
BOSULIF tabletti, kalvopäällysteinen 100 mg, 400 mg, 500 mg
Vaikuttavat aineet ja niiden määrät
Bosulif 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää bosutinibimonohydraattia vastaten 100 mg bosutinibia.
Bosulif 400 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää bosutinibimonohydraattia vastaten 400 mg bosutinibia.
Bosulif 500 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää bosutinibimonohydraattia vastaten 500 mg bosutinibia.
Bosulif 50 mg kovat kapselit
Yksi kova kapseli sisältää bosutinibimonohydraattia vastaten 50 mg bosutinibia.
Bosulif 100 mg kovat kapselit
Yksi kova kapseli sisältää bosutinibimonohydraattia vastaten 100 mg bosutinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti.
Kova kapseli
Kliiniset tiedot
Käyttöaiheet
Bosulif on tarkoitettu
- aikuisille ja vähintään 6 vuoden ikäisille pediatrisille potilaille äskettäin todetun Philadelphia-kromosomipositiivisen kroonisen myelooisen leukemian (Ph+ KML) kroonisen vaiheen hoitoon
- aikuisille ja vähintään 6 vuoden ikäisille pediatrisille potilaille Philadelphia-kromosomipositiivisen kroonisen myelooisen leukemian (Ph+ KML) kroonisen vaiheen hoitoon, silloin kun potilas on aiemmin saanut hoitoa yhdellä tai useammalla tyrosiinikinaasin estäjällä (TKI) ja kun imatinibin, nilotinibin ja dasatinibin ei katsota olevan tarkoituksenmukaisia hoitovaihtoehtoja
- aikuisille potilaille Ph+ KML:n akseleraatiovaiheen ja blastikriisivaiheen hoitoon, silloin kun potilas on aiemmin saanut hoitoa tyrosiinikinaasin estäjällä (TKI) ja kun imatinibin, nilotinibin ja dasatinibin ei katsota olevan tarkoituksenmukaisia hoitovaihtoehtoja.
Ehto
Hoidon saa aloittaa kroonisen myelooisen leukemian (KML) diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Annostus
Aikuiset potilaat, joilla on äskettäintodetun Ph+ KML:n krooninen vaihe
Bosutinibin suositeltu annos on 400 mg kerran vuorokaudessa.
Aikuiset potilaat, joilla on Ph+ KML:n krooninen vaihe, akseleraatiovaihe tai blastikriisivaihe ja kun potilas on resistentti tai intolerantti aiemmalle hoidolle
Bosutinibin suositeltu annos on 500 mg kerran vuorokaudessa.
Kummankin käyttöaiheen kliinisissä tutkimuksissa bosutinibihoitoa jatkettiin taudin etenemiseen saakka tai kunnes potilas ei enää sietänyt hoitoa.
Pediatriset potilaat, joilla on äskettäintodetun Ph+ KML:n krooninen vaihe tai joilla on Ph+ KML:n krooninen vaihe ja potilas on resistentti tai intolerantti aiemmalle hoidolle
Bosutinibin suositeltu annos pediatrisille potilaille, joiden sairaus on äskettäin todettu, on 300 mg/m2 kehon pinta-alasta laskettuna suun kautta kerran vuorokaudessa ja suositeltu annos pediatrisille potilaille, jotka ovat resistenttejä tai intolerantteja aiemmille hoidoille, on 400 mg/m2 kehon pinta-alasta laskettuna suun kautta kerran vuorokaudessa; annossuositukset esitetään taulukossa 1. Haluttu annos voidaan tarvittaessa toteuttaa yhdistämällä eri vahvuisia bosutinibia sisältäviä kalvopäällysteisiä tabletteja ja/tai kovia kapseleita.
Taulukko 1 – Bosutinibiannostus pediatrisille potilaille, joilla on äskettäin todetun Ph+ KML:n krooninen vaihe tai joilla on Ph+ KML:n krooninen vaihe ja potilas on resistentti tai intolerantti aiemmalle hoidolle
Kehon pinta-ala | Suositeltu annos, äskettäin todettu sairaus | Suositeltu annos, resistentti tai intolerantti potilas |
0,55 – < 0,63 m2 | 200 mg | 250 mg |
0,63 – < 0,75 m2 | 200 mg | 300 mg |
0,75 – < 0,9 m2 | 250 mg | 350 mg |
0,9 – < 1,1 m2 | 300 mg | 400 mg |
≥ 1,1 m2 | 400 mg* | 500 mg* |
* suurin aloitusannos (vastaa suurinta aloitusannosta aikuisten käyttöaiheessa)
Lyhenteet: KML = krooninen myelooinen leukemia; Ph+ = Philadelphia-kromosomipositiivinen.
Annoksen muuttaminen
Aikuisilla KML-potilailla, jotka ovat resistenttejä tai intolerantteja aiemmalle hoidolle, annos voidaan suurentaa 600 mg:aan, jos vaste ei ole ollut tyydyttävä tai jos on viitteitä sairauden etenemisestä ja jos ei ole vaikeusasteen 3 tai 4 haittavaikutuksia eikä pitkittyviä vaikeusasteen 2 haittavaikutuksia.
Aikuisille potilaille, joilla on äskettäin todetun KML:n krooninen vaihe, annosta voidaan suurentaa 100 mg:n lisäyksinä enintään 600 mg:aan kerran vuorokaudessa, jos potilaan BCR-ABL (breakpoint cluster region Abelson) ‑transkriptimäärä ei ole ≤ 10 % kolmen kuukauden kohdalla eikä hänellä ole annoksen suurentamisajankohtana vaikeusasteen 3 tai 4 haittavaikutusta ja jos kaikki vaikeusasteen 2 ei-hematologiset toksisuudet ovat lieventyneet vähintään vaikeusasteeseen 1.
Pediatrisille potilaille, joiden kehon pinta-ala (BSA) on < 1,1 m2 ja joiden vaste on kolmen kuukauden jälkeen riittämätön, harkitaan annoksen suurentamista 50 mg:n lisäyksinä enintään maksimiannokseen, joka on korkeintaan100 mg kehon pinta-alan mukaista annosta suurempi. Pediatrisille potilaille, joiden kehon pinta-ala on ≥ 1,1 m2 ja joiden vaste on kolmen kuukauden jälkeen riittämätön, harkitaan annoksen suurentamista aikuisia koskevaa suositusta vastaavasti 100 mg:n lisäyksinä. Jos kliininen vaste on riittämätön eikä pediatrisen potilaan annosta voida enää suurentaa, hoito lopetetaan.
Maksimiannos pediatrisille aiemmin hoitoa saaneille KML-potilaille on 600 mg kerran vuorokaudessa ja äskettäin todettua KML:ää sairastaville pediatrisille potilaille 500 mg kerran vuorokaudessa.
Yli 600 mg:n vuorokausiannoksia ei ole tutkittu eikä niitä pitäisi siksi antaa.
Annosmuutokset haittavaikutusten vuoksi
Bosutinibihoito tulee keskeyttää, jos potilaalle kehittyy kliinisesti merkittävää keskivaikeaa tai vaikeaa ei-hematologista toksisuutta. Hoitoa voidaan jatkaa 100 mg pienemmällä vuorokausiannoksella, kun toksisuus on korjaantunut. Annoksen suurentamista takaisin pienentämistä edeltäneeseen kerran vuorokaudessa otettavaan annokseen tulee harkita, jos se on kliinisesti perusteltua (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaille on käytetty alle 300 mg:n vuorokausiannoksia, mutta tehoa ei ole varmistettu.
Kohonneet maksan transaminaasit: Maksan transaminaasien noustessa > 5-kertaiseksi viitevälin ylärajaan nähden, bosutinibihoito tulee keskeyttää, kunnes transaminaasit laskevat ≤ 2,5-kertaiseksi viitevälin ylärajaan nähden. Tämän jälkeen hoitoa voidaan jatkaa annoksella 400 mg kerran vuorokaudessa. Bosutinibihoidon lopettamista on harkittava, jos transaminaasien korjaantuminen vie kauemmin kuin 4 viikkoa. Bosutinibihoito tulee lopettaa, jos transaminaasit nousevat ≥ 3-kertaiseksi viitevälin ylärajaan nähden, mikäli tähän liittyy samanaikainen bilirubiinin nousu > 2-kertaiseksi viitevälin ylärajaan nähden ja alkalinen fosfataasi on < 2-kertainen viitevälin ylärajaan nähden (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ripuli: Bosutinibihoito tulee keskeyttää, jos potilaalla ilmenee vaikeusasteen 3–4 ripulia NCI Common Terminology Criteria for Adverse Events (CTCAE) -vaikeusasteluokituksen mukaan. Hoitoa voidaan jatkaa annoksella 400 mg kerran vuorokaudessa sen jälkeen, kun ripuli on lievittynyt vaikeusasteelle ≤ 1 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatrisille potilaille voidaan tehdä annosmuutokset ei-hematologisen toksisuuden vuoksi vastaavasti kuin aikuisille, mutta annosta pienennettäessä kertamuutos voi olla toisenlainen. Pediatrisille potilaille, joiden kehon pinta-ala on < 1,1 m2, harkitaan annoksen pienentämistä aluksi 50 mg:lla, minkä jälkeen sitä pienennetään 50 mg kerrallaan taulukon 2 suositusten mukaisesti, jos haittavaikutus jatkuu. Jos pediatrisen potilaan kehon pinta-ala on ≥ 1,1 m2, annosta pienennetään vastaavasti kuin aikuisilla.
Hematologiset haittavaikutukset
Vaikea-asteisessa tai pitkittyvässä neutropeniassa ja trombosytopeniassa annosta suositellaan pienentämään taulukossa 2 kuvatun mukaisesti.
Taulukko 2 – Aikuisten ja pediatristen potilaiden annoksen muuttaminen neutropenian ja trombosytopenian ilmaantuessa
B-Neut < 1,0 x 109/l ja/tai B-Trom < 50 x 109/l | Keskeytä bosutinibihoito, kunnes B-Neut on ≥ 1,0 x 109/l ja B-Trom ≥ 50 x 109/l. Aloita bosutinibihoito uudelleen samalla annoksella, jos arvot korjaantuvat enintään 2 viikon hoitotauon aikana. Jos veriarvot pysyvät alhaisina yli 2 viikkoa, arvojen korjaannuttua pienennä aikuisten potilaiden ja pediatristen potilaiden, joiden kehon pinta-ala on ≥ 1,1 m2, annosta 100 mg tai pediatristen potilaiden, joiden kehon pinta-ala on < 1,1 m2, annosta 50 mg, ja jatka hoitoa. Jos sytopenia uusiutuu, pienennä veriarvojen korjaannuttua aikuisten potilaiden ja pediatristen potilaiden, joiden kehon pinta-ala on ≥ 1,1 m2, annosta vielä 100 mg tai pediatristen potilaiden, joiden kehon pinta-ala on < 1,1 m2, annosta vielä 50 mg, ja jatka hoitoa. Alle 300 mg:n vuorokausiannoksia on käytetty aikuisille potilaille ja pediatrisille potilaille, joiden kehon pinta-ala on ≥ 1,1 m2, mutta tehoa ei ole varmistettu. Alle 300 mg:n/m2 annoksia on käytetty pediatrisille potilaille, mutta tehoa ei ole varmistettu. |
Annoksen jääminen ottamatta
Jos annoksen ottamatta jäämisestä on yli 12 tuntia, potilaalle ei pidä antaa lisäannosta. Potilaan pitää ottaa tavanomainen määrätty annos seuraavana päivänä.
Erityispotilasryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Iäkkäät potilaat eivät tarvitse erityisiä annossuosituksia. Koska iäkkäiden hoidosta ei ole riittävästi tietoa, varovaisuutta tulee noudattaa hoidettaessa näitä potilaita.
Munuaisten vajaatoiminta
KML-tutkimuksiin ei otettu mukaan potilaita, joiden seerumin kreatiniini oli > 1,5 kertaa viitevälin ylärajaa suurempi. Tutkimuksissa keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla havaittiin altistuksen (pitoisuus-aikakuvaajan alle jäävä pinta-ala [AUC]) suurentumista.
Äskettäintodetun Ph+ KML:n krooninen vaihe
Jos aikuisella potilaalla on keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma [ClCr] 30–50 ml/min, laskettuna Cockroft-Gaultin kaavalla), bosutinibin suositeltu annostus on 300 mg vuorokaudessa ruoan kanssa otettuna (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Jos aikuisella potilaalla on vaikea munuaisten vajaatoiminta (ClCr < 30 ml/min, laskettuna Cockroft-Gaultin kaavalla), bosutinibin suositeltu annostus on 200 mg vuorokaudessa ruoan kanssa otettuna (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Keskivaikeaa munuaisten vajaatoimintaa sairastavilla aikuisilla potilailla annoksen suurentamista 400 mg:aan vuorokaudessa ruoan kanssa otettuna ja vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla 300 mg:aan vuorokaudessa ruoan kanssa otettuna voidaan harkita, jos potilailla ei ole vaikeita eikä pitkään jatkuvia keskivaikeita haittavaikutuksia ja jos heillä ei saavuteta riittävää hematologista, sytogeneettistä tai molekulaarista vastetta.
Ph+ KML:n krooninen vaihe, akseleraatiovaihe tai blastikriisivaihe potilailla, jotka ovat resistenttejä tai intolerantteja aiemmalle hoidolle
Keskivaikeaa munuaisten vajaatoimintaa sairastavilla aikuisilla potilailla (kreatiniinipuhdistuma [ClCr] 30–50 ml/min, laskettuna Cockroft-Gaultin kaavalla) bosutinibin suositeltu annostus on 400 mg vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (ClCr < 30 ml/min, laskettuna Cockroft-Gaultin kaavalla) bosutinibin suositeltu annostus on 300 mg vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Keskivaikeaa munuaisten vajaatoimintaa sairastavilla aikuisilla potilailla annoksen suurentamista 500 mg:aan kerran vuorokaudessa ruoan kanssa otettuna ja vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla 400 mg:aan kerran vuorokaudessa ruoan kanssa otettuna voidaan harkita, jos potilailla ei ole vaikeita eikä pitkään jatkuvia keskivaikeita haittavaikutuksia ja jos heillä ei saavuteta riittävää hematologista, sytogeneettistä tai molekulaarista vastetta.
Sydänsairaudet
Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli huonossa hoitotasapainossa oleva tai merkittävä sydänsairaus (esim. äskettäinen sydäninfarkti, kongestiivinen sydämen vajaatoiminta tai epästabiilii angina pectoris). Varovaisuutta tulee noudattaa potilaalla, jolla on merkittävä sydänsairaus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Äskettäinen tai jatkuva kliinisesti merkittävä maha-suolikanavan toiminnan häiriö
Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli äskettäin ollut tai oli parhaillaan kliinisesti merkittäviä maha-suolikanavan toiminnan häiriöitä (esim. vaikea-asteista oksentelua ja/tai ripulia). Varovaisuutta tulee noudattaa potilaalla, jolla on äskettäin ollut tai on edelleen kliinisesti merkittäviä maha-suolikanavan toiminnan häiriöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Bosutinibin turvallisuutta ja tehoa alle 1 vuoden ikäisten pediatristen potilaiden äskettäin todetun tai resistentin tai intolerantin Ph+ KML:n kroonisen vaiheen hoidossa ei ole varmistettu. Tietoja ei ole saatavilla. Alle 6 vuoden ikäisistä pediatrisista potilaista saatavilla olevan tiedon perusteella ei voida antaa suosituksia annostuksesta (ks. kohta Farmakodynamiikka).
Antotapa
Bosulif otetaan suun kautta ruoan kanssa kerran vuorokaudessa (ks. kohta Farmakokinetiikka).
Kalvopäällysteiset tabletit pitää niellä kokonaisina. Kalvopäällysteisiä tabletteja ei saa paloitella, murskata, rikkoa eikä pureskella.
Kovat kapselit voidaan niellä kokonaisina. Jos potilas ei kykene nielemään kovaa kapselia kokonaisena / kovia kapseleita kokonaisina, kukin kova kapseli voidaan avata ja sen sisältö sekoittaa omenasoseeseen tai jogurttiin. Kovan kapselin sisällön sekoittaminen omenasoseeseen tai jogurttiin ei korvaa kunnollista ateriaa, ja annos pitää ottaa ruoan kanssa siedettävyyden parantamiseksi maha-suolikanavassa.
Jos valmiste sekoitetaan omenasoseeseen tai jogurttiin, potilaan pitää ottaa seos kokonaan välittömästi pureskelematta. Seosta ei pidä säilyttää myöhempää käyttöä varten. Jos kaikkea valmistettua seosta ei niellä, lisäannosta ei pidä antaa, vaan on odotettava seuraavaan päivään, jolloin hoitoa jatketaan. Antamisen helpottamiseksi suositellut omenasose- tai jogurttimäärät ovat taulukossa 3.
Taulukko 3 – Bosutinibiannos ja pehmeän ruoan tilavuudet käytettäessä kovia kapseleita
Annos | Omenasose- tai jogurttitilavuus |
200 mg | 20 ml (4 teelusikallista) |
250 mg | 25 ml (5 teelusikallista) |
300 mg | 30 ml (6 teelusikallista) |
350 mg | 30 ml (6 teelusikallista) |
400 mg | 35 ml (7 teelusikallista) |
500 mg | 45 ml (9 teelusikallista) |
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Maksan vajaatoiminta (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Varoitukset ja käyttöön liittyvät varotoimet
Maksan toiminnan poikkeavuudet
Bosutinibihoitoon voi aikuisilla ja pediatrisilla potilailla liittyä seerumin transaminaasien (alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT]) kohoamista.
Transaminaasien kohoamista on tavallisesti esiintynyt hoitojakson alkuvaiheessa (> 80 %:lla niistä potilaista, joilla esiintyi jonkinasteista transaminaasien kohoamista, arvojen kohoaminen todettiin ensimmäisen kerran 3 kuukauden sisällä bosutinibihoidon aloittamisesta). Bosutinibia saavista potilaista tulee ottaa maksan toimintakokeet ennen hoidon aloittamista ja kuukausittain 3 ensimmäisen hoitokuukauden ajan sekä kliinisen tarpeen mukaan.
Transaminaasien kohoaminen saattaa edellyttää bosutinibihoidon tilapäistä keskeyttämistä (hoidon jatkamista pienemmällä annoksella voidaan harkita, kun arvot ovat korjautuneet vaikeusasteeseen 1 tai lähtötilanteeseen) ja/tai pysyvää lopettamista. Transaminaasien kohoaminen erityisesti samanaikaisen bilirubiinin nousun kanssa saattaa olla varhainen merkki lääkeaineen aiheuttamasta maksavauriosta. Tällaiset potilaat tulee hoitaa asianmukaisesti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Ripuli ja oksentelu
Bosutinibihoitoon voi aikuisilla ja pediatrisilla potilailla liittyä ripulia ja oksentelua. Siksi potilaiden, joilla on äskettäin ollut tai on parhaillaan kliinisesti merkittäviä maha-suolikanavan toiminnan häiriöitä, tulee käyttää tätä lääkevalmistetta varoen ja ainoastaan huolellisen hyöty-riskisuhteen arvioinnin jälkeen, koska tällaisia potilaita ei otettu mukaan kliinisiin tutkimuksiin. Potilaita, joilla ilmenee ripulia ja oksentelua, tulee hoitaa tavanomaisin keinoin, kuten ripuli- tai pahoinvointilääkevalmisteella ja/tai nestekorvaushoidolla. Lisäksi ripulin ja oksentelun hoito voi edellyttää bosutinibin tilapäistä keskeyttämistä, annoksen pienentämistä ja/tai hoidon lopettamista (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Pahoinvointilääke domperidoni voi voimistaa QT-ajan (QTc) pitenemistä ja aiheuttaa kääntyvien kärkien (torsade de pointes) takykardiaa, joten domperidonin samanaikaista käyttöä on vältettävä. Sitä tulee käyttää vain, jos muut lääkkeet eivät tehoa. Tällaisissa tilanteissa riskien ja hyötyjen tapauskohtainen arviointi on välttämätöntä ja potilasta on seurattava QTc-ajan pitenemisen havaitsemiseksi.
Myelosuppressio
Bosutinibihoitoon voi aikuisilla ja pediatrisilla potilailla liittyä myelosuppressiota, jolle tyypillistä on anemia, neutropenia ja trombosytopenia. Täydellinen verenkuva tulee ottaa viikottain ensimmäisen hoitokuukauden ajan ja sen jälkeen kuukausittain, tai kliinisen tarpeen mukaan. Myelosuppression hoito saattaa edellyttää bosutinibin tilapäistä keskeyttämistä, annoksen pienentämistä ja/tai hoidon lopettamista (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Nesteretentio
Bosutinibihoitoon saattaa aikuisilla potilailla liittyä nesteen kerääntymistä elimistöön, mukaan lukien perikardiumeffuusio, pleuraeffuusio, keuhkoödeema ja/tai perifeerinen edeema. Bosutinibihoitoon saattaa pediatrisilla potilailla liittyä lieväasteista perikardiumeffuusiota ja perifeeristä edeemaa.
Potilaita tulee seurata ja hoitaa tavanomaisilla menetelmillä.
Nesteretention hoito saattaa lisäksi edellyttää bosutinibin tilapäistä keskeyttämistä, annoksen pienentämista ja/tai hoidon lopettamista (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Seerumin lipaasi
Kohonnutta seerumin lipaasia on havaittu. Varovaisuutta suositellaan noudattamaan hoidettaessa potilaita, joilla on aiemmin ollut haimatulehdus. Jos kohonneeseen lipaasiin liittyy vatsaoireita, bosutinibihoito tulee keskeyttää ja harkita asianmukaisia diagnostisia toimenpiteitä haimatulehduksen poissulkemiseksi (ks. kohta Annostus ja antotapa).
Infektiot
Bosutinibi saattaa altistaa potilaan bakteeri-, sieni-, virus- tai alkueläininfektioille.
Kardiovaskulaarinen toksisuus
Bosulif voi aiheuttaa kardiovaskulaarista toksisuutta, mukaan lukien sydämen vajaatoimintaa ja iskeemisiä sydäntapahtumia. Sydämen vajaatoimintatapahtumia esiintyi yleisemmin aiemmin hoidetuilla potilailla kuin potilailla, joilla oli äskettäin todettu krooninen myelooinen leukemia. Ne olivat yleisempiä iäkkäillä potilailla tai potilailla, joilla oli riskitekijöitä, kuten anamneesissa sydämen vajaatoiminta. Iskeemisiä sydäntapahtumia esiintyi sekä aiemmin hoidetuilla potilailla että potilailla, joilla oli äskettäin todettu krooninen myelooinen leukemia. Ne olivat yleisempiä potilailla, joilla oli sepelvaltimotaudin riskitekijöitä, kuten anamneesissa diabetes, painoindeksi yli 30, hypertensio tai verisuonisairauksia.
Potilaita on seurattava sydämen vajaatoiminnan ja sydänlihasiskemian merkkien ja oireiden havaitsemiseksi ja hoidettava kliinisen tarpeen mukaan. Kardiovaskulaarista toksisuutta voidaan hoitaa myös keskeyttämällä hoito, pienentämällä annosta ja/tai lopettamalla bosutinibihoito.
Rytmihäiriöitä aiheuttava potentiaali
QTc-ajan pitenemistä ilman siihen liittyviä rytmihäiriöitä on havaittu. Varovaisuutta tulee noudattaa annettaessa bosutinibia potilaille, joilla on aiemmin esiintynyt tai joilla on alttius QTc-ajan pitenemiseen, joilla on huonossa hoitotasapainossa oleva tai merkittävä sydänsairaus, kuten äskettäinen sydäninfarkti, kongestiivinen sydämen vajaatoiminta, epästabiili angina pectoris tai kliinisesti merkittävä bradykardia, tai jotka käyttävät QTc-aikaa tunnetusti pidentäviä lääkevalmisteita (esim. rytmihäiriölääkkeitä ja muita QTc-aikaa mahdollisesti pidentäviä lääkeaineita, ks. kohta Yhteisvaikutukset). Hypokalemia ja hypomagnesemia saattavat voimistaa tätä vaikutusta.
QTc-aikaan kohdistuvia vaikutuksia tulee seurata ja lähtötilanteen sydänfilmi (EKG) on suositeltavaa rekisteröidä ennen bosutinibin aloittamista sekä kliinisen tarpeen mukaan. Hypokalemia tai hypomagnesemia on korjattava ennen bosutinibin antoa, ja kalium- ja magnesiumarvoja tulee seurata määräajoin hoidon aikana.
Munuaisten vajaatoiminta
Bosutinibihoito voi aiheuttaa munuaisten toiminnan kliinisesti merkittävän heikentymisen kroonista myelooista leukemiaa (KML) sairastavilla aikuisilla ja pediatrisilla potilailla. Kliinisissä lääketutkimuksissa bosutinibilla hoidetuilla potilailla on havaittu laskennallisen glomerulusten suodatusnopeuden (eGFR) pienenemistä ajan kuluessa (ks. kohta Haittavaikutukset).
On tärkeää arvioida munuaisten toiminta ennen hoidon aloittamista ja seurata sitä tarkoin bosutinibihoidon aikana. Erityistä huomiota tulee kiinnittää potilaisiin, joiden munuaisten toiminta on jo ennalta heikentynyt tai joilla on munuaisten toimintahäiriön riskitekijöitä, mukaan lukien sellaisten lääkevalmisteiden samanaikainen käyttö, jotka voivat mahdollisesti aiheuttaa munuaistoksisuutta, kuten diureetit, angiotensiinikonvertaasientsyymin estäjät (ACE-estäjät), angiotensiinireseptorin salpaajat ja tulehduskipulääkkeet (NSAIDit).
Munuaisten vajaatoimintaa selvittävässä tutkimuksessa bosutinibialtistukset kasvoivat tutkittavilla, joiden munuaisten toiminta oli kohtalaisesti tai vaikeasti heikentynyt. Annoksen pienentämistä suositellaan potilaille, joilla on keskivaikea tai vaikea munuaisten vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
KML-tutkimuksiin ei otettu mukaan potilaita, joiden seerumin kreatiniini oli > 1,5 kertaa viitevälin ylärajaa suurempi (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Kliinisiä tietoja on hyvin niukasti (n = 3) sellaisista KML-potilaista, joilla on keskivaikea munuaisten vajaatoiminta ja jotka saivat bosutinibia suurennetulla 600 mg:n annostuksella.
Aasialaiset potilaat
Populaatiofarmakokineettisten analyysien perusteella aasialaisilla puhdistuma oli pienempi, mikä johti lisääntyneeseen altistukseen. Siksi näitä potilaita tulee seurata tarkoin haittavaikutusten varalta erityisesti, jos annostusta suurennetaan.
Vakavat ihoreaktiot
Bosutinibi voi aiheuttaa vakavia ihoreaktioita, kuten Stevens-Johnsonin oireyhtymää ja toksista epidermaalista nekrolyysia. Bosutinibin käyttö on lopetettava pysyvästi potilaalla, jolla ilmenee vakava ihoreaktio hoidon aikana.
Tuumorilyysioireyhtymä
Koska tuumorilyysioireyhtymän kehittyminen on mahdollista, on suositeltavaa, että kliinisesti merkittävä nestevajaus ja suurentuneet virtsahappopitoisuudet korjataan ennen bosutinibihoidon aloittamista (ks. kohta Haittavaikutukset).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n (HBV) uudelleen aktivoitumista on tapahtunut kyseisen viruksen pysyvillä kantajilla sen jälkeen, kun potilas on saanut BCR-ABL-tyrosiinikinaasin estäjiä. Tämä aiheutti joissakin tapauksissa akuuttia maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan.
Potilaat on testattava HBV-infektion varalta ennen bosutinibihoidon aloittamista. Maksasairauksien ja HBV:n hoitoon perehtyneitä asiantuntijoita on kuultava ennen hoidon aloittamista, jos potilaan HBV-serologia on positiivinen (mukaan lukien potilaat, joilla sairaus on aktiivinen) ja jos potilas saa positiivisen HBV-testituloksen hoidon aikana. HBV:n kantajia, jotka tarvitsevat bosutinibihoitoa, on seurattava tarkasti aktiivisen HBV-infektion oireiden varalta koko hoidon ajan ja useita kuukausia hoidon jälkeen (ks. kohta Haittavaikutukset).
Valoherkkyys
Altistumista suoralle auringonvalolle tai ultravioletti (UV) -säteilylle tulisi välttää tai minimoida, koska bosutinibihoitoon liittyy valoherkkyyden riski. Potilaita tulee neuvoa käyttämään peittävää vaatetusta ja aurinkovoidetta, jossa on korkea auringonsuojakerroin (SPF, Sun Protection Factor).
Sytokromi P450 (CYP)3A:n estäjät
Bosutinibin samanaikaista käyttöä voimakkaiden tai kohtalaisten CYP3A:n estäjien kanssa tulee välttää, koska bosutinibipitoisuus plasmassa suurenee (ks. kohta Yhteisvaikutukset).
Jos mahdollista, samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei estä CYP3A:ta tai estää sitä mahdollisimman vähän.
Jos bosutinibihoidon aikana on välttämätöntä käyttää voimakasta tai kohtalaista CYP3A:n estäjää, bosutinibihoidon keskeyttämistä tai sen annoksen pienentämistä tulisi harkita.
CYP3A:n induktorit
Bosutinibin samanaikaista käyttöä voimakkaiden tai kohtalaisten CYP3A:n induktorien kanssa tulee välttää, koska bosutinibipitoisuus plasmassa pienenee (ks. kohta Yhteisvaikutukset).
Ruoan vaikutus
Greippiä sisältäviä tuotteita, kuten greippimehua, ja muita CYP3A:ta tunnetusti estäviä ruoka-aineita tulee välttää (ks. kohta Yhteisvaikutukset).
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 100 mg:n, 400 mg:n tai 500 mg:n kalvopäällysteinen tabletti tai per 50 mg:n tai 100 mg:n kova kapseli. Potilaille, joilla on ruokavalion natriumrajoitus, voidaan sanoa, että tämä lääkevalmiste on ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus bosutinibiin
CYP3A:n estäjät
Bosutinibin samanaikaista käyttöä voimakkaiden CYP3A:n estäjien (mm. itrakonatsoli, ketokonatsoli, posakonatsoli, vorikonatsoli, klaritromysiini, telitromysiini, nefatsodoni, mibefradiili, indinaviiri, lopinaviiri/ritonaviiri, nelfinaviiri, ritonaviiri, sakinaviiri, bosepreviiri, telapreviiri, greippihedelmävalmisteet, mukaan lukien greippimehu) tai kohtalaisten CYP3A:n estäjien (mm. flukonatsoli, siprofloksasiini, erytromysiini, diltiatseemi, verapamiili, amprenaviiri, atatsanaviiri, darunaviiri/ritonaviiri, fosamprenaviiri, aprepitantti, kritsotinibi, imatinibi) kanssa tulisi välttää, koska bosutinibipitoisuus plasmassa suurenee.
Varovaisuutta on noudatettava heikkojen CYP3A:n estäjien ja bosutinibin samanaikaisessa käytössä.
Jos mahdollista, samanaikaiseen käyttöön suositellaan valitsemaan lääkevalmiste, joka ei estä CYP3A-entsyymejä tai estää niitä mahdollisimman vähän.
Jos bosutinibihoidon aikana on välttämätöntä käyttää voimakasta tai kohtalaista CYP3A:n estäjää, bosutinibin annon keskeyttämistä tai sen annoksen pienentämistä tulisi harkita.
Kun tutkimuksessa 24 terveelle koehenkilölle annettiin paastotilassa viisi 400 mg:n vuorokausiannosta ketokonatsolia (voimakas CYP3A:n estäjä) samanaikaisesti yhden 100 mg:n bosutinibikerta-annoksen kanssa, ketokonatsoli suurensi bosutinibin huippupitoisuuden (Cmax) 5,2‑kertaiseksi ja bosutinibin AUC-arvon plasmassa 8,6‑kertaiseksi verrattuna bosutinibin käyttöön yksinään.
Kun tutkimuksessa 20 terveelle koehenkilölle annettiin aterian jälkeen yksi 125 mg:n kerta-annos aprepitanttia (kohtalainen CYP3A:n estäjä) samanaikaisesti yhden 500 mg:n bosutinibikerta-annoksen kanssa, aprepitantti suurensi bosutinibin huippupitoisuuden (Cmax) 1,5‑kertaiseksi ja bosutinibin AUC-arvon plasmassa 2,0‑kertaiseksi verrattuna bosutinibin käyttöön yksinään.
CYP3A:n induktorit
Bosutinibin samanaikaista käyttöä voimakkaiden CYP3A:n induktorien (mm. karbamatsepiini, fenytoiini, rifampisiini, mäkikuisma) tai kohtalaisten CYP3A:n induktorien (mm. bosentaani, efavirentsi, etraviriini, modafiniili, nafsilliini) kanssa tulisi välttää, koska bosutinibin pitoisuus plasmassa pienenee.
Kun bosutinibia annettiin samanaikaisesti rifampisiinin kanssa, bosutinibialtistus väheni huomattavasti. Siksi bosutinibin annoksen suurentaminen samanaikaisessa käytössä voimakkaiden tai kohtalaisten CYP3A-induktorien kanssa ei todennäköisesti kompensoi riittävästi vähentynyttä altistusta.
Varovaisuutta on noudatettava heikkojen CYP3A:n induktorien ja bosutinibin samanaikaisessa käytössä.
Kun bosutinibikerta-annos annettiin samanaikaisesti kuuden rifampisiinin 600 mg:n vuorokausiannoksen kanssa, 24 terveen koehenkilön ruokailun jälkeinen bosutinibialtistus (plasman Cmax ja AUC) väheni 14 %:iin (Cmax) ja 6 %:iin (AUC) arvoista, jotka todettiin annettaessa 500 mg:n bosutinibiannoksia yksinään.
Protonipumpun estäjät
Bosutinibin ja protonipumpun estäjien samanaikaisessa käytössä tulee noudattaa varovaisuutta. Vaihtoehtoina protonipumpun estäjille tulee harkita lyhytvaikutteisia antasideja. Lisäksi bosutinibi ja antasidit tulee ottaa eri aikaan (esim. bosutinibi otetaan aamulla ja antasidit illalla) aina, kun mahdollista. Bosutinibin vesiliukoisuus on pH-riippuvainen in vitro. Kun tutkimuksessa annettiin 24 terveelle koehenkilölle paastotilassa suun kautta kerta-annos bosutinibia (400 mg) samanaikaisesti suun kautta annettujen toistuvien lansopratsoliannosten (60 mg) kanssa, bosutinibin Cmax pieneni 54 %:iin ja AUC pieneni 74 %:iin arvoista, jotka todettiin annettaessa bosutinibia (400 mg) yksinään.
Bosutinibin vaikutus muihin lääkevalmisteisiin
Kun tutkimuksessa 27 terveelle koehenkilölle annettiin aterian jälkeen yksi 500 mg:n kerta-annos bosutinibia samanaikaisesti yhden 150 mg:n dabigatraanieteksilaattimesilaattikerta-annoksen (P-glykoproteiinin [P-gp:n] substraatti) kanssa, bosutinibi ei suurentanut dabigatraanin huippupitoisuutta (Cmax) tai AUC-arvoa plasmassa verrattuna dabigatraanieteksilaattimesilaatin käyttöön yksinään. Tutkimustulokset osoittavat, että bosutinibilla ei ole kliinisesti merkittävää P-gp:n estovaikutusta.
In vitro -tutkimuksen perusteella on epätodennäköistä, että bosutinibi aiheuttaisi terapeuttisina pitoisuuksina CYP1A2:n, CYP2B6:n, CYP2C9:n, CYP2C19:n ja CYP3A4:n induktiota ja siitä johtuvia lääkeaineyhteisvaikutuksia.
In vitro -tutkimusten perusteella on epätodennäköistä, että bosutinibi estäisi terapeuttisina pitoisuuksina CYP1A2:n, CYP2A6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n, CYP2D6:n tai CYP3A4/5:n lääkeainemetaboliaa ja aiheuttaisi siitä johtuvia lääkeaineyhteisvaikutuksia.
In vitro -tutkimusten perusteella bosutinibin rintasyövän resistenssiproteiinia (BCRP, systeemisesti), orgaanisten anionien kuljettajapolypeptidia (OATP)1B1, OATP1B3:a, orgaanisten anionien kuljettajaa (OAT)1, OAT3:a tai orgaanisten kationien kuljettajaa (OCT)2 estävä potentiaali kliinisesti merkittävinä pitoisuuksina on vähäinen. Bosutinibi mahdollisesti estää BCRP:tä maha-suolikanavassa ja OCT1:tä.
Rytmihäiriölääkkeet ja muut QT-aikaa mahdollisesti pidentävät aineet
Bosutinibia tulee käyttää varoen potilailla, joille on kehittynyt tai saattaa kehittyä QT-ajan pitenemistä, mukaan lukien potilaat, jotka käyttävät rytmihäiriölääkkeitä (esim. amiodaronia, disopyramidia, prokaiiniamidia, kinidiiniä ja sotalolia) tai muita mahdollisesti QT-ajan pitenemistä aiheuttavia lääkevalmisteita (esim. klorokiini, halofantriini, klaritromysiini, domperidoni, haloperidoli, metadoni ja moksifloksasiini) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/ehkäisy
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä bosutinibihoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen jälkeen ja välttämään raskaaksi tuloa bosutinibihoidon aikana. Lisäksi potilaalle tulee kertoa, että oksentelu tai ripuli saattaa heikentää ehkäisytablettien tehoa, koska ne eivät imeydy täydellisesti.
Raskaus
On vain vähän tietoja bosutinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Bosutinibin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä. Jos bosutinibia käytetään raskauden aikana tai jos potilas tulee raskaaksi hoidon aikana, hänelle on kerrottava sikiölle mahdollisesti aiheutuvasta vaarasta.
Imetys
Ei tiedetä, erittyvätkö bosutinibi ja sen metaboliitit ihmisen rintamaitoon. Rotilla tehty tutkimus [14C]-radioisotooppileimatulla bosutinibilla osoitti bosutinibista peräisin olevaa radioaktiivisuutta erittyneen nisämaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imeväiseen kohdistuvia riskejä ei voida poissulkea. Rintaruokinta on lopetettava bosutinibihoidon ajaksi.
Hedelmällisyys
Bosutinibi saattaa non-kliinisten löydösten perusteella heikentää ihmisen lisääntymistoimintoja ja hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta). Miehet tulisi ohjeistaa hakeutumaan siemennesteen talteenottoa koskevaan neuvontaan ennen kuin bosutinibihoito aloitetaan, koska hoito voi heikentää hedelmällisyyttä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Bosutinibilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Jos bosutinibia käyttävällä potilaalla esiintyy huimausta, väsymystä, näkökyvyn heikkenemistä tai muita haittavaikutuksia, jotka saattavat vaikuttaa auton ajamiseen ja koneiden käyttämiseen turvallisesti, hänen on vältettävä näitä toimia kunnes haittavaikutus on hävinnyt.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yhteensä 1 372 aikuista leukemiapotilasta sai vähintään yhden bosutinibikerta-annoksen. Hoidon mediaanikesto oli 26,30 kuukautta (vaihteluväli: 0,03–170,49 kuukautta). Näillä potilailla oli joko äskettäin todetun KML:n krooninen vaihe tai he eivät sietäneet aiempaa KML:n krooniseen vaiheeseen, akseleraatiovaiheeseen tai blastikriisivaiheeseen tai Ph+ akuuttiin lymfaattiseen leukemiaan (ALL) annettua hoitoa tai heidän tautinsa ei vastannut aiempaan hoitoon. Turvallisuusanalyyseihin sisältyi tietoja päättyneestä jatkotutkimuksesta.
