvaus raportoitiin kumpikin yhdellä potilaalla (0,3 %). Aksitinibilla tehdyissä monoterapiatutkimuksissa (N = 850) valtimoiden tromboembolisia ja tromboottisia haittavaikutuksia (mukaan lukien ohimeneviä aivoverenkiertohäiriöitä, sydäninfarkteja ja aivohalvauksia) raportoitiin 5,3 %:lla aksitinibia saaneista potilaista.
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa valtimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 2,8 %:lla aksitinibia saaneista potilaista. Asteen 3 valtimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 1,2 %:lla potilaista. Asteen 4 valtimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 1,3 %:lla potilaista. Kuolemaan johtaneita valtimoiden tromboembolisia ja tromboottisia tapahtumia raportoitiin 0,3 %:lla aksitinibia saaneista potilaista.
Polysytemia (ks. Hemoglobiini- tai hematokriittiarvojen nousu kohdassa Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa raportoitiin polysytemiaa 1,4 %:lla aksitinibia saaneista potilaista. Rutiiniluonteisissa laboratoriokokeissa havaittiin hemoglobiiniarvojen suurentuneen viitearvojen ylärajaa suuremmaksi 9,7 %:lla aksitinibia saaneista potilaista. Neljässä munuaissolukarsinoomaa sairastavilla potilailla tehdyssä kliinisessä aksitinibitutkimuksessa (N = 537) hemoglobiiniarvot suurenivat viitearvojen ylärajaa suuremmaksi 13,6 %:lla aksitinibia saaneista potilaista.
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa polysytemiaa raportoitiin 1,5 %:lla aksitinibia saaneista potilaista.
Verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa raportoitiin verenvuotoon liittyviä haittavaikutuksia 21,4 %:lla aksitinibia saaneista potilaista. Tutkimukseen ei otettu mukaan potilaita, joilla oli hoitamaton aivometastaasi. Verenvuotoon liittyneitä haittavaikutuksia aksitinibia saaneilla potilailla olivat muun muassa nenäverenvuoto (7,8 %), verivirtsaisuus (3,6 %), veriyskä (2,5 %), verenvuoto peräsuolesta (2,2 %), verenvuoto ikenistä (1,1 %), mahalaukun verenvuoto (0,6 %), aivoverenvuoto (0,3 %) ja alemman ruoansulatuskanavan verenvuoto (0,3 %). Asteen ≥ 3 verenvuotoon liittyneitä haittavaikutuksia raportoitiin 3,1 %:lla aksitinibia saaneista potilaista (mukaan lukien aivoverenvuoto, mahalaukun ja alemman ruoansulatuskanavan verenvuoto ja veriyskä). Kuolemaan johtanutta verenvuotoa raportoitiin yhdellä aksitinibia saaneella potilaalla (0,3 %) (mahalaukun verenvuoto). Aksitinibilla tehdyissä monoterapiatutkimuksissa (N = 850) raportoitiin veriyskää 3,9 %:lla potilaista; vaikeusasteeltaan asteen ≥ 3 veriyskää raportoitiin 0,5 %:lla potilaista.
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa verenvuototapahtumia raportoitiin 25,7 %:lla aksitinibia saaneista potilaista. Asteen 3 verenvuotoon liittyneitä haittavaikutuksia raportoitiin 3 %:lla potilaista. Asteen 4 verenvuotoon liittyneitä haittavaikutuksia raportoitiin 1 %:lla potilaista ja kuolemaan johtaneita verenvuotoja 0,4 %:lla aksitinibia saaneista potilaista.
Ruoansulatuskanavan perforaatio ja fistelimuodostus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa raportoitiin ruoansulatuskanavan perforaation tyyppisiä tapahtumia 1,7 %:lla aksitinibia saaneista potilaista, mukaan lukien anaalifisteleitä (0,6 %), fisteleitä (0,3 %) ja ruoansulatuskanavan perforaatioita (0,3 %). Aksitinibilla tehdyissä monoterapiatutkimuksissa (N = 850) ruoansulatuskanavan perforaation tyyppisiä tapahtumia raportoitiin 1,9 %:lla potilaista ja yhdellä potilaalla raportoitiin kuolemaan johtanut ruoansulatuskanavan perforaatio (0,1 %).
Munuaissolukarsinoomaa sairastavilla potilailla (N = 672) tehtyjen aksitinibin kliinisten tutkimusten yhdistetyissä tiedoissa ruoansulatuskanavan perforaatiota ja fistelimuodostusta raportoitiin 1,9 %:lla aksitinibia saaneista potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Aksitinibin yliannokseen ei ole spesifistä hoitoa.
Munuaissolukarsinoomaa sairastavilla potilailla tehdyssä aksitinibin kontrolloidussa kliinisessä tutkimuksessa yksi potilas sai epähuomiossa annoksen 20 mg kaksi kertaa vuorokaudessa neljän vuorokauden ajan, ja hänellä esiintyi heitehuimausta (aste 1).
Aksitinibilla tehdyssä kliinisessä annoshakututkimuksessa aloitusannoksia 10 mg kaksi kertaa vuorokaudessa tai 20 mg kaksi kertaa vuorokaudessa saaneilla potilailla esiintyi haittavaikutuksena hypertensiota, kouristuskohtauksia, joihin liittyi hypertensiota, ja kuolemaan johtanutta veriyskää.
Yliannosepäilyn yhteydessä aksitinibilääkitys on lopetettava ja aloitettava elintoimintoja tukeva hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä:Solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EK01
Vaikutusmekanismi
Aksitinibi on endoteelikasvutekijäreseptoreiden (VEGFR)-1, VEGFR-2 ja VEGFR-3 tyrosiinikinaasin voimakas ja selektiivinen estäjä. Nämä reseptorit osallistuvat patologiseen angiogeneesiin, kasvainten kasvuun ja syövän metastaattiseen etenemiseen. Aksitinibin on osoitettu estävän tehokkaasti endoteelisolujen endoteelikasvutekijävälitteistä lisääntymistä ja eloonjääntiä. Aksitinibi esti VEGFR-2:n fosforylaatiota vieraslajisiirrekasvainten verisuonissa, jotka ilmensivät kohdetta in vivo, ja hidasti kasvaimen kasvua, aiheutti kasvaimen regressiota ja esti etäpesäkkeiden muodostumista useissa kokeellisissa syöpämalleissa.
Vaikutus QTc-aikaan
Satunnaistetussa, kaksisuuntaisessa ristikkäistutkimuksessa 35 terveelle tutkimuspotilaalle annettiin suun kautta kerta-annos aksitinibia (5 mg) 7 vuorokauden ajan yhdessä 400 mg:n ketokonatsoliannoksen kanssa tai ilman sitä. Tämän tutkimuksen tulokset osoittivat, että enimmillään kaksinkertainen plasman aksitinibialtistus verrattuna 5 mg:n annosta normaalisti seuraavaan altistukseen ei aiheuttanut kliinisesti merkittävää QT-ajan pitenemistä.
Kliininen teho ja turvallisuus
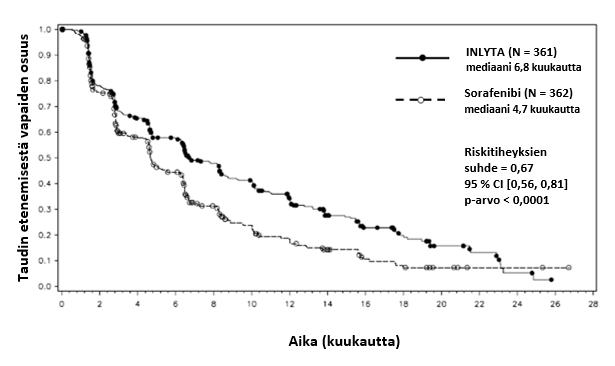
Aksitinibin turvallisuutta ja tehoa tutkittiin satunnaistetussa, avoimessa, vaiheen 3 monikeskustutkimuksessa. Edennyttä munuaissolukarsinoomaa sairastavat potilaat (N = 723), joiden sairaus oli edennyt yhden aiemman systeemisen hoidon (kuten sunitinibia, bevasitsumabia, temsirolimuusia tai sytokiinejä sisältävän hoidon) aikana tai sen jälkeen, satunnaistettiin (1:1) saamaan aksitinibia (N = 361) tai sorafenibia (N = 362). Ensisijainen päätetapahtuma, taudin etenemisestä vapaa elinaika (progression-free survival, PFS), arvioitiin sokkoutettua, riippumatonta keskitettyä arviointia käyttäen. Toissijaisia päätetapahtumia olivat objektiivisen hoitovasteen saaneiden potilaiden osuus (objective response rate, ORR) ja kokonaiselinaika (overall survival, OS).
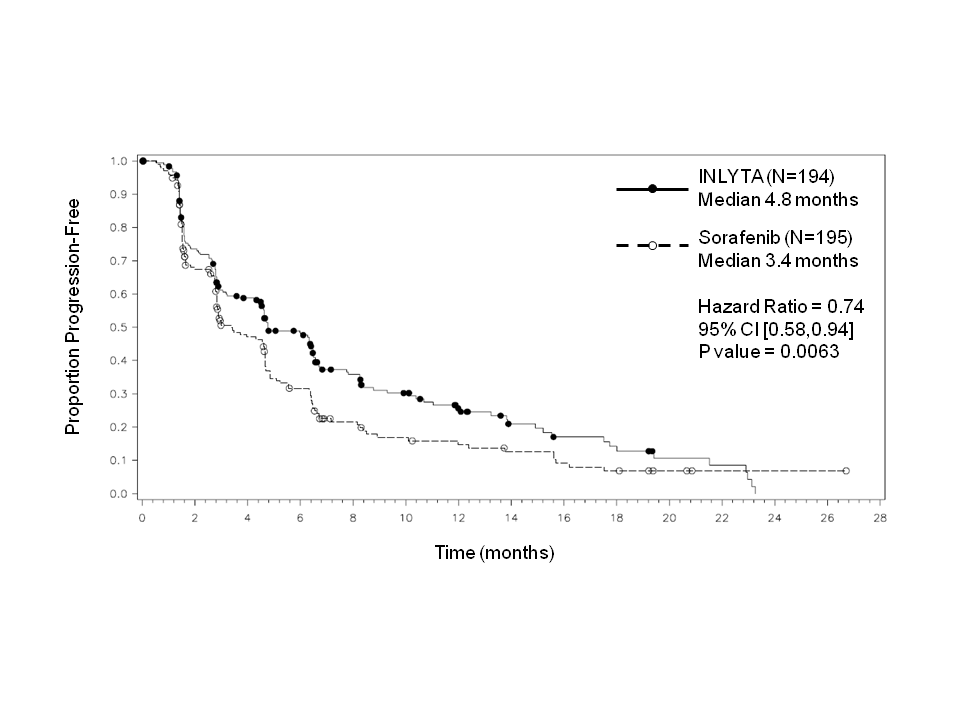
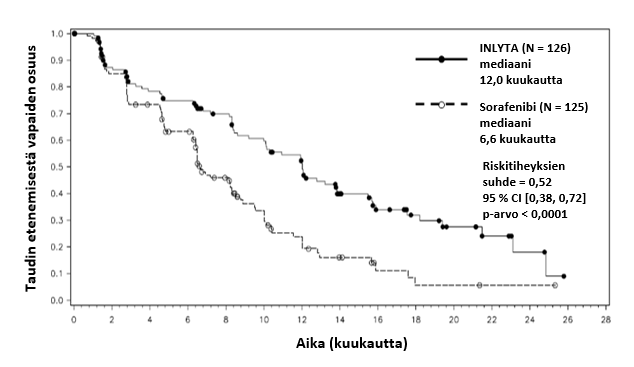
Tähän tutkimukseen mukaan otetuista potilaista 389 potilasta (53,8 %) oli saanut yhtä aiempaa sunitinibipohjaista hoitoa, 251 potilasta (34,7 %) oli saanut yhtä aiempaa sytokiinipohjaista hoitoa (interleukiini-2:ta tai alfa-interferonia), 59 potilasta (8,2 %) oli saanut yhtä aiempaa bevasitsumabipohjaista hoitoa ja 24 potilasta (3,3 %) oli saanut yhtä aiempaa temsirolimuusipohjaista hoitoa. Lähtötilanteen demografiset ja sairauteen liittyvät ominaisuudet olivat aksitinibi- ja sorafenibiryhmissä iän, sukupuolen, rodun, Eastern Cooperative Oncology Group (ECOG) ‑syöpätutkimusjärjestön toimintakykyluokituksen, maantieteellisen alueen ja aiemman hoidon suhteen samankaltaiset.
Aksitinibilla todettiin tilastollisesti merkitsevä hyöty ensisijaisen päätetapahtuman eli taudin etenemisestä vapaan elinajan (PFS) suhteen sorafenibiin verrattuna koko potilasjoukossa ja kahdessa tärkeimmässä alaryhmässä (aiempi sunitinibi- tai sytokiinihoito) (ks. taulukko 2 ja kuvat 1, 2 ja 3). Vaikutuksen suuruus mitattuna taudin etenemisestä vapaan elinajan mediaanina oli erilainen aiemman hoidon mukaisissa alaryhmissä. Kaksi alaryhmistä oli liian pieniä luotettavien tulosten saamiseksi (aiempi temsirolimuusi- tai bevasitsumabihoito). Hoitohaarojen välillä ei ollut tilastollisesti merkitsevää eroa kokonaiselinajassa koko potilasjoukossa tai aiemman hoidon mukaisissa alaryhmissä.
Taulukko 2. Tehoa koskevat tulokset
Päätetapahtuma / tutkimuspotilasjoukko | aksitinibi | sorafenibi | HR (95 % CI) | p-arvo |
Koko potilasjoukko (ITT) | N = 361 | N = 362 | ||
Mediaani PFSa,b kuukausina (95 % CI) Mediaani OSd kuukausina (95 % CI) ORRb,e % (95 % CI) | 6,8 (6,4, 8,3) 20,1 (16,7, 23,4) 19,4 (15,4, 23,9) | 4,7 (4,6, 6,3) 19,2 (17,5, 22,3) 9,4 (6,6, 12,9) | 0,67 (0,56, 0,81) 0,97 (0,80, 1,17) 2,06f (1,41, 3,00) | < 0,0001c NS 0,0001g |
Aiempi sunitinibihoito | N = 194 | N = 195 | ||
Mediaani PFSa,b kuukausina (95 % CI) Mediaani OSd kuukausina (95 % CI) ORRb,e % (95 % CI) | 4,8 (4,5, 6,5) 15,2 (12,8, 18,3) 11,3 (7,2, 16,7) | 3,4 (2,8, 4,7) 16,5 (13,7, 19,2) 7,7 (4,4, 12,4) | 0,74 (0,58, 0,94) 1,00 (0,78, 1,27) 1,48f (0,79, 2,75) | 0,0063h NS NS |
Aiempi sytokiinihoito | N = 126 | N = 125 | ||
Mediaani PFSa,b kuukausina (95 % CI) Mediaani OSd kuukausina (95 % CI) ORRb,e % (95 % CI) | 12,0 (10,1, 13,9) 29,4 (24,5, NE) 32,5 (24,5, 41,5) | 6,6 (6,4, 8,3) 27,8 (23,1, 34,5) 13,6 (8,1, 20,9) | 0,52 (0,38, 0,72) 0,81 (0,56, 1,19) 2,39f (1,43–3,99) | < 0,0001h NS 0,0002i |
CI = luottamusväli (confidence interval), HR = riskitiheyksien suhde (hazard ratio) (aksitinibi/sorafenibi); ITT: Intent-to-treat; NE: ei arvioitavissa; NS: ei tilastollisesti merkitsevä; ORR: objektiivisen hoitovasteen saaneiden potilaiden osuus (objective response rate); OS: kokonaiselinaika (overall survival); PFS: taudin etenemisestä vapaa elinaika (progression-free survival)
a Aika satunnaistamisesta taudin etenemiseen tai potilaan mistä tahansa syystä tapahtuneeseen kuolemaan, kumpi näistä tapahtuu ensin. Tiedonkeruu katkaistu 3.6.2011.
b Riippumaton radiologinen arvio Response Evaluation Criteria in Solid Tumours (RECIST) -luokituksen mukaan.
c ECOG-toimintakykyluokan ja aiemman hoidon mukaan ositetun hoidon log-rank-testin yksitahoinen p-arvo.
d Tiedonkeruu katkaistu 1.11.2011.
e Tiedonkeruu katkaistu 31.8.2010.
f Objektiiviseen hoitovasteeseen käytetään riskisuhdetta. Riskisuhde > 1 viittaa suurempaan hoitovasteen todennäköisyyteen aksitinibiryhmässä; riskisuhde < 1 viittaa suurempaan hoitovasteen todennäköisyyteen sorafenibiryhmässä.
g ECOG-toimintakykyluokan ja aiemman hoidon mukaan ositetun hoidon Cochran-Mantel-Haenszel-testin yksitahoinen p-arvo.
h ECOG-toimintakykyluokan mukaan ositetun hoidon log-rank-testin yksitahoinen p-arvo
i ECOG-toimintakykyluokan mukaan ositetun hoidon Cochran-Mantel-Haenszel-testin yksitahoinen p-arvo.
Kuva 1. Taudin etenemisestä vapaan elinajan Kaplan-Meier-käyrä perustuen koko potilasjoukosta tehtyyn riippumattomaan arviointiin
Kuva 2. Taudin etenemisestä vapaan elinajan Kaplan-Meier-käyrä perustuen aiempaa sunitinibihoitoa saaneesta alaryhmästä tehtyyn riippumattomaan arviointiin
Kuva 3. Taudin etenemisestä vapaan elinajan Kaplan-Meier-käyrä perustuen aiempaa sytokiinihoitoa saaneesta alaryhmästä tehtyyn riippumattomaan arviointiin
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset aksitinibin käytöstä munuais- ja munuaisaltaan karsinooman (nefroblastoomaa, nefroblastomatoosia, kirkassolukarsinoomaa, mesoblastista nefroomaa, medullaarista munuaiskarsinoomaa ja munuaisen rhabdoidikasvainta lukuun ottamatta) hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Absoluuttinen biologinen hyötyosuus on suun kautta otettujen aksitinibitablettien jälkeen keskimäärin 58 % verrattuna laskimoon tapahtuvaan antoon. Aksitinibin puoliintumisaika plasmassa on 2,5−6,1 tuntia. Aksitinibin käyttö 5 mg:n annoksina kaksi kertaa vuorokaudessa johti lääkeaineen vähäisempään kuin kaksinkertaiseen kumulaatioon elimistöön verrattuna kerta-annoksen antoon. Perustuen aksitinibin lyhyeen puoliintumisaikaan vakaa tila odotetaan saavutettavan 2–3 vuorokauden kuluessa aloitusannoksen ottamisesta.
Imeytyminen ja jakautuminen
Aksitinibin huippupitoisuus plasmassa saavutetaan yleensä 4 tunnin kuluessa aksitinibin ottamisesta suun kautta, mediaaniaika huippupitoisuuden saavuttamiselle (Tmax) on 2,5–4,1 tuntia. Aksitinibin antaminen kohtalaisen rasvaisen aterian yhteydessä johti altistuksen pienenemiseen 10 % verrattuna paastotilaan (paasto yön yli). Runsasrasvainen, runsaskalorinen ateria johti altistuksen suurenemiseen 19 % verrattuna paastotilaan (paasto yön yli). Aksitinibi voidaan ottaa aterian yhteydessä tai tyhjään mahaan (ks. kohta Annostus ja antotapa).
Keskimääräinen Cmax- ja AUC-arvo suurenivat suhteessa annokseen aksitinibin annosalueella 5−10 mg. Aksitinibista sitoutuu in vitro ihmisen plasman proteiineihin > 99 %, jolloin ensisijainen sitoutumiskohta on albumiini, mutta sitoutumista tapahtuu kohtalaisesti myös α1-hapan glykoproteiiniin. Kun edennyttä munuaissolukarsinoomaa sairastavat potilaat ottivat ruokailun jälkeen annoksen 5 mg kaksi kertaa vuorokaudessa, keskimääräinen geometrinen huippupitoisuus plasmassa oli 27,8 ng/ml ja 24 tunnin AUC-arvo oli 265 ng.h/ml. Suun kautta otetun valmisteen keskimääräinen geometrinen puhdistuma oli 38 l/h ja näennäinen jakautumistilavuus oli 160 l.
Biotransformaatio ja eliminaatio
Aksitinibi metaboloituu pääasiassa maksassa CYP3A4/5-isoentsyymin välityksellä ja vähäisemmässä määrin isoentsyymien CYP1A2 ja CYP2C19 sekä UGT1A1:n välityksellä.
Kun suun kautta otettiin 5 mg:n radioaktiivinen annos aksitinibia, 30–60 % radioaktiivisuudesta havaittiin ulosteissa ja 23 % radioaktiivisuudesta havaittiin virtsassa. Muuttumaton aksitinibi, joka vastasi 12 %:a annoksesta, oli ulosteissa todettu pääasiallinen yhdiste. Muuttumatonta aksitinibia ei havaittu virtsassa, vaan suurin osa virtsassa havaitusta radioaktiivisuudesta oli karboksyylihappo- ja sulfoksidimetaboliitteja. Plasmassa pääasiallinen radioaktiivinen yhdiste oli N-glukuronidimetaboliitti (50 % verenkierrossa olevasta radioaktiivisuudesta), ja muuttumatonta aksitinibia ja sulfoksidimetaboliittia oli kumpaakin noin 20 % verenkierrossa olevasta radioaktiivisuudesta.
Sulfoksidi- ja N-glukuronidimetaboliitit ovat teholtaan in vitro noin 400 ja 8000 kertaa heikompia VEGFR-2-reseptorin estäjiä aksitinibiin verrattuna.
Erityispotilasryhmät
Iäkkäät, sukupuoli ja rotu
Edennyttä syöpää (mukaan lukien edennyttä munuaissolukarsinoomaa) sairastavista potilaista ja terveistä vapaaehtoisista koehenkilöistä tehdyt populaatiofarmakokineettiset analyysit osoittavat, ettei iällä, sukupuolella, painolla, rodulla, munuaisten toiminnalla, UGT1A1-genotyypillä tai CYP2C19-genotyypillä ole kliinisesti merkityksellistä vaikutusta.
Pediatriset potilaat
Aksitinibia ei ole tutkittu alle 18-vuotiailla potilailla.
Maksan vajaatoiminta
In vitro- ja in vivo ‑tiedot osoittavat, että aksitinibi metaboloituu pääasiassa maksassa.
Lievää maksan vajaatoimintaa (Child-Pugh-luokka A) sairastavien potilaiden systeeminen altistus aksitinibikerta-annoksen jälkeen oli samankaltainen kuin koehenkilöillä, joiden maksan toiminta oli normaali, mutta kohtalaista maksan vajaatoimintaa (Child-Pugh-luokka B) sairastavilla potilailla altistus oli suurempi (noin kaksinkertainen). Aksitinibia ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla potilailla eikä sitä pidä käyttää tälle potilasryhmälle (ks. kohdasta Annostus ja antotapa suositukset annoksen muuttamiseen).
Munuaisten vajaatoiminta
Virtsassa ei havaittu muuttumatonta aksitinibia.
Aksitinibia ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla. Munuaissolukarsinoomaa sairastavilla potilailla tehtyihin aksitinibin kliinisiin tutkimuksiin ei otettu mukaan potilaita, joiden seerumin kreatiniinipitoisuus oli > 1,5-kertainen viitearvojen ylärajaan nähden tai joiden laskettu kreatiniinipuhdistuma oli < 60 ml/min. Populaatiofarmakokineettisten analyysien mukaan aksitinibin puhdistuma ei muuttunut munuaisten vajaatoiminnassa, eikä aksitinibiannosta tarvitse muuttaa.
Prekliiniset tiedot turvallisuudesta
Toistuvien annosten toksisuus
Hiirille ja koirille enintään 9 kuukauden ajan toistuvasti annettujen annosten jälkeen voimakkainta toksisuutta todettiin ruoansulatuskanavassa, hematopoieettisessa järjestelmässä, lisääntymistoiminnoissa, luustossa ja hampaissa. Haitaton annos (No Observed Adverse Effect Levels, NOAEL) oli suunnilleen vastaava tai pienempi (AUC-arvojen perusteella) kuin suositellusta kliinisestä aloitusannoksesta ihmiselle odotettavissa oleva altistus.
Karsinogeenisuus
Aksitinibilla ei ole tehty karsinogeenisuustutkimuksia.
Genotoksisuus
Aksitinibi ei ollut mutageeninen eikä klastogeeninen tavanomaisissa genotoksisuusmäärityksissä in vitro. Polyploidian huomattavaa lisääntymistä havaittiin in vitro pitoisuuksilla > 0,22 µg/ml, ja mikrotumaisten polykromaattisten erytrosyyttien määrän lisääntymistä havaittiin in vivo, kun suurin annos, jolla ei ole havaittavaa vaikutusta (NOEL), oli 69 kertaa ihmisen odotettavissa oleva altistus. Genotoksisuuslöydöksiä ei pidetä kliinisesti merkityksellisinä ihmisellä havaituilla altistustasoilla.
Lisääntymis- ja kehitystoksisuus
Aksitinibiin liittyviä löydöksiä kiveksissä ja lisäkiveksissä olivat elimen painon väheneminen, atrofia tai degeneraatio, sukusolujen määrän väheneminen, hypospermia tai siittiöiden muodon poikkeavuudet sekä siittiötiheyden ja ‑määrän väheneminen. Tämä havaittiin hiirillä noin 12-kertaisella altistuksella ihmisen odotettavissa olevaan altistukseen verrattuna ja koirilla ihmisen odotettavissa olevaa altistusta pienemmillä altistuksilla. Uroshiirten parittelussa tai hedelmällisyydessä ei esiintynyt vaikutuksia altistuksilla, jotka olivat noin 57 kertaa ihmisen odotettavissa oleva altistus. Naarailla havaittuja löydöksiä olivat sukukypsyyden viivästymisen merkit, keltarauhasten väheneminen tai puuttuminen, kohdun painon väheneminen ja kohdun atrofia suunnilleen ihmisen odotettavissa olevaa altistusta vastaavilla altistuksilla. Naarashiirillä havaittiin hedelmällisyyden ja sikiöiden elinkyvyn heikkenemistä kaikilla tutkituilla annoksilla; pienimmällä annoksella altistus oli noin 10-kertainen ihmisen odotettavissa olevaan altistukseen verrattuna.
Tiineiden hiirien altistuttua aksitinibille havaittiin useammin suulakihalkioepämuodostumia ja luuston poikkeavuuksia, kuten luutumisen hidastumista, ihmisen odotettavissa olevaa altistusta pienemmillä altistuksilla. Peri- ja postnataalista kehitystoksisuutta koskevia tutkimuksia ei ole tehty.
Toksisuuslöydökset epäkypsillä eläimillä
Hiirillä ja koirilla, jotka saivat aksitinibia vähintään yhden kuukauden ajan, havaittiin korjautuvaa kasvuruston dysplasiaa, kun altistus oli noin kuusinkertainen ihmisen odotettavissa olevaan altistukseen verrattuna. Yli yhden kuukauden ajan aksitinibia saaneilla hiirillä havaittiin osittain korjaantuvaa hammaskariesta, kun altistus vastasi ihmisen odotettavissa olevaa altistusta. Muuta pediatristen potilaiden hoidon kannalta mahdollisesti olennaista toksisuutta ei ole tutkittu nuorilla eläimillä.
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Mikrokiteinen selluloosa
Laktoosimonohydraatti
Kroskarmelloosinatrium
Magnesiumstearaatti
Tabletin kalvopäällyste
Hypromelloosi 2910 (15 mPa·s)
Titaanidioksidi (E 171)
Laktoosimonohydraatti
Triasetiini (E 1518)
Punainen rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
INLYTA tabletti, kalvopäällysteinen
1 mg (L:kyllä) 56 fol (106,42 €)
5 mg (L:kyllä) 56 fol (1074,57 €)
PF-selosteen tieto
Inlyta 1 mg kalvopäällysteiset tabletit
Alumiini/alumiini -läpipainopakkaus, jossa 14 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 28 tai 56 kalvopäällysteistä tablettia.
HDPE-purkki, jossa silikageelikuivausainetta ja polypropeeninen suljin, ja joka sisältää 180 kalvopäällysteistä tablettia.
Inlyta 5 mg kalvopäällysteiset tabletit
Alumiini/alumiini -läpipainopakkaus, jossa 14 kalvopäällysteistä tablettia. Yksi pakkaus sisältää 28 tai 56 kalvopäällysteistä tablettia.
HDPE-purkki, jossa silikageelikuivausainetta ja polypropeeninen suljin, ja joka sisältää 60 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Inlyta 1 mg: Punainen, soikea kalvopäällysteinen tabletti, jonka toiselle puolelle on kaiverrettu ”Pfizer” ja vastakkaiselle puolelle ”1 XNB”.
Inlyta 5 mg: Punainen, kolmion muotoinen kalvopäällysteinen tabletti, jonka toiselle puolelle on kaiverrettu ”Pfizer” ja vastakkaiselle puolelle ”5 XNB”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
INLYTA tabletti, kalvopäällysteinen
1 mg 56 fol
5 mg 56 fol
- Ylempi erityiskorvaus (100 %). Aksitinibi: Edennyttä munuaissolukarsinoomaa sairastavien aikuispotilaiden hoito erityisin edellytyksin (168).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Aksitinibi: Edennyttä munuaissolukarsinoomaa sairastavien aikuispotilaiden hoito erityisin edellytyksin (361).
ATC-koodi
L01EK01
Valmisteyhteenvedon muuttamispäivämäärä
29.07.2021
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com