
CAPRELSA tabletti, kalvopäällysteinen 100 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Terveydenhuollon ammattilainen
Yleinen
Vaikuttavat aineet ja niiden määrät
Caprelsa 100 mg tabletti
Yksi kalvopäällystetty tabletti sisältää 100 mg vandetanibia.
Caprelsa 300 mg tabletti
Yksi kalvopäällystetty tabletti sisältää 300 mg vandetanibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti)
Kliiniset tiedot
Käyttöaiheet
Caprelsa on tarkoitettu aggressiivista ja oireista RET-mutaation (Rearranged during Transfection) omaavaa medullaarista kilpirauhaskarsinoomaa sairastaville potilaille, joiden paikallisesti edennyttä tai metastaattista sairautta ei voi hoitaa leikkauksella.
Caprelsa on tarkoitettu aikuisille ja vähintään 5-vuotiaille lapsille ja nuorille.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa medullaarisen kilpirauhaskarsinooman hoitoon ja syövän lääkehoitoon perehtyneen lääkärin valvonnassa, jolla on kokemusta EKG:n arvioinnista.
Annostus ja antotapa
Hoito tulee aloittaa ja hoitoa tulee jatkaa medullaarisen kilpirauhaskarsinooman hoitoon ja syövän lääkehoitoon perehtyneen lääkärin valvonnassa, jolla on kokemusta EKG:n arvioinnista.
Rearranged during Transfection (RET) ‑mutaatiostatus
Saatavilla olevien tietojen perusteella Caprelsa-valmisteen ei katsota toimivan riittävän hyvin potilailla, joilla ei ole tunnistettua RET-mutaatiota. Tästä syystä ennen Caprelsa-hoidon aloittamista on vahvistettava validoidulla testausmenetelmällä, että potilaalla on RET-mutaatio. RET-mutaatiostatusta määritettäessä kudosnäytteet on mahdollisuuksien mukaan otettava hoidon aloittamishetkellä diagnoosihetken sijasta.
Annostus medullaarista kilpirauhaskarsinoomaa sairastaville aikuispotilaille
Suositeltu annos on 300 mg kerran päivässä otettuna ruoan kanssa tai ilman ruokaa suunnilleen samaan aikaan joka päivä.
Jos annos unohtuu, se tulee ottaa heti muistettaessa. Jos seuraavan annoksen ottamiseen on alle 12 tuntia, annos jätetään väliin. Potilaiden ei tule ottaa kaksinkertaista annosta (kahta annosta samalla kertaa) unohtuneen annoksen korvaamiseksi.
Annoksen muuttaminen aikuispotilaiden medullaarisen kilpirauhaskarsinooman hoidossa
Korjattu QT-aika on arvioitava huolellisesti ennen hoidon aloittamista. Jos haittavaikutukset ovat CTCAE-luokituksen (Common Terminology Criteria for Adverse Events) mukaan 3 astetta tai korkeampaa toksisuutta tai EKG:n QT-aika on pidentynyt, vandetanibin anto on keskeytettävä ainakin tilapäisesti ja jatkettava pienemmällä annoksella, kun toksisuus on hävinnyt tai lieventynyt 1 CTCAE-asteeseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). 300 mg vuorokausiannos voidaan pienentää 200 mg:aan (kahteen 100 mg:n tablettiin) ja sen jälkeen tarvittaessa 100 mg:aan. Potilasta on seurattava asianmukaisesti. Koska puoliintumisaika on 19 vuorokautta, haittavaikutukset, mm. pidentynyt QT-aika, eivät välttämättä korjaannu nopeasti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus medullaarista kilpirauhaskarsinoomaa sairastaville pediatrisille potilaille
Valmisteen annostus pediatrisille potilaille perustuu kehon pinta-alaan ja ilmoitetaan yksikkönä mg/m2. Caprelsa-hoitoa saaville pediatrisille potilaille ja heidän huoltajilleen on hoidon määräämisen yhteydessä ja jatkossa jokaisen annosmuutoksen yhteydessä annettava annostusohje sekä tietoa oikeasta annoksesta. Suositellut annokset ja annosmuutokset on esitetty taulukossa 1.
Taulukko 1: Annostusohje medullaarista kilpirauhaskarsinoomaa sairastaville pediatrisille potilaille
| Kehon pinta-ala (m2) | Aloitusannos (mg)a | Annoksen suurentaminen (mg)b, jos potilas on sietänyt aloitusannosta hyvin 8 viikon ajan | Annoksen pienentäminen (mg)c |
| 0,7 ‑ < 0,9 | 100 joka toinen päivä | 100 vuorokaudessa | - |
| 0,9 ‑ < 1,2 | 100 vuorokaudessa | 7 päivän jakso: 100-200-100-200-100-200-100 | 100 joka toinen päivä |
| 1,2 ‑ < 1,6 | 7 päivän jakso: 100-200-100-200-100-200-100 | 200 vuorokaudessa | 100 vuorokaudessa |
| ≥ 1,6 | 200 vuorokaudessa | 300 vuorokaudessa | 7 päivän jakso: 100-200-100-200-100-200-100 |
| aAloitusannos on annos, jolla hoito aloitetaan. b Pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa ei ole käytetty yli 150 mg/m2:n vandetanibiannoksia. c Jos potilaalla todetaan annoksen pienentämistä edellyttävä haittavaikutus, vandetanibin ottaminen on lopetettava vähintään viikon ajaksi. Valmisteen käyttö voidaan aloittaa uudelleen pienennetyllä annoksella, kun haittavaikutukset ovat täysin hävinneet. | |||
Annoksen muuttaminen medullaarista kilpirauhaskarsinoomaa sairastaville pediatrisille potilaille
- Jos potilaalla ilmenee 3 CTCAE-asteen tai sitä korkeampaa toksisuutta tai EKG:n QT-aika on pidentynyt, vandetanibin anto on ainakin tilapäisesti keskeytettävä ja jatkettava pienemmällä annoksella, kun toksisuus on hävinnyt tai lieventynyt 1 CTCAE-asteeseen.
- Aloitusannosta saaville potilaille (a taulukossa 1) hoito aloitetaan uudelleen pienemmällä annoksella (c taulukossa 1).
- Suurennettua annosta saavien potilaiden (b taulukossa 1) hoito aloitetaan uudelleen aloitusannoksella (a taulukossa 1). Jos CTCAE-luokituksen (Common Terminology Criteria for Adverse Events) mukaan 3 asteen tai korkeampaa toksisuutta tai EKG:n QT-ajan pidentymistä ilmenee toisen kerran, Caprelsa-valmisteen anto on keskeytettävä ainakin tilapäisesti ja jatkettava pienemmällä annoksella (c taulukossa 1), kun toksisuus on hävinnyt tai lieventynyt 1 CTCAE-asteeseen.
- Jos CTCAE-luokituksen mukaan 3 asteen tai korkeampaa toksisuutta tai EKG:n QT-ajan pidentymistä ilmenee vielä uudelleen, vandetanibin käyttö on lopetettava pysyvästi.
Potilasta on seurattava asianmukaisesti. Koska puoliintumisaika on 19 vuorokautta, haittavaikutukset, mukaan lukien pidentynyt QT-aika, eivät välttämättä korjaannu nopeasti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kesto
Vandetanibia voidaan antaa taudin etenemiseen saakka tai kunnes hoidon jatkamisen hyödyt eivät enää ole suuremmat kuin siitä aiheutuvat riskit, jolloin on otettava huomioon haittavaikutusten vakavuus (ks. kohta Haittavaikutukset) suhteessa kasvaimen statuksen kliinisen vakauttamisen asteeseen.
Erityispotilasryhmät
Pediatriset potilaat
Caprelsa-valmistetta ei pidä antaa alle 5-vuotiaille lapsille. Caprelsa-valmisteen turvallisuutta ja tehoa alle 5-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Käytöstä alle 9-vuotiaille perinnöllistä medullaarista kilpirauhaskarsinoomaa sairastaville lapsipotilaille ei ole kokemusta (ks. kohta Farmakodynamiikka). 5-18-vuotiaiden potilaiden annokset määräytyvät taulukon 1 mukaisesti. Pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa ei ole käytetty yli 150 mg/m2:n vandetanibiannoksia.
Iäkkäät potilaat
Aloitusannosta ei ole tarpeen muuttaa iäkkäille potilaille. Vandetanibin käytöstä yli 75-vuotiaille medullaarista kilpirauhaskarsinoomaa sairastaville potilaille on rajallisesti kliinistä tietoa.
