
JAKAVI tabletti 5 mg, 10 mg, 15 mg, 20 mg
Vaikuttavat aineet ja niiden määrät
Jakavi 5 mg tabletti
Yksi tabletti sisältää 5 mg ruksolitinibia (fosfaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 71,45 mg laktoosimonohydraattia.
Jakavi 10 mg tabletti
Yksi tabletti sisältää 10 mg ruksolitinibia (fosfaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 142,90 mg laktoosimonohydraattia.
Jakavi 15 mg tabletti
Yksi tabletti sisältää 15 mg ruksolitinibia (fosfaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 214,35 mg laktoosimonohydraattia.
Jakavi 20 mg tabletti
Yksi tabletti sisältää 20 mg ruksolitinibia (fosfaattina).
Apuaine, jonka vaikutus tunnetaan
Yksi tabletti sisältää 285,80 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti.
Kliiniset tiedot
Käyttöaiheet
Myelofibroosi (MF)
Jakavi on tarkoitettu primaarista myelofibroosia (krooninen idiopaattinen myelofibroosi), polysytemia veran jälkeistä myelofibroosia tai essentiellin trombosytoosin jälkeistä myelofibroosia sairastavien aikuispotilaiden oireiden tai sairauteen liittyvän splenomegalian hoitoon.
Polysytemia vera (PV)
Jakavi on tarkoitettu sellaisten polysytemia veraa sairastavien aikuispotilaiden hoitoon, jotka ovat resistenttejä hydroksiureahoidolle tai jotka eivät siedä tätä hoitoa.
Käänteishyljintä (GvHD)
Akuutti käänteishyljintä
Jakavi on tarkoitettu akuutin käänteishyljinnän hoitoon aikuisilla ja vähintään 28 vrk:n ikäisillä pediatrisilla potilailla, joilla kortikosteroidihoito tai muu systeeminen hoito ei ole tuottanut riittävää vastetta (ks. kohta Farmakodynamiikka).
Krooninen käänteishyljintä
Jakavi on tarkoitettu kroonisen käänteishyljinnän hoitoon aikuisilla ja vähintään 6 kk:n ikäisillä pediatrisilla potilailla, joilla kortikosteroidihoito tai muu systeeminen hoito ei ole tuottanut riittävää vastetta (ks. kohta Farmakodynamiikka).
Ehto
Hoitavan lääkärin tulee olla perehtynyt syövän hoitoon.
Annostus ja antotapa
Jakavi-hoidon saa aloittaa vain syöpälääkevalmisteiden käyttöön perehtynyt lääkäri.
Ennen Jakavi-hoidon aloittamista tutkitaan täydellinen verenkuva, johon kuuluu valkosolujen erittelylaskenta.
Täydellistä verenkuvaa, johon kuuluu valkosolujen erittelylaskenta, on seurattava 2–4 viikon välein, kunnes Jakavi-annos vakiintuu, ja tämän jälkeen kliinisen tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Aloitusannos
Myelofibroosi (MF)
Jakavin suositeltava aloitusannos myelofibroosin hoidossa perustuu trombosyyttiarvoon (ks. taulukko 1):
Taulukko 1 Aloitusannokset myelofibroosin hoidossa
| Trombosyyttiarvo | Aloitusannos |
| Yli 200 x 109/l | 20 mg kahdesti vuorokaudessa |
| 100–200 x 109/l | 15 mg kahdesti vuorokaudessa |
| 75 – < 100 x 109/l | 10 mg kahdesti vuorokaudessa |
| 50 – < 75 x 109/l | 5 mg kahdesti vuorokaudessa |
Polysytemia vera (PV)
Jakavin suositeltava aloitusannos polysytemia veran hoidossa on 10 mg kahdesti vuorokaudessa.
Käänteishyljintä
Jakavin suositeltava aloitusannos akuutin ja kroonisen käänteishyljinnän hoidossa perustuu ikään (ks. taulukot 2 ja 3):
Taulukko 2 Aloitusannokset akuutin käänteishyljinnän hoidossa
| Ikäryhmä | Aloitusannos |
| Vähintään 12 vuotta | 10 mg kahdesti vuorokaudessa |
| 6 vuotta – < 12 vuotta | 5 mg kahdesti vuorokaudessa |
| 28 vrk – < 6 vuotta | 8 mg/m2 kahdesti vuorokaudessa |
Taulukko 3 Aloitusannokset kroonisen käänteishyljinnän hoidossa
| Ikäryhmä | Aloitusannos |
| Vähintään 12 vuotta | 10 mg kahdesti vuorokaudessa |
| 6 vuotta – < 12 vuotta | 5 mg kahdesti vuorokaudessa |
| 6 kuukautta – < 6 vuotta | 8 mg/m2 kahdesti vuorokaudessa |
Aloitusannos käänteishyljinnän hoidossa voidaan antaa joko tablettina potilaille, jotka pystyvät nielemään tabletin kokonaisena, tai oraaliliuoksena.
Jakavi voidaan lisätä kortikosteroidi- ja/tai kalsineuriiniestäjähoidon rinnalle.
Annosmuutokset
Annosta voidaan titrata tehon ja turvallisuuden mukaan.
Myelofibroosi ja polysytemia vera
Jos teho katsotaan riittämättömäksi ja veriarvot ovat riittävät, annosta voidaan suurentaa enintään 5 mg kahdesti vuorokaudessa kerrallaan aina kahdesti vuorokaudessa annettavaan 25 mg enimmäisannokseen saakka.
Aloitusannosta ei saa suurentaa ensimmäisten neljän hoitoviikon aikana, eikä annosta saa tämän jälkeen suurentaa tiheämmin kuin 2 viikon välein.
Hoito on lopetettava, jos potilaan trombosyyttiarvo on alle 50 x 109/l tai absoluuttinen neutrofiiliarvo on alle 0,5 x 109/l. PV:ssä hoito on myös keskeytettävä, jos hemoglobiiniarvo laskee alle 80 g/l. Kun veriarvot ovat korjautuneet tätä tasoa suuremmiksi, valmisteen annostelu voidaan aloittaa uudelleen käyttäen 5 mg annoksia kahdesti vuorokaudessa, ja annosta voidaan suurentaa vähitellen täydellisen verenkuvan huolellisen seurannan perusteella. Täydelliseen verenkuvaan on kuuluttava myös valkosolujen erittelylaskenta.
Annoksen pienentämistä on harkittava, jos trombosyyttiarvo laskee hoidon aikana taulukossa 4 mainittuihin arvoihin, jotta hoitoa ei jouduttaisi keskeyttämään trombosytopenian vuoksi.
Taulukko 4 Suositeltavat annostukset MF-potilailla trombosytopenian ilmetessä
| Annostus trombosyyttiarvon laskiessa | |||||
| 25 mg 2 x / vrk | 20 mg 2 x / vrk | 15 mg 2 x / vrk | 10 mg 2 x / vrk | 5 mg 2 x / vrk | |
| Trombosyyttiarvo | Uusi annos | ||||
| 100 – < 125 x 109/l | 20 mg 2 x / vrk | 15 mg 2 x / vrk | Ei muutosta | Ei muutosta | Ei muutosta |
| 75 – < 100 x 109/l | 10 mg 2 x / vrk | 10 mg 2 x / vrk | 10 mg 2 x / vrk | Ei muutosta | Ei muutosta |
| 50 – < 75 x 109/l | 5 mg 2 x / vrk | 5 mg 2 x / vrk | 5 mg 2 x / vrk | 5 mg 2 x / vrk | Ei muutosta |
| Alle 50 x 109/l | Keskeytä | Keskeytä | Keskeytä | Keskeytä | Keskeytä |
PV:ssä annoksen pienentämistä on myös harkittava, jos hemoglobiiniarvo laskee alle 120 g/l ja annoksen pienentämistä suositellaan, jos kyseinen arvo laskee alle 100 g/l.
Käänteishyljintä
Jos käänteishyljintää sairastavalla potilaalla on trombosytopenia, neutropenia tai kohonnut kokonaisbilirubiiniarvo, tavanomaisen tukihoidon (mm. kasvutekijöiden, infektiolääkkeiden ja verensiirtojen) antamisen jälkeen voi olla tarpeen pienentää annosta tai keskeyttää hoito tilapäisesti. On suositeltavaa pienentää annosta yhden annostason verran (10 mg:sta kahdesti vuorokaudessa 5 mg:aan kahdesti vuorokaudessa tai 5 mg:sta kahdesti vuorokaudessa 5 mg:aan kerran vuorokaudessa). Jos potilas ei siedä Jakavi-hoitoa annoksella 5 mg kerran vuorokaudessa, hoito on keskeytettävä. Tarkat annostussuositukset esitetään taulukossa 5.
Taulukko 5 Suositeltavat annostukset ruksolitinibihoidon aikana käänteishyljintää sairastavilla potilailla trombosytopenian, neutropenian tai kohonneen kokonaisbilirubiiniarvon ilmetessä
| Laboratorioarvo | Suositeltava annostus |
| Trombosyyttiarvo < 20 x 109/l | Pienennä Jakavi-annosta yhden annostason verran. Jos trombosyyttiarvo suurenee tasolle ≥ 20 x 109/l seitsemän vuorokauden kuluessa, annosta voidaan suurentaa alkuperäiselle annostasolle. Muussa tapauksessa jatketaan pienennetyn annoksen käyttöä. |
| Trombosyyttiarvo < 15 x 109/l | Keskeytä Jakavi-hoito, kunnes trombosyyttiarvo on ≥ 20 x 109/l, ja aloita hoito sitten uudelleen yhtä annostasoa pienemmällä annoksella. |
| Absoluuttinen neutrofiiliarvo ≥ 0,5 – < 0,75 x 109/l | Pienennä Jakavi-annosta yhden annostason verran. Aloita hoito uudelleen alkuperäisellä annostasolla, jos absoluuttinen neutrofiiliarvo on > 1,0 x 109/l. |
| Absoluuttinen neutrofiiliarvo < 0,5 x 109/l | Keskeytä Jakavi-hoito, kunnes absoluuttinen neutrofiiliarvo on > 0,5 x 109/l, ja aloita hoito sitten uudelleen yhtä annostasoa pienemmällä annoksella. Jos absoluuttinen neutrofiiliarvo on > 1,0 x 109/l, hoito voidaan aloittaa uudelleen alkuperäisellä annostasolla. |
| Kokonaisbilirubiiniarvo kohonnut ilman käänteishyljinnän vaikutusta, ei maksaan kohdistuvaa käänteishyljintää | > 3,0–5,0 x viitearvojen yläraja (ULN): Jatka Jakavi-hoitoa yhtä annostasoa pienemmällä annoksella, kunnes arvo on ≤ 3,0 x ULN. |
| > 5,0 – 10,0 x ULN: Keskeytä Jakavi-hoito enintään 14 vuorokauden ajaksi, kunnes kokonaisbilirubiiniarvo on ≤ 3,0 x ULN. Jos kokonaisbilirubiiniarvo on ≤ 3,0 x ULN, hoito voidaan aloittaa uudelleen samalla annoksella. Jos arvo ei ole ≤ 3,0 x ULN, kun 14 vuorokautta on kulunut, aloita hoito uudelleen yhtä annostasoa pienemmällä annoksella. | |
| > 10,0 x ULN: Keskeytä Jakavi-hoito, kunnes kokonaisbilirubiiniarvo on ≤ 3,0 x ULN, ja aloita sitten hoito uudelleen yhtä annostasoa pienemmällä annoksella. | |
| Kokonaisbilirubiiniarvo kohonnut käänteishyljinnän vaikutuksesta, potilaalla maksaan kohdistuvaa käänteishyljintää | > 3,0 x ULN: Jatka Jakavi-hoitoa yhtä annostasoa pienemmällä annoksella, kunnes kokonaisbilirubiiniarvo on ≤ 3,0 x ULN. |
Annosmuutokset, jos potilas käyttää samanaikaisesti voimakkaita CYP3A4:n estäjiä tai sekä CYP2C9- että CYP3A4-entsyymien estäjiä
Jos ruksolitinibia annetaan yhdessä voimakkaan CYP3A4:n estäjän tai sekä CYP2C9- että CYP3A4-entsyymejä estävän lääkkeen (esim. flukonatsolin) kanssa, ruksolitinibin kerta-annosta pienennetään noin 50 % ja lääke otetaan kahdesti vuorokaudessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset). Ruksolitinibin samanaikaista käyttöä yli 200 mg/vrk flukonatsoliannosten kanssa on vältettävä.
Erityisryhmät
Munuaisten vajaatoiminta
Erityisiin annosmuutoksiin ei ole tarvetta hoidettaessa lievää tai kohtalaista munuaisten vajaatoimintaa sairastavia potilaita.
Jos potilaalla on vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma alle 30 ml/min), MF-, PV- ja käänteishyljintäpotilaiden trombosyyttiarvoon perustuvaa suositeltavaa aloitusannosta pienennetään noin 50 % ja lääke annetaan kahtena osa-annoksena vuorokaudessa. Hoidon turvallisuutta ja tehoa on seurattava huolellisesti ruksolitinibihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hemodialyysihoitoa saavien loppuvaiheen munuaisten vajaatoimintapotilaiden (ESRD) kohdalla parhaista annosteluvaihtoehdoista on vain rajallisesti tietoa. Tästä potilasryhmästä saatavilla oleviin tietoihin perustuvat farmakokineettiset/farmakodynaamiset mallinnukset viittaavat siihen, että hemodialyysihoitoa saavien ESRD-potilaiden aloitusannos MF:ssä on 15–20 mg kerta-annos tai kaksi 10 mg annosta 12 tunnin välein hemodialyysin jälkeen ja vain hemodialyysipäivänä. 15 mg kerta-annosta suositellaan MF-potilaille, joiden trombosyyttiarvo on 100–200 x 109/l. 20 mg kerta-annosta tai kahta 10 mg annosta 12 tunnin välein suositellaan MF-potilaille, joiden trombosyyttiarvo on > 200 x 109/l. Myöhemmät annokset (kerta-annos tai kaksi 10 mg annosta 12 tunnin välein) annetaan vain hemodialyysipäivinä aina kunkin dialyysikerran jälkeen.
Hemodialyysihoitoa saavien ESRD-potilaiden suositeltu aloitusannos PV:ssä on 10 mg kerta-annos tai kaksi 5 mg annosta 12 tunnin välein dialyysihoidon jälkeen ja ainoastaan hemodialyysipäivänä. Nämä annossuositukset perustuvat mallinnuksiin ja ESRD-potilailla potilaskohtaista turvallisuutta ja tehoa on seurattava huolellisesti kaikkien annosmuutosten yhteydessä. Peritoneaalidialyysihoitoa tai jatkuvaa venovenoosista hemofiltraatiohoitoa saavien potilaiden kohdalla annostelusta ei ole tietoa (ks. kohta Farmakokinetiikka).
Käänteishyljintää sairastavista potilaista, joilla on loppuvaiheen munuaisten vajaatoiminta, ei ole tietoja.
Maksan vajaatoiminta
Jos MF-potilaalla on maksan vajaatoiminta (asteesta riippumatta), trombosyyttiarvoon perustuvaa suositeltavaa aloitusannosta pienennetään noin 50 % ja lääke annetaan kahtena osa-annoksena vuorokaudessa. Myöhempiä annoksia muutetaan huolellisen turvallisuus- ja tehoseurannan perusteella. Suositeltava aloitusannos PV:n hoidossa on 5 mg kahdesti vuorokaudessa. Ruksolitinibiannosta voidaan titrata sytopeniariskin minimoimiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos potilaalla on käänteishyljintään liittymätön lievä, kohtalainen tai vaikea maksan vajaatoiminta, ruksolitinibin aloitusannosta pienennetään 50 % (ks. kohta Farmakokinetiikka).
Jos potilaalla on maksaan kohdistuvaa käänteishyljintää ja kokonaisbilirubiiniarvo on kohonnut tasolle >3 x ULN, veriarvoja on seurattava tiheämmin toksisuuden varalta, ja annoksen pienentämistä yhden annostason verran suositellaan.
Iäkkäät potilaat (≥ 65 vuotta)
Iäkkäille potilaille ei suositella muita annosmuutoksia.
Pediatriset potilaat
Jakavin turvallisuutta ja tehoa enintään 18 vuoden ikäisten myelofibroosia tai polysytemia veraa sairastavien lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla (ks. kohta Farmakodynamiikka).
Hoidon lopettaminen
Myelofibroosin ja polysytemia veran hoitoa voidaan jatkaa niin kauan kuin hyöty-riskiarvio pysyy positiivisena. Hoito on kuitenkin lopetettava 6 kuukauden kuluttua, jos pernan koko ei ole pienentynyt tai oireet eivät ole helpottuneet hoidon aloittamisen jälkeen.
Ruksolitinibihoito suositellaan lopetettavaksi, jos potilaalla on nähty jonkinasteista kliinisen tilan kohentumista, mutta hänen pernansa jää 40 % leveämmäksi verrattuna lähtötasoon (vastaa karkeasti arvioiden pernan tilavuuden kasvua noin 25 %:lla), eikä hänellä enää nähdä konkreettista sairauteen liittyvien oireiden paranemista.
Käänteishyljintää hoidettaessa voidaan harkita Jakavi-annoksen pienentämistä vähitellen, jos potilas on saavuttanut vasteen ja kortikosteroidihoito on lopetettu. On suositeltavaa pienentää Jakavi-annosta 50 % kahden kuukauden välein. Jos käänteishyljinnän oireet ja löydökset uusiutuvat Jakavi-annoksen vähittäisen pienentämisen aikana tai sen jälkeen, on harkittava annoksen suurentamista uudelleen.
Antotapa
Jakavi otetaan suun kautta joko ruoan kanssa tai tyhjään mahaan.
Jos annos jää väliin, potilaan ei pidä ottaa ylimääräistä annosta, vaan seuraava annos otetaan tavanomaiseen tapaan lääkemääräyksen mukaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Raskaus ja imetys.
Varoitukset ja käyttöön liittyvät varotoimet
Myelosuppressio
Jakavi-hoito voi aiheuttaa hematologisia haittavaikutuksia kuten trombosytopeniaa, anemiaa ja neutropeniaa. Ennen Jakavi-hoidon aloittamista tutkitaan täydellinen verenkuva, johon kuuluu valkosolujen erittelylaskenta. Hoito on lopetettava, jos MF-potilaan trombosyyttiarvo on alle 50 x 109/l tai absoluuttinen neutrofiiliarvo on alle 0,5 x 109/l (ks. kohta Annostus ja antotapa).
On todettu, että trombosytopenian kehittyminen hoidon aikana on todennäköisempää, jos MF-potilaan trombosyyttiarvot ovat hoidon alussa alhaiset (< 200 x 109/l).
Trombosytopenia on yleensä korjautuvaa, ja sitä hoidetaan yleensä pienentämällä annosta tai keskeyttämällä Jakavi-hoito tilapäisesti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Trombosyyttisiirrot voivat kuitenkin olla aiheellisia kliinisen tarpeen mukaan.
Jos potilaalle kehittyy anemia, verensiirto voi olla tarpeen. Myös annosmuutoksia tai hoidon keskeyttämistä voi olla tarpeen harkita, jos potilaalle kehittyy anemia.
Potilailla, joiden hemoglobiiniarvo on alle 100 g/l hoidon aloitusvaiheessa, on suurempi riski hemoglobiinipitoisuuden putoamiselle alle 80 g/l hoidon aikana verrattuna potilaisiin, joiden hemoglobiinipitoisuus lähtötasossa on tätä korkeampi (79,3 % vs. 30,1 %). Tavallista tiheämpää veriarvojen sekä Jakaviin liittyvien haittavaikutusten kliinisten löydösten ja oireiden seurantaa suositellaan hoidettaessa potilaita, joiden hemoglobiiniarvo on lähtötasossa alle 100 g/l.
Neutropenia (absoluuttinen neutrofiiliarvo < 0,5 x 109/l) oli yleensä korjautuvaa, ja sitä hoidettiin keskeyttämällä Jakavi-hoito tilapäisesti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Täydellistä verenkuvaa on seurattava kliinisen tarpeen mukaan, ja annosta on muutettava tarvittavaan tapaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Infektiot
Jakavilla hoidetuilla potilailla on esiintynyt vakavia bakteeri-, mykobakteeri-, sieni-, virus- ja muita opportunistisia infektioita. Vakavien infektioiden kehittymisriskiä potilaille on arvioitava. Lääkärin on seurattava Jakavi-hoitoa saavia potilaita huolellisesti infektioiden oireiden ja löydösten varalta ja aloitettava asianmukainen hoito ripeästi. Jakavi-hoitoa ei saa aloittaa, ennen kuin aktiiviset vakavat infektiot ovat korjautuneet.
Tuberkuloosia on raportoitu Jakavia saaneilla potilailla. Ennen hoidon aloittamista potilaat tulee arvioida aktiivisen ja inaktiivisen (”piilevän”) tuberkuloosin varalta paikallisten suositusten mukaisesti. Arviointiin voivat kuulua sairaushistoria, mahdollinen aiempi tuberkuloosialtistus ja/tai soveltuvat tutkimukset kuten keuhkojen röntgentutkimus, tuberkuliinitesti ja/tai gammainterferonierityksen määritys. Lääkäreitä muistutetaan virheellisen negatiivisen tuberkuliini-ihotestituloksen riskistä, erityisesti potilailla, jotka ovat vaikeasti sairaita tai joilla on heikentynyt immuunivaste.
Hepatiitti B -viruskuorman (HBV-DNA-tiitterin) kasvua, joko alaniiniaminotransferaasi- ja aspartaattiaminotransferaasiarvojen nousun kera tai ilman sitä, on raportoitu kroonista HBV-infektiota sairastavilla, Jakavi-hoitoa saavilla potilailla. HBV-infektion seulontaa suositellaan ennen Jakavi-hoidon aloittamista. Potilaita, joilla on krooninen HBV-infektio, on hoidettava ja seurattava kliinisten hoitosuositusten mukaisesti.
Vyöruusu
Lääkärin on kerrottava potilaille vyöruusun varhaisoireista ja ‑löydöksistä ja kerrottava heille, että hoitoon on hakeuduttava mahdollisimman varhain.
Progressiivinen multifokaalinen leukoenkefalopatia
Progressiivista multifokaalista leukoenkefalopatiaa (PML) on raportoitu käytettäessä Jakavia. Lääkäreiden tulee olla erityisen valppaana sellaisten PML:ään viittaavien oireiden suhteen, joita potilaat eivät välttämättä huomaa (esim. kognitiiviset, neurologiset tai psykiatriset oireet tai merkit). Potilaita on seurattava uusien tai pahenevien oireiden tai merkkien varalta. Jos näitä oireita/merkkejä havaitaan, on harkittava neurologin konsultointia sekä asianmukaisia PML-diagnoosiin liittyviä toimenpiteitä. Jos PML:ää epäillään, hoito on keskeytettävä kunnes PML on poissuljettu.
Lipidiarvojen poikkeavuudet/kohoaminen
Jakavi-hoitoon on liittynyt lipidiarvojen, mukaan lukien kokonaiskolesteroli-, HDL-kolesteroli-, LDL-kolesteroli- ja triglyseridiarvojen, nousua. Lipidiarvojen seurantaa ja dyslipidemian hoitoa suositellaan hoitosuositusten mukaisesti.
Merkittävät sydän- ja verisuonitapahtumat (MACE)
Suuressa satunnaistetussa, aktiivisesti kontrolloidussa tutkimuksessa, jossa tutkittiin tofasitinibin (toinen JAK:n estäjä) käyttöä nivelreuman hoidossa 50-vuotiailla ja sitä vanhemmilla potilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitapahtumien riskitekijä, havaittiin suurempi merkittävien sydän- ja verisuonitapahtumien, eli sydän- ja verisuoniperäisten kuolemien, ei-fataalien sydäninfarktien ja ei-fataalien aivoinfarktien ilmaantuvuus tofasitinibia saaneilla potilailla verrattuna tuumorinekroositekijän (TNF) estäjiä saaneisiin.
Jakavia saaneilla potilailla on ilmoitettu merkittäviä sydän- ja verisuonitapahtumia. Ennen Jakavi-hoidon aloitusta tai jatkamista on huomioitava hoitoon liittyvät hyödyt ja riskit kyseiselle potilaalle erityisesti, jos potilas on täyttänyt 65 vuotta tai jos hän tupakoi tai on aiemmin tupakoinut pitkäaikaisesti, tai jos hänellä on ennestään ateroskleroottinen sydän- ja verisuonitauti tai muita kardiovaskulaarisia riskitekijöitä.
Tromboosi
Suuressa satunnaistetussa, aktiivisesti kontrolloidussa tutkimuksessa, jossa tutkittiin tofasitinibin (toinen JAK:n estäjä) käyttöä nivelreuman hoidossa 50-vuotiailla ja sitä vanhemmilla potilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitapahtumien riskitekijä, havaittiin tofasitinibia saaneilla potilailla annosriippuvaisesti enemmän tromboembolisia laskimotapahtumia (VTE), mukaan lukien syvä laskimotromboosi (DVT) ja keuhkoembolia (PE), verrattuna TNF:n estäjiä saaneisiin.
Jakavia saaneilla potilailla on ilmoitettu syviä laskimotrombooseja (DVT) ja keuhkoembolioita (PE). Kliinisissä tutkimuksissa Jakavilla hoidetuilla MF- ja PV-potilailla tromboembolisten tapahtumien ilmaantuvuus oli vastaava kuin verrokkiryhmässä.
Ennen Jakavi-hoidon aloitusta tai jatkamista on huomioitava hoitoon liittyvät hyödyt ja riskit kyseiselle potilaalle erityisesti, jos potilaalla on sydän- ja verisuonitapahtumien riskitekijöitä (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet ”Merkittävät sydän- ja verisuonitapahtumat (MACE)”).
Jos potilaalle ilmaantuu tromboosiin viittaavia oireita, hänet on tutkittava viipymättä ja hoidettava asianmukaisesti.
Toinen primaarimaligniteetti
Suuressa satunnaistetussa, aktiivisesti kontrolloidussa tutkimuksessa, jossa tutkittiin tofasitinibin (toinen JAK:n estäjä) käyttöä nivelreuman hoidossa 50-vuotiailla ja sitä vanhemmilla potilailla, joilla oli lisäksi vähintään yksi sydän- ja verisuonitapahtumien riskitekijä, havaittiin tofasitinibia saaneilla potilailla enemmän maligniteetteja, erityisesti keuhkosyöpiä, lymfoomia ja ei-melanoomatyyppisiä ihosyöpiä (NMSC) verrattuna TNF:n estäjiä saaneisiin.
JAK:n estäjiä, myös Jakavia, saaneilla potilailla on ilmoitettu lymfoomia ja muita maligniteetteja.
Ei-melanoomatyyppisiä ihosyöpätapauksia (NMSC), mukaan lukien tyvisolu-, okasolu- ja merkelinsolukarsinooma, on raportoitu ruksolitinibihoitoa saaneilla potilailla. Useimmat näistä MF- ja PV-potilaista olivat saaneet pitkäaikaista hydroksiureahoitoa ja heillä oli jo aikaisemmin esiintynyt NMSC:tä tai premaligneja ihovaurioita. Säännönmukaisesti toistuvien ihotutkimusten suorittamista suositellaan potilaille, joilla on suurentunut ihosyövän riski.
Erityisryhmät
Munuaisten vajaatoiminta
Jakavi-aloitusannosta on pienennettävä, jos potilaalla on vaikea munuaisten vajaatoiminta. Hemodialyysihoitoa saavilla loppuvaiheen munuaisten vajaatoimintapotilailla aloitusannos MF:n hoidossa perustuu trombosyyttiarvoon, kun taas PV:n hoidossa suositeltava aloitusannos on 10 mg:n kerta-annos (ks. kohta Annostus ja antotapa). Myöhemmät annokset (20 mg kerta-annos tai kaksi 10 mg annosta 12 tunnin välein MF-potilailla; 10 mg kerta-annos tai kaksi 5 mg annosta 12 tunnin välein PV-potilailla) annetaan vain hemodialyysipäivinä aina kunkin dialyysikerran jälkeen. Muiden annosmuutosten yhteydessä turvallisuutta ja tehoa on seurattava huolellisesti. Käänteishyljintää sairastaville potilaille, joilla on vaikea munuaisten vajaatoiminta, Jakavin aloitusannosta on pienennettävä noin 50 % (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Maksan vajaatoiminta
Jakavi-aloitusannosta on pienennettävä noin 50 % MF:n ja PV:n hoidossa, jos potilaalla on maksan vajaatoiminta. Muiden annosmuutosten on perustuttava lääkevalmisteen turvallisuuteen ja tehoon. Käänteishyljintää sairastavilla potilailla, joilla on käänteishyljintään liittymätön maksan vajaatoiminta, Jakavin aloitusannosta on pienennettävä noin 50 % (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Jos potilaalla todetaan ruksolitinibihoidon aikana maksan vajaatoiminta, täydellistä verenkuvaa (johon kuuluu valkosolujen erittelylaskenta) on seurattava 1–2 viikon välein tai tätä tiiviimmin ensimmäisten kuuden ruksolitinibihoitoviikon ajan ja sittemmin kliinisen tarpeen mukaan, kunnes maksan toiminta ja verenkuva ovat vakiintuneet.
Yhteisvaikutukset
Jos Jakavia aiotaan käyttää yhdessä voimakkaan CYP3A4:n estäjän tai sekä CYP3A4- että CYP2C9-entsyymejä estävän lääkkeen (esim. flukonatsolin) kanssa, Jakavin kerta-annosta pienennetään noin 50 % ja lääke otetaan kahdesti vuorokaudessa (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
On suositeltavaa seurata veriarvoja ja ruksolitinibiin liittyvien haittavaikutusten kliinisiä oireita ja löydöksiä aiempaa tiiviimmin (esim. kahdesti viikossa), kun potilas käyttää voimakasta CYP3A4:n estäjää tai sekä CYP2C9- että CYP3A4-entsyymejä estävää lääkettä.
Sytoreduktiivisten hoitojen ja Jakavin samanaikainen käyttö oli yhteydessä sytopenioihin, jotka olivat hoidettavissa (ks. kohta Annostus ja antotapa annosmuutokset sytopenioiden aikana).
Hoidon lopetusoireet
Jakavi-hoidon keskeyttämisen tai lopettamisen jälkeen MF:n oireet voivat uusiutua noin yhden viikon kuluessa. Joillakin Jakavi-hoidon lopettaneilla potilailla on esiintynyt vaikeita haittatapahtumia, etenkin, jos heillä on ollut samanaikaisesti jokin akuutti sairaus. Jakavi-hoidon äkillisen lopettamisen mahdollista osuutta kyseisiin tapahtumiin ei ole selvitetty. Jos hoitoa ei tarvitse lopettaa äkillisesti, voidaan harkita Jakavi-annoksen pienentämistä vähitellen. Annoksen vähittäisen pienentämisen hyötyjä ei tosin ole osoitettu.
Apuaineet, joiden vaikutus tunnetaan
Jakavi sisältää laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Ruksolitinibi poistuu elimistöstä CYP3A4:n ja CYP2C9:n katalysoiman metabolian kautta. Kyseisiä entsyymejä estävät lääkevalmisteet voivat siten lisätä potilaan altistusta ruksolitinibille.
Ruksolitinibiannoksen pienentämiseen johtavat yhteisvaikutukset
CYP3A4:n estäjät
Voimakkaat CYP3A4:n estäjät (esim. (mutta ei ainoastaan) bosepreviiri, klaritromysiini, indinaviiri, itrakonatsoli, ketokonatsoli, lopinaviiri/ritonaviiri, ritonaviiri, mibefradiili, nefatsodoni, nelfinaviiri, posakonatsoli, sakinaviiri, telapreviiri, telitromysiini, vorikonatsoli)
Terveillä henkilöillä ruksolitinibin (10 mg kerta-annos) anto yhdessä voimakkaan CYP3A4:n estäjän (ketokonatsolin) kanssa suurensi ruksolitinibin Cmax-arvoa 33 % ja sen AUC-arvoa 91 % verrattuna tilanteeseen, jossa ruksolitinibia käytettiin yksinään. Ketokonatsolin samanaikainen anto pidensi puoliintumisaikaa 3,7 tunnista 6,0 tuntiin.
Jos ruksolitinibia käytetään yhdessä voimakkaan CYP3A4:n estäjän kanssa, ruksolitinibin kerta-annosta pienennetään noin 50 % ja lääke otetaan kahdesti vuorokaudessa.
Potilaita seurataan huolellisesti (esim. kahdesti viikossa) sytopenioiden varalta, ja annosta titrataan turvallisuuden ja tehon mukaan (ks. kohta Annostus ja antotapa).
Sekä CYP2C9- että CYP3A4-entsyymejä estävät lääkkeet
Terveillä henkilöillä ruksolitinibin (10 mg kerta‑annos) anto yhdessä sekä CYP2C9- että CYP3A4-entsyymejä estävän flukonatsolin kanssa suurensi ruksolitinibin Cmax-arvoa 47 % ja sen AUC-arvoa 232 % verrattuna tilanteeseen, jossa ruksolitinibia käytettiin yksinään.
Annoksen pienentämistä 50 %:lla on harkittava käytettäessä lääkkeitä, jotka ovat sekä CYP2C9:n että CYP3A4:n estäjiä (esim. flukonatsoli). Ruksolitinibin samanaikaista käyttöä yli 200 mg/vrk flukonatsoliannosten kanssa on vältettävä.
Entsyymejä indusoivat aineet
CYP3A4:n indusorit (esim. [mutta ei ainoastaan] avasimibi, karbamatsepiini, fenobarbitaali, fenytoiini, rifabutiini, rifampiini [rifampisiini], mäkikuisma [Hypericum perforatum])
Potilaita on seurattava huolellisesti, ja annosta säädettävä turvallisuuden ja tehon mukaan (ks. kohta Annostus ja antotapa).
Kun terveille henkilöille annettiin ruksolitinibia (50 mg kerta-annos), kun he olivat ensin käyttäneet voimakasta CYP3A4:n indusorina toimivaa rifampisiinia (600 mg vuorokausiannos 10 päivän ajan), ruksolitinibin AUC-arvo oli 70 % pienempi verrattuna tilanteeseen, jossa ruksolitinibihoitoa käytettiin yksinään. Altistus ruksolitinibin aktiivisille metaboliiteille ei muuttunut. Ruksolitinibin farmakodynaaminen aktiivisuus pysyi kaiken kaikkiaan ennallaan, mikä viittaa siihen, että CYP3A4:n induktiolla oli hyvin vähäinen vaikutus farmakodynamiikkaan. Yhteisvaikutus voi johtua suuresta ruksolitinibiannoksesta, jolloin farmakodynaamisia vaikutuksia voi esiintyä lähellä Emax-arvoa. On mahdollista, että yksittäisen potilaan ruksolitinibiannosta on suurennettava, jos hänelle aloitetaan hoito voimakkaalla entsyymi-induktorilla.
Muut huomioon otettavat, ruksolitinibihoitoon vaikuttavat yhteisvaikutukset
Heikot tai kohtalaiset CYP3A4:n estäjät (esim. [mutta ei ainoastaan] siprofloksasiini, erytromysiini, amprenaviiri, atatsanaviiri, diltiatseemi, simetidiini)
Terveillä henkilöillä ruksolitinibin (10 mg kerta-annos) anto yhdessä erytromysiinin kanssa (500 mg erytromysiiniä kahdesti vuorokaudessa 4 päivän ajan) suurensi ruksolitinibin Cmax-arvoa 8 % ja sen AUC-arvoa 27 % verrattuna tilanteeseen, jossa ruksolitinibia käytettiin yksinään.
Annosmuutoksia ei suositella, kun ruksolitinibia käytetään yhdessä heikon tai kohtalaisen CYP3A4:n estäjän (esim. erytromysiinin) kanssa. Potilaita on kuitenkin seurattava huolellisesti sytopenioiden varalta, kun kohtalaisen CYP3A4:n estäjän käyttö aloitetaan.
Ruksolitinibin vaikutukset muihin lääkkeisiin
P-glykoproteiinin ja muiden kuljettajaproteiinien kuljettamat aineet
Ruksolitinibi saattaa estää P-glykoproteiinin (P-gp:n) ja rintasyövän resistenssiproteiinin (BCRP:n) toimintaa suolessa. Tämä voi lisätä systeemistä altistusta näiden kuljettajaproteiinien substraateille, kuten dabigatraanieteksilaatille, siklosporiinille, rosuvastatiinille sekä mahdollisesti digoksiinille. Tällaisten lääkeaineiden osalta suositellaan hoidon seurantaa (therapeutic drug monitoring, TDM) tai kliinistä seurantaa.
On mahdollista, että P-gp:n ja BCRP:n toiminnan mahdollinen estyminen suolessa voidaan minimoida pitämällä eri lääkkeiden annon välinen aika mahdollisimman pitkänä.
Terveillä vapaaehtoisilla koehenkilöillä suoritetun tutkimuksen perusteella ruksolitinibi ei estä CYP3A4:n substraattina toimivan, suun kautta otetun midatsolaamin metaboliaa. Näin ollen kasvanutta altistusta CYP3A4:n substraateille ei oleteta esiintyvän, jos niitä käytetään yhdessä ruksolitinibin kanssa. Toisen terveillä vapaaehtoisilla koehenkilöillä suoritetun tutkimuksen perusteella ruksolitinibi ei vaikuta suun kautta otettavan, etinyyliestradiolia ja levonorgestreelia sisältävän ehkäisyvalmisteen farmakokinetiikkaan. Näin ollen samanaikaisesti käytetyn ruksolitinibin ei oleteta heikentävän edellä mainitun yhdistelmän ehkäisytehoa.
Raskaus ja imetys
Raskaus
Jakavin käytöstä raskaana oleville naisille ei ole olemassa tietoja.
Eläimillä tehdyissä tutkimuksissa on havaittu, että ruksolitinibi on alkio- ja sikiötoksinen. Teratogeenisuutta ei ole havaittu kanilla eikä rotalla. Altistusmarginaalit olivat kuitenkin pienet suhteessa suurimpaan kliinisesti käytettävään annokseen, joten tulosten sovellettavuus käyttöön ihmisillä on siten rajallinen (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tunneta. Varmuuden vuoksi Jakavin käyttö raskauden aikana on vasta-aiheista (ks. kohta Vasta-aiheet).
Naiset, jotka voivat tulla raskaaksi / Ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä Jakavi-hoidon aikana. Jos raskaus alkaa Jakavi-hoidon aikana, on tehtävä potilaskohtainen riski-hyötyarvio ja potilaalle on annettava huolellista neuvontaa sikiöön mahdollisesti kohdistuvista riskeistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Jakavia ei pidä käyttää imetyksen aikana (ks. kohta Vasta-aiheet) ja imetys on lopetettava, kun hoito aloitetaan. Ei tiedetä, erittyvätkö ruksolitinibi ja/tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet ruksolitinibin ja sen metaboliittien erittyvän maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
Ruksolitinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläimillä tehdyissä tutkimuksissa ei ole havaittu hedelmällisyyteen kohdistuvia vaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Jakavi ei ole sedatiivinen. Jos potilasta huimaa Jakavin ottamisen jälkeen, hänen on kuitenkin vältettävä ajamista ja koneiden käyttämistä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Myelofibroosi
Yleisimmin ilmoitettuja haittavaikutuksia olivat trombosytopenia ja anemia.
Hematologisia haittavaikutuksia (kaikki Common Terminology Criteria for Adverse Events [CTCAE] -luokituksen vaikeusasteet) olivat anemia (83,8 %), trombosytopenia (80,5 %) ja neutropenia (20,8 %).
Anemia, trombosytopenia ja neutropenia ovat annosriippuvaisia.
Kolme yleisintä ei-hematologista haittavaikutusta olivat mustelmanmuodostus (33,3 %), muut verenvuodot (mukaan lukien nenäverenvuoto, toimenpiteiden jälkeiset verenvuodot ja verivirtsaisuus) (24,3 %) ja huimaus (21,9 %).
Kolme yleisintä ei-hematologista laboratorioarvojen poikkeavuutta, jotka todettiin hoidon haittavaikutuksina, olivat kohonnut ALAT-arvo (40,7 %), kohonnut ASAT-arvo (31,5 %) ja hypertriglyseridemia (25,2 %). MF-potilailla suoritetuissa vaiheen 3 tutkimuksissa ei havaittu CTCAE-luokituksen asteen 3 tai 4 hypertriglyseridemiaa tai kohonneita ASAT-arvoja eikä CTCAE-luokituksen asteen 4 kohonneita ALAT-arvoja eikä hyperkolesterolemiaa.
30,0 % potilaista keskeytti hoidon mistä tahansa syystä johtuneen haittatapahtuman vuoksi.
Polysytemia vera
Yleisimmin ilmoitettuja haittavaikutuksia olivat anemia ja kohonnut ALAT-arvo.
Hematologisina haittavaikutuksina (mitä tahansa CTCAE-luokituksen vaikeusastetta olevia) todettiin anemiaa (61,8 %), trombosytopeniaa (25,0 %) ja neutropeniaa (5,3 %). CTCAE-luokituksen asteen 3 tai 4 anemiaa ja trombosytopeniaa raportoitiin 2,9 %:lla ja 2,6 %:lla potilaista.
Kolme yleisintä ei-hematologista haittavaikutusta olivat painon nousu (20,3 %), huimaus (19,4 %) ja päänsärky (17,9 %).
Kolme yleisintä ei-hematologista laboratorioarvopoikkeavuutta (mitä tahansa CTCAE-luokituksen vaikeusastetta olevia), jotka todettiin hoidon haittavaikutuksina, olivat kohonnut ALAT-arvo (45,3 %), kohonnut ASAT-arvo (42,6 %) ja hyperkolesterolemia (34,7 %). Potilailla ei havaittu CTCAE-luokituksen asteen 4 kohonneita ALAT-arvoja eikä hyperkolesterolemiaa, mutta yksi CTCAE-luokituksen asteen 4 ASAT-arvon kohoamistapaus havaittiin.
19,4 % potilaista keskeytti hoidon mistä tahansa syystä johtuneen haittatapahtuman vuoksi.
Akuutti käänteishyljintä
Yleisimmin ilmoitettuja haittavaikutuksia REACH2-tutkimuksessa (aikuisilla ja nuorilla potilailla) olivat trombosytopenia, anemia, neutropenia, kohonnut ALAT-arvo ja kohonnut ASAT-arvo. Yleisimmin ilmoitettuja haittavaikutuksia pediatristen potilaiden yhdistetyssä aineistossa (REACH2-tutkimuksen nuoret potilaat ja REACH4-tutkimuksen pediatriset potilaat) olivat anemia, neutropenia, kohonnut ALAT-arvo, hyperkolesterolemia ja trombosytopenia.
Hematologisten laboratorioarvojen poikkeavuuksia, jotka todettiin hoidon haittavaikutuksina REACH2-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja pediatristen potilaiden yhdistetyssä aineistossa (REACH2-tutkimus ja REACH4-tutkimus), olivat trombosytopenia (85,2 % ja 55,1 %), anemia (75,0 % ja 70,8 %) ja neutropenia (65,1 % ja 70,0 %). Asteen 3 anemiaa ilmoitettiin 47,7 %:lla REACH2-tutkimuksen potilaista ja 45,8 %:lla potilaista pediatristen potilaiden yhdistetyssä aineistossa. REACH2-tutkimuksessa asteen 3 trombosytopeniaa ilmoitettiin 31,3 %:lla potilaista ja asteen 4 trombosytopeniaa 47,7 %:lla potilaista, ja pediatristen potilaiden yhdistetyssä aineistossa asteen 3 trombosytopeniaa ilmoitettiin 14,6 %:lla potilaista ja asteen 4 trombosytopeniaa 22,4 %:lla potilaista. REACH2-tutkimuksessa asteen 3 neutropeniaa ilmoitettiin 17,9 %:lla potilaista ja asteen 4 neutropeniaa 20,6 %:lla potilaista, ja pediatristen potilaiden yhdistetyssä aineistossa asteen 3 neutropeniaa ilmoitettiin 32,0 %:lla potilaista ja asteen 4 neutropeniaa 22,0 %:lla potilaista.
Yleisimmät ei-hematologiset haittavaikutukset REACH2-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja pediatristen potilaiden yhdistetyssä aineistossa (REACH2-tutkimus ja REACH4-tutkimus) olivat sytomegaloviruksen (CMV) aiheuttama infektio (32,3 % ja 31,4 %), sepsis (25,4 % ja 9,8 %), virtsatieinfektiot (17,9 % ja 9,8 %), hypertensio (13,4 % ja 17,6 %) ja pahoinvointi (16,4 % ja 3,9 %).
Yleisimmät ei-hematologiset laboratorioarvojen poikkeavuudet, jotka todettiin hoidon haittavaikutuksina REACH2-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja pediatristen potilaiden yhdistetyssä aineistossa (REACH2-tutkimus ja REACH4-tutkimus), olivat kohonnut ALAT-arvo (54,9 % ja 63,3 %), kohonnut ASAT-arvo (52,3 % ja 50,0 %) ja hyperkolesterolemia (49,2 % ja 61,2 %). Useimmat poikkeavuudet olivat astetta 1 tai 2, mutta asteen 3 kohonnut ALAT-arvo ilmoitettiin REACH2-tutkimuksessa 17,6 %:lla potilaista ja pediatristen potilaiden yhdistetyssä aineistossa 27,3 %:lla potilaista.
Hoidon keskeytti mistä tahansa syystä johtuneen haittatapahtuman vuoksi 29,4 % REACH2-tutkimuksen potilaista ja 21,6 % pediatristen potilaiden yhdistetyn aineiston potilaista.
Krooninen käänteishyljintä
Yleisimmin ilmoitettuja haittavaikutuksia REACH3-tutkimuksessa (aikuisilla ja nuorilla potilailla) olivat anemia, hyperkolesterolemia ja kohonnut ASAT-arvo. Yleisimmin ilmoitettuja haittavaikutuksia pediatristen potilaiden yhdistetyssä aineistossa (REACH3-tutkimuksen nuoret potilaat ja REACH5-tutkimuksen pediatriset potilaat) olivat neutropenia, hyperkolesterolemia ja kohonnut ALAT-arvo.
Hematologisten laboratorioarvojen poikkeavuuksia, jotka todettiin hoidon haittavaikutuksina REACH3-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja pediatristen potilaiden yhdistetyssä aineistossa (REACH3-tutkimus ja REACH5-tutkimus), olivat anemia (68,6 % ja 49,1 %), neutropenia (36,2 % ja 59,3 %) ja trombosytopenia (34,4 % ja 35,2 %). Asteen 3 anemiaa ilmoitettiin 14,8 %:lla REACH3-tutkimuksen potilaista ja 17,0 %:lla pediatristen potilaiden yhdistetyn aineiston potilaista. REACH3-tutkimuksessa asteen 3 neutropeniaa ilmoitettiin 9,5 %:lla potilaista ja asteen 4 neutropeniaa 6,7 %:lla potilaista, ja pediatristen potilaiden yhdistetyssä aineistossa asteen 3 neutropeniaa ilmoitettiin 17,3 %:lla potilaista ja asteen 4 neutropeniaa 11,1 %:lla potilaista. REACH3-tutkimuksen aikuisilla ja nuorilla potilailla asteen 3 trombosytopeniaa ilmoitettiin 5,9 %:lla potilaista ja asteen 4 trombosytopeniaa 10,7 %:lla. Pediatristen potilaiden yhdistetyssä aineistossa asteen 3 trombosytopeniaa ilmoitettiin 7,7 %:lla potilaista ja asteen 4 trombosytopeniaa 11,1 %:lla potilaista.
Yleisimmät ei-hematologiset haittavaikutukset REACH3-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja pediatristen potilaiden yhdistetyssä aineistossa (REACH3-tutkimus ja REACH5-tutkimus) olivat hypertensio (15,0 % ja 14,5 %) ja päänsärky (10,2 % ja 18,2 %).
Yleisimmät ei-hematologiset laboratorioarvojen poikkeavuudet, jotka todettiin hoidon haittavaikutuksina, olivat REACH3-tutkimuksessa (aikuisilla ja nuorilla potilailla) ja yhdistetyssä pediatristen potilaiden aineistossa (REACH3-tutkimus ja REACH5-tutkimus) hyperkolesterolemia (52,3 % ja 54,9 %), kohonnut ASAT-arvo (52,2 % ja 45,5 %) ja kohonnut ALAT-arvo (43,1 % ja 50,9 %). Useimmat poikkeavuudet olivat astetta 1 tai 2, mutta pediatristen potilaiden yhdistetyssä aineistossa ilmoitettiin seuraavia laboratorioarvojen asteen 3 poikkeavuuksia: kohonnut ALAT-arvo (14,9 %) ja kohonnut ASAT-arvo (11,5 %).
Hoidon keskeytti mistä tahansa syystä johtuneen haittatapahtuman vuoksi 18,1 % REACH3-tutkimuksen potilaista ja 14,5 % pediatristen potilaiden yhdistetyn aineiston potilaista.
Haittavaikutustaulukko
Jakavin turvallisuutta MF-potilailla arvioitiin kahden vaiheen 3 tutkimuksen (COMFORT-I ja COMFORT-II) pitkäaikaisseurantatietojen perusteella. Mukana oli tietoja potilaista, jotka oli aluksi satunnaistettu ruksolitinibihoitoon (n = 301), ja potilaista, jotka saivat ruksolitinibia siirryttyään vertailuhoidosta ruksolitinibihoitoon (n = 156). Mediaanialtistus, johon MF-potilailla todettujen haittavaikutusten yleisyysluokitus perustuu, oli 30,5 kk (vaihteluväli 0,3–68,1 kk).
Jakavin turvallisuutta PV-potilailla arvioitiin kahden vaiheen 3 tutkimuksen (RESPONSE ja RESPONSE 2) pitkäaikaisseurantatietojen perusteella. Mukana oli tietoja potilaista, jotka oli aluksi satunnaistettu ruksolitinibihoitoon (n = 184), ja potilaista, jotka saivat ruksolitinibia siirryttyään vertailuhoidosta ruksolitinibihoitoon (n = 156). Mediaanialtistus, johon PV-potilailla todettujen haittavaikutusten yleisyysluokitus perustuu, oli 41,7 kk (vaihteluväli 0,03–59,7 kk).
Jakavin turvallisuutta akuutin käänteishyljinnän hoidossa arvioitiin vaiheen 3 REACH2-tutkimuksessa ja vaiheen 2 REACH4-tutkimuksessa. REACH2-tutkimuksen tiedot koskivat 201:tä ≥ 12-vuotiasta potilasta, jotka oli aluksi satunnaistettu Jakavi-hoitoon (n = 152) tai saivat Jakavia siirryttyään parhaasta saatavilla olevasta hoidosta Jakavi-hoitoon (n = 49). Mediaanialtistus, johon haittavaikutusten yleisyysluokitus perustuu, oli 8,9 viikkoa (vaihteluväli 0,3–66,1 viikkoa). Pediatristen vähintään 2-vuotiaiden potilaiden yhdistetyssä aineistossa (6 potilasta REACH2-tutkimuksesta ja 45 potilasta REACH4-tutkimuksesta) mediaanialtistus oli 16,7 viikkoa (vaihteluväli 1,1–48,9 viikkoa).
Jakavin turvallisuutta kroonisen käänteishyljinnän hoidossa arvioitiin vaiheen 3 REACH3-tutkimuksessa ja vaiheen 2 REACH5-tutkimuksessa. REACH3-tutkimuksen tiedot koskivat 226:ta ≥ 12-vuotiasta potilasta, jotka oli aluksi satunnaistettu Jakavi-hoitoon (n = 165) tai saivat Jakavia siirryttyään parhaasta saatavilla olevasta hoidosta Jakavi-hoitoon (n = 61). Mediaanialtistus, johon haittavaikutusten yleisyysluokitus perustuu, oli 41,4 viikkoa (vaihteluväli 0,7–127,3 viikkoa). Pediatristen vähintään 2-vuotiaiden potilaiden yhdistetyssä aineistossa (10 potilasta REACH3-tutkimuksesta ja 45 potilasta REACH5-tutkimuksesta) mediaanialtistus oli 57,1 viikkoa (vaihteluväli 2,1–155,4 viikkoa).
Kliinisessä tutkimusohjelmassa haittavaikutusten vaikeusaste arvioitiin CTCAE-luokituksen perusteella ja käyttäen seuraavia määritelmiä: aste 1 = lievä, aste 2 = keskivaikea, aste 3 = vaikea, aste 4 = henkeä uhkaava tai invalidisoiva ja aste 5 = kuolema.
Kliinisissä tutkimuksissa MF:n ja PV:n hoidon yhteydessä ilmoitetut haittavaikutukset (taulukko 6) ja akuutin ja kroonisen käänteishyljinnän hoidon yhteydessä ilmoitetut haittavaikutukset (taulukko 7) esitetään MedDRA-elinjärjestelmäluokittain. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa niiden yleisyyden mukaan yleisimmistä alkaen. Kunkin haittavaikutuksen kohdalla mainittava yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 6 Vaiheen 3 tutkimuksissa MF:n ja PV:n hoidon yhteydessä raportoitujen haittavaikutusten yleisyysluokitukset
| Haittavaikutus | Yleisyysluokitus MF-potilailla | Yleisyysluokitus PV-potilailla |
| Infektiot | ||
| Virtsatietulehduksetd | Hyvin yleinen | Hyvin yleinen |
| Vyöruusud | Hyvin yleinen | Hyvin yleinen |
| Keuhkokuume | Hyvin yleinen | Yleinen |
| Sepsis | Yleinen | Melko harvinainen |
| Tuberkuloosi | Melko harvinainen | Tuntematone |
| HBV:n reaktivaatio | Tuntematone | Melko harvinainen |
| Veri ja imukudosa,d | ||
| Anemiaa | ||
CTCAEc-luokituksen aste 4 (< 65 g/l) | Hyvin yleinen | Melko harvinainen |
CTCAEc-luokituksen aste 3 (< 80–65 g/l) | Hyvin yleinen | Yleinen |
| Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Trombosytopeniaa | ||
CTCAEc-luokituksen aste 4 (< 25x 109/l) | Yleinen | Melko harvinainen |
CTCAEc-luokituksen aste 3 (50–25 x 109/l) | Hyvin yleinen | Yleinen |
| Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Neutropeniaa | ||
CTCAEc-luokituksen aste 4 (< 0,5 x 109/l) | Yleinen | Melko harvinainen |
CTCAEc-luokituksen aste 3 (< 1,0–0,5 x 109/l) | Yleinen | Melko harvinainen |
| Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Yleinen |
| Pansytopeniaa,b | Yleinen | Yleinen |
| Verenvuoto (mikä tahansa verenvuoto, mukaan lukien kallonsisäiset ja ruoansulatuskanavan verenvuodot, mustelmat ja muut verenvuodot) | Hyvin yleinen | Hyvin yleinen |
| Mustelmat | Hyvin yleinen | Hyvin yleinen |
| Ruoansulatuskanavan verenvuoto | Hyvin yleinen | Yleinen |
| Kallonsisäinen verenvuoto | Yleinen | Melko harvinainen |
| Muut verenvuodot (mukaan lukien nenäverenvuoto, toimenpiteiden jälkeiset verenvuodot ja verivirtsaisuus) | Hyvin yleinen | Hyvin yleinen |
| Aineenvaihdunta ja ravitsemus | ||
Hyperkolesterolemiaa Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
Hypertriglyseridemiaa Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Painon nousu | Hyvin yleinen | Hyvin yleinen |
| Hermosto | ||
| Huimaus | Hyvin yleinen | Hyvin yleinen |
| Päänsärky | Hyvin yleinen | Hyvin yleinen |
| Ruoansulatuselimistö | ||
| Kohonnut lipaasiarvo, mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Ummetus | Hyvin yleinen | Hyvin yleinen |
| Ilmavaivat | Yleinen | Yleinen |
| Maksa ja sappi | ||
| Kohonnut ALAT-arvoa | ||
CTCAEc-luokituksen aste 3 (> 5 x - 20 x ULN) | Yleinen | Yleinen |
| Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Kohonnut ASAT-arvoa | ||
| Mikä tahansa CTCAEc-luokituksen aste | Hyvin yleinen | Hyvin yleinen |
| Verisuonisto | ||
| Hypertensio | Hyvin yleinen | Hyvin yleinen |
| a Esiintymistiheys perustuu lähtötasoon verrattuna uusiin tai vaikeutuneisiin laboratorioarvojen poikkeavuuksiin. | ||
| b Pansytopenia määritellään seuraavasti: laboratoriokokeissa samanaikaisesti hemoglobiini < 100 g/l, trombosyytit < 100 x 109/l ja neutrofiilit < 1,5 x 109/l (tai neutrofiiliarvon puuttuessa matalat veren valkosoluarvot, aste 2) | ||
| c Common Terminology Criteria for Adverse Events ‑luokituksen (CTCAE) versio 3.0; aste 1 = lievä, aste 2 = keskivaikea, aste 3 = vaikea, aste 4 = henkeä uhkaava | ||
| d Näistä haittavaikutuksista kerrotaan tekstiosassa. | ||
| e Markkinoilletulon jälkeen ilmoitettu haittavaikutus. | ||
Hoidon lopettamisen yhteydessä MF:n oireet kuten uupumus, luukipu, kuume, kutina, yöhikoilu, oireinen splenomegalia ja painon lasku saattavat uusiutua MF-potilailla. Kliinisissä MF-tutkimuksissa MF:n kokonaisoirepistemäärä palautui vähitellen lähtötasolle 7 päivän kuluessa hoidon lopettamisesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 7 Käänteishyljintää koskeneissa kliinisissä tutkimuksissa raportoitujen haittavaikutusten yleisyysluokitus
| Akuutti käänteishyljintä (REACH2) | Akuutti käänteishyljintä (pediatrinen yhdistetty aineisto) | Krooninen käänteishyljintä (REACH3) | Krooninen käänteishyljintä (pediatrinen yhdistetty aineisto) | |
| Haittavaikutus | Yleisyysluokitus | Yleisyysluokitus | Yleisyysluokitus | Yleisyysluokitus |
| Infektiot | ||||
| CMV-infektiot | Hyvin yleinen | Hyvin yleinen | Yleinen | Yleinen |
| CTCAE3-luokituksen aste ≥ 3 | Hyvin yleinen | Yleinen | Yleinen | Ei oleellinen5 |
| Sepsis | Hyvin yleinen | Yleinen | -6 | -6 |
| CTCAE-luokituksen aste ≥ 34 | Hyvin yleinen | Yleinen | -6 | -6 |
| Virtsatieinfektiot | Hyvin yleinen | Yleinen | Yleinen | Yleinen |
| CTCAE-luokituksen aste ≥ 3 | Yleinen | Yleinen | Yleinen | Yleinen |
| BK-viruksen aiheuttamat infektiot | -6 | -6 | Yleinen | Yleinen |
| CTCAE-luokituksen aste ≥ 3 | -6 | -6 | Melko harvinainen | Ei oleellinen5 |
| Veri ja imukudos | ||||
| Trombosytopenia1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Hyvin yleinen | Hyvin yleinen | Yleinen | Yleinen |
| CTCAE-luokituksen aste 4 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Anemia1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Neutropenia1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Hyvin yleinen | Hyvin yleinen | Yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 4 | Hyvin yleinen | Hyvin yleinen | Yleinen | Hyvin yleinen |
| Pansytopenia1,2 | Hyvin yleinen | Hyvin yleinen | -6 | -6 |
| Aineenvaihdunta ja ravitsemus | ||||
| Hyperkolesterolemia1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Yleinen | Ei oleellinen5 | Yleinen | Yleinen |
| CTCAE-luokituksen aste 4 | Yleinen | Ei oleellinen5 | Melko harvinainen | Yleinen |
| Painonnousu | -6 | -6 | Yleinen | Yleinen |
| CTCAE-luokituksen aste ≥ 3 | -6 | -6 | Ei oleellinen5 | Yleinen |
| Hermosto | ||||
| Päänsärky | Yleinen | Yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste ≥ 3 | Melko harvinainen | Ei oleellinen5 | Yleinen | Yleinen |
| Verisuonisto | ||||
| Hypertensio | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste ≥ 3 | Yleinen | Hyvin yleinen | Yleinen | Yleinen |
| Ruoansulatuselimistö | ||||
| Kohonnut lipaasiarvo1 | -6 | -6 | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | -6 | -6 | Yleinen | Yleinen |
| CTCAE-luokituksen aste 4 | -6 | -6 | Melko harvinainen | Yleinen |
| Kohonnut amylaasiarvo1 | -6 | -6 | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | -6 | -6 | Yleinen | Yleinen |
| CTCAE-luokituksen aste 4 | -6 | -6 | Yleinen | Ei oleellinen5 |
| Pahoinvointi | Hyvin yleinen | Yleinen | -6 | -6 |
| CTCAE-luokituksen aste ≥ 3 | Melko harvinainen | Ei oleellinen5 | -6 | -6 |
| Ummetus | -6 | -6 | Yleinen | Yleinen |
| CTCAE-luokituksen aste ≥ 3 | -6 | -6 | Ei oleellinen5 | Ei oleellinen5 |
| Maksa ja sappi | ||||
| Kohonnut ALAT-arvo1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Hyvin yleinen | Hyvin yleinen | Yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 4 | Yleinen | Ei oleellinen5 | Melko harvinainen | Yleinen |
| Kohonnut ASAT-arvo1 | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | Yleinen | Yleinen | Yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 4 | Ei oleellinen5 | Ei oleellinen5 | Melko harvinainen | Ei oleellinen5 |
| Luusto, lihakset ja sidekudos | ||||
| Kohonnut veren kreatiinifosfokinaasiarvo1 | -6 | -6 | Hyvin yleinen | Hyvin yleinen |
| CTCAE-luokituksen aste 3 | -6 | -6 | Yleinen | Ei oleellinen5 |
| CTCAE-luokituksen aste 4 | -6 | -6 | Yleinen | Ei oleellinen5 |
| Munuaiset ja virtsatiet | ||||
| Kohonnut veren kreatiniiniarvo1 | -6 | -6 | Hyvin yleinen | Yleinen |
| CTCAE-luokituksen aste 3 | -6 | -6 | Yleinen | Ei oleellinen5 |
| CTCAE-luokituksen aste 4 | -6 | -6 | Ei oleellinen5 | Ei oleellinen5 |
1 Esiintymistiheys perustuu lähtötasoon verrattuna uusiin tai vaikeutuneisiin laboratorioarvojen poikkeavuuksiin. 2 Pansytopenia määritellään seuraavasti: laboratoriokokeissa samanaikaisesti hemoglobiini < 100 g/l, trombosyytit < 100 x 109/l ja neutrofiilit < 1,5 x 109/l (tai neutrofiiliarvon puuttuessa matalat veren valkosoluarvot, aste 2). 3 CTCAE-luokituksen versio 4.03. 4 Asteen ≥ 3 sepsis sisältää 20 (10 %) asteen 5 tapahtumaa REACH2-tutkimuksesta. Pediatristen potilaiden yhdistetyssä aineistossa ei todettu asteen 5 tapahtumia. 5 Ei oleellinen: ei ilmoitettuja tapauksia 6 ” -”: ei todettu haittavaikutus tässä käyttöaiheessa. | ||||
Tiettyjen haittavaikutusten kuvaus
Anemia
Vaiheen 3 kliinisissä MF-tutkimuksissa ensimmäisen vähintään CTCAE-astetta 2 edustavan anemian alkamiseen kulunut mediaaniaika oli 1,5 kk. Yksi potilas (0,3 %) lopetti hoidon anemian vuoksi.
Ruksolitinibihoitoa saaneiden potilaiden hemoglobiiniarvon keskilasku saavutti matalimman arvonsa (noin 10 g/l alle lähtötason) 8–12 hoitoviikon jälkeen, minkä jälkeen arvo korjautui vähitellen uudelle vakaalle tasolle noin 5 g/l lähtötasoarvojen alapuolelle. Tätä ilmiötä todettiin riippumatta siitä, saiko potilas hoidon aikana verensiirron.
Satunnaistetussa, lumekontrolloidussa COMFORT-I-tutkimuksessa 60,6 % Jakavi-hoitoa saaneista ja 37,7 % lumehoitoa saaneista MF-potilaista sai punasolusiirtoja satunnaistetun hoidon aikana. COMFORT-II-tutkimuksessa 53,4 % Jakavi-ryhmän potilaista ja 41,1 % parhaan saatavilla olevan hoidon ryhmän potilaista sai punasolutiivistesiirtoja.
Avaintutkimusten satunnaistetuissa vaiheissa anemiaa ilmeni harvemmin PV-potilailla kuin MF-potilailla (40,8 % vs 82,4 %). PV-populaatiossa CTCAE-luokituksen asteen 3 ja 4 tapahtumia raportoitiin 2,7 %:lla potilaista vastaavan esiintyvyyden ollessa 42,56 % MF-potilailla.
Akuuttia (REACH2) ja kroonista (REACH3) käänteishyljintää koskevissa vaiheen 3 tutkimuksissa (minkä tahansa asteen) anemiaa ilmoitettiin 75,0 %:lla potilaista, joilla oli akuutti käänteishyljintä, ja 68,6 %:lla potilaista, joilla oli krooninen käänteishyljintä, ja CTCAE-luokituksen asteen 3 anemiaa ilmoitettiin 47,7 %:lla potilaista, joilla oli akuutti käänteishyljintä, ja 14,8 %:lla potilaista, joilla oli krooninen käänteishyljintä. Pediatrisilla potilailla (minkä tahansa asteen) anemiaa ilmoitettiin 70,8 %:lla potilaista, joilla oli akuutti käänteishyljintä, ja 49,1 %:lla potilaista, joilla oli krooninen käänteishyljintä, ja CTCAE-luokituksen asteen 3 anemiaa ilmoitettiin 45,8 %:lla potilaista, joilla oli akuutti käänteishyljintä, ja 17,0 %:lla potilaista, joilla oli krooninen käänteishyljintä.
Trombosytopenia
Vaiheen 3 kliinisissä MF-tutkimuksissa mahdollisen asteen 3 tai 4 trombosytopenian alkuun kulunut mediaaniaika oli noin 8 viikkoa. Trombosytopenia korjautui yleensä, kun annosta pienennettiin tai hoito keskeytettiin. Trombosyyttiarvojen korjautumiseen yli 50 x 109/l kulunut mediaaniaika oli 14 vrk. Tutkimusten satunnaistetun vaiheen aikana trombosyyttisiirtoja annettiin 4,7 %:lle ruksolitinibiryhmän potilaista ja 4,0 %:lle vertailuryhmien potilaista. 0,7 % ruksolitinibiryhmän potilaista ja 0,9 % vertailuryhmien potilaista lopetti hoidon trombosytopenian vuoksi. Asteen 3 tai 4 trombosytopeniaa esiintyi useammin potilailla, joiden trombosyyttiarvo oli ennen ruksolitinibihoidon alkua 100–200 x 109/l, kuin niillä, joiden trombosyyttiarvo oli > 200 x 109/l (esiintymistiheydet 64,2 % ja 38,5 %).
Avaintutkimusten satunnaistetuissa vaiheissa trombosytopeniaa kokeneiden potilaiden osuus oli pienempi PV-potilailla (16,8 %) kuin MF-potilailla (69,8 %). Vaikea-asteisen (eli CTCAE-luokituksen asteen 3 ja 4) trombosytopenian esiintyvyys oli alhaisempi PV-potilailla (2,7 %) kuin MF-potilailla (11,6 %).
Akuuttia käänteishyljintää koskevassa vaiheen 3 tutkimuksessa (REACH2) asteen 3 trombosytopeniaa ilmoitettiin 31,3 %:lla potilaista ja asteen 4 trombosytopeniaa 47,7 %:lla potilaista. Kroonista käänteishyljintää koskevassa vaiheen 3 tutkimuksessa (REACH3) asteen 3 ja 4 trombosytopenian esiintyvyys oli pienempi (asteen 3 trombosytopeniaa esiintyi 5,9 %:lla potilaista ja asteen 4 trombosytopeniaa 10,7 %:lla potilaista) kuin akuuttia käänteishyljintää sairastavilla. Asteiden 3 ja 4 trombosytopenian esiintyvyydet (14,6 % ja 22,4 %) olivat akuuttia käänteishyljintää sairastavilla pediatrisilla potilailla pienemmät kuin REACH2-tutkimuksessa. Kroonista käänteishyljintää sairastavilla pediatrisilla potilailla asteiden 3 ja 4 trombosytopenian esiintyvyydet (7,7 % ja 11,1 %) olivat pienemmät kuin akuuttia käänteishyljintää sairastavilla pediatrisilla potilailla.
Neutropenia
Vaiheen 3 kliinisissä MF-tutkimuksissa mahdollisen asteen 3 tai 4 neutropenian alkuun kulunut mediaaniaika oli 12 viikkoa. Tutkimusten satunnaistetun vaiheen aikana hoito keskeytettiin tai annosta pienennettiin neutropenian vuoksi 1,0 %:lla potilaista, ja 0,3 % potilaista lopetti hoidon neutropenian vuoksi.
PV-potilailla suoritettujen vaiheen 3 tutkimusten satunnaistetussa vaiheessa neutropeniaa raportoitiin 1,6 %:lla ruksolitinibille altistuneista potilaista ja 7 %:lla vertailuhoidoille altistuneista. Ruksolitinibiryhmässä yhdelle potilaalle kehittyi CTCAE-luokituksen asteen 4 neutropenia. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana kahdella potilaalla raportoitiin CTCAE-luokituksen asteen 4 neutropeniaa.
Akuuttia käänteishyljintää koskevassa vaiheen 3 tutkimuksessa (REACH2) asteen 3 neutropeniaa ilmoitettiin 17,9 %:lla potilaista ja asteen 4 neutropeniaa 20,6 %:lla potilaista. Kroonista käänteishyljintää koskevassa vaiheen 3 tutkimuksessa (REACH3) asteiden 3 ja 4 neutropenian esiintyvyydet (9,5 % ja 6,7 %) olivat pienemmät kuin akuuttia käänteishyljintää sairastavilla. Akuuttia käänteishyljintää sairastavilla pediatrisilla potilailla asteen 3 neutropeniaa esiintyi 32,0 %:lla ja asteen 4 neutropeniaa 22,0 %:lla potilaista ja kroonista käänteishyljintää sairastavilla pediatrisilla potilailla asteen 3 neutropeniaa esiintyi 17,3 %:lla ja asteen 4 neutropeniaa 11,1 %:lla potilaista.
Verenvuoto
Vaiheen 3 MF-avaintutkimuksissa ilmoitettiin verenvuototapahtumia (mukaan lukien kallonsisäiset ja ruoansulatuskanavan verenvuodot, mustelmanmuodostus ja muut verenvuototapahtumat) 32,6 %:lla ruksolitinibille altistuneista potilaista ja 23,2 %:lla vertailuhoidoille altistuneista (vertailuhoitona oli lume tai paras saatavilla oleva hoito). Asteen 3–4 tapahtumien esiintymistiheys oli ruksolitinibiryhmässä samaa luokkaa kuin vertailuhoitoja saaneilla (4,7 % vs 3,1 %). Valtaosa potilaista, joilla ilmeni hoidonaikaisia verenvuototapahtumia, ilmoitti mustelmanmuodostusta (65,3 %). Mustelmanmuodostusta ilmoitettiin ruksolitinibiryhmässä yleisemmin kuin vertailuhoitoja saaneilla (21,3 % vs 11,6 %). Kallonsisäistä verenvuotoa raportoitiin 1 %:lla ruksolitinibia saaneista potilaista ja 0,9 %:lla vertailuhoitoja saaneista. Ruoansulatuskanavan verenvuotoa raportoitiin 5,0 %:lla ruksolitinibia saaneista potilaista, kun vastaava luku vertailuhoitoja saaneilla oli 3,1 %. Muita verenvuototapahtumia (mukaan lukien nenäverenvuoto-, toimenpiteiden jälkeiset verenvuoto- ja verivirtsaisuustapahtumat) ilmoitettiin 13,3 %:lla ruksolitinibihoitoa saaneista potilaista ja 10,3 %:lla vertailuhoitoja saaneista.
MF-potilailla suoritettujen vaiheen 3 kliinisten tutkimusten pitkäaikaisseurannan aikana verenvuototapahtumien kumulatiivinen esiintymistiheys suureni suhteessa seuranta-ajan pitenemiseen. Mustelmanmuodostus oli yleisimmin raportoitu verenvuototapahtuma (33,3 %). Kallonsisäistä verenvuotoa raportoitiin 1,3 %:lla potilaista ja ruoansulatuskanavan verenvuotoa 10,1 %:lla potilaista.
PV-potilailla suoritettujen vaiheen 3 tutkimusten vertailevassa vaiheessa verenvuototapahtumia (mukaan lukien kallonsisäiset ja ruoansulatuskanavan verenvuodot, mustelmat ja muut verenvuototapahtumat) raportoitiin 16,8 %:lla ruksolitinibihoitoa saaneista potilaista, 15,3 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE-tutkimuksen potilaista ja 12,0 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE 2 ‑tutkimuksen potilaista. Mustelmia raportoitiin 10,3 %:lla ruksolitinibihoitoa saaneista potilaista, 8,1 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE-tutkimuksen potilaista ja 2,7 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE 2 ‑tutkimuksen potilaista. Kallonsisäistä verenvuotoa tai ruoansulatuskanavan verenvuototapahtumia ei raportoitu lainkaan ruksolitinibihoitoa saaneilla potilailla. Yksi ruksolitinibihoitoa saanut potilas koki asteen 3 verenvuototapahtuman (toimenpiteen jälkeinen verenvuoto); yhtäkään asteen 4 verenvuototapahtumaa ei raportoitu. Muita verenvuototapahtumia (mukaan lukien esim. nenäverenvuodot, toimenpiteiden jälkeiset verenvuodot, ikenien verenvuodot) raportoitiin 8,7 %:lla ruksolitinibihoitoa saaneista potilaista, 6,3 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE-tutkimuksen potilaista ja 6,7 %:lla parasta saatavilla olevaa hoitoa saaneista RESPONSE 2 ‑tutkimuksen potilaista.
PV-potilailla suoritettujen vaiheen 3 kliinisten tutkimusten pitkäaikaisseurannan aikana verenvuototapahtumien kumulatiivinen esiintymistiheys suureni suhteessa seuranta-ajan pitenemiseen. Mustelmanmuodostus oli yleisimmin raportoitu verenvuototapahtuma (17,4 %). Kallonsisäistä verenvuotoa raportoitiin 0,3 %:lla potilaista ja ruoansulatuskanavan verenvuotoa 3,5 %:lla potilaista.
Akuuttia käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH2) vertailevassa vaiheessa verenvuototapahtumia raportoitiin 25,0 %:lla ruksolitinibihoitoa saaneista ja 22,0 %:lla parasta saatavilla olevaa hoitoa saaneista. Verenvuototapahtumien alaryhmät olivat yleisesti samanlaiset eri hoitoryhmien välillä: mustelmanmuodostusta (5,9 %:lla ruksolitinibihoitoa saaneista ja 6,7 %:lla parasta saatavilla olevaa hoitoa saaneista), ruoansulatuskanavan verenvuotoa (9,2 %:lla ruksolitinibihoitoa saaneista ja 6,7 %:lla parasta saatavilla olevaa hoitoa saaneista) ja muita verenvuototapahtumia (13,2 %:lla ruksolitinibihoitoa saaneista ja 10,7 %:lla parasta saatavilla olevaa hoitoa saaneista). Kallonsisäistä verenvuotoa raportoitiin 0,7 %:lla parasta saatavilla olevaa hoitoa saaneista potilaista, ja ruksolitinibihoitoa saaneilla potilailla sitä ei raportoitu lainkaan. Pediatrisista potilaista 23,5 %:lla esiintyi verenvuototapahtumia. Tapahtumat, joita raportoitiin ≥ 5 %:lla potilaista, olivat hemorraginen kystiitti (5,9 %) ja nenäverenvuoto (5,9 %). Pediatrisilla potilailla ei raportoitu kallonsisäisiä verenvuotoja.
Kroonista käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH3) vertailevassa vaiheessa verenvuototapahtumia raportoitiin 11,5 %:lla ruksolitinibihoitoa saaneista ja 14,6 %:lla parasta saatavilla olevaa hoitoa saaneista. Verenvuototapahtumien alaryhmät olivat yleisesti samanlaiset eri hoitoryhmien välillä: mustelmanmuodostusta (4,2 %:lla ruksolitinibihoitoa saaneista ja 2,5 %:lla parasta saatavilla olevaa hoitoa saaneista), ruoansulatuskanavan verenvuotoa (1,2 %:lla ruksolitinibihoitoa saaneista ja 3,2 %:lla parasta saatavilla olevaa hoitoa saaneista) ja muita verenvuototapahtumia (6,7 %:lla ruksolitinibihoitoa saaneista ja 10,1 %:lla parasta saatavilla olevaa hoitoa saaneista). Pediatrisista potilaista 9,1 %:lla esiintyi verenvuototapahtumia. Raportoidut tapahtumat olivat nenäverenvuoto, veriuloste, hematooma, toimenpiteen jälkeinen verenvuoto ja ihon verenvuoto (1,8 % kutakin). Kallonsisäistä verenvuotoa ei raportoitu kroonista käänteishyljintää sairastavilla.
Infektiot
MF:n vaiheen 3 avaintutkimuksissa ilmoitettiin asteen 3 tai 4 virtsatieinfektioita 1,0 %:lla potilaista, vyöruusua 4,3 %:lla ja tuberkuloosia 1,0 %:lla. Vaiheen 3 kliinisissä tutkimuksissa sepsistä ilmoitettiin 3,0 %:lla potilaista. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana ei nähty viitteitä siitä, että sepsiksen esiintyvyys lisääntyisi ajan kuluessa.
Vaiheen 3 tutkimusten satunnaistetussa vaiheessa PV-potilailla raportoitiin yksi (0,5 %) CTCAE-luokituksen asteen 3, mutta ei ainuttakaan asteen 4 virtsatieinfektiota. Vyöruusun esiintyvyys oli samankaltainen PV-potilailla (4,3 %) kuin MF-potilailla (4,0 %). PV-potilailla raportoitiin yksi CTCAE-luokituksen asteen 3 postherpeettinen neuralgia -tapaus. Keuhkokuumetta raportoitiin 0,5 %:lla ruksolitinibihoitoa saaneista potilaista ja 1,6 %:lla vertailuhoitoja saaneista potilaista. Ruksolitinibiryhmän potilailla ei raportoitu sepsistä eikä tuberkuloosia.
PV-potilailla suoritettujen vaiheen 3 kliinisten tutkimusten pitkäaikaisseurannan aikana usein raportoituja infektioita olivat virtsatieinfektiot (11,8 %), vyöruusu (14,7 %) ja keuhkokuume (7,1 %). Sepsistä raportoitiin 0,6 %:lla potilaista. Tuberkuloosia ei raportoitu pitkäaikaisseurannan aikana.
Akuuttia käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH2) vertailevassa vaiheessa raportoitiin virtsatieinfektioita 9,9 %:lla (asteen ≥ 3 tasoisina 3,3 %:lla) potilaista ruksolitinibiryhmässä ja 10,7 %:lla (asteen ≥ 3 tasoisina 6,0 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. CMV-infektioita raportoitiin 28,3 %:lla (asteen ≥ 3 tasoisina 9,3 %:lla) potilaista ruksolitinibiryhmässä ja 24,0 %:lla (asteen ≥ 3 tasoisina 10,0 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. Sepsistapahtumia raportoitiin 12,5 %:lla (asteen ≥ 3 tasoisina 11,1 %:lla) potilaista ruksolitinibiryhmässä ja 8,7 %:lla (asteen ≥ 3 tasoisina 6,0 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. BK-viruksen aiheuttamia infektioita raportoitiin vain ruksolitinibiryhmässä, jossa 3 potilaalla todettiin kullakin yksi asteen 3 tapahtuma. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana virtsatieinfektioita raportoitiin 17,9 %:lla (asteen ≥ 3 tasoisina 6,5 %:lla) potilaista ja CMV-infektioita 32,3 %:lla (asteen ≥ 3 tasoisina 11,4 %:lla). Eri elimiä affisioivia CMV-infektioita todettiin hyvin harvoilla potilailla; kun kaikki vaikeusasteet otetaan huomioon, CMV-koliittia raportoitiin 4 potilaalla, CMV-enteriittiä 2 potilaalla ja CMV-infektio ruoansulatuskanavassa 1 potilaalla. Minkä tahansa vaikeusasteen sepsistapahtumia, mukaan lukien septinen sokki, raportoitiin 25,4 %:lla potilaista (asteen ≥ 3 tasoisina 21,9 %:lla). Akuuttia käänteishyljintää sairastavilla pediatrisilla potilailla virtsatieinfektioita ja sepsistapahtumia raportoitiin vähemmän (9,8 % kumpaakin) kuin aikuisilla ja nuorilla potilailla. CMV-infektioita raportoitiin 31,4 %:lla (asteen 3 tasoisina 5,9 %:lla) pediatrisista potilaista.
Kroonista käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH3) vertailevassa vaiheessa raportoitiin virtsatieinfektioita 8,5 %:lla (asteen ≥ 3 tasoisina 1,2 %:lla) potilaista ruksolitinibiryhmässä ja 6,3 %:lla (asteen ≥ 3 tasoisina 1,3 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. BK-viruksen aiheuttamia infektioita raportoitiin 5,5 %:lla (asteen ≥ 3 tasoisina 0,6 %:lla) potilaista ruksolitinibiryhmässä ja 1,3 %:lla parasta saatavilla olevaa hoitoa saaneessa ryhmässä. CMV-infektioita raportoitiin 9,1 %:lla (asteen ≥ 3 tasoisina 1,8 %:lla) potilaista ruksolitinibiryhmässä ja 10,8 %:lla (asteen ≥ 3 tasoisina 1,9 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. Sepsistapahtumia raportoitiin 2,4 %:lla (asteen ≥ 3 tasoisina 2,4 %:lla) potilaista ruksolitinibiryhmässä ja 6,3 %:lla (asteen ≥ 3 tasoisina 5,7 %:lla) parasta saatavilla olevaa hoitoa saaneessa ryhmässä. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana virtsatieinfektioita raportoitiin 9,3 %:lla (asteen ≥ 3 tasoisina 1,3 %:lla) ja BK-viruksen aiheuttamia infektioita 4,9 %:lla (asteen ≥ 3 tasoisina 0,4 %:lla). CMV-infektioita raportoitiin 8,8 %:lla potilaista (asteen ≥ 3 tasoisina 1,3 %:lla) ja sepsistapahtumia 3,5 %:lla potilaista (asteen ≥ 3 tasoisina 3,5 %:lla). Kroonista käänteishyljintää sairastavista pediatrisista potilaista 5,5 %:lla raportoitiin virtsatieinfektioita (asteen 3 tasoisena 1,8 %:lla) ja 1,8 %:lla raportoitiin BK-viruksen aiheuttama infektio (astetta ≥ 3 ei raportoitu). CMV-infektio ilmaantui 7,3 %:lle potilaista (astetta ≥ 3 ei raportoitu).
Kohonneet lipaasiarvot
RESPONSE-tutkimuksen satunnaistetussa vaiheessa lipaasiarvojen huononeminen oli ruksolitinibiryhmässä yleisempää kuin vertailuryhmässä. Tämä johtui pääosin asteen 1 kohoamistapahtumien erosta (18,2 % vs. 8,1 %). Asteen ≥ 2 kohoamistapahtumien määrä oli hoitoryhmissä samaa luokkaa. RESPONSE 2 ‑tutkimuksessa esiintymistiheys oli samaa luokkaa sekä ruksolitinibiryhmässä että vertailuryhmässä (10,8 % vs. 8 %). Vaiheen 3 PV-tutkimusten pitkäaikaisseurannan aikana asteen 3 lipaasiarvojen kohoamista raportoitiin 7,4 %:lla potilaista ja asteen 4 lipaasiarvojen kohoamista 0,9 %:lla potilaista. Kyseisillä potilailla kohonneiden lipaasiarvojen yhteydessä ei raportoitu samanaikaisia haimatulehduksen oireita eikä löydöksiä.
Vaiheen 3 MF-tutkimuksissa kohonneita lipaasiarvoja raportoitiin 18,7 %:lla ruksolitinibiryhmän potilaista ja 16,6 %:lla vertailuryhmän potilaista COMFORT‑I-tutkimuksessa sekä 19,3 %:lla ruksolitinibiryhmän potilaista ja 14,0 %:lla vertailuryhmän potilaista COMFORT‑II-tutkimuksessa. Kyseisillä potilailla kohonneiden lipaasiarvojen yhteydessä ei raportoitu samanaikaisia haimatulehduksen oireita eikä löydöksiä.
Akuuttia käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH2) vertailevassa vaiheessa raportoitiin uutta tai vaikeutunutta lipaasiarvojen kohoamista 19,7 %:lla ruksolitinibiryhmän potilaista ja 12,5 %:lla parasta saatavilla olevaa hoitoa saaneen ryhmän potilaista; asteen 3 kohoamisen (3,1 % vs. 5,1 %) ja asteen 4 kohoamisen (0 % vs. 0,8 %) esiintymistiheys oli samaa luokkaa. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana raportoitiin lipaasiarvojen kohoamista 32,2 %:lla potilaista; asteen 3 lipaasiarvojen kohoamista raportoitiin 8,7 %:lla potilaista ja asteen 4 lipaasiarvojen kohoamista 2,2 %:lla potilaista. Kohonneita lipaasiarvoja raportoitiin 20,4 %:lla pediatrisista potilaista (aste 3 8,5 %:lla ja aste 4 4,1 %:lla).
Kroonista käänteishyljintää sairastavilla potilailla suoritetun vaiheen 3 tutkimuksen (REACH3) vertailevassa vaiheessa raportoitiin uutta tai vaikeutunutta lipaasiarvojen kohoamista 32,1 %:lla ruksolitinibiryhmän potilaista ja 23,5 %:lla parasta saatavilla olevaa hoitoa saaneen ryhmän potilaista; asteen 3 kohoamisen (10,6 % vs. 6,2 %) ja asteen 4 kohoamisen (0,6 % vs. 0 %) esiintymistiheys oli samaa luokkaa. Ruksolitinibihoitoa saaneiden potilaiden pidennetyn seurantajakson aikana raportoitiin lipaasiarvojen kohoamista 35,9 %:lla potilaista; asteen 3 lipaasiarvojen kohoamista raportoitiin 9,5 %:lla potilaista ja asteen 4 lipaasiarvojen kohoamista 0,4 %:lla potilaista. Pediatrisilla potilailla kohonneita lipaasiarvoja raportoitiin harvemmin (20,4 %:lla; aste 3 3,8 %:lla ja aste 4 1,9 %:lla).
Systolisen verenpaineen nousu
Vaiheen 3 kliinisissä MF-avaintutkimuksissa todettiin ainakin yhden tutkimuskäynnin yhteydessä systolisen verenpaineen ≥ 20 mmHg:n nousu lähtötasoon nähden 31,5 %:lla potilaista, verrattuna 19,5 %:iin vertailuhoitoa saaneista potilaista. COMFORT-I-tutkimuksessa (MF-potilailla) systolisen verenpaineen keskimääräinen muutos lähtötasosta oli 0–2 mmHg:n nousu ruksolitinibiryhmässä vs 2–5 mmHg:n lasku lumelääkeryhmässä. Tutkimuksessa COMFORT-II keskiarvot ruksolitinibihoitoa saaneiden ja vertailuhoitoa saaneiden MF-potilasryhmien osalta eivät juurikaan eronneet toisistaan.
PV-potilailla suoritetun avaintutkimuksen satunnaistetussa vaiheessa systolinen verenpaine nousi ruksolitinibihaarassa keskimäärin 0,65 mmHg ja laski BAT-haarassa keskimäärin 2 mmHg.
Erityiset potilasryhmät
Pediatriset potilaat
Turvallisuuden arviointi perustui tietoihin yhteensä 106 potilaasta, joilla oli käänteishyljintää ja joiden ikä oli 2 vuodesta alle 18 vuoteen. Näistä 51 potilasta osallistui akuutin käänteishyljinnän tutkimuksiin (45 potilasta REACH4-tutkimukseen ja 6 potilasta REACH2-tutkimukseen) ja 55 potilasta kroonisen käänteishyljinnän tutkimuksiin (45 potilasta REACH5-tutkimukseen ja 10 potilasta REACH3-tutkimukseen). Ruksolitinibia saaneilla pediatrisilla potilailla todettu turvallisuusprofiili oli samankaltainen kuin aikuispotilailla.
Iäkkäät potilaat
Turvallisuuden arviointi perustui tietoihin ruksolitinibihoitoa saaneista yhteensä 29 potilaasta REACH2-tutkimuksesta ja 25 potilaasta REACH3-tutkimuksesta ja joiden ikä oli yli 65 vuotta. Uusia turvallisuuteen liittyviä tekijöitä ei tunnistettu ja turvallisuusprofiili yli 65-vuotiailla potilailla on tavallisesti samankaltainen kuin 18–65-vuotiailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Jakavin yliannokselle ei tunneta vastalääkettä. Akuutti siedettävyys on ollut hyväksyttävää enintään 200 mg kerta-annoksilla. Suositusannoksia suurempien annosten toistuvaan antoon liittyy myelosuppression lisääntymistä, mm. leukopeniaa, anemiaa ja trombosytopeniaa. Potilaalle annetaan asianmukaista elintoimintoja tukevaa hoitoa.
Hemodialyysi ei todennäköisesti tehosta ruksolitinibin eliminaatiota.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EJ01
Vaikutusmekanismi
Ruksolitinibi on Janus- eli JAK-kinaasien JAK1 ja JAK2 selektiivinen estäjä (IC50-pitoisuus JAK1-entsyymin kohdalla 3,3 nM ja JAK2-entsyymin kohdalla 2,8 nM). Nämä kinaasit välittävät useiden hematopoieesin ja immuunitoiminnan kannalta keskeisten sytokiinien ja kasvutekijöiden signalointia.
MF ja PV ovat pahanlaatuisia myeloproliferatiivisia tauteja, joiden tiedetään olevan yhteydessä JAK1- ja JAK2-signaloinnin säätelyhäiriöön. Säätelyhäiriön oletetaan perustuvan mm. suuriin kiertäviin sytokiinipitoisuuksiin, jotka aktivoivat JAK-STAT-reittiä, aktivoiviin gain-of-function-mutaatioihin (esim. JAK2V617F) ja negatiivisten säätelymekanismien vaimentumiseen. MF-potilailla todetaan JAK-signaloinnin säätelyhäiriöitä JAK2V617F-mutaatiostatuksesta riippumatta. JAK2:sta aktivoivia mutaatioita (V617F- tai eksoni 12 -mutaatiot) ilmenee > 95 %:lla PV-potilaista.
Ruksolitinibi estää JAK-STAT-signalointia ja solujen proliferaatiota verisyöpien sytokiiniriippuvaisissa solumalleissa ja Ba/F3-soluissa, joista mutatoituneen JAK2V617F-proteiinin ilmentyminen on tehnyt sytokiineista riippumattomia. Sen IC50-pitoisuus on 80–320 nM.
JAK-STAT-signaalireitit osallistuvat useiden käänteishyljinnän patogeneesin kannalta tärkeiden immuunisolutyyppien kehityksen, proliferaation ja aktivaation säätelyyn.
Farmakodynaamiset vaikutukset
Ruksolitinibi estää sytokiinien indusoimaa STAT3-fosforylaatiota terveiden henkilöiden, MF-potilaiden ja PV-potilaiden kokoveressä. Ruksolitinibin STAT3-fosforylaatioon kohdistuva estovaikutus oli suurimmillaan 2 tunnin kuluttua annostelusta ja palautui 8 tuntiin mennessä lähes lähtötasoarvoihin sekä terveillä henkilöillä että MF-potilailla. Tämä viittaa siihen, että sen paremmin kanta-aine kuin aktiiviset metaboliititkaan eivät kumuloidu elimistöön.
MF-potilailla todettiin, että yleisoireisiin liittyvien tulehdusmerkkiaineiden (esim. TNFα, IL-6 ja CRP) kohonneet lähtötasoarvot pienenivät ruksolitinibihoidon jälkeen. Potilaiden MF ei muuttunut ajan mittaan resistentiksi ruksolitinibihoidon farmakodynaamisille vaikutuksille. Myös PV-potilailla esiintyi kohonneita tulehdusmerkkiaineiden lähtötasoarvoja, jotka pienenivät ruksolitinibihoidon myötä.
Terveiden henkilöiden tarkassa QT-aikatutkimuksessa ei havaittu viitteitä siitä, että ruksolitinibi olisi pidentänyt QT-/QTc-aikaa, kun sitä annettiin enintään hoitotasoa suurempina 200 mg:n kerta-annoksina. Tämä viittaa siihen, että ruksolitinibi ei vaikuta sydämen repolarisaatioon.
Kliininen teho ja turvallisuus
Myelofibroosi
MF-potilailla (primaarinen MF tai polysytemia veran tai essentiaalisen trombosytemian jälkeinen MF) tehtiin kaksi satunnaistettua vaiheen 3 tutkimusta (COMFORT-I ja COMFORT-II). Molempien tutkimusten potilailla oli palpoituva splenomegalia (perna vähintään 5 cm kylkikaaren alapuolella), ja he kuuluivat IWG-työryhmän (International Working Group) konsensuskriteerien mukaan riskiluokkaan keskikorkea-2 tai suuren riskin luokkaan. Jakavi-hoidon aloitusannos perustui trombosyyttiarvoon. Potilaat, joiden trombosyyttiarvot olivat ≤ 100 x 109/l eivät soveltuneet COMFORT-tutkimuksiin, mutta 69 potilasta otettiin EXPAND-tutkimukseen, joka on Ib-vaiheen avoin, annostuksen löytämiseen tähtäävä tutkimus potilailla, joilla on MF (primaari MF, polysytemia veran jälkeinen MF tai essentiaalisen trombosytemian jälkeinen MF) ja joiden lähtötason trombosyyttiarvot ovat ≥ 50 ja < 100 x 109/l.
COMFORT-I oli kaksoissokkoutettu, satunnaistettu, lumekontrolloitu tutkimus, johon osallistuneilla 309 potilaalla oli hoitoresistentti tauti tai saatavilla olevat hoidot eivät soveltuneet heille. Ensisijainen tehon päätetapahtuma oli niiden tutkimushenkilöiden osuus, joilla pernan tilavuus oli viikolla 24 tehdyssä magneetti- (MRI) tai tietokonetomografia (TT) -kuvauksessa ≥ 35 % lähtötasoarvoa pienempi.
Toissijaisia päätetapahtumia olivat aika, jonka pernan tilavuus pysyi ≥ 35 % lähtötasoarvoa pienempänä; niiden potilaiden osuus, joiden muokatulla MFSAF-päiväkirjalla (MF Symptom Assessment Form, versio 2.0) mitatut kokonaisoirepisteet pienenivät viikkoon 24 mennessä ≥ 50 % lähtötasoarvoista; kokonaisoirepisteiden muutos lähtötasosta viikkoon 24 mennessä; ja kokonaiselossaolo.
COMFORT-II oli avoin, satunnaistettu tutkimus, johon osallistui 219 potilasta. Heidät satunnaistettiin suhteessa 2:1 saamaan joko ruksolitinibihoitoa tai parasta saatavilla olevaa hoitoa. Parhaan saatavilla olevan hoidon ryhmässä 47 % potilaista sai hydroksiureaa ja 16 % glukokortikoideja. Ensisijainen tehon päätetapahtuma oli niiden tutkimushenkilöiden osuus, joilla pernan tilavuus oli viikolla 48 tehdyssä magneetti- tai TT-kuvauksessa ≥ 35 % lähtötasoarvoa pienempi.
Toissijaisiin päätetapahtumiin kuuluivat niiden potilaiden osuus, joilla pernan tilavuus viikolla 24 oli ≥ 35 % lähtötasoarvoa pienempi sekä aika, jonka pernan tilavuus pysyi ≥ 35 % lähtötasoarvoja pienempänä.
COMFORT-I- ja COMFORT-II-tutkimusten eri hoitoryhmien potilaat olivat verrattavissa lähtötilanteen demografisten tietojen ja tautipiirteiden suhteen.
Taulukko 8 Potilaat, joiden pernan tilavuus pieneni ≥ 35 % lähtötasoarvosta viikkoon 24 mennessä COMFORT-I-tutkimuksessa ja viikkoon 48 mennessä COMFORT-II-tutkimuksessa (hoitoaikomuspopulaatio [ITT]; % potilaista)
| COMFORT-I | COMFORT-II | |||
Jakavi (N=155) | Lume (N=153) | Jakavi (N=144) | Paras saatavilla oleva hoito (N=72) | |
| Ajankohta | Viikko 24 | Viikko 48 | ||
| Potilaat, joiden pernan tilavuus pieneni ≥ 35 %; lukumäärä (%) | 65 (41,9) | 1 (0,7) | 41 (28,5) | 0 |
| 95 % luottamusvälit | 34,1; 50,1 | 0; 3,6 | 21,3; 36,6 | 0,0; 5,0 |
| p-arvo | < 0,0001 | < 0,0001 | ||
Jakavi-ryhmässä oli merkitsevästi yleisempää, että pernan tilavuus pieneni ≥ 35 % lähtötasoarvosta (taulukko 8). Ero ei riippunut siitä, oliko potilaalla JAK2V617F-mutaatiota (taulukko 9) tai mikä taudin alatyyppi oli kyseessä (primaarinen MF tai polysytemia veran tai essentiaalisen trombosytemian jälkeinen MF).
Taulukko 9Sellaisten potilaiden prosentuaaliset osuudet, joiden pernan tilavuus pieneni ≥ 35 %, jaoteltuina potilaiden JAK-mutaatioluokituksen mukaan (turvallisuuspopulaatio)
| COMFORT-I | COMFORT-II | |||||||
| Jakavi | Lume | Jakavi | Paras saatavilla oleva hoito | |||||
| JAK-mutaatio-tilanne | Positiivi- set (N = 113) n (%) | Negatiivi-set (N = 40) n (%) | Positiivi-set (N = 121) n (%) | Negatiivi-set (N = 27) n (%) | Positiivi-set (N = 110) n (%) | Negatiivi-set (N = 35) n (%) | Positiivi-set (N = 49) n (%) | Negatii-viset (N = 20) n (%) |
| Potilaat, joiden pernan tilavuus pieneni ≥ 35 %; lukumäärä (%) | 54 (47,8) | 11 (27,5) | 1 (0,8) | 0 | 36 (32,7) | 5 (14,3) | 0 | 0 |
| Ajankohta | 24 viikon jälkeen | 48 viikon jälkeen | ||||||
Todennäköisyys pernavasteen säilymiseksi (≥ 35 % pienentyminen) vähintään 24 viikon ajan Jakavi-hoidolla oli 89 % COMFORT-I-tutkimuksessa ja 87 % COMFORT-II-tutkimuksessa. 52 %:lla pernavaste säilyi vähintään 48 viikon ajan COMFORT-II-tutkimuksessa.
COMFORT-I-tutkimuksessa 45,9 % Jakavi-ryhmään kuuluneista potilaista saavutti ≥ 50 % MFSAF-päiväkirjalla (Myelofibrosis Symptom Assessment Form, versio 2.0) mitattujen kokonaisoirepisteiden pienentymisen suhteessa lähtötasoon viikkoon 24 mennessä. Vastaava osuus lumelääkeryhmässä oli 5,3 % (p < 0,0001 khiin neliötestillä). Keskimääräinen muutos kokonaisterveydentilassa viikolla 24 (mitattu EORTC QLQ-C30 -kyselyn avulla) oli +12,3 Jakavi-ryhmässä ja -3,4 lumelääkeryhmässä (p < 0,0001).
COMFORT-I-tutkimuksessa kuolleisuusluku 34,3 kuukauden mediaaniseuranta-ajan jälkeen oli ruksolitinibihoitoon satunnaistettujen potilaiden keskuudessa 27,1 %. Vastaava luku lumelääkeryhmässä oli 35,1 % (riskisuhde HR: 0,687; 95 %:n luottamusväli 0,459; 1,029; p = 0,0668).
COMFORT-I-tutkimuksessa todettiin 61,7 kuukauden mediaaniseuranta-ajan jälkeen, että ruksolitinibihoitoon satunnaistettujen potilaiden kuolleisuusluku oli 44,5 % (69 potilasta 155:stä) ja lumelääkehoitoon satunnaistettujen kuolleisuusluku 53,2 % (82 potilasta 154:stä). Ruksolitinibiryhmässä kuoleman riski oli 31 % pienempi kuin lumelääkeryhmässä (riskisuhde HR 0,69; 95 %:n luottamusväli 0,50; 0,96; p=0,025).
COMFORT-II-tutkimuksessa kuolleisuusluku 34,7 kuukauden mediaaniseuranta-ajan jälkeen oli ruksolitinibihoitoon satunnaistettujen potilaiden keskuudessa 19,9 %. Vastaava luku parasta saatavilla olevaa hoitoa (BAT) saaneiden ryhmään satunnaistettujen potilaiden keskuudessa oli 30,1 % (riskisuhde HR: 0,48; 95 %:n luottamusväli 0,28; 0,85; p = 0,009). Molemmissa tutkimuksissa todettu pienempi kuolleisuus ruksolitinibihoitoa saaneiden ryhmässä oli pääasiassa seurausta polysytemia veran jälkeistä myelofibroosia ja essentiellin trombosytoosin jälkeistä myelofibroosia sairastavien potilaiden alaryhmistä saaduista tuloksista.
COMFORT-II-tutkimuksessa todettiin 55,9 kuukauden mediaaniseuranta-ajan jälkeen, että ruksolitinibihoitoon satunnaistettujen potilaiden kuolleisuusluku oli 40,4 % (59 potilasta 146:sta) ja parhaan saatavilla olevan hoidon ryhmään satunnaistettujen kuolleisuusluku 47,9 % (35 potilasta 73:sta). Ruksolitinibiryhmässä kuoleman riski oli 33 % pienempi kuin parhaan saatavilla olevan hoidon ryhmässä (riskisuhde HR 0,67; 95 %:n luottamusväli 0,44; 1,02; p = 0,062).
Polysytemia vera
Satunnaistettu, avoin, aktiivikontrolloitu vaiheen 3 tutkimus (RESPONSE) suoritettiin 222 PV-potilaalla, jotka olivat Euroopan leukemiaverkoston (European LeukemiaNet, ELN) kansainvälisen työryhmän julkaisemien kriteerien perusteella resistenttejä hydroksiureahoidolle tai eivät sietäneet hydroksiureaa. Näistä 110 potilasta satunnaistettiin tutkimuksen ruksolitinibihaaraan ja 112 potilasta BAT-haaraan. Jakavin aloitusannos oli 10 mg kahdesti vuorokaudessa. Annoksia säädettiin tämän jälkeen yksilöllisesti potilaiden sietokyvyn ja lääkkeen tehon mukaan siten, että enimmäisannos oli 25 mg kahdesti vuorokaudessa. Tutkija valitsi BAT-hoidon potilaskohtaisesti. Vaihtoehtoihin kuuluivat hydroksiurea (59,5 %), interferoni/pegyloitu interferoni (11,7 %), anagrelidi (7,2 %), pipobromaani (1,8 %) sekä seuranta (15,3 %).
Lähtötason demografia ja tautiominaisuudet olivat toisiinsa verrannolliset molemmissa hoitohaaroissa. Osallistujien mediaani-ikä oli 60 vuotta (vaihteluväli: 33–90 vuotta). Ruksolitinibiryhmään kuuluvien potilaiden PV-diagnoosista oli keskimäärin (mediaani) kulunut 8,2 vuotta ja he olivat aiemmin saaneet hydroksiureaa keskimäärin (mediaani) noin 3 vuoden ajan. Useimmat potilaat (> 80 %) olivat läpikäyneet ainakin kaksi flebotomiaa viimeksi kuluneiden 24 viikon aikana ennen seulontaa. Vertailevaa tietoa pitkäaikaiseloonjäämisestä ja tautikomplikaatioiden ilmaantuvuudesta ei ole.
Ensisijainen yhdistetty päätemuuttuja oli sellaisten potilaiden osuus, jotka saavuttivat sekä flebotomiakelpoisuuden puuttumisen (hematokriitin, eli HCT-kontrollin saavuttaminen) että pernatilavuuden ≥ 35 % pienenemisen lähtötasosta viikkoon 32 mennessä. Flebotomiakelpoisuus määriteltiin vahvistettuna > 45 % hematokriittiarvona, eli ainakin 3 prosenttiyksikköä lähtötasoa korkeampana HCT-arvona tai > 48 % HCT-arvona, riippuen siitä, kumpi näistä oli alhaisempi. Tärkeimpiin toissijaisiin päätemuuttujiin kuuluivat sellaisten potilaiden osuus, jotka saavuttivat ensisijaisen päätemuuttujan ja joilla tauti ei ollut edennyt viikkoon 48 mennessä, sekä sellaisten potilaiden osuus, jotka olivat saavuttaneet täydellisen hematologisen remission viikolla 32.
Tutkimuksessa saavutettiin ensisijaiset tavoitteet, ja suurempi osuus Jakavia saaneista potilaista saavutti yhdistetyn ensisijaisen päätemuuttujan sekä sen jokaisen osakomponentin verrattuna vertailuhoitoon. Merkitsevästi useampi Jakavi-hoitoa saanut potilas (23 %) saavutti ensisijaisen vasteen (p < 0,001) verrattuna BAT-hoitoa saaneisiin potilaisiin (0,9 %). Hematokriittikontrolli saavutettiin 60 %:lla Jakavia saaneista potilaista verrattuna 18,8 %:iin BAT-hoitohaaran potilaista. Pernatilavuuden pieneneminen ≥ 35 % saavutettiin puolestaan 40 %:lla Jakavi-haaran potilaista ja 0,9 %:lla BAT-hoitoa saaneista potilaista (Kuvaaja 1).
Tutkimuksessa saavutettiin myös molemmat tärkeimmät sekundaariset päätemuuttujat. Täydellisen hematologisen remission saavuttaneiden potilaiden osuudet olivat 23,6 % Jakavi-haarassa ja 8,0 % BAT-haarassa (p = 0,0013) ja sellaisten potilaiden osuudet, jotka olivat saavuttaneet vakaan primaarivasteen viikolla 48, olivat 20 % Jakavin osalta ja 0,9 % BAT-hoidon osalta (p < 0,0001).
Kuvaaja 1 Primaarisen päätemuuttujan ja sen osakomponenttien saavuttaneiden potilaiden osuudet viikolla 32
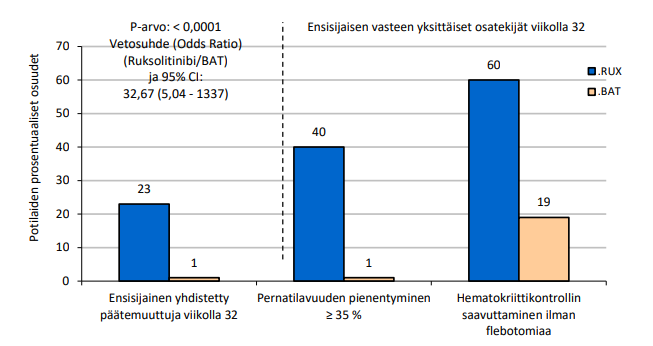
Potilaiden oirekuormaa arvioitiin elektroniseen potilaspäiväkirjaan tehtyjen merkintöjen (14 kysymystä) perusteella laskettujen MPN-SAF-kokonaispisteiden (TSS) avulla. Viikolla 32 ruksolitinibihoitoa saaneista potilaista 49 % saavutti ≥ 50 % TSS-14-pisteiden alenemisen ja 64 % ≥ 50 % TSS-5-pisteiden alenemisen verrattuna vain 5 %:iin ja 11 %:iin BAT-hoitoa saaneista potilaista.
Tuntemukset hoidosta saatavasta hyödystä mitattiin Patient Global Impression of Change (PGIC) ‑kyselyn avulla. 66 % ruksolitinibihoitoa saaneista ja 19 % BAT-hoitoa saaneista potilaista ilmoitti tilanteensa kohentumisesta jo niinkin varhain kuin neljän viikon kuluttua hoidon aloittamisesta. Ruksolitinibia saaneiden ryhmässä hoidosta saatavan hyödyn tuntemukset olivat lisäksi kasvaneet enemmän kuin BAT-ryhmässä viikolla 32 (78 % vs 33 %).
RESPONSE-tutkimuksen tiedoista tehtiin lisäanalyysejä, joilla arvioitiin vasteen säilymistä viikolla 80 ja viikolla 256 satunnaistamisen jälkeen. Niistä 25 potilaasta, jotka olivat saavuttaneet primaarisen vasteen viikolla 32, 3 potilaalla todettiin etenemistä viikkoon 80 mennessä ja 6 potilaalla viikkoon 256 mennessä. Todennäköisyys, että vaste säilyi viikosta 32 viikkoon 80, oli 92 %, ja todennäköisyys, että vaste säilyi viikosta 32 viikkoon 256, oli 74 % (ks. taulukko 10).
Taulukko 10 Primaarisen vasteen säilyminen RESPONSE-tutkimuksessa
| Viikko 32 | Viikko 80 | Viikko 256 | |
Primaarinen vaste saavutettu viikon 32 kohdalla* n/N (%) | 25/110 (23 %) | ei oleellinen | ei oleellinen |
| Potilaat, joilla primaarinen vaste säilyi | ei oleellinen | 22/25 | 19/25 |
| Primaarisen vasteen säilymisen todennäköisyys | ei oleellinen | 92 % | 74 % |
| * Primaarisen vasteen yhdistettyä päätemuuttujaa koskevien kriteerien mukaisesti: sekä flebotomiakelpoisuuden puuttuminen (HCT-kontrollin saavuttaminen) että pernatilavuuden pieneneminen ≥ 35 % lähtötasosta. | |||
Toiseen satunnaistettuun, avoimeen, aktiivikontrolloituun vaiheen 3b tutkimukseen (RESPONSE 2) osallistui 149 PV-potilasta, jotka olivat resistenttejä hydroksiureahoidolle tai eivät sietäneet sitä mutta joilla ei ollut palpoituvaa splenomegaliaa. Ensisijainen päätetapahtuma määriteltiin niiden potilaiden osuudeksi, jotka saavuttivat HCT-kontrollin (ei flebotomiakelpoisuutta) viikolla 28, ja se saavutettiin (62,2 % Jakavi-ryhmässä ja 18,7 % BAT-ryhmässä). Tärkein toissijainen päätetapahtuma määriteltiin niiden potilaiden osuudeksi, jotka saavuttivat täydellisen hematologisen remission viikolla 28; tämäkin päätetapahtuma saavutettiin (23,0 % Jakavi-ryhmässä ja 5,3 % BAT-ryhmässä).
Käänteishyljintä
Kahdessa satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa Jakavia arvioitiin sellaisten vähintään 12-vuotiaiden potilaiden hoidossa, joilla oli akuutti käänteishyljintä (REACH2) tai krooninen käänteishyljintä (REACH3) allogeenisen hematopoieettisen kantasolusiirron jälkeen ja joiden vaste kortikosteroideille ja/tai muille systeemisille hoidoille oli riittämätön. Jakavin aloitusannos oli 10 mg kaksi kertaa vuorokaudessa.
Akuutti käänteishyljintä
REACH2-tutkimuksessa 309 potilasta, joilla oli asteen II–IV akuutti kortikosteroidihoitoon reagoimaton käänteishyljintä, satunnaistettiin suhteessa 1:1 saamaan joko Jakavi-hoitoa tai parasta saatavilla olevaa hoitoa. Potilaat ositettiin sen perusteella, mikä oli akuutin käänteishyljinnän vaikeusaste satunnaistamishetkellä. Käänteishyljintä määriteltiin kortikosteroidihoitoon reagoimattomaksi, jos se eteni vähintään 3 hoitopäivän jälkeen, jos vastetta ei ollut saatu 7 päivän kuluttua tai jos kortikosteroidihoidon vähittäinen pienentäminen epäonnistui.
Tutkija valitsi parhaan saatavilla olevan hoidon potilaskohtaisesti. Vaihtoehtoihin kuuluivat antitymosyyttiglobuliini (ATG), kehonulkoinen fotofereesi (ECP), mesenkymaaliset kantasolut (MSC), pieniannoksinen metotreksaatti (MTX), mykofenolaattimofetiili (MMF), mTORin estäjät (everolimuusi tai sirolimuusi), etanersepti ja infliksimabi.
Jakavi-hoidon tai parhaan saatavilla olevan hoidon lisäksi potilaat olivat saattaneet saada tavanomaista allogeenisen kantasolusiirron tukihoitoa, kuten infektiolääkkeitä ja verensiirtoja. Ruksolitinibi lisättiin kortikosteroidien ja/tai kalsineuriinin estäjien, kuten siklosporiinin tai takrolimuusin, ja/tai paikallisesti käytettävien tai inhaloitavien kortikosteroidien rinnalle ja käytön jatkaminen tehtiin hoitoyksikön ohjeiden mukaisesti.
Tutkimukseen voitiin ottaa mukaan potilaita, jotka olivat saaneet akuuttiin käänteishyljintään yhtä muuta aiempaa systeemistä hoitoa kuin kortikosteroideja tai kalsineuriinin estäjiä. Muiden akuutin käänteishyljinnän systeemiseen hoitoon tarkoitettujen lääkevalmisteiden kuin kortikosteroidien ja kalsineuriinin estäjien aiemman käytön jatkaminen oli sallittua vain, jos valmisteita käytettiin akuutin käänteishyljinnän estohoitoon (eli hoito oli aloitettu ennen akuutin käänteishyljinnän toteamista) tavanomaisen hoitokäytännön mukaisesti.
Potilaat, jotka saivat parasta saatavilla olevaa hoitoa, saivat siirtyä käyttämään ruksolitinibia päivän 28 jälkeen, jos seuraavat ehdot täyttyivät:
- ensisijaisen päätemuuttujan määritelmän mukaista vastetta (täydellinen vaste [CR] tai osittainen vaste [PR]) ei saavutettu päivään 28 mennessä, TAI
- vaste hävisi myöhemmin, ja etenemisen, haittatapahtumien komplisoiman vasteen (mixed response) tai vasteen puuttumisen kriteerit täyttyivät, minkä seurauksena tarvittiin uutta akuutin käänteishyljinnän systeemistä immunosuppressiivista lisähoitoa, JA
- kroonisen käänteishyljinnän oireita ja löydöksiä ei ollut.
Jakavi-annoksen vähittäinen pienentäminen oli sallittua päivän 56 käynnin jälkeen, jos potilaalla oli vaste hoidolle.
Lähtötilanteen demografiset tiedot ja sairauden ominaispiirteet olivat samankaltaiset molemmissa hoitoryhmissä. Potilaiden mediaani-ikä oli 54 vuotta (vaihteluväli 12–73 vuotta). Tutkimukseen osallistuneista potilaista 2,9 % oli nuoria, 59,2 % oli miehiä ja 68,9 % oli valkoihoisia. Suurimmalla osalla osallistujista oli pahanlaatuinen perussairaus.
Akuutin käänteishyljinnän vaikeusaste oli II 34 %:lla Jakavi-ryhmän potilaista ja 34 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista, III 46 %:lla Jakavi-ryhmän potilaista ja 47 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista ja IV 20 %:lla Jakavi-ryhmän potilaista ja 19 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista.
Syyt potilaiden riittämättömään vasteeseen kortikosteroidihoidolle Jakavi-ryhmässä ja parhaan saatavilla olevan hoidon ryhmässä olivat i) vasteen jääminen saavuttamatta, kun kortikosteroidihoitoa oli annettu 7 päivän ajan (46,8 %:lla Jakavi-ryhmässä ja 40,6 %:lla parhaan saatavilla olevan hoidon ryhmässä), ii) kortikosteroidiannoksen vähittäisen pienentämisen epäonnistuminen (30,5 %:lla Jakavi-ryhmässä ja 31,6 %:lla parhaan saatavilla olevan hoidon ryhmässä) tai iii) sairauden eteneminen 3 hoitopäivän jälkeen (22,7 %:lla Jakavi-ryhmässä ja 27,7 %:lla parhaan saatavilla olevan hoidon ryhmässä).
Koko potilasjoukossa akuutin käänteishyljinnän yleisimmät kohde-elimet olivat iho (54,0 %) ja alempi ruoansulatuskanava (68,3 %). Jakavi-ryhmän potilailla akuutti käänteishyljintä kohdistui useammin ihoon (60,4 %:lla) ja maksaan (23,4 %:lla) verrattuna parhaan saatavilla olevan hoidon ryhmään (ihoon kohdistuvaa 47,7 %:lla ja maksaan kohdistuvaa 16,1 %:lla).
Yleisimpiä aiemmin käytettyjä akuutin käänteishyljinnän systeemisiä hoitoja olivat kortikosteroidien ja kalsineuriinin estäjien yhdistelmät (49,4 %:lla Jakavi-ryhmän ja 49,0 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista).
Ensisijainen päätemuuttuja oli kokonaisvasteosuus (ORR) päivänä 28. Se määriteltiin niiden potilaiden osuudeksi kussakin ryhmässä, joilla saavutettiin täydellinen vaste (CR) tai osittainen vaste (PR) ilman sellaista systeemisten lisähoitojen tarvetta, joka olisi johtunut aiemmasta etenemisestä, haittatapahtumien komplisoimasta vasteesta (mixed response) tai vasteen puuttumisesta. Vasteen arvioi tutkija Harrisin ym. (2016) kriteerien perusteella.
Keskeinen toissijainen päätemuuttuja oli sellaisten potilaiden osuus, jotka olivat saavuttaneet CR- tai PR-vasteen päivään 28 mennessä ja joilla CR- tai PR-vaste säilyi päivään 56 asti.
REACH2-tutkimuksen ensisijainen tavoite saavutettiin. Kokonaisvasteosuus hoitopäivänä 28 oli Jakavi-ryhmässä suurempi (62,3 %) kuin parhaan saatavilla olevan hoidon ryhmässä (39,4 %). Hoitoryhmien välinen ero oli tilastollisesti merkitsevä (ositettu Cochrane–Mantel–Haenszelin testi p < 0,0001, kaksitahoinen; kerroinsuhde 2,64; 95 %:n luottamusväli 1,65; 4,22).
Lisäksi täydellisen vasteen saaneiden osuus oli Jakavi-ryhmässä suurempi (34,4 %) kuin parhaan saatavilla olevan hoidon ryhmässä (19,4 %).
Kokonaisvasteosuus päivänä 28 oli Jakavi-ryhmässä 76 % potilailla, joilla oli asteen II käänteishyljintä, 56 % potilailla, joilla oli asteen III käänteishyljintä, ja 53 % potilailla, joilla oli asteen IV käänteishyljintä. Parhaan saatavilla olevan hoidon ryhmässä vastaava osuus oli 51 % potilailla, joilla oli asteen II käänteishyljintä, 38 % potilailla, joilla oli asteen III käänteishyljintä, ja 23 % potilailla, joilla oli asteen IV käänteishyljintä.
Sairaus oli edennyt 2,6 %:lla niistä Jakavi-ryhmän potilaista ja 8,4 %:lla niistä parhaan saatavilla olevan hoidon ryhmän potilaista, jotka eivät olleet saaneet vastetta päivän 28 kohdalla.
Kokonaistulokset on esitetty taulukossa 11.
Taulukko 11 Kokonaisvasteosuus päivän 28 kohdalla REACH2-tutkimuksessa
Jakavi N = 154 | Paras saatavilla oleva hoito N = 155 | |||
| n (%) | 95 %:n luottamusväli | n (%) | 95 %:n luottamusväli | |
| Vasteen saaneet yhteensä | 96 (62,3) | 54,2; 70,0 | 61 (39,4) | 31,6; 47,5 |
| Kerroinsuhde (95 %:n luottamusväli) | 2,64 (1,65; 4,22) | |||
| p-arvo (kaksitahoinen) | p < 0,0001 | |||
| Täydellinen vaste | 53 (34,4) | 30 (19,4) | ||
| Osittainen vaste | 43 (27,9) | 31 (20,0) | ||
Tietojen primaarianalyysin perusteella tutkimuksen keskeinen toissijainen päätetapahtuma saavutettiin. Pitkäkestoisten vasteiden kokonaisosuus päivän 56 kohdalla oli 39,6 % (95 %:n luottamusväli 31,8; 47,8) Jakavi-ryhmässä ja 21,9 % (95 %:n luottamusväli 15,7; 29,3) parhaan saatavilla olevan hoidon ryhmässä. Hoitoryhmien välillä oli tilastollisesti merkitsevä ero (kerroinsuhde 2,38; 95 %:n luottamusväli 1,43; 3,94; p = 0,0007). Täydellisen vasteen saaneiden potilaiden osuus oli 26,6 % Jakavi-ryhmässä ja 16,1 % parhaan saatavilla olevan hoidon ryhmässä. Yhteensä 49 alun perin parhaan saatavilla olevan hoidon ryhmään satunnaistettua potilasta (31,6 %) siirtyi Jakavi-ryhmään.
Krooninen käänteishyljintä
REACH3-tutkimuksessa 329 potilasta, joilla oli keskivaikea tai vaikea kortikosteroidihoitoon reagoimaton krooninen käänteishyljintä, satunnaistettiin suhteessa 1:1 saamaan joko Jakavi-hoitoa tai parasta saatavilla olevaa hoitoa. Potilaat ositettiin sen perusteella, mikä oli kroonisen käänteishyljinnän vaikeusaste satunnaistamishetkellä. Käänteishyljintä määriteltiin kortikosteroidihoitoon reagoimattomaksi, jos vastetta ei ollut saatu tai sairaus eteni 7 päivän jälkeen, jos sairaus jatkui 4 viikkoa tai jos kortikosteroidiannoksen vähittäinen pienentäminen epäonnistui kahdesti.
Tutkija valitsi parhaan saatavilla olevan hoidon potilaskohtaisesti. Vaihtoehtoihin kuuluivat kehonulkoinen fotofereesi (ECP), pieniannoksinen metotreksaatti (MTX), mykofenolaattimofetiili (MMF), mTORin estäjät (everolimuusi tai sirolimuusi), infliksimabi, rituksimabi, pentostatiini, imatinibi ja ibrutinibi.
Jakavi-hoidon tai parhaan saatavilla olevan hoidon lisäksi potilaat olivat saattaneet saada tavanomaista allogeenisen kantasolusiirron tukihoitoa, kuten infektiolääkkeitä ja verensiirtoja. Kortikosteroidien ja kalsineuriinin estäjien, kuten siklosporiinin tai takrolimuusin, sekä paikallisesti käytettävien tai inhaloitavien kortikosteroidien käytön jatkaminen oli sallittua hoitoyksikön ohjeiden mukaisesti.
Tutkimukseen voitiin ottaa mukaan potilaita, jotka olivat saaneet krooniseen käänteishyljintään yhtä muuta aiempaa systeemistä hoitoa kuin kortikosteroideja ja/tai kalsineuriinin estäjiä. Muiden kroonisen käänteishyljinnän systeemiseen hoitoon tarkoitettujen lääkevalmisteiden kuin kortikosteroidien ja kalsineuriinin estäjien aiemman käytön jatkaminen oli sallittua vain, jos valmisteita käytettiin kroonisen käänteishyljinnän estohoitoon (eli hoito oli aloitettu ennen kroonisen käänteishyljinnän toteamista) tavanomaisen hoitokäytännön mukaisesti.
Parasta saatavilla olevaa hoitoa saaneet potilaat saivat siirtyä käyttämään ruksolitinibia päivänä 169 tai sen jälkeen sairauden etenemisen, haittatapahtumien komplisoiman vasteen (mixed response), vasteen muuttumattomuuden, parhaan saatavilla olevan hoidon aiheuttaman toksisuuden tai kroonisen käänteishyljinnän uudelleenaktivoitumisen vuoksi.
Valmisteen tehosta sellaisten potilaiden hoidossa, joilla aktiivinen akuutti käänteishyljintä muuttuu krooniseksi käänteishyljinnäksi ilman kortikosteroidihoidon ja mahdollisen systeemisen hoidon annosten vähittäistä pienentämistä, ei ole tietoa. Tehosta luovuttajan lymfosyytti-infuusion (DLI) jälkeisen akuutin tai kroonisen käänteishyljinnän hoidossa tai potilailla, jotka eivät sietäneet steroidihoitoa, ei ole tietoa.
Jakavi-annoksen vaiheittainen pienentäminen oli sallittua päivän 169 käynnin jälkeen.
Lähtötilanteen demografiset tiedot ja sairauden ominaispiirteet olivat samankaltaiset molemmissa hoitoryhmissä. Potilaiden mediaani-ikä oli 49 vuotta (vaihteluväli 12–76 vuotta). Tutkimukseen osallistuneista potilaista 3,6 % oli nuoria, 61,1 % oli miehiä ja 75,4 % oli valkoihoisia. Suurimmalla osalla osallistujista oli pahanlaatuinen perussairaus.
Kortikosteroidihoitoon reagoimattoman kroonisen käänteishyljinnän vaikeusaste toteamishetkellä oli samankaltainen molemmissa hoitoryhmissä. Käänteishyljintä oli keskivaikea 41 %:lla Jakavi-ryhmän ja 45 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista ja vaikea 59 %:lla Jakavi-ryhmän ja 55 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista.
Potilaiden riittämättömälle vasteelle kortikosteroidihoitoon Jakavi-ryhmässä ja parhaan saatavilla olevan hoidon ryhmässä olivat ominaisia seuraavat tekijät: i) vasteen puuttuminen tai taudin eteneminen vähintään 7 päivää kestäneen, prednisoniannosta 1 mg/kg/vrk vastanneen kortikosteroidihoidon jälkeen (37,6 % Jakavi-ryhmässä ja 44,5 % parhaan saatavilla olevan hoidon ryhmässä), ii) sairauden jatkuminen 4 viikkoa kestäneen, annoksella 0,5 mg/kg/vrk käytetyn hoidon jälkeen (35,2 % Jakavi-ryhmässä ja 25,6 % parhaan saatavilla olevan hoidon ryhmässä) tai iii) kortikosteroidiriippuvuus (27,3 % Jakavi-ryhmässä ja 29,9 % parhaan saatavilla olevan hoidon ryhmässä).
Koko potilasjoukossa iho-oireita oli 73 %:lla Jakavi-ryhmän potilaista ja 69 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista ja keuhko-oireita 45 %:lla Jakavi-ryhmän potilaista ja 41 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista.
Yleisimpiä aiemmin käytettyjä kroonisen käänteishyljinnän systeemisiä hoitoja olivat pelkät kortikosteroidit (43 %:lla Jakavi-ryhmän ja 49 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista) ja kortikosteroidien ja kalsineuriinin estäjien yhdistelmät (41 %:lla Jakavi-ryhmän ja 42 %:lla parhaan saatavilla olevan hoidon ryhmän potilaista).
Ensisijainen päätemuuttuja oli kokonaisvasteosuus päivänä 169. Se määriteltiin niiden potilaiden osuudeksi kummassakin ryhmässä, joilla saavutettiin täydellinen vaste tai osittainen vaste ilman sellaista lisähoitojen tarvetta, joka olisi johtunut aiemmasta etenemisestä, haittatapahtumien komplisoimasta vasteesta (mixed response) tai vasteen puuttumisesta. Vasteen arvioi tutkija National Institutes of Health -tutkimuslaitoksen (NIH) kriteerien perusteella.
Keskeinen toissijainen päätemuuttuja oli elossaolo ilman hoidon epäonnistumista (failure free survival, FFS), aika tapahtumaan ‑yhdistelmäpäätemuuttuja, johon sisältyi ensimmäisenä ilmennyt seuraavista tapahtumista: i) perussairauden paheneminen tai uusiutuminen tai perussairaudesta johtuva kuolema, ii) muuhun kuin perussairauden pahenemiseen liittyvä kuolema tai iii) kroonisen käänteishyljinnän uuden systeemisen hoidon lisääminen tai aloittaminen.
REACH3-tutkimuksen ensisijainen tavoite saavutettiin. Primaarianalyysin ajankohtana (tiedonkeruun katkaisupäivä 8.5.2020) kokonaisvasteosuus viikon 24 kohdalla oli Jakavi-ryhmässä suurempi (49,7 %) kuin parhaan saatavilla olevan hoidon ryhmässä (25,6 %). Hoitoryhmien välinen ero oli tilastollisesti merkitsevä (ositettu Cochrane–Mantel–Haenszelin testi p < 0,0001, kaksitahoinen; kerroinsuhde 2,99; 95 %:n luottamusväli 1,86; 4,80). Tulokset on esitetty taulukossa 12.
Sairaus oli edennyt 2,4 %:lla niistä Jakavi-ryhmän potilaista ja 12,8 %:lla niistä parhaan saatavilla olevan hoidon ryhmän potilaista, jotka eivät olleet saaneet vastetta päivän 169 kohdalla.
Taulukko 12 Kokonaisvasteosuus päivän 169 kohdalla REACH3-tutkimuksessa
Jakavi N = 165 | Paras saatavilla oleva hoito N = 164 | |||
| n (%) | 95 %:n luottamusväli | n (%) | 95 %:n luottamusväli | |
| Vasteen saaneet yhteensä | 82 (49,7) | 41,8; 57,6 | 42 (25,6) | 19,1; 33,0 |
| Kerroinsuhde (95 %:n luottamusväli) | 2,99 (1,86; 4,80) | |||
| p-arvo (kaksitahoinen) | p < 0,0001 | |||
| Täydellinen vaste | 11 (6,7) | 5 (3,0) | ||
| Osittainen vaste | 71 (43,0) | 37 (22,6) | ||
Keskeisen toissijaisen päätemuuttujan, elossaolon ilman hoidon epäonnistumista (failure free survival, FFS), osoitettiin olevan Jakavi-ryhmässä tilastollisesti merkitsevästi 63 % pidempi kuin parhaan saatavilla olevan hoidon ryhmässä (riskisuhde [HR] 0,370; 95 %:n luottamusväli 0,268; 0,510; p < 0.0001). 6 kuukauden kohdalla suurin osa FFS-tapahtumista kuului luokkaan ”kroonisen käänteishyljinnän uuden systeemisen hoidon lisääminen tai aloittaminen” ja tämän tapahtuman todennäköisyys oli 13,4 % Jakavi-ryhmässä ja 48,5 % parhaan saatavilla olevan hoidon ryhmässä. Todennäköisyys tapahtumalle ”perussairauden paheneminen” oli 2,46 % Jakavi-ryhmässä ja 2,57 % parhaan saatavilla olevan hoidon ryhmässä. Todennäköisyys tapahtumalle ”muuhun kuin perussairauden pahenemiseen liittyvä kuolema” oli 9,19 % Jakavi-ryhmässä ja 4,46 % parhaan saatavilla olevan hoidon ryhmässä. Kumulatiivisissa esiintymistiheyksissä ei ollut eroja hoitojen välillä keskityttäessä vain muuhun kuin perussairauden pahenemiseen liittyvään kuolemaan.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Jakavin käytöstä MF:n ja PV:n hoidossa kaikissa pediatrisissa potilasryhmissä. Jakavin turvallisuus ja teho yli 2-vuotiaiden pediatristen käänteishyljintää sairastavien potilaiden hoidossa on osoitettu satunnaistetuissa vaiheen 3 tutkimuksissa REACH2 ja REACH3, sekä avoimissa yksiryhmäisissä vaiheen 2 tutkimuksissa REACH4 ja REACH5 (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa). Yksiryhmäisyyden vuoksi ruksolitinibin osuutta kokonaistehoon ei voida eritellä.
Akuutti käänteishyljintä
REACH4-tutkimuksessa Jakavin turvallisuuden, tehon ja farmakokinetiikan arvioimiseksi 45 pediatrista potilasta, joilla oli asteen II–IV akuutti käänteishyljintä, sai Jakavia ja kortikosteroidihoitoa kalsineuriininestäjähoidon kanssa tai ilman sitä. Potilaat otettiin tutkimukseen neljään iänmukaiseen ryhmään (ryhmä 1 [≥ 12 – < 18 vuotta, N = 18], ryhmä 2 [≥ 6 – < 12 vuotta, N = 12], ryhmä 3 [≥ 2 – < 6 vuotta, N = 15] ja ryhmä 4 [≥ 28 vrk – < 2 vuotta, N = 0]. Käytetyt annokset olivat 10 mg kahdesti vuorokaudessa ryhmässä 1, 5 mg kahdesti vuorokaudessa ryhmässä 2 ja 4 mg/m2 kahdesti vuorokaudessa ryhmässä 3. Potilaat saivat hoitoa 24 viikon ajan tai hoidon lopettamiseen asti. Alle 12-vuotiaille pediatrisille potilaille Jakavi annettiin joko 5 mg:n tablettina tai kapselina/oraaliliuoksena.
Tutkimukseen otettiin potilaita, joiden tauti oli steroidihoitoon reagoimaton tai jotka eivät olleet saaneet aiempaa hoitoa. Tauti määriteltiin steroidihoitoon reagoimattomaksi joko hoitoyksikön kriteerien mukaan tai, jos hoitoyksikön kriteerejä ei ollut saatavilla, lääkärin päätöksen mukaan; potilaat olivat saaneet saada kortikosteroidien lisäksi enintään yhtä muuta aiempaa systeemistä hoitoa akuuttiin käänteishyljintään. Aiemmin hoitamattoman määritelmä oli, että potilas ei ollut saanut mitään aiempaa systeemistä hoitoa akuuttiin käänteishyljintään (poikkeuksena enintään 72 tuntia kestänyt aiempi systeeminen kortikosteroidihoito metyyliprednisolonilla tai vastaavalla valmisteella akuutin käänteishyljinnän alkamisen jälkeen). Jakavin lisäksi potilaat saivat systeemisiä kortikosteroideja ja/tai kalsineuriinin estäjiä (siklosporiini tai takrolimuusi). Lisäksi hoitoyksikön ohjeiden mukaiset paikallisesti käytettävät kortikosteroidihoidot oli sallittu. REACH4-tutkimuksessa 40 potilasta (88,9 %) sai samanaikaista kalsineuriinin estäjähoitoa. Lisäksi potilaat olivat saattaneet saada tavanomaista allogeenisen kantasolusiirron tukihoitoa, kuten infektiolääkkeitä ja verensiirtoja. Jakavi-hoito täytyi lopettaa, jos vastetta akuutin käänteishyljinnän hoidossa ei ollut saavutettu päivänä 28.
Jakavi-annoksen vähittäinen pienentäminen oli sallittua päivän 56 tutkimuskäynnin jälkeen.
Potilaista 62,2 % (n = 28) oli miespuolisia ja 37,8 % (n = 17) oli naispuolisia. 27 potilaalla (60,0 %) oli pahanlaatuinen perussairaus, yleisimmin leukemia (26 potilaalla, 57,8 %). REACH4-tutkimukseen otetusta 45 pediatrisesta potilaasta 13:lla (28,9 %) oli akuutti aiemmin hoitamaton käänteishyljintä, ja 32:lla (71,1 %) oli steroidihoitoon reagoimaton akuutti käänteishyljintä. Lähtötilanteessa akuutin käänteishyljinnän vaikeusaste oli II 64,4 %:lla potilaista, III 26,7 %:lla potilaista ja IV 8,9 %:lla potilaista.
Kokonaisvasteosuus (ORR, overall response rate) päivänä 28 (ensisijainen tehon päätemuuttuja) oli REACH4-tutkimuksessa 84,4 % (90 %:n luottamusväli 72,8; 92,5) kaikilla potilailla; täydellinen vaste (CR) saavutettiin 48,9 %:lla potilaista ja osittainen vaste (PR) 35,6 %:lla potilaista. Hoitoa edeltäneen tilan mukaisesti jaoteltuna kokonaisvasteosuus päivänä 28 oli 90,6 % potilailla, joilla oli steroidihoitoon reagoimaton käänteishyljintä.
Pitkäkestoisten vasteiden kokonaisosuus päivänä 56 (keskeinen toissijainen päätemuuttuja) eli niiden potilaiden osuus, jotka olivat saavuttaneet täydellisen tai osittaisen vasteen päivänä 28 ja joilla vaste säilyi päivänä 56, oli 66,7 % kaikilla REACH4-tutkimuksen potilailla ja 68,8 % potilailla, joilla oli steroidihoitoon reagoimaton käänteishyljintä.
Krooninen käänteishyljintä
REACH5-tutkimuksessa Jakavin turvallisuuden, tehon ja farmakokinetiikan arvioimiseksi 45 pediatrista potilasta, joilla oli keskivaikea tai vaikea krooninen käänteishyljintä, sai Jakavia ja kortikosteroidihoitoa kalsineuriininestäjähoidon kanssa tai ilman sitä. Potilaat otettiin tutkimukseen neljään iänmukaiseen ryhmään (ryhmä 1 [≥ 12 – < 18 vuotta, N = 22], ryhmä 2 [≥ 6 – < 12 vuotta, N = 16], ryhmä 3 [≥ 2 – < 6 vuotta, N = 7] ja ryhmä 4 [≥ 28 vrk – < 2 vuotta, N = 0]. Käytetyt annokset olivat 10 mg kahdesti vuorokaudessa ryhmässä 1, 5 mg kahdesti vuorokaudessa ryhmässä 2 ja 4 mg/m2 kahdesti vuorokaudessa ryhmässä 3. Potilaat saivat hoitoa 39 hoitojakson / 156 viikon ajan tai hoidon lopettamiseen asti. Alle 12-vuotiaille pediatrisille potilaille Jakavi annettiin joko 5 mg:n tablettina tai oraaliliuoksena.
Tutkimukseen otettiin potilaita, joiden tauti oli steroidihoitoon reagoimaton tai jotka eivät olleet saaneet aiempaa hoitoa. Tauti määriteltiin steroidihoitoon reagoimattomaksi joko hoitoyksikön kriteerien mukaan tai, jos hoitoyksikön kriteerejä ei ollut saatavilla, lääkärin päätöksen mukaan; potilaat olivat saaneet saada kortikosteroidien lisäksi muuta aiempaa systeemistä hoitoa krooniseen käänteishyljintään. Aiemmin hoitamattoman määritelmä oli, että potilas ei ollut saanut mitään aiempaa systeemistä hoitoa krooniseen käänteishyljintään (poikkeuksena enintään 72 tuntia kestänyt aiempi systeeminen kortikosteroidihoito metyyliprednisolonilla tai vastaavalla valmisteella kroonisen käänteishyljinnän alkamisen jälkeen). Jakavin lisäksi potilaat saivat edelleen systeemisiä kortikosteroideja ja/tai kalsineuriinin estäjiä (siklosporiini tai takrolimuusi). Lisäksi hoitoyksikön ohjeiden mukaiset paikallisesti käytettävät kortikosteroidihoidot oli sallittu. REACH5-tutkimuksessa 23 potilasta (51,1 %) sai samanaikaista kalsineuriinin estäjähoitoa. Lisäksi potilaat olivat saattaneet saada tavanomaista allogeenisen kantasolusiirron tukihoitoa, kuten infektiolääkkeitä ja verensiirtoja. Jakavi-hoito täytyi lopettaa, jos vastetta kroonisen käänteishyljinnän hoidossa ei ollut saavutettu päivänä 169.
Jakavin annoksen vähittäinen pienentäminen oli sallittua päivän 169 tutkimuskäynnin jälkeen.
Potilaista 64,4 % (n = 29) oli miespuolisia ja 35,6 % (n = 16) oli naispuolisia, ja 30 potilaalla (66,7 %) oli siirtoa edeltänyt pahanlaatuinen perussairaus, yleisimmin leukemia (27 potilaalla, 60 %).
REACH5-tutkimukseen otetuista 45 pediatrisesta potilaasta 17:llä (37,8 %) oli krooninen käänteishyljintä, johon potilas ei ollut saanut aiempaa hoitoa, ja 28:lla (62,2 %) oli steroidihoitoon reagoimaton krooninen käänteishyljintä. Tauti oli vaikea 62,2 %:lla potilaista ja keskivaikea 37,8 %:lla potilaista. 31 potilaalla (68,9 %) oli taudin ilmentymiä ihossa, 18 potilaalla (40 %) suussa ja 14 potilaalla (31,1 %) keuhkoissa.
Kokonaisvasteosuus (ORR) päivänä 169 (ensisijainen tehon päätemuuttuja) oli REACH5-tutkimuksessa 40 % (90 %:n luottamusväli: 27,7; 53,5) kaikilla pediatrisilla potilailla ja 39,3 % potilailla, joilla oli steroidihoitoon reagoimaton käänteishyljintä.
Farmakokinetiikka
Imeytyminen
Ruksolitinibi kuuluu biofarmaseuttisen BCS-luokituksen luokkaan 1. Sen läpäisevyys ja liukoisuus ovat suuret ja jakaantuminen nopeaa. Kliinisissä tutkimuksissa suun kautta otettu ruksolitinibi imeytyy nopeasti, ja maksimipitoisuudet plasmassa (Cmax) saavutetaan noin 1 tunnin kuluttua annoksesta. Ihmisillä tehdyn massatasapainotutkimuksen perusteella suun kautta otettu ruksolitinibi imeytyy vähintään 95-prosenttisesti joko kanta-aineena tai ensikierron metabolian aikana muodostuvina metaboliitteina. Ruksolitinibin Cmax-keskiarvo ja kokonaisaltistuksen (AUC) keskiarvo suurenivat suhteessa annokseen 5–200 mg kerta-annoksilla. Ruksolitinibin farmakokinetiikka ei muuttunut kliinisesti merkittävässä määrin, kun lääke otettiin runsasrasvaisen aterian yhteydessä. Cmax-keskiarvo pieneni kohtalaisesti (24 %), kun taas AUC-keskiarvo pysyi lähes ennallaan (4 % suurenema), kun lääke otettiin runsasrasvaisen aterian yhteydessä.
Jakautuminen
Keskimääräinen vakaan tilan jakautumistilavuus on MF- ja PV-potilailla noin 75 litraa, akuuttia käänteishyljintää sairastavilla nuorilla ja aikuisilla potilailla noin 67,5 litraa ja kroonista käänteishyljintää sairastavilla nuorilla ja aikuisilla potilailla noin 60,9 litraa. Keskimääräinen vakaan tilan jakautumistilavuus on noin 30 litraa akuuttia tai kroonista käänteishyljintää sairastavilla pediatrisilla potilailla, joiden kehon pinta-ala (BSA, body surface area) on alle 1 m2. Kliinisesti relevantteina pitoisuuksina ruksolitinibi sitoutuu noin 97-prosenttisesti plasman proteiineihin, lähinnä albumiiniin, in vitro. Rotilla tehdyssä koko kehon autoradiografiatutkimuksessa todettiin, että ruksolitinibi ei läpäise veri-aivoestettä.
Biotransformaatio
Ruksolitinibi metaboloituu pääosin CYP3A4:n kautta (> 50 %). Lisäksi metaboliaan osallistuu CYP2C9. Ihmisen plasmassa esiintyy eniten kanta-ainetta, jota on noin 60 % verenkierron lääkeperäisestä materiaalista. Plasmassa esiintyy kahta merkittävää aktiivista metaboliittia, joiden AUC-arvot olivat 25 % ja 11 % kanta-aineen AUC-arvosta. Näiden metaboliittien JAK-kinaasiin kohdistuva farmakologinen aktiivisuus on 1/2 ‑ 1/5 kanta-aineen vastaavasta aktiivisuudesta. Kaikki aktiiviset metaboliitit aiheuttavat yhdessä 18 % ruksolitinibin kokonaisfarmakodynamiikasta. In vitro ‑tutkimusten perusteella kliinisesti relevantit ruksolitinibipitoisuudet eivät estä CYP1A2-, CYP2B6-, CYP2C8-, CYP2C9-, CYP2C19-, CYP2D6-entsyymejä eikä CYP3A4:ää, eivätkä aikaansaa voimakasta CYP1A2-, CYP2B6- tai CYP3A4-induktiota. In vitro -tiedot viittaavat siihen, että ruksolitinibi saattaa estää P-gp:tä ja BCRP:tä.
Eliminaatio
Ruksolitinibi eliminoituu lähinnä metaboloitumalla. Ruksolitinibin eliminaation puoliintumisajan keskiarvo on noin 3 tuntia. Kun terveille aikuisille annettiin kerta-annos [14C]-leimattua ruksolitinibia suun kautta, aine eliminoitui lähinnä metaboloitumalla. 74 % radioaktiivisuudesta erittyi virtsaan ja 22 % ulosteeseen. Alle 1 % erittyneestä kokonaisradioaktiivisuudesta erittyi muuttumattoman kanta-aineen muodossa.
Lineaarisuus/ei-lineaarisuus
Sekä kerta-annostutkimuksissa että toistuvaisannostutkimuksissa todettiin lineaarisuus suhteessa annokseen.
Erityisryhmät
Iän, sukupuolen tai etnisen taustan vaikutus
Terveillä henkilöillä suoritettujen tutkimusten perusteella sukupuolen ja etnisen taustan ei todettu vaikuttavan olennaisesti ruksolitinibin farmakokinetiikkaan.
Populaatiofarmakokinetiikka
MF-potilailla tehdyssä populaatiofarmakokinetiikan tutkimuksessa ei todettu ilmeistä yhteyttä peroraalisen puhdistuman ja iän tai etnisen taustan välillä. Ennustettu peroraalinen puhdistuma oli naisilla 17,7 l/h ja miehillä 22,1 l/h. Yksilöllinen vaihtelu MF-potilailla oli 39 %. PV-potilailla puhdistuma oli 12,7 l/h. Yksilöllinen vaihtelu oli 42 % eikä selkeää suhdetta oraalisen puhdistuman ja sukupuolen, potilaan iän tai etnisen taustan välillä havaittu PV-potilailla suoritetun populaatiofarmakokineettisen arvioinnin perusteella. Puhdistuma oli 10,4 l/h nuorilla ja aikuisilla potilailla, joilla oli akuutti käänteishyljintä, ja 7,8 l/h nuorilla ja aikuisilla potilailla, joilla oli krooninen käänteishyljintä. Tutkittavien välinen vaihtelu oli 49 %. Akuuttia tai kroonista käänteishyljintää sairastavilla pediatrisilla potilailla, joiden kehon pinta-ala oli alle 1 m2, puhdistuma oli 6,5–7 l/h. Selkeää suhdetta oraalisen puhdistuman ja sukupuolen, potilaan iän tai etnisen taustan välillä ei havaittu käänteishyljintää sairastavilla potilailla suoritetun populaatiofarmakokineettisen arvioinnin perusteella. Annoksella 10 mg kahdesti vuorokaudessa altistus suureni käänteishyljintää sairastavilla potilailla, joilla oli alhainen kehon pinta-ala. Keskimääräinen ennustettu altistuminen (AUC) oli 31 %, 22 % ja 12 % suurempi potilailla, joilla kehon pinta-alat olivat vastaavasti 1 m2, 1,25 m2 ja 1,5 m2 tässä järjestyksessä, verrattaessa tyypilliseen aikuiseen (1,79 m2).
Pediatriset potilaat
Jakavin farmakokinetiikkaa alle 18-vuotiaiden lasten MF:n tai PV:n hoidossa ei ole varmistettu.
Kuten käänteishyljintää sairastavilla aikuispotilailla, ruksolitinibi imeytyi nopeasti peroraalisen annon jälkeen myös käänteishyljintää sairastavilla pediatrisilla potilailla. Annoksella 5 mg kahdesti vuorokaudessa 6–11-vuotiaille lapsille saavutettiin vastaava altistus kuin annoksella 10 mg kahdesti vuorokaudessa nuorille ja aikuisille akuuttia tai kroonista käänteishyljintää sairastaville potilaille, mikä vahvisti, että altistus vastasi ekstrapoloitua altistusta. 2–5-vuotiaille akuuttia tai kroonista käänteishyljintää sairastaville lapsille annos vastaavan altistuksen saavuttamiseksi oli ekstrapoloinnin perusteella 8 mg/m2 kahdesti vuorokaudessa.
Ruksolitinibia ei ole arvioitu akuuttia tai kroonista käänteishyljintää sairastavien alle 2-vuotiaiden pediatristen potilaiden hoidossa, joten nuorempien potilaiden altistuksen ennustamiseen on käytetty ikään liittyviä tekijöitä huomioivaa mallinnusta, joka perustuu aikuispotilaista saatuihin tietoihin.
Akuuttia tai kroonista käänteishyljintää sairastavien pediatristen potilaiden yhdistettyjen populaatiofarmakokinetiikan analyysien perusteella ruksolitinibin puhdistuma pieneni kehon pinta-alan pienentyessä. Kun kehon pinta-alan vaikutus korjattiin, muut demografiset tekijät, kuten ikä, paino ja painoindeksi, eivät vaikuttaneet kliinisesti merkitsevästi ruksolitinibialtistukseen.
Munuaisten vajaatoiminta
Munuaisten toimintaa arvioitiin sekä MDRD:n (Modification of Diet in Renal Disease) että virtsaan erittyvän kreatiniinin avulla. 25 mg ruksolitinibikerta-annoksen jälkeen ruksolitinibialtistus oli eriasteista munuaisten vajaatoimintaa sairastavilla samaa luokkaa kuin henkilöillä, joiden munuaistoiminta oli normaali. Ruksolitinibin metaboliittien AUC-arvoilla plasmassa oli kuitenkin taipumus suurentua munuaisten vajaatoiminnan vaikeutuessa, ja ne suurenivat eniten henkilöillä, joilla oli vaikea munuaisten vajaatoiminta. Ei tiedetä, onko lisääntyneellä metaboliittialtistuksella merkitystä lääkehoidon turvallisuuden kannalta. Annoksen muuttaminen on suositeltavaa, jos potilaalla on vaikea munuaisten vajaatoiminta ja loppuvaiheen munuaissairaus (ks. kohta Annostus ja antotapa). Ainoastaan dialyysipäivinä tapahtuva annostus vähentää altistumista metaboliiteille, mutta myös farmakodynaamista vaikutusta etenkin dialyysikertojen välisinä päivinä.
Maksan vajaatoiminta
25 mg ruksolitinibikerta-annoksen jälkeen ruksolitinibin AUC-keskiarvo suureni eriasteista maksan vajaatoimintaa sairastavilla potilailla 87 % (lievä vajaatoiminta), 28 % (keskivaikea vajaatoiminta) ja 65 % (vaikea vajaatoiminta) verrattuna henkilöihin, joiden maksan toiminta oli normaali. AUC-arvon ja Child–Pugh-pisteillä mitatun maksan vajaatoiminnan asteen välillä ei ollut selvää yhteyttä. Terminaalinen eliminaation puoliintumisaika oli maksan vajaatoimintapotilailla pidempi kuin terveillä verrokeilla (4,1–5,0 h ja 2,8 h). On suositeltavaa pienentää annosta noin 50 %, jos MF- tai PV-potilaalla on maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
Käänteishyljintäpotilailla, joilla on käänteishyljintään liittymätön maksan vajaatoiminta, ruksolitinibin aloitusannosta on pienennettävä 50 %.
Prekliiniset tiedot turvallisuudesta
Ruksolitinibia on arvioitu farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta ja lisääntymistoksisuutta koskevissa tutkimuksissa ja yhdessä karsinogeenisuustutkimuksessa. Toistuvaisannostutkimuksissa ruksolitinibin farmakologisen vaikutuksen kohde-elimiä ovat luuydin, ääreisveri ja imukudos. Koirilla havaittiin infektioita, joihin liittyi yleensä immunosuppressiota. Koiralla tehdyssä telemetriatutkimuksessa todettiin verenpaineen haitallista laskua ja sykkeen nopeutumista, ja hengitystutkimuksessa rotalla havaittiin hengityksen minuuttitilavuuden haitallista pienenemistä. (Vapaan lääkeaineen Cmax-arvoihin perustuvat) haitattoman pitoisuuden raja-arvot olivat koiratutkimuksissa 15,7 kertaa ja rottatutkimuksissa 10,4 kertaa suuremmat kuin ihmisen suurimmat suositusannokset (25 mg kahdesti vuorokaudessa). Ruksolitinibin neurofarmakologisten vaikutusten arvioinnissa ei havaittu mitään vaikutuksia.
Nuorilla rotilla tehdyissä tutkimuksissa ruksolitinibin anto vaikutti kasvua ja luustoa kuvaaviin mittareihin. Eläimillä todettiin luuston kasvun heikkenemistä, kun ≥ 5 mg/kg/vrk annosten anto aloitettiin päivänä 7 syntymän jälkeen (verrattavissa vastasyntyneeseen ihmiseen) ja ≥ 15 mg/kg/vrk annosten anto aloitettiin päivänä 14 tai päivänä 21 syntymän jälkeen (verrattavissa ihmisellä pikkulapseen, 1–3-vuotiaaseen). Kun ≥ 30 mg/kg/vrk annosten anto aloitettiin päivänä 7 syntymän jälkeen, todettiin murtumia ja rottien varhaista lopettamista. Vapaan lääkeaineen AUC-arvon perusteella laskettu NOAEL-altistustaso (taso, jolla ei todeta mitään haittavaikutuksia) nuorilla rotilla, joiden hoito aloitettiin aikaisintaan päivänä 7 syntymän jälkeen, oli 0,3‑kertainen verrattuna aikuisiin potilaisiin, jotka saavat 25 mg:n annoksia kahdesti vuorokaudessa. Luuston kasvun heikkenemistä ja murtumia todettiin, kun altistus oli 1,5-kertainen (luuston kasvun heikkeneminen) tai 13-kertainen (murtumat) verrattuna aikuiseen potilaaseen, joka saa 25 mg:n annoksia kahdesti vuorokaudessa. Yleisesti ottaen vaikutukset olivat vaikeampia, kun valmisteen anto aloitettiin varhemmin syntymän jälkeen. Luuston kehitystä lukuun ottamatta ruksolitinibin vaikutukset olivat nuorilla rotilla samankaltaiset kuin aikuisilla rotilla. Nuoret rotat ovat aikuisia rottia herkempiä ruksolitinibin aiheuttamalle toksisuudelle.
Eläintutkimuksissa ruksolitinibi pienensi sikiöpainoa ja lisäsi jo kiinnittyneiden alkioiden menetystä. Teratogeenisuudesta ei havaittu minkäänlaisia merkkejä rotilla eikä kaneilla. Altistusmarginaalit olivat kuitenkin pienet suhteessa suurimpaan kliinisesti käytettävään annokseen, joten tulosten sovellettavuus käyttöön ihmisillä on siten rajallinen. Hedelmällisyyteen kohdistuvaa vaikutusta ei havaittu. Pre- ja postnataalista kehitystä koskeneessa tutkimuksessa havaittiin, että tiineyden kesto piteni hieman, implantaatiokohtien määrä väheni ja syntyneiden poikasten määrä väheni. Poikasilta havaittiin alentunutta alkupainon keskiarvoa ja lyhytaikaista keskipainon nousun hidastumista. Ruksolitinibi ja/tai sen metaboliitit erittyivät imettävien rottien maitoon pitoisuuksina, jotka olivat 13 kertaa emon plasman lääkepitoisuuksia suuremmat. Ruksolitinibi ei ollut mutageeninen eikä klastogeeninen. Ruksolitinibi ei ollut karsinogeeninen siirtogeenisessä Tg.rasH2-hiirimallissa.
Farmaseuttiset tiedot
Apuaineet
Mikrokiteinen selluloosa
Magnesiumstearaatti
Vedetön kolloidinen piidioksidi
Natriumtärkkelysglykolaatti (tyyppi A)
Povidoni K30
Hydroksipropyyliselluloosa, 300–600 cps
Laktoosimonohydraatti
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Säilytä alle 30°C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JAKAVI tabletti
5 mg (L:kyllä) 56 fol (1778,27 €)
10 mg (L:kyllä) 56 fol (3348,61 €)
15 mg (L:kyllä) 56 fol (3348,61 €)
20 mg (L:kyllä) 56 fol (3348,61 €)
PF-selosteen tieto
PVC/PE/PVDC/alumiini-läpipainopakkaukset, joissa 14 tai 56 tablettia, tai monipakkaukset, joissa 168 (kolme 56 tabletin pakkausta) tablettia.
Kaikkia pakkauskokoja tai -tyyppejä ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Jakavi 5 mg tabletti
Pyöreä, kaareva, valkoinen tai luonnonvalkoinen, halkaisijaltaan noin 7,5 mm:n kokoinen tabletti, jonka toisella puolella on kaiverrus ”NVR” ja toisella puolella kaiverrus ”L5”.
Jakavi 10 mg tabletti
Pyöreä, kaareva, valkoinen tai luonnonvalkoinen, halkaisijaltaan noin 9,3 mm:n kokoinen tabletti, jonka toisella puolella on kaiverrus ”NVR” ja toisella puolella kaiverrus ”L10”.
Jakavi 15 mg tabletti
Soikea, pyöristetty, valkoinen tai luonnonvalkoinen, noin 15,0 x 7,0 mm:n kokoinen tabletti, jonka toisella puolella on kaiverrus ”NVR” ja toisella puolella kaiverrus ”L15”.
Jakavi 20 mg tabletti
Pitkänomainen, pyöristetty, valkoinen tai luonnonvalkoinen, noin 16,5 x 7,4 mm:n kokoinen tabletti, jonka toisella puolella on kaiverrus ”NVR” ja toisella puolella kaiverrus ”L20”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JAKAVI tabletti
5 mg 56 fol
10 mg 56 fol
15 mg 56 fol
20 mg 56 fol
- Ylempi erityiskorvaus (100 %). Momelotinibi ja ruksolitinibi: Myelofibroosin hoito erityisin edellytyksin (174).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Momelotinibi ja ruksolitinibi: Primaarista myelofibroosia, polysytemia veran jälkeistä myelofibroosia tai essentiellin trombosytoosin jälkeistä myelofibroosia sairastavien aikuispotilaiden oireiden tai sairauteen liittyvän splenomegalian hoito (374).
ATC-koodi
L01EJ01
Valmisteyhteenvedon muuttamispäivämäärä
10.11.2025
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com