1 349 potilaalla (98,3 %) raportoitiin vähintään yksi minkä tahansa vaikeusasteen haittavaikutus. Yleisimmin (≥ 20 %:lla potilaista) raportoituja haittavaikutuksia olivat ripuli (80,4 %), pahoinvointi (41,5 %), vatsakipu (35,6 %), trombosytopenia (34,4 %), oksentelu (33,7 %), ihottuma (32,8 %), suurentunut ALAT (28,0 %), anemia (27,2 %), kuume (23,4 %), suurentunut ASAT (22,5 %), väsymys (32,0 %) ja päänsärky (20,3 %). 943 potilaalla (68,7 %) raportoitiin vähintään yksi vaikeusasteen 3 tai 4 haittavaikutus. Vaikeusasteen 3 tai 4 haittavaikutuksia, joita raportoitiin ≥ 5 %:lla potilaista, olivat trombosytopenia (19,7 %), suurentunut ALAT (14,6 %), neutropenia (10,6 %), ripuli (10,6 %), anemia (10,3 %), suurentunut lipaasi (10,1 %), suurentunut ASAT (6,7 %) ja ihottuma (5,0 %).
Haittavaikutustaulukko
Haittavaikutukset on lueteltu elinjärjestelmän ja esiintyvyyden mukaan. Esiintymistiheyksiksi on määritelty: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4 – Bosutinibin haittavaikutukset | |
Infektiot | |
Hyvin yleinen | Nasofaryngiitti Hengitystieinfektioa |
Yleinen | Influenssab Keuhkokuumec Keuhkoputkitulehdus |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
Melko harvinainen | Tuumorilyysioireyhtymä** |
Veri ja imukudos | |
Hyvin yleinen | Trombosytopeniad Anemiae Neutropeniaf |
Yleinen | Leukopeniag |
Melko harvinainen | Kuumeinen neutropenia Granulosytopenia |
Immuunijärjestelmä | |
Yleinen | Lääkeaineyliherkkyys |
Melko harvinainen | Anafylaktinen sokki |
Aineenvaihdunta ja ravitsemus | |
Hyvin yleinen | Heikentynyt ruokahalu |
Yleinen | Hypofosfatemiah Elimistön kuivuminen Hyperkalemiai |
Hermosto | |
Hyvin yleinen | Päänsärky Huimaus |
Yleinen | Makuaistin häiriöt |
Kuulo ja tasapainoelin | |
Yleinen | Tinnitus |
Sydän | |
Yleinen | Perikardiumeffuusio, sydämen vajaatoiminta j, iskeeminen sydäntapahtumak |
Melko harvinainen | Perikardiitti |
Verisuonisto | |
Yleinen | Hypertensio j |
Hengityselimet, rintakehä ja välikarsina | |
Hyvin yleinen | Yskä Hengenahdistus Pleuraeffuusio |
Yleinen | Keuhkoverenpainetautim |
Melko harvinainen | Hengitysvajaus Akuutti keuhkoedeeman |
Tuntematon | Interstitiaalinen keuhkosairaus |
Ruoansulatuselimistö | |
Hyvin yleinen | Ripuli Pahoinvointi Vatsakipuo Oksentelu |
Yleinen | Gastriitti Maha-suolikanavan verenvuotop Akuutti haimatulehdusq |
Maksa ja sappi | |
Yleinen | Epänormaali maksan toimintar Maksatoksisuuss |
Melko harvinainen | Maksavauriot |
Iho ja ihonalainen kudos | |
Hyvin yleinen | Ihottumau |
Yleinen | Kutina Akne Urtikaria Valoherkkyysreaktiov |
Melko harvinainen | Lääkeaineihottuma Hilseilevä ihottuma Erythema multiforme Ihovaskuliitti** |
Tuntematon | Stevens‑Johnsonin oireyhtymä**, toksinen epidermaalinen nekrolyysi** |
Luusto, lihakset ja sidekudos | |
Hyvin yleinen | Nivelkipu, selkäkipu |
Yleinen | Lihaskipu |
Munuaiset ja virtsatiet | |
Yleinen | Akuutti munuaisten vajaatoiminta Munuaisten vajaatoiminta Munuaisfunktion huononeminen |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleinen | Väsymysw Kuume Turvotusx |
Yleinen | Rintakipuy Kipu |
Tutkimukset | |
Hyvin yleinen | Suurentunut alaniiniaminotransferaasiz Suurentunut aspartaattiaminotransferaasi Suurentunut lipaasiaa Suurentunut veren kreatiniini |
Yleinen | Suurentunut amylaasibb Suurentunut veren kreatiinikinaasi Suurentunut veren bilirubiinicc Suurentunut gammaglutamyylitransferaasi EKG:ssä todettu pidentynyt QT‑aikadd |
a Hengitystieinfektio käsittää seuraavat: alahengitystieinfektio, hengitystieinfektio, hengitysteiden virusinfektio, ylähengitystieinfektio, virusperäinen ylähengitystieinfektio
b Influenssa käsittää seuraavat: H1N1-influenssa, influenssa
c Keuhkokuume käsittää seuraavat: atyyppinen keuhkokuume, keuhkokuume, bakteerikeuhkokuume, sienikeuhkokuume, nekrotisoiva keuhkokuume, streptokokkikeuhkokuume
d Trombosytopenia käsittää seuraavat: trombosyyttimäärän pieneneminen, trombosytopenia
e Anemia käsittää seuraavat: anemia, hemoglobiiniarvon pieneneminen, punasolujen määrän pieneneminen
f Neutropenia käsittää seuraavat: neutropenia, neutrofiilimäärän pieneneminen
g Leukopenia käsittää seuraavat: leukopenia, valkosolumäärän pieneneminen
h Hypofosfatemia käsittää seuraavat: pienentynyt veren fosforipitoisuus, hypofosfatemia
i Hyperkalemia käsittää seuraavat: suurentunut veren kaliumpitoisuus, hyperkalemia
j Sydämen vajaatoiminta käsittää seuraavat: sydämen vajaatoiminta, akuutti sydämen vajaatoiminta, krooninen sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, sydänperäinen sokki, kardiorenaalinen oireyhtymä, pienentynyt ejektiofraktio, sydämen vasemman kammion vajaatoiminta
k Iskeemiset sydäntapahtumat käsittää seuraavat: akuutti sepelvaltimo-oireyhtymä, akuutti sydäninfarkti, angina pectoris, epästabiili angina pectoris, sepelvaltimokovettuma, sepelvaltimotauti , sepelvaltimotukos, sepelvaltimon ahtauma, sydäninfarkti, sydänlihasiskemia, suurentunut troponiinipitoisuus
l Hypertensio käsittää seuraavat: kohonnut verenpaine, kohonnut systolinen verenpaine, primaarinen hypertensio, hypertensio, hypertensiivinen kriisi
m Keuhkoverenpainetauti käsittää seuraavat: keuhkovaltimoiden verenpainetauti, kohonnut keuhkovaltimoiden verenpaine, keuhkoverenpainetauti
n Akuutti keuhkoedeema käsittää seuraavat: akuutti keuhkoedeema, keuhkoedeema
o Vatsakipu käsittää seuraavat: epämukava tunne vatsassa, vatsakipu, alavatsakipu, ylävatsakipu, vatsan arkuus, ruoansulatuselimistön kipu
p Maha-suolikanavan verenvuoto käsittää seuraavat: verenvuoto peräaukosta, mahalaukun verenvuoto, maha-suolikanavan verenvuoto, suoliston verenvuoto, alemman maha-suolikanavan verenvuoto, peräsuolen verenvuoto, ylemmän maha-suolikanavan verenvuoto
q Akuutti haimatulehdus käsittää seuraavat: haimatulehdus, akuutti haimatulehdus
r Epänormaali maksan toiminta käsittää seuraavat: suurentuneet maksaentsyymit, epänormaali maksan toiminta, poikkeavuudet maksan toimintakokeissa, maksan toimintakoearvojen suureneminen, suurentuneet transaminaasit
s Maksatoksisuus käsittää seuraavat: hepatiitti, toksinen hepatiitti, maksatoksisuus, maksahäiriö
t Maksavaurio käsittää seuraavat: lääkkeen aiheuttama maksavaurio, maksasoluvaurio, maksavaurio
u Ihottuma käsittää seuraavat: ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiseva ihottuma
v Valoherkkyysreaktio käsittää seuraavat: valoherkkyysreaktio, monimuotoinen valoihottuma
w Väsymys käsittää seuraavat: astenia, väsymys, huonovointisuus
x Turvotus käsittää seuraavat: silmäluomen turvotus, kasvojen turvotus, yleistynyt turvotus, paikallinen turvotus, turvotus, raajojen turvotus, periorbitaalinen turvotus, periorbitaalinen turpoaminen, raajojen turpoaminen, turpoaminen, silmäluomen turpoaminen
y Rintakipu käsittää seuraavat: epämukava tunne rinnassa, rintakipu
z Suurentunut alaniiniaminotransferaasi käsittää seuraavat: poikkeava alaniiniaminotransferaasi, suurentunut alaniiniaminotransferaasi
aa Suurentunut lipaasi käsittää seuraavat: hyperlipasemia, suurentunut lipaasi
bb Suurentunut amylaasi käsittää seuraavat: suurentunut amylaasi, hyperamylasemia
cc Suurentunut veren bilirubiini käsittää seuraavat: suurentunut konjugoitunut bilirubiini, suurentunut veren bilirubiini, suurentunut veren konjugoitumaton bilirubiini, hyperbilirubinemia
dd EKG:ssä todettu pidentynyt QT-aika käsittää seuraavat: EKG:ssä todettu pidentynyt QT-aika, pitkän QT-ajan oireyhtymä
** Haittavaikutus tunnistettu aikuisilla markkinoille tulon jälkeen.
Pediatriset potilaat
Yhteensä 55 iältään ≥ 1-vuotiasta pediatrista potilasta sai faasin I/II kansainvälisessä, yhden hoitohaaran avoimessa BCHILD-monikeskustutkimuksessa vähintään yhden bosutinibiannoksen. Hoidon mediaanikesto oli 13,5 kuukautta (vaihteluväli: 0,2–60,9 kuukautta). Näillä potilailla oli joko äskettäin todetun Ph+ KML:n krooninen vaihe tai resistentti tai intolerantti Ph+ KML:n krooninen vaihe tai KML:n akseleraatiovaihe.
Pediatrisista potilaista 54:llä (98,2 %) raportoitiin vähintään yksi minkä tahansa vaikeusasteen toksisuutta koskeva haittavaikutus. Yleisimmin raportoituja haittavaikutuksia olivat ripuli (82 %), vatsakipu (65 %), oksentelu (56 %), pahoinvointi (51 %), ihottuma (36 %), väsymys (35 %), trombosytopenia (35 %), päänsärky (33 %), kuume (33 %), suurentunut ALAT-arvo (29 %) ja heikentynyt ruokahalu (24 %).
Yleisimmin raportoituja vaikeusasteen 3 tai vaikeusasteen 4 haittavaikutuksia olivat trombosytopenia (18 %), suurentunut ALAT-arvo (15 %) ja ripuli (13 %).
Veri ja imukudos
BCHILD-tutkimuksessa, johon osallistui 55 pediatrista potilasta, ilmenneitä vereen ja imukudokseen liittyneitä tapahtumia olivat trombosytopenia 19 potilaalla (34,5 %), anemia 10 potilaalla (18,2 %) ja neutropenia 7 potilaalla (12,7 %). Yksi potilas lopetti hoidon vaikeusasteen 4 neutropenian vuoksi. Niistä potilaista, joilla oli vereen ja imukudokseen liittyneitä tapahtumia, 37,5 % hoidettiin keskeyttämällä hoito ja 16,7 %:lla annosta oli tarpeen pienentää. Niistä potilaista, joiden hoito keskeytettiin, yhdelläkään tapahtuma ei uusiutunut, kun bosutinibihoitoa jatkettiin. Mediaaniaika tapahtuman ensimmäiseen ilmaantumiskertaan oli 13 vuorokautta (vaihteluväli: 1–757 vuorokautta), ja vaikeusasteen 3/4 tapahtumien kumulatiivinen mediaanikestoaika oli 16,0 (vaihteluväli: 4–47) vuorokautta.
Maksa ja sappi
55 tutkittavalla laboratoriokokeiden tulosten perusteella ALAT- ja ASAT-arvojen suurenemisen ilmaantuvuus oli 67,3 % (ALAT) ja 63,6 % (ASAT), ja 43 tutkittavalla (78,2 %) joko ALAT- tai ASAT-arvo suureni. Transaminaasiarvo kohosi useimmiten hoidon varhaisvaiheessa; niistä tutkittavista, joilla oli minkä tahansa vaikeusasteen kohonnut transaminaasiarvo, 83,7 %:lla ensimmäinen tapahtuma ilmeni kolmen kuukauden kuluessa. Mediaaniaika suurentuneen ALAT- ja ASAT-arvon ilmaantumiseen oli ALAT-arvon osalta 22,0 vuorokautta (vaihteluväli: 9–847 vuorokautta) ja ASAT-arvon osalta 18,5 vuorokautta (vaihteluväli: 9–169 vuorokautta). Vaikeusasteen 3/4 tapahtumien mediaanikestoaika oli suurentuneen ALAT-arvon osalta 18,0 vuorokautta (vaihteluväli: 2–132 vuorokautta) ja suurentuneen ASAT-arvon osalta 12 vuorokautta (vaihteluväli: 5–19 vuorokautta).
Ruoansulatuselimistö
BCHILD-tutkimuksessa todettuja ruoansulatuselimistön haittoja olivat ripuli 81,8 %:lla, oksentelu 56,4 %:lla ja pahoinvointi 50,9 %:lla 55:stä bosutinibihoitoa saaneesta pediatrisesta potilaasta. Kolme (5,5 %) potilasta lopetti bosutinibihoidon ripulin (n = 3), vatsakivun (n = 2), pahoinvoinnin (n = 1) ja/tai oksentelun (n = 1) vuoksi. Niistä pediatrisista potilaista, joilla oli ruoansulatuselimistön haittoja, 9 (19 %) potilasta hoidettiin keskeyttämällä hoito ja neljän (8,3 %) potilaan annosta oli tarpeen pienentää. Niistä 9 potilaasta, joiden hoito oli tarpeen keskeyttää, 8 (88,9 %) altistettiin hoidolle uudelleen. Näistä 55,6 %:lla altistaminen uudelleen onnistui. Mediaaniaika ripulin ilmaantumiseen oli 2 vuorokautta, ja kaikkien vaikeusasteiden ripulin mediaanikestoaika oli 2 vuorokautta.
Munuaiset
Pediatrisilla potilailla tehdyssä tutkimuksessa 45 potilaalla (82 %) yhteensä 55 potilaasta oli lähtötilanteessa normaali eGFR-arvo (≥ 90 ml/min/1,73 m2) arvioituna Bedside Schwartzin yhtälön perusteella lähtötilanteessa. Näistä 45 potilaasta 19 potilaalla (34,5 %) eGFR-arvo oli pienentynyt 13,47 kuukauden kohdalla vaikeusasteeseen 1 (60 – < 90 ml/min/1,73 m2) ja 1 potilaalla (1,8 %) vaikeusasteeseen 2 (30 – < 60 ml/min/1,73 m2). Lähtötilanteen arvoista riippumatta yhdenkään tutkittavan eGFR-arvo ei ollut lähtötilanteen jälkeen < 45 ml/min/1,73 m2.
Valikoitujen haittavaikutusten kuvaus
Veri ja imukudos
Anemiaa raportoitiin haittavaikutuksena 372 aikuisella potilaalla (27,1 %), joista 6 lopetti bosutinibihoidon anemian vuoksi. Enintään vaikeusasteen 1 toksisuutta esiintyi 95 potilaalla (25,5 %), vaikeusasteen 2 toksisuutta 135 potilaalla (36,3 %), vaikeusasteen 3 toksisuutta 113 potilaalla (30,4 %) ja vaikeusasteen 4 toksisuutta 29 potilaalla (7,8 %). Näillä potilailla mediaaniaika tapahtuman ensimmäiseen ilmaantumiskertaan oli 29 vuorokautta (vaihteluväli: 1−3 999 vuorokautta) ja mediaanikesto esiintymiskertaa kohden oli 22 vuorokautta (vaihteluväli: 1−3 682 vuorokautta).
Neutropeniaa raportoitiin haittavaikutuksena 209 aikuisella potilaalla (15,2 %), joista 19 lopetti bosutinibihoidon neutropenian vuoksi. Enintään vaikeusasteen 1 toksisuutta esiintyi 19 potilaalla (9,1 %), vaikeusasteen 2 toksisuutta 45 potilaalla (21,5 %), vaikeusasteen 3 toksisuutta 95 potilaalla (45,5 %) ja vaikeusasteen 4 toksisuutta 50 potilaalla (23,9 %). Näillä potilailla mediaaniaika tapahtuman ensimmäiseen ilmaantumiskertaan oli 56 vuorokautta (vaihteluväli: 1–1 769 vuorokautta) ja mediaanikesto esiintymiskertaa kohden oli 15 vuorokautta (vaihteluväli: 1–913 vuorokautta).
Trombosytopeniaa raportoitiin haittavaikutuksena 472 aikuisella potilaalla (34,4 %), joista 42 lopetti bosutinibin trombosytopenian vuoksi. Enintään vaikeusasteen 1 toksisuutta esiintyi 114 potilaalla (24,2 %), vaikeusasteen 2 toksisuutta 88 potilaalla (18,6 %), vaikeusasteen 3 toksisuutta 172 potilaalla (36,4 %) ja vaikeusasteen 4 toksisuutta 98 potilaalla (20,8 %). Näillä potilailla mediaaniaika tapahtuman ensimmäiseen ilmaantumiskertaan oli 28 vuorokautta (vaihteluväli: 1–1 688 vuorokautta) ja mediaanikesto esiintymiskertaa kohden oli 15 vuorokautta (vaihteluväli: 1–3 921 vuorokautta).
Maksa ja sappi
Aikuisilla potilailla, joilla raportoitiin haittavaikutuksena minkä tahansa vaikeusasteen ALAT- tai ASAT-arvon kohoamista, mediaaniaika arvon kohoamiseen oli 29 vuorokautta (vaihteluväli molempien osalta 1–3 995 vuorokautta). ALAT-arvot pysyivät koholla 17 vuorokautta (mediaani, vaihteluväli:1–1 148 vuorokautta) ja ASAT-arvot pysyivät koholla 15 vuorokautta (mediaani, vaihteluväli:1–803 vuorokautta).
Kaksi tapausta, jotka sopivat lääkkeen aiheuttamaan maksavaurioon (määritelmä ALAT- tai ASAT-arvon samanaikainen suureneminen ≥ 3‑kertaiseksi, kokonaisbilirubiinin suureneminen > 2‑kertaiseksi, ja alkalinen fosfataasi < 2‑kertainen viitevälin ylärajaan nähden), on ilmennyt ilman muuta syytä 2 aikuisella potilaalla 1 711:stä bosutinibia saaneesta potilaasta (0,1 %).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on ilmoitettu BCR-ABL-tyrosiinikinaasin estäjien käytön yhteydessä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ruoansulatuselimistö
Ripulin sai 1 103 potilasta (80,4 %), joista 14 lopetti bosutinibihoidon tämän haittatapahtuman vuoksi. Ripulin hoitoon annettiin samanaikaisia lääkevalmisteita 756 potilaalle (68,5 %). Vaikeusasteen 1 toksisuutta esiintyi 575 potilaalla (52,1 %), vaikeusasteen 2 toksisuutta 383 potilaalla (34,7 %), vaikeusasteen 3 toksisuutta 144 potilaalla (13,1 %) ja vaikeusasteen 4 haittatapahtuma yhdellä potilaalla (0,1 %). Mediaaniaika tapahtuman ensimmäiseen ilmaantumiskertaan oli 2 vuorokautta (vaihteluväli: 1–2 702 vuorokautta) ja kaikkien vaikeusasteiden ripulin mediaanikestoaika oli 2 vuorokautta (vaihteluväli: 1–4 247 vuorokautta).
Ripulin saaneista 1 103 potilaasta 218 potilaan (19,8 %) ripuli hoidettiin keskeyttämällä hoito, ja näistä 208 potilasta (95,4 %) sai uudelleen bosutinibia. Bosutinibia uudelleen saaneista potilaista 201:llä (96,6 %) ei enää esiintynyt ripulia tai heidän bosutinibihoitoaan ei keskeytetty ripulin uusiutumisen vuoksi.
Sydän
Sydämen vajaatoimintaa ilmeni 1 372 potilaan joukossa 50 (3,6 %) potilaalla ja iskeemisiä sydäntapahtumia 57 (4,2 %) potilaalla.
Seitsemällä potilaalla (0,5 %) esiintyi QTcF-ajan pitenemistä (yli 500 ms). Yhdellätoista potilaalla (0,8 %) esiintyi QTcF-ajan pitenemistä lähtötasosta > 60 ms. Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli huonossa hoitotasapainossa oleva tai merkittävä sydän- ja verisuonitauti, kuten QTc-ajan piteneminen (ks. kohdat Farmakodynamiikka ja Prekliiniset tiedot turvallisuudesta).
Munuaiset
Äskettäin todettua KML:n kroonista vaihetta sairastavilla potilailla, jotka saivat 400 mg:n annoksia, potilaiden hoidonaikaisen eGRF-arvon (laskettuna MDRD (modification of diet in renal disease) -yhtälöllä) pienenemisen mediaani lähtötilanteesta oli 11,1 ml/min/1,73 m2 yhden vuoden kohdalla ja 14,1 ml/min/1,73 m2 viiden vuoden kohdalla. Aiemmin hoitamattomilla KML-potilailla, jotka saivat 500 mg:n annoksia, potilaiden hoidonaikaisen eGFR-arvon pienenemisen mediaani oli 9,2 ml/min/1,73 m2 yhden vuoden kohdalla, 12,0 ml/min/1,73 m2 viiden vuoden kohdalla ja 16,6 ml/min/1,73 m2 kymmenen vuoden kohdalla. Aiemmin hoitoa saaneilla KML:n kroonista vaihetta ja edennyttä vaihetta sairastavilla potilailla, jotka saivat 500 mg:n annoksia, potilaiden hoidonaikaisen eGFR-arvon pienenemisen mediaani oli 7,6 ml/min/1,73 m2 yhden vuoden kohdalla, 12,3 ml/min/1,73 m2 viiden vuoden kohdalla ja 15,9 ml/min/1,73 m2 kymmenen vuoden kohdalla. Ph+ KML ‑potilailla, jotka olivat aiemmin saaneet hoitoa yhdellä tai useammalla tyrosiinikinaasin estäjällä ja jotka saivat 500 mg:n annoksia, potilaiden hoidonaikaisen eGFR-arvon pienenemisen mediaani lähtötilanteesta oli 9,2 ml/min/1,73 m2 yhden vuoden kohdalla ja 14,5 ml/min/1,73 m2 neljän vuoden kohdalla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kokemus bosutinibin yliannoksesta rajoittui kliinisissä tutkimuksissa yksittäisiin tapauksiin. Bosutinibin yliannostustapauksessa potilasta on seurattava ja hänelle on annettava asianmukaista elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EA04.
Vaikutusmekanismi
Bosutinibi kuuluu kinaasin estäjiksi kutsuttujen lääkevalmisteiden farmakologiseen luokkaan. Bosutinibi estää poikkeavaa BCR-ABL-kinaasia, joka edistää kroonista myelooista leukemiaa. Mallinnustutkimukset osoittavat, että bosutinibi sitoo BCR-ABL:n kinaasidomeenin. Bosutinibi on myös Scr-kinaasiperheen (mukaan lukien Scr, Lyn ja Hck) estäjä. Bosutinibi estää hyvin vähäisessä määrin verihiutalekasvutekijän (PDGF) reseptoria ja KIT-proto-onkogeenia (c-Kit).
Bosutinibi esti in vitro -tutkimuksissa todennettujen KML-solulinjojen, Ph+ ALL ‑solulinjojen ja potilaasta peräisin olevien primaaristen primitiivisten KML-solujen proliferaatiota ja eloonjääntiä. Bosutinibi esti 16 imatinibille resistenttiä BCR‑ABL-muotoa ilmentävää hiiren myelooista solulinjaa kaikkiaan 18:sta tällaisesta solulinjasta. Bosutinibi pienensi nude-hiirten KML-kasvainten kokoa ja esti imatinibille resistenttien BCR‑ABL-muotoa ilmentävien hiiren myelooisten kasvainten kasvua. Bosutinibi estää myös reseptorityrosiinikinaaseja c‑Fms ja EphA, B‑reseptoreja, Trk‑kinaasiperhettä, Axl‑kinaasiperhettä, Tec‑kinaasiperhettä, joitakin ErbB‑perheeseen kuuluvia kinaaseja, ei-reseptorityrosiinikinaasia Csk, Ste20-perheen seriini-treoniinikinaaseja ja kahta kalmoduliiniriippuvaista proteiinikinaasia.
Farmakodynaamiset vaikutukset
500 mg:n bosutinibiannoksen vaikutusta QTc-aikaan selvitettiin satunnaistetussa, kerta-annoksella toteutetussa, bosutinibin suhteen kaksoissokkoutetussa, lumekontrolloidussa ja avoimessa moksifloksasiinilla kontrolloidussa ristikkäistutkimuksessa terveillä tutkimushenkilöillä.
Tästä tutkimuksesta saadut tiedot osoittavat, että bosutinibi ei pidennä terveiden tutkimushenkilöiden QTc-aikaa käytettäessä 500 mg:n vuorokausiannosta ruoan kanssa, eikä myöskään olosuhteissa, joissa pitoisuus plasmassa suurenee terapeuttisia pitoisuuksia suuremmaksi. Terveille tutkimushenkilöille suun kautta annetun 500 mg:n bosutinibikerta-annoksen (terapeuttinen annos) jälkeen ja 500 mg:n bosutinibiannoksen ja 400 mg:n ketokonatsoliannoksen yhdistelmän (bosutinibin terapeuttista pitoisuutta suuremman pitoisuuden saavuttamiseksi) jälkeen QTc-ajan keskimuutoksen yksisuuntaisen 95 %:n luottamusvälin yläraja oli alle 10 ms jokaisena annoksen jälkeisenä ajankohtana eikä QTc-ajan pitenemiseen viittaavia haittatapahtumia havaittu.
Maksan vajaatoimintaa sairastavilla potilailla tehdyssä tutkimuksessa havaittiin, että maksan toiminnan heikentyessä QTc-ajan pitenemistä arvoon > 450 ms esiintyi useammin. Aiempaa hoitoa saaneilla Ph+‑leukemiaa sairastavilla potilailla, joita hoidettiin 500 mg:lla bosutinibia vaiheen 1/2 kliinisessä tutkimuksessa, QTcF-ajan pitenemistä > 60 ms lähtötilanteesta havaittiin 9 potilaalla (1,6 %) 570 potilaasta. Äskettäin todettua KML:n kroonista vaihetta sairastavilla potilailla, joita hoidettiin 400 mg:lla bosutinibia vaiheen 3 kliinisessä tutkimuksessa, QT‑aika ei pidentynyt > 60 ms lähtötilanteesta yhdelläkään bosutinibiryhmän potilaista (N = 268). Äskettäin todettua Ph+ KML:n kroonista vaihetta sairastavilla potilailla, joita hoidettiin 500 mg:lla bosutinibia vaiheen 3 kliinisessä tutkimuksessa, QTcF-ajan pitenemistä > 60 ms lähtötilanteesta havaittiin 2 potilaalla (0,8 %) 248 bosutinibia saaneesta potilaasta. Aiempaa hoitoa yhdellä tai useammalla tyrosiinikinaasin estäjällä saaneilla Ph+ KML -potilailla, joita hoidettiin 500 mg:lla bosutinibia vaiheen 4 kliinisessä tutkimuksessa (N = 163), QTcF‑aika ei pidentynyt > 60 ms lähtötilanteesta yhdelläkään potilaalla. Bosutinibin proarytmistä potentiaalia ei voida sulkea pois.
Kliininen teho
Kliininen tutkimus aikuispotilailla, joilla oli aiemmin hoitamaton kroonisen vaiheen KML
Bosutinibi 400 mg ‑tutkimus
Lääkeaineiden paremmuutta arvioineessa kaksihaaraisessa vaiheen 3 avoimessa monikeskustutkimuksessa verrattiin kerran vuorokaudessa annetun 400 mg:n bosutinibiannoksen turvallisuutta ja tehoa kerran vuorokaudessa annettuun 400 mg:n imatinibiannokseen aikuispotilailla, joilla oli äskettäin todetun Ph+ KML:n krooninen vaihe. Tutkimuksessa satunnaistettiin 536 potilasta (268 kummassakin hoitoryhmässä), joilla oli joko äskettäin todetun Ph+ tai Ph‑ KML:n krooninen vaihe (hoitoaikeen mukainen potilasjoukko, intent‑to‑treat population [ITT]). Tässä potilasjoukossa oli mukana 487 Ph+ KML:aa sairastavaa potilasta, jotka ilmensivät b2a2‑ ja/tai b3a2-transkripteja ja joilla oli lähtötilanteessa BCR‑ABL-kopioita > 0 (modifioitu hoitoaikeen mukainen [mITT] ‑potilasjoukko).
Tehon ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla voitiin osoittaa merkittävä molekulaarinen vaste (major molecular response, MMR) 12 kuukauden (48 viikon) kohdalla bosutinibiryhmässä verrattuna imatinibiryhmään mITT-potilasjoukossa. MMR:n määritelmä oli ≤ 0,1 %:n BCR‑ABL/ABL-suhde kansainvälisellä asteikolla (IS) (vastaa ≥ 3 logaritmiyksikön pienenemää vakioidusta lähtötilanteesta) ja vähintään 3 000 ABL-transkriptia keskuslaboratorion määrityksen mukaan.
Keskeiset toissijaiset päätetapahtumat olivat CCyR (täydellinen sytogeneettinen vaste) 12. kuukauteen mennessä, CCyR:n kesto, MMR:n kesto, tapahtumavapaa elinaika (event-free survival, EFS) ja kokonaiselinaika (overall survival, OS). CCyR 12. kuukauteen mennessä määriteltiin Ph+‑metafaasien puuttumiseksi raitavärjäysanalyysissa, jossa oli mukana ≥ 20 luuydinaspiraatista saatua metafaasia, tai MMR:ksi, jos riittävää sytogeneettistä arviota ei ollut saatavilla. Vain seuraavien päätetapahtumien p-arvot korjattiin monitestausta varten: MMR 12 kuukauden kohdalla ja CCyR 12. kuukauteen mennessä.
mITT-potilasjoukon taustatiedot olivat vertailukelpoisia tutkimuksen kahden hoitoryhmän välillä seuraavien ominaisuuksien suhteen: ikä (bosutinibiryhmässä mediaani-ikä oli 52 vuotta ja imatinibiryhmässä 53 vuotta, bosutinibiryhmässä 19,5 % potilaista ja imatinibiryhmässä 17,4 % potilaista oli vähintään 65‑vuotiaita), sukupuoli (naisia 42,3 % ja 44,0 %), etninen tausta (valkoihoisia 78,0 % ja 77,6 %, aasialaisia 12,2 % ja 12,4 %, tummaihoisia tai afroamerikkalaisia 4,1 % ja 4,1 %, muita 5,7 % ja 5,4 % ja 1 tuntematon imatinibiryhmässä) ja Sokal-pisteet (pieni riski 35,0 % ja 39,4 %, keskiriski 43,5 % ja 38,2 %, suuri riski 21,5 % ja 22,4 %).
Kun mITT-potilasjoukon seurantaa oli jatkettu 60 kuukautta, 60,2 % bosutinibilla hoidetuista potilaista (N = 246) ja 59,8 % imatinibilla hoidetuista potilaista (N = 239) sai edelleen ensilinjan hoitoa.
Kun mITT-potilasjoukon seurantaa oli jatkettu 60 kuukautta, 0,8 % bosutinibilla hoidetuista potilaista ja 1,7 % imatinibilla hoidetuista potilaista keskeytti hoidon KML:n edettyä akseleraatiovaiheeseen tai blastikriisivaiheeseen. KML transformoitui kuudella bosutinibipotilaalla (2,4 %) ja seitsemällä imatinibipotilaalla (2,9 %) akseleraatiovaiheeseen tai blastikriisivaiheeseen. Hoidon keskeytti suboptimaalisen vasteen tai hoidon epäonnistumisen vuoksi tutkijan arvion mukaan 5,3 % bosutinibiryhmän potilaista ja 15,5 % imatinibiryhmän potilaista. Kaksitoista bosutinibiryhmän potilasta (4,9 %) ja 14 imatinibiryhmän potilasta (5,8 %) kuoli tutkimuksen aikana. Hoitoaikeen mukaisessa (ITT, intention to treat) potilasjoukossa ei tapahtunut lisätransformoitumisia, ja ITT-potilasjoukon bosutinibiryhmässä kuoli lisäksi kaksi potilasta.
Taulukossa 5 on yhteenveto tehoa koskevista tuloksista (MMR ja CCyR).
Taulukko 5 – Yhteenveto: MMR kuukausien 12 ja 18 kohdalla ja CCyR 12. kuukauteen mennessä mITT-potilasjoukossa hoitoryhmittäin
Vaste | Bosutinibi (N = 246) | Imatinibi (N = 241) | Vetokertoimien suhde (odds ratio) (95 % CI)a |
Merkittävä molekulaarinen vaste MMR kuukauden 12 kohdalla, n (%) (95 % CI) | 116 (47,2)b (40,9, 53,4) | 89 (36,9) (30,8, 43,0) | 1,55 (1,07, 2,23) |
Yksisuuntainen p-arvo | 0,0100b | ||
MMR kuukauden 18 kohdalla, n (%) (95 % CI) | 140 (56,9) (50,7, 63,1) | 115 (47,7) (41,4, 54,0) | 1,45 (1,02, 2,07) |
Yksisuuntainen p-arvo | 0,0208c | ||
Täydellinen sytogeneettinen vaste CCyR 12. kuukauteen mennessä, n (%) (95 % CI) | 190 (77,2)b (72,0, 82,5) | 160 (66,4) (60,4, 72,4) | 1,74 (1,16, 2,61) |
Yksisuuntainen p-arvo | 0,0037b | ||
Huomaa: MMR:n määritelmä oli ≤ 0,1 %:n BCR-ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 3 000 ABL-transkriptia keskuslaboratorion arvion mukaan. Täydellisen sytogeneettisen vasteen määritelmä oli Ph+‑metafaasien puuttuminen raitavärjäysanalyysissa, jossa oli ≥ 20 luuydinaspiraatista saatua metafaasia, tai MMR, jos riittävää sytogeneettistä arviota ei ollut saatavilla.
Lyhenteet: BCR-ABL = breakpoint cluster region‑Abelson, CI = luottamusväli, CMH = Cochran‑Mantel‑Haenszel, CCyR = täydellinen sytogeneettinen vaste, mITT = modified intent-to-treat, modifioitu hoitoaikeen mukainen, MMR = merkittävä molekulaarinen vaste, N = potilaiden lukumäärä, Ph+ = Philadelphia-kromosomipositiivinen.
a Korjattu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden mukaan.
b Tilastollisesti merkitsevä vertailu ennalta määritetyllä merkitsevyystasolla; perustuu CMH-testiin, joka
on stratifioitu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden mukaan.
c Perustuu CMH-testiin, joka on stratifioitu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden
mukaan.
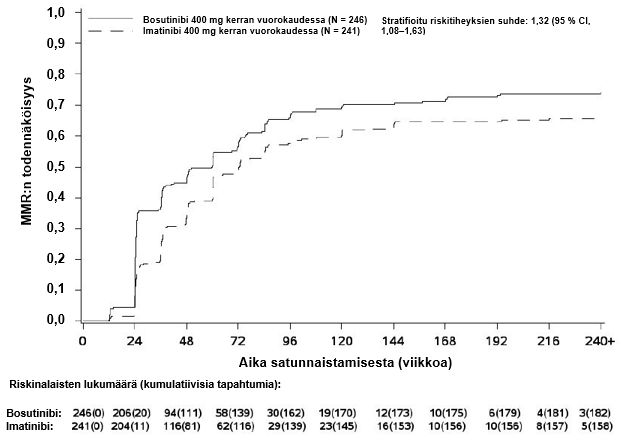
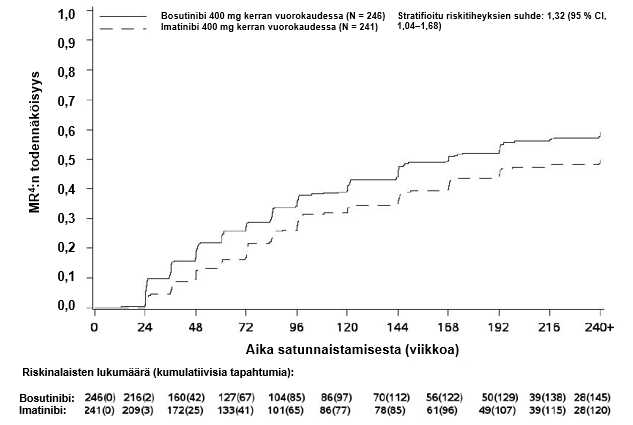
Kuukauden 12 kohdalla MR4:n saavuttaneiden osuus (määritelmä: ≤ 0,01 % BCR‑ABL [vastaa ≥ 4 logaritmiyksikön pienenemää vakioidusta lähtöarvosta] ja vähintään 9 800 ABL-transkriptia) oli mITT-potilasjoukossa suurempi bosutinibiryhmässä kuin imatinibiryhmässä (20,7 % [95 % CI: 15,7 %, 25,8 %] vs 12,0 % [95 % CI: 7,9 %, 16,1 %], vetokertoimien suhde (OR) 1,88 [95 % CI: 1,15, 3,08], yksisuuntainen p‑arvo = 0,0052).
Kuukausien 3, 6 ja 9 kohdalla MMR:n saavuttaneiden osuus oli suurempi bosutinibiryhmässä kuin imatinibiryhmässä (taulukko 6).
Taulukko 6 - MMR:n vertailu hoitoryhmittäin kuukausien 3, 6 ja 9 kohdalla mITT‑potilasjoukossa
Ajankohta | MMR:n saavuttaneiden tutkittavien lukumäärä (%) | Vetokertoimien suhde (95 % CI)a | |
Bosutinibi (N = 246) | Imatinibi (N = 241) | ||
Kuukausi 3 (95 % CI) | 10 (4,1) (1,6, 6,5) | 4 (1,7) (0,0, 3,3) | 2,48 (0,77, 7,98) |
Yksisuuntainen p-arvob | 0,0578 | ||
Kuukausi 6 (95 % CI) | 86 (35,0) (29,0, 40,9) | 44 (18,3) (13,4, 23,1) | 2,42 (1,59, 3,69) |
Yksisuuntainen p-arvob | < 0,0001 | ||
Kuukausi 9 (95 % CI) | 104 (42,3) (36,1, 48,4) | 71 (29,5) (23,7, 35,2) | 1,78 (1,22, 2,60) |
Yksisuuntainen p-arvob | 0,0015 | ||
Huomaa: Prosentuaaliset osuudet perustuivat kummankin hoitoryhmän potilaiden lukumäärään. MMR:n määritelmä oli ≤ 0,1 %:n BCR-ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 3 000 ABL-transkriptia keskuslaboratorion arvion mukaan.
Lyhenteet: BCR-ABL = breakpoint cluster region‑Abelson, CI = luottamusväli, CMH = Cochran‑Mantel‑Haenszel, mITT = modified intent-to-treat, modifioitu hoitoaikeen mukainen, MMR = merkittävä molekulaarinen vaste, N = potilaiden lukumäärä.
a Korjattu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden mukaan.
b Perustuu CMH-testiin, joka on stratifioitu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden
mukaan.
60. kuukauteen mennessä MMR:n, MR4:n ja MR4.5:n saavuttaneiden osuus mITT-potilasjoukossa oli suurempi bosutinibiryhmässä kuin imatinibiryhmässä (taulukko 7). Taulukossa 8 on yhteenveto MMR-vasteista 60. kuukauteen mennessä Sokal-riskiryhmittäin.
Taulukko 7 – Yhteenveto: molekulaarinen vaste mITT-potilasjoukossa 60. kuukauteen mennessä
Vaste | Bosutinibi (N = 246) | Imatinibi (N = 241) | Vetokertoimien suhde (95 % CI)a |
Molekulaarinen vaste 60. kuukauteen mennessä, n (%) (95 % CI) | |||
MMR | 182 (74,0) (68,5, 79,5) | 158 (65,6) (59,6, 71,6) | 1,52 (1,02, 2,25) |
MR4 | 145 (58,9) (52,8, 65,1) | 120 (49,8) (43,5, 56,1) | 1,46 (1,02, 2,09) |
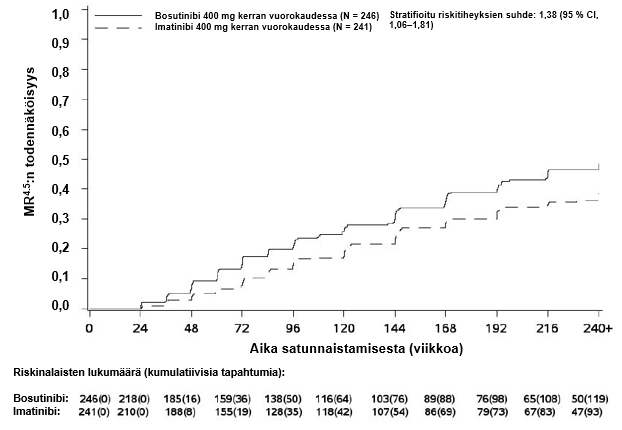
MR4.5 | 119 (48,4) (42,1, 54,6) | 93 (38,6) (32,4, 44,7) | 1,50 (1,05, 2,16) |
Huomaa: MMR:n/MR4:n /MR4.5:n määritelmä oli ≤ 0,1/0,01/0,0032 %:n BCR-ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3/4/4,5 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 3 000/9 800/30 990 ABL-transkriptia keskuslaboratorion arvion mukaan.
Lyhenteet: BCR-ABL = breakpoint cluster region‑Abelson, CI = luottamusväli, mITT = modified intent-to-treat, modifioitu hoitoaikeen mukainen, MMR = merkittävä molekulaarinen vaste, MR = molekulaarinen vaste, N = potilaiden lukumäärä.
a Korjattu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden mukaan.
Taulukko 8 – Yhteenveto: MMR 60. kuukauteen mennessä Sokal-riskipisteittäin mITT-potilasjoukossa
Vaste | Bosutinibi | Imatinibi | Vetokertoimien suhde (95 % CI) |
Sokal ̵̵ pieni riski MMR, n (%) (95 % CI) | N = 86 67 (77,9) (69,1, 86,7) | N = 95 68 (71,6) (62,5, 80,6) | 1,40 (0,71, 2,76) |
Sokal ̵̵ keskiriski MMR, n (%) (95 % CI) | N = 107 79 (73,8) (65,5, 82,2) | N = 92 62 (67,4) (57,8, 77,0) | 1,37 (0,74, 2,52) |
Sokal ̵̵ suuri riski MMR, n (%) (95 % CI) | N = 53 36 (67,9) (55,4, 80,5) | N = 54 28 (51,9) (38,5, 65,2) | 1,97 (0,90, 4,32) |
Huomaa: Prosentuaaliset osuudet perustuivat kummankin hoitoryhmän potilaiden lukumäärään. MMR:n määritelmä oli ≤ 0,1 %:n BCR‑ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 3 000 ABL-transkriptia keskuslaboratorion arvion mukaan.
Lyhenteet: BCR‑ABL = breakpoint cluster region‑Abelson, CI = luottamusväli, mITT = modified intent-to-treat, modifioitu hoitoaikeen mukainen, MMR = merkittävä molekulaarinen vaste, N = potilaiden lukumäärä.
CCyR:n kumulatiivinen ilmaantuvuus, joka on korjattu kilpailevan riskin eli hoidon keskeyttämisen ilman CCyR:n saavuttamista suhteen, oli suurempi bosutinibiryhmässä kuin imatinibiryhmässä mITT-potilasjoukossa (83,3 % [95 % CI: 78,1 %, 87,4 %] vs 76,8 % [95 % CI: 70,9 %, 81,6 %] kuukautena 60; stratifioituun suhteellisten subdistributionaalisten riskitiheyksien malliin perustuva riskitiheyksien suhde [HR]: 1,35 [95 % CI: 1,11, 1,64]). Mediaaniaika CCyR:n saavuttamiseen (vain hoitoon vastanneilla) oli 24,0 viikkoa (vaihteluväli: 11,4–120,7) bosutinibiryhmässä ja 24,3 viikkoa (vaihteluväli: 11,4–96,6) imatinibiryhmässä.
Mediaaniaika MMR:n saavuttamiseen oli 36,1 viikkoa (vaihteluväli: 11,9–241,9), MR4:n saavuttamiseen 83,7 viikkoa (vaihteluväli: 12,4–244,3) ja MR4.5:n saavuttamiseen 108,0 viikkoa (vaihteluväli: 24,1–242,1) bosutinibiryhmässä verrattuna imatinibiryhmään, jossa vastaavat luvut 47,7 viikkoa (vaihteluväli: 12,1–216,1), 84,4 viikkoa (vaihteluväli: 23,6–241,9) ja 120,4 viikkoa (vaihteluväli: 24,6–240,7) mITT-potilasryhmässä (vain hoitoon vastanneilla).
MMR:n, MR4:n ja MR4.5:n kumulatiivinen ilmaantuvuus, joka on korjattu kilpailevan riskin eli hoidon keskeyttämisen ilman tapahtumaa suhteen, oli suurempi bosutinibiryhmässä kuin imatinibiryhmässä, kuten kuvat 1–3 osoittavat.
Kuva 1 – Kumulatiiviset MMR-vasteet (mITT-potilasjoukko)
Kuva 2 – Kumulatiiviset MR4-vasteet (mITT-potilasjoukko)
Kuva 3 – Kumulatiiviset MR4.5-vasteet (mITT-potilasjoukko)
mITT-potilasjoukossa CCyR:n saavuttaneilla potilailla vasteen säilymisen Kaplan-Meierin estimaatti vuoden 4 kohdalla oli bosutinibiryhmässä 97,4 % (95 % CI: 93,9 %, 98,9 %) ja imatinibiryhmässä 93,7 % (95 % CI: 88,9 %, 96,5 %) (HR 0,39 [95 % CI: 0,14, 1,13]). MMR:n saavuttaneilla potilailla vasteen säilymisen Kaplan-Meierin estimaatti vuoden 4 kohdalla oli bosutinibiryhmässä 92,2 % (95 % CI: 86,8 %, 95,4 %) ja imatinibiryhmässä 92,0 % (95 % CI: 85,9 %, 95,5 %) (HR 1,09 [95 % CI: 0,49, 2,44]).
60. kuukauteen mennessä mITT-potilasjoukossa bosutinibihoitoa saaneista potilaista 43,9 %:lla (95 % CI: 37,7 %, 50,1 %) ja imatinibihoitoa saaneista potilaista 38,6 %:lla (95 % CI: 32,4 %, 44,7 %) (OR 1,24 [95 % CI: 0,87, 1,78]) oli jatkuva MR4, joka määriteltiin seuraavien kriteerien mukaan: vähintään 3 vuoden hoito ja vähintään MR4 kaikissa 1 vuoden ajanjaksolla tehdyissä arvioissa.
Hoidonaikaista tapahtumavapaata elinaikaa koskevien tapahtumien kumulatiivinen ilmaantuvuus kuukauden 60 kohdalla mITT-potilasjoukossa oli bosutinibiryhmässä 6,9 % (95 % CI: 4,2 %, 10,5 %) ja imatinibiryhmässä 10,4 % (95 % CI: 6,9 %, 14,6 %) (HR 0,64, 95 % CI: 0,35, 1,17).
Kokonaiselinajan Kaplan‑Meierin estimaatti oli mITT-potilasjoukossa kuukauden 60 kohdalla bosutinibipotilailla 94,9 % (95 % CI: 91,1 %, 97,0 %) ja imatinibipotilailla 94,0 % (95 % CI: 90,1 %, 96,4 %) (HR 0,80, 95 % CI: 0,37, 1,73).
Retrospektiivisessä analyysissä ITT-potilasjoukon arvioitavissa olleista potilaista useampi bosutinibiryhmässä 200/248 (80,6 %) saavutti varhaisen molekulaarisen vasteen (BCR-ABL-transkripteja ≤ 10 % kuukauden 3 kohdalla) verrattuna imatinibiryhmän potilaisiin 153/253 (60,5 %), OR 2,72 (95 % CI: 1,82, 4,08). Taulukossa 9 on yhteenveto MMR:sta ja tapahtumavapaasta elinajasta kuukauden 60 kohdalla bosutinibipotilailla, jotka saavuttivat varhaisen molekulaarisen vasteen tai eivät saavuttaneet sitä.
Taulukko 9 – Hoitotulokset kuukauden 60 kohdalla bosutinibipotilaista, joilla BCR-ABL ≤ 10 % vs > 10 % kuukauden 3 kohdalla (ITT-potilasjoukko)
Bosutinibi (N = 248) | Potilaat, joilla BCR‑ABL ≤ 10 % kuukauden 3 kohdalla (N = 200) | Potilaat, joilla BCR‑ABL > 10 % kuukauden 3 kohdalla (N = 48) | Riskitiheyksien suhde (95 % CI)a |
Kumulatiiviset MMR-vasteet, % (95 % CI) | 84,0 (78,1, 88,4) | 56,5 (41,1, 69,4) | 2,67 (1,90, 3,75) |
Kumulatiiviset tapahtumavapaan elinajan (EFS) tapahtumat, % (95 % CI) | 5,5 (2,9, 9,3) | 12,5 (5,1, 23,4) | 0,40 (0,14, 1,17) |
Lyhenteet: BCR-ABL = breakpoint cluster region‑Abelson, CI = luottamusväli, ITT = hoitoaikeen mukainen, intent‑to‑treat, MMR = merkittävä molekulaarinen vaste, EFS = tapahtumavapaa elinaika, N = potilaiden lukumäärä, joilla ≥ 3 000 ABL-kopiota kuukauden 3 kohdalla.
a Korjattu maantieteellisen alueen ja satunnaistamishetken Sokal-pisteiden mukaan.
Bosutinibiryhmässä havaittiin kuukauden 60 kohdalla vähemmän uusia mutaatioita mITT-potilasjoukossa [6 potilaalla (2,4 %) bosutinibiryhmässä ja 12 potilaalla (5,0 %) imatinibiryhmässä].
Vaiheen 1/2 kliininen tutkimus KML:n kroonista vaihetta, akseleraatiovaihetta tai blastikriisivaihetta sairastavilla potilailla, jotka olivat imatinibille resistenttejä tai intolerantteja
Yksihaaraisessa vaiheen 1/2 avoimessa monikeskustutkimuksessa arvioitiin bosutinibin 500 mg kerran päivässä tehoa ja turvallisuutta KML-potilailla, jotka olivat imatinibille resistenttejä tai intolerantteja. Potilaat oli jaettu eri kohortteihin sen perusteella, sairastivatko he kroonista vaihetta, akseleraatiovaihetta vai blastikriisivaihetta ja olivatko he saaneet aiemmin hoitoa yhdellä tyrosiinikinaasin estäjällä (imatinibi) vai useammalla kuin yhdellä tyrosiinikinaasin estäjällä (imatinibi, jonka jälkeen dasatinibi ja/tai nilotinibi).
Tässä tutkimuksessa bosutinibia sai 570 potilasta mukaan lukien KML:n kroonisen vaiheen potilaat, jotka olivat saaneet aiemmin hoitoa vain yhdellä tyrosiinikinaasin estäjällä (imatinibi) tai jotka olivat aiemmin saaneet imatinibia sekä ainakin yhtä muuta tyrosiinikinaasin estäjää (dasatinibi ja/tai nilotinibi), KML:n akseleraatiovaiheen tai blastikriisivaiheen potilaat, jotka olivat aiemmin saaneet hoitoa ainakin yhdellä tyrosiinikinaasin estäjällä (imatinibi), sekä Ph+ ALL:aa sairastavat potilaat, jotka olivat aiemmin saaneet hoitoa ainakin yhdellä tyrosiinikinaasin estäjällä (imatinibi).
Tutkimuksessa tehon ensisijainen päätetapahtuma oli merkittävän sytogeneettisen vasteen (MCyR) osuus viikolla 24 imatinibille resistenteillä KML:n kroonista vaihetta sairastavilla potilailla, jotka olivat saaneet aiemmin hoitoa vain yhdellä tyrosiinikinaasin estäjällä (imatinibi). Muita tehon päätetapahtumia olivat kumulatiiviset sytogeneettiset ja molekulaariset vasteet, aika sytogeneettisten ja molekulaaristen vasteiden saavuttamiseen ja vasteiden kestot, vaste lähtötilanteen mutaatioissa, taudin transformoituminen akseleraatio-/blastikriisivaiheeksi, taudin etenemisestä vapaa elinaika sekä kaikkien kohorttien OS.
Potilaat, jotka saivat edelleen bosutinibia vaiheen 1/2 tutkimuksen lopussa ja jotka tutkijan arvion mukaan hyötyivät bosutinibihoidosta, sekä potilaat, jotka olivat jo lopettaneet bosutinibihoidon osana vaiheen 1/2 tutkimusta ja jotka olivat eloonjäämisen pitkäaikaisseurannassa tai jotka olivat saattaneet vaiheen 1/2 tutkimuksen loppuun, olivat soveltuvia osallistumaan jatkotutkimukseen. Kukin potilas pysyi jatkotutkimuksessa joko bosutinibihoidossa tai eloonjäämisen pitkäaikaisseurannassa, kunnes viimeinen potilas saavutti 10 vuoden seuranta-ajan, joka laskettiin siitä päivästä, jolloin hänelle annettiin ensimmäinen bosutinibiannos vaiheen 1/2 tutkimuksessa.
Jatkotutkimuksessa tehon päätetapahtumia olivat sytogeneettisten ja molekulaaristen vasteiden kestot, taudin transformoituminen akseleraatio-/blastikriisivaiheeksi, taudin etenemisestä vapaa elinaika ja OS.
Tehoa koskevat analyysit sisältävät tiedot tästä päättyneestä jatkotutkimuksesta.
KML-potilaiden krooninen vaihe
Tehoa koskevat tulokset Ph+ KML:n kroonisen vaiheen potilaista, jotka olivat aiemmin saaneet imatinibia ja ainakin yhtä muuta tyrosiinikinaasin estäjää (seuranta-aika vähintään 120 kuukautta, hoidon mediaanikesto 9 kuukautta (vaihteluväli: 0,23–164,28 kuukautta), ja 20,2 % sai edelleen hoitoa 60 kuukauden kohdalla ja 7,6 % 120 kuukauden kohdalla), sekä tulokset Ph+ KML:n kroonisen vaiheen potilaista, jotka olivat aiemmin saaneet vain imatinibia (seuranta-aika vähintään 120 kuukautta, hoidon mediaanikesto 26 kuukautta (vaihteluväli: 0,16–170,49 kuukautta), ja 40,5 % sai edelleen hoitoa 60 kuukauden kohdalla ja 19,4 % 120 kuukauden kohdalla), on esitetty taulukossa 9.
KML-potilaiden akseleraatio- ja blastikriisivaihe
Tehoa koskevat tulokset Ph+ KML:n akseleraatiovaihetta (seuranta-aika vähintään 120 kuukautta, hoidon mediaanikesto 10 kuukautta (vaihteluväli: 0,10–156,15 kuukautta), ja 12,7 % sai edelleen hoitoa 60 kuukauden kohdalla ja 7,6 % 120 kuukauden kohdalla) ja blastikriisivaihetta (seuranta-aika vähintään 120 kuukautta, hoidon mediaanikesto 2,8 kuukautta (vaihteluväli: 0,03–71,38 kuukautta), ja 3,1 % sai edelleen hoitoa 60 kuukauden kohdalla ja 0 % 120 kuukauden kohdalla) sairastavista potilaista on esitetty taulukossa 10.
Taulukko 10 – Tehoa koskevat tulokset aiemmin hoitoa saaneilla KML:n kroonista ja edennyttä vaihetta sairastavilla potilailla*
Ph+ KML:n krooninen vaihe: aiempi hoito ainoastaan imatinibilla | Ph+ KML:n krooninen vaihe: Aiempi hoito imatinibilla ja dasatinibilla tai nilotinibilla | Akseleraatio-vaihe: Aiempi hoito ainakin imatinibilla | Blastikriisi-vaihe: Aiempi hoito ainakin imatinibilla | |
Kumulatiivinen sytogeneettinen vastea MCyR, % (95 % CI) CCyR, % (95 % CI) | N = 262 59,9 (53,7, 65,9) 49,6 (43,4, 55,8) | N = 112 42,0 (32,7, 51,7) 32,1 (23,6, 41,6) | N = 72 40,3 (28,9, 52,5) 30,6 (20,2, 42,5) | N = 54 37,0 (24,3, 51,3) 27,8 (16,5, 41,6) |
Kumulatiivinen molekulaarinen vastea MMR, % (95 % CI) MR4, % (95 % CI) | N = 197 42,1 (35,1, 49,4) 37,1 (30,3, 44,2) | N = 107 17,8 (11,0, 26,3) 15,0 (8,8, 23,1) | N = 54 16,7 (7,9, 29,3) 13,0 (5,4, 24,9) | N = 48 10,4 (3,5, 22,7) 10,4 (3,5, 22,7) |
Aika MCyR:n saavuttamiseen vain vasteen saaneillab, mediaani (vaihteluväli), viikkoa | 12,3 (4,0, 346,0) | 12,3 (3,9, 550,6) | 12,0 (3,9, 144,7) | 8,2 (3,9, 25,1) |
MCyR:n kestob K‑M 5 vuoden kohdalla, % (95 % CI) K‑M 10 vuoden kohdalla, % (95 % CI) Mediaani, viikkoa (95 % CI) | N = 157 70,7 (63,1, 78,3) 65,3 (56,6, 74,0) N/R | N = 47 66,6 (51,5, 81,7) 55,3 (36,3, 74,4) N/R | N = 29 40,8 (20,9, 60,7) 40,8 (20,9, 60,7) 84,0 (24,0, N/E) | N = 20 21,2 (0,1, 42,3) N/E 29,1 (11,9, 38,3) |
Aika CCyR:n saavuttamiseen vain vasteen saaneillab, mediaani (vaihteluväli), viikkoa | 24,0 (7,7, 240,6) | 24,0 (11,6, 216,0) | 23,8 (4,1, 120,0) | 8,4 (3,9, 25,1) |
CCyR:n kestob K‑M 5 vuoden kohdalla, % (95 % CI) K‑M 10 vuoden kohdalla, % (95 % CI) Mediaani, viikkoa (95 % CI) | N = 130 69,7 (61,3, 78,2) 63,4 (54,0, 72,8) N/R | N = 36 54,4 (36,7, 72,1) 40,8 (22,0, 59,6) 252,0 (24,0, N/E) | N = 22 40,0 (18,5, 61,5) 40,0 (18,5, 61,5) 72,0 (36,1, N/E) | N = 15 24,9 (0,9, 48,9) N/E 20,0 (9,1, 29,6) |
Aika MMR:n saavuttamiseen vain vasteen saaneillab, mediaani (vaihteluväli), viikkoa | 35,6 (3,1, 367,1) | 12,4 (4,0, 171,7) | 36,1 (12,1, 144,1) | 4,7 (3,9, 168,9) |
MMR:n kestob K‑M 5 vuoden kohdalla, % (95 % CI) K‑M 10 vuoden kohdalla, % (95 % CI) Mediaani, viikkoa (95 % CI) | N = 83 74,1 (64,2, 83,9) 63,4 (50,2, 76,6) N/R | N = 19 70,0 (47,5, 92,5) 70,0 (47,5, 92,5) N/R | N = 9 66,7 (35,9, 97,5) 66,7 (35,9, 97,5) N/R | N = 5 60,0 (17,1, 100,0) N/E N/R |
Aika MR4:n saavuttamiseen vain vasteen saaneillab, mediaani (vaihteluväli), viikkoa | 28,0 (3,1, 583,1) | 23,8 (4,0, 240,1) | 24,1 (22,9, 96,0) | 4,7 (3,9, 284,9) |
MR4:n kestob,e K‑M 5 vuoden kohdalla, % (95 % CI) K‑M 10 vuoden kohdalla, % (95 % CI) Mediaani, viikkoa (95 % CI) | N = 73 74,7 (64,2, 85,2) 60,8 (46,1, 75,4) N/R | N/A | N/A | N/A |
Transformoituminen akseleraatio-/ blastikriisivaiheeseenc Hoidon aikana, n | N = 284 15 | N = 119 5 | N = 79 3 | N/A |
Taudin etenemisestä vapaa elinaikac KumIlm 5 vuoden kohdalla, % (95 % CI)d KumIlm 10 vuoden kohdalla, % (95 % CI)d | N = 284 19,7 (15,6, 24,9) 23,9 (19,5, 29,5) | N = 119 24,4 (17,8, 33,4) 26,9 (20,0, 36,2) | N = 79 41,8 (32,2, 54,2) 41,8 (32,2, 54,2) | N = 64 67,2 (56,6, 79,7) N/E |
Kokonaiselinaikac K‑M 5 vuoden kohdalla, % (95 % CI) K‑M 10 vuoden kohdalla, % (95 % CI) Mediaani, kuukautta (95 % CI) | N = 284 83,5 (78,7, 88,3) 71,5 (64,4, 78,7) N/R | N = 119 74,1 (64,8, 83,4) 60,4 (47,2, 73,7) N/R | N = 79 58,5 (46,9, 70,2) 50,7 (36,5, 65,0) N/R | N = 64 22,5 (7,1, 37,9) 22,5 (7,1, 37,9) 10,9 (8,7, 19,7) |
Tietojen keruupäivämäärä: Vaiheen 1/2 tutkimus 2. lokakuuta 2015, jatkotutkimus 2. syyskuuta 2020.
Sytogeneettisen vasteen kriteerit: MCyR sisälsi täydellisen (0 % Ph+‑metafaaseja luuytimestä tai positiivisia soluja < 1 % FISH-tutkimuksessa (fluoresenssi in situ hybridisaatio) tai osittaisen (1–35 %) sytogeneettisen vasteen. Sytogeneettinen vaste perustuu Ph+‑metafaasien prosenttiosuuteen ≥ 20 metafaasisoluista kussakin luuydinnäytteessä. FISH-analyysia (≥ 200 solua) voidaan käyttää lähtötilanteen jälkeen tehtäviin sytogeneettisiin arvioihin, jos ≥ 20 metafaasia ei ole saatavissa. Jatkotutkimuksessa CCyR imputoitiin MMR:stä, jos käyttökelpoista sytogeneettistä arviointia ei ollut saatavana tietyltä päivältä.
Molekulaarisen vasteen kriteerit: Vaiheen 1/2 tutkimuksessa MMR/MR4 määriteltiin seuraavasti: ≤ 0,1/0,01 % BCR-ABL-transkriptejä keskuslaboratorion arvioimana (ei kansainvälisellä asteikolla). Jatkotutkimuksessa vasteen saaneilla MMR/MR4 merkittiin tapausraporttilomakkeeseen paikallisen laboratorion arvioinnin perusteella.
Lyhenteet: Ph+ = Philadelphia-kromosomipositiivinen, KML = krooninen myelooinen leukemia, K‑M = Kaplan-Meierin estimaatti, N = potilaiden lukumäärä, N/A = ei käytettävissä, N/R = ei saavutettu vähimmäisseurannassa, N/E = ei arvioitavissa, CI = luottamusväli, MCyR = merkittävä sytogeneettinen vaste, CCyR = täydellinen sytogeneettinen vaste, KumIlm = kumulatiivinen ilmaantuvuus, MMR = merkittävä molekulaarinen vaste, BCR-ABL = breakpoint cluster region/Abelson
a Sisältää potilaat (N), joista on olemassa käypä lähtötilanteen arvio sytogenetiikkaa ja molekulaarista vastetta varten lukuun ottamatta potilaita Kiinasta, Etelä-Afrikasta, Intiasta tai Venäjältä, koska näistä maista ei voitu lähettää näytteitä molekulaarista arviointia varten. Lähtötilanteessa vasteen saaneet, joiden vaste säilyi lähtötilanteen jälkeen, voivat olla analyysissa vasteen saaneita. Vähimmäisseuranta-aika (aika viimeisen potilaan ensimmäisestä annoksesta tietojen keruupäivämäärään saakka) oli kroonisen vaiheen potilailla 120 kuukautta.
b Sisältää potilaat (N), jotka saavuttivat vasteen tai joilla se säilyi.
c Sisältää potilaat (N), jotka saivat vähintään yhden bosutinibiannoksen.
d Kumulatiivinen ilmaantuvuus -analyysi, joka on korjattu kilpailevan riskin eli hoidon keskeyttämisen ilman tapahtumaa suhteen.
e Ei analysoitu ryhmille, joissa rajallinen määrä potilaita.
Kokonaiselinaika kroonisen vaiheen, akseleraatiovaiheen ja blastikriisivaiheen kohorteissa on esitetty graafisesti kuvassa 4.
Kuva 4 - Kaplan-Meierin estimaatti kokonaiselinajalle (OS) kroonisen vaiheen 2. linjan hoidossa (CP2L), kroonisen vaiheen 3. linjan hoidossa (CP3L), akseleraatiovaiheessa (AP) ja blastikriisivaiheessa (BP)
Vaiheen 1/2 tutkimuksen rajallisten kliinisten tietojen perusteella on saatu jonkin verran näyttöä kliinisestä aktiivisuudesta eri BCR‑ABL-mutaatiostatuksilla (ks. taulukko 11).
Taulukko 11 – KML:n kroonista vaihetta sairastavien arvioitavissa olevien potilaiden vasteet lähtötilanteen BCR-ABL-mutaatiostatuksen mukaisesti: aikaisempi hoito imatinibi ja dasatinibi ja/tai nilotinibi (3. linjan hoito)
Lähtötilanteen BCR-ABL-mutaatiostatus | Esiintymistiheys lähtötilanteessa n (%)a | MCyR saavutettu tai säilytettyResp/Evalb (%) N = 112 |
Mutaatio määritetty | 98 (100,0) | 36/92 (39,1) |
Ei mutaatiota | 59 (60,2) | 23/55 (41,8) |
Vähintään 1 mutaatio | 39 (39,8) | 13/37 (35,1) |
Dasatinibille resistentit mutaatiot | 10 (10,2) | 1/9 (11,1) |
E255K/V | 2 (2,0) | 0/2 |
F317L | 8 (8,2) | 1/7 (14,3) |
Nilotinibille resistentit mutaatiotc | 13 (13,3) | 8/13 (61,5) |
Y253H | 6 (6,1) | 5/6 (83,3) |
E255K/V | 2 (2,0) | 0/2 |
F359C/I/V | 7 (7,1) | 5/7 (71,4) |
Tietojen keruupäivämäärä: Vaiheen 1/2 tutkimus 2. lokakuuta 2015, jatkotutkimus 2. syyskuuta 2020
Huom.: Lähtötilanteen mutaatiot määritettiin ennen kuin potilas sai ensimmäisen annoksen tutkimuslääkettä. Lyhenteet: BCR‑ABL = breakpoint cluster region-Abelson, KML = krooninen myelooinen leukemia, MCyR = merkittävä sytogeneettinen vaste, N/n = potilaiden lukumäärä, Resp = vasteen saaneet potilaat, Eval = arvioitavissa olevat potilaat.
a Prosenttiluku perustuu niiden potilaiden lukumäärään, joiden mutaatiot määritettiin lähtötilanteessa.b Arvioitavissa oleva potilasjoukko sisältää potilaat, joista on olemassa käypä sairauden arviointi lähtötilanteessa.
c 2 potilaalla oli useampi kuin yksi tähän luokkaan kuuluva mutaatio.
Yksi aikaisempaa nilotinibihoitoa saanut potilas, jolla oli E255V‑mutaatio, sai parhaana vasteena täydellisen hematologisen vasteen (CHR).
In vitro ‑testaus osoitti, että bosutinibilla oli vain vähän aktiivisuutta T315I‑ ja V299L‑mutaatiota vastaan. Siksi kliinistä aktiivisuutta ei ole odotettavissa potilailla, joilla on näitä mutaatioita.
Vaiheen 4 kliininen tutkimus Ph+ KML -potilailla, joita oli aikaisemmin hoidettu yhdellä tai useammalla tyrosiinikinaasin estäjällä
Yksihaaraisessa vaiheen 4 avoimessa ei-satunnaistetussa monikeskustutkimuksessa arvioitiin bosutinibin 500 mg kerran päivässä tehoa ja turvallisuutta KML-potilailla, jotka olivat resistenttejä tai intolerantteja tyrosiinikinaasin estäjille. Potilaat oli jaettu eri kohortteihin sen perusteella, sairastivatko he kroonista vaihetta, akseleraatiovaihetta vai blastikriisivaihetta ja olivatko he saaneet aiemmin hoitoa yhdellä vai useammalla tyrosiinikinaasin estäjällä.
Tässä tutkimuksessa 163 potilasta sai bosutinibia mukaan lukien 46 Ph+ KML:n kroonisen vaiheen potilasta, joita oli aikaisemmin hoidettu yhdellä tyrosiinikinaasin estäjällä (imatinibi tai dasatinibi tai nilotinibi), 61 Ph+ KML:n kroonisen vaiheen potilasta, joita oli aikaisemmin hoidettu kahdella tyrosiinikinaasin estäjällä (imatinibi ja/tai dasatinibi ja/tai nilotinibi), 49 Ph+ KML:n kroonisen vaiheen potilasta, joita oli aikaisemmin hoidettu kolmella tyrosiinikinaasin estäjällä (imatinibi ja dasatinibi ja nilotinibi), 4 Ph+ KML:n akseleraatiovaiheen potilasta, joita oli aikaisemmin hoidettu vähintään yhdellä tyrosiinikinaasin estäjällä (2 potilasta kahdella tyrosiinikinaasin estäjällä ja 2 potilasta kolmella tyrosiinikinaasin estäjällä) ja 3 Ph- KML -potilasta, joita oli aikaisemmin hoidettu vähintään yhdellä tyrosiinikinaasin estäjällä.
Tehon ensisijainen päätetapahtuma oli kumulatiivinen vahvistettu MCyR 1. vuoteen (viikkoon 52) mennessä Ph+ KML:n kroonisen vaiheen potilailla, joita oli aikaisemmin hoidettu yhdellä tai kahdella tyrosiinikinaasin estäjällä, ja Ph+ KML:n kroonisen vaiheen potilailla, joita oli aikaisemmin hoidettu kolmella tyrosiinikinaasin estäjällä. Ph+ KML:n akseleraatiovaiheen ja blastikriisivaiheen potilailla, joita oli hoidettu millä tahansa tyrosiinikinaasin estäjällä, tehon ensisijainen päätetapahtuma oli kumulatiivinen vahvistettu hematologinen kokonaisvaste (OHR) 1. vuoteen (viikkoon 52) mennessä. Muita tehon päätetapahtumia Ph+ KML:n kroonisen vaiheen potilailla olivat kumulatiivinen sytogeneettinen ja molekulaarinen vaste, sytogeneettisten ja molekulaaristen vasteiden kestot, vaste lähtötilanteen mutaatioissa, taudin transformoituminen akseleraatio-/blastikriisivaiheeksi, PFS ja OS. Muita päätetapahtumia Ph+ akseleraatiovaiheen/blastikriisivaiheen potilaiden kohortissa olivat sytogeneettisten ja molekulaaristen vasteiden kumulatiiviset osuudet, PFS ja OS.
KML-potilaiden krooninen vaihe
Ensisijainen päätetapahtuma kumulatiivinen vahvistettu MCyR (95 % CI) 1. vuoteen (viikkoon 52) mennessä oli 76,5 % (66,9, 84,5) potilailla, joita oli aikaisemmin hoidettu yhdellä tai kahdella tyrosiinikinaasin estäjällä, ja 62,2 % (46,5, 76,2) potilailla, joita oli aikaisemmin hoidettu kolmella tyrosiinikinaasin estäjällä.
Muut tehoa koskevat tulokset Ph+ KML:n kroonisen vaiheen potilailla tutkimuksen päätyttyä vähintään 3 vuoden seurannan jälkeen on esitetty taulukossa 12. Potilaita oli hoidettu yhdellä tyrosiinikinaasin estäjällä (hoidon mediaanikesto 47,5 kuukautta (vaihteluväli: 0,9–50,1 kuukautta) ja 60,9 % edelleen hoidossa), kahdella tyrosiinikinaasin estäjällä (hoidon mediaanikesto 41,9 kuukautta (vaihteluväli: 0,4–48,9 kuukautta) ja 45,9 % edelleen hoidossa) ja kolmella tyrosiinikinaasin estäjällä (hoidon mediaanikesto 20,0 kuukautta (vaihteluväli: 0,2–48,9 kuukautta) ja 38,8 % edelleen hoidossa).
Taulukko 12 – Tehoa koskevat tulokset aiemmin hoitoa saaneilla Ph+ KML:n kroonisen vaiheen potilailla
Ph+ KML:n krooninen vaihe: aiempi hoito 1 TKI:lla | Ph+ KML:n krooninen vaihe: aiempi hoito 2 TKI:lla | Ph+ KML:n krooninen vaihe: aiempi hoito 3 TKI:lla | Ph+ KML:n krooninen vaihe: koko kohortti | |
Kumulatiivinen vahvistettu | N = 43 | N = 55 | N = 45 | N = 143 |
MCyRa 1. vuoteen mennessä, % (95 % CI) | 83,7 (69,3, 93,2) | 70,9 (57,1, 82,4) | 62,2 (46,5, 76,2) | 72,0 (63,9, 79,2) |
Kumulatiivinen sytogeneettinen vastea,b | N = 43 | N = 55 | N = 45 | N = 143 |
MCyR, % (95 % CI) | 88,4 (74,9, 96,1) | 85,5 (73,3, 93,5) | 77,8 (62,9, 88,8) | 83,9 (76,9, 89,5) |
CCyR, % (95 % CI) | 86,0 (72,1, 94,7) | 83,6 (71,2, 92,2) | 73,3 (58,1, 85,4) | 81,1 (73,7, 87,2) |
Kumulatiivinen molekulaarinen vastea,b | N = 46 | N = 55 | N = 48 | N = 149 |
MMR, % (95 % CI) | 82,6 (68,6, 92,2) | 76,4 (63,0, 86,8) | 56,3 (41,2, 70,5) | 71,8 (63,9, 78,9) |
MR4, % (95 % CI) | 73,9 (58,9, 85,7) | 63,6 (49,6, 76,2) | 41,7 (27,6, 56,8) | 59,7 (51,4, 67,7) |
MR4.5, % (95 % CI) | 58,7 (43,2, 73,0) | 50,9 (37,1, 64,6) | 35,4 (22,2, 50,5) | 48,3 (40,1, 56,6) |
Aika sytogeneettiseen vasteeseen vain vasteen saaneillab, mediaani (vaihteluväli), kuukautta | ||||
MCyR | 3,0 (1,0, 11,8) | 2,9 (0,3, 6,4) | 3,0 (1,8, 8,8) | 3,0 (0,3, 11,8) |
CCyR | 3,0 (1,0, 17,6) | 2,9 (0,3, 6,4) | 3,0 (1,8, 8,8) | 3,0 (0,3, 17,6) |
Sytogeneettisen vasteen kestob | ||||
MCyR, K-M 3 vuoden kohdalla, % (95 % CI) | 96,6 (77,9, 99,5) | 94,4 (79,2, 98,6) | 96,9 (79,8, 99,6) | 95,6 (88,7, 98,4) |
CCyR, K-M 3 vuoden kohdalla, % (95 % CI) | 96,4 (77,2, 99,5) | 94,4 (79,2, 98,6) | 100,0 (100,0, 100,0) | 96,5 (89,5, 98,9) |
Aika molekulaariseen vasteeseen vain vasteen saaneillab, mediaani (vaihteluväli), kuukautta | ||||
MMR | 3,0 (2,8, 23,3) | 3,0 (1,0, 35,9) | 3,1 (1,8, 9,3) | 3,0 (1,0, 35,9) |
MR4 | 6,0 (2,8, 47,4) | 3,1 (1,0, 36,1) | 3,2 (1,8, 47,9) | 5,5 (1,0, 47,9) |
MR4.5 | 9,2 (2,8, 47,6) | 6,0 (2,8, 36,2) | 5,8 (1,8, 18,0) | 6,0 (1,8, 47,6) |
Molekulaarisen vasteen kestob | ||||
MMR, K-M 3 vuoden kohdalla, % (95 % CI) | 90,7 (73,9, 96,9) | 81,5 (63,2, 91,3) | 90,2 (65,9, 97,5) | 87,2 (78,0, 92,7) |
MR4, K-M 3 vuoden kohdalla, % (95 % CI) | 89,5 (70,9, 96,5) | 68,7 (48,0, 82,5) | 85,2 (51,9, 96,2) | 80,7 (69,4, 88,1) |
Tietojen keruupäivämäärä: 23. marraskuuta 2020. Lyhenteet: Ph+ = Philadelphia-kromosomipositiivinen, KML= krooninen myelooinen leukemia, TKI = tyrosiinikinaasin estäjä, K‑M = Kaplan-Meierin estimaatti, N = potilaiden lukumäärä, CI = luottamusväli, MCyR = merkittävä sytogeneettinen vaste, CCyR = täydellinen sytogeneettinen vaste, MMR = merkittävä molekulaarinen vaste, MR4 = ≥ 4 logaritmiyksikön pienenemä BCR‑ABL-transkripteissa vakioidusta lähtöarvosta, MR4.5 = ≥ 4,5 logaritmiyksikön pienenemä BCR‑ABL-transkripteissa vakioidusta lähtöarvosta. Kumulatiivisen vahvistetun MCyR:n kriteerit: Vaste vahvistetaan kahdella peräkkäisellä arvioinnilla, joiden välissä on vähintään 28 vuorokautta. Jotta potilasta voidaan pitää vasteen saaneena, potilaan lähtötilanteen vasteen tulee olla säilynyt vähintään 52 viikkoa tai parantunut lähtötilanteeseen nähden. Potilaiden, joilla on osittainen sytogeneettinen vaste (PCyR) lähtötilanteessa, tulee saavuttaa CCyR hoidon aikana, jotta heitä voidaan pitää sytogeneettisen vasteen saaneina. Potilaat, joilla on vähintään MMR ja syvempi molekulaarinen vaste lähtötilanteeseen nähden, lasketaan vahvistetun CCyR:n saaneiksi. Kumulatiivisen sytogeneettisen vasteen kriteerit: Merkittävä sytogeneettinen vaste sisälsi täydellisen (0 % Ph+‑metafaaseja luuytimestä tai positiivisia soluja < 1 % FISH-tutkimuksessa (fluoresenssi in situ hybridisaatio) tai osittaisen (1–35 %) sytogeneettisen vasteen. Sytogeneettinen vaste perustuu Ph+‑metafaasien prosenttiosuuteen ≥ 20 metafaasisolusta kussakin luuydinnäytteessä. FISH-analyysia (≥ 200 solua) voidaan käyttää CCyR:n arviointiin, jos ≥ 20 metafaasia ei ole saatavissa. Potilaat, joista ei ollut saatavilla käyttökelpoista luuydinnäytettä tai FISH-arviointia, ja joilla on vähintään MMR, lasketaan CCyR:n saaneiksi. Kumulatiivisen molekulaarisen vasteen kriteerit: MMR:n/MR4:n/MR4.5:n määritelmä oli ≤ 0,1 %, ≤ 0,01 % ja ≤ 0,0032 % BCR-ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3, ≥ 4 ja ≥ 4,5 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 10 000, 10 000 ja 32 000 ABL-transkriptia keskuslaboratorion arvion mukaan. a Sisältää potilaat (N), joista on olemassa käypä lähtötilanteen arvio. Vähimmäisseuranta-aika (aika viimeisen potilaan ensimmäisestä annoksesta tietojen keruupäivämäärään saakka) 36 kuukautta. b Sisältää potilaat (N), jotka saavuttivat vasteen tai joilla se säilyi. | ||||
MMR:n, MR4:n ja MR4.5:n kumulatiivinen ilmaantuvuus, joka on korjattu kilpailevan riskin eli hoidon keskeyttämisen ilman tapahtumaa suhteen, on esitetty kuvassa 5.
Kuva 5 – Kumulatiiviset molekulaariset vasteet (MR) (kroonisen vaiheen arvioitavissa olevat potilaat)
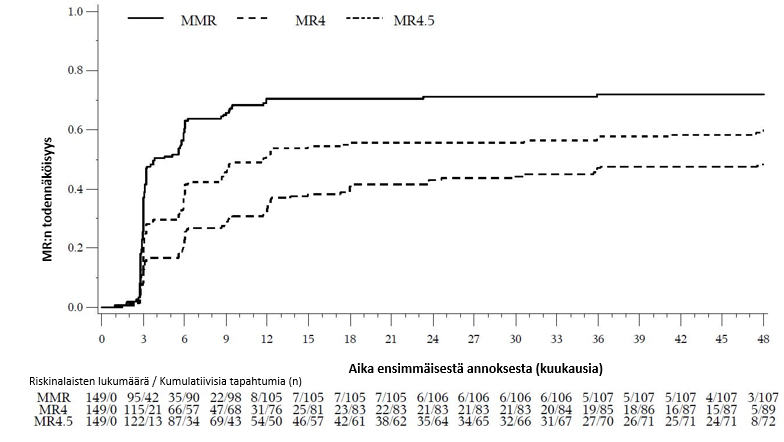
Saavutetut molekulaariset vasteet hoitolinjan mukaisesti on esitetty taulukossa 13.
Taulukko 13 – Saavutetut molekulaariset vasteet
Ph+ KML:n krooninen vaihe: aiempi hoito 1 TKI:lla | Ph+ KML:n krooninen vaihe: aiempi hoito 2 TKI:lla | Ph+ KML:n krooninen vaihe: aiempi hoito 3 TKI:lla | Ph+ KML:n krooninen vaihe: koko kohortti | |
Potilaat ilman MMR:ää lähtötilanteessaa | N = 25 | N = 28 | N = 26 | N = 79 |
MMR, % (95 % CI) | 76,0 (54,9, 90,6) | 64,3 (44,1, 81,4) | 38,5 (20,2, 59,4) | 59,5 (47,9, 70,4) |
Potilaat ilman MR4:ää lähtötilanteessaa | N = 37 | N = 38 | N = 37 | N = 112 |
MR4, % (95 % CI) | 70,3 (53,0, 84,1) | 55,3 (38,3, 71,4) | 32,4 (18,0, 49,8) | 52,7 (43,0, 62,2) |
Potilaat ilman MR4.5:tä lähtötilanteessaa | N = 42 | N = 46 | N = 43 | N = 131 |
MR4.5, % (95 % CI) | 54,8 (38,7, 70,2) | 43,5 (28,9, 58,9) | 30,2 (17,2, 46,1) | 42,7 (34,1, 51,7) |
Potilaat, joilla MMR lähtötilanteessaa | N = 21 | N = 27 | N = 22 | N = 70 |
Syvempi MR, % (95 % CI) | 85,7 (63,7, 97,0) | 66,7 (46,0, 83,5) | 63,6 (40,7, 82,8) | 71,4 (59,4, 81,6) |
Tietojen keruupäivämäärä: 23. marraskuuta 2020.
Lyhenteet: Ph+ = Philadelphia-kromosomipositiivinen, KML= krooninen myelooinen leukemia, TKI = tyrosiinikinaasin estäjä, N = potilaiden lukumäärä, CI = luottamusväli, MMR = merkittävä molekulaarinen vaste, MR = molekulaarinen vaste, MR4 = ≥ 4 logaritmiyksikön pienenemä BCR‑ABL-transkripteissa vakioidusta lähtöarvosta, MR4.5 = ≥ 4,5 logaritmiyksikön pienenemä BCR‑ABL-transkripteissa vakioidusta lähtöarvosta.
a Sisältää potilaat (N), joista on olemassa käypä lähtötilanteen arvio. Jotta potilasta voidaan pitää vasteen saaneena, potilaan vasteen tulee olla parantunut lähtötilanteeseen nähden. Molekulaarisen vasteen kriteerit: MMR:n/MR4:n /MR4.5:n määritelmä oli ≤ 0,1 %, ≤ 0,01 % ja ≤ 0,0032 % BCR-ABL/ABL-suhde IS-asteikolla (vastaa ≥ 3, ≥ 4 ja ≥ 4,5 logaritmiyksikön pienenemää vakioidusta lähtöarvosta) ja vähintään 10 000, 10 000 ja 32 000 ABL-transkriptia keskuslaboratorion arvion mukaan.
Kroonisen vaiheen potilailla ei tapahtunut hoidon aikana taudin etenemistä KML:n akseleraatio- tai blastikriisivaiheeseen.
KML-potilaiden akseleraatiovaihe
Ph+ KML:n akseleraatiovaiheen potilailla hoidon mediaanikesto oli 22,1 kuukautta (vaihteluväli: 1,6–50,1 kuukautta), kumulatiivinen vahvistettu OHR 1. vuoteen (viikkoon 52) mennessä oli 75,0 % (95 % CI: 19,4, 99,4), kuten oli myös CCyR:n kumulatiivinen osuus, kaikki 3 potilasta säilyttivät CCyR:n hoidon aikana.
Vaste BCR-ABL-mutaatioiden perusteella lähtötilanteessa
Kymmenellä potilaalla kroonisen vaiheen kohortissa oli mutaatioita lähtötilanteessa (A365V, E453K, E255K, E255V, Q252H, L298V [n = 1 kutakin], Y253F ja G250E [n = 2 kutakin]). Yhdellä potilaalla kroonisen vaiheen kohortissa oli F359I-mutaatio, joka tunnistettiin tutkimuspäivänä 8. Yhdellä potilaalla akseleraatiovaiheen kohortissa oli 2 mutaatiota (F311L ja L387F) lähtötilanteessa. Kroonisen vaiheen kohortissa potilailla, joilla oli mutaatioita, molekulaarisia vasteita todettiin 4 potilaalla 11:sta (36,4 %), 1 potilas, jolla oli E255V-mutaatio, saavutti MMR:n, ja 3 potilasta, joilla oli vastaavasti F359I, Y253F ja A365V, saavutti MR4.5:n. Akseleraatiovaiheen kohortissa oleva potilas, jolla oli mutaatioita, ei saanut mitään vastetta.
Pediatriset potilaat
Bosulif-valmisteen tehoa pediatrisilla potilailla arvioitiin BCHILD-tutkimuksessa ”Vaiheen I/II bosutinibitutkimus äskettäin todetun Ph+ KML:n kroonista vaihetta tai resistenttiä/intoleranttia Ph+ KML:aa sairastavilla pediatrisilla potilailla”.
BCHILD-tutkimus on vaiheen I/II kansainvälinen, yhden hoitohaaran avoin monikeskustutkimus, joka tehtiin suositellun kerran vuorokaudessa suun kautta otettavan bosutinibiannoksen määrittämiseksi pediatrisille potilaille (ikä 1 – < 18 vuotta), joilla on äskettäin todetun Ph+ KML:n krooninen vaihe tai Ph+ KML ja jotka ovat saaneet aiemmin hoitoa vähintään yhdellä tyrosiinikinaasin estäjällä (resistentti/intolerantti KML), sekä turvallisuuden, siedettävyyden ja tehon arvioimiseksi alustavasti ja bosutinibin farmakokinetiikan arvioimiseksi tässä potilasjoukossa.
Äskettäin todettua Ph+ KML:n kroonista vaihetta sairastavat pediatriset potilaat
Bosulif-valmisteen tehoa äskettäin todettua Ph+ KML (CP1L):n kroonista vaihetta sairastaville pediatrisille potilaille arvioitiin osana BCHILD-tutkimusta. Faasin II annoslaajennusosiossa 30 äskettäin todettua KML:ää sairastavaa potilasta sai bosutinibia annoksena 300 mg/m2 kerran vuorokaudessa. Kokonaiselinajan seurannan mediaanikestoaika koko äskettäin todettua KML:ää sairastavassa kohortissa (N = 30) oli 21,91 (1,08, 45,11) kuukautta ja hoidon mediaanikestoaika oli 13,68 (0,20, 43,70). Yhteenveto äskettäin todettua KML:ää sairastavilla potilailla missä tahansa vaiheessa todetuista kumulatiivisista sytogeneettisistä ja molekulaarisista vasteista esitetään taulukossa 15. Vasteen saaneista yhden potilaan täydellinen sytogeneettinen vaste ja merkittävä sytogeneettinen vaste hävisi.
Arvioitavissa olleista äskettäin todettua sairautta sairastavista potilaista (≥ 10 000 ABL-kopiota), 81,08 %:lla (95 %:n luottamusväli: 64,2; 97,7) BCR-ABL-suhde oli ≤ 10 % kolmen kuukauden kohdalla ja 62,5 %:lla (95 %:n luottamusväli: 38,8; 86,2) BCR-ABL-suhde oli ≤ 1 % 6 kuukauden kohdalla.
Äskettäin todettua sairautta sairastavien potilaiden kohortissa ei ollut kuolemia, eikä etenemistä akseleraatiovaiheeseen tai blastikriisivaiheeseen todettu.
Kroonista imatinibille resistenttiä tai intoleranttia Ph+ KML:ää sairastavat pediatriset potilaat
Bosulifin tehoa pediatrisilla potilailla, joilla oli resistentti tai intoleranssi Ph+ KML, arvioitiin osana BCHILD-tutkimusta.
Vaiheen I annoksen suurentamisosassa 28 potilasta, joilla oli resistentti tai intolerantti KML, saivat bosutinibia annoksina 300–400 mg/m2 kerran vuorokaudessa. Faasin II osioon otettiin mukaan 6 potilasta (400 mg/m2).
Taulukko 14 – KML:ää sairastavien potilaiden demografiset ominaisuudet
Faasi 1 (300 mg/m2) (N = 6) | Faasi 1 (350 mg/m2) (N = 11) | Faasi 1 (400 mg/m2) (N = 11) | Faasi 2 CP1L (300 mg/m2) (N = 30) | Faasi 2 R/I (400 mg/m2) (N = 6) | |
Ikä (vuotta), n (%) | |||||
>= 1 – < 6 | 2 (33,3) | 2 (18,2) | 0 | 2 (6,7) | 0 |
>= 6 – < 12 | 3 (50,0) | 4 (36,4) | 3 (27,3) | 10 (33,3) | 1 (16,7) |
>= 12 – < 18 | 1 (16,7) | 5 (45,5) | 8 (72,7) | 18 (60,0) | 5 (83,3) |
Mediaani (vaihtelu-väli) | 8,50 (1, 17) | 11,00 (4, 17) | 15,00 (6, 17) | 12,50 (5, 17) | 14,50 (11, 16) |
Sukupuoli, n (%) | |||||
Poika | 5 (83,3) | 4 (36,4) | 7 (63,6) | 18 (60,0) | 4 (66,7) |
Tyttö | 1 (16,7) | 7 (63,6) | 4 (36,4) | 12 (40,0) | 2 (33,3) |
Etninen tausta, n (%) | |||||
Valko-ihoinen | 0 | 5 (45,5) | 7 (63,6) | 22 (73,3) | 4 (66,7) |
Musta-ihoinen tai afro-amerik-kalainen | 0 | 1 (9,1) | 1 (9,1) | 5 (16,7) | 1 (16,7) |
Aasia-lainen | 0 | 1 (9,1) | 3 (27,3) | 1 (3,3) | 1 (16,7) |
Amerikan intiaani tai Alaskan alkuperäis-kansa | 0 | 0 | 0 | 0 | 0 |
Havaijin tai muun Tyynen-meren saaren alkuperäis-kansa | 0 | 0 | 0 | 2 (6,7) | 0 |
Ei tiedossa | 6 (100,0) | 4 (36,4) | 0 | 0 | 0 |
Etnisyys, n (%) | |||||
Latinalais-amerik-kalainen | 0 | 0 | 2 (18,2) | 7 (23,3) | 0 |
Ei latinalais-amerik-kalainen | 0 | 8 (72,7) | 9 (81,8) | 23 (76,7) | 6 (100,0) |
Ei tiedossa | 6 (100,0) | 3 (27,3) | 0 | 0 | 0 |
Kokonaiselinajan seurannan mediaanikestoaika koko faasin 1 osiossa (N = 28) oli 29,27 kuukautta (15,21, 85,88) ja faasin 2 osiossa (N = 6) se oli 9,66 kuukautta (2,00, 15,54). Hoidon mediaanikestoaika faasin 1 osiossa oli 17,26 kuukautta (vaihteluväli 0,30–60,85) ja faasin 2 osiossa se oli 9,64 kuukautta (1,97, 15,54).
Yhteenveto KML:ää sairastavilla potilailla missä tahansa vaiheessa ilmenneistä kumulatiivisista sytogeneettisistä ja molekulaarisista vasteista esitetään taulukossa 15. Faasi 1:ssä vasteen saaneista kolmella potilaalla täydellinen sytogeneettinen vaste hävisi ja kahdella potilaalla merkittävä sytogeneettinen vaste hävisi. Faasin 1 osiossa merkittävän molekulaarisen vasteen säilymisen todennäköisyys 18 kuukauden kohdalla oli 92,3 % (95 %:n luottamusväli: 56,6, 98,8).
Etenemistä akseleraatiovaiheeseen tai blastikriisivaiheeseen ei todettu.
Taulukko 15 – Tehoa koskevat tulokset resistenttiä tai intoleranttia Ph+ KML:aa sairastavilla pediatrisilla potilailla
Faasi 1 yhteensä (R/I) (N = 28) | Faasi 2 CP1L (300 mg/m2) (N = 30) | Faasi 2 R/I (400 mg/m2) (N = 6) | |
Kumulatiivinen MCyR, % (95 % CI) | 24 (85,7) (67,3, 96,0) | 26 (86,7) (69,3, 96,2) | 6 (100,0) (54,1, 100,0) |
Kumulatiivinen CCyR, % (95 % CI) | 23 (82,1) (63,1, 93,9) | 25 (83,3) (65,3, 94,4) | 6 (100,0) (54,1, 100,0) |
Potilaat, joilla ei MCyR:a lähtötilanteessa, N | 4 | N/A | 1 |
MCyR, % (95 % CI) | 3 (75,0) (19,4, 99,4 | N/A | 1 (100,0) (2,5, 100,0) |
Potilaat, joilla ei CCyR:a lähtötilanteessa, N | 9 | N/A | 2 |
CCyR, % (95 % CI) | 7 (77,8) (40,0, 97,2) | N/A | 2 (100,0) (15,8, 100,0) |
Kumulatiivinen MMR, % (95 % CI) | 16 (57,1) (37,2, 75,5) | 13 (43,3) (25,5, 62,6) | 4 (66,7) (22,3, 95,7) |
Kumulatiivinen MR4, % (95 % CI) | 6 (21,4) (8,3, 41,0) | 5 (16,7) (5,6, 34,7) | 1 (16,7) (0,4, 64,1) |
Kumulatiivinen MR4.5, % (95 % CI) | 5 (17,9) (6,1, 36,9) | 0 (0,0) (0,0, 11,6) | 0 (0,0) (0,0, 45,9) |
Lyhenteet: CCyR = täydellinen sytogeneettinen vaste; CI = luottamusväli, KML = krooninen myelooinen leukemia; MCyR = merkittävä sytogeneettinen vaste; MMR = merkittävä molekulaarinen vaste; MR = molekulaarinen vaste; N/n = potilaiden lukumäärä; Ph = Philadelphia-kromosomipositiivinen; R/I = resistentti tai intolerantti. | |||
Farmakokinetiikka
Bosutinibin farmakokinetiikkaa arvioitiin, kun bosutinibia oli annettu aikuisille KML-potilaille suun kautta ruokailun yhteydessä, ja ellei muuta ole mainittu, se esitettiin geometrisenä keskiarvona (variaatiokerroin, %).
Imeytyminen
Terveille koehenkilöille ruoan kanssa annetun bosutinibin kerta-annoksen (500 mg) absoluuttinen biologinen hyötyosuus oli 34 %. Imeytyminen oli suhteellisen hidasta: mediaaniaika huippupitoisuuden (tmax) saavuttamiseen kesti yli 6 tuntia. Bosutinibin AUC suurenee suhteessa annokseen annosvälillä 100 – 600 mg. Bosutinibin farmakokineettiset parametrit aikuisilla saatiin populaatiofarmakokineettisestä analyysistä, jossa käytettiin eri tutkimusten yhdistettyjä tietoja. Useiden suun kautta annettujen 400 mg:n Bosulif-annosten jälkeen bosutinibin vakaan tilan Cmax oli 127 ng/ml (31 %), Ctrough oli 68 ng/ml (39 %) ja AUC oli 2 370 ng•h/ml (34 %); useiden suun kautta annettujen 500 mg:n Bosulif-annosten jälkeen bosutinibin vakaan tilan Cmax oli 171 ng/ml (38 %), Ctrough oli 91 ng/ml (42 %) ja AUC oli 3 150 ng•h/ml (38 %).
Bosutinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun ruokailun jälkeen annettiin samansuuruinen bosutinibiannos joko tablettina tai avaamattomana kovana kapselina. Farmakokinetiikka oli verrannollinen annettaessa bosutinibia sisältävien kovien kapselien sisältö omenasoseeseen tai jogurttiin sekoitettuna tai annettaessa bosutinibi avaamattomana kovana kapselina terveille aikuisille tutkittaville ruokailun jälkeen.
Bosutinibin liukoisuus riippuu pH:sta ja imeytyminen heikkenee mahanesteen pH:n noustessa (ks. kohta Yhteisvaikutukset).
Ruoan vaikutus
Kun bosutinibitabletteja annettiin terveille tutkittaville runsasrasvaisen aterian yhteydessä, bosutinibin Cmax suureni 1,8-kertaiseksi ja AUC suureni 1,7-kertaiseksi verrattuna niiden antamiseen paastotilassa. Erillisessä tutkimuksessa bosutinibia sisältävät kovat kapselit annettiin ruokailun jälkeen, jolloin altistus oli noin 1,5–1,6 kertaa suurempi kuin annettaessa ne paastotilassa.
Jakautuminen
Kun terveille tutkittaville annettiin bosutinibin 120 mg:n kerta-annos laskimonsisäisesti, jakautumistilavuuden keskiarvo (variaatiokerroin, CV %) oli 2331 (32) l, mikä viittaa siihen, että bosutinibi jakautuu laajasti verisuoniston ulkopuoliseen kudokseen.
Bosutinibi sitoutui voimakkaasti ihmisen plasman proteiineihin in vitro (94 %) ja terveillä koehenkilöillä ex vivo (96 %) eikä sitoutuminen ollut pitoisuudesta riippuvaista.
Biotransformaatio
In vitro- ja in vivo ‑tutkimukset viittasivat siihen, että bosutinibi (kanta-aine) käy ihmisellä läpi pääasiassa maksametabolian. Kerta-annoksena tai toistuvina annoksina ihmiselle annetun bosutinibin (400 tai 500 mg) tärkeimmät verenkierrossa esiintyvät metaboliitit näyttäisivät olevan oksideklorinoitunut (M2) ja N‑desmetyloitunut (M5) bosutinibi. Bosutinibi-N‑oksidi-metaboliittia (M6) esiintyi verenkierrossa vähäisemmässä määrin. Systeeminen altistus N‑desmetyloituneelle metaboliitille oli 25 % kanta-aineesta ja altistus oksideklorinoituneelle metaboliitille oli 19 % kanta-aineesta. Kaikkien kolmen metaboliitin aktiivisuus oli Src‑transformoidun fibroblastin kiinnittymisestä riippumattomassa proliferaatiomäärityksessä ≤ 5 % bosutinibin aktiivisuudesta. Bosutinibi ja N‑desmetyylibosutinibi olivat ulosteessa pääasialliset lääkkeeseen liittyneet yhdisteet. In vitro ‑tutkimukset ihmisen maksan mikrosomeilla osoittivat, että bosutinibin metaboliaan osallistuva pääasiallinen sytokromi P450 -isoentsyymi on CYP3A4. Lääkkeiden yhteisvaikutustutkimukset osoittivat, että ketokonatsolilla ja rifampisiinilla on merkittävä vaikutus bosutinibin farmakokinetiikkaan (ks. kohta Yhteisvaikutukset). CYP-entsyymien 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ja 3A5 ei havaittu metaboloivan bosutinibia.
Eliminaatio
Kun terveille tutkittaville annettiin bosutinibin 120 mg:n kerta-annos laskimonsisäisesti, terminaalisen eliminaation puoliintumisajan keskiarvo (CV %) oli 35,5 (24) tuntia ja puhdistuman keskiarvo (CV %) oli 61,9 (26) l/h. Massatasapainotutkimuksessa suun kautta otetulla bosutinibilla keskimäärin 94,6 % annetusta kokonaisannoksesta havaittiin 9 vuorokauden kuluessa; uloste (91,3 % annoksesta) oli tärkein erittymisreitti, ja 3,29 % annoksesta havaittiin virtsassa. 75 % annoksesta todettiin 96 tunnin kuluessa. Muuttumattoman bosutinibin erittyminen virtsaan oli vähäistä, noin 1 % annetusta annoksesta sekä terveillä koehenkilöillä että potilailla, joilla oli pitkälle edennyt kiinteä syöpäkasvain.
Erityispotilasryhmät
Maksan vajaatoiminta
Ruoan kanssa otettua 200 mg:n bosutinibiannosta arvioitiin 18 maksan vajaatoimintaa sairastavan potilaan (Child‑Pugh-luokat A, B ja C) kohortilla ja 9 kaltaistetulla terveellä koehenkilöllä. Bosutinibin Cmax plasmassa suureni Child‑Pugh-luokan A potilailla 2,4‑kertaiseksi, Child‑Pugh-luokan B potilailla 2‑kertaiseksi ja Child‑Pugh-luokan C potilailla 1,5‑kertaiseksi, ja bosutinibin AUC plasmassa suureni Child‑Pugh-luokan A potilailla 2,3‑kertaiseksi, Child‑Pugh-luokan B potilailla 2‑kertaiseksi ja Child‑Pugh-luokan C potilailla 1,9‑kertaiseksi. Bosutinibin t1/2 suureni maksan vajaatoimintaa sairastavilla potilailla verrattuna terveisiin koehenkilöihin.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa selvittävässä tutkimuksessa 26:lle lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavalle sekä 8 kaltaistetulle terveelle vapaaehtoiselle tutkittavalle annettiin ruoan kanssa kerta-annoksena 200 mg bosutinibia. Munuaisten vajaatoiminnan määritelmä perustui Cockcroft-Gaultin kaavalla laskettuun kreatiniinipuhdistumaan (ClCr): vaikea munuaisten vajaatoiminta < 30 ml/min, keskivaikea munuaisten vajaatoiminta 30–49 ml/min ja lievä munuaisten vajaatoiminta 50–79 ml/min. Keskivaikeaa munuaisten vajaatoimintaa sairastavilla AUC-arvo suureni 35 % ja vaikeaa munuaisten vajaatoimintaa sairastavilla 60 % verrattuna terveisiin vapaaehtoisiin. Suurin bosutinibialtistus (Cmax) kasvoi 28 % keskivaikeaa vajaatoimintaa sairastavien ryhmässä ja 34 % vaikeaa vajaatoimintaa sairastavien ryhmässä. Bosutinibialtistus ei lisääntynyt lievää munuaisten vajaatoimintaa sairastavilla. Bosutinibin eliminaation puoliintumisaika munuaisten vajaatoimintaa sairastavilla oli vastaava kuin terveillä tutkittavilla.
Munuaisten vajaatoiminnan vuoksi tehdyt annosmuutokset perustuivat edellä mainitun tutkimuksen tuloksiin ja bosutinibin tunnetusti lineaariseen farmakokinetiikkaan annosvälillä 200–600 mg.
Ikä, sukupuoli ja etninen tausta
Näiden demografisten tekijöiden vaikutuksen arvioimiseksi ei ole tehty varsinaisia tutkimuksia. Populaatiofarmakokineettiset analyysit Ph+‑leukemiaa sairastavista potilaista tai potilaista, joilla oli kiinteä syöpäkasvain, sekä terveistä tutkittavista osoittavat, ettei iällä, sukupuolella tai painolla ole kliinisesti merkittävää vaikutusta. Populaatiofarmakokineettiset analyysit osoittivat, että aasialaisilla puhdistuma oli 18 % pienempi, mikä vastaa bosutinibialtistuksen (AUC) lisääntymistä noin 25 %.
Pediatriset potilaat
Bosutinibin farmakokinetiikkaa arvioitiin 41:llä äskettäin todettua tai resistenttiä/intoleranttia sairautta sairastavalla iältään 1- – < 18-vuotiaalla pediatrisella potilaalla annosvälillä 300–400 mg/m2, kun annos annettiin suun kautta kerran vuorokaudessa ruokailun yhteydessä. Tmax-arvon mediaani todettiin pediatrisilla potilailla noin 3 tuntia annoksen jälkeen (vaihteluväli 1–8 tuntia annoksen jälkeen). Altistukset suurenivat suhteessa annokseen välillä 100–600 mg, ja AUCtau:n geometrinen keskiarvo 300–400 mg/m2 ‑kohorteissa vastasi vastaavan äskettäin todetun ja resistentin tai intolerantin Ph+ KML:n käyttöaiheiden aikuisia koskevan annostason AUCtau:n geometristä keskiarvoa (+/- 20 %), mutta pediatrisilla potilailla Cmax ja puhdistuma olivat suuremmat ja Cmin oli pienempi kuin aikuisilla.
Prekliiniset tiedot turvallisuudesta
Bosutinibia on tutkittu farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, lisääntymistoksisuutta ja fototoksisuutta arvioineissa tutkimuksissa.
Farmakologinen turvallisuus
Bosutinibi ei vaikuttanut hengitysfunktioihin. Keskushermostovaikutuksia selvittäneessä tutkimuksessa bosutinibihoitoa saaneilla rotilla esiintyi silmän mustuaisten supistumista ja kävelyn heikentymistä. Silmän mustuaisen koon kannalta vaikutuksetonta annosta (no observed effect level, NOEL) ei määritetty, mutta kävelyn osalta NOEL todettiin altistuksella, joka oli noin 11‑kertainen verrattuna ihmisen saamaan altistukseen 400 mg:n kliinisestä annoksesta ja 8‑kertainen verrattuna ihmiseen saamaan altistukseen 500 mg:n kliinisestä annoksesta (kyseisten lajien vapaan lääkeaineen Cmax -arvon perusteella). Bosutinibin aktiivisuus hERG-määrityksissä in vitro viittasi sydänkammioiden repolarisaation (QTc-ajan) pidentymisen mahdollisuuteen. Koirilla tehdyssä tutkimuksessa suun kautta annettu bosutinibi ei aiheuttanut verenpaineen muutoksia, poikkeavia eteis- tai kammioperäisiä rytmihäiriötä, PR‑, QRS‑ tai QTc‑ajan pitenemistä EKG:ssä, kun altistus oli enintään 3‑kertainen verrattuna ihmisen saamaan altistukseen 400 mg:n kliinisestä annoksesta ja 2‑kertainen verrattuna ihmisen saamaan altistukseen 500 mg:n kliinisestä annoksesta (kyseisten lajien vapaan lääkeaineen Cmax -arvon perusteella). Viivästynyttä sydämen syketaajuuden kiihtymistä havaittiin. Koirille laskimoon annetulla valmisteella tehdyssä tutkimuksessa havaittiin tilapäistä sydämen syketaajuuden kiihtymistä ja verenpaineen alenemista sekä hyvin vähäistä QTc-ajan pitenemistä (< 10 ms) altistuksilla, jotka olivat noin 6–20‑kertaisia verrattuna ihmisen saamaan altistukseen 400 mg:n kliinisestä annoksesta ja 4–15‑kertaisia verrattuna ihmisen saamaan altistukseen 500 mg:n kliinisestä annoksesta (kyseisten lajien vapaan lääkeaineen Cmax ‑arvon perusteella). Havaittujen vaikutusten ja lääkehoidon välinen suhde ei varmistunut.
Toistuvan altistuksen toksisuus
Toistuvan altistuksen toksisuutta selvittäneissä tutkimuksissa, jotka kestivät rotilla enintään 6 kuukautta ja koirilla enintään 9 kuukautta, todettiin ruoansulatuselimistön olevan bosutinibin toksisuuden ensisijainen kohde-elin. Toksisuuden kliinisiä oireita olivat ulosteeseen liittyneet muutokset sekä vähentynyt ruoan kulutus ja painon aleneminen, jotka joissakin tapauksissa johtivat kuolemaan tai elektiiviseen eutanasiaan.
Histopatologisessa tutkimuksessa havaittiin luumenin laajenemista, pikarisolujen hyperplasiaa, suoliston verenvuotoja ja eroosiota sekä turvotusta, sekä sinusten erytrosytoosia ja suoliliepeen imusolmukkeiden verenvuotoja. Myös maksa tunnistettiin toksisuuden kohde-elimeksi rotilla. Toksisuudelle oli tunnusomaista maksan painon lisääntyminen, joka korreloi hepatosellulaariseen hypertrofiaan ja ilmeni ilman maksaentsyymien nousua tai mikroskooppisia merkkejä hepatosellulaarisesta sytotoksisuudesta. Merkitystä ihmisille ei tunneta. Eri lajien altistuksen vertailu osoittaa, että rotilla tehdyssä 6 kuukautta kestäneessä tutkimuksessa ja koirilla tehdyssä 9 kuukautta kestäneessä tutkimuksessa altistukset, joista ei aiheutunut haittatapahtumia, olivat samankaltaisia kuin ihmisen saama altistus 400 mg:n tai 500 mg:n kliinisen annoksen jälkeen (kyseisten lajien vapaan lääkeaineen AUC-arvon perusteella).
Genotoksisuus
Genotoksisuustutkimuksissa bakteereilla in vitro ‑menetelmillä sekä nisäkkäillä in vitro- ja in vivo ‑menetelmillä, joihin saattoi liittyä metabolinen aktivaatio, ei todettu näyttöä bosutinibin mutageenisuudesta.
Lisääntymistoksisuus ja kehitystoksisuus
Rotilla tehdyssä hedelmällisyystutkimuksessa urosten hedelmällisyys oli hieman heikentynyt. Naarailla havaittiin lisääntyneitä alkion resorptioita sekä alkion kiinnittymisen ja elinkykyisten alkioiden vähenemistä. Hedelmällisyyden suhteen haitattomiksi annoksiksi uroksilla (30 mg/kg/vrk) ja naarailla (3 mg/kg/vrk) todettujen annosten altistus oli uroksilla 0,6‑kertainen ja naarailla 0,3‑kertainen verrattuna ihmiseen saamaan altistukseen 400 mg:n kliinisestä annoksesta ja uroksilla 0,5‑kertainen ja naarailla 0,2‑kertainen verrattuna ihmisen saamaan altistukseen 500 mg:n kliinisestä annoksesta (kyseisten lajien vapaan lääkeaineen AUC-arvon perusteella). Vaikutusta miehen hedelmällisyyteen ei voida sulkea pois (ks. kohta Raskaus ja imetys).
Sikiön altistuminen bosutinibista peräisin olevalle radioaktiivisuudelle tiineyden aikana osoitettiin tiineillä Sprague-Dawley-rotilla tehdyssä lääkkeiden kulkeutumista istukan kautta selvittäneessä tutkimuksessa. Rotilla tehdyssä pre- ja postnataalisessa kehitystutkimuksessa poikasia syntyi lukumääräisesti vähemmän annoksella ≥ 30 mg/kg/vrk. Koko poikueen menetyksen ilmaantuvuus suureni ja jälkeläisten syntymänjälkeinen kasvu heikkeni annoksella 70 mg/kg/vrk. Annos, jolla ei havaittu mitään haitallisia kehitysvaikutuksia (10 mg/kg/vrk), johti rotilla altistukseen, joka vastasi 1,3‑kertaisesti ihmisen saamaa altistusta 400 mg:n annoksesta ja 1,0‑kertaisesti ihmisen saamaa altistusta 500 mg:n annoksesta (kyseisten lajien vapaan lääkeaineen AUC-arvon perusteella). Kaniinilla emolle toksisilla annoksilla tehdyssä kehitystoksisuustutkimuksessa todettiin sikiön poikkeavuuksia (kehittymässä olevan rintalastan yhteensulautuma ja kahdella sikiöllä oli erilaisia viskeraalisia löydöksiä) sekä vähäistä sikiön painon laskua. Altistus suurimmalla kaniinilla tutkitulla annoksella (10 mg/kg/vrk), josta ei aiheutunut sikiölle haittavaikutuksia, oli 0,9‑kertainen verrattuna ihmisen saamaan altistukseen 400 mg:n annoksesta ja 0,7‑kertainen verrattuna ihmisen saamaan altistukseen 500 mg:n annoksesta (kyseisten lajien vapaan lääkeaineen AUC-arvon perusteella).
Kun imettäville Sprague-Dawley-rotille annettiin oraalinen [14C]‑radioisotooppileimattu bosutinibikerta-annos (10 mg/kg), radioaktiivisuus erittyi nopeasti nisämaitoon jo puoli tuntia valmisteen annon jälkeen. Radioaktiivisuuspitoisuus oli maidossa 8‑kertainen plasmaan verrattuna. Tämän vuoksi radioaktiivisuutta esiintyi mitattavina pitoisuuksina imetettyjen poikasten plasmassa.
Karsinogeenisuus
Bosutinibi ei ollut karsinogeeninen rotilla tehdyssä 2 vuotta kestäneessä ja rasH2‑hiirillä tehdyssä 6 kuukautta kestäneessä karsinogeenisuustutkimuksessa.
Fototoksisuus
Bosutinibin on osoitettu voivan sitoa UV‑B- ja UV‑A-valoa, joka jakautuu pigmentillisten rottien ihoon ja silmän suonikalvostoon. Bosutinibilla ei kuitenkaan osoitettu ihoon tai silmiin kohdistuvaa fototoksisuutta altistettaessa pigmentilliset rotat bosutinibille UV‑säteilyssä, kun bosutinibialtistus oli enintään 3‑kertainen verrattuna ihmisen saamaan altistukseen 400 mg:n annoksesta ja 2‑kertainen verrattuna ihmisen saamaan altistukseen 500 mg:n annoksesta (kyseisten lajien vapaan lääkeaineen Cmax -arvon perusteella).
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Mikrokiteinen selluloosa (E 460)
Kroskarmelloosinatrium (E 468)
Poloksameerit 188
Povidoni (E 1201)
Magnesiumstearaatti (E 470b)
Kalvopäällyste
Bosulif 100 mg kalvopäällysteiset tabletit
Polyvinyylialkoholi
Titaanidioksidi (E 171)
Makrogoli 3350
Talkki (E 553b)
Keltainen rautaoksidi (E 172)
Bosulif 400 mg kalvopäällysteiset tabletit
Polyvinyylialkoholi
Titaanidioksidi (E 171)
Makrogoli 3350
Talkki (E 553b)
Keltainen rautaoksidi (E 172)
Punainen rautaoksidi (E 172)
Bosulif 500 mg kalvopäällysteiset tabletit
Polyvinyylialkoholi
Titaanidioksidi (E 171)
Makrogoli 3350
Talkki (E 553b)
Punainen rautaoksidi (E 172)
Bosulif kovat kapselit
Mannitoli (E 421)
Mikrokiteinen selluloosa (E 460)
Kroskarmelloosinatrium (E 468)
Poloksameerit 188
Povidoni (E 1201)
Magnesiumstearaatti (E 470b)
Bosulif kovien kapseleiden kuori
Liivate
Titaanidioksidi (E 171)
Keltainen rautaoksidi (E 172)
Punainen rautaoksidi (E 172)
Kovien kapseleiden painomuste
Shellakka (E 904)
Propyleeniglykoli (E 1520)
Ammoniakkiliuos, väkevä (E 527)
Musta rautaoksidi (E 172)
Kaliumhydroksidi (E 525)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Kalvopäällysteiset tabletit
4 vuotta.
Kovat kapselit
3 vuotta.
Säilytys
Kalvopäällysteiset tabletit
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Kovat kapselit
Säilytä alle 30 °C. Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BOSULIF tabletti, kalvopäällysteinen
100 mg (L:ei) 28 fol (330,75 €)
400 mg (L:ei) 28 fol (1182,16 €)
500 mg (L:ei) 28 fol (1454,57 €)
PF-selosteen tieto
Kalvopäällysteiset tabletit
Valkoinen, läpinäkymätön, 3‑kerroksinen PVC/polyklorotrifluoroetyleeni/PVC-läpipainolevy, joka on taustapuolella sinetöity läpipainofoliolla ja joka sisältää 14 tai 15 tablettia.
Bosulif 100 mg kalvopäällysteiset tabletit
Yksi kartonkikotelo sisältää 28, 30 tai 112 tablettia.
Bosulif 400 mg kalvopäällysteiset tabletit
Yksi kartonkikotelo sisältää 28 tai 30 tablettia.
Bosulif 500 mg kalvopäällysteiset tabletit
Yksi kartonkikotelo sisältää 28 tai 30 tablettia.
Kovat kapselit
Suurtiheyspolyeteenipurkki (HDPE-purkki) ja polypropeenisuljin (PP-suljin) sekä kuumainduktiosinetti (HIS).
Bosulif 50 mg kovat kapselit
Kartonkikotelossa yksi purkki, joka sisältää 30 kovaa kapselia.
Bosulif 100 mg kovat kapselit
Kartonkikotelossa yksi purkki, joka sisältää 150 kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Bosulif 100 mg kalvopäällysteiset tabletit
Keltainen, soikea (leveys: 5,6 mm; pituus: 10,7 mm), kaksoiskupera, kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”100”.
Bosulif 400 mg kalvopäällysteiset tabletit
Oranssi, soikea (leveys: 8,8 mm; pituus: 16,9 mm), kaksoiskupera, kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”400”.
Bosulif 500 mg kalvopäällysteiset tabletit
Punainen, soikea (leveys: 9,5 mm; pituus: 18,3 mm), kaksoiskupera, kalvopäällysteinen tabletti, jonka toisella puolella on merkintä ”Pfizer” ja vastakkaisella puolella ”500”.
Bosulif 50 mg kovat kapselit
Valkoinen runko-osa / oranssi kansiosa (pituus noin 18 mm); runko-osassa painatus ”BOS 50” ja kansiosassa painatus ”Pfizer” mustalla musteella.
Bosulif 100 mg kovat kapselit
Valkoinen runko-osa / ruskehtavanpunainen kansiosa (pituus noin 22 mm); runko-osassa painatus ”BOS 100” ja kansiosassa painatus ”Pfizer” mustalla musteella.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BOSULIF tabletti, kalvopäällysteinen
100 mg 28 fol
400 mg 28 fol
500 mg 28 fol
- Ylempi erityiskorvaus (100 %). Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117).
- Peruskorvaus (40 %).
ATC-koodi
L01EA04
Valmisteyhteenvedon muuttamispäivämäärä
11.12.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com