Munuaisten vajaatoiminta medullaarista kilpirauhaskarsinoomaa sairastavilla aikuispotilailla
Farmakokineettinen tutkimus lievää, keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavilla vapaaehtoisilla tutkittavilla osoitti, että kerta-annoksen jälkeinen vandetanibialtistus kasvoi 1,5-, 1,6- ja 2-kertaiseksi potilailla, joilla oli lievä, keskivaikea (kreatiniinipuhdistuma välillä ≥ 30 ja < 50 ml/min) tai vaikea (kreatiniinipuhdistuma alle 30 ml/min) munuaisten vajaatoiminta lähtötilanteessa (ks. kohta Farmakokinetiikka). Kliiniset tiedot viittaavat siihen, että lievää munuaisten vajaatomintaa sairastavien potilaiden aloitusannosta ei tarvitse muuttaa. On vain vähän tietoa 300 mg:n annoksesta potilailla, joilla on keskivaikea munuaisten vajaatoiminta: Annosta oli pienennettävä 200 mg:aan viidellä potilaalla kuudesta, koska haittavaikutuksena ilmeni QT-ajan pidentymistä. Aloitusannos on pienennettävä 200 mg:aan potilaille, jotka sairastavat keskivaikeaa munuaisten vajaatoimintaa; tehoa ja turvallisuutta ei ole kuitenkaan varmistettu 200 mg:n annoksella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Vandetanibia ei suositella potilaille, jotka sairastavat vaikeaa munuaisten vajaatoimintaa, koska vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on vain vähän tietoa eikä turvallisuutta ja tehoa ole varmistettu.
Munuaisten vajaatoiminta medullaarista kilpirauhaskarsinoomaa sairastavilla pediatrisilla potilailla
Vandetanibin käytöstä munuaisten vajaatoimintaa sairastaville pediatrisille potilaille ei ole kokemusta. Munuaisten vajaatoimintaa sairastavista aikuispotilaista saatavilla olevan tiedon perusteella:
- Lievää munuaisten vajaatoimintaa sairastavien pediatristen potilaiden aloitusannoksen muuttamista ei suositella.
- Taulukon 1 mukaista pienennettyä annosta on käytettävä keskivaikeaa munuaisten vajaatoimintaa sairastaville pediatrisille potilaille. Hoito edellyttää yksilöllistä seurantaa lääkärin valvonnassa. Tämä koskee erityisesti sellaisia pediatrisia potilaita, joiden kehon pinta-ala on pieni.
- Vandetanibia ei suositella vaikeaa munuaisten vajaatoimintaa sairastaville pediatrisille potilaille.
Maksan vajaatoiminta
Vandetanibin käyttöä ei suositella maksan vajaatoimintaa sairastaville aikuisille tai pediatrisille potilaille (seerumin bilirubiini yli 1,5-kertainen viitealueen ylärajaan nähden; tämä kriteeri ei koske potilaita, joilla on Gilbertin oireyhtymä, tai potilaita, joiden alaniiniaminotransferaasin (ALAT), aspartaattiaminotransferaasin (ASAT) tai alkalisen fosfataasin (AFOS) arvot ovat yli 2,5-kertaisia viitealueen ylärajaan nähden tai jos ne ovat yli 5,0‑kertaisia viitealueen ylärajaan nähden ja lääkäri on katsonut arvojen kohoamisen liittyvän maksaetäpesäkkeisiin), koska tietoja maksan vajaatoimintapotilaista on vähän eikä turvallisuutta ja tehoa ole varmistettu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vapaaehtoisilla tehtyjen farmakokineettisten tutkimusten tulokset viittaavat siihen, että aloitusannosta ei tarvitse muuttaa potilaille, joilla on lievä, keskivaikea tai vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Antotapa
Caprelsa otetaan suun kautta. Jos potilaalla on nielemisvaikeuksia, vandetanibitabletit voidaan liuottaa puoleen lasilliseen hiilihapotonta vettä. Muita nesteitä ei saa käyttää. Tabletti pudotetaan veteen murskaamatta, ja nestettä sekoitetaan, kunnes tabletti on hajonnut (kestää noin 10 minuuttia). Dispersioliuos juodaan välittömästi. Lasiin jääneet lääkejäännökset sekoitetaan puoleen lasiin vettä ja neste juodaan. Liuos voidaan antaa myös nenä-mahaletkun tai mahalaukkuavanneletkun kautta.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille
- Synnynnäinen pitkä QT-oireyhtymä
- Potilaat, joiden korjattu QT-aika on yli 480 ms
- Vandetanibin samanaikainen käyttö seuraavien lääkevalmisteiden kanssa, joiden tiedetään myös pidentävän QT-aikaa ja/tai aiheuttavan kääntyvien kärkien takykardiaa: arseeni, sisapridi, laskimoon annettava (i.v.) erytromysiini, toremifeeni, mitsolastiini, moksifloksasiini, luokan IA ja III rytmihäiriölääkkeet (ks. kohta Yheisvaikutukset)
- Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Vandetanibin käyttöön liittyvien riskien vuoksi on tärkeää rajoittaa vandetanibihoito potilaisiin, jotka todella tarvitsevat hoitoa ja joilla on symptomaattinen aggressiivinen sairaudenkulku. Joko oireinen tai aggressiivinen sairaus ei ole yksinään riittävä syy vandetanibihoidon aloitukseen. Muutosnopeus biomarkkeritasoissa kuten kalsitoniinissa ja/tai karsinoembryonaalisessa antigeenissa kuten myös muutosnopeus kasvaimen tilavuudessa odottavan seurannan aikana, voi auttaa tunnistamaan sekä potilaat, jotka tarvitsevat hoitoa, että myös optimaalisen hetken vandetanibihoidon aloittamiseen.
Pidentynyt QT-aika ja kääntyvien kärkien takykardia Vandetanibihoitoa ei saa aloittaa potilailla, joiden EKG:n QT-aika on pidempi kuin 480 ms. Vandetanibia ei pidä antaa potilaille, joille on aiemmin ilmennyt kääntyvien kärkien takykardiaa. Vandetanibia ei ole tutkittu potilailla, joilla ilmenee kammioarytmioita tai joilla on hiljattain ollut sydäninfarkti. EKG, seerumin kalium-, kalsium- ja magnesiumtasot sekä kilpirauhasta säätelevän hormonin (TSH) tasot on mitattava lähtötilanteessa, hoidon aloittamisen jälkeen viikoilla 1, 3, 6 ja 12 ja tämän jälkeen 3 kuukauden välein vähintään vuoden ajan. Vastaavaat mittaukset on tehtävä, kun annosta on pienennetty pidentyneen QT-ajan takia tai lääkkeen antamisessa on ollut yli kahden viikon tauko. EKG-tutkimukset on tehtävä ja verikokeet on otettava myös kliinisen tarpeen mukaan hoidon aikana ja hoidon lopettamisen jälkeen. Säännöllistä QT-ajan EKG-seurantaa on jatkettava. Seerumin kalium-, magnesium- ja kalsiumtason on pidettävä normaalialueella, jotta EKG:n QT-ajan pidentymisen riskiä voidaan vähentää. QT-ajan, elektrolyyttipitoisuuden ja munuaistoiminnan lisäseurantaa tarvitaan varsinkin ripulin alkamisen, pahenemisen/nestevajauksen, elektrolyyttitasapainon häiriön ja/tai munuaisten vajaatoiminnan vuoksi. Jos korjattu QT-aika pitenee merkittävästi mutta pysyy 500 ms:n alapuolella, on konsultoitava kardiologia. Vandetanibin annostelu on vasta-aiheista tai sitä ei suositella valmisteiden kanssa, joiden tiedetään pidentävän EKG:n QT-aikaa (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset). Vandetanibin samanaikaista käyttöä ei suositella ondansetronin kanssa (ks. kohta Yhteisvaikutukset). Vandetanibin käyttö on lopetettava potilailla, joilla todetaan yksittäinen korjattu QT-aika ≥ 500 ms. Annostusta voidaan jatkaa pienennetyllä annoksella sen jälkeen, kun korjattu QT-aika on laskenut hoitoa edeltävälle tasolle ja mahdollinen elektrolyyttitasapainon häiriö on korjattu. |
Posteriorinen reversiibeli enkefalopatiaoireyhtymä, PRES (reversiibeli posteriorinen leukoenkefalopatiaoireyhtymä, RPLS)
PRES:ia, joka on aivojen magneettikuvauksella diagnosoitu subkortikaalinen verisuoniperäinen turvotus, on todettu harvoissa tapauksissa potilailla, joita hoidetaan samanaikaisesti vandetanibilla ja kemoterapialla. PRES:iä on todettu myös vandetanibia monoterapiana saaneilla potilailla. Oireyhtymän mahdollisuutta on harkittava potilailla, joilla ilmenee kouristuksia, päänsärkyä, näköhäiriöitä, sekavuutta tai henkisen toimintakyvyn muutosta. Aivojen magneettikuvaus on tehtävä potilaalle, jolla ilmenee kouristuksia, sekavuutta tai henkisen toimintakyvyn muutosta.
Vaikeat ihoreaktiot ja muut ihoreaktiot
Vandetanibihoidon yhteydessä on ilmoitettu vaikeita ihoreaktioita, mukaan lukien toksinen epidermaalinen nekrolyysi ja Stevens–Johnsonin oireyhtymä, jotka saattavat olla henkeä uhkaavia tai johtaa kuolemaan. Valmisteen määräämisen yhteydessä potilaille on kerrottava tämän tilan merkeistä ja oireista, ja heidän vointiaan on seurattava huolellisesti ihoreaktioiden varalta. Jos Stevens–Johnsonin oireyhtymää tai toksista epidermaalista nekrolyysia epäillään, vandetanibihoito on keskeytettävä ja potilas ohjattava erikoissairaanhoidon piiriin arviointia ja hoitoa varten. Jos Stevens–Johnsonin oireyhtymä tai toksinen epidermaalinen nekrolyysi vahvistetaan, vandetanibihoito on lopetettava pysyvästi ja vaihtoehtoista hoitoa on harkittava (tarpeen mukaan).
Vandetanibihoitoa saaneilla potilailla on havaittu valoherkkyysreaktioita. Auringonvaloaltistukselta on suojauduttava käyttämällä suojaavia vaatteita ja/tai käyttämällä aurinkovoiteita vandetanibihoitoon liittyvän mahdollisen fototoksisen reaktion riskin vuoksi.
Lieviä ja keskivaikeita ihoreaktioita voidaan hoitaa joko oireenmukaisesti tai pienentämällä annosta tai keskeyttämällä hoito.
Ripuli
Ripuli on sekä sairauteen liittyvä oire että vandetanibin tunnettu haittavaikutus. Ripulin hoitoon suositellaan tavanomaisia ripulilääkkeitä. QT-aikaa ja seerumin elektrolyyttejä on seurattava säännöllisemmin. Jos vaikeaa (3-4 CTCAE-asteen) ripulia ilmenee, vandetanibihoito on keskeytettävä, kunnes ripuli lievittyy. Kun ripuli on parantunut, hoitoa on jatkettava pienennetyllä annoksella (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Verenvuoto
Varovaisuutta on noudatettava annettaessa vandetanibia potilaille, joilla on aivojen etäpesäkkeitä, koska kallonsisäisiä verenvuotoja on raportoitu.
Sydämen vajaatoiminta
Vandetanibia saavilla potilailla on havaittu sydämen vajaatoimintaa. Sydämen vajaatoimintapotilailla hoito saatetaan joutua keskeyttämään tilapäisesti tai lopullisesti. Sydämen vajaatoiminta ei ehkä korjaannu lopettamalla vandetanibihoito. Muutama tapaus on johtanut kuolemaan.
Kohonnut verenpaine
Kohonnutta verenpainetta, mukaan lukien hypertensiivistä kriisiä, on todettu vandetanibilla hoidetuilla potilailla. Potilaita on seurattava kohonneen verenpaineen varalta ja tilaa on hoidettava tarvittaessa. Vandetanibihoitoa ei pidä aloittaa uudelleen, ennen kuin verenpaine on saatu hoidolla hallintaan. Annosta voidaan joutua pienentämään (ks. kohta Haittavaikutukset).
Haavojen paranemiseen liittyvät komplikaatiot
Vandetanibin vaikutusta haavojen paranemiseen ei ole arvioitu muodollisissa tutkimuksissa. Haavojen paraneminen voi häiriytyä, jos potilas saa VEGF-signaalireittiä estäviä lääkkeitä. Ilmiötä on raportoitu vandetanibia saaneilla potilailla. On hyvin niukasti näyttöä siitä, miten pitkäksi ajaksi hoito on ihanteellista keskeyttää ennen suunniteltua leikkausta. On kuitenkin aiheellista harkita vandetanibihoidon keskeyttämistä vähintään 4 viikoksi ennen elektiivistä leikkausta yksilöllisen hyöty-riskiarvioinnin perusteella. Päätös vandetanibihoidon jatkamisesta suuren leikkauksen jälkeen tehdään haavan riittävää paranemista koskevan kliinisen arvioinnin perusteella.
Osteonekroosi
Vandetanibihoitoa saaneilla potilailla on raportoitu osteonekroositapauksia, mukaan lukien leukaluun osteonekroosia. Joitakin tapauksia on raportoitu potilailla, jotka ovat saaneet aiemmin tai samanaikaisesti luun resorptiota estävää hoitoa. Suun tutkimus tulee suorittaa ennen vandetanibihoidon aloittamista ja säännöllisesti hoidon aikana. Potilaille tulee antaa ohjeistusta suuhygienian hoidosta. Jos mahdollista, vandetanibihoito tulee keskeyttää vähintään 4 viikkoa ennen suunniteltua hammaskirurgiaa tai invasiivisia hammastoimenpiteitä erityisesti potilailla, jotka saavat osteonekroosiin liittyviä lääkkeitä, kuten bisfosfonaatteja. Vandetanibihoidon lopettamista tulee harkita potilailla, joille kehittyy osteonekroosi (ks. kohta Haittavaikutukset).
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen vandetanibihoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Munuaisten vajaatoiminta
Vandetanibihoitoa saaneilla potilailla on ilmoitettu munuaisten vajaatoimintaa (ks. kohta Haittavaikutukset). Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen (ks. kohta Annostus ja antotapa).
Vandetanibialtistus suurenee potilailla, joilla on munuaisten vajaatoiminta. Potilailla, joilla on keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma välillä ≥ 30 ja < 50 ml/min), vandetanibin aloitusannos on pienennettävä 200 mg:aan ja QT-aikaa on seurattava tarkasti.
Vandetanibin käyttöä ei suositella vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma alle 30 ml/min) sairastaville potilaille (ks. kohdat Annostus ja antotapa, Farmakodynamiikka ja Farmakokinetiikka). Tietoja ei ole saatavilla potilaista, joilla on dialyysihoitoa vaativa loppuvaiheen munuaissairaus.
Potilaat, joilla on maksan vajaatoiminta
Vandetanibin käyttöä ei suositella maksan vajaatoimintaa sairastaville potilaille (seerumin bilirubiini yli 1,5 kertaa normaaliin ylärajaan nähden), koska tietoja maksan vajaatoimintapotilaista on vähän eikä turvallisuutta ja tehoa ole varmistettu. Vapaaehtoisilla saadut farmakokineettiset tulokset viittaavat siihen, että aloitusannosta ei tarvitse muuttaa potilailla, joilla on lievä, keskivaikea tai vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Alaniiniaminotransferaasin pitoisuuden nousu
Alaniiniaminotransferaasin pitoisuuden nousua ilmenee yleisesti vandetanibihoitoa saavilla potilailla. Useimmissa tapauksissa nousu korjaantuu hoidon jatkuessa. Muissa tapauksessa tila korjaantuu tavallisesti 1-2 viikkoa hoidon keskeyttämisen jälkeen. Alaniiniaminotransferaasin pitoisuuden ajoittainen seuranta on suositeltavaa.
Interstitiaalinen keuhkosairaus
Vandetanibihoitoa saavilla potilailla on todettu interstitiaalista keuhkosairautta (ILD), joka on muutamissa tapauksissa johtanut kuolemaan. Jos potilaalle ilmaantuu hengitystieoireita, kuten hengenahdistusta, yskää ja kuumetta, vandetanibihoito on keskeytettävä ja potilas on tutkittava viipymättä. Jos ILD-diagnoosi on vahvistettu, vandetanibihoito on lopetettava lopullisesti ja potilasta on hoidettava asianmukaisesti.
CYP3A4-indusorit
Vandetanibin samanaikaista käyttöä voimakkaiden CYP3A4-indusorien (kuten rifampisiinin, mäkikuisman, karbamatsepiinin ja fenobarbitaalin) kanssa on vältettävä (ks. kohta Yhteisvaikutukset).
Kalsitoniinipitoisuus alle 500 pg/ml
Vandetanibista saatavaa hyötyä potilaille, joiden kalsitoniinipitoisuus on alle 500 pg/ml, ei ole määritetty. Käyttöä on sen vuoksi harkittava huolellisesti vandetanibihoidosta aiheutuvien riskien takia, jos potilaan kalsitoniinipitoisuus on < 500 pg/ml.
Pediatriset potilaat
Pediatrisilla potilailla tehdyssä tutkimuksessa kaikkien lasten ja nuorten todettiin kasvavan lineaarisesti vandetanibihoidon aikana kaikkien vastaanottokäyntien pituusmittausten perusteella. Tietoja ei kuitenkaan ole saatavilla pitkän aikavälin turvallisuudesta pediatrisilla potilailla.
Potilaskortti
Kaikkien Caprelsa-valmistetta määräävien lääkäreiden on tunnettava lääkäreille tarkoitettu informaatio ja toimintaohjeet. Lääkärin on keskusteltava Caprelsa-hoidon riskeistä potilaan kanssa. Potilaalle on annettava potilaskortti jokaisen lääkemääräyksen yhteydessä.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
Vandetanibin vaikutus muihin lääkevalmisteisiin
Terveillä tutkittavilla altistus midatsolaamille (CYP3A4:n substraatti) ei muuttunut, kun sitä annettiin vandetanibin 800 mg:n kerta-annoksen kanssa.
Vandetanibi on orgaanisten kationien kuljettaja 2:n (OCT2) estäjä. Terveillä tutkittavilla, joilla oli villin tyypin OCT2, metformiinin (OCT2:n substraatti) AUC(0-t) suureni 74 % ja Cmax suureni 50 % ja metformiinin CLR munuaispuhdistuma pieneni 52 %, kun sitä annettiin yhdessä vandetanibin kanssa. Samanaikaisesti metformiinia ja vandetanibia saaville potilaille suositellaan asianmukaista kliinistä ja/tai laboratorioseurantaa ja tällaisten potilaiden metformiiniannosta on mahdollisesti pienennettävä.
Terveillä tutkittavilla digoksiinin (P-glykoproteiinin P-gp:n substraatti) AUC(0-t) suureni 23 % ja Cmax suureni 29 %, kun sitä annettiin samanaikaisesti vandetanibin kanssa, koska vandetanibi estää P-glykoproteiinia. Lisäksi digoksiinin bradykardinen vaikutus saattaa suurentaa riskiä, että vandetanibi pidentää QT-aikaa ja aiheuttaa kääntyvien kärkien takykardiaa. Sen vuoksi samanaikaisesti digoksiinia ja vandetanibia saaville potilaille suositellaan asianmukaista kliinistä (esim. EKG) ja/tai laboratorioseurantaa ja tällaisten potilaiden digoksiiniannosta on mahdollisesti pienennettävä. (Ks. vandetanibiseuranta kohdasta Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Muiden P-glykoproteiinin substraattien, kuten dabigatraanin, suhteen suositellaan kliinistä seurantaa, jos niitä annetaan yhdessä vandetanibin kanssa.
Muiden lääkevalmisteiden vaikutus vandetanibiin
Terveillä tutkittavilla vandetanibin (300 mg:n kerta-annos) ja voimakkaan CYP3A4:n estäjän, itrakonatsolin (toistuvat 200 mg:n annokset kerran vuorokaudessa), välillä ei todettu kliinisesti merkittävää yhteisvaikutusta. Terveillä miespuolisilla tutkittavilla altistus vandetanibille pieneni 40 %, kun sitä annettiin yhdessä rifampisiinin kanssa, joka on tehokas CYP3A4:n indusoija. Vandetanibin antamista voimakkaiden CYP3A4:n indusoijien kanssa on vältettävä.
Terveillä tutkittavilla vandetanibin Cmax pieneni 15 %, kun taas vandetanibin AUC(0-t) ei muuttunut, kun sitä annettiin samanaikaisesti omepratsolin kanssa. Sekä vandetanibin Cmax että AUC(0-t) pysyivät muuttumattomina, kun vandetanibia annettiin samanaikaisesti ranitidiinin kanssa. Sen vuoksi vandetanibin annosta ei tarvitse muuttaa, kun sitä annetaan samanaikaisesti joko omepratsolin tai ranitidiinin kanssa.
Farmakodynaamiset yhteisvaikutukset
Vandetanibia erittyy muuttumattomana sappinesteeseen, mikä on yksi vandetanibin erittymisreiteistä. Vandetanibi ei ole monilääkeresistentin proteiini 2:n (MRP2:n), P-glykoproteiinin (P-gp:n) tai BCRP-kuljetusproteiinin substraatti.
Tunnetusti QT-aikaa pidentävät lääkkeet
Vandetanibin on osoitettu pidentävän EKG:n QT-aikaa; kääntyvien kärkien takykardiaa on raportoitu melko harvoin. Siksi vandetanibin käyttö samanaikaisesti QT-aikaa pidentävien ja/tai kääntyvien kärkien takykardiaa aiheuttavien lääkkeiden kanssa on joko vasta-aiheista tai sitä ei suositella riippuen olemassa olevista korvaavista hoidoista.
- Vasta-aiheiset yhdistelmät (ks. kohta Vasta-aiheet): sisapridi, laskimoon annettava (i.v.) erytromysiini, toremifeeni, mitsolastiini, moksifloksasiini, arseeni, luokan IA ja III rytmihäiriölääkkeet
- Yhdistelmät, joita ei suositella: metadoni, haloperidoli, amisulpiridi, klooripromatsiini, sulpiridi, tsuklopentiksoli, halofantriini, pentamidiini ja lumefantriini.
Jos soveltuvaa korvaavaa hoitoa ei ole, vandetanibin kanssa voidaan käyttää ei-suositeltuja yhdistelmiä lisättäessä EKG:n QT-ajan seurantaa ja elektrolyyttitasojen arviointia ja jatkettaessa kontrollia ripulin ilmetessä tai sen pahentuessa.
Farmakodynaamisen ja farmakokineettisen yhteisvaikutustutkimuksen tulokset osoittivat, että samanaikainen ondansetronin käyttö terveillä potilailla vaikutti vain vähän vandetanibin farmakokinetiikkaan. Sillä oli kuitenkin pieni, noin 10 millisekunnin, lisäävä vaikutus QT-ajan pidentymiseen. Siksi vandetanibin ja ondansetronin samanaikainen käyttö ei ole suositeltavaa. Jos ondansetronia annetaan samanaikaisesti vandetanibin kanssa, seerumin elektrolyyttejä ja EKG:tä on seurattava huolellisesti ja mahdollista poikkeavuutta hoidettava aggressiivisesti.
K-vitamiinin antagonistit
Syöpäpotilaiden kohonneen verisuonitukosriskin vuoksi käytössä on usein antikoagulantteja. Koska vaste antikoagulanttihoitoon vaihtelee yksilöllisesti ja koska K-vitamiinin antagonisteilla ja kemoterapia-aineilla saattaa olla yhteisvaikutuksia, INR (International Normalised Ratio) -seurantavälejä on suositeltavaa lyhentää, jos potilaalle päätetään antaa K-vitamiinin antagonisteja.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / raskauden ehkäisy miehillä ja naisilla
Hedelmällisessä iässä olevien naisten ja miesten on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään neljä kuukautta viimeisen annoksen jälkeen.
Raskaus
Vandetanibin käytöstä raskauden aikana on vain vähän tietoa. Kuten farmakologisten vaikutusten perusteella on oletettu, vandetanibilla on osoitettu olevan merkittäviä vaikutuksia naarasrottien lisääntymiseen sen kaikissa vaiheissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Jos vandetanibia käytetään raskauden aikana tai potilas tulee raskaaksi vandetanibihoidon aikana, on potilaalle kerrottava mahdollisesta sikiön epämuodostumien tai raskauden keskeytymisen riskistä. Raskaana olevan naisen hoitoa voi jatkaa vain, jos äidille koituva hyöty arvioidaan sikiölle koituvaa riskiä suuremmaksi.
Imetys
Tietoa vandetanibin käytöstä imetyksen aikana ei ole saatavilla. Vandetanibi ja/tai sen metaboliitit erittyvät rotilla maitoon, ja sitä on löytynyt lääkeainetta saaneiden imettävien rottien poikasten plasmasta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys on lopetettava vandetanibihoidon ajaksi.
Hedelmällisyys
Vandetanibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläimillä tehtyjen tutkimusten tulokset osoittavat, että vandetanibi voi heikentää miesten ja naisten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutuksista vandetanibihoitoa saaneiden pediatristen potilaiden hedelmällisyyteen ei ole tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia vandetanibin vaikutuksista ajokykyyn tai koneiden käyttökykyyn ei ole tehty. Väsymystä ja näön hämärtymistä on kuitenkin raportoitu. Tällaisista oireista kärsivien potilaiden on noudatettava varovaisuutta autoa ajaessaan tai koneita käyttäessään.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia ovat ripuli, ihottuma, pahoinvointi, kohonnut verenpaine ja päänsärky.
