
MYLOTARG kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 5 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten sisältää 5 mg gemtutsumabi-otsogamisiinia.
Käyttökuntoon saattamisen jälkeen (ks. kohta Käyttö- ja käsittelyohjeet) konsentraattiliuos sisältää 1 mg/ml gemtutsumabi-otsogamisiinia.
Gemtutsumabi-otsogamisiini on vasta-ainekonjugoitu solunsalpaaja (antibody-drug conjugate, ADC). Se koostuu CD33-antigeeniin kohdentuvasta monoklonaalisesta vasta-aineesta (hP67.6; yhdistelmä-DNA-tekniikalla valmistettu IgG4:n kappa-isotyypin humanisoitu immunoglobuliini, joka on valmistettu nisäkässoluviljelmän NS0-soluissa), joka on yhdistetty kovalenttisesti sytotoksiseen N‑asetyyli-gamma-kalikeamisiiniin.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos (kuiva-aine välikonsentraattia varten).
Kliiniset tiedot
Käyttöaiheet
MYLOTARG on tarkoitettu käytettäväksi yhdessä daunorubisiinin ja sytarabiinin kanssa 15-vuotiaille tai tätä vanhemmille potilaille aiemmin hoitamattoman de novo CD33‑positiivisen akuutin myelooisen leukemian (AML) hoitoon lukuun ottamatta akuuttia promyelosyyttista leukemiaa (APL) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Ehto
Valmistetta tulee käyttää vain syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
MYLOTARG tulee antaa syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa hoitopaikassa, jossa täydellinen elvytysvälineistö on heti saatavilla.
MYLOTARG-valmistetta tulee käyttää vain intensiiviseen induktiosolunsalpaajahoitoon soveltuville potilaille.
Infuusioon liittyvien oireiden lieventämiseksi suositellaan esilääkitystä kortikosteroidilla, antihistamiinilla ja parasetamolilla 1 tunti ennen tämän valmisteen antoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tuumorilyysioireyhtymään liittyvän hyperurikemian kehittyminen on pyrittävä estämään asianmukaisin toimin, joita ovat esimerkiksi nesteytys ja hyperurikemiaa estävän lääkkeen tai muiden hyperurikemian hoitoon tarkoitettujen aineiden anto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Induktio
Suositeltu MYLOTARG-annos on 3 mg/m2/annos (enintään yksi 5 mg:n injektiopullo) 2 tunnin infuusiona päivinä 1, 4 ja 7. Potilaalle annetaan myös 60 mg/m2/vrk daunorubisiinia 30 minuutin infuusiona päivinä 1–3 ja 200 mg/m2/vrk sytarabiinia jatkuvana infuusiona päivinä 1–7.
Jos tarvitaan toinen induktio, MYLOTARG-valmistetta ei tule antaa toisen induktiohoidon aikana. Toisen induktiosyklin aikana annetaan vain daunorubisiinia ja sytarabiinia seuraavina suositusannostuksina: 35 mg/m2/vrk daunorubisiinia päivinä 1 ja 2 ja 1 g/m2 sytarabiinia 12 tunnin välein päivinä 1–3.
Konsolidaatio
Jos potilas saavuttaa induktion jälkeen täydellisen remission (complete remission [CR] = alle 5 % blasteja normosellulaarisessa luuytimessä, absoluuttinen neutrofiilimäärä [B-Neut] yli 1,0 × 109 solua/l, perifeerisen veren verihiutalemäärä vähintään 100 × 109/l ilman verensiirtoa), suositellaan enintään 2 konsolidaatiohoitoa seuraavasti: laskimonsisäinen daunorubisiini (60 mg/m2 päivänä 1 [ensimmäinen hoitokuuri] tai päivinä 1 ja 2 [toinen hoitokuuri]), laskimonsisäinen sytarabiini (1 g/m2 12 tunnin välein 2 tunnin infuusiona päivinä 1–4) ja laskimonsisäinen MYLOTARG (3 mg/m2/annos 2 tunnin infuusiona enintään enimmäisannoksena [yksi 5 mg:n injektiopullo] päivänä 1).
Taulukko 1. MYLOTARG-annostusohjelmat yhdessä solunsalpaajahoidon kanssa
Hoito | MYLOTARG | daunorubisiini | sytarabiini |
Induktioa | 3 mg/m2/annos (enintään yksi 5 mg:n injektiopullo) päivinä 1, 4 ja 7 | 60 mg/m2/vrk päivinä 1–3 | 200 mg/m2/vrk päivinä 1–7 |
Toinen induktio (tarvittaessa) | MYLOTARG-valmistetta ei tule antaa toisen induktiohoidon aikana. | 35 mg/m2/vrk päivinä 1–2 | 1 g/m2 12 tunnin välein päivinä 1–3 |
1. konsolidaatioa,b | 3 mg/m2/annos (enintään yksi 5 mg:n injektiopullo) päivänä 1 | 60 mg/m2/vrk päivänä 1 | 1 g/m2 12 tunnin välein päivinä 1–4 |
2. konsolidaatioa,b | 3 mg/m2/annos (enintään yksi 5 mg:n injektiopullo) päivänä 1 | 60 mg/m2/vrk päivinä 1–2 | 1 g/m2 12 tunnin välein päivinä 1–4 |
a. Ks. tietoa annosmuutoksista taulukoista 3 ja 4. b. Jos potilas saavuttaa täydellisen remission (CR) induktion jälkeen. | |||
Annoksen ja hoito-ohjelman muutokset
Hoito-ohjelman muuttaminen hyperleukosytoosin vuoksi
Jos AML:aan liittyy hyperleukosytoosi (leukosyyttimäärä ≥ 30 x 109/l), suositellaan perifeerisen veren valkosolumäärän pienentämiseksi sytoreduktiota joko leukafereesillä, suun kautta annettavalla hydroksiurealla tai sytarabiinilla, johon voidaan yhdistää hydroksiurea. Sytoreduktio tulee tehdä 48 tuntia ennen MYLOTARG-valmisteen antoa.
Jos leukoreduktio tehdään joko sytarabiinin ja hydroksiurean yhdistelmällä tai pelkällä sytarabiinilla aiemmin hoitamatonta de novo hyperleukosyyttista AML:aa sairastavalle potilaalle, joka saa MYLOTARG-valmistetta osana yhdistelmähoitoa, hoito-ohjelmaa tulee muuttaa seuraavan taulukon 2 mukaisesti:
Taulukko 2. Hoito-ohjelman muuttaminen hoidettaessa hyperleukosytoosia sytarabiinilla
Hoito | MYLOTARG | daunorubisiini | sytarabiini | hydroksiurea |
Induktioa | 3 mg/m2/annos (enintään yksi 5 mg:n injektiopullo) päivinä 3, 6 ja 9 | 60 mg/m2/vrk päivinä 3–5 | 200 mg/m2/vrk päivinä 1–7 | Päivä 1 (tavanomaisen hoitokäytännön mukaan) |
Ks. taulukosta 1 konsolidaatiohoidon annossuositukset. a. Ks. lisätietoa annosmuutoksista taulukoista 3 ja 4. | ||||
Annosmuutos haittavaikutusten vuoksi
Yksittäisen potilaan MYLOTARG-annosta on suositeltavaa muuttaa turvallisuus‑ ja siedettävyyssyistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Joidenkin haittavaikutusten hoito voi vaatia annostelun tilapäistä keskeyttämistä tai MYLOTARG-hoidon pysyvää lopettamista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukossa 3 on esitetty ohjeet annosmuutoksiin hematologisen toksisuuden vuoksi ja taulukossa 4 ei-hematologisen toksisuuden vuoksi.
Taulukko 3. Annosmuutokset hematologisen toksisuuden vuoksi
Hematologinen toksisuus | Annosmuutokset |
Pitkittynyt trombosytopenia (Verihiutalemäärä < 100 × 109/l konsolidaatiohoidon suunniteltuna aloituspäivänä) |
|
Pitkittynyt neutropenia |
|
Taulukko 4. Annosmuutokset ei-hematologisen toksisuuden vuoksi
Ei-hematologinen toksisuus | Annosmuutokset |
VOD | Keskeytä MYLOTARG (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Bilirubiini > 2 × ULN ja ASAT ja/tai ALAT > 2,5 × ULN | Siirrä MYLOTARG-hoitoa, kunnes bilirubiini on jälleen ≤ 2 × ULN ja ASAT ja ALAT ≤ 2,5 × ULN ennen jokaista annosta. Harkitse suunnitellun annoksen jättämistä väliin, jos arvojen korjaantuminen peräkkäisten infuusioiden välillä viivästyy yli 2 päivää. |
Infuusioon liittyvät reaktiot | Keskeytä infuusio ja aloita asianmukainen lääketieteellinen hoito oireiden vaikeusasteen mukaan. Potilasta on tarkkailtava, kunnes merkit ja oireet ovat hävinneet kokonaan ja infuusiota voidaan jatkaa. Harkitse hoidon pysyvää lopettamista, jos infuusioreaktio on vaikea tai henkeä uhkaava (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Muut vaikeat tai henkeä uhkaavat ei-hematologiset toksisuudet | Siirrä MYLOTARG-hoitoa, kunnes toksisuuden vaikeusaste lievittyy korkeintaan vaikeusasteelle lievä. Harkitse suunnitellun annoksen jättämistä väliin, jos arvojen korjaantuminen peräkkäisten infuusioiden välillä viivästyy yli 2 päivää. |
Lyhenteet: ALAT = alaniiniaminotransferaasi, ASAT = aspartaattiaminotransferaasi, ULN = normaalin vaihteluvälin yläraja, VOD = maksan veno-okklusiivinen tauti. | |
Erityisryhmät
Maksan vajaatoiminta
Aloitusannosta ei tarvitse muuttaa maksan vajaatoimintaa sairastavalle potilaalle, jos bilirubiini ≤ 2 × normaalin vaihteluvälin yläraja (ULN) ja aspartaattiaminotransferaasi (ASAT)/ alaniiniaminotransferaasi (ALAT) ≤ 2,5 × ULN. Siirrä MYLOTARG-hoitoa, kunnes bilirubiini on korjaantunut ≤ 2 × ULN ja ASAT ja ALAT ≤ 2,5 × ULN ennen jokaista annosta (ks. taulukko 4, kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavalle potilaalle. MYLOTARG-hoitoa ei ole tutkittu potilailla, joilla on vaikea munuaisten vajaatoiminta. MYLOTARG ei poistu elimistöstä munuaisten kautta. Sen farmakokinetiikkaa vaikeaa munuaisten vajaatoimintaa sairastavilla ei tunneta (ks. kohta Farmakokinetiikka).
Iäkkäät
Annosta ei tarvitse muuttaa iäkkäille (≥ 65‑vuotiaille) potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
MYLOTARG-valmisteen turvallisuutta ja tehoa alle 15 vuoden ikäisten potilaiden hoidossa ei ole varmistettu. Tällä hetkