
ROACTEMRA injektioneste, liuos, esitäytetty kynä 162 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tämä potilaskortti sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen RoActemra-hoidon aloittamista ja hoidon aikana.
Vaikuttavat aineet ja niiden määrät
Jokainen esitäytetty kynä sisältää 162 mg RoActemraa (tosilitsumabia) 0,9 ml:ssa liuosta.
RoActemra on rekombinantti, humanisoitu monoklonaalinen immunoglobuliinin G1 (IgG1) vasta-aine.
Apuaine, jonka vaikutus tunnetaan:
Jokainen 162 mg/0,9 ml:n esitäytetty kynä sisältää 0,18 mg (0,2 mg/ml) polysorbaattia 80.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste), esitäytetty kynä (ACTPen).
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
RoActemra on tarkoitettu yhdessä metotreksaatin (MTX) kanssa:
- vaikean, aktiivisen ja etenevän nivelreuman hoitoon aikuisille, joita ei aiemmin ole hoidettu MTX:lla
- aikuispotilaiden kohtalaisen tai vaikean aktiivisen nivelreuman hoitoon, kun aikaisempi hoito yhdellä tai useammalla tautiprosessia hidastavalla reumalääkkeellä (disease modifying anti-rheumatic drugs, DMARD) tai tuumorinekroositekijän (TNF) estäjällä ei ole tuottanut riittävää hoitovastetta tai potilas ei siedä niitä.
RoActemraa voidaan antaa monoterapiana näille potilaille, jos he eivät siedä MTX:a tai, jos jatkuva MTX-hoito ei sovi heille.
RoActemran on röntgentutkimuksissa havaittu vähentävän nivelvaurioiden etenemistä ja parantavan fyysistä toimintakykyä, kun sitä annetaan yhdessä MTX:n kanssa.
Yleisoireinen lastenreuma
RoActemra on tarkoitettu 12-vuotiaiden ja sitä vanhempien lasten aktiivisen yleisoireisen lastenreuman hoitoon, kun aikaisempi hoito tulehduskipulääkkeillä (NSAID-lääkkeillä) ja systeemisillä kortikosteroideilla ei ole tuottanut riittävää hoitovastetta.
RoActemraa voidaan antaa monoterapiana (jos potilas ei siedä MTX:a tai MTX-hoito ei sovi hänelle) tai yhdessä MTX:n kanssa.
Lasten polyartriitti
RoActemra on tarkoitettu yhdessä metotreksaatin (MTX) kanssa 12-vuotiaiden ja sitä vanhempien lasten polyartriitin hoitoon (reumatekijä positiivinen tai negatiivinen ja laajentunut oligoartriitti), kun aikaisempi hoito MTX:lla ei ole tuottanut riittävää hoitovastetta.
RoActemraa voidaan antaa monoterapiana, jos potilas ei siedä MTX:a tai jos jatkuva MTX-hoito ei sovi hänelle.
Jättisoluarteriitti
RoActemra on tarkoitettu aikuispotilaille jättisoluarteriitin hoitoon.
Ehto
Hoidon saavat aloittaa valmisteyhteenvedossa mainittuun indikaatioon perehtyneet lääkärit.
Annostus ja antotapa
Tosilitsumabin ihon alle annettava lääkemuoto annetaan kertakäyttöisellä esitäytetyllä kynällä. Hoidon saa aloittaa nivelreuman, aktiivisen yleisoireisen lastenreuman, lasten polyartriitin ja/tai jättisoluarteriitin diagnosointiin ja hoitoon perehtynyt lääkäri.
Alle 12-vuotiaat pediatriset potilaat eivät saa käyttää esitäytettyä kynää, koska heillä ihonalainen kudoskerros on ohuempi, mistä saattaa aiheutua riski, että injektio injisoidaan lihakseen.
Ensimmäinen pistos on annettava pätevän terveydenhuollon ammattilaisen valvonnassa. Potilas tai potilaan vanhempi/huoltaja voi pistää tämän lääkevalmisteen vain silloin, jos lääkäri katsoo sen tarkoituksenmukaiseksi, potilas tai potilaan vanhempi/huoltaja käy seurannassa tarvittavin väliajoin ja potilaalle on opastettu oikea pistostekniikka.
Jos potilas siirtyy ihon alle annettavaan tosilitsumabihoitoon laskimoon annettavasta tosilitsumabihoidosta, ensimmäinen annos ihon alle on annettava pätevän terveydenhuollon ammattilaisen valvonnassa hoito-ohjelman mukaisen seuraavan laskimoon annettavan annoksen antoajankohtana.
Kaikille RoActemra-hoitoa saaville potilaille on annettava potilaskortti.
Varmista potilaan tai hänen vanhempansa/huoltajansa soveltuvuus ihon alle annettavana hoitona toteutettavaan kotihoitoon. Potilasta tai hänen vanhempaansa/huoltajaansa tulee ohjeistaa kertomaan terveydenhuollon ammattilaiselle ennen seuraavan annoksen antamista, jos potilaalle ilmaantuu allergisen reaktion oireita. Potilaan on hakeuduttava välittömästi lääkäriin, jos hänelle ilmaantuu vakavien allergisten reaktioiden oireita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Nivelreumaa sairastavat potilaat
Suositeltu annostus on 162 mg ihon alle kerran viikossa.
Potilaan hoidon vaihtamisesta tosilitsumabin laskimoon annettavasta lääkemuodosta tosilitsumabin vakioannoksena ihon alle annettavaan lääkemuotoon on vähän tietoa. Antoväliä kerran viikossa pitää noudattaa.
Laskimoon annettavasta lääkemuodosta ihon alle annettavan lääkemuodon käyttöön siirtyvien potilaiden ensimmäinen annos ihon alle on annettava hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa hoito-ohjelman mukaisesti seuraavana antoajankohtana.
Jättisoluarteriittia sairastavat potilaat
Suositeltu annostus on 162 mg ihon alle kerran viikossa yhdistelmänä asteittain vähennettävän glukokortikoidihoidon kanssa. Tätä lääkevalmistetta voidaan käyttää yksinään glukokortikoidihoidon lopettamisen jälkeen.
Tosilitsumabimonoterapiaa ei saa käyttää akuuttien relapsien hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jättisoluarteriitti on luonteeltaan krooninen sairaus. Hoidon jatkamisen 52 viikkoa pidempään pitää perustua sairauden aktiivisuuteen, lääkärin harkintaan ja potilaan valintaan.
Nivelreumaa ja jättisoluarteriittia sairastavat potilaat
Annoksen muuttaminen poikkeavien laboratorioarvojen takia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Poikkeavat maksaentsyymit
| Laboratorioarvo | Toimenpide |
> 1–3 × viitevälin yläraja (ULN) (Upper Limit of Normal = viitevälin yläraja) | Muuta samanaikaisesti annettavaa DMARD-annosta (nivelreuma) tai immuunivastetta muuntavan lääkeaineen annosta (jättisoluarteriitti), jos tarkoituksenmukaista. Jos arvon nousu on pitkäaikaista, vähennä tosilitsumabiannosta antoväliin kerran kahdessa viikossa tai keskeytä hoito, kunnes ALAT- (alaniiniaminotransferaasi) ja ASAT- (aspartaattiaminotransferaasi) arvot ovat normalisoituneet. Aloita hoito uudelleen kerran viikossa tai kerran kahdessa viikossa annettavina injektioina, kliinisen tilan mukaan. |
| > 3–5 × viitevälin yläraja | Keskeytä hoito, kunnes arvo on < 3 × viitevälin yläraja ja noudata yllä annettuja suosituksia arvolle > 1–3 × viitevälin yläraja. Jos arvot nousevat uudestaan > 3 × viitevälin yläraja (varmistetaan toistetulla mittauksella, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), lopeta hoito. |
| > 5 × viitevälin yläraja | Lopeta hoito. |
- Matala absoluuttinen neutrofiilien määrä (ANC)
Aiemmin tosilitsumabilla hoitamattomille potilaille ei suositella hoidon aloitusta, jos absoluuttinen neutrofiilien määrä on < 2 × 109/l.
| Laboratorioarvo (solumäärä × 109/l) | Toimenpide |
| ANC > 1 | Jatka samalla annoksella. |
| ANC 0,5–1 | Keskeytä tosilitsumabihoito. Kun ANC nousee > 1 × 109/l, aloita hoito uudelleen kerran kahdessa viikossa annettavina injektioina kliinisen tilan mukaan. |
| ANC < 0,5 | Lopeta hoito. |
- Pieni trombosyyttien määrä
| Laboratorioarvo (solumäärä × 103/μl) | Toimenpide |
| 50–100 | Keskeytä tosilitsumabihoito. Kun trombosyyttien määrä nousee > 100 × 103/μl, aloita hoito uudelleen kerran kahdessa viikossa annettavilla injektioilla kliinisen tilan mukaan. |
| < 50 | Lopeta hoito. |
Nivelreumaa ja jättisoluarteriittia sairastavat potilaat
Annoksen unohtaminen
Jos potilas unohtaa viikoittaisen ihon alle annettavan tosilitsumabiannoksensa eikä aikataulun mukaisesta antoajankohdasta ole kulunut yli 7 vuorokautta, potilasta on neuvottava ottamaan unohtunut annos seuraavana hoitoaikataulun mukaisena päivänä. Jos potilas unohtaa kerran kahdessa viikossa annettavan tosilitsumabiannoksensa eikä aikataulun mukaisesta antoajankohdasta ole kulunut yli 7 vuorokautta, potilasta on neuvottava ottamaan unohtunut annos heti ja seuraava annos seuraavana hoitoaikataulun mukaisena päivänä.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen yli 65-vuotiaille potilaille.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievää tai kohtalaista munuaisten vajaatoimintaa sairastaville potilaille. Tosilitsumabin käyttöä ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka). Munuaisten toimintaa on seurattava tarkasti näillä potilailla.
Maksan vajaatoiminta
Tosilitsumabin käyttöä ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Siksi annossuosituksia ei voida antaa.
Pediatriset potilaat
Tosilitsumabin vakioannoksena ihon alle annettavan lääkemuodon turvallisuutta ja tehoa lasten (vastasyntyneistä alle 1 vuoden ikäisiin) hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Annosmuutosten pitää perustua vain potilaan painossa ajan mittaan tapahtuvaan pysyvään muutokseen.
Tosilitsumabia voidaan käyttää yksinään tai yhdistelmänä metotreksaatin kanssa.
Yleisoireista lastenreumaa sairastavat potilaat
Yli 12-vuotiaille potilaille suositeltu annostus on 162 mg ihon alle kerran viikossa, jos potilas painaa vähintään 30 kg, tai 162 mg ihon alle kerran kahdessa viikossa, jos potilas painaa alle 30 kg.
Esitäytettyä kynää ei pidä käyttää alle 12-vuotiaiden pediatristen potilaiden hoitoon.
Potilaiden painon on oltava vähintään 10 kg, jotta heille voidaan antaa tosilitsumabihoitoa ihon alle.
Lasten polyartriittia sairastavat potilaat
Yli 12-vuotiaille potilaille suositeltu annostus on 162 mg ihon alle kerran kahdessa viikossa, jos potilas painaa vähintään 30 kg, tai 162 mg ihon alle kerran kolmessa viikossa, jos potilas painaa alle 30 kg.
Esitäytettyä kynää ei pidä käyttää alle 12-vuotiaiden pediatristen potilaiden hoitoon.
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavat potilaat
Annosmuutokset laboratoriotulosten poikkeavuuksien vuoksi
Samanaikaisesti annetun MTX:n ja/tai muiden lääkevalmisteiden annoksia on tarvittaessa muutettava tai annostus on lopetettava ja tosilitsumabin antaminen on keskeytettävä, kunnes potilaan kliininen tila on selvitetty. Koska monet samanaikaiset sairaudet voivat vaikuttaa yleisoireista lastenreumaa tai lasten polyartriittia sairastavien potilaiden laboratorioarvoihin, päätöksen tosilitsumabihoidon lopettamisesta poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin.
- Poikkeavat maksaentsyymit
| Laboratorioarvo | Toimenpide |
> 1–3 × viitevälin yläraja (Upper Limit of Normal = viitevälin yläraja) | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Jos arvon nousu on pitkäaikaista, keskeytä tosilitsumabihoito, kunnes ALAT- (alaniiniaminotransferaasi) ja ASAT (aspartaattiaminotransferaasi) -arvot ovat normalisoituneet. |
| > 3 × viitevälin yläraja – 5 × viitevälin yläraja | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Keskeytä tosilitsumabihoito, kunnes arvo on < 3 × viitevälin yläraja ja noudata yllä annettuja suosituksia arvolle > 1–3 × viitevälin yläraja. |
| > 5 × viitevälin yläraja | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa tai lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Matala absoluuttinen neutrofiilien määrä (ANC)
| Laboratorioarvo (solumäärä × 109/l) | Toimenpide |
| ANC > 1 | Jatka samalla annoksella. |
| ANC 0,5–1 | Keskeytä tosilitsumabihoito. Kun ANC nousee > 1 × 109/l, aloita hoito uudelleen. |
| ANC < 0,5 | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa tai lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
- Pieni trombosyyttien määrä
| Laboratorioarvo (solumäärä × 103/μl) | Toimenpide |
| 50–100 | Muuta samanaikaisesti annettu MTX-annos, jos tarkoituksenmukaista. Keskeytä tosilitsumabihoito. Kun trombosyyttien määrä nousee > 100 × 103/μl, aloita hoito uudelleen |
| < 50 | Lopeta tosilitsumabihoito. Päätöksen hoidon lopettamisesta yleisoireista lastenreumaa tai lasten polyartriittia sairastavilla potilailla poikkeavien laboratorioarvojen vuoksi on perustuttava yksittäisen potilaan lääketieteelliseen arviointiin. |
Tosilitsumabin antotiheyden harventamista poikkeavien laboratorioarvojen vuoksi ei ole tutkittu yleisoireista lastenreumaa tai lasten polyartriittia sairastavilla potilailla.
Tosilitsumabin ihon alle annettavan lääkemuodon turvallisuutta ja tehoa lapsille, jotka sairastavat jotakin muuta sairautta kuin yleisoireista lastenreumaa tai lasten polyartriittia, ei ole tutkittu.
Laskimoon annettavasta lääkemuodosta saatavissa olevat tiedot viittaavat siihen, että kliinistä paranemista voidaan havaita 12 viikon kuluessa tosilitsumabihoidon aloittamisesta. Jos tämän ajanjakson kuluessa ei havaita tilan paranemista, hoidon jatkamista on harkittava tarkoin uudelleen.
Annoksen unohtaminen
Jos yleisoireista lastenreumaa sairastavan potilaan ihon alle viikoittain annettava tosilitsumabipistos unohtuu eikä hoito-ohjelman mukaisesta antoajankohdasta ole kulunut yli 7 vuorokautta, potilasta pitää ohjeistaa ottamaan unohtunut annos tavanomaisen hoito-ohjelman seuraavana lääkkeenottoajankohtana. Jos potilas unohtaa ihon alle kerran kahdessa viikossa annettavan tosilitsumabipistoksen eikä hoito-ohjelman mukaisesta antoajankohdasta ole kulunut yli 7 vuorokautta, potilasta pitää ohjeistaa ottamaan unohtunut annos heti, kun sen unohtuminen huomataan, ja seuraava annos tavanomaisen hoito-ohjelman seuraavana lääkkeenottoajankohtana.
Jos lasten polyartriittia sairastavan potilaan ihon alle annettava tosilitsumabipistos unohtuu eikä hoito-ohjelman mukaisesta antoajankohdasta ole kulunut yli 7 vuorokautta, potilaan on otettava unohtunut annos heti, kun sen unohtuminen huomataan, ja ottaa seuraava annos tavanomaisen hoito-ohjelman seuraavana lääkkeenottoajankohtana. Jos potilas unohtaa ihon alle annettavan tosilitsumabi-injektion ja aikataulun mukaisesta antoajankohdasta on kulunut yli 7 vuorokautta tai jos potilas on epävarma siitä, milloin pistos pitää ottaa, hänen on otettava yhteyttä lääkäriin tai apteekkihenkilökuntaan.
Antotapa
Tämä lääkevalmiste on tarkoitettu annettavaksi ihon alle.
Kun potilaalle on annettu riittävä opastus pistostekniikasta, potilas voi pistää tämän lääkevalmisteen itse, jos lääkäri katsoo sen asianmukaiseksi. Esitäytetyn kynän koko sisältö (0,9 ml) pitää antaa injektiona ihon alle. Suositeltuja injektiokohtia (vatsa, reisi ja olkavarsi) pitää vaihdella eikä injektiota saa koskaan pistää luomiin, arpiin eikä ihoalueelle, jolla on aristusta, mustelmia, punoitusta, kovettuma tai iho ei ole ehjä.
Esitäytettyä kynää ei saa ravistaa.
Tarkemmat ohjeet RoActemran esitäytetyn kynän käyttöön on esitetty pakkausselosteessa. Ks. ohjeet kohdasta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiiviset vaikeat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
RoActemran ihon alle annettava lääkemuoto ei ole tarkoitettu annettavaksi laskimoon.
RoActemran ihon alle annettava lääkemuoto ei ole tarkoitettu alle 10 kg painaville yleisoireista lastenreumaa sairastaville lapsille.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Kaikki käyttöaiheet
Infektiot
Vakavia ja joskus kuolemaan johtaneita infektioita on raportoitu potilailla, jotka ovat saaneet immunosuppressiivisia lääkkeitä mukaan lukien tosilitsumabia (ks. kohta Haittavaikutukset). Hoitoa ei saa aloittaa, jos potilaalla on aktiivinen infektio (ks. kohta Vasta-aiheet). Jos potilaalle kehittyy vakava infektio, tosilitsumabihoito on keskeytettävä, kunnes infektio on saatu hallintaan (ks. kohta Haittavaikutukset). Tämän lääkevalmisteen käyttöä harkitessaan lääkärin on noudatettava varovaisuutta, jos potilaalla on aikaisemmin esiintynyt uusiutuvia tai kroonisia infektioita tai jos hänellä on infektioalttiutta lisäävä perussairaus (esim. divertikuliitti, diabetes ja interstitiaalinen keuhkosairaus).
Käytettäessä immunosuppressiivisia lääkkeitä, kuten tosilitsumabia, potilaiden tilaa on seurattava tarkoin, jotta mahdolliset vakavat infektiot havaitaan ajoissa, sillä akuutin vaiheen reaktion estyminen voi heikentää akuutin tulehduksen oireita. Potilaan mahdollista infektiota arvioitaessa on huomioitava tosilitsumabin vaikutukset C‑reaktiiviseen proteiiniin (CRP), neutrofiileihin ja infektion oireisiin. Potilaita (kuten yleisoireista lastenreumaa tai lasten polyartriittia sairastavat pikkulapset, jotka eivät välttämättä osaa kertoa oireistaan) ja yleisoireista lastenreumaa tai lasten polyartriittia sairastavien potilaiden vanhempia/huoltajia pitää ohjeistaa ottamaan heti yhteyttä terveydenhoitohenkilöstöön, jos he havaitsevat mitä tahansa infektioon viittaavia oireita, jotta tutkimukset ja hoito voidaan aloittaa mahdollisimman nopeasti.
Tuberkuloosi
Kuten muitakin biologisia lääkkeitä käytettäessä latentin tuberkuloosin poissulkemista seulontatutkimusten avulla suositellaan kaikilla potilailla ennen tosilitsumabihoidon aloittamista. Jos latentti tuberkuloosi todetaan, se on hoidettava tavanomaisilla mykobakteerilääkkeillä ennen kuin hoito aloitetaan. Lääkettä määrääviä lääkäreitä muistutetaan tuberkuliinikokeen ja verinäytteestä määritettävän IGRA-testin väärän negatiivisen tuloksen riskistä, varsinkin vaikeasti sairailla tai immuunipuutteisilla potilailla.
Potilaita ja yleisoireista lastenreumaa tai lasten polyartriittia sairastavien potilaiden vanhempia/huoltajia tulisi kehottaa ottamaan yhteyttä lääkäriin, jos tällä lääkevalmisteella annettavan hoidon aikana tai sen jälkeen ilmenee tuberkuloosi-infektioon viittaavia merkkejä tai oireita (esim. itsepintainen yskä, kuihtuminen/painon lasku, lievä lämmönnousu).
Virustautien uudelleen aktivoituminen
Virusten uudelleen aktivoitumista (esim. hepatiitti B -virus) on raportoitu biologisilla nivelreumalääkkeillä hoidetuilla potilailla. Tosilitsumabin kliinisiin tutkimuksiin ei ole hyväksytty hepatiittipositiivisia potilaita.
Divertikuliitin komplikaatiot
Melko harvinaisena divertikuliitin komplikaationa on raportoitu divertikkelien perforaatioita tosilitsumabihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Tätä lääkevalmistetta tulisi antaa varoen potilaille, joilla on aikaisemmin esiintynyt suoliston haavaumia tai divertikuliittia. Jos potilaalle ilmaantuu komplisoituneeseen divertikuliittiin viittaavia oireita, kuten vatsakipua, verenvuotoa ja/tai selittämättömiä muutoksia suolen toiminnassa ja kuumetta, potilas on tutkittava heti, jotta divertikuliitti ja siihen mahdollisesti liittyvä suolen puhkeaminen havaitaan mahdollisimman varhaisessa vaiheessa.
Yliherkkyysreaktiot
Vakavia yliherkkyysreaktioita, anafylaksiaa mukaan lukien, on raportoitu tosilitsumabin käytön yhteydessä (ks. kohta Haittavaikutukset). Reaktiot voivat olla vaikeampia ja mahdollisesti fataaleja niillä potilailla, jotka ovat kokeneet yliherkkyysreaktioita aikaisemman tosilitsumabihoidon yhteydessä, vaikka heille annetaan esilääkityksenä steroideja ja antihistamiineja. Anafylaktisen tai muun vakavan yliherkkyysreaktion ilmaantuessa, tosilitsumabin anto on heti keskeytettävä, asianmukainen hoito on aloitettava ja hoito on lopetettava pysyvästi.
Aktiivinen maksasairaus ja maksan vajaatoiminta
Tosilitsumabihoitoon voi liittyä kohonneita aminotransferaasiarvoja, varsinkin jos sitä annetaan yhdessä MTX:n kanssa. Siksi on noudatettava varovaisuutta, jos harkitaan hoidon aloittamista potilaille, joilla on aktiivinen maksasairaus tai maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Maksatoksisuus
Ohimenevää tai jaksottaista lievää tai kohtalaista maksan aminotransferaasiarvojen nousua on raportoitu yleisesti tosilitsumabihoidon yhteydessä (ks. kohta Haittavaikutukset). Kohonneita arvoja havaittiin enemmän, kun tosilitsumabihoitoon yhdistettiin jokin mahdollisesti maksatoksinen lääkevalmiste (esim. MTX). Muita maksantoimintakokeissa ilmenevien yhdisteiden määritystä, kuten bilirubiini, on harkittava kliinisen tarpeen mukaan.
Tosilitsumabihoitoa saaneilla potilailla on havaittu vakavia lääkkeestä aiheutuneita maksavaurioita, mukaan lukien akuutti maksan vajaatoiminta, hepatiitti ja ikterus (ks. kohta Haittavaikutukset). Vakava maksavaurio ilmeni kahdesta viikosta yli viiteen vuoteen hoidon aloittamisesta. Maksansiirron vaativia maksan vajaatoimintatapauksia on raportoitu. Potilaita on neuvottava ottamaan yhteyttä lääkäriin heti, jos maksavaurion oireita ilmaantuu.
Varovaisuutta on noudatettava harkittaessa hoidon aloittamista potilaille, joiden kohonneet ALAT- tai ASAT-arvot ovat > 1,5 × viitevälin yläraja. Hoitoa ei suositella potilaille, joiden ALAT- tai ASAT-arvot hoidon alussa ovat > 5 × viitevälin yläraja.
Nivelreuma- ja jättisoluarteriittipotilaiden sekä yleisoireista lastenreumaa tai lasten polyartriittia sairastavien potilaiden ALAT- ja ASAT-arvoja on seurattava joka 4.–8. viikko hoidon kuuden ensimmäisen kuukauden aikana ja sen jälkeen joka 12. viikko. Suositukset annoksen muuttamisesta aminotransferaasiarvojen perusteella, mukaan lukien tosilitsumabihoidon lopettamista, ks. kohta Annostus ja antotapa. Jos ASAT- tai ALAT-arvojen nousu on > 3–5 × viitevälin yläraja, hoito on keskeytettävä.
Hematologiset poikkeamat
Neutrofiili- ja trombosyyttiarvon laskua on esiintynyt, kun tosilitsumabia on annettu 8 mg/kg yhdessä MTX:n kanssa (ks. kohta Haittavaikutukset). Neutropenian riski voi olla suurentunut, jos potilasta on aikaisemmin hoidettu TNF-estäjällä.
Aiemmin tosilitsumabilla hoitamattomille potilaille ei suositella hoidon aloitusta, jos absoluuttinen neutrofiilien määrä on < 2 × 109/l. Varovaisuutta on noudatettava harkittaessa hoidon aloittamista potilaille, joiden trombosyyttiarvo on alentunut (trombosyyttiarvo alle 100 × 103/μl). Hoidon jatkamista ei suositella, jos potilaan ANC on < 0,5 × 109/l tai trombosyyttiarvo < 50 × 103/μl.
Vaikeaan neutropeniaan voi liittyä vakavien infektioiden kohonnut riski. Tosilitsumabin kliinisissä tutkimuksissa ei ole tähän mennessä ilmennyt selvää yhteyttä alentuneiden neutrofiiliarvojen ja vakavien infektiotapausten välillä.
Nivelreuma- ja jättisoluarteriittipotilailla neutrofiili- ja trombosyyttiarvoja on seurattava 4–8 viikon ajan hoidon alkamisesta ja myöhemmin normaalin kliinisen käytännön mukaan. Suositukset annoksen muuttamisesta ANC- ja trombosyyttiarvojen mukaan, ks. kohta Annostus ja antotapa.
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavilla lapsipotilailla neutrofiili- ja trombosyyttiarvoja on seurattava toisen antokerran yhteydessä ja sen jälkeen hyvän hoitokäytännön mukaan (ks. kohta Annostus ja antotapa).
Veren rasva-arvot
Veren rasva-arvojen, kuten kokonaiskolesterolin, LDL- ja HDL-kolesterolin sekä triglyseridien, nousua havaittiin tosilitsumabilla hoidetuilla potilailla (ks. kohta Haittavaikutukset). Suurimmalla osalla potilaista ei havaittu aterogeenisen vaikutuksen lisääntymistä. Kohonneet kokonaiskolesteroliarvot saatiin yleensä hallintaan kolesterolia alentavilla lääkkeillä.
Kaikkien potilaiden rasva-arvot on määritettävä 4–8 viikon kuluttua hoidon aloittamisesta. Potilaita tulisi hoitaa hyperlipidemian paikallisten hoitosuositusten mukaisesti.
Neurologiset häiriöt
Lääkäreiden tulisi kiinnittää erityistä huomiota oireisiin, jotka voivat viitata keskushermoston myeliinikadon puhkeamiseen. Toistaiseksi ei tiedetä, liittyykö tosilitsumabihoitoon keskushermoston myeliinikatoa.
Maligniteetti
Nivelreumapotilailla on lisääntynyt pahanlaatuisten kasvainten riski. Immunomodulatoriset lääkkeet voivat suurentaa tätä riskiä. Kliiniset tiedot eivät ole riittäviä, jotta voitaisiin arvioida pahanlaatuisten kasvainten mahdollista ilmaantuvuutta tosilitsumabialtistuksen jälkeen. Pitkäaikainen turvallisuusarviointi on meneillään.
Rokotukset
Eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältäviä rokotteita ei tulisi antaa tällä lääkevalmisteella annettavan hoidon aikana, sillä niiden kliinistä turvallisuutta ei ole osoitettu. Avoimessa satunnaistetussa tutkimuksessa tosilitsumabin ja MTX:n yhdistelmällä hoidetut aikuiset nivelreumapotilaat saavuttivat tehokkaan vasteen sekä 23-valenttiselle pneumokokkipolysakkaridi- että jäykkäkouristusrokotteelle. Vaste oli verrattavissa pelkkää MTX-hoitoa saaneiden potilaiden vasteeseen. Kaikille potilaille, etenkin lapsille tai iäkkäille, suositellaan kaikkien ajantasaisten rokotusten antamista voimassa olevien rokotussuositusten mukaisesti ennen hoidon aloittamista. Eläviä taudinaiheuttajia sisältävien rokotteiden antamisen ja hoidon aloittamisen välillä on pidettävä tauko, jonka kesto on nykyisten immunosuppressantteja koskevien rokotussuositusten mukainen.
Sydän- ja verisuonitautiriski
Nivelreumapotilailla on suurentunut sydän- ja verisuonisairauksien riski, ja riskitekijöiden (esim. hypertensio, hyperlipidemia) hallinnan on oltava osa näiden potilaiden tavanomaista perushoitoa.
Yhteiskäyttö TNF-estäjien kanssa
Tosilitsumabin käytöstä yhdessä TNF-estäjien tai muiden biologisten reumalääkkeiden kanssa ei ole kokemuksia nivelreumaa sairastavilla potilailla. Tämän lääkevalmisteen käyttöä yhdessä muiden biologisten reumalääkkeiden kanssa ei suositella.
Jättisoluarteriittia sairastavat potilaat
Tosilitsumabimonoterapiaa ei saa käyttää akuuttien relapsien hoitoon, koska sen tehoa niiden hoitoon ei ole varmistettu. Potilaalle pitää antaa glukokortikoideja lääkärin harkinnan ja hoitokäytännön mukaan.
Yleisoireista lastenreumaa sairastavat potilaat
Makrofagiaktivaatio-oireyhtymä (MAS) on vakava henkeä uhkaava tila, joka voi kehittyä yleisoireista lastenreumaa sairastaville potilaille. Kliinisissä tutkimuksissa tosilitsumabia ei ole tutkittu aktiivisen makrofagiaktivaatio-oireyhtymän oireita saaneilla potilailla.
Polysorbaatti 80 (E 433)
Tämä lääkevalmiste sisältää 0,18 mg polysorbaattia 80 per 162 mg/0,9 ml:n esitäytetty kynä, mikä vastaa 0,2 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Tiedossa olevat potilaan allergiat on otettava huomioon.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Tosilitsumabi 10 mg/kg kerta-annoksena annettuna ei vaikuttanut kliinisesti merkitsevästi MTX-altistukseen MTX-hoidon (10–25 mg kerran viikossa) aikana.
Populaatiofarmakokineettisessä analyysissä havaittiin, etteivät MTX, steroideihin kuulumattomat tulehduskipulääkkeet (NSAID-lääkkeet) eivätkä kortikosteroidit vaikuttaneet nivelreumapotilailla tosilitsumabin puhdistumaan. Jättisoluarteriittipotilailla kumulatiivisten kortikosteroidiannosten ei havaittu vaikuttaneen tosilitsumabialtistukseen.
Kroonista tulehdusta vahvistavat sytokiinit, kuten IL-6, estävät maksan CYP450-entsyymien ilmentymistä. CYP450-entsyymien ilmentyminen voi siis palautua, kun aloitetaan voimakas sytokiinia estävä lääkitys, esimerkiksi tosilitsumabi.
In vitro ‑tutkimukset viljellyillä ihmisen maksasoluilla osoittivat, että IL-6 pienensi CYP1A2-, CYP2C9-, CYP2C19- ja CYP3A4-entsyymien ilmentymää. Tosilitsumabi normalisoi näiden entsyymien ilmentymää.
Tutkimuksessa nivelreumapotilailla simvastatiinipitoisuudet (CYP3A4) olivat laskeneet 57 % viikon kuluttua tosilitsumabikerta-annoksen antamisesta ja olivat vastaavat tai hieman korkeammat kuin terveillä koehenkilöillä mitatut pitoisuudet.
Potilaita on seurattava tarkoin tosilitsumabihoitoa aloitettaessa tai lopetettaessa, jos he saavat CYP450-entsyymien 3A4, 1A2 tai 2C9 välityksellä metaboloituvia lääkkeitä (esim. metyyliprednisoloni, deksametasoni [oraalisen glukokortikoidihoidon lopettamisoireiden mahdollisuus], atorvastatiini, kalsiuminestäjät, teofylliini, varfariini, fenprokumoni, fenytoiini, siklosporiini tai bentsodiatsepiinit). Näiden lääkkeiden annokset määritellään yksilöllisesti ja annosten suurentaminen saattaa olla tarpeen hoitotehon säilyttämiseksi. Eliminaation pitkän puoliintumisajan (t1/2) takia tosilitsumabin vaikutus CYP450-entsyymin toimintaan saattaa jatkua vielä useita viikkoja hoidon päättymisen jälkeen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä luotettavaa ehkäisyä hoidon aikana ja vähintään kolmen kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole riittävästi tietoja tosilitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu spontaanien keskenmenojen / alkio- ja sikiökuolemien lisääntymistä suuria annoksia annettaessa (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta.
RoActemraa ei pidä käyttää raskauden aikana, ellei se ole ehdottoman välttämätöntä.
Imetys
Ei tiedetä, erittyykö tosilitsumabi ihmisillä äidinmaitoon. Tosilitsumabin erittymistä äidinmaitoon ei ole tutkittu eläimillä. On päätettävä, lopetetaanko imetys vai pidättäydytäänkö RoActemra‑hoidosta ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Käytettävissä olevat ei-kliiniset tiedot eivät viittaa tosilitsumabihoidon vaikuttavan hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
RoActemralla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn, esim. huimaus (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Turvallisuusprofiili perustuu 4 510 potilaan altistumiseen tosilitsumabille kliinisissä tutkimuksissa. Valtaosa näistä potilaista osallistui aikuisten nivelreumaa (n = 4 009) koskeneisiin tutkimuksiin, ja muu kokemus on saatu jättisoluarteriittia (n = 149), lasten polyartriittia (n = 240) ja yleisoireista lastenreumaa (n = 112) koskeneista tutkimuksista. Tosilitsumabin turvallisuusprofiili on näissä käyttöaiheissa samankaltainen eikä siinä ole havaittavissa eroja.
Yleisimmät haittavaikutukset olivat ylähengitystieinfektiot, nasofaryngiitti, päänsärky, hypertensio ja ALAT-arvon nousu.
Vakavimmat haittavaikutukset olivat vakavat infektiot, divertikuliitin komplikaatiot ja yliherkkyysreaktiot.
Taulukoitu lista haittavaikutuksista
Taulukossa 1 luetellaan haittavaikutukset, jotka on todettu kliinisissä tutkimuksissa ja/tai tosilitsumabin markkinoille tulon jälkeisessä käytössä spontaanien tapausselostusten, kirjallisuudessa raportoitujen tapausten ja ei-interventiotutkimusohjelmissa todettujen tapausten perusteella. Haittavaikutukset luetellaan MedDRA-elinjärjestelmän (SOC) mukaan. Vastaava yleisyysluokka perustuu seuraavaan esitystapaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Lista tosilitsumabihoitoa saaneilla potilailla esiintyneistä haittavaikutuksista
| MedDRA-elinjärjestelmä | Yleisyysluokka suositellulla termillä | ||||
| Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen | |
| Infektiot | Ylähengitystie-infektiot | Selluliitti, keuhkokuume, suun herpes simplex ‑infektio, vyöruusu | Divertikuliitti | ||
| Veri ja imukudos | Leukopenia, neutropenia, hypofibrinogenemia | ||||
| Immuunijärjestelmä | Anafylaksia (fataali)1, 2, 3 | ||||
| Umpieritys | Kilpirauhasen vajaatoiminta | ||||
| Aineenvaihdunta ja ravitsemus | Hyperkoleste-rolemia* | Hypertriglyseridemia | |||
| Hermosto | Päänsärky, huimaus | ||||
| Silmät | Sidekalvotulehdus | ||||
| Verisuonisto | Hypertensio | ||||
| Hengityselimet, rintakehä ja välikarsina | Yskä, hengenahdistus | ||||
| Ruoansulatuselimistö | Vatsakipu, suun haavaumat, gastriitti | Suutulehdus, mahahaava | |||
| Maksa ja sappi | Lääkkeestä aiheutunut maksavaurio, hepatiitti ja ikterus. | Maksan vajaatoiminta | |||
| Iho ja ihonalainen kudos | Ihottuma, kutina, nokkosihottuma | Stevens-Johnsonin oireyhtymä3 | |||
| Munuaiset ja virtsatiet | Munuaiskivitauti | ||||
| Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan reaktiot | Perifeerinen ödeema, yliherkkyysreaktiot | |||
| Tutkimukset | Maksan aminotransferaasiarvojen nousu, painon nousu, kokonaisbilirubiiniarvon nousu* | ||||
*Sisältää tavanomaisessa laboratorioseurannassa kerätyt kohonneet arvot (ks. alempana oleva teksti)
1 Katso kohta Vasta-aiheet
2 Katso kohta Varoitukset ja käyttöön liittyvät varotoimet
3 Tämä haittavaikutus havaittiin markkinoille tulon jälkeen, mutta sitä ei ole raportoitu kontrolloiduissa kliinisissä tutkimuksissa. Yleisyysluokka arvioitiin käyttämällä 95 prosentin luottamusvälin ylärajaa ja se perustuu kliinisissä tutkimuksissa tosilitsumabille altistuneiden potilaiden kokonaismäärään.
Valikoitujen haittavaikutusten kuvaus (ihonalainen annostelu)
Nivelreumaa sairastavat potilaat
Ihon alle annosteltavan tosilitsumabin turvallisuutta nivelreumapotilaille on tutkittu kaksoissokkoutetussa, kontrolloidussa monikeskustutkimuksessa, SC-I:ssä. Tutkimus tehtiin vertailukelpoisuuden (non-inferiority) osoittamiseksi, ja siinä verrattiin ihon alle annosteltavan (162 mg kerran viikossa) ja laskimoon annosteltavan (8 mg/kg) tosilitsumabin tehoa ja turvallisuutta. Tutkimukseen osallistui 1 262 potilasta. Kaikki potilaat saivat peruslääkityksenä ei-biologisia tautiprosessia hidastavia reumalääkkeitä (DMARD). Ihon alle annosteltavan tosilitsumabin turvallisuus ja immunogeenisuus oli yhtenevä laskimoon annosteltavan tosilitsumabin turvallisuusprofiilin kanssa eikä uusia tai odottamattomia haittavaikutuksia havaittu (ks. taulukko 1). Pistoskohdan reaktioita havaittiin yleisemmin ihon alle annosteltua tosilitsumabin saaneilla potilailla verrattuna kontrolliryhmään (laskimoon tosilitsumabin saaneiden ryhmä), jossa lumelääkettä annosteltiin ihon alle.
Pistoskohdan reaktiot
Tutkimuksessa SC-I kuusi kuukautta kestäneen kontrolloidun jakson aikana pistoskohdan reaktioita ilmaantui 10,1 %:lle (64/631) ihon alle annosteltua tosilitsumabia saaneista potilaista ja vastaavasti 2,4 %:lle (15/631) ihon alle kerran viikossa annosteltua lumelääkettä saaneista potilaista. Pistoskohdan reaktiot (ihon punoitus, kutina, kipu ja hematooma) olivat vaikeusasteeltaan lieviä tai kohtalaisia. Suurin osa reaktioista hävisi ilman hoitoa eikä hoitoa jouduttu keskeyttämään pistoskohdan reaktioiden vuoksi.
Immunogeenisuus
Anti-tosilitsumabi-vasta-aineet testattiin tutkimuksessa SC-I kuuden kuukauden kontrolloidun jakson aikana yhteensä 625 tosilitsumabia 162 mg:n viikkoannoksena saaneelta potilaalta. Viidelle potilaalle (0,8 %) kehittyi anti-tosilitsumabi-vasta-aineita, ja kaikille näille potilaille kehittyi neutraloivia anti-tosilitsumabi-vasta-aineita. Yksi potilas reagoi positiivisesti IgE-isotyyppitestiin (0,2 %).
Anti-tosilitsumabi-vasta-aineet testattiin tutkimuksessa SC-II kuuden kuukauden kontrolloidun jakson aikana yhteensä 434 potilaalta, jotka olivat saaneet tosilitsumabia annoksella 162 mg joka toinen viikko. Seitsemälle potilaalle (1,6 %) kehittyi anti-tosilitsumabi-vasta-aineita, ja näistä kuudelle potilaalle (1,4 %) kehittyi neutraloivia anti- tosilitsumabi-vasta-aineita. Neljä potilasta reagoi positiivisesti IgE-isotyyppitestiin (0,9 %).
Lääkevasta-aineiden muodostumisen ja kliinisen hoitovasteen tai haittatapahtumien välillä ei havaittu yhteyttä.
Neutrofiilit
Kuusi kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen SC-I tavanomaisessa laboratorioseurannassa neutrofiiliarvojen laskua tason 1 × 109/l alapuolelle ilmaantui 2,9 %:lle potilaista, joille annosteltiin tosilitsumabia viikoittain ihon alle.
Neutrofiiliarvojen laskun tason 1 × 109/l alapuolelle ja vakavien infektioiden ilmaantumisen välillä ei ollut selkeää yhteyttä.
Trombosyytit
Kuusi kuukautta kestäneen kliinisen tosilitsumabitutkimuksen SC-I tavanomaisessa laboratorioseurannassa yhdelläkään ihon alle annoksen viikoittain saaneista potilaista ei esiintynyt trombosyyttiarvon laskua tasoon ≤ 50 × 103/μl.
Maksan aminotransferaasiarvojen nousu
Kuusi kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen SC-I tavanomaisessa laboratorioseurannassa todettiin maksan aminotransferaasiarvojen nousua tasolle ≥ 3 × viitevälin yläraja 6,5 %:lla (ALAT) ja 1,4 %:lla (ASAT) annoksen ihon alle viikoittain saaneista potilaista.
Veren rasva-arvot
Kuusi kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen SC-I tavanomaisessa laboratorioseurannassa tosilitsumabia viikoittain ihon alle saaneista potilaista 19 %:lla veren kokonaiskolesteroliarvo nousi pitkäkestoisesti arvoon > 6,2 mmol/l (240 mg/dl) ja 9 %:lla potilaista LDL-kolesteroliarvo nousi pitkäkestoisesti arvoon ≥ 4,1 mmol/l (160 mg/dl).
Yleisoireista lastenreumaa (sJIA) sairastavat potilaat
Ihon alle annosteltavan tosilitsumabin turvallisuusprofiilia arvioitiin 51 yleisoireista lastenreumaa sairastavalla (1–17-vuotiaalla) lapsipotilaalla. Yleisoireista lastenreumaa sairastavilla potilailla havaitut haittavaikutukset olivat yleensä luonteeltaan samankaltaisia kuin nivelreumapotilailla on todettu (ks. kohta Haittavaikutukset).
Infektiot
Infektioiden esiintyvyys oli verrannollinen tosilitsumabia ihon alle ja laskimoon saaneilla yleisoireista lastenreumaa sairastavilla lapsipotilailla.
Injektiokohdan reaktiot
Ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa (WA28118) yhteensä 41,2 %:lla (21/51) yleisoireista lastenreumaa sairastavista potilaista oli injektiokohdan reaktioita, kun tosilitsumabi annettiin ihon alle. Yleisimpiä injektiokohdan reaktioita olivat punoitus, kutina, kipu ja injektiokohdan turvotus. Valtaosa raportoiduista injektiokohdan reaktioista oli graduksen 1 tapahtumia, yksikään raportoiduista injektiokohdan reaktioista ei ollut vakava eivätkä ne vaatineet yhdenkään potilaan hoidon lopettamista tai keskeyttämistä.
Immunogeenisuus
Ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa (WA28118) 90,2 %:lla (46/51) potilaista, joilta määritettiin anti-tosilitsumabi-vasta-aineet, oli vähintään yksi seulontamääritystulos hoidon aloittamisen jälkeen. Kenellekään ei kehittynyt positiivisia anti-tosilitsumabivasta-aineita tutkimushoidon aikana.
Laboratorioarvojen poikkeavuudet
52 viikkoa kestäneessä avoimessa ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa (WA28118) neutrofiiliarvojen laskua arvon 1 × 109/l alapuolelle ilmaantui 23,5 %:lle ihon alle annosteltavaa tosilitsumabia saaneista potilaista. Trombosyyttiarvon laskua arvon 100 × 103/μl alapuolelle ilmaantui 2 %:lle ihon alle annosteltavaa tosilitsumabia saaneista potilaista. Ihon alle annosteltavaa tosilitsumabia saaneiden potilaiden ALAT-arvo suureni ≥ 3 × viitearvojen ylärajan 9,8 %:lla potilaista ja ASAT-arvo suureni ≥ 3 × viitearvojen ylärajan 4,0 %:lla potilaista.
Veren rasva-arvot
52 viikkoa kestäneessä avoimessa ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa (WA28118) LDL-kolesterolipitoisuus suureni jossakin vaiheessa tutkimushoidon aikana arvoon ≥ 130 mg/dl 23,4 %:lla potilaista ja kokonaiskolesterolipitoisuus suureni arvoon ≥ 200 mg/dl 35,4 %:lla potilaista.
Lasten polyartriittia (pJIA) sairastavat potilaat
Ihon alle annosteltavan tosilitsumabin turvallisuusprofiilia arvioitiin myös 52 lasten polyartriittia sairastavalla potilaalla. Lasten polyartriittia sairastavassa potilasjoukossa kokonaisaltistus tosilitsumabille jakautui seuraavasti: 184,4 potilasvuotta laskimoon annetulle tosilitsumabille ja 50,4 potilasvuotta ihon alle annetulle tosilitsumabille. Lasten polyartriittia sairastavilla potilailla havaittu turvallisuusprofiili oli yleisesti yhdenmukainen tosilitsumabin tunnetun turvallisuusprofiilin kanssa injektiokohdan reaktioita lukuun ottamatta (ks. taulukko 1). Injektiokohdan reaktioita esiintyi ihon alle annettuja injektioita saaneilla lasten polyartriittia sairastavilla potilailla yleisemmin kuin aikuisilla nivelreumapotilailla.
Infektiot
Ihon alle tapahtuvaa antoa koskeneessa tosilitsumabitutkimuksessa infektioiden esiintyvyys oli verrannollinen tosilitsumabia ihon alle ja laskimoon saaneilla lasten polyartriittia sairastavilla potilailla.
Injektiokohdan reaktiot
Lasten polyartriittia sairastavista potilaista yhteensä 28,8 %:lla (15/52) oli injektiokohdan reaktioita, kun tosilitsumabi annettiin ihon alle. Injektiokohdan reaktioita esiintyi 44 %:lla ≥ 30 kg:n painoisista potilaista verrattuna 14,8 %:iin alle 30 kg:n painoisista potilaista. Yleisimpiä injektiokohdan reaktioita olivat injektiokohdan punoitus, turvotus, hematooma, kipu ja kutina. Raportoidut injektiokohdan reaktiot eivät olleet vakavia (gradus 1) eikä yhdenkään potilaan injektiokohdan reaktio vaatinut hoidon lopettamista tai keskeyttämistä.
Immunogeenisuus
Ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa 5,8 %:lle (3/52) potilaista kehittyi neutraloivia anti-tosilitsumabi-vasta-aineita, mutta vakavaa tai kliinisesti merkittävää yliherkkyysreaktiota ei kehittynyt. Näistä kolmesta potilaasta yksi vetäytyi myöhemmin tutkimuksesta. Vasta-aineiden kehittymisen ja kliinisen vasteen tai haittavaikutusten välillä ei havaittu korrelaatiota.
Laboratorioarvojen poikkeavuudet
Koko tosilitsumabille altistuneen potilasjoukon tavanomaisessa laboratorioseurannassa neutrofiilien määrä väheni 15,4 %:lla tosilitsumabia ihon alle saaneista potilaista alle arvon 1 × 109/l. Tosilitsumabia ihon alle saaneiden potilaiden ALAT-arvo suureni ≥ 3 × viitearvojen ylärajan 9,6 %:lla potilaista ja ASAT-arvo suureni ≥ 3 × viitearvojen ylärajan 3,8 %:lla potilaista. Yhdenkään tosilitsumabia ihon alle saaneen potilaan trombosyyttimäärä ei laskenut arvoon ≤ 50 × 103/μl.
Veren rasva-arvot
Ihon alle tapahtuvaa antoa koskeneessa tutkimuksessa LDL-kolesterolipitoisuus suureni jossakin vaiheessa tutkimushoidon aikana 14,3 %:lla potilaista arvoon ≥ 130 mg/dl ja kokonaiskolesterolipitoisuus suureni 12,8 %:lla potilaista arvoon ≥ 200 mg/dl.
Jättisoluarteriittia sairastavat potilaat
Ihon alle annosteltavan tosilitsumabin turvallisuutta on tutkittu yhdessä vaiheen III tutkimuksessa (WA28119), jossa oli mukana 251 jättisoluarteriittia sairastavaa potilasta. Altistuksen kesto (potilasvuosina) tutkimuksen 12 kuukauden kaksoissokkoutetun, lumekontrolloidun vaiheen aikana tosilitsumabille altistuneissa kaikissa potilasryhmissä oli yhteensä 138,5 potilasvuotta. Tosilitsumabihoitoryhmissä havaittu kokonaisturvallisuusprofiili oli yhdenmukainen tosilitsumabin tunnetun turvallisuusprofiilin kanssa (ks. taulukko 1).
Infektiot
Infektioiden tai vakavien infektiotapahtumien esiintyvyys oli tasapainossa tosilitsumabia viikoittain saaneen ryhmän (200,2/9,7 tapahtumaa 100 potilasvuotta kohden) sekä lumevalmisteen ja 26 viikon aikana asteittain lopetetun prednisonihoidon yhdistelmää (156,0/4,2 tapahtumaa 100 potilasvuotta kohden) ja lumevalmisteen ja 52 viikon aikana asteittain lopetetun hoidon yhdistelmää (210,2/12,5 tapahtumaa 100 potilasvuotta kohden) saaneen ryhmän välillä.
Pistoskohdan reaktiot
Ihon alle annosteltua tosilitsumabia viikoittain saaneessa ryhmässä yhteensä 6 % (6/100) potilaista raportoi ihon alle annetun pistoksen antokohdassa jonkin haittavaikutuksen. Yhtään injektiokohdan reaktiota ei raportoitu vakavana haittatapahtumana eivätkä ne vaatineet hoidon lopettamista.
Immunogeenisuus
Tosilitsumabia ihon alle viikoittain saaneessa ryhmässä yhdelle potilaalle (1,1 %, 1/95) kehittyi neutraloivia anti-tosilitsumabi-vasta-aineita, jotka eivät kuitenkaan olleet IgE-isotyyppiä. Tälle potilaalle ei kehittynyt yliherkkyysreaktiota eikä pistoskohdan reaktiota.
Neutrofiilit
Kaksitoista kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen tavanomaisessa laboratorioseurannassa neutrofiiliarvojen laskua tason 1 × 109/l alapuolelle ilmaantui 4 %:lle potilaista, joille annosteltiin tosilitsumabia viikoittain ihon alle. Tällaista ei havaittu kummassakaan lumevalmisteen ja asteittain lopetettavan prednisonihoidon yhdistelmää saaneessa ryhmässä.
Trombosyytit
Kaksitoista kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen tavanomaisessa laboratorioseurannassa tosilitsumabia viikoittain ihon alle saaneen ryhmän yhden potilaan (1 %, 1/100) trombosyyttiarvo laski kerran tilapäisesti tason 100 × 103/μl alapuolelle eikä siihen liittynyt verenvuototapahtumia. Trombosyyttimäärän laskua alle tason 100 × 103/μl ei havaittu kummassakaan lumevalmisteen ja asteittain lopetettavan prednisonihoidon yhdistelmää saaneessa ryhmässä.
Maksan aminotransferaasiarvojen nousu
Kaksitoista kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen tavanomaisessa laboratorioseurannassa ALAT-arvon kohoamista ≥ 3 × viitevälin yläraja esiintyi 3 %:lla tosilitsumabia ihon alle viikoittain saaneen ryhmän potilaista verrattuna 2 %:iin lumevalmisteen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneessa ryhmässä, mutta ei yhdelläkään lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneen ryhmän potilaista. ASAT-arvon kohoamista > 3 × viitevälin yläraja esiintyi 1 %:lla tosilitsumabia ihon alle viikoittain saaneen ryhmän potilaista, mutta ei yhdelläkään kummankaan lumevalmisteen ja asteittain lopetettavan prednisonihoidon yhdistelmää saaneen ryhmän potilaista.
Veren rasva-arvot
Kaksitoista kuukautta kestäneen kontrolloidun kliinisen tosilitsumabitutkimuksen tavanomaisessa laboratorioseurannassa 34 %:lla potilaista kokonaiskolesteroliarvo nousi pitkäkestoisesti arvoon > 6,2 mmol/l (240 mg/dl), ja 15 %:lla tosilitsumabia ihon alle viikoittain saaneen ryhmän potilaista LDL-kolesteroliarvo nousi pitkäkestoisesti arvoon ≥ 4,1 mmol/l (160 mg/dl).
Valikoitujen haittavaikutusten kuvaus (laskimonsisäinen annostelu)
Nivelreumaa sairastavat potilaat
Tosilitsumabin turvallisuutta on tutkittu viidessä vaiheen III kaksoissokkoutetussa, kontrolloidussa tutkimuksessa ja niiden jatkotutkimuksissa (ks. kohta Farmakodynamiikka).
Koko vertailtu potilasjoukko käsittää kaikki kaksoissokkoutetun vaiheen potilaat kustakin ydintutkimuksesta satunnaistamisesta joko ensimmäiseen hoito-ohjelmaan tehtyyn muutokseen saakka tai kunnes hoito-ohjelmaa oli jatkettu kaksi vuotta. Vertailujakso oli neljässä tutkimuksessa kuusi kuukautta ja yhdessä tutkimuksessa enintään kaksi vuotta. Kaksoissokkoutetuissa, kontrolloiduissa tutkimuksissa 774 potilasta sai 4 mg/kg tosilitsumabia yhdistelmänä MTX:n kanssa, 1 870 potilasta sai 8 mg/kg tosilitsumabia yhdistelmänä MTX:n tai muun DMARDin kanssa ja 288 potilasta sai 8 mg/kg tosilitsumabia monoterapiana.
Koko altistunut potilasjoukko käsittää kaikki potilaat, jotka saivat vähintään yhden tosilitsumabiannoksen tutkimusten joko kaksoissokkoutetun vertailujakson tai avoimen jatkovaiheen aikana. Tämän potilasjoukon 4 009 potilaasta 3 577 potilasta sai hoitoa vähintään kuuden kuukauden ajan, 3 296 potilasta sai hoitoa vähintään yhden vuoden ajan, 2 806 potilasta sai hoitoa vähintään kahden vuoden ajan ja 1 222 potilasta vähintään kolmen vuoden ajan.
Infektiot
Kuusi kuukautta kestäneissä vertailututkimuksissa infektioiden esiintyvyys oli 127 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat 8 mg/kg tosilitsumabia yhdessä DMARDien kanssa, ja 112 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat lumevalmistetta ja DMARDeja. Populaatiossa, jossa altistuminen tosilitsumabille oli pitkäaikaista, infektioiden kokonaisesiintyvyys oli 108 tapahtumaa 100 potilasvuotta kohti.
Kuusi kuukautta kestäneissä kliinisissä vertailututkimuksissa vakavien infektioiden esiintyvyys oli 5,3 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat 8 mg/kg tosilitsumabia yhdessä DMARDien kanssa ja 3,9 tapahtumaa 100 potilasvuotta kohti potilailla, jotka saivat lumevalmistetta ja DMARDeja. Monoterapiatutkimuksessa vakavien infektioiden esiintyvyys oli 3,6 tapahtumaa 100 potilasvuotta kohti tosilitsumabiryhmässä ja 1,5 tapahtumaa 100 potilasvuotta kohti MTX-ryhmässä.
Populaatiossa, jossa altistuminen oli pitkäaikaista, vakavien infektioiden (bakteeri-, virus- tai sieni-infektio) kokonaisesiintyvyys oli 4,7 tapahtumaa 100 potilasvuotta kohti. Raportoituja vakavia infektioita, joista jotkut johtivat kuolemaan, olivat aktiivinen tuberkuloosi (intra- tai ekstrapulmonaalinen), invasiiviset keuhkoinfektiot, mukaan lukien kandidiaasi, aspergilloosi, koksidioidomykoosi ja pneumocystis jirovecii -infektio, keuhkokuume, selluliitti, vyöruusu, gastroenteriitti, divertikuliitti, sepsis ja bakteeriartriitti. Opportunistisia infektioita on myös raportoitu.
Interstitiaalinen keuhkosairaus
Keuhkojen heikentynyt toimintakyky voi lisätä infektioriskiä. Interstitiaalista keuhkosairautta on raportoitu markkinoille tulon jälkeen (mukaan lukien keuhkotulehdus ja keuhkofibroosi). Näistä tapauksista muutama on ollut kuolemaan johtava.
Ruoansulatuskanavan perforaatiot
Kuusi kuukautta kestäneissä kontrolloiduissa kliinisissä tutkimuksissa ruoansulatuskanavan perforaatioiden kokonaisesiintyvyys tosilitsumabihoidon aikana oli 0,26 tapahtumaa 100 potilasvuotta kohti. Populaatiossa, jossa altistuminen oli pitkäaikaista, kokonaisesiintyvyys oli 0,28 tapahtumaa 100 potilasvuotta kohti. Ruoansulatuskanavan perforaatioita tosilitsumabihoidon yhteydessä raportoitiin ensisijaisesti divertikuliitin komplikaatioina mukaan lukien yleistynyt märkäinen peritoniitti, ruoansulatuskanavan alaosan perforaatio, fistelit ja absessi.
Infuusion liittyvät reaktiot
Kuusi kuukautta kestäneissä tutkimuksissa infuusioon liittyviä haittatapahtumia (valikoidut tapahtumat, jotka ilmaantuivat infuusion aikana tai 24 tunnin kuluessa sen päättymisestä) esiintyi 6,9 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa, ja 5,1 %:lla potilaista, jotka saivat lumevalmistetta ja DMARDeja. Infuusion aikana raportoidut tapahtumat olivat pääasiassa hypertensioepisodeja. Tapahtumia, joita raportoitiin 24 tunnin aikana infuusion päättymisen jälkeen, olivat päänsärky ja ihoreaktiot (ihottuma, nokkosihottuma). Nämä tapahtumat eivät olleet hoitoa rajoittavia.
Anafylaktisten reaktioiden esiintyvyys (yhteensä 8 tapausta / 4 009 potilasta, 0,2 %) oli moninkertainen annoksen ollessa 4 mg/kg verrattuna annokseen 8 mg/kg. Hoidon keskeyttämistä vaatineita kliinisesti merkitseviä tosilitsumabihoitoon liittyneitä yliherkkyysreaktioita todettiin yhteensä 56:lla (1,4 %) niistä 4 009 potilaasta, jotka saivat hoitoa vertailututkimuksissa ja avoimissa kliinisissä tutkimuksissa. Nämä reaktiot ilmaantuivat yleensä 2.–5. tosilitsumabi-infuusion aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myyntiluvan myöntämisen jälkeen on raportoitu yksi fataali anafylaksiatapaus tosilitsumabihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Anti-tosilitsumabi-vasta-aineet määritettiin kuusi kuukautta kestäneissä kliinisissä vertailututkimuksissa yhteensä 2 876 potilaalta. Näistä 46 potilaalle (1,6 %) kehittyi anti-tosilitsumabi-vasta-aineita, ja heistä kuudella esiintyi lääketieteellisesti merkitsevä yliherkkyysreaktio, jonka seurauksena viidellä potilaalla hoito lopetettiin pysyvästi. Neutraloivia vasta-aineita kehittyi 30 potilaalla (1,1 %).
Neutrofiilit
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa neutrofiiliarvo laski tason 1 × 109/l alapuolelle 3,4 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg ja DMARDeja verrattuna < 0,1 %:iin potilaista, jotka saivat lumevalmistetta ja DMARDeja. Potilaista, joiden absoluuttinen neutrofiiliarvo laski tasolle < 1 × 109/l, noin puolella lasku todettiin kahdeksan viikon kuluessa hoidon alkamisesta. Tason 0,5 × 109/l alapuolelle laskeneita arvoja todettiin 0,3 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa. Infektioita, joihin liittyi neutropenia, on raportoitu.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin neutrofiiliarvojen laskeneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Trombosyytit
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa trombosyyttiarvo laski tason 100 × 103/μl alapuolelle 1,7 %:lla tosilitsumabia 8 mg/kg ja DMARDeja saaneista potilaista verrattuna < 1 %:lla lumevalmistetta ja DMARDeja saaneisiin potilaisiin. Näihin muutoksiin ei liittynyt verenvuototapahtumia.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin trombosyyttiarvojen laskeneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Myyntiluvan myöntämisen jälkeisessä seurannassa on hyvin harvoin raportoitu pansytopeniaa.
Maksan aminotransferaasiarvojen nousu
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa ohimenevä ALAT-/ASAT-arvojen nousu > 3 × viitevälin yläraja todettiin 2,1 %:lla tosilitsumabia 8 mg/kg saaneista ja 4,9 %:lla MTX:a saaneista potilaista sekä 6,5 %:lla potilaista, jotka saivat tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa, sekä 1,5 %:lla lumevalmistetta ja DMARDeja saaneista potilaista.
Kohonneet arvot yleistyivät, kun tosilitsumabimonoterapiahoitoon lisättiin jokin mahdollisesti maksatoksinen lääkevalmiste (esim. MTX). ALAT-/ASAT-arvojen nousu > 5 × viitevälin yläraja todettiin 0,7 %:lla pelkkää tosilitsumabia saaneista potilaista ja 1,4 %:lla potilaista, jotka saivat tosilitsumabia yhdessä DMARDien kanssa. Suurimmalla osalla näistä potilaista tosilitsumabi hoito keskeytettiin pysyvästi. Kaksoissokkoutetun kontrollivaiheen tavanomaisessa laboratorioseurannassa 6,2 %:lla tosilitsumabia 8 mg/kg ja DMARDeja saaneista potilaista todettiin konjugoitumattoman bilirubiinin pitoisuuksia, jotka olivat korkeammat kuin viitevälin yläraja. Konjugoitumattoman bilirubiinin pitoisuus nousi tasolle > 1–2 × viitevälin yläraja yhteensä 5,8 %:lla potilaista ja 0,4 %:lla nousu oli > 2 × viitevälin yläraja.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin ALAT-/ASAT-arvojen nousseen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Veren rasva-arvot
Kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa on raportoitu yleisesti veren rasva-arvojen, kuten kokonaiskolesterolin, triglyseridien, LDL- ja/tai HDL-kolesterolin, nousua. Tavanomaisessa laboratorioseurannassa noin 24 %:lla potilaista, jotka saivat tosilitsumabia kliinisissä tutkimuksissa, todettiin pysyvästi kohonneita kokonaiskolesteroliarvoja (≥ 6,2 mmol/l). 15 %:lla potilaista havaittiin pysyvästi kohonneita LDL-kolesteroliarvoja (≥ 4,1 mmol/l). Kohonneet veren rasva-arvot saatiin hallintaan kolesterolia alentavilla lääkkeillä.
Kaksoissokkoutetussa kontrollivaiheessa ja pitkäaikaisessa altistumisessa havaittiin veren rasva-arvojen kohonneen vastaavalla tavalla ja ilmaantuvuudella kuin kuusi kuukautta kestäneissä kontrolloiduissa tutkimuksissa.
Ihoreaktiot
Myyntiluvan myöntämisen jälkeisessä seurannassa on harvoin raportoitu Stevens-Johnsonin oireyhtymää.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tietoja tosilitsumabin yliannostuksesta on rajoitetusti. Yksi tahaton yliannostustapaus on raportoitu. Siinä multippelia myeloomaa sairastava potilas sai kerta-annoksena laskimoon 40 mg/kg. Haittavaikutuksia ei havaittu.
Vakavia haittavaikutuksia ei havaittu, kun terveille koehenkilöille annettiin kerta-annoksena enintään 28 mg/kg, mutta annostusta rajoittavaa neutropeniaa todettiin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressiiviset lääkeaineet, interleukiinin estäjät, ATC-koodi: L04AC07.
Vaikutusmekanismi
Tosilitsumabi sitoutuu spesifisesti sekä liukoisiin että kalvoon sitoutuneisiin IL‑6-reseptoreihin (sIL‑6 ja mIL‑6R). Tosilitsumabin on osoitettu estävän sIL‑6R- ja mIL‑6R-reseptorien kautta tapahtuvaa signaalinvälitystä. IL-6 on pleiotrooppinen tulehdusta vahvistava sytokiini, jota tuottavat useat eri solutyypit, kuten T- ja B-solut, monosyytit ja fibroblastit. IL-6 osallistuu erilaisiin fysiologisiin tapahtumaketjuihin, joita ovat esimerkiksi T-solun aktivoituminen, immunoglobuliinierityksen käynnistyminen, akuutin vaiheen proteiinisynteesin käynnistyminen maksassa ja hematopoieesin stimuloituminen. IL-6 on yhdistetty erilaisten sairauksien, kuten tulehdussairauksien, osteoporoosin ja kasvainten, patogeneesiin.
Farmakodynaamiset vaikutukset
Tosilitsumabilla tehdyissä kliinisissä nivelreumatutkimuksissa havaittiin CRP:n (C-reaktiivisen proteiinin), laskon (La), seerumin amyloidi A:n (SAA) ja fibrinogeenin nopea lasku. Akuutin vaiheen proteiineihin kohdistuvan vaikutuksen mukaisesti tosilitsumabihoitoon liittyi myös trombosyyttiarvon lasku normaalialueen sisällä. Hemoglobiiniarvon nousua havaittiin, sillä tosilitsumabi heikentää IL-6:n vaikutuksia hepsidiinin tuotantoon, mikä lisää raudan saatavuutta. Hoitoa saaneilla potilailla CRP-arvon lasku normaalialueelle havaittiin jo toisella hoitoviikolla, ja lasku säilyi koko hoidon ajan.
Kliinisessä jättisoluarteriittitutkimuksessa (WA28119) havaittiin samankaltainen CRP:n ja laskon nopea lasku sekä keskimääräisen hemoglobiinipitoisuuden lievää suurenemista. Kun tosilitsumabia annettiin annoksilla 2–28 mg/kg laskimoon tai 81–162 mg ihon alle terveille vapaaehtoisille koehenkilöille, heidän neutrofiiliarvonsa laskivat alimmalle tasolleen 2–5 päivän kuluttua annostelusta. Sen jälkeen neutrofiiliarvot palautuivat hoitoa edeltävälle tasolle annosriippuvaisesti.
Nivelreuma- ja jättisoluarteriittipotilailla havaittiin neutrofiiliarvojen laskevan vastaavalla tavalla kuin terveillä vapaaehtoisilla tosilitsumabin annon jälkeen (ks. kohta Haittavaikutukset).
Ihonalainen annostelu
Nivelreumaa sairastavat potilaat
Kliininen teho
Ihon alle annosteltavan tosilitsumabin tehoa nivelreuman oireiden lieventämisessä ja radiologisen vasteen saamisessa on tutkittu kahdessa satunnaistetussa kaksoissokkoutetussa kontrolloidussa monikeskustutkimuksessa. Tutkimuksessa I (SC-I) potilaiden piti olla iältään > 18-vuotiaita ja sairastaa keskivaikeaa tai vaikeaa aktiivista nivelreumaa, joka oli diagnosoitu ACR-kriteerien mukaan. Lisäksi heillä piti olla lähtötilanteessa vähintään 4 arkaa ja 4 turvonnutta niveltä. Kaikki potilaat saivat peruslääkityksenä yhtä tai useampaa tautiprosessia hidastavaa reumalääkettä (DMARD). Tutkimuksessa II (SC-II) potilaiden piti olla iältään > 18-vuotiaita ja sairastaa keskivaikeaa tai vaikeaa aktiivista nivelreumaa, joka oli diagnosoitu ACR-kriteerien mukaan. Lisäksi heillä piti olla lähtötilanteessa vähintään 8 arkaa ja 6 turvonnutta niveltä.
Potilaan altistuminen lääkkeelle muuttuu vaihdettaessa laskimonsisäisestä annostelusta (8 mg/kg joka neljäs viikko) ihonalaiseen annosteluun (162 mg ihon alle kerran viikossa). Altistumisen suuruus vaihtelee potilaan painon mukaan (lisääntyy keveämmillä potilailla ja vähenee painavammilla potilailla), mutta kliininen vaste on yhdenmukainen laskimonsisäistä hoitoa saaneiden potilaiden kanssa.
Kliininen vaste
Tutkimuksessa SC-I oli mukana keskivaikeaa tai vaikeaa aktiivista nivelreumaa sairastavia potilaita, joiden kliininen hoitovaste senhetkiseen hoitoon, johon kuului yksi tai useampi DMARD, oli riittämätön ja joista noin 20 % ei ollut aiemmin saanut riittävää vastetta yhdellä tai useammalla TNF:n estäjällä. Tutkimuksessa SC-I satunnaistettiin 1 262 potilasta suhteessa 1:1 saamaan tosilitsumabia ihon alle annoksena 162 mg viikoittain tai tosilitsumabia laskimoon annoksena 8 mg/kg neljän viikon välein yhdistelmänä yhden tai useamman ei-biologisen DMARD-lääkkeen kanssa. Tutkimuksen ensisijainen päätetapahtuma oli ero niiden potilaiden osuudessa, jotka saivat ACR 20-vasteen viikolla 24. Tutkimuksen SC-I tulokset esitetään taulukossa 2.
Taulukko 2. ACR-vasteosuudet tutkimuksessa SC-I (% potilaista) viikolla 24
| SC-Ia | ||
TCZ ihon alle viikoittain annoksena 162 mg + DMARD n = 558 | TCZ laskimoon annoksena 8 mg/kg + DMARD n = 537 | |
| ACR 20 viikolla 24 | 69,4 % | 73,4 % |
| Painotettu ero (95 % CI) | -4,0 (-9,2, 1,2) | |
| ACR 50 viikolla 24 | 47,0 % | 48,6 % |
| Painotettu ero (95 % CI) | -1,8 (-7,5, 4,0) | |
| ACR 70 viikolla 24 | 24,0 % | 27,9 % |
| Painotettu ero (95 % CI) | -3,8 (-9,0, 1,3) | |
DMARD = tautiprosessia hidastava reumalääke
TCZ = tosilitsumabi
a = Per Protocol ‑potilasjoukko
Tutkimuksessa SC-I mukana olleiden potilaiden DAS28-indeksin keskiarvo tutkimuksen alkaessa oli 6,6 tosilitsumabia ihon alle saaneiden ryhmässä ja 6,7 tosilitsumabia laskimoon saaneiden ryhmässä. Viikolla 24 havaittiin kummassakin hoitohaarassa DAS28-indeksin merkitsevä 3,5 yksikön lasku lähtötilanteesta (keskimääräinen paraneminen), ja yhtäläinen osuus potilaista tosilitsumabia ihon alle (38,4 %) ja laskimoon (36,9 %) saaneiden ryhmässä oli saavuttanut DAS28-indeksillä osoitetun kliinisen remission (DAS28 < 2,6).
Radiologinen hoitovaste
Radiologista hoitovastetta ihon alle annettuun tosilitsumabiin arvioitiin kaksoissokkoutetussa, kontrolloidussa, monikeskustutkimuksessa aktiivista nivelreumaa sairastavilla potilailla (SC-II). Tutkimuksessa SC-II arvioitiin keskivaikeaa tai vaikeaa aktiivista nivelreumaa sairastavia potilaita, joiden kliininen vaste senhetkiseen reumahoitoon, johon kuului yksi tai useampi DMARD, oli riittämätön ja joista noin 20 % ei ollut saanut riittävää vastetta yhdellä tai useammalla TNF-estäjällä. Potilaiden piti olla iältään > 18-vuotiaita ja sairastaa ACR-kriteerien mukaan diagnosoitua aktiivista nivelreumaa. Lisäksi heillä piti olla lähtötilanteessa vähintään 8 arkaa ja 6 turvonnutta niveltä. Tutkimuksessa SC-II satunnaistettiin 656 potilasta suhteessa 2:1 saamaan joko tosilitsumabia ihon alle annoksella 162 mg kerran kahdessa viikossa tai lumevalmistetta yhdistelmänä yhden tai useamman ei-biologisen DMARD-lääkkeen kanssa.
Tutkimuksessa SC-II niveltuhon estymistä arvioitiin radiologisesti ja se ilmaistiin mTSS-pisteiden (van der Heijde modified mean total Sharp score) muutoksena lähtötilanteesta. Viikolla 24 tosilitsumabia ihon alle saaneilla potilailla osoitettiin merkitsevästi vähemmän radiologista etenemistä verrattuna lumevalmistetta saaneisiin potilaisiin (keskimääräiset mTSS-pisteet 0,62 vs. 1,23, p = 0,0149 (van Elteren). Nämä tulokset ovat yhdenmukaiset tosilitsumabia laskimoon saaneilla potilailla todettujen tulosten kanssa.
Tutkimuksessa SC-II tosilitsumabia ihon alle kerran kahdessa viikossa saaneiden potilaiden ACR-vasteet viikolla 24 olivat ACR 20 60,9 %, ACR 50 39,8 % ja ACR 70 19,7 %. Lumevalmistetta saaneiden ryhmässä vastaavat luvut olivat ACR 20 31,5 %, ACR 50 12,3 % ja ACR 70 5,0 %. Potilaiden DAS28-indeksin keskiarvo tutkimuksen alkaessa oli 6,7 tosilitsumabia ihon alle saaneiden ryhmässä ja 6,6 lumevalmistetta saaneiden ryhmässä. Viikolla 24 havaittiin tosilitsumabia ihon alle saaneiden hoitohaarassa DAS28-indeksin merkitsevä 3,1 yksikön lasku lähtötilanteesta, kun lumevalmistetta saaneiden haarassa lasku oli 1,7 yksikköä. Tosilitsumabia ihon alle saaneiden haarassa 32,0 % potilaista saavutti DAS28 < 2,6 ja lumevalmistetta saaneiden haarassa osuus oli 4,0 %.
Terveydentilaa ja elämänlaatua kuvaavat tulokset
Viikkoon 24 mennessä HAQ-DI laski tutkimuksessa SC-I keskimäärin 0,6 yksikköä lähtötilanteesta tosilitsumabia ihon alle ja laskimoon saaneiden ryhmissä. Myös niiden potilaiden osuus, joiden HAQ-DI:n tulos osoitti kliinisesti merkitsevää parannusta viikolla 24 (muutos lähtötilanteesta ≥ 0,3 yksikköä) oli yhtäläinen valmistetta ihon alle (65,2 %) ja laskimoon (67,4 %) saaneiden ryhmissä. Osuuksien painotettu ero oli ‑2,3 % (95 %:n CI ‑8,1, 3,4). SF-36-kyselyssä keskimuutos lähtötilanteesta viikolla 24 psyykkisen osion pisteissä oli 6,22 pistettä valmistetta ihon alle saaneiden ryhmässä ja 6,54 pistettä valmistetta laskimoon saaneiden ryhmässä. Myös fyysisen osion pisteet olivat samankaltaiset, 9,49 pistettä valmistetta ihon alle saaneiden ryhmässä ja 9,65 pistettä valmistetta laskimoon saaneiden ryhmässä.
Viikkoon 24 mennessä HAQ-DI laski tutkimuksessa SC-II huomattavasti enemmän potilailla, joita hoidettiin ihonalaisella tosilitsumabilla kerran kahdessa viikossa (0,4) verrattuna lumevalmisteella hoidettuihin potilaisiin (0,3). Niiden potilaiden osuus, joiden HAQ-DI:n tulos osoitti kliinisesti merkitsevää parannusta viikolla 24 (muutos lähtötilanteesta ≥ 0,3 yksikköä) oli suurempi ryhmässä, joka sai ihonalaista hoitoa kerran kahdessa viikossa (58 %) verrattuna lumevalmistetta saaneiden ryhmään (46,8 %). SF-36 elämänlaatumittarilla (keskimääräinen muutos psyykkisissä ja fyysisissä elämänlaadun osa-alueissa) saatu tulos oli huomattavasti parempi ryhmässä, joka sai ihonalaista tosilitsumabia (6,5 ja 5,3) kuin lumevalmistetta saaneiden ryhmässä (3,8 ja 2,9).
Ihonalainen annostelu
Yleisoireista lastenreumaa sairastavat potilaat
Kliininen teho
52 viikkoa kestäneessä avoimessa farmakokinetiikkaa, farmakodynamiikkaa ja turvallisuutta koskeneessa monikeskustutkimuksessa (WA28118) oli mukana iältään 1–17‑vuotiaita yleisoireista lastenreumaa sairastavia lapsipotilaita. Tutkimuksessa määritettiin sopiva ihon alle annettava tosilitsumabiannos, jonka farmakokinetiikka ja farmakodynamiikka sekä turvallisuusprofiili ovat verrannolliset valmisteen laskimoon antoon nähden.
Tutkimukseen mukaan soveltuneet potilaat saivat painonmukaisia tosilitsumabiannoksia: ≥ 30 kg:n painoiset potilaat (n = 26) saivat 162 mg tosilitsumabia joka viikko ja alle 30 kg:n painoiset potilaat (n = 25) saivat 162 mg tosilitsumabia 10 päivän välein (n = 8) tai joka toinen viikko (n = 17) 52 viikon ajan. Näistä 51 potilaasta 26 (51 %) potilasta ei ollut saanut tosilitsumabia aiemmin ja 25 (49 %) potilasta oli saanut tosilitsumabia laskimoon ja siirtyi lähtötilanteessa ihon alle annettavaan tosilitsumabihoitoon.
Eksploratiiviset tehon tulokset osoittivat, että ihon alle annettu tosilitsumabi paransi kaikkia eksploratiivisia tehoa koskevia parametreja, mukaan lukien lastenreuman aktiivisuutta osoittavia pisteitä (Juvenile Arthritis Disease Activity Score [JADAS]-71) potilailla, jotka eivät olleet saaneet tosilitsumabia aikaisemmin, ja piti kaikki eksploratiiviset tehoa koskevat parametrit ennallaan koko hoitojakson ajan kummassakin painoryhmässä (alle 30 kg ja ≥ 30 kg) niillä potilailla, jotka siirtyivät laskimoon annetusta hoidosta ihon alle annettavaan hoitoon.
Ihonalainen annostelu
Lasten polyartriittia sairastavat potilaat
Kliininen teho
52 viikkoa kestäneessä avoimessa farmakokinetiikkaa, farmakodynamiikkaa ja turvallisuutta koskeneessa monikeskustutkimuksessa oli mukana iältään 1–17‑vuotiaita lasten polyartriittia sairastavia potilaita. Tutkimuksessa määritettiin sopiva ihon alle annettava tosilitsumabiannos, jonka farmakokinetiikka ja farmakodynamiikka sekä turvallisuusprofiili ovat verrannolliset valmisteen laskimoon antoon nähden.
Tutkimukseen mukaan soveltuneet potilaat saivat painonmukaisia tosilitsumabiannoksia: ≥ 30 kg:n painoiset potilaat (n = 25) saivat 162 mg tosilitsumabia joka toinen viikko ja alle 30 kg:n painoiset potilaat (n = 27) saivat 162 mg tosilitsumabia joka kolmas viikko 52 viikon ajan. Näistä 52 potilaasta 37 (71 %) potilasta ei ollut saanut hoitoa aiemmin ja 15 (29 %) potilasta oli saanut laskimoon annettavaa hoitoa ja siirtyi lähtötilanteessa ihon alle annettavaan hoitoon.
Kun ihon alle annettavaa tosilitsumabia annetaan alle 30 kg:n painoisille potilaille 162 mg joka kolmas viikko ja ≥ 30 kg:n painoisille potilaille 162 mg joka toinen viikko, tehoa ja turvallisuutta koskevat hoitotulokset ovat farmakokineettisen altistuksen ja farmakodynaamisten vasteiden perusteella samankaltaiset kuin lasten polyartriitin hoitoon hyväksytyllä laskimoon annettavalla tosilitsumabilla.
Eksploratiiviset tehon tulokset osoittivat, että ihon alle annettu tosilitsumabi paransi lastenreuman aktiivisuutta osoittavien pisteiden (Juvenile Arthritis Disease Activity Score [JADAS]-71) mediaania potilailla, jotka eivät olleet saaneet hoitoa aikaisemmin, ja piti JADAS-71-pisteiden mediaanin ennallaan koko hoitojakson ajan kummassakin painoryhmässä (alle 30 kg ja ≥ 30 kg) niillä potilailla, jotka siirtyivät laskimoon annetusta hoidosta ihon alle annettavaan hoitoon.
Ihonalainen annostelu
Jättisoluarteriittia sairastavat potilaat
Kliininen teho
Tosilitsumabin tehoa ja turvallisuutta jättisoluarteriitin hoidossa on tutkittu vaiheen III satunnaistetussa kaksoissokkoutetussa lumekontrolloidussa monikeskustutkimuksessa (WA28119). Tutkimuksessa selvitettiin myös hoidon paremmuutta.
Tutkimukseen osallistui yhteensä 251 vastadiagnosoitua tai relapsoitunutta potilasta. Potilaat satunnaistettiin neljään tutkimushaaraan. Tutkimus käsitti 52 viikon pituisen sokkoutetun jakson (osa 1), jota seurasi 104 viikon pituinen avoin jatkotutkimus (osa 2). Osassa 2 määritettiin hoidon pitkäaikaisturvallisuutta ja tehon säilymistä 52 viikon tosilitsumabihoidon jälkeen sekä relapsien osuutta, mahdollista pidemmän hoidon tarvetta ja hoidon mahdollisia vaikutuksia steroidien pitkäaikaisen käytön vähentämiseen.
Tosilitsumabin kahta ihon alle annettavaa annostusta (162 mg joka viikko ja 162 mg joka toinen viikko) verrattiin kahteen erilliseen lumekontrolloituun ryhmään. Potilaat oli satunnaistettu eri ryhmiin suhteessa 2:1:1:1.
Kaikki potilaat saivat perushoitona glukokortikoideja (prednisonia). Kummassakin tosilitsumabiryhmässä ja toisessa lumeryhmässä prednisonihoito lopetettiin asteittain ennakolta määritellysti 26 viikon aikana. Toisessa lumeryhmässä prednisonihoito lopetettiin asteittain ennakolta määritellysti 52 viikon aikana.
Glukokortikoidihoidon kesto oli seulontavaiheessa ja ennen tosilitsumabihoidon (tai lumehoidon) aloittamista samankaltainen kaikissa neljässä hoitoryhmissä (ks. taulukko 3).
Taulukko 3. Kortikosteroidihoidon kesto tutkimuksen WA28119 seulonnan aikana
Lume + 26 viikon aikana asteittain lopetettava prednisonihoito n = 50 | Lume + 52 viikon aikana asteittain lopetettava prednisonihoito n = 51 | Tosilitsumabi 162 mg s.c. kerran viikossa + 26 viikon aikana asteittain lopetettava prednisonihoito n = 100 | Tosilitsumabi 162 mg s.c. joka toinen viikko + 26 viikon aikana asteittain lopetettava prednisonihoito n = 49 | |
| Kesto (vrk) | ||||
| Keskiarvo (keskihajonta) | 35,7 (11,5) | 36,3 (12,5) | 35,6 (13,2) | 37,4 (14,4) |
| Mediaani | 42,0 | 41,0 | 41,0 | 42,0 |
| Min - Max | 6–63 | 12–82 | 1–87 | 9–87 |
s.c. = ihon alle
Tehon ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat pitkäkestoisen remission ilman steroidihoitoa viikolla 52, kun tosilitsumabihoidon ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää verrattiin lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään. Tutkimuksen ensisijainen päätetapahtuma saavutettiin (taulukko 4).
Tehon toissijaisena päätetapahtumana oli niiden potilaiden osuus, jotka saavuttivat pitkäkestoisen remission viikolla 52, kun tosilitsumabi-hoidon ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää verrattiin lumevalmisteen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään. Myös tutkimuksen toissijainen päätetapahtuma saavutettiin (taulukko 4).
Hoidon tehoa tarkasteltaessa havaittiin, että tosilitsumabi oli tilastollisesti merkitsevästi lumevalmistetta parempi remission saavuttamisessa ilman steroidihoitoa viikolla 52, kun tosilitsumabin ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää verrattiin lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään sekä lumelääkkeen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään.
Pitkäkestoisen remission viikolla 52 saavuttaneiden potilaiden prosenttiosuudet esitetään taulukossa 4.
Toissijaiset päätetapahtumat
Arvioitaessa aikaa jättisoluarteriitin ensimmäiseen pahenemisvaiheeseen havaittiin, että pahenemisvaiheen riski oli merkittävästi pienempi tosilitsumabia ihon alle viikoittain saaneessa ryhmässä verrattuna lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään sekä lumevalmisteen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään. Riski oli samoin pienempi tosilitsumabia ihon alle joka toinen viikko saaneessa ryhmässä verrattuna lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneeseen ryhmään (merkitsevyystasolla 0,01 verrattuna). Tosilitsumabiannos ihon alle viikoittain pienensi pahenemisvaiheen riskiä kliinisesti merkittävästi verrattuna lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmään myös potilailla, joilla oli tutkimukseen tullessaan relapsoitunut jättisoluarteriitti, samoin kuin potilailla, joilla sairaus oli äskettäin diagnosoitu (taulukko 4).
Kumulatiivinen glukokortikoidiannos
Kumulatiivinen prednisoniannos oli viikolla 52 huomattavasti pienempi kahdessa tosilitsumabia saaneessa ryhmässä verrattuna kahteen lumeryhmään (taulukko 3). Erillisessä analyysissä potilaista, jotka saivat jättisoluarteriitin äkillisen pahenemisvaiheen hoitoon prednisonia ensimmäisten 52 viikon aikana, kumulatiivinen prednisoniannos vaihteli huomattavasti. Pahenemisvaiheeseen hoitoa saaneiden potilaiden annosten mediaani oli tosilitsumabia viikoittain saaneilla potilailla 3 129,75 mg ja tosilitsumabia joka toinen viikko saaneilla potilailla 3 847 mg. Kumpikin on huomattavasti pienempi kuin lumevalmistetta ja 26 viikon aikana asteittain lopetettavaa prednisonihoitoa saaneessa ryhmässä (4 023,5 mg) tai lumevalmistetta ja 52 viikon aikana asteittain lopetettavaa prednisonihoitoa saaneessa ryhmässä (5 389,5 mg).
Taulukko 4. Tutkimuksen WA28119 tehoa koskevat tulokset
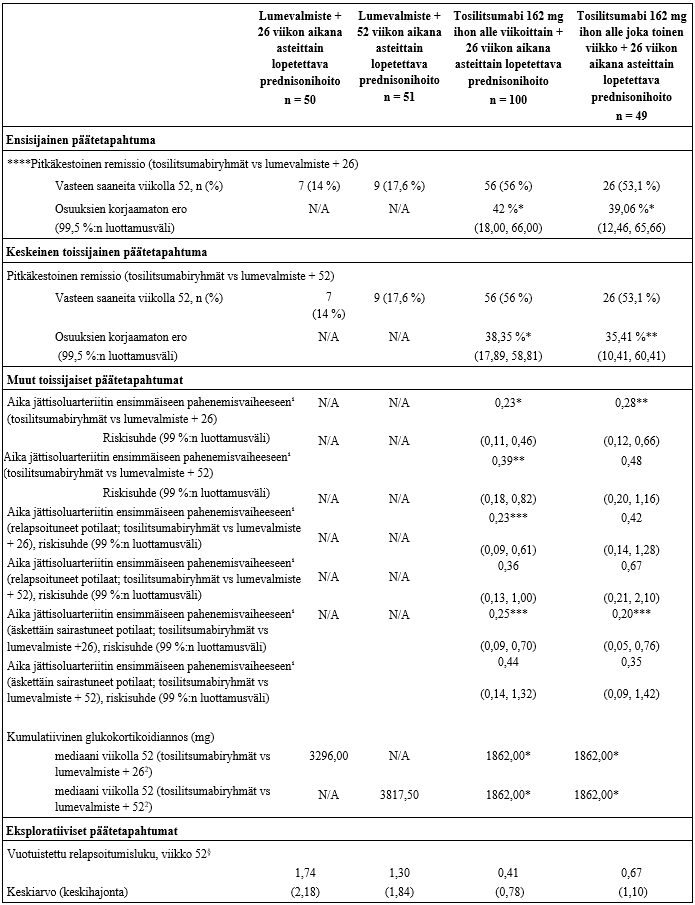
* p < 0,0001
** p < 0,005 (paremmuuden osoittavien ensisijaisten ja keskeisten toissijaisten testien merkitsevyyden raja-arvo)
*** deskriptiivinen p-arvo < 0,005
****Sairauden pahenemisvaihe: jättisoluarteriitin toistuvia oireita ja löydöksiä ja/tai lasko ≥ 30 mm/h, prednisoniannosta tarpeen suurentaa
Remissio: ei sairauden pahenemisvaihetta ja CRP normalisoitunut
Pitkäkestoinen remissio: remissio viikosta 12 viikkoon 52, potilaiden on noudatettava tutkimussuunnitelmassa määriteltyä prednisonihoidon asteittaista lopettamista
¹ analyysi kliinisen remission ja sairauden ensimmäisen pahenemisvaiheen välisestä ajasta (päivää)
2 parametrittömien tietojen p-arvot määriteltiin Van Elterenin analyysillä
§ tilastollisia analyysejä ei ole tehty
N/A = ei oleellinen (Not applicable)
Elämänlaatua koskevat päätetapahtumat
Tutkimuksen (WA28119) SF-36-tuloksissa eriteltiin fyysisen (PCS) ja mentaalisen (MCS) osa-alueen yhdistelmäpisteet. Fyysisen osa-alueen pisteiden keskimääräinen muutos lähtötilanteesta viikkoon 52 oli suurempi (osoittaa suurempaa paranemista) tosilitsumabia viikoittain ja joka toinen viikko saaneissa ryhmissä (viikoittain: 4,10; joka toinen viikko: 2,76) kuin kahdessa lumeryhmässä (lumevalmiste ja 26 viikon aikana asteittain lopetettu hoito: ‑0,28; lumevalmiste ja 52 viikon aikana asteittain lopetettu hoito: ‑1,49), mutta vain vertailussa tosilitsumabin viikoittain ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää ja lumevalmisteen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneiden ryhmän (5,59, 99 %:n luottamusväli: 8,6, 10,32) välillä todettiin tilastollisesti merkitsevä ero (p = 0,0024). Mentaalisen osa-alueen pisteiden keskimääräinen muutos lähtötilanteesta viikkoon 52 sekä tosilitsumabia viikoittain että joka toinen viikko saaneissa ryhmissä (viikoittain: 7,28; joka toinen viikko: 6,12) oli suurempi kuin lumevalmisteen ja 52 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneessa ryhmässä (2,84) (mutta ero ei ollut tilastollisesti merkitsevä [viikoittain: p = 0,0252; joka toinen viikko: p = 0,1468]) ja samankaltainen kuin lumevalmisteen ja 26 viikon aikana asteittain lopetettavan prednisonihoidon yhdistelmää saaneessa ryhmässä (6,67).
Sairauden aktiivisuuden kokonaisarvio (Patient’s Global Assessment) tehtiin 0–100 mm:n VAS-asteikolla (Visual Analogue Scale). VAS-asteikkoon perustuvan potilaan kokonaisarvion keskimääräinen muutos lähtötilanteesta viikkoon 52 oli pienempi (osoittaa suurempaa paranemista) tosilitsumabia viikoittain (‑19,0) ja joka toinen viikko (‑25,3) saaneissa ryhmissä kuin kummassakaan lumevalmistetta (lumevalmiste ja 26 viikon aikana asteittain lopetettava hoito: ‑3,4, lumevalmiste ja 52 viikon aikana asteittain lopetettava hoito: -7,2) saaneessa ryhmässä, mutta vain tosilitsumabia joka toinen viikko yhdistelmänä 26 viikon aikana asteittain lopetettavan prednisonihoidon kanssa saaneilla osoitettiin tilastollisesti merkitsevä ero lumehoitoon verrattuna [lumevalmisteen ja 26 viikon aikana asteittain lopetettavan hoidon yhdistelmän p = 0,0059, ja lumevalmisteen ja 52 viikon aikana asteittain lopetettavan hoidon yhdistelmän p = 0,0081].
FACIT-F-mittarin (FACIT-fatigue) muutosta lähtötilanteesta viikkoon 52 osoittavat indeksiluvut laskettiin kaikista ryhmistä. Indeksilukujen muutoksen keskiarvot (keskihajonta) olivat seuraavat: tosilitsumabi viikoittain ja 26 viikon aikana asteittain lopetettava hoito: 5,61 (10,115); tosilitsumabi joka toinen viikko ja 26 viikon aikana asteittain lopetettava hoito: 1,81 (8,836); lumevalmiste ja 26 viikon aikana asteittain lopetettava hoito: 0,26 (10,702); ja lumevalmiste ja 52 viikon aikana asteittain lopetettava hoito: ‑1,63 (6,753).
EQ-5D-mittarin indeksiluvun muutos lähtötilanteesta viikkoon 52 oli tosilitsumabi viikoittain yhdistelmänä 26 viikon aikana asteittain lopetettavan hoidon kanssa saaneilla 0,10 (0,198), tosilitsumabia joka toinen viikko yhdistelmänä 26 viikon aikana asteittain lopetettavan hoidon kanssa saaneilla 0,05 (0,215), lumevalmistetta yhdistelmänä 26 viikon aikana asteittain lopetettavan hoidon kanssa saaneilla 0,07 (0,293) sekä lumevalmistetta yhdistelmänä 52 viikon aikana asteittain lopetettavan hoidon kanssa saaneilla -0,02 [0,159].
Sekä FACIT-F- että EQ-5D-mittarin suurempi indeksiluku osoittaa tilan paranemista.
Laskimonsisäinen annostelu
Nivelreumaa sairastavat potilaat
Kliininen teho
Tosilitsumabin tehoa arvioitiin viidessä satunnaistetussa kaksoissokkoutetussa monikeskustutkimuksessa nivelreuman oireiden lievittymisen perusteella. Tutkimuksiin I–V otettiin ≥ 18-vuotiaita potilaita, joilla oli ACR-kriteerien (American College of Rheumatology) mukaan diagnosoitu aktiivinen nivelreuma ja lähtötilanteessa vähintään kahdeksan aristavaa ja kuusi turvonnutta niveltä.
Tutkimuksessa I tosilitsumabi annettiin laskimoon neljän viikon välein monoterapiana. Tutkimuksissa II, III ja V tosilitsumabi annettiin laskimoon neljän viikon välein yhdessä MTX:n kanssa. Vertailuna käytettiin lumevalmisteen ja MTX:n yhdistelmää. Tutkimuksessa IV tosilitsumabi annettiin laskimoon neljän viikon välein yhdessä muiden DMARDien kanssa. Vertailuna käytettiin lumevalmisteen ja muiden DMARDien yhdistelmää. Kaikissa viidessä tutkimuksessa ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat ACR 20‑vasteen viikkoon 24 mennessä.
Tutkimuksessa I arvioitiin 673 potilasta, jotka eivät olleet saaneet MTX:a satunnaistamista edeltäneiden kuuden kuukauden aikana ja joiden aikaisempaa MTX-hoitoa ei oltu keskeytetty kliinisesti merkittävien toksisten vaikutusten tai hoidon tehottomuuden vuoksi. Valtaosa (67 %) potilaista ei ollut aikaisemmin saanut MTX:a. Tosilitsumabia annettiin 8 mg/kg neljän viikon välein monoterapiana. Vertailuryhmä sai MTX:a viikon välein (viikkoannos titrattiin 7,5 mg:sta enintään 20 mg:aan kahdeksan viikon jakson aikana).
Tutkimus II kesti kaksi vuotta, ja siihen kuului suunniteltu analyysi viikkojen 24, 52 ja 104 kohdalla. Tutkimuksessa arvioitiin 1 196 potilasta, joille MTX ei ollut tuottanut riittävää hoitovastetta. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin sokkoutetusti neljän viikon välein 52 viikon ajan yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa). Avoimessa vaiheessa, viikon 52 jälkeen, kaikilla potilailla oli mahdollisuus saada tosilitsumabihoitoa 8 mg/kg. Niistä potilaista, jotka olivat mukana tutkimuksen loppuun asti ja jotka oli satunnaistettu saamaan lumevalmistetta ja MTX:a, 86 % sai avoimessa vaiheessa tosilitsumabia 8 mg/kg tutkimuksen toisena vuonna. Ensisijainen päätetapahtuma 24 viikon kohdalla tehdyssä analyysissä oli ACR 20‑vasteen kriteerit täyttävien potilaiden suhteellinen osuus. Viikoilla 52 ja 104 muut ensisijaiset päätetapahtumat olivat nivelvaurioiden estyminen ja fyysisen toimintakyvyn paraneminen.
Tutkimuksessa III arvioitiin 623 potilasta, joille MTX ei ollut tuonut riittävää hoitovastetta. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa).
Tutkimuksessa IV arvioitiin 1 220 potilasta, jotka eivät olleet saavuttaneet riittävää hoitovastetta nykyisellä reumalääkityksellään, johon kuului vähintään yksi DMARD. Tosilitsumabiannos 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä DMARDien vakaaseen annokseen.
Tutkimuksessa V arvioitiin 499 potilasta, joille yksi tai useampi TNF-estäjä ei ollut tuonut riittävää hoitovastetta tai jotka eivät sietäneet näitä lääkkeitä. Hoito TNF-estäjillä lopetettiin ennen satunnaistamista. Tosilitsumabiannos 4 mg/kg tai 8 mg/kg tai lumevalmiste annettiin neljän viikon välein yhdistettynä vakaaseen MTX-annokseen (10–25 mg viikossa).
Kliininen vaste
Kaikissa tutkimuksissa kuuden kuukauden ACR 20-, ACR 50- ja ACR 70-vasteet olivat tilastollisesti merkitsevästi suuremmat tosilitsumabia 8 mg/kg saaneiden potilaiden ryhmissä kuin vertailuryhmissä (taulukko 5). Tutkimuksessa I tosilitsumabi 8 mg/kg todettiin tehokkaammaksi kuin vaikuttava vertailuaine MTX.
Hoitoteho oli sama riippumatta potilaiden reumatekijästatuksesta, iästä, sukupuolesta, etnisestä taustasta, aikaisempien hoitojen lukumäärästä tai taudin tilasta. Vaikutus alkoi nopeasti (jo toisella hoitoviikolla), ja vaste suureni jatkuvasti hoidon keston myötä. Avoimissa jatkotutkimuksissa I–V vasteen on todettu kestävän yli 3 vuotta.
Kaikissa tutkimuksissa tosilitsumabiannosta 8 mg/kg saaneilla potilailla todettiin merkitsevää paranemista ACR-vasteen kaikissa yksittäisissä osatekijöissä (aristavien ja turvonneiden nivelten lukumäärä, potilaan ja lääkärin yleisarvio, toimintakykyä mittaava pistearvo, kivun arviointi ja CRP-arvo) verrattuna potilaisiin, jotka saivat lumevalmistetta yhdessä MTX:in tai muiden DMARDien kanssa.
Tutkimuksissa I–V potilaiden keskimääräinen DAS28 (Disease Activity Score) -indeksi hoidon alussa oli 6,5–6,8. DAS28-indeksin merkitsevää laskua (keskimääräinen parannus) havaittiin tosilitsumabilla hoidetuilla potilailla verrattuna kontrolliryhmän potilaisiin. Tosilitsumabilla hoidetuilla potilailla DAS28 laski 3,1–3,4 yksikköä ja kontrolliryhmässä 1,3–2,1 yksikköä. Tosilitsumabihoitoa saaneista potilaista 28–34 % saavutti DAS28-remission viikolla 24 (DAS28 < 2,6). Osuus oli huomattavasti suurempi kuin kontrolliryhmässä, jossa vastaava luku oli 1–12 %. Tutkimuksessa II, 65 % potilaista saavutti DAS28 < 2,6 viikolla 104. Vastaava luku viikolla 52 oli 48 % ja viikolla 24 33 %.
Tutkimusten II, III ja IV yhdistetyssä analyysissä ACR 20-, ACR 50- ja ACR 70-vasteiden saavuttaneiden potilaiden osuus oli merkitsevästi suurempi ryhmässä, joka sai tosilitsumabia 8 mg/kg yhdessä DMARDien kanssa (59 %, 37 % ja 18 %), kuin ryhmässä, joka sai tosilitsumabia 4 mg/kg yhdessä DMARDien kanssa (50 %, 27 % ja 11 %; p < 0,03). Vastaavasti DAS28-remission (DAS28 < 2,6) saavuttaneiden potilaiden osuus oli merkitsevästi suurempi tosilitsumabia 8 mg/kg ja DMARDeja saaneessa ryhmässä (31 %) kuin tosilitsumabia 4 mg/kg ja DMARDeja saaneessa ryhmässä (16 %, p < 0.0001).
Taulukko 5. ACR-vasteet kontrolloiduissa tutkimuksissa, joissa vertailuaineina olivat lumevalmiste / MTX / DMARDit, (% potilaista)
Tutkimus I AMBITION | Tutkimus II LITHE | Tutkimus III OPTION | Tutkimus IV TOWARD | Tutkimus V RADIATE | ||||||
| Vko | TCZ 8 mg/kg | MTX | TCZ 8 mg/kg + MTX | Lume + MTX | TCZ 8 mg/kg + MTX | Lume + MTX | TCZ 8 mg/kg + DMARD | Lume+ DMARD | TCZ 8 mg/kg + MTX | Lume + MTX |
n = 286 | n = 284 | n = 398 | n = 393 | n = 205 | n = 204 | n = 803 | n = 413 | n = 170 | n = 158 | |
| ACR20 | ||||||||||
| 24 | 70 %*** | 52 % | 56 %*** | 27 % | 59 %*** | 26 % | 61 %*** | 24 % | 50 %*** | 10 % |
| 52 | 56 %*** | 25 % | ||||||||
| ACR50 | ||||||||||
| 24 | 44 %** | 33 % | 32 %*** | 10 % | 44 %*** | 11 % | 38 %*** | 9 % | 29 %*** | 4 % |
| 52 | 36 %*** | 10 % | ||||||||
| ACR70 | ||||||||||
| 24 | 28 %** | 15 % | 13 %*** | 2 % | 22 %*** | 2 % | 21 %*** | 3 % | 12 %** | 1 % |
| 52 | 20 %*** | 4 % | ||||||||
TCZ - tosilitsumabi
MTX - metotreksaatti
DMARD - tautiprosessia hidastava reumalääke (disease modifying anti-rheumatic drug)
** - p < 0.01, tosilitsumabi vs. lumevalmiste + MTX / DMARD
*** - p < 0.0001, tosilitsumabi vs. lumevalmiste + MTX / DMARD
Merkittävä kliininen vaste
Tosilitsumabia yhdessä MTX:n kanssa saaneista potilaista 14 % saavutti merkittävän kliinisen vasteen, kun hoitoa oli annettu kaksi vuotta (ACR 70-vaste säilyi 24 viikkoa tai pitempään).
Radiologinen vaste
Tutkimukseen II osallistui potilaita, joiden aikaisempi hoito MTX:lla ei tuonut riittävää hoitovastetta. Rakenteellisten nivelvaurioiden estymistä arvioitiin radiologisesti ja tulos ilmaistiin modifioidun Sharp-indeksin ja sen osatekijöiden, eroosioasteen ja nivelraon kaventumisen muutoksena. Rakenteellisen nivelvaurion estyminen näkyi merkitsevästi hitaampana radiologisena etenemisenä tosilitsumabihoitoa saaneilla potilailla kuin vertailuryhmän potilailla (taulukko 6).
Avoimessa jatkotutkimuksessa (tutkimus II) niveltuhon etenemistä estävä vaikutus säilyi hoidon toisena vuonna tosilitsumabia yhdessä MTX:n kanssa saaneilla potilailla. Sharp-Genant-kokonaispistearvon keskimääräinen muutos lähtötilanteesta viikolla 104 oli huomattavasti pienempi potilailla, jotka oli satunnaistettu saamaan tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa (p < 0,0001) kuin niillä potilailla, jotka oli satunnaistettu saamaan lumevalmistetta yhdessä MTX:n kanssa.
Taulukko 6. Radiologiset muutokset (keskiarvo) 52 viikon aikana tutkimuksessa II
Lume + MTX (+TCZ viikosta 24 alkaen) n = 393 | TCZ 8 mg/kg + MTX n = 398 | |
| Sharp-Genant-kokonaispistearvo | 1,13 | 0,29* |
| Eroosioindeksi | 0,71 | 0,17* |
| Nivelraon kaventuminen | 0,42 | 0,12** |
MTX - metotreksaatti
TCZ - tosilitsumabi
* - p ≤ 0,0001, kun tosilitsumabia verrattiin lumevalmisteen + MTX:n yhdistelmään.
** - p ≤ 0,005, kun tosilitsumabia verrattiin lumevalmisteen + MTX:n yhdistelmään.
Vuoden kestäneen hoidon jälkeen tosilitsumabia yhdessä MTX:n kanssa saaneista potilaista 85 %:lla (n = 348) ei havaittu niveltuhon etenemistä Sharp-Genant-kokonaispistearvon muutoksena mitattuna (muutos 0 tai vähemmän). Vastaava tulos lumevalmistetta yhdessä MTX:n kanssa saaneilla potilailla oli 67 % (n = 290) (p ≤ 0,001). Tulos säilyi samanlaisena kaksi vuotta kestäneen hoidon jälkeen (83 %; n = 353). Viikkojen 52 ja 104 välillä 93 %:lla potilaista (n = 271) ei havaittu niveltuhon etenemistä.
Terveydentilaa ja elämänlaatua kuvaavat tulokset
Raportit tosilitsumabihoitoa saaneilta potilailta osoittivat paranemista kaikissa potilaan arviointiin perustuvissa mittareissa (HAQ-DI = Health Assessment Questionnaire Disability Index, Short Form ‑36- ja FACIT = Functional Assessment of Chronic Illness Therapy -kyselylomakkeet). Fyysistä toimintakykyä mittaava HAQ-DI-tulos parani tilastollisesti merkitsevästi tosilitsumabihoitoa saaneiden potilaiden ryhmässä DMARDeja saaneisiin potilaisiin verrattuna. Tutkimuksen II avoimessa vaiheessa havaittu parannus fyysisessä toimintakyvyssä säilyi jopa kahden vuoden ajan. Viikolla 52 keskimääräinen muutos HAQ-DI:ssä oli -0,58 niillä potilailla, jotka saivat tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa. Lumevalmistetta yhdessä MTX:n kanssa saaneilla potilailla vastaava tulos oli 0,39. Keskimääräinen muutos HAQ-DI:ssä säilyi viikon 104 loppuun asti tosilitsumabia 8 mg/kg yhdessä MTX:n kanssa saaneilla potilailla (-0.61).
Hemoglobiiniarvot
Tosilitsumabihoitoa saaneiden potilaiden hemoglobiiniarvot olivat parantuneet tilastollisesti merkitsevästi viikolla 24 verrattuna DMARDeja saaneiden potilaiden arvoihin (p < 0.0001). Hemoglobiiniarvojen keskiarvot nousivat viikkoon kaksi mennessä ja pysyivät normaalialueella viikkoon 24 asti.
Tosilitsumabin vertailu adalimumabiin monoterapiassa
Tutkimuksessa VI (WA19924), joka oli 24 viikon pituinen kaksoissokkoutettu tosilitsumabimonoterapiaa ja adalimumabimonoterapiaa vertaileva tutkimus, oli mukana 326 nivelreumapotilasta, jotka eivät sietäneet MTX-hoitoa tai joille MTX-hoidon jatkamista ei katsottu tarkoituksenmukaiseksi (mukaan lukien potilaat, jotka eivät saaneet riittävää vastetta MTX-hoitoon). Tosilitsumabiryhmän potilaat saivat tosilitsumabia (8 mg/kg) infuusiona laskimoon neljän viikon välein ja lumevalmistetta ihon alle kahden viikon välein. Adalimumabiryhmän potilaat saivat adalimumabia (40 mg) injektiona ihon alle kahden viikon välein sekä lumevalmistetta infuusiona laskimoon neljän viikon välein.
Tosilitsumabihoidon osoitettiin olevan adalimumabia tilastollisesti merkitsevästi tehokkaampi tautiaktiivisuuden hallinnassa sekä ensisijaisen päätetapahtuman osalta (DAS28-indeksin muutos lähtötilanteesta viikolle 24) että kaikkien toissijaisten päätetapahtumien osalta (taulukko 7).
Taulukko 7. Tutkimuksen VI (WA19924) tehon tulokset
ADA + lume (i.v.) n = 162 | TCZ + lume (s.c.) n = 163 | p-arvo(a) | |
| Ensisijainen päätetapahtuma: keskimuutos lähtötilanteesta viikkoon 24 | |||
| DAS28 (korjattu keskiarvo) | -1,8 | -3,3 | |
| Korjatun keskiarvon ero (95 %:n luottamusväli) | -1,5 (-1,8, -1,1) | < 0,0001 | |
| Toissijaiset päätetapahtumat: vasteen saaneiden prosenttiosuus viikolla 24 (b) | |||
| DAS28 < 2,6, n (%) | 17 (10,5) | 65 (39,9) | < 0,0001 |
| DAS28 ≤ 3,2, n (%) | 32 (19,8) | 84 (51,5) | < 0,0001 |
| ACR 20-vaste, n (%) | 80 (49,4) | 106 (65,0) | 0,0038 |
| ACR 50-vaste, n (%) | 45 (27,8) | 77 (47,2) | 0,0002 |
| ACR 70-vaste, n (%) | 29 (17,9) | 53 (32,5) | 0,0023 |
a p-arvo on korjattu potilaiden maantieteellisen sijainnin ja nivelreuman kestoajan suhteen kaikkien päätetapahtumien osalta sekä lisäksi kaikkien jatkuvien päätetapahtumien lähtötilanteen arvon osalta.b Hoitoon vastaamattomien potilaiden puuttuvien tietojen paikkaus. Monivertailun hallintaan käytetty Bonferroni-Holmin menetelmää
i.v. = laskimoon
s.c. = ihon alle
ADA = adalimumabi
TCZ = tosilitsumabi
Tosilitsumabin ja adalimumabin kliiniset haittatapahtumaprofiilit olivat yleisesti ottaen samankaltaiset. Niiden potilaiden osuus, joilla esiintyi vakavia haittatapahtumia, oli samankaltainen kummassakin hoitoryhmässä (tosilitsumabi 11,7 % vs. adalimumabi 9,9 %). Tosilitsumabihaarassa havaitut haittavaikutukset olivat luonteeltaan yhdenmukaisia tosilitsumabin tunnetun turvallisuusprofiilin kanssa ja ne ilmenivät vastaavalla frekvenssillä kuin taulukossa 1 luetellut haittavaikutukset. Infektioita ja infestaatioita ilmeni enemmän tosilitsumabihaarassa (48 % vs. 42 %), vakavien infektioiden ilmaantuvuus oli samanlainen (3,1 %). Kumpikin tutkimushoito aiheutti samantyyppisiä turvallisuuteen liittyvien laboratorioarvojen muutoksia (neutrofiili- ja trombosyyttimäärän vähenemistä, ALAT- ja ASAT- sekä lipidiarvojen suurenemista), mutta muutosten suuruus ja huomattavien poikkeavuuksien esiintymistiheys oli tosilitsumabiryhmässä suurempi kuin adalimumabiryhmässä. Neljällä (2,5 %) potilaalla tosilitsumabiryhmässä ja kahdella (1,2 %) potilaalla adalimumabiryhmässä esiintyi CTC-luokituksen mukaista gradus 3 tai 4 neutrofiilimäärän vähenemistä. Yhdellätoista (6,8 %) potilaalla tosilitsumabiryhmässä ja viidellä (3,1 %) potilaalla adalimumabiryhmässä esiintyi CTC-luokituksen mukaista gradus 2 tai vaikeampiasteista ALAT-arvon suurenemista. LDL-arvo suureni lähtötilanteesta tosilitsumabiryhmässä keskimäärin 0,64 mmol/l (25 mg/dl) ja adalimumabiryhmässä 0,19 mmol/l (7 mg/dl). Turvallisuuden havaittiin olleen tosilitsumabiryhmässä yhdenmukainen tosilitsumabin tunnetun turvallisuusprofiilin kanssa eikä uusia tai odottamattomia haittavaikutuksia havaittu (ks. taulukko 1).
Farmakokinetiikka
Tosilitsumabin farmakokinetiikalle tyypillistä on epälineaarinen eliminaatio, joka on lineaarisen puhdistuman ja Michaelis-Mentenin eliminaation yhdistelmä. Eliminaation epälineaarinen osuus johtaa altistuksen lisääntymiseen enemmän kuin suhteessa annokseen. Tosilitsumabin farmakokineettiset parametrit eivät muutu ajan mittaan. Koska kokonaispuhdistuma on riippuvainen tosilitsumabin pitoisuudesta seerumissa, myös tosilitsumabin puoliintumisaika on pitoisuudesta riippuvainen ja vaihtelee seerumissa olevan pitoisuuden mukaan. Minkään tähän mennessä testatun potilasjoukon populaatiofarmakokineettiset analyysit eivät osoita, että näennäisen puhdistuman ja vasta-aineiden esiintymisen lääkkeelle välillä olisi suhde.
Laskimonsisäinen annostelu
Nivelreumaa sairastavat potilaat
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 3 552 nivelreumapotilaan tietokannasta. Potilaat saivat tosilitsumabia 4 mg/kg tai 8 mg/kg tunnin kestävänä infuusiona neljän viikon välein 24 viikon ajan, tai 162 mg ihon alle joko kerran viikossa tai kerran kahdessa viikossa 24 viikon ajan.
Seuraavat parametrit (odotettu keskiarvo ± SD) arvioitiin tosilitsumabiannokselle 8 mg/kg, joka annettiin neljän viikon välein: vakaan tilan AUC = 38 000 ± 13 000 h × µg/ml, minimipitoisuus (Cmin) = 15,9 ± 13,1 μg/ml ja maksimipitoisuus (Cmax) = 182 ± 50,4 µg/ml. AUC- ja Cmax-arvojen kumulaatiosuhteet olivat pienet: AUC 1,32 ja Cmax 1,09. Kumulaatiosuhde oli suurempi Cmin-arvojen osalta (2,49), mikä oli odotettua johtuen epälineaarisesta puhdistumasta alhaisilla pitoisuustasoilla. Cmax-arvon osalta vakaa tila saavutettiin ensimmäisen annoksen jälkeen, AUC:n osalta 8 viikon kuluttua ja Cmin-arvon osalta 20 viikon kuluttua. Tosilitsumabin AUC-, Cmin- ja Cmax-arvot nousivat potilaan painon mukaan. Jos potilas painoi 100 kg tai enemmän, tosilitsumabin vakaan tilan AUC-arvo (odotettu keskiarvo ± SD) oli 50 000 ± 16 800 μg × h/ml, Cmin-arvo 24,4 ± 17,5 μg/ml, ja Cmax-arvo 226 ± 50,3 μg/ml. Arvot ovat korkeammat kuin edellä analysoidun potilaspopulaation keskimääräiset arvot (ts. kaikki painoryhmät). Tosilitsumabin annosvastekäyrä tasaantuu suuremmilla altistuksilla siten, että lisäykset pitoisuudessa eivät samassa suhteessa anna lisähyötyä tehossa. Kliinisesti merkittävää parannusta tehossa ei havaittu potilailla, joita hoidettiin yli 800 mg:n tosilitsumabiannoksilla. Siksi ei suositella kerta-annoksia, jotka ylittävät 800 mg (ks. kohta Annostus ja antotapa).
Jakautuminen
Nivelreumapotilailla sentraalinen jakautumistilavuus oli 3,72 l ja perifeerinen jakautumistilavuus 3,35 l, joten jakautumistilavuus oli vakaan tilan aikana 7,07 l.
Eliminaatio
Laskimoon annettu tosilitsumabi poistuu verenkierrosta kaksivaiheisesti. Tosilitsumabin kokonaispuhdistuma oli pitoisuudesta riippuva, ja se on lineaarisen ja epälineaarisen puhdistuman summa. Lineaarinen puhdistuma arvioitiin populaatiofarmakokineettisen analyysin parametrina, ja se oli 9,5 ml/h. Pitoisuudesta riippuva epälineaarinen puhdistuma on ratkaisevassa asemassa, kun tosilitsumabipitoisuus on alhainen. Tosilitsumabipitoisuuden suurentuessa epälineaarinen puhdistumareitti kyllästyy, minkä jälkeen puhdistuma määräytyy pääasiassa lineaarisen puhdistuman perusteella.
Tosilitsumabin puoliintumisaika (t1/2) riippui pitoisuudesta. Kun vakaa tila oli saavutettu annoksen ollessa 8 mg/kg neljän viikon välein, efektiivinen t1/2 lyheni pitoisuuden pienentyessä annosvälin aikana 18 vuorokaudesta 6 vuorokauteen.
Lineaarisuus
Tosilitsumabin farmakokineettiset parametrit eivät muuttuneet ajan myötä. AUC- ja Cmin-arvojen havaittiin suurenevan annossuhdetta enemmän annoksen ollessa 4 mg/kg tai 8 mg/kg neljän viikon välein. Cmax nousi samassa suhteessa kuin annos. Annoksen ollessa 8 mg/kg arvioitu vakaan tilan AUC-arvo oli 3,2-kertainen verrattuna annokseen 4 mg/kg. Vastaava vakaan tilan Cmin-arvo oli 30-kertainen.
Ihonalainen annostelu
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 3 552 nivelreumapotilaan tietokannasta. Potilaat olivat saaneet tosilitsumabia 162 mg ihon alle viikoittain, 162 mg ihon alle kerran kahdessa viikossa tai 4 mg/kg tai 8 mg/kg laskimoon neljän viikon välein 24 viikon ajan.
Tosilitsumabin farmakokineettiset parametrit eivät muuttuneet ajan myötä. Viikoittain annettujen 162 mg:n annosten yhteydessä tosilitsumabin vakaan tilan AUC1 viikon -arvo (odotettu keskiarvo ± SD) oli 7 970 ± 3 432 μg × h/ml, Cmin-arvo oli 43,0 ± 19,8 μg/ml ja Cmax-arvo oli 49,8 ± 21,0 μg/ml. Kumulaatiosuhde oli AUC-arvon osalta 6,32, Cmin-arvon osalta 6,30 ja Cmax-arvon osalta 5,27. AUC-, Cmin- ja Cmax-arvojen vakaa tila saavutettiin 12 viikon jälkeen.
Kerran kahdessa viikossa annettujen 162 mg:n annosten yhteydessä tosilitsumabin vakaan tilan AUC2 viikon -arvo (odotettu keskiarvo ± SD) oli 3 430 ± 2 660 μg × h/ml, Cmin-arvo oli 5,7 ± 6,8 μg/ml ja Cmax-arvo oli 13,2 ± 8,8 μg/ml. Kumulaatiosuhde oli AUC-arvon osalta 2,67, Cmin-arvon osalta 6,02 ja Cmax-arvon osalta 2,12. AUC- ja Cmin-arvojen vakaa tila saavutettiin 12 viikon jälkeen ja Cmax-arvon vakaa tila 10 viikon jälkeen.
Imeytyminen
Ihon alle annostellun tosilitsumabin tmax-arvo (huippupitoisuuden ajankohta) nivelreumapotilailla oli noin 2,8 vuorokautta. Ihon alle annosteltavan lääkemuodon biologinen hyötyosuus oli 79 %.
Eliminaatio
Nivelreumapotilailla tosilitsumabin pitoisuusriippuvainen näennäinen puoliintumisaika (t1/2) oli jopa 13 vuorokautta vakaassa tilassa potilailla, jotka saivat tosilitsumabia 162 mg viikossa ihon alle ja enintään viisi vuorokautta potilailla, jotka saivat tosilitsumabia 162 mg kerran kahdessa viikossa.
Ihonalainen annostelu
Yleisoireista lastenreumaa sairastavat potilaat
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 140 yleisoireista lastenreumaa sairastavan potilaan potilasjoukosta. Potilaat olivat saaneet tosilitsumabia 8 mg/kg laskimoon joka toinen viikko (≥ 30 kg painavat potilaat), 12 mg/kg laskimoon joka toinen viikko (< 30 kg painavat potilaat), 162 mg ihon alle joka viikko (≥ 30 kg painavat potilaat), 162 mg ihon alle 10 päivän välein tai joka toinen viikko (< 30 kg painavat potilaat).
Ihon alle annetun tosilitsumabin altistuksesta yleisoireista lastenreumaa sairastaville alle 2-vuotiaille ja alle 10 kg:n painaville lapsipotilaille on vähän tietoja.
Yleisoireista lastenreumaa sairastavien potilaiden painon on oltava vähintään 10 kg, jotta heille voidaan antaa tosilitsumabihoitoa ihon alle (ks. kohta Annostus ja antotapa).
Taulukko 8. Yleisoireista lastenreumaa sairastaville lapsipotilaille ihon alle annetun hoidon farmakokineettisten parametrien ennakoidut vakaan tilan keskiarvot ± keskihajonnat
Tosilitsumabin farmakokineettiset parametrit | 162 mg joka viikko ≥ 30 kg | 162 mg joka 2. viikko alle 30 kg |
Cmax (µg/ml) | 99,8 ± 46,2 | 134 ± 58,6 |
Cmin (µg/ml) | 79,2 ± 35,6 | 65,9 ± 31,3 |
Cmean (µg/ml) | 91,3 ± 40,4 | 101 ± 43,2 |
Kumuloitumisen Cmax | 3,66 | 1,88 |
Kumuloitumisen Cmin | 4,39 | 3,21 |
Kumuloitumisen Cmean tai AUCτ* | 4,28 | 2,27 |
*τ = kaksi hoito-ohjelmaa: kerran viikossa ja joka toinen viikko ihon alle
Noin 90 % potilaista saavutti vakaan tilan ihon alle annettujen annosten 162 mg kerran viikossa ja joka toinen viikko yhteydessä viikkoon 12 mennessä.
Imeytyminen
Yleisoireista lastenreumaa sairastavien lapsipotilaiden ihon alle annettuna imeytymisen puoliintumisaika oli noin 2 vuorokautta ja yleisoireista lastenreumaa sairastavien lapsipotilaiden ihon alle annettavaksi tarkoitetun lääkemuodon biologinen hyötyosuus oli 95 %.
Jakautuminen
Yleisoireista lastenreumaa sairastavien lapsipotilaiden keskusjakautumistilavuus oli 1,87 l ja ääreisjakautumistilavuus oli 2,14 l, joten vakaan tilan jakautumistilavuus oli 4,01 l.
Eliminaatio
Tosilitsumabin kokonaispuhdistuma oli pitoisuudesta riippuvainen ja se on lineaarisen ja epälineaarisen puhdistuman summa. Lineaarista puhdistumaa arvioitiin populaatiofarmakokineettisen analyysin parametrina, ja se oli yleisoireista lastenreumaa sairastavilla lapsipotilailla 5,7 ml/h. Yleisoireista lastenreumaa sairastavien lapsipotilaiden ihon alle annetun tosilitsumabin efektiivinen t½ on antovälin aikana vakaassa tilassa enintään 14 vuorokautta riippumatta siitä, annetaanko annos 162 mg kerran viikossa vai joka toinen viikko.
Ihonalainen annostelu
Lasten polyartriittia sairastavat potilaat
Tosilitsumabin farmakokinetiikka määritettiin populaatiofarmakokineettisellä analyysillä 237 lasten polyartriittia sairastavan potilaan potilasjoukosta. Potilaat olivat saaneet tosilitsumabia 8 mg/kg laskimoon joka neljäs viikko (≥ 30 kg painavat potilaat), 10 mg/kg laskimoon joka neljäs viikko (< 30 kg painavat potilaat), 162 mg ihon alle joka toinen viikko (≥ 30 kg painavat potilaat), 162 mg ihon alle joka kolmas viikko (< 30 kg painavat potilaat).
Taulukko 9. Lasten polyartriittia sairastaville lapsipotilaille ihon alle annetun hoidon farmakokineettisten parametrien ennakoidut vakaan tilan keskiarvot ± keskihajonnat
Tosilitsumabin farmakokineettiset parametrit | 162 mg joka 2. viikko ≥ 30 kg | 162 mg joka 3. viikko alle 30 kg |
Cmax (µg/ml) | 29,4 ± 13,5 | 75,5 ± 24,1 |
Cmin (µg/ml) | 11,8 ± 7,08 | 18,4 ± 12,9 |
Cavg (µg/ml) | 21,7 ± 10,4 | 45,5 ± 19,8 |
Kumuloitumisen Cmax | 1,72 | 1,32 |
Kumuloitumisen Cmin | 3,58 | 2,08 |
Kumuloitumisen Cmean tai AUCτ * | 2,04 | 1,46 |
*τ = kaksi hoito-ohjelmaa: joka 2. viikko tai joka 3. viikko ihon alle
Noin 90 % potilaista saavutti vakaan tilan laskimoon annettujen annosten 10 mg/kg (paino < 30 kg) yhteydessä viikkoon 12 mennessä ja annosten 8 mg/kg (paino ≥ 30 kg) yhteydessä viikkoon 16 mennessä. Noin 90 % potilaista kummassakin ryhmässä (antotiheys joka toinen viikko ja joka kolmas viikko) saavutti vakaan tilan ihon alle annettujen 162 mg:n annosten yhteydessä viikkoon 12 mennessä.
Imeytyminen
Lasten polyartriittia sairastavien potilaiden ihon alle annettuna imeytymisen puoliintumisaika oli noin 2 vuorokautta, ja lasten polyartriittia sairastavien potilaiden ihon alle annettavaksi tarkoitetun lääkemuodon biologinen hyötyosuus oli 96 %.
Jakautuminen
Lasten polyartriittia sairastavien potilaiden keskusjakautumistilavuus oli 1,97 l ja ääreisjakautumistilavuus oli 2,03 l, joten vakaan tilan jakautumistilavuus oli 4,0 l.
Eliminaatio
Populaatiofarmakokineettinen analyysi lasten polyartriittia sairastavista potilaista osoitti, että kehon koko vaikuttaa lineaariseen puhdistumaan ja siksi painoon perustuvaa annostusta tulisi harkita (ks. taulukko 9).
Lasten polyartriittia sairastavien potilaiden ihon alle annetun tosilitsumabin efektiivinen t½ on antovälin aikana vakaassa tilassa < 30 kg:n painoisilla potilailla enintään 10 vuorokautta (162 mg ihon alle kerran kolmessa viikossa) ja ≥ 30 kg:n painoisilla potilailla enintään 7 vuorokautta (162 mg ihon alle kerran kahdessa viikossa). Laskimoon annettu tosilitsumabi poistuu verenkierrosta kaksivaiheisesti. Tosilitsumabin kokonaispuhdistuma oli pitoisuudesta riippuvainen ja se on lineaarisen ja epälineaarisen puhdistuman summa. Lineaarista puhdistumaa arvioitiin populaatiofarmakokineettisen analyysin parametrina, ja se oli 6,25 ml/h. Pienten tosilitsumabipitoisuuksien yhteydessä pitoisuudesta riippuvaisella epälineaarisella puhdistumalla on suuri merkitys. Kun epälineaarinen puhdistumareitti on suurempien tosilitsumabipitoisuuksien yhteydessä saturoitunut, puhdistuma määräytyy pääasiassa lineaarisen puhdistuman mukaan.
Ihonalainen annostelu
Jättisoluarteriittia sairastavat potilaat
Tosilitsumabin farmakokinetiikkaa jättisoluarteriittia sairastavilla potilailla selvitettiin analysoitavasta tietueesta populaatiofarmakokineettisen mallin avulla. Tietueessa oli mukana 149 jättisoluarteriittipotilasta, jotka saivat hoitona 162 mg ihon alle viikoittain tai 162 mg ihon alle joka toinen viikko. Kehitetyn mallin rakenne oli sama kuin aiemmin nivelreumapotilaiden tietojen perusteella kehitetty farmakokineettinen malli (ks. taulukko 10).
Taulukko 10. Jättisoluarteriittipotilaiden ihon alle annostelun jälkeisen vakaan tilan ennustettujen farmakokineettisten parametrien keskiarvot ± keskihajonta
Ihonalainen annostelu | ||
Tosilitsumabin farmakokineettiset parametrit | 162 mg joka toinen viikko | 162 mg viikoittain |
Cmax (µg/ml) | 19,3 ± 12,8 | 73 ± 30,4 |
Ctrough (µg/ml) | 11,1 ± 10,3 | 68,1 ± 29,5 |
Cmean (µg/ml) | 16,2 ± 11,8 | 71,3 ± 30,1 |
Kumuloitumisen Cmax | 2,18 | 8,88 |
Kumuloitumisen Ctrough | 5,61 | 9,59 |
Kumuloitumisen Cmean tai AUCτ* | 2,81 | 10,91 |
*τ = kaksi hoito-ohjelmaa: joka toinen viikko tai kerran viikossa ihon alle
Tosilitsumabin viikoittaisten annosten vakaan tilan profiili oli lähes tasainen, sillä pienimpien pitoisuuksien ja huippupitoisuuksien välillä oli vain hyvin pientä vaihtelua, kun taas joka toinen viikko annettujen tosilitsumabiannosten yhteydessä todettiin huomattavaa vaihtelua. Noin 90 % vakaasta tilasta (AUCτ) saavutettiin joka toinen viikko tapahtuneen annostelun yhteydessä viikkoon 14 mennessä ja viikoittain tapahtuneen annostelun yhteydessä viikkoon 17 mennessä.
Farmakokinetiikkaa koskevien tämänhetkisten tietojen perusteella tosilitsumabin vakaan tilan pienin pitoisuus on tässä potilasjoukossa 50 % suurempi verrattuna nivelreumapotilaista saadussa laajassa tietoaineistossa oleviin keskimääräisiin pitoisuuksiin. Näiden erojen syitä ei tiedetä. Farmakokineettisiin eroihin ei liity merkittäviä eroja farmakodynaamisissa parametreissa, joten niiden kliinistä merkitystä ei tunneta.
Jättisoluarteriittipotilailla huomattiin suurempi altistuminen niillä potilailla, jotka painavat vähemmän. Annoksella 162 mg kerran viikossa, vakaan tilan Cavg oli 51 % korkeampi potilailla, joiden paino oli alle 60 kg, verrattuna potilaisiin, joiden paino oli 60–100 kg. Annoksella 162 mg joka toinen viikko, vakaan tilan Cavg oli 129 % korkeampi potilailla, joiden paino oli alle 60 kg, verrattuna potilaisiin, joiden paino oli 60–100 kg. Yli 100 kg painavista potilaista on vain vähän tietoa (n = 7).
Imeytyminen
Jättisoluarteriittia sairastavien potilaiden ihon alle annettuna imeytymisen t½ oli noin 4 vuorokautta. Ihon alle annettavan lääkemuodon biologinen hyötyosuus oli 0,8. Tmax-arvojen mediaani oli tosilitsumabin viikoittaisen annon jälkeen 3 vuorokautta ja tosilitsumabiannoksen joka toinen viikko annon jälkeen 4,5 vuorokautta.
Jakautuminen
Jättisoluarteriittipotilaiden keskustilan jakautumistilavuus oli 4,09 l, ääreistilan jakautumistilavuus oli 3,37 l, jolloin vakaan tilan jakautumistilavuudeksi saadaan 7,46 l.
Eliminaatio
Tosilitsumabin kokonaispuhdistuma oli pitoisuudesta riippuvainen ja se on lineaarisen puhdistuman ja epälineaarisen puhdistuman summa. Lineaarinen puhdistuma arvioitiin populaatiofarmakokineettisen analyysin parametrina, ja jättisoluarteriittipotilailla se oli 6,7 ml/h.
Jättisoluarteriittipotilailla tosilitsumabin efektiivinen t½ vaihteli vakaassa tilassa 162 mg:n annosten viikoittaisen annon yhteydessä 18,3 vuorokaudesta 18,9 vuorokauteen ja 162 mg:n annosten joka toinen viikko annon yhteydessä 4,2 vuorokaudesta 7,9 vuorokauteen. Kun pitoisuus seerumissa on suuri ja tosilitsumabin kokonaispuhdistumassa vallitsevana on lineaarinen puhdistuma, noin 32 vuorokauden efektiivinen t1/2 perustui potilasjoukon parametrien estimaatteihin.
Erityisryhmät
Munuaisten vajaatoiminta
Varsinaista tutkimusta munuaisten vajaatoiminnan vaikutuksesta tosilitsumabin farmakokinetiikkaan ei ole tehty. Nivelreumaa ja jättisoluarteriittia sairastavilla potilailla tehtyjen tutkimusten populaatiofarmakokineettisessä analyysissä mukana olleista potilaista useimmilla oli normaali munuaistoiminta tai lievä munuaisten vajaatoiminta. Lievä munuaisten vajaatoiminta (Cockcroft-Gaultin kaavaan perustuva laskennallinen kreatiniinipuhdistuma) ei vaikuttanut tosilitsumabin farmakokinetiikkaan.
Jättisoluarteriittia koskeneeseen tutkimukseen osallistuneista potilaista noin kolmanneksella oli lähtötilanteessa kohtalaista munuaisten vajaatoimintaa (laskennallinen kreatiniinipuhdistuma 30–59 ml/min). Sen ei todettu vaikuttavan näiden potilaiden altistukseen tosilitsumabille.
Lievää tai kohtalaista munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa.
Maksan vajaatoiminta
Varsinaista tutkimusta maksan vajaatoiminnan vaikutuksesta tosilitsumabin farmakokinetiikkaan ei ole tehty.
Ikä, sukupuoli ja etninen tausta
Nivelreuma- ja jättisoluarteriittipotilaiden populaatiofarmakokineettiset analyysit osoittivat, etteivät ikä, sukupuoli ja etninen tausta vaikuttaneet tosilitsumabin farmakokinetiikkaan.
Yleisoireista lastenreumaa ja lasten polyartriittia sairastavien potilaiden populaatiofarmakokineettisen analyysin tulokset vahvistivat, että kehon koko on ainoa kovariaatti, joka vaikuttaa oleellisesti tosilitsumabin farmakokinetiikkaan, eliminaatio ja imeytyminen mukaan lukien. Siksi painoon perustuvaa annostusta tulisi harkita (ks. taulukot 8 ja 9).
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten prekliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuustutkimuksia ei ole tehty, koska monoklonaalisia IgG1-vasta-aineita ei pidetä luontaisina karsinogeeneinä.
Käytettävissä olevat ei-kliiniset tiedot osoittivat, että IL-6 vaikuttaa tiettyjen syöpätyyppien etenemiseen ja apoptoosiresistenssiin. Nämä tiedot eivät viittaa siihen, että tosilitsumabihoitoon liittyisi merkittävää syövän kehittymisen ja etenemisen riskiä. Pitkäaikaisessa kuuden kuukauden toksisuustutkimuksessa ei havaittu proliferoivia leesioita jaavanmakakeilla (cynomolgus-apinoilla) eikä IL-6-defisienteillä hiirillä.
Käytettävissä olevat ei-kliiniset tiedot eivät viittaa hedelmällisyyteen kohdistuviin vaikutuksiin tosilitsumabihoidon aikana. Jaavanmakakeilla tehdyssä toksisuustutkimuksessa ei havaittu endokriinisesti aktiivisiin elimiin eikä lisääntymiselimiin kohdistuneita vaikutuksia. Lisääntymistoimintoihin kohdistuvia vaikutuksia ei havaittu myöskään IL-6-defisienteillä hiirillä. Tosilitsumabilla ei havaittu olevan suoraa eikä epäsuoraa haitallista vaikutusta tiineyteen eikä alkion- tai sikiönkehitykseen, kun sitä annettiin jaavanmakakeille tiineyden alkuvaiheessa. Keskenmenojen / alkio- ja sikiökuolemien vähäistä lisääntymistä havaittiin kuitenkin suurta annostusta 50 mg/kg/vrk saaneiden ryhmässä, jossa systeeminen altistus oli suuri (> 100-kertainen ihmisen altistukseen verrattuna), lumeryhmään ja muihin pientä annosta saaneiden ryhmiin verrattuna. Vaikka IL-6 ei näytä olevan ratkaisevan tärkeä sytokiini sikiön kasvun kannalta eikä emon ja sikiön immunologisessa vuorovaikutuksen säätelyssä, tämän löydöksen yhteyttä tosilitsumabihoitoon ei voida sulkea pois.
Hoito hiiren analogeilla ei aiheuttanut lisääntynyttä toksisuutta nuorilla hiirillä. Häiriöitä ei havaittu etenkään luuston kehityksessä, immuniteetissä eikä seksuaalisessa kypsymisessä.
Tosilitsumabin non-kliininen turvallisuusprofiili cynomolgus-apinoilla ei viittaa siihen, että laskimonsisäisen ja ihonalaisen antoreitin välillä olisi eroa.
Farmaseuttiset tiedot
Apuaineet
L-histidiini (pH:n säätöön)
L-histidiinimonohydrokloridimonohydraatti (pH:n säätöön)
L-arginiini/L-arginiinihydrokloridi
L-metioniini
Polysorbaatti 80 (E 433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä. Kun esitäytetty kynä on otettu jääkaapista, se voidaan säilyttää enintään 30 °C:n lämpötilassa korkeintaan 2 viikkoa.
Pidä esitäytetyt kynät ulkopakkauksessa. Herkkä valolle. Herkkä kosteudelle.
Pakkauksen jääkaapista pois ottamisen päivämäärä ja kellonaika on kirjoitettava pahvikoteloon. Hävitä kynä, jos se on ollut poissa jääkaapista yli 2 viikkoa. Älä käytä esitäytetyn kynän lämmittämiseen ulkoisia lämmönlähteitä, kuten kuumaa vettä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ROACTEMRA injektioneste, liuos, esitäytetty kynä
162 mg (L:ei) 4 x 1 kpl (162 mg/0,9 ml) (767,77 €)
PF-selosteen tieto
RoActemra 162 mg esitäytetty kynä sisältää 0,9 ml liuosta esitäytetyssä ruiskussa (tyypin I lasia), jossa on valmiiksi kiinnitetty neula. Ruiskussa on jäykkä neulansuojus (elastomeerisuljin, jossa polypropeenikuori) ja männänpysäytin (fluororesiinipäällysteistä butyylikumia).
Pakkauskoot: 4 esitäytettyä kynää. 12 esitäytetyn kynän kerrannaispakkaus, joka sisältää 3 pakkausta, joissa kussakin 4 esitäytettyä kynää. Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai hieman kellertävä liuos, jonka pH on 5,5–6,5 ja jonka osmolaalisuus on 200–372 mosm/kg.
Käyttö- ja käsittelyohjeet
RoActemra on pakattu kertakäyttöön tarkoitettuun esitäytettyyn kynään. Kun esitäytetty kynä on otettu jääkaapista, esitäytetyn kynän on annettava lämmetä huoneenlämpöiseksi (18 °C – 28 °C) 45 minuutin ajan ennen injektion antamista. Kynää ei saa ravistaa. Injektion antaminen on aloitettava 3 minuutin kuluessa siitä, kun neulan suojakorkki on poistettu, jotta lääkevalmiste ei kuivu ja tuki neulaa. Jos esitäytettyä kynää ei käytetä 3 minuutin kuluessa siitä, kun suojakorkki on poistettu, kynä on laitettava pistävälle ja viiltävälle jätteelle tarkoitettuun astiaan, ja käyttöön on otettava uusi esitäytetty kynä.
Jos violetinvärinen osoitin ei liiku aktivointipainikkeen painamisen jälkeen, sinun on laitettava esitäytetty kynä pistävälle ja viiltävälle jätteelle tarkoitettuun astiaan. Älä yritä käyttää esitäytettyä ruiskua uudelleen. Älä ota uutta pistosta uudella esitäytetyllä kynällä. Soita terveydenhuollon ammattilaiselle ja kysy neuvoa.
Älä käytä kynää, jos lääkevalmiste on sameaa tai siinä on hiukkasia tai jos liuos ei ole väritöntä tai hieman kellertävää tai jos esitäytetyn kynän jokin osa vaikuttaa vialliselta.
Kattavat ohjeet RoActemran esitäytetyn kynän käyttämiseen on esitetty pakkausselosteessa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ROACTEMRA injektioneste, liuos, esitäytetty kynä
162 mg 4 x 1 kpl
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313).
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L04AC07
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com