Haittavaikutustaulukko
Seuraavat haittavaikutukset on havaittu kliinisissä tutkimuksissa potilailla, jotka saivat vandetanibia medullaarisen kilpirauhaskarsinooman hoitoon, sekä valmisteen markkinoilletulon jälkeen. Haittavaikutusten yleisyys on esitetty taulukossa 2 luokiteltuna Council for International Organizations of Medical Sciences (CIOMS III)-yleisyysluokituksen mukaan lueteltuna MedDRA-elinjärjestelmän ja Preferred Term -luokituksen mukaisesti ja yleisyysluokituksen perusteella. Haittavaikutusten esiintymistiheys on määritetty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
| Taulukko 2 Haittavaikutukset ja elinjärjestelmä | ||||
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Tuntematon |
| Infektiot | Nenänielun tulehdus, keuhkoputkentulehdus, ylähengitysteiden infektiot, virtsatieinfektiot | Keuhkokuume, sepsis, influenssa, rakkotulehdus, sivuontelotulehdus, kurkunpäätulehdus, karvatupen tulehdus, paise, sieni-infektio, munuaisaltaan tulehdus | Umpilisäkkeen tulehdus, stafylokokki-infektio, umpipussitulehdus, selluliitti, vatsapeitteen märkäpesäke | |
| Umpieritys | Kilpirauhasen vajaatoiminta | |||
| Aineenvaihdunta ja ravitsemus | Vähentynyt ruokahalu, hypokalsemia | Hypokalemia, hyperkalsemia, hyperglykemia, kuivuminen, hyponatremia | Aliravitsemus | |
| Psyykkiset häiriöt | Unettomuus, masennus | Ahdistuneisuus | ||
| Hermosto | Päänsärky, tuntoharha, tuntohäiriö, huimaus | Vapina, letargia, tajunnan menetys, tasapainohäiriöt, makuhäiriö | Kouristus, klonus, aivoedeema | |
| Silmät | Näön hämärtyminen, sarveiskalvon rakenteellinen muutos (mukaan lukien sarveiskalvon kertymät ja samentumat) | Näön heikkeneminen, valorenkaiden näkeminen, fotopsia, glaukooma, sidekalvotulehdus, kuivasilmäisyys, keratopatia | Kaihi, silmän akkommodaatiohäiriöt | |
| Sydän | EKG:n QT-ajan pidentyminen (*)(**) | Sydämen vajaatoiminta, akuutti sydämen vajaatoiminta, syke- ja rytmihäiriöt, sydämen johtumishäiriöt, kammion arytmia ja sydämenpysähdys | ||
| Verisuonisto | Kohonnut verenpaine | Hypertensiivinen kriisi, iskeemiset aivoverisuonitapahtumat | Aneurysmat ja valtimon dissekaatiot | |
| Hengityselimet, rintakehä ja välikarsina | Nenäverenvuoto, veriyskös, keuhkotulehdus | Hengitysvajaus, aspiraatiokeuhkokuume | ||
| Ruoansulatuselimistö | Vatsakipu, ripuli, pahoinvointi, oksentelu, ylävatsavaivat | Koliitti, suun kuivuminen, suutulehdus, nielemishäiriöt, ummetus, mahatulehdus, ruoansulatuskanavan verenvuoto | Haimatulehdus, vatsakalvotulehdus, ileus, suolen puhkeama, ulosteinkontinenssi | |
| Maksa ja sappi | Sappikivitauti | |||
| Iho ja ihonalainen kudos | Valoherkistyneisyysreaktio, ihottuma ja muut ihoreaktiot (mukaan lukien akne, ihon kuivuminen, ihotulehdus, kutina), kynsisairaus | Palmoplantaarinen erytrodysestesia, hiustenlähtö | Rakkulainen ihotulehdus | Stevens–Johnsonin oireyhtymä / toksinen epidermaalinen nekrolyysi(***), erythema multiforme |
| Luusto, lihakset ja sidekudos | Osteonekroosi, leuan osteonekroosi | |||
| Munuaiset ja virtsatiet | Valkuaisvirtsaisuus, munuaiskivitauti | Dysuria, verivirtsaisuus, munuaisten vajaatoiminta, tihentynyt virtsaamistarve, virtsaamispakko | Kromaturia, anuria | |
| Yleisoireet ja antopaikassa todettavat haitat | Voimattomuus, väsymys, kipu, edeema | Pyreksia | Heikentynyt paraneminen | |
| Tutkimukset | EKG:n pidentynyt QT-aika | Seerumin ALAT- tai ASAT-arvon nousu, painon lasku, veren kreatiniiniarvon nousu | Hemoglobiiniarvon nousu, seerumin amylaasiarvon nousu | |
| * vandetanibipotilaista 13,4 %:lla ilmeni vähintään 500 ms:n korjattu QT-aika (Bazett) verrattuna 1,0 %:iin lumelääkettä saaneista. Korjattu QT-aika (F) pidentyi > 20 ms:a yli 91 %:lla potilaista, > 60 ms:a 35 %:lla, > 100 ms:a 1,7 %:lla. Kahdeksalla prosentilla potilaista annosta pienennettiin QT-ajan pidentymisen vuoksi. ** mukaan lukien kaksi kuolemantapausta potilailla, joilla korjattu QT-aika oli > 550 ms:a (toinen sepsiksen vuoksi ja toinen sydämen vajaatoiminnan vuoksi) *** Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet | ||||
Valikoitujen haittavaikutusten kuvaus
Kääntyvien kärkien takykardiaa, interstitiaalista keuhkosairautta (joskus kuolemaan johtavaa) ja PRES:iä (RPLS) on ilmennyt potilailla, jotka ovat saaneet vandetanibia monoterapiana. Näiden haittavaikutusten voidaan olettaa olevan melko harvinaisia potilailla, jotka saavat vandetanibia medullaarisen kilpirauhaskarsinooman hoitoon.
Silmätapahtumat, kuten näön hämärtyminen, olivat yleisiä potilailla, jotka saivat vandetanibia medullaariseen kilpirauhaskarsinooman hoitoon. Hoidettujen potilaiden suunnitellut rakovalotutkimukset ovat paljastaneet sarveiskalvon samentumaa (vortex keratopatia). Rutiininomaiset rakovalotutkimukset eivät kuitenkaan ole pakollisia vandetanibia saaville potilaille.
Vandetanibilla hoidettujen potilaiden hemoglobiinitasojen mediaanit nousivat eripituisilla lääkealtistusjaksoilla 0,5-1,5 g/dl perustasoon verrattuna.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus -ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Pediatriset potilaat
Lääkekehityksen aikana saadut pediatrisen kliinisen tutkimuksen tulokset vandetanibin käytöstä medullaarista kilpirauhaskarsinoomaa sairastaville potilaille rajoittuvat 16 potilaaseen, jotka olivat iältään 9–17-vuotiaita ja jotka sairastivat perinnöllistä medullaarista kilpirauhaskarsinoomaa (IRUSZACT0098-tutkimus). Vaikka tutkimus oli kooltaan suppea lapsilla ilmenevän medullaarisen kilpirauhaskarsinooman harvinaisuuden vuoksi, sitä pidetään kohdepopulaatiota edustavana. Tutkimuksessa tehdyt turvallisuutta koskevat havainnot ovat yhdenmukaiset medullaarista kilpirauhaskarsinoomaa sairastavien aikuisten hoidossa käytetyn vandetanibin turvallisuusprofiilin kanssa. Tietoja ei ole saatavilla pitkän aikavälin turvallisuudesta pediatrisilla potilailla.
Yliannostus
Vandetanibin yliannostukseen ei ole spesifistä hoitoa, eikä yliannostuksen mahdollisia oireita ole määritetty. Joidenkin haittavaikutusten, kuten ihottuman, ripulin ja verenpaineen nousun, yleisyyden ja vaikeusasteen havaittiin nousseen lukuisten 300 mg ja sitä suurempien annosten jälkeen terveillä vapaaehtoisilla tehdyissä tutkimuksissa ja potilailla. Lisäksi QT-ajan pitenemisen ja kääntyvien kärkien takykardian mahdollisuus on otettava huomioon. Kliinisissä tutkimuksissa ei ole käytetty yli 150 mg/m2:n vandetanibiannoksia pediatrisilla potilailla.
Yliannostukseen liittyviä haittavaikutuksia on hoidettava oireenmukaisesti, ja erityisesti vaikeaa ripulia on hoidettava asianmukaisesti. Yliannostustapauksessa uusien annosten ottaminen keskeytetään ja tarvittaviin toimiin on ryhdyttävä sen varmistamiseksi, ettei haittavaikutuksia ole ilmennyt esimerkiksi EKG on mitattava 24 tunnin kuluessa QT-ajan pitenemisen määrittämiseksi. Yliannostukseen liittyvät haittavaikutukset voivat pitkittyä vandetanibin pitkän puoliintumisajan takia (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EX04
Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Vandetanibi on voimakas verisuonten endoteelikasvutekijän reseptori-2:n (VEGFR-2, joka tunnetaan myös reseptorin [KDR] sisältävänä kinaasin I-domeenina), epidermaalisen kasvutekijän reseptorin (EGFR) ja RET (Rearranged during Transfection) -tyrosiinikinaasien inhibiittori. Vandetanibi on myös verisuonten endoteelisen reseptori 3:n tyrosiinikinaasin submikromolaarinen inhibiittori.
Vandetanibi estää VEGF:n stimuloiman endoteelisolun migraatiota, proliferaatiota, eloonjäämistä ja uuden verisuonen muodostumista angiogeneesin in vitro -malleissa. Lisäksi vandetanibi estää epidermaalisen kasvutekijän (EGF) stimuloiman EGF-reseptorin tyrosiinikinaasia kasvain- ja endoteelisoluissa. Vandetanibi estää EGFR-riippuvaista soluproliferaatiota ja solun eloonjäämistä in vitro -tutkimuksissa. Vandetanibi estää myös sekä RET:in villityyppiä että suurinta osaa sen mutatoituneista, aktiivisista muodoista ja se estää merkittävästi medullaarisen kilpirauhaskarsinooman solulinjojen proliferaatiota in vitro.
Vandetanibin antaminen in vivo vähensi kasvainsolun aiheuttamaa angiogeneesia, kasvaimen verisuonten läpäisevyyttä ja kasvaimen mikroverisuonitiheyttä sekä esti kasvaimen kasvua atyymisiin hiiriin siirretyissä ihmisen kasvaimen ksenograftimalleissa. Vandetanibi esti myös medullaarisen kilpirauhaskarsinooman ksenograftikasvainten kasvun in vivo.
Vandetanibin tarkkaa vaikutusmekanismia paikallisesti edenneessä tai etäpesäkkeisessä medullaarisessa kilpirauhaskarsinoomassa ei tunneta.
Kliininen teho aikuisilla
Medullaarisen kilpirauhaskarsinooman tutkimuksen kliiniset tiedot
Satunnaistetulla, kaksoissokkoutetulla lumekontrolloidulla tutkimuksella (tutkimus 58) pyrittiin osoittamaan vandetanibin 300 mg:n annoksen turvallisuus ja teho lumelääkkeeseen verrattuna. Tutkimukseen osallistui 331 potilasta, joilla oli paikallisesti edennyt tai metastaattinen medullaarinen kilpirauhaskarsinooma, jota ei voitu hoitaa leikkauksella. Vain potilaat, joiden kalsitoniiniarvo oli ≥ 500 pg/ml (konventionaalinen yksikkö) tai ≥ 146,3 pmol/l (kansainvälistä yksikköä) otettiin tutkimukseen. Tutkimukseen osallistuneista vandetanibia saaneista potilaista 10:n ja lumelääkettä saaneista 4:n (4 % kaikista potilaista) WHO PS (World Health Organization Performance Status) -pisteytys oli ≥ 2 ja vandetanibia saaneista 28:lla (12,1 %) ja lumelääkettä saaneista 10:llä (10,1 %) ilmeni sydämen toimintakyvyn heikkenemistä. Sydämen toimintakyvyn heikkenemiseksi katsottiin potilaalla aiemmin ilmennyt sydän- ja verenkiertoelimistön poikkeava toiminta.
Tutkimuksen ensisijaisena tavoitteena oli osoittaa etenemisvapaan elinajan piteneminen vandetanibihoidolla lumelääkkeeseen verrattuna. Toissijaisina päätetapahtumina arvioitiin yleinen hoitovaste (ORR), taudin hallinta (DCR) määritettynä osittaisena vasteena (PR) tai täydellisenä vasteena (CR) tai stabiilina sairautena (SD) vähintään viikolle 24, vasteen kesto (DOR), aika kivun pahenemiseen perustuen Brief Pain Inventory (BPI)-pahimman kivun asteikkoon ja kokonaiseloonjääminen (OS). Ensisijainen päätetapahtuma eli etenemisvapaa elinaika, yleinen hoitovaste ja taudin hallinta perustuivat keskitettyyn, riippumattomaan kuvantamistietojen sokkoarviointiin. Toissijaisena päätetapahtumana arvioitiin myös vandetanibin biokemiallista vastetta lumelääkkeeseen verrattuna mittaamalla kalsitoniinin (CTN) ja karsinoembryonaalisen antigeenin (CEA) pitoisuuksia.
Potilaita hoidettiin vandetanibilla tai lumelääkkeellä taudin objektiiviseen etenemiseen saakka. Kun tutkijan arviointiin perustuva taudin objektiivinen eteneminen alkoi, potilaat keskeyttivät sokkoutetun tutkimuksen ja heille tarjottiin mahdollisuutta osallistua avoimeen vandetanibitutkimukseen. Kaksikymmentäkahdeksan 231:stä vandetanibia saaneesta potilaasta (12,1 %) ja kolme 99:stä lumelääkettä saaneesta (3,0 %) keskeytti hoidon haittatapahtuman vuoksi. Haittatapahtuman vuoksi vandetanibin keskeyttäneistä 28:sta potilaasta 14 (50 %) keskeytti hoidon ilman annoksen pienentämistä. Keskivaikeaa munuaisten vajaatoimintaa sairastaneilla vandetanibia saaneilla potilailla annos pienennettiin 200 mg:aan viidellä potilaalla kuudesta (83 %); yhdellä potilaalla annosta tarvitsi pienentää vielä 100 mg:aan.
Ensisijainen etenemisvapaan elinajan analyysi osoitti lumelääkkeeseen verrattaessa tilastollisesti merkitsevää etenemisvapaan elinajan pitenemistä potilailla, jotka oli satunnaistettu saamaan vandetanibia (riskisuhde(HR) = 0,46; 95 %:n luottamusväli (CI) = 0,31–0,69; p = 0,0001).
Vandetanibihoitoon satunnaistettujen potilaiden etenemisvapaan elinajan mediaania ei saavutettu, mutta tilastollisen mallin mukaan 43prosenttipisteeseen asti saatavilla olevien havaintojen avulla etenemisvapaan elinajan mediaaniksi voidaan ennustaa 30,5 kuukautta ja 95 %:n luottamusvälillä 25,5-36,5 kuukautta. Lumelääkeryhmään satunnaistettujen potilaiden etenemisvapaan elinajan mediaani oli 19,3 kuukautta. Tutkimuksen kuukauden 12 kohdalla elossa olevien etenemisvapaiden potilaiden osuus oli 192 (83 %) vandetanibiryhmään satunnaistetuilla ja 63 (63 %) lumelääkeryhmään satunnaistetuilla. Vandetanibiryhmässä tauti eteni kaikkiaan 73:lla potilaalla (32 %): 64:llä (28 %) ilmeni kiinteän kasvaimen etenemiskriteerin (RECIST) täyttävä vaste ja 9 (4 %) kuoli ilman taudin etenemistä. Jäljellä olevat 158 potilasta (68 %) otettiin mukaan etenemisvapaan elinajan analyysiin. Taudin etenemistä ilmeni lumelääkeryhmässä 51:llä potilaalla (51 %): 46:lla (46 %) ilmeni RECIST-kriteeristön mukainen eteneminen ja 5 potilasta (5 %) kuoli ilman taudin etenemistä. Jäljellä olevat 49 potilasta (49 %) otettiin mukaan etenemisvapaan elinajan analyysiin.
Kuva 1: Kaplan-Meier-käyrä (PFS)
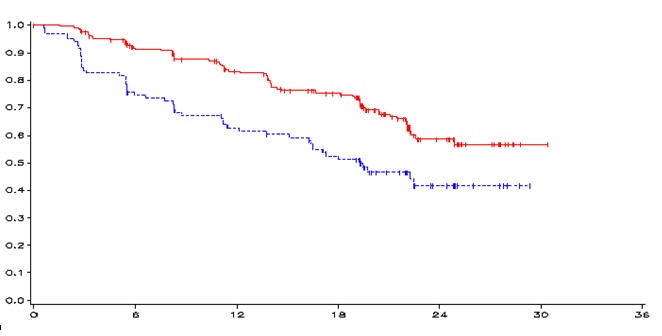
| kuukaudet | 0 | 6 | 12 | 18 | 24 | 30 | 36 |
| n-vandetanibi | 231 | 196 | 169 | 140 | 40 | 1 | 0 |
| n-lumelääke | 100 | 71 | 57 | 45 | 13 | 0 | 0 |
____ vandetanibi 300 mg, ------ lumelääke, Y-akseli=PFS, X-akseli=aika kuukausina,
n-vandetanibi=riskipotilaiden määrä-vandetanibi, n-lumelääke=riskipotilaiden määrä-lumelääke
HR = 0,46; 95 % CI (0,36-0,69), p = 0.0001
| PFS | N | Mediaani PFS | HR | 95 % CI | p-arvo |
| Vandetanibi 300 mg | 73/231 (32 %) | Ei saavutettu (oletettu 30,5 kuukautta) | 0,46 | 0,31; 0,69 | 0,0001 |
| Lumelääke | 51/100 (51 %) | 19,3 kuukautta |
Elossaolo ja lopullisen kokonaiseloonjäämisen mediaani (81,6 kuukautta vandetanibiryhmässä ja 80,4 kuukautta lumelääkeryhmässä) olivat samanlaiset molemmissa hoitoryhmissä. Lopullisessa kokonaiseloonjäämisessä ei ollut tilastollisesti merkitsevää eroa (riskisuhde (HR) 0,99, 95,002 %:n luottamusväli 0,72; 1,38, p = 0,9750). Tulosten tulkinnassa on noudatettava varovaisuutta lumelääkeryhmästä avoimeen vandetanibitutkimukseen siirtyneiden potilaiden suuren osuuden vuoksi (79,0 % [79/100] potilaista).
Useimmilla (95 %:lla potilaista) oli etäpesäkkeinen sairaus. Neljällätoista vandetanibilla hoidetuista potilaista ja 3:lla lumelääkepotilaista oli vain paikallisesti edennyt sairaus, jota ei voitu hoitaa leikkauksella. Kliinistä kokemusta on vähän vandetanibia saaneista potilaista, joiden paikallisesti edennyttä sairautta ei voi leikata ja joilla ei ole etäpesäkkeitä.
Vandetanibilla todettiin tilastollisesti merkitseviä etuja toissijaisten päätetapahtumien osalta hoitovasteessa, taudin hallinnassa ja biokemiallisessa vasteessa.
Taulukko 3:Yhteenveto tutkimuksen 58 muista tehoa koskevista löydöksistä
| YLEINEN HOITOVASTEa | N | Hoitovaste | ORb | 95 %:n luottamusväli | p-arvo |
| Vandetanibi 300 mg | 104/231 | 45 % | 5,48 | 2,99; 10,79 | < 0,0001 |
| Lumelääke | 13/100 | 13 % | |||
| TAUDIN HALLINTAa | N | Hoitovaste | ORb | 95 %:n luottamusväli | p-arvo |
| Vandetanibi 300 mg | 200/231 | 87 % | 2,64 | 1,48; 4,69 | 0,001 |
| Lumelääke | 71/100 | 71 % | |||
| CTN-vaste | N | Hoitovaste | ORb | 95 %:n luottamusväli | p-arvo |
| Vandetanibi 300 mg | 160/231 | 69 % | 72,9 | 26,2; 303,2 | < 0,0001 |
| Lumelääke | 3/100 | 3 % | |||
| CEA-vaste | N | Hoitovaste | ORb | 95 %:n luottamusväli | p-arvo |
| Vandetanibi 300 mg | 119/231 | 52 % | 52,0 | 16,0; 320,3 | < 0,0001 |
| Lumelääke | 2/100 | 2 % | |||
| KOKONAISELOON- JÄÄMINEN | N | Kokonais- eloonjäämisen mediaani | HRc | 95 %:n luottamusväli | p-arvo |
| Vandetanibi 300 mg | 116/231 | 81,6 kuukautta | 0,99 | 0,72; 1,38 | 0,9750 |
| Lumelääke | 52/100 | 80,4 kuukautta | |||
| a Yleinen hoitovaste = täydelliset + osittaiset vasteet. Taudin hallinta = hoitovaste + stabiili tauti viikolla 24. Intent to treat (ITT)-analyysissa olivat mukana potilaat, jotka osallistuivat avoimeen vandetanibitutkimukseen ennen keskitettyä, riippumatonta etenemisen toteamista. b OR = Odds Ratio, todennäköisyyssuhde. Arvo > 1 suosii vandetanibia. Analyysissa käytettiin logistista regressiomallia, jossa hoito oli ainoana tekijänä. c HR = Riskisuhde. Arvo < 1 suosii vandetanibia. Analyysissa käytettiin log rank -testiä, jossa hoito oli ainoana tekijänä. N = tapahtumien / satunnaistettujen potilaiden määrä | |||||
Vandetanibilla todettiin tilastollisesti merkitsevä etu toissijaisessa päätetapahtumassa, joka oli kivun pahenemiseen kulunut aika (yhdistelmäpäätetapahtuma, joka on johdettu pahimman kivun BPI–asteikosta ja potilaan ilmoittamasta opioidianalgeettien käytöstä) (vandetanibi 49 %, lumelääke 57 %, HR 0,61, 97,5 % CI: 0,43 - 0,87, p < 0.006: 8 vs. 3 kuukautta). Tilastollisesti merkitseviä eroja ei havaittu ripulin eksploratiivisissa päätepisteissä (raportoitiin ulostustiheytenä).
RET-mutaatiostatus
RET-mutaatiostatuksen uudelleenanalysointi tutkimuksessa 58
Tutkimuksessa 58 kaikilta satunnaistetuilta potilailta, joiden DNA:ta oli saatavilla (297/298) testattiin RET-mutaatio käyttäen aluksi polymeraasiketjureaktioon (PCR) perustuvaa Amplification Refractory Mutation System (ARMS) -menetelmää M918T-mutaatioon ja suoraa DNA:n sekvensointimenetelmää mutaatioille eksoneissa 10, 11, 13, 14, 15 ja 16 (M918T-mutaation kohta). Näytteet, joissa ei ollut M918T-mutaatiota, analysoitiin uudelleen siten, että RET-sekvenssit rikastettiin mukautetulla Agilent SureSelect ‑reagenssilla ja sekvensoitiin Illumina-sekvensointilaitteella. Tietojen käsittelyyn ja RET-varianttien automaattiseen tunnistamiseen käytettiin Broad Genome Analysis ToolKit (GATK) ‑työnkulkua, ja hankalat tapaukset käytiin läpi manuaalisesti Broad Integrative Genomics Viewer (IGV) ‑työkalun avulla.
79 potilaalta ei aluksi tunnistettu M918T-mutaatiota. Näistä 79 potilaasta 69:stä oli riittävän paljon kudosnäytettä, jotta RET-mutaatiostatus voitiin analysoida jälkikäteen uudelleen käyttäen saatavilla olevia uusia testejä. Suurin osa potilaista (52/69) todettiin uudelleen luokiteltaessa RET-mutaatiopositiivisiksi. 17 potilaalla 69:stä (11:llä vandetanibiryhmässä ja 6:lla lumeryhmässä) ei havaittu mitään RET-mutaatiota (M918T-mutaatiota tai muuta mutaatiota). RET-mutaatiopositiivisiksi uudelleenluokiteltujen potilaiden tiedot yhdistettiin alun perin RET-mutaatiopositiivisiksi luokiteltujen 187 potilaan tietojen kanssa, jolloin RET-mutaatiopositiivisia potilaita oli yhteensä 239 (joista 172 sai vandetanibia ja 67 lumelääkettä). Tulokset perustuivat kuvantamistutkimusten tulosten sokkoutettuun keskitettyyn arviointiin.
Taulukko 4: Tehon päätetapahtumat RET-mutaatiopositiivisilla potilailla
| Tehon päätetapahtuma (vandetanibi vs. lumelääke) | Potilaat, joilla on RET-mutaatio (n = 239) |
| Objektiivinen hoitovaste | 51,7 % vs 14,9 % |
Tehon päätetapahtuma PFS HR (95 %:n luottamusväli) | 0,46 (0,29; 0,74) |
| 2 vuoden PFS-osuus | 55,7 % vs 40,1 % |
Kliininen teho pediatrisilla potilailla:
Vandetanibin vaikutuksia arvioitiin yhdessä tutkimuskeskuksessa toteutetussa vaiheen I/II avoimessa yhden hoitoryhmän tutkimuksessa (IRUSZACT0098-tutkimus), johon osallistui 16 potilasta, jotka eivät soveltuneet leikkaushoitoon ja joilla oli paikallisesti edennyt tai levinnyt perinnöllinen medullaarinen kilpirauhaskarsinooma. Potilaiden ominaisuudet tutkimuksen alussa olivat: keskimääräinen ikä 14,2 vuotta (vaihteluväli 9–17 vuotta), 50 % naispuolisia, 50 % miespuolisia, 93,8 % valkoihoisia, 26,7 % latinalaisamerikkalaisia ja 6,3 % mustaihoisia. Useimpien potilaiden (81,3 %) kilpirauhanen oli poistettu leikkauksella joko osittain tai kokonaan ennen tutkimuksen alkua. Vandetanibin aloitusannos oli kaikilla potilailla 100 mg/m2 vuorokaudessa lukuun ottamatta yhtä potilasta, jonka hoito aloitettiin annoksella 150 mg/m2/vrk. Kun potilaat olivat sietäneet hoitoa hyvin ensimmäiset 1 tai 2 hoitojaksoa (1 hoitojakso = 28 päivää), jäljellä olevat potilaat jatkoivat hoitoa annoksella 100 mg/m2. Ensisijainen tehon päätemuuttuja oli RECIST v. 1.0 ‑mittarilla arvioitu objektiivinen vaste (ORR). ORR oli 43,8 %, ja kaikki vasteet olivat osittaisia. Potilaista 31,3 %:lla tauti pysyi vakaana vähintään 8 viikkoa. Vasteen saaneiden määrä (Disease Control Rate, DCR), joka sisälsi parhaan vasteen saavuttaneet ja potilaat, joiden tauti pysyi vakaana yli 24 viikon ajan, oli 75,0 %. Tässä tutkimuksessa Caprelsa-valmistetta ei käytetty 5–8‑vuotiaiden potilaiden hoitoon.
Farmakokinetiikka
Imeytyminen
Suun kautta otettuna vandetanibin imeytyminen on hidasta, ja huippupitoisuus plasmassa saavutetaan tyypillisesti keskimäärin 6 tunnissa (vaihteluväli 4-10 tuntia) annoksesta. Vandetanibi kertyy noin 8–kertaisesti moniannostuksessa, ja vakaa tilaa saavutetaan noin 2 kuukaudessa.
Jakautuminen
Vandetanibi sitoutuu ihmisen seerumin albumiiniin ja happamaan alfa-1-glykoproteiiniin in vitro - proteiiniin sitoutumisen ollessa noin 90 %. Kolorektaalisyöpää sairastavien ex vivo -plasmanäytteissä vakaan tilan altistuksessa 300 mg:n päivittäisellä kerta-annoksella proteiiniin sitoutumisen keskimääräinen prosenttiosuus oli 93,7 %:a (vaihtelu 92,2 %:sta 95,7 %:iin). Vandetanibin 300 mg:n annoksen farmakokinetiikkaa kuvaa noin 7 450 l:n jakautumistilavuus.
Biotransformaatio
Suun kautta otetusta 14C-vandetanibista on mitattu muuttumatonta vandetanibia ja vandetanibin N-oksidi- ja N-desmetyylimetaboliitteja plasmasta, virtsasta ja ulosteista. Glukuronidikonjugaattia on esiintynyt vähäisenä metaboliittina vain kuonaeritteissä. N-desmetyylivandetanibia tuottaa ensisijaisesti CYP3A4, ja vandetanibi-N-oksidia muodostavat flaviinia sisältävät mono-oksigenaasientsyymit FMO1 ja FMO3. N-desmetyylivandetanibin ja vandetanibi-N-oksidin verenkierrossa olevat pitoisuudet ovat noin 11 %:a ja 1,4 %:a vandetanibin pitoisuudesta.
Eliminaatio
Vandetanibin 300 mg:n annoksen farmakokinetiikkaa kuvastavat seuraavat arvot: puhdistuma noin 13,2 l/h ja puoliintumisaika plasmassa noin 19 päivää. 21 päivän kerääntymisjakson kuluessa 14C-vandetanibin kerta-annoksesta noin 69 % voitiin jäljittää, 44 % ulosteista ja 25 % virtsasta. Annoksen erittyminen on hidasta, ja plasman puoliintumisajan perusteella erittymistä voidaan odottaa tapahtuvan vielä 21 päivän jälkeenkin.
Erityisryhmät
Munuaisten vajaatoiminta
Vapaaehtoisilla kerta-annoksena toteutettu farmakokineettinen tutkimus osoitti, että altistus vandetanibille lisääntyy (1,5-, 1,6- ja 2-kertaiseksi) lievää, keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavilla verrattuna henkilöihin, joilla on normaali munuaisten toiminta (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Maksan vajaatoiminta
Vapaaehtoisilla toteutettu farmakokineettinen kerta-annostutkimus osoitti, että maksan vajaatoiminta ei vaikuta vandetanibialtistukseen. Tietoja käytöstä maksan vajaatoimintapotilailla (seerumin bilirubiini yli 1,5 kertaa normaaliin ylärajaan nähden) on vain vähän (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Ruoan vaikutus
Ruoka ei vaikuta vandetanibialtistukseen.
Farmakokinetiikka pediatrisilla potilailla
Vandetanibin farmakokineettiset parametrit olivat 9–17‑vuotiailla medullaarista kilpirauhaskarsinoomaa sairastavilla pediatrisilla potilailla vastaavat kuin aikuisilla. Glioomaan liittyvissä käyttöaiheissa 5–8‑vuotiaiden lasten vandetanibialtistus oli verrannollinen 9–18‑vuotiaiden medullaarista kilpirauhaskarsinoomaa sairastavien potilaiden altistumiseen. Valmisteen käyttöaiheen mukainen annostus 100 mg/m2/vrk (kehon pinta-alan mukaan) aiheuttaa pediatrisille potilaille vastaavan altistuksen kuin 300 mg:n vuorokausiannos aikuisille.
Prekliiniset tiedot turvallisuudesta
Vandetanibilla ei ole osoitettu olevan mutageenisia tai klastogeenisia vaikutuksia.
Toistuvan annoksen 9 kuukautta kestäneissä toksisuustutkimuksissa havaittuja vaikutuksia olivat oksentelu, painonlasku ja ripuli koirilla sekä kasvuhäiriöt nuorilla koirilla ja rotilla, joilla kasvulevyt olivat avoimet. Rotilla havaittiin vaikutuksia hampaissa, munuaisissa ja ihossa. Näitä löydöksiä esiintyi kliinisesti merkittävien plasmapitoisuuksien yhteydessä ja niistä suurin osa oli ohimeneviä 4 viikon kuluessa annostelun lopettamisesta, ja ne johtuivat verisuonten endoteelisen kasvutekijäreseptorin (VEGFR) tai EGFR-reseptorin estosta.
Muissa tutkimuksissa havaittuja vaikutuksia ovat olleet hERG-kanavan (human ether- á-go-go related gene) esto ja QT-ajan pidentyminen koirilla. Systolisen ja diastolisen verenpaineen nousua havaittiin rotilla ja koirilla. Hiirillä vandetanibin havaittiin hidastavan mutta ei estävän haavojen paranemista. In vitro sytotoksisuusanalyysissa vandetanibilla osoitettiin olevan fototoksisia ominaisuuksia. Haavan paranemista tutkineessa eläinmallissa vandetanibia saaneilla hiirillä todettiin ihon rikkoutumisherkkyyden lisääntymistä verrokkeihin verrattaessa. Tämä viittaa siihen, että vandetanibi hidastaa mutta ei estä haavojen paranemista. Ei ole määritetty, miten pitkä aika tarvitaan vandetanibihoidon keskeyttämisen ja elektiivisen kirurgisen toimenpiteen välillä haavan heikentyneen paranemisen riskin välttämiseksi. Muutamille kliinisiin tutkimuksiin osallistuneille potilaille tehtiin kirurginen toimenpide vandetanibihoidon aikana, eikä haavan paranemiseen liittyviä komplikaatioita raportoitu.
Lisääntymistoksisuus
Urosrottien hedelmällisyystutkimuksessa ei havaittu vaikutusta paritteluun eikä hedelmällisyyslukuun, kun hoitamattomat naaraat parittelivat vandetanibia saaneiden urosten kanssa. Samassa tutkimuksessa havaittiin kuitenkin elävien alkioiden määrän vähäistä pienenemistä ja ennen kiinnittymistä tapahtuvien alkionmenetysten lisääntymistä. Naarasrottien hedelmällisyystutkimuksessa havaittiin kiimakierron epäsäännöllistymistä, raskauksien pientä vähentymistä ja alkion kiinnittymisen vähentymistä. Rottien toistuvan annoksen toksisuustutkimuksissa havaittiin munasarjojen keltarauhasten määrän vähentymistä rotilla, joille annettiin vandetanibia 1 kuukauden ajan.
Rotilla alkio-/sikiötoksisuus ilmeni sikiön menetyksenä, sikiön kehityksen hidastumisena, sydämen verisuonten anomalioina ja kallon luiden ennenaikaisena luutumisena. Rottien pre- ja postnataalia kehitystä selvittäneessä tutkimuksessa vandetanibi lisäsi sikiökuolemia ja hidasti poikasten kasvua syntymän jälkeen, kun tiineyden ja/tai imetyksen aikana käytettiin emon toksisuutta aiheuttaneita annoksia. Vandetanibi erittyi rotilla maitoon ja sitä esiintyi poikasten plasmassa imettävän emon saadessa lääkettä.
Karsinogeenisuus
Vandetanibi ei ole osoittanut mitään karsinogeenisiä ominaisuuksia rasH2-transgeenisillä hiirillä tehdyssä 6 kuukauden karsinogeenisuustutkimuksessa. Kahden vuoden karsinogeenisuustutkimusta rotilla heikensi pieni eloonjääneiden osuus suuria annoksia saaneiden naarasrottien ryhmässä ja vandetanibille altistuneiden eläimien pieni määrä; mitään karsinogeenisiä vaikutuksia ei kuitenkaan havaittu jäljelle jääneissä eläimissä
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kalsiumvetyfosfaattidihydraatti
Mikrokiteinen selluloosa
Krospovidoni (tyyppi A)
Povidoni (K 29–32)
Magnesiumstearaatti
Kalvopäällyste
Hypromelloosi
Makrogoli (300)
Titaanidioksidi (E171)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Säilytä alle 30 °C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CAPRELSA tabletti, kalvopäällysteinen
100 mg (L:ei) 30 fol (2974,27 €)
PF-selosteen tieto
PVC/PVDC-alumiinikalvoläpipainopakkaukset, jotka sisältävät 30 kalvopäällysteistä tablettia.
Valmisteen kuvaus:
Caprelsa 100 mg tabletti
Caprelsa 100 mg tabletti on pyöreä, kaksoiskupera, valkoinen kalvopäällysteinen tabletti, jossa toisella puolella painatus ″Z100″.
Caprelsa 300 mg tabletti
Caprelsa 300 mg tabletti on soikean muotoinen, kaksoiskupera, valkoinen kalvopäällysteinen tabletti, jossa toisella puolella painatus ”Z300”.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia.
Korvattavuus
CAPRELSA tabletti, kalvopäällysteinen
100 mg 30 fol
- Ei korvausta.
ATC-koodi
L01EX04
Valmisteyhteenvedon muuttamispäivämäärä
13.11.2025
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi