

EMEND kapseli, kova 125 mg+80 mg, 80 mg, 125 mg
Vaikuttavat aineet ja niiden määrät
Yksi 125 mg:n kapseli sisältää 125 mg aprepitanttia. Yksi 80 mg:n kapseli sisältää 80 mg aprepitanttia.
Apuaine, jonka vaikutus tunnetaan
Yksi kapseli sisältää 125 mg sakkaroosia (125 mg:n kapselissa).
Apuaine, jonka vaikutus tunnetaan
Yksi kapseli sisältää 80 mg sakkaroosia (80 mg:n kapselissa).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Kliiniset tiedot
Käyttöaiheet
Pahoinvoinnin ja oksentelun ehkäisy syöpäsairauksien hoitoon käytettävän voimakkaasti tai kohtalaisesti pahoinvointia aiheuttavan solunsalpaajalääkityksen yhteydessä aikuisilla ja nuorilla 12 vuoden iästä lähtien.
EMEND 125 mg/80 mg annetaan yhdistelmähoidon osana (ks. kohta Annostus ja antotapa).
Annostus ja antotapa
Annostus
Aikuiset
EMEND-kapseleita annetaan kolmen vuorokauden ajan osana hoito-ohjelmaa, johon kuuluvat myös kortikosteroidi ja 5-HT3-antagonisti. Suositeltu annos on 125 mg suun kautta kerran vuorokaudessa yksi tunti ennen solunsalpaajahoidon aloittamista ensimmäisenä päivänä sekä 80 mg suun kautta kerran vuorokaudessa toisena ja kolmantena päivänä aamuisin.
Seuraavia hoito-ohjelmia suositellaan aikuisille pahoinvoinnin ja oksentelun ehkäisyyn pahoinvointia aiheuttavan syöpäsairauksien solunsalpaajalääkityksen yhteydessä:
Voimakkaasti pahoinvointia aiheuttava solunsalpaajahoito
| 1. päivä | 2. päivä | 3. päivä | 4. päivä | |
| EMEND | 125 mg suun kautta | 80 mg suun kautta | 80 mg suun kautta | ei lainkaan |
| Deksametasoni | 12 mg suun kautta | 8 mg suun kautta | 8 mg suun kautta | 8 mg suun kautta |
| 5-HT3-antagonistit | Vakioannos 5-HT3-antagonisteja. Ks. sopiva annos valitun 5-HT3-antagonistin tuotetiedoista | ei lainkaan | ei lainkaan | ei lainkaan |
Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä sekä aamuisin 2. - 4. päivänä. Deksametasoniannoksessa on huomioitu lääkeaineiden yhteisvaikutukset.
Kohtalaisesti pahoinvointia aiheuttava solunsalpaajahoito
| 1. päivä | 2. päivä | 3. päivä | |
| EMEND | 125 mg suun kautta | 80 mg suun kautta | 80 mg suun kautta |
| Deksametasoni | 12 mg suun kautta | ei lainkaan | ei lainkaan |
| 5-HT3-antagonistit | Vakioannos 5-HT3-antagonisteja. Ks. sopiva annos valitun 5-HT3-antagonistin tuotetiedoista | ei lainkaan | ei lainkaan |
Deksametasoni annetaan 30 minuuttia ennen solunsalpaajahoitoa ensimmäisenä päivänä. Deksametasoniannoksessa on huomioitu lääkeaineiden yhteisvaikutukset.
Pediatriset potilaat
Nuoret (12–17-vuotiaat)
EMEND-kapseleita annetaan 3 vuorokauden ajan osana hoito-ohjelmaa, johon sisältyy 5‑HT3-antagonisti. EMEND-kapseleiden suositusannos on 125 mg suun kautta päivänä 1 ja 80 mg suun kautta päivinä 2 ja 3. EMEND annetaan suun kautta yksi tunti ennen solunsalpaajahoitoa päivinä 1, 2 ja 3. Jos solunsalpaajaa ei anneta päivinä 2 ja 3, EMEND annetaan aamuisin. Katso valitun 5‑HT3-antagonistin valmisteyhteenvedosta tarkoituksenmukaiset annostustiedot. Jos kortikosteroidia, kuten deksametasonia, annetaan samanaikaisesti EMENDin kanssa, annettava kortikosteroidin annos on 50 % tavanomaisesta annoksesta (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka).
80 mg:n ja 125 mg:n kapselien turvallisuutta ja tehoa alle 12-vuotiaiden lasten hoidossa ei ole osoitettu. Tietoja ei ole saatavilla. Katso oraalisuspensiota varten tarkoitetun jauheen valmisteyhteenvedosta tarkoituksenmukainen annostus imeväisille, pikkulapsille ja 6 kuukauden – alle 12 vuoden ikäisille lapsille.
Yleistä
Yhteiskäytön tehosta muiden kortikosteroidien ja 5-HT3-antagonistien kanssa on vain vähän tietoja. Lisätietoa yhteiskäytöstä kortikosteroidien kanssa, ks. kohta Yhteisvaikutukset. Tutustu samanaikaisesti käytettävien 5-HT3-antagonistien valmisteyhteenvetoihin.
Erityisryhmät
Iäkkäät (≥ 65-vuotiaat)
Annoksen muuttaminen ei ole tarpeen iäkkäitä potilaita hoidettaessa (ks. kohta Farmakokinetiikka).
Sukupuoli
Annoksen muuttaminen sukupuolen perusteella ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen hoidettaessa potilaita, joilla on munuaisten vajaatoiminta tai hemodialyysihoitoa vaativa munuaissairaus (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievässä maksan vajaatoiminnassa. Kohtalaista maksan vajaatoimintaa sairastavien potilaiden hoidosta on vain vähän tietoja ja vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta ei ollenkaan. Aprepitantin käytössä on noudatettava varovaisuutta näissä potilasryhmissä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Antotapa
Kovat kapselit niellään kokonaisina.
EMEND voidaan ottaa aterian yhteydessä tai tyhjään mahaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Samanaikainen käyttö pimotsidin, terfenadiinin, astemitsolin ja sisapridin kanssa (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavat potilaat
Kohtalaista maksan vajaatoimintaa sairastavien potilaiden hoidosta on käytettävissä vähän tietoja ja vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta ei lainkaan. EMENDin käytössä on noudatettava varovaisuutta näitä potilaita hoidettaessa (ks. kohta Farmakokinetiikka).
CYP3A4‑yhteisvaikutukset
EMENDin käytössä on noudatettava varovaisuutta, jos potilas saa samanaikaisesti suun kautta muita lääkkeitä, jotka metaboloituvat ensisijaisesti CYP3A4-entsyymin välityksellä ja joilla on kapea terapeuttinen alue, kuten siklosporiinia, takrolimuusia, sirolimuusia, everolimuusia, alfentaniilia, ergotamiinijohdoksia, fentanyyliä ja kinidiiniä (ks. kohta Yhteisvaikutukset). On myös syytä olla erityisen varovainen annettaessa samanaikaisesti irinotekaania, koska yhteiskäyttö voi lisätä sen toksisuutta.
Yhteiskäyttö varfariinin (CYP2C9‑substraatin) kanssa
Pitkäaikaista varfariinihoitoa saavien potilaiden INR-arvoa (International Normalized Ratio) on seurattava tarkoin EMEND-hoidon aikana ja 14 vuorokauden ajan jokaisen kolmen vuorokauden EMEND-hoitojakson jälkeen (ks. kohta Yhteisvaikutukset).
Yhteiskäyttö hormonaalisten ehkäisyvalmisteiden kanssa
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä EMEND-hoidon aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä EMEND-hoidon aikana ja kahden kuukauden ajan viimeisen EMEND-annoksen jälkeen (ks. kohta Yhteisvaikutukset).
Apuaineet
EMEND-kapselit sisältävät sakkaroosia. Potilaiden, joilla on harvinainen perinnöllinen fruktoosi-intoleranssi, glukoosi-galaktoosi imeytymishäiriö tai sakkaroosi-isomaltaasin vajaatoiminta, ei tule käyttää tätä valmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kapseli eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Aprepitantti (125 mg/80 mg) on CYP3A4:n substraatti, kohtalainen estäjä ja indusoija. Aprepitantti on myös CYP2C9:n indusoija. EMEND-hoidon aikana CYP3A4-entsyymin toiminta on estynyt. EMEND aiheuttaa hoidon lopettamisen jälkeen ohimenevän, lievän CYP2C9- ja CYP3A4-entsyymin ja glukuronidaation induktion. Aprepitantilla ei näytä olevan yhteisvaikutuksia kuljetusproteiinin, P-glykoproteiinin, kanssa, mistä on osoituksena se, ettei sillä ole yhteisvaikutuksia digoksiinin kanssa.
Aprepitantin vaikutus muiden lääkeaineiden farmakokinetiikkaan
CYP3A4:n esto
Kohtalaisena CYP3A4:n estäjänä aprepitantti (125 mg/80 mg) voi suurentaa muiden samanaikaisesti annettujen CYP3A4-entsyymin välityksellä metaboloituvien lääkeaineiden pitoisuuksia plasmassa. Suun kautta annettujen CYP3A4:n substraattien aikaansaama kokonaisaltistus voi nousta jopa kolminkertaiseksi kolmen päivän EMEND-hoidon aikana. Aprepitantilla on todennäköisesti vähäisempi vaikutus laskimoon annettujen CYP3A4:n substraattien pitoisuuteen plasmassa. EMENDiä ei saa käyttää samanaikaisesti pimotsidin, terfenadiinin, astemitsolin eikä sisapridin kanssa (ks. kohta Vasta-aiheet). Aprepitantin aiheuttama CYP3A4:n toiminnan estyminen voi suurentaa näiden lääkeaineiden pitoisuuksia plasmassa, mikä saattaa johtaa vakaviin tai hengenvaarallisiin reaktioihin. Varovaisuutta on noudatettava annettaessa samanaikaisesti EMENDiä ja suun kautta annettavia lääkkeitä, jotka metaboloituvat ensisijaisesti CYP3A4-entsyymin välityksellä ja joilla on kapea terapeuttinen alue, kuten siklosporiinia, takrolimuusia, sirolimuusia, everolimuusia, alfentaniilia, dihydroergotamiinia, ergotamiinia, fentanyyliä ja kinidiiniä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kortikosteroidit
Deksametasoni: Tavanomaista deksametasonin suun kautta otettavaa annosta tulisi pienentää noin 50 %, kun sitä annetaan yhdessä EMENDin (125 mg/80 mg) kanssa. Kliinisissä solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyä koskevissa tutkimuksissa yhteisvaikutusten mahdollisuus otettiin huomioon deksametasoniannosta valittaessa (ks. kohta Annostus ja antotapa). Hoito-ohjelmassa annettiin EMENDiä 125 mg ja deksametasonia 20 mg suun kautta ensimmäisenä päivänä ja EMENDiä 80 mg/vrk ja deksametasonia 8 mg suun kautta 2. - 5. päivänä. Tällöin deksametasonin, CYP3A4:n substraatin, AUC-arvo nousi 2,2-kertaiseksi ensimmäisenä ja viidentenä hoitopäivänä.
Metyyliprednisoloni:Metyyliprednisolonin tavanomaista laskimoon annettavaa annosta tulisi pienentää noin 25 % ja suun kautta otettavaa annosta noin 50 %, kun sitä annetaan yhdessä EMENDin (125 mg/80 mg) kanssa. EMENDiä annettiin ensimmäisenä päivänä 125 mg sekä 2. - 3. päivänä 80 mg/vrk ja metyyliprednisolonia annettiin ensimmäisenä päivänä 125 mg laskimoon sekä 2. - 3. päivänä 40 mg suun kautta. EMENDin vaikutuksesta metyyliprednisolonin, CYP3A4:n substraatin, AUC-arvo nousi 1,3-kertaiseksi ensimmäisenä ja 2,5-kertaiseksi kolmantena hoitopäivänä.
Yhtäjaksoisesti annetun metyyliprednisolonin AUC-arvo voi hoidon myöhäisemmässä vaiheessa pienentyä kahden viikon kuluessa EMEND-annoksesta, johtuen aprepitantin CYP3A4:ä indusoivasta vaikutuksesta. Tämän vaikutuksen oletetaan olevan selvempi annettaessa metyyliprednisolonia suun kautta.
Solunsalpaajat
Kun EMENDiä annettiin farmakokineettisissä tutkimuksissa 125 mg ensimmäisenä päivänä ja 80 mg/vrk toisena ja kolmantena päivänä, se ei vaikuttanut laskimoon ensimmäisenä päivänä annetun doketakselin eikä laskimoon ensimmäisenä tai kahdeksantena päivänä annetun vinorelbiinin farmakokinetiikkaan. EMENDin vaikutus suun kautta annettujen CYP3A4:n substraattien farmakokinetiikkaan on suurempi kuin laskimoon annettujen CYP3A4:n substraattien farmakokinetiikkaan. Siksi yhteisvaikutusta suun kautta annettujen, pääasiassa tai osittain CYP3A4-entsyymin välityksellä metaboloituvien solunsalpaajien (esim. etoposidi, vinorelbiini) kanssa ei voida sulkea pois. Pääasiassa tai osittain CYP3A4:n välityksellä metaboloituvia lääkevalmisteita saavien potilaiden hoidossa on syytä noudattaa varovaisuutta ja heidän tilaansa tulisi seurata tavanomaista tarkemmin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myyntiluvan myöntämisen jälkeen on ilmoitettu neurotoksisia tapahtumia, jotka ovat mahdollisia ifosfamidin haittavaikutuksia, kun aprepitanttia ja ifosfamidia on annettu samanaikaisesti.
Immunosuppressiiviset aineet
Solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyyn annetun kolmen vuorokauden aprepitanttihoidon aikana altistuminen CYP3A4-entsyymin välityksellä metaboloituville immunosuppressiivisille lääkkeille (esim. siklosporiinille, takrolimuusille, everolimuusille ja sirolimuusille) saattaa ensin suurentua kohtalaisesti ja ohimenevästi ja pienentyä sitten jonkin verran. Kolmen vuorokauden hoidon lyhyen keston vuoksi ja koska altistuksen muutokset ovat vähäisiä ja ajasta riippuvia, immunosuppressiivisen lääkkeen annostuksen pienentämistä ei suositella kolmen vuorokauden EMEND-hoidon aikana
Midatsolaami
Kun midatsolaamia tai muita CYP3A4-entsyymin välityksellä metaboloituvia bentsodiatsepiineja (alpratsolaamia, triatsolaamia) annetaan yhdessä EMENDin (125 mg/80 mg) kanssa, niiden pitoisuus plasmassa saattaa suurentua, minkä mahdolliset vaikutukset on syytä ottaa huomioon.
Kun EMENDiä annettiin ensimmäisenä päivänä 125 mg ja 2. - 5. päivänä 80 mg/vrk ja midatsolaamia annettiin 2 mg kerta-annoksena suun kautta EMEND-hoito-ohjelman ensimmäisenä ja viidentenä päivänä, EMEND nosti herkän CYP3A4:n substraatin, midatsolaamin, AUC-arvon 2,3-kertaiseksi ensimmäisenä ja 3,3-kertaiseksi viidentenä hoitopäivänä.
Toisessa tutkimuksessa midatsolaamia annettiin 2 mg laskimoon ennen kolmipäiväisen EMEND-hoidon alkamista sekä 4., 8. ja 15. hoitopäivänä. EMENDiä annettiin ensimmäisenä päivänä 125 mg sekä toisena ja kolmantena päivänä 80 mg/vrk. EMEND nosti midatsolaamin AUC-arvoa neljäntenä päivänä 25 %, mutta laski midatsolaamin AUC-arvoa kahdeksantena päivänä 19 % ja 15. päivänä 4 %. Näillä vaikutuksilla ei katsottu olevan kliinistä merkitystä.
Kolmannessa tutkimuksessa, jossa annettiin midatsolaamia laskimoon ja suun kautta, annettiin EMENDiä 125 mg ensimmäisenä päivänä ja 80 mg toisena ja kolmantena päivänä. Lisäksi annettiin ondansetronia 32 mg ensimmäisenä päivänä sekä deksametasonia 12 mg ensimmäisenä päivänä ja 8 mg 2. - 4. päivänä. Tämä yhdistelmä (ts. EMEND, ondansetroni ja deksametasoni) laski suun kautta annetun midatsolaamin AUC-arvoa 16 % 6. päivänä, 9 % 8. päivänä, 7 % 15. päivänä ja 17 % 22. päivänä. Näillä vaikutuksilla ei katsottu olevan kliinistä merkitystä.
Lisäksi yhdessä tutkimuksessa annettiin midatsolaamia laskimoon ja EMENDiä. 2 mg midatsolaamia annettiin laskimoon yksi tunti sen jälkeen, kun EMENDiä oli annettu 125 mg:n kerta-annos suun kautta. Midatsolaamin AUC-arvo plasmassa nousi 1,5-kertaiseksi. Tällä vaikutuksella ei katsottu olevan kliinistä merkitystä.
Induktio
Heikkona CYP2C9:n, CYP3A4:n ja glukuronidaation indusoijana aprepitantti voi vähentää näiden reittien kautta eliminoituvien substraattien pitoisuutta plasmassa kahden viikon aikana hoidon alkamisesta. Vaikutus voi näkyä vasta kolme vuorokautta kestävän EMEND-hoidon lopettamisen jälkeen. CYP2C9:n ja CYP3A4:n substraattien induktio on ohimenevä ja maksimaalinen vaikutus saavutetaan 3-5 vuorokautta kolme vuorokautta kestävän EMEND-hoidon lopettamisen jälkeen. Vaikutus kestää muutaman vuorokauden, heikkenee hitaasti sen jälkeen ja on kliinisesti merkityksetön kahden viikon kuluttua EMEND-hoidon lopettamisen jälkeen. Glukuronidaation lievä induktio on myös nähtävissä, kun aprepitanttia on annettu suun kautta 80 mg seitsemän päivän ajan. Vaikutuksesta CYP2C8- ja CYP2C19-entsyymeihin ei ole tietoa. Varovaisuutta on syytä noudattaa, mikäli varfariinia, asenokumarolia, tolbutamidia, fenytoiinia tai muita lääkeaineita, joiden tiedetään metaboloituvan CYP2C9-entsyymin välityksellä, annetaan tämän jakson aikana.
Varfariini
Pitkäaikaista varfariinihoitoa saavien potilaiden tromboplastiiniaikaa (INR) on seurattava tarkoin solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyyn annettavan EMEND-hoidon aikana ja kahden viikon ajan jokaisen kolmen vuorokauden EMEND-hoitojakson jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kun terveille koehenkilöille, joiden pitkäaikainen varfariinihoito oli tasapainossa, annettiin EMENDiä ensimmäisenä päivänä 125 mg kerta-annoksena sekä toisena ja kolmantena päivänä 80 mg/vrk, EMEND ei vaikuttanut kolmantena hoitopäivänä määritettyyn R(+)- eikä S(-)-varfariinin AUC-arvoon plasmassa. S(-)-varfariinin (CYP2C9:n substraatti) ennen seuraavaa annosta mitattu minimipitoisuus (trough) oli kuitenkin pienentynyt 34 % ja INR lyhentynyt 14 % viiden vuorokauden kuluttua EMEND-hoidon päättymisestä.
Tolbutamidi
Kun EMENDiä annettiin ensimmäisenä päivänä 125 mg sekä toisena ja kolmantena päivänä 80 mg/vrk, tolbutamidin (CYP2C9:n substraatti) AUC-arvo pieneni neljäntenä päivänä 23 %, kahdeksantena päivänä 28 % ja 15. päivänä 15 %, kun sitä annettiin 500 mg:n kerta-annos suun kautta ennen kolmen vuorokauden EMEND-hoidon alkamista sekä 4., 8. ja 15. päivänä.
Hormonaaliset ehkäisyvalmisteet
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä EMEND-hoidon aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä EMEND-hoidon aikana ja kahden kuukauden ajan viimeisen EMEND-annoksen jälkeen.
Kliinisessä tutkimuksessa EMEND-hoidon aikana annettiin etinyyliestradiolia ja noretisteronia sisältävä ehkäisytabletti 1. - 21. hoitopäivänä. EMENDiä annettiin 8. päivänä 125 mg ja 9. - 10. päivänä 80 mg/vrk. Lisäksi annettiin ondansetronia 32 mg laskimoon 8. päivänä sekä deksametasonia suun kautta 8. päivänä 12 mg ja 9. - 11. päivänä 8 mg/vrk. Tutkimuksessa etinyyliestradiolin minimipitoisuudet (trough) pienenivät 9. - 21. päivänä jopa 64 % ja noretisteronin vastaavasti jopa 60 %.
5-HT3-antagonistit
Kliinisissä yhteisvaikutustutkimuksissa aprepitantilla ei ollut kliinisesti merkittäviä vaikutuksia ondansetronin, granisetronin eikä hydrodolasetronin (dolasetronin aktiivinen metaboliitti) farmakokinetiikkaan.
Muiden lääkevalmisteiden vaikutus aprepitantin farmakokinetiikkaan
Jos EMENDiä annetaan yhdessä CYP3A4:n toimintaa estävien lääkeaineiden (esim. ketokonatsolin, itrakonatsolin, vorikonatsolin, posakonatsolin, klaritromysiinin, telitromysiinin, nefatsodonin ja proteaasinestäjien) kanssa, on noudatettava varovaisuutta, koska yhteiskäyttö saattaa suurentaa aprepitantin pitoisuutta plasmassa moninkertaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
EMENDin samanaikaista antoa yhdessä CYP3A4:n toimintaa voimakkaasti indusoivien lääkeaineiden (esim. rifampisiinin, fenytoiinin, karbamatsepiinin, fenobarbitaalin) kanssa tulisi välttää, koska nämä pienentävät aprepitantin pitoisuutta plasmassa ja saattavat siten heikentää EMEND-hoidon tehoa. EMENDin ja mäkikuismaa (Hypericum perforatum) sisältävien rohdosvalmisteiden yhteiskäyttöä ei suositella.
Ketokonatsoli
Kun ketokonatsolia, voimakasta CYP3A4:n estäjää, annettiin 400 mg/vrk kymmenen päivän ajan ja aprepitanttia annettiin 125 mg kerta-annoksena ketokonatsolihoidon viidentenä päivänä, aprepitantin AUC-arvo nousi noin viisinkertaiseksi ja aprepitantin terminaalisen puoliintumisajan keskiarvo noin kolminkertaiseksi.
Rifampisiini
Kun rifampisiinia, voimakasta CYP3A4:n indusoijaa, annettiin 600 mg/vrk 14 päivän ajan ja aprepitanttia annettiin 375 mg kerta-annoksena tämän hoitojakson yhdeksäntenä päivänä, aprepitantin AUC-arvo pieneni 91 % ja aprepitantin terminaalisen puoliintumisajan keskiarvo pieneni 68 %.
Pediatriset potilaat
Yhteisvaikutustutkimuksia on tehty vain aikuisilla.
Raskaus ja imetys
Ehkäisy miehille ja naisille
Hormonaalisten ehkäisyvalmisteiden teho saattaa heikentyä EMEND-hoidon aikana ja 28 päivän ajan hoidon jälkeen. Vaihtoehtoisia ei-hormonaalisia täydentäviä ehkäisymenetelmiä on käytettävä EMEND-hoidon aikana ja kahden kuukauden ajan viimeisen EMEND-annoksen jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Raskaus
Aprepitantin käytöstä raskauden aikana ei ole kliinistä tietoa. Aprepitantin mahdollista toksista vaikutusta lisääntymiseen ei ole täysin selvitetty, koska eläinkokeissa ei ole onnistuttu saavuttamaan suurempaa altistusta, kuin mitä saadaan aikaan ihmisessä käytettäessä terapeuttista annosta 125 mg/80 mg. Näissä tutkimuksissa ei ole havaittu viitteitä välittömistä eikä välillisistä haitallisista vaikutuksista tiineyteen, alkion-/sikiönkehitykseen, synnytykseen eikä postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Neurokiniinisäätelyn muutosten mahdollisia vaikutuksia lisääntymiseen ei tunneta. EMENDiä ei pitäisi käyttää raskauden aikana, ellei se ole aivan välttämätöntä.
Imetys
Aprepitantti erittyy imettävien rottien maitoon. Koska ei tiedetä, erittyykö aprepitantti äidinmaitoon, ei imettämistä suositella EMEND-hoidon aikana.
Hedelmällisyys
Aprepitantin mahdollisia vaikutuksia hedelmällisyyteen ei ole täysin selvitetty, koska eläinkokeissa ei onnistuttu saavuttamaan suurempaa altistusta kuin saadaan aikaan ihmisessä käytettäessä terapeuttista annosta. Näissä hedelmällisyystutkimuksissa ei havaittu viitteitä välittömistä eikä välillisistä haitallisista vaikutuksista paritteluun, hedelmällisyyteen, alkion-/sikiönkehitykseen eikä siittiöiden lukumäärään ja liikkuvuuteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
EMENDillä voi olla vähäinen vaikutus ajokykyyn, pyöräilyyn ja koneidenkäyttökykyyn. EMENDin käytön yhteydessä voi esiintyä heitehuimausta ja uupumusta (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Aprepitantin turvallisuutta on arvioitu noin 6500 aikuisen aineistossa yli 50:ssä tutkimuksessa ja 184 lapsen ja nuoren aineistossa kahdessa kontrolloidussa pediatrisessa kliinisessä tutkimuksessa.
Kun aikuiset olivat saaneet voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (Highly Emetogenic Chemotherapy (HEC)), yleisimmät haittavaikutukset, joita raportoitiin aprepitanttihoito-ohjelmaa saaneilla potilailla useammin kuin tavanomaista hoitoa saaneilla, olivat: nikottelu (4,6 % vs. 2,9 %), ALAT-arvon kohoaminen (2,8 % vs. 1,1 %), ruoansulatushäiriöt (2,6 % vs. 2,0 %), ummetus (2,4 % vs. 2,0 %), päänsärky (2,0 % vs. 1,8 %) ja vähentynyt ruokahalu (2,0 % vs. 0,5 %). Kun potilaat olivat saaneet kohtalaisesti pahoinvointia aiheuttavaa solunsalpaajalääkitystä (Moderately Emetogenic Chemotherapy (MEC)), yleisin haittavaikutus, joka raportoitiin aprepitanttihoito-ohjelmaa saaneilla potilailla useammin kuin tavanomaista hoitoa saaneilla, oli uupumus (1,4 % vs. 0,9 %).
Yleisimpiä haittavaikutuksia, joita raportoitiin enemmän aprepitanttihoitoa saaneilla pediatrisilla potilailla kuin verrokkivalmistetta saaneilla potilailla, jotka saivat syöpäsairauksien hoitoon käytettävää, pahoinvointia aiheuttavaa solunsalpaajalääkitystä, olivat hikka (3,3 % vs. 0,0 %) ja kasvojen punoitus (1,1 % vs. 0,0 %).
Haittavaikutustaulukko
Seuraavia haittavaikutuksia todettiin HEC- ja MEC-tutkimusten yhdistetyssä analyysissä useammin aprepitanttia saaneilla potilailla kuin tavanomaista hoitoa saaneilla aikuisilla tai pediatrisilla potilailla tai lääkkeen markkinoille tulon jälkeen. Taulukossa annetut esiintymistiheydet pohjautuvat aikuisilla potilailla tehtyihin tutkimuksiin; pediatrisissa tutkimuksissa havaitut esiintymistiheydet olivat joko samankaltaiset tai niitä ilmeni vähemmän, mikäli niitä ei ole erikseen mainittu taulukossa. Joitakin aikuisten harvinaisempia haittavaikutuksia ei todettu pediatrisissa tutkimuksissa.
Esiintymistiheydet on määritelty seuraavasti: Hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1000, <1/100), harvinainen (≥1/10 000, <1/1000) ja hyvin harvinainen (<1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
| Elinjärjestelmä | Haittavaikutus | Esiintymistiheys |
| Infektiot | kandidiaasi, stafylokokki-infektiot | harvinainen |
| Veri ja imukudos | kuumeinen neutropenia, anemia | melko harvinainen |
| Immuunijärjestelmä | yliherkkyysreaktiot mukaan lukien anafylaktiset reaktiot | tuntematon |
| Aineenvaihdunta ja ravitsemus | vähentynyt ruokahalu | yleinen |
| jatkuva jano | harvinainen | |
| Psyykkiset häiriöt | ahdistuneisuus | melko harvinainen |
| ajan ja paikan tajun hämärtyminen, euforinen mieliala | harvinainen | |
| Hermosto | päänsärky | yleinen |
| heitehuimaus, uneliaisuus | melko harvinainen | |
| kognitiiviset häiriöt, horros, makuaistin häiriöt | harvinainen | |
| Silmät | sidekalvotulehdus | harvinainen |
| Kuulo ja tasapainoelin | korvien soiminen | harvinainen |
| Sydän | sydämentykytys | melko harvinainen |
| bradykardia, sydän- ja verisuonihäiriöt | harvinainen | |
| Verisuonisto | punoitus/kasvojen punoitus | melko harvinainen |
| Hengityselimet, rintakehä ja välikarsina | nikottelu | yleinen |
| suunielun kipu, aivastelu, yskä, lima nielussa, kurkun ärsytys | harvinainen | |
| Ruoansulatuselimistö | ummetus, ruoansulatushäiriöt | yleinen |
| röyhtäily, pahoinvointi†, oksentelu†, gastroesofageaalinen refluksitauti, vatsakivut, suun kuivuminen, ilmavaivat | melko harvinainen | |
| puhjennut pohjukaissuolihaava, suutulehdus, vatsan pingotus, kova uloste, neutropeeninen koliitti | harvinainen | |
| Iho ja ihonalainen kudos | ihottuma, akne | melko harvinainen |
| valoherkkyysreaktio, voimakas hikoilu, talivuoto, ihon haavaumat, kutiava ihottuma, Stevens-Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi | harvinainen | |
| kutina, nokkosihottuma | tuntematon | |
| Luusto, lihakset ja sidekudos | lihasheikkous, lihasspasmit | harvinainen |
| Munuaiset ja virtsatiet | dysuria | melko harvinainen |
| tiheä virtsaamistarve | harvinainen | |
| Yleisoireet ja antopaikassa todettavat haitat | uupumus | yleinen |
| voimattomuus, yleinen huonovointisuus | melko harvinainen | |
| edeema, epämukava tunne rintakehässä, kävelyhäiriöt | tuntematon | |
| Tutkimukset | ALAT-arvon nousu | yleinen |
| ASAT-arvon nousu, alkalisen fosfataasin nousu veressä | melko harvinainen | |
| punasoluja virtsassa, vähentynyt veren natriumpitoisuus, painon lasku, neutrofiilimäärän pieneneminen, glukoosia virtsassa, lisääntynyt virtsamäärä | harvinainen | |
| †Pahoinvointi ja oksentelu olivat tehon muuttujia ensimmäisen viiden päivän ajan solunsalpaajahoidon jälkeen ja ne ilmoitettiin haittavaikutuksina vasta sen jälkeen. | ||
Valittujen haittavaikutusten kuvaus
HEC- ja MEC-tutkimusten jatkohoitotutkimuksessa, jossa annettiin vielä kuusi jaksoa solunsalpaajahoitoa, haittavaikutukset olivat aikuisilla yleisesti samanlaisia kuin ensimmäisen hoitojakson aikana.
Aktiivikontrolloidussa kliinisessä lisätutkimuksessa, johon osallistui 1169 aprepitanttia ja voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saavaa aikuispotilasta, haittavaikutusprofiili oli yleisesti samanlainen kuin muissa aprepitantilla tehdyissä HEC-tutkimuksissa.
Muut kuin solunsalpaajahoidon aiheuttamaa pahoinvointia ja oksentelua koskevat tutkimukset
Aikuispotilailla, jotka saivat 40 mg:n kerta-annoksen aprepitanttia leikkauksen jälkeisen pahoinvoinnin ja oksentelun ehkäisyyn, todettiin lisäksi seuraavia haittavaikutuksia, joita esiintyi yleisemmin kuin ondansetronia saaneilla potilailla: ylävatsakipu, epänormaalit äänet suolistosta, ummetus*, dysartria, hengenahdistus, hypestesia, unettomuus, mioosi, pahoinvointi, tuntohäiriöt, vatsavaivat, subileus*, heikentynyt näontarkkuus, hengityksen vinkuminen.
*Raportoitu potilailla, jotka käyttivät suurempia annoksia aprepitanttia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa EMEND-hoito on keskeytettävä ja potilaalle on annettava yleistä elintoimintoja tukevaa hoitoa. Potilaan tilaa on tarkkailtava. Aprepitantin antiemeettisen vaikutuksen vuoksi oksennuttaminen lääkkeiden avulla ei ehkä tehoa.
Aprepitanttia ei voida poistaa hemodialyysin avulla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Pahoinvointilääkkeet, ATC-koodi: A04AD12.
Aprepitantti on selektiivinen antagonisti, jolla on voimakas affiniteetti ihmisen substanssi P:n neurokiniini 1 (NK1) -reseptoreihin.
Kolmen päivän aprepitanttihoito-ohjelma aikuisilla
Kahdessa satunnaistetussa kaksoissokkotutkimuksessa oli mukana yhteensä 1094 aikuispotilasta, joiden solunsalpaajahoito sisälsi sisplatiinia ≥ 70 mg/m2. Niissä verrattiin aprepitanttia ja ondansetronia/deksametasonia sisältävää hoito-ohjelmaa (ks. kohta Annostus ja antotapa) tavanomaiseen hoito-ohjelmaan (plasebo + ondansetroni 32 mg laskimoon ensimmäisenä päivänä + deksametasoni 20 mg suun kautta ensimmäisenä päivänä ja 8 mg suun kautta kahdesti päivässä 2.–4. päivänä). Vaikka ondansetronin 32 mg:n annosta laskimoon käytettiin kliinisissä tutkimuksissa, ei tätä annosta enää suositella käytettäväksi. Katso valitun 5-HT3-antagonistin tuotetiedoista sopiva annostus.
Tehon arviointi perustui seuraavaan yhdistettyyn kriteeriin: täydellinen hoitovaste (ei oksenteluepisodeja eikä varalääkkeiden käyttöä) pääasiassa ensimmäisen solunsalpaajahoitojakson aikana. Kummankin tutkimuksen tuloksia arvioitiin erikseen ja yhdistettyinä.
Taulukossa 1 on yhteenveto tärkeimmistä tutkimustuloksista, kun tulokset oli arvioitu yhdistettyinä.
Taulukko 1.
Voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneet aikuispotilaat,
Vasteprosentti hoitoryhmittäin eri vaiheissa – Ensimmäinen solunsalpaajahoitojakso
| YHDISTETYT KRITEERIT | Aprepitantti- hoito-ohjelma (n= 521)† % | Tavanomainen hoito (n= 524)† % | Ero* | |
| % | (95% CI) | |||
| Täydellinen hoitovaste (ei oksentelua eikä varalääkitystä) | ||||
| Yhteensä (0-120h) | 67,7 | 47,8 | 19,9 | (14,0; 25,8) |
| 0-24h | 86,0 | 73,2 | 12,7 | (7,9; 17,6) |
| 25-120h | 71,5 | 51,2 | 20,3 | (14,5; 26,1) |
| YKSITTÄISET KRITEERIT | ||||
| Ei oksentelua (ei oksenteluepisodeja varalääkkeiden käytöstä riippumatta) | ||||
| Yhteensä (0-120h) | 71,9 | 49,7 | 22,2 | (16,4; 28,0) |
| 0-24h | 86,8 | 74,0 | 12,7 | (8,0; 17,5) |
| 25-120h | 76,2 | 53,5 | 22,6 | (17,0; 28,2) |
| Ei merkittävää pahoinvointia (enintään < 25 mm VAS-asteikolla 0–100 mm janalla) | ||||
| Yhteensä (0-120h) | 72,1 | 64,9 | 7,2 | (1,6; 12,8) |
| 25-120h | 74,0 | 66,9 | 7,1 | (1,5; 12,6) |
| * Luottamusvälejä laskettaessa ei ole huomioitu sukupuolta eikä samanaikaista solunsalpaajahoitoa, jotka olivat mukana kerroinsuhteiden (odds ratio) ja logististen mallien primaarianalyyseissä. † Aprepitanttihoito-ohjelmassa yhdeltä potilaalta saatiin tulokset vain akuutin vaiheen osalta. Tämän potilaan tulokset suljettiin pois analyyseistä, joihin otettiin mukaan kaikki tulokset ja viivästyneen vaiheen tulokset; tavanomaista hoitoa saaneiden ryhmässä yhdeltä potilaalta saatiin tulokset vain viivästyneen vaiheen osalta. Tämän potilaan tulokset suljettiin pois analyyseistä, joihin otettiin mukaan kaikki tulokset ja akuutin vaiheen tulokset. | ||||
Ensimmäisen pahoinvointikohtauksen arvioitu ilmaantumisajankohta yhteisanalyysissä on kuvattuna Kaplan-Meierin käyrällä kuvassa 1.
Kuva 1.
Niiden voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä saaneiden aikuispotilaiden prosentuaalinen osuus ajan funktiona, joilla ei ollut pahoinvointia – Ensimmäinen solunsalpaajahoitojakso
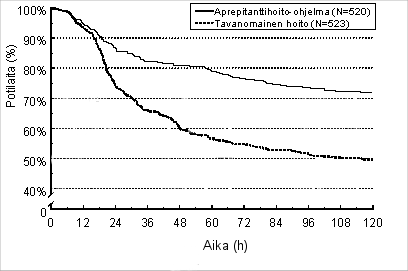
Tilastollisesti merkitsevät tehon erot todettiin kummassakin tutkimuksessa myös erikseen.
Näiden kahden kliinisen tutkimuksen aikuispotilaista 851 osallistui jatkohoitotutkimukseen, jossa heille annettiin vielä viisi solunsalpaajahoitojaksoa. Aprepitanttihoidon teho todennäköisesti säilyi kaikkien jaksojen ajan.
Satunnaistetussa kaksoissokkotutkimuksessa oli mukana yhteensä 866 aikuispotilasta (864 naista, 2 miestä), joiden solunsalpaajahoito sisälsi syklofosfamidia 750-1500 mg/m2 tai syklofosfamidia 500-1500 mg/m2 ja doksorubisiinia (≤ 60 mg/m2) tai epirubisiinia (≤ 100 mg/m2). Tutkimuksessa verrattiin aprepitanttia ja ondansetronia/deksametasonia sisältävää hoito-ohjelmaa (ks. kohta Annostus ja antotapa) tavanomaiseen hoito-ohjelmaan (plasebo + ondansetroni 8 mg suun kautta (kaksi kertaa ensimmäisenä päivänä ja 12 tunnin välein toisena ja kolmantena päivänä) + deksametasoni 20 mg suun kautta ensimmäisenä päivänä).
Tehon arviointi perustui seuraavaan yhdistettyyn kriteeriin: täydellinen hoitovaste (ei oksenteluepisodeja eikä varalääkkeiden käyttöä) pääasiassa ensimmäisen solunsalpaajahoitojakson aikana.
Taulukossa 2 on yhteenveto tärkeimmistä tutkimustuloksista.
Taulukko 2.
Aikuispotilaiden vasteprosentti hoitoryhmittäin eri vaiheissa – Ensimmäinen solunsalpaajahoitojakso
Kohtalaisesti pahoinvointia aiheuttava solunsalpaajalääkitys
| YHDISTETYT KRITEERIT | Aprepitantti- hoito-ohjelma (n= 433)† % | Tavanomainen hoito (n= 424) % | Ero* | |
| % | (95% CI) | |||
| Täydellinen hoitovaste (ei oksentelua eikä varalääkitystä) | ||||
| Yhteensä (0-120h) | 50,8 | 42,5 | 8,3 | (1,6; 15,0) |
| 0-24h | 75,7 | 69,0 | 6,7 | (0,7; 12,7) |
| 25-120h | 55,4 | 49,1 | 6,3 | (-0,4; 13,0) |
| YKSITTÄISET KRITEERIT | ||||
| Ei oksentelua (ei oksenteluepisodeja varalääkkeiden käytöstä riippumatta) | ||||
| Yhteensä (0-120h) | 75,7 | 58,7 | 17,0 | (10,8; 23,2) |
| 0-24h | 87,5 | 77,3 | 10,2 | (5,1; 15,3) |
| 25-120h | 80,8 | 69,1 | 11,7 | (5,9; 17,5) |
| Ei merkittävää pahoinvointia (enintään < 25 mm VAS-asteikolla 0–100 mm janalla) | ||||
| Yhteensä (0-120h) | 60,9 | 55,7 | 5,3 | (-1,3; 11,9) |
| 0-24 h | 79,5 | 78,3 | 1,3 | (-4,2; 6,8) |
| 25-120h | 65,3 | 61,5 | 3,9 | (-2,6; 10,3) |
| * Luottamusvälejä laskettaessa ei ole huomioitu ikäryhmää (< 55-vuotiaat, ≥ 55-vuotiaat) eikä tutkijaryhmää, jotka olivat mukana kerroinsuhteiden (odds ratio) ja logististen mallien primaarianalyyseissä. † Aprepitanttihoito-ohjelmassa yhdeltä potilaalta saatiin tulokset vain akuutin vaiheen osalta. Tämän potilaan tulokset suljettiin pois analyyseistä, joihin otettiin mukaan kaikki tulokset ja viivästyneen vaiheen tulokset. | ||||
Saman kliinisen tutkimuksen 744 aikuispotilasta osallistui jatkohoitotutkimukseen, jossa heille annettiin vielä kolme solunsalpaajahoitojaksoa. Aprepitanttihoidon teho todennäköisesti säilyi kaikkien jaksojen ajan.
Toisessa satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmissä tehdyssä kliinisessä monikeskustutkimuksessa, jossa aprepitanttihoito-ohjelmaa verrattiin tavanomaiseen hoitoon, oli mukana 848 aikuispotilasta (652 naista ja 196 miestä), joiden solunsalpaajahoito-ohjelma sisälsi erisuuruisina laskimoon annettuina annoksina oksaliplatiinia, karboplatiinia, epirubisiinia, idarubisiinia, ifosfamidia, irinotekaania, daunorubisiinia, doksorubisiinia; syklofosfamidia (< 1500 mg/m2 laskimoon) tai sytarabiinia (> 1 g/m2 laskimoon). Aprepitanttihoito-ohjelmaa saavat potilaat saivat solunsalpaajahoitoa eri syöpätyyppeihin, kuten rintasyöpään (52 %), ruoansulatuselimistön syöpiin (21 %), joihin kuului myös kolorektaalisyöpä, keuhkosyöpään (13 %) ja gynekologisiin syöpiin (6 %). Aprepitanttia ja ondansetronia/deksametasonia sisältävää hoito-ohjelmaa (ks. kohta Annostus ja antotapa) verrattiin tavanomaiseen hoito-ohjelmaan (plasebo + ondansetroni 8 mg suun kautta (kaksi kertaa ensimmäisenä päivänä ja 12 tunnin välein toisena ja kolmantena päivänä) + deksametasoni 20 mg suun kautta ensimmäisenä päivänä).
Tehon arviointi perustui seuraavien ensisijaisten ja tärkeimpien toissijaisten päätetapahtumien arviointiin: ei oksentelua koko arviointijakson aikana (0-120 tuntia solunsalpaajahoidon jälkeen), aprepitanttihoito-ohjelman turvallisuuden ja siedettävyyden arviointi solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyssä ja täydellinen vaste (ei oksentelua eikä varalääkkeiden käyttöä) koko arviointijakson aikana (0-120 tuntia solunsalpaajahoidon jälkeen). Lisäksi "ei merkittävää pahoinvointia" arvioitiin tutkimuksellisena päätetapahtumana koko arviointijakson aikana (0-120 tuntia solunsalpaajahoidon jälkeen) ja post-hoc-analyysinä akuutin ja viivästyneen vaiheen aikana.
Taulukossa 3 on tiivistelmä tärkeimmistä tutkimustuloksista.
Taulukko 3.
Aikuispotilaiden vasteprosentti hoitoryhmittäin eri vaiheissa tutkimuksessa 2 – Ensimmäinen solunsalpaajahoitojakso
Kohtalaisesti pahoinvointia aiheuttava solunsalpaajalääkitys
| Aprepitanttihoito-ohjelma (n = 425) % | Tavanomainen hoito (n = 406) % | Ero* | ||
| % | (95 % CI) | |||
| Täydellinen hoitovaste (ei oksentelua eikä varalääkitystä) | ||||
| Yhteensä (0-120 h) | 68,7 | 56,3 | 12,4 | (5,9; 18,9) |
| 0-24 h | 89,2 | 80,3 | 8,9 | (4,0; 13,8) |
| 25-120 h | 70,8 | 60,9 | 9,9 | (3,5; 16,3) |
| Ei oksentelua (ei oksenteluepisodeja varalääkkeiden käytöstä riippumatta) | ||||
| Yhteensä (0-120 h) | 76,2 | 62,1 | 14,1 | (7,9; 20,3) |
| 0-24 h | 92,0 | 83,7 | 8,3 | (3,9; 12,7) |
| 25-120 h | 77,9 | 66,8 | 11,1 | (5,1; 17,1) |
| Ei merkittävää pahoinvointia (enintään < 25 mm VAS-asteikolla 0–100 mm janalla) | ||||
| Yhteensä (0-120 h) | 73,6 | 66,4 | 7,2 | (1,0; 13,4) |
| 0-24 h | 90,9 | 86,3 | 4,6 | (0,2; 9,0) |
| 25-120 h | 74,9 | 69,5 | 5,4 | (-0,7; 11,5) |
| *Luottamusvälejä laskettaessa ei ole huomioitu sukupuolta eikä aluetta, jotka olivat mukana primaarisessa analyysissä, jossa käytettiin logistisia malleja. | ||||
Aprepitanttiyhdistelmähoidon hyöty koko tutkimuspopulaatiossa perustui pääasiassa niiden potilaiden, esimerkiksi naisten, tuloksiin, jotka saivat huonon vasteen tavanomaisella hoito-ohjelmalla. Aprepitanttiryhmässä tulokset olivat kuitenkin numeerisesti paremmat kuin tavanomaista hoitoa saaneessa ryhmässä iästä, kasvaintyypistä tai sukupuolesta riippumatta. Täydellinen hoitovaste saavutettiin aprepitanttihoito-ohjelmalla 209/324 (65 %) naisella ja 83/101 (82 %) miehellä ja tavanomaisella hoito-ohjelmalla 161/320 (50 %) naisella ja 68/87 (78 %) miehellä.
Pediatriset potilaat
Satunnaistetussa, kaksoissokkoutetussa, aktiivista vertailuvalmistetta sisältäneessä kliinisessä tutkimuksessa, johon osallistui 302 lasta ja nuorta (iältään 6 kuukautta ⎼ 17 vuotta), jotka saivat kohtalaisesti tai voimakkaasti pahoinvointia aiheuttavaa solunsalpaajalääkitystä, aprepitanttihoitoa verrattiin vertailulääkitykseen solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun ehkäisyssä. Aprepitanttihoidon tehoa arvioitiin yhden hoitosyklin (hoitosykli 1) aikana. Potilailla oli mahdollisuus saada aprepitanttia avoimessa vaiheessa myöhempien hoitosyklien aikana (valinnaiset hoitosyklit 2⎼6), mutta tehoa ei arvioitu näiden valinnaisten hoitosyklien aikana. 12⎼17-vuotiaiden nuorten (n = 47) aprepitanttilääkitys sisälsi 125 mg:n annokset suun kautta annettavia EMEND-kapseleita päivänä 1 ja 80 mg/vrk päivinä 2 ja 3 yhdessä ondansetronin kanssa päivänä 1. 6 kuukauden ⎼ alle 12 vuoden ikäisten lasten (n = 105) aprepitanttilääkitys sisälsi 3,0 mg/kg EMEND-jauhetta oraalisuspensiota varten (enintään 125 mg) suun kautta päivänä 1 ja 2,0 mg/kg (enintään 80 mg) suun kautta päivinä 2 ja 3 yhdessä ondansetronin kanssa päivänä 1. Iältään 12⎼17-vuotiaiden nuorten (n = 48) ja 6 kuukauden – alle 12 vuoden ikäisten lasten (n = 102) vertailulääkitys koostui aprepitanttia vastaavasta lumelääkkeestä päivinä 1, 2 ja 3 yhdessä ondansetronin kanssa päivänä 1. EMEND tai lumelääke yhdessä ondansetronin kanssa annettiin yksi tunti tai 30 minuuttia ennen solunsalpaajalääkityksen aloittamista. Laskimoon annettava deksametasoni oli sallittu molemmissa pediatristen potilaiden ikäryhmissä osana pahoinvointilääkitystä lääkärin harkinnan mukaan. Deksametasonin annoksen pienentämistä (50 %) edellytettiin aprepitanttia saavien pediatristen potilaiden kohdalla. Annoksen pienentämistä ei edellytetty niiden pediatristen potilaiden kohdalla, jotka saivat vertailuhoitoa. 29 % aprepitanttilääkitystä saaneista ja 28 % vertailuhoitoa saaneista pediatrisista potilaista käytti deksametasonia osana lääkitystä hoitosyklin 1 aikana.
EMENDin pahoinvointia ehkäisevää vaikutusta arvioitiin 5 päivän (120 tunnin) aikana solunsalpaajahoidon aloittamisesta päivänä 1. Ensisijainen päätemuuttuja oli täydellinen vaste viivästyneessä vaiheessa (25⎼120 tuntia solunsalpaajahoidon aloittamisen jälkeen) hoitosyklin 1 aikana. Yhteenveto keskeisistä tutkimustuloksista on esitetty taulukossa 4.
Taulukko 4.
Täydellisen vasteen saaneet pediatriset potilaat (%), joilla ei ilmennyt oksentelua, hoitoryhmittäin ja vaiheen mukaan – hoitosykli 1 (Intent to treat -potilaat)
| Aprepitanttilääkitys n/m (%) | Vertailulääkitys n/m (%) | |
| ENSISIJAINEN PÄÄTEMUUTTUJA | ||
| Täydellinen vaste* – viivästynyt vaihe | 77/152 (50,7)† | 39/150 (26,0) |
| MUUT ENNALTA MÄÄRITELLYT PÄÄTEMUUTTUJAT | ||
| Täydellinen vaste* – akuutti vaihe | 101/152 (66,4)‡ | 78/150 (52,0) |
| Täydellinen vaste* – kokonaisvaihe | 61/152 (40,1)† | 30/150 (20,0) |
| Ei oksentelua§ – kokonaisvaihe | 71/152 (46,7)† | 32/150 (21,3) |
| *Täydellinen vaste = Ei oksentelua tai yökkäilyä tai yökkäämistä ilman oksennusta eikä hätälääkkeiden käyttöä. †p < 0,01 verrattuna vertailulääkitykseen. ‡p < 0,05 verrattuna vertailulääkitykseen. §Ei oksentelua = Ei oksentelua, yökkäilyä eikä yökkäämistä ilman oksennusta. n/m = Niiden potilaiden määrä, joilla oli toivottu vaste / ajankohtaan sisällytettyjen potilaiden määrä. Akuutti vaihe: 0-24 tuntia solunsalpaajahoidon aloittamisesta. Viivästynyt vaihe: 25-120 tuntia solunsalpaajahoidon aloittamisesta. Kokonaisvaihe: 0-120 tuntia solunsalpaajahoidon aloittamisesta. | ||
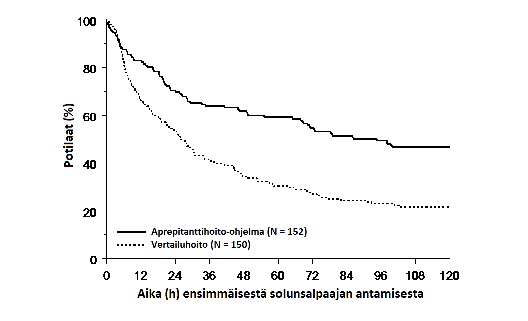
Arvioitu aika ensimmäiseen oksentelukohtaukseen solunsalpaajahoidon aloittamisen jälkeen oli pidempi aprepitanttilääkityksellä (arvioitu ensimmäiseen oksentelukohtaukseen kuluneen ajan mediaani oli 94,5 tuntia) verrattuna vertailulääkettä saaneiden ryhmään (arvioitu ensimmäiseen oksentelukohtaukseen kuluneen ajan mediaani oli 26,0 tuntia), kuten Kaplan‑Meier-käyrissä kuvassa 2 on esitetty.
Kuva 2.
Pediatristen potilaiden aika ensimmäiseen oksentelukohtaukseen solunsalpaajahoidon antamisen aloittamisesta kokonaisvaiheessa ‑ hoitosykli 1 (Intent to treat ‑potilaat)
Hoitosyklin 1 potilaiden alaryhmissä havaitun tehon analyysi osoitti, että riippumatta ikäryhmästä, sukupuolesta, deksametasonin käytöstä pahoinvoinnin estohoitona ja siitä, miten voimakkaasti solunsalpaajahoito aiheuttaa pahoinvointia, aprepitanttihoidolla saavutettiin parempi hallinta kuin vertailuhoidolla täydelliseen vasteeseen liittyvien päätemuuttujien suhteen.
Farmakokinetiikka
Aprepitantin farmakokinetiikka on epälineaarinen. Sekä puhdistuma että absoluuttinen hyötyosuus pienenevät annoksen suurentuessa.
Imeytyminen
Suun kautta otetun aprepitantin absoluuttisen hyötyosuuden keskiarvo on 80 mg:n kapselin jälkeen 67 % ja 125 mg:n kapselin jälkeen 59 %. Aprepitantin huippupitoisuuden keskiarvo plasmassa (Cmax) mitattiin noin neljän tunnin kuluttua (tmax). Aprepitantin AUC-arvo suureni jopa 40 %, kun kapseli otettiin suun kautta noin 800 kcal sisältävän standardiaamiaisen yhteydessä. Tätä ei pidetä kliinisesti merkittävänä.
Aprepitantin farmakokinetiikka on epälineaarinen koko kliinisellä annosalueella. AUC0-∞ -arvo oli 26 % suurempi kuin annosten suhde, kun terveille nuorille aikuisille annettiin 80 mg:n ja 125 mg:n kerta-annokset aterian yhteydessä.
Kun EMENDiä annettiin ensimmäisenä päivänä 125 mg kerta-annoksena suun kautta sekä toisena ja kolmantena päivänä 80 mg kerran päivässä, AUC0-24h (keskiarvo ± S.D.) oli ensimmäisenä päivänä noin 19,6±2,5 mikrog•h/ml ja kolmantena päivänä 21,2±6,3 mikrog•h/ml. Cmax -arvo oli ensimmäisenä päivänä 1,6±0,36 mikrog/ml ja kolmantena päivänä 1,4±0,22 mikrog/ml.
Jakautuminen
Aprepitantti sitoutuu voimakkaasti proteiineihin. Sitoutumisaste on keskimäärin 97 %. Näennäisen jakautumistilavuuden geometrinen keskiarvo vakaan tilan aikana (Vdss) on ihmisellä noin 66 l.
Biotransformaatio
Aprepitantti metaboloituu tehokkaasti. Kun terveille nuorille aikuisille annettiin [14C]-merkittyä fosaprepitanttia (aprepitantin aihiolääkettä) 100 mg:n kerta-annoksena laskimoon, aprepitantin osuus oli noin 19 % plasmassa tavatusta radioaktiivisuudesta 72 tunnin kuluessa annoksesta. Tämä osoittaa, että plasmassa oli huomattava määrä metaboliitteja. Ihmisen plasmasta on tunnistettu 12 aprepitantin metaboliittia. Aprepitantti metaboloituu suurelta osin morfoliinirenkaan ja sen sivuketjujen oksidaation kautta. Nämä metaboliitit ovat vain heikosti aktiivisia. Ihmisen maksan mikrosomeissa tehdyt in vitro -tutkimukset ovat osoittaneet, että aprepitantti metaboloituu pääasiassa CYP3A4-entsyymin välityksellä ja mahdollisesti vähäisessä määrin CYP1A2:n ja CYP2C19:n välityksellä.
Eliminaatio
Aprepitantti ei erity muuttumattomana virtsaan. Metaboliitit erittyvät virtsaan ja sapen kautta ulosteeseen. Kun terveille koehenkilöille annettiin [14C]-merkittyä fosaprepitanttia (aprepitantin aihiolääkettä) 100 mg:n kerta-annoksena laskimoon, 57 % radioaktiivisuudesta todettiin virtsassa ja 45 % ulosteessa.
Aprepitantin plasmapuhdistuma on annoksesta riippuvainen. Se pienenee annoksen suurentuessa ja on terapeuttisella annosalueella noin 60–72 ml/min. Terminaalinen puoliintumisaika on noin 9 - 13 tuntia.
Farmakokinetiikka erityisryhmissä
Iäkkäät: Kun aprepitanttia annettiin suun kautta ensimmäisenä päivänä 125 mg kerta-annoksena ja 2. – 5. päivänä 80 mg kerran päivässä, aprepitantin AUC0-24h oli ensimmäisenä päivänä 21 % ja viidentenä päivänä 36 % suurempi iäkkäillä (≥ 65-vuotiailla) kuin nuoremmilla aikuisilla. Cmax oli iäkkäillä potilailla ensimmäisenä päivänä 10 % suurempi ja viidentenä päivänä 24 % suurempi kuin nuoremmilla aikuisilla. Näiden erojen ei katsota olevan kliinisesti merkittäviä. EMENDin annosta ei tarvitse muuttaa iäkkäitä potilaita hoidettaessa.
Sukupuoli: Kun aprepitanttia annetaan suun kautta 125 mg kerta-annoksena, aprepitantin Cmax-arvo on 16 % suurempi naisilla kuin miehillä. Aprepitantin puoliintumisaika on naisilla 25 % pienempi kuin miehillä ja tmax on suunnilleen sama. Näiden erojen ei katsota olevan kliinisesti merkittäviä. EMEND-annoksen muuttaminen ei ole tarpeen sukupuolen perusteella.
Maksan vajaatoiminta: Lievä maksan vajaatoiminta (Child-Pugh-luokka A) ei vaikuta aprepitantin farmakokinetiikkaan kliinisesti merkittävästi. Annosta ei tarvitse muuttaa hoidettaessa lievää maksan vajaatoimintaa sairastavia potilaita. Saatavilla olevien tietojen perusteella ei voida vetää johtopäätöksiä keskivaikean maksan vajaatoiminnan (Child-Pugh-luokka B) vaikutuksista aprepitantin farmakokinetiikkaan. Käytettävissä ei ole kliinisiä eikä farmakokineettisiä tietoja vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavien potilaiden hoidosta.
Munuaisten vajaatoiminta: Aprepitanttia annettiin 240 mg kerta-annoksena potilaille, joilla oli vaikea munuaisten vajaatoiminta (CrCl < 30 ml/min) ja potilaille, joilla oli hemodialyysihoitoa vaativa munuaissairaus.
Vaikeassa munuaisten vajaatoiminnassa aprepitantin kokonaispitoisuuden (sitoutumattoman ja proteiiniin sitoutuneen) AUC0-∞ oli 21 % pienempi ja Cmax oli 32 % pienempi kuin terveillä koehenkilöillä. Hemodialyysihoitoa saavilla munuaistautia sairastavilla potilailla aprepitantin kokonaispitoisuuden AUC0-∞ oli 42 % pienempi ja Cmax oli 32 % pienempi. Koska aprepitantin sitoutuminen proteiiniin heikkenee vain vähän munuaisten vajaatoiminnan aikana, farmakologisesti aktiivisen sitoutumattoman aprepitantin AUC ei poikennut munuaisten vajaatoimintaa sairastaneilla potilailla merkitsevästi terveiden koehenkilöiden vastaavista arvoista. Hemodialyysihoito, jota annettiin 4 tai 48 tuntia annoksen jälkeen, ei vaikuttanut merkitsevästi aprepitantin farmakokinetiikkaan – dialysaatissa tavattiin alle 0,2 % annoksesta.
EMENDin annoksen muuttaminen ei ole tarpeen hoidettaessa potilaita, joilla on munuaisten vajaatoiminta tai hemodialyysihoitoa vaativa munuaissairaus.
Pediatriset potilaat: Osana 3 vuorokauden hoitoa aprepitanttikapseleiden annostuksella (125/80/80 mg) nuorille potilaille (12–17-vuotiaille) saavutettiin AUC0-24h-arvo, joka oli yli 17 µg•h/ml päivänä 1, ja pitoisuudet (Cmin) päivien 2 ja 3 päättyessä olivat yli 0,4 µg/ml suurimmalla osalla potilaista. Huippupitoisuuden mediaani plasmassa (Cmax) oli suunnilleen 1,3 µg/ml päivänä 1 ja se saavutettiin suunnilleen 4 tunnissa. Osana 3 vuorokauden hoitoa oraalisuspensiota varten tarkoitetun aprepitanttijauheen annostuksella (3/2/2 mg/kg) iältään 6 kuukauden - alle 12 vuoden ikäisille potilaille saavutettiin AUC0-24h-arvo, joka oli yli 17 µg•h/ml päivänä 1, ja pitoisuudet (Cmin) päivien 2 ja 3 päättyessä olivat yli 0,1 µg/ml suurimmalla osalla potilaista. Huippupitoisuuden mediaani plasmassa (Cmax) oli suunnilleen 1,2 µg/ml päivänä 1 ja se saavutettiin suunnilleen 5-7 tunnissa.
Aprepitantin populaatiofarmakokineettinen analyysi pediatristen (6 kuukauden - 17 vuoden ikäisten) potilaiden suhteen viittaa siihen, että sukupuolella ja rodulla ei ole kliinisesti merkityksellistä vaikutusta aprepitantin farmakokinetiikkaan.
Pitoisuuden suhde tehoon
Terveille nuorille miehille tehdyissä positroniemissiotomografiatutkimuksissa (PET) on osoitettu erittäin spesifisen NK1-reseptorin merkkiaineen avulla, että aprepitantti kulkeutuu aivoihin ja kiinnittyy NK1-reseptoreihin tavalla, joka on suhteessa annokseen ja lääkeaineen pitoisuuteen plasmassa. Aikuisilla kolmen vuorokauden EMEND-hoidon aikana saavutettavien plasman aprepitanttipitoisuuksien arvioidaan sitovan aivojen NK1-reseptorit yli 95-prosenttisesti.
Prekliiniset tiedot turvallisuudesta
Prekliiniset tiedot eivät viittaa mihinkään erityiseen vaaraan ihmisillä käytettäessä. Tiedot perustuvat kerta-annosten ja toistuvaisannosten toksisuutta sekä genotoksisuutta, karsinogeenisuutta, lisääntymistoksisuutta ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tuloksiin. On kuitenkin syytä ottaa huomioon, että jyrsijöissä systeeminen altistus oli sama tai jopa pienempi kuin terapeuttisten annosten aikaansaama altistus ihmisessä annoksen ollessa 125 mg/80 mg. Vaikka lisääntymistutkimuksissa ei todettu haittavaikutuksia eläimissä ihmisen altistumistasoihin verrattavilla altistuksilla, eivät nämä tulokset ole riittävät vaaran arvioimiseen ihmisellä.
Nuorilla rotilla tehdyssä toksisuustutkimuksessa aprepitantin anto syntymän jälkeen päivästä 10 päivään 63 saakka aiheutti varhaisempaa emättimen avautumista naarailla annoksesta 250 mg/kg kahdesti vuorokaudessa alkaen ja viivästynyttä esinahan separaatiota uroksilla annoksesta 10 mg/kg kahdesti vuorokaudessa alkaen. Kliinisesti merkitykselliselle altistukselle ei ollut asetettu marginaaleja. Hoitoon liittyviä vaikutuksia paritteluun, hedelmällisyyteen tai alkioiden/sikiöiden selviytymiseen ei ollut eikä lisääntymiselimissä todettu patologisia muutoksia. Nuorilla koirilla tehdyssä toksisuustutkimuksessa, jossa annettiin valmistetta syntymän jälkeen päivästä 14 päivään 42 saakka, havaittiin uroksilla kivesten painon ja Leydigin solujen pienenemistä annoksella 6 mg/kg/vrk ja naarailla kohdun painon suurenemista, kohdun ja kohdunkaulan hypertrofiaa ja emätinkudosten turvotusta annoksesta 4 mg/kg/vrk lähtien. Kliinisesti merkitykselliselle aprepitanttialtistukselle ei ollut asetettu marginaaleja. Suositellun annostusohjelman mukaisen lyhytkestoisen hoidon yhteydessä katsotaan, että nämä löydökset eivät todennäköisesti ole kliinisesti merkityksellisiä.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Sakkaroosi
Mikrokiteinen selluloosa (E 460)
Hydroksipropyyliselluloosa (E 463)
Natriumlauryylisulfaatti
Kapselikuori (125 mg)
Liivate
Titaanidioksidi (E 171)
Punainen rautaoksidi (E 172)
Keltainen rautaoksidi (E 172
Kapselikuori (80 mg)
Liivate
Titaanidioksidi (E 171)
Painomuste
Shellakka
Kaliumhydroksidi
Musta rautaoksidi (E 172).
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EMEND kapseli, kova
125 mg+80 mg 1x125 mg + 2x80 mg (41,87 €)
80 mg 2 fol (28,59 €)
125 mg 5 x 1 fol (194,80 €)
PF-selosteen tieto
Eri pakkauskokoja ja eri vahvuuksia on saatavana.
Alumiiniläpipainopakkaus, joka sisältää yhden 80 mg:n kapselin.
Alumiiniläpipainopakkaus, joka sisältää kaksi 80 mg:n kapselia.
5 alumiiniläpipainopakkausta, joista kukin sisältää yhden 80 mg:n kapselin.
Alumiiniläpipainopakkaus, joka sisältää yhden 125 mg:n kapselin.
5 alumiiniläpipainopakkausta, joista kukin sisältää yhden 125 mg:n kapselin.
Alumiiniläpipainopakkaus, joka sisältää yhden 125 mg:n kapselin ja kaksi 80 mg:n kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
125 mg:n kapseli on läpinäkymätön, alaosa on valkoinen ja yläosa vaaleanpunainen. Kapselin alaosaan on painettu mustalla painovärillä merkinnät “462” ja “125 mg”. 80 mg:n kapselit ovat läpinäkymättömiä, ala- ja yläosa on valkoinen. Kapselin alaosaan on painettu mustalla painovärillä merkinnät “461” ja “80 mg”.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Korvattavuus
EMEND kapseli, kova
125 mg+80 mg 1x125 mg + 2x80 mg
80 mg 2 fol
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Gynekologiset syövät (128), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
EMEND kapseli, kova
125 mg 5 x 1 fol
- Ei korvausta.
ATC-koodi
A04AD12
Valmisteyhteenvedon muuttamispäivämäärä
19.08.2020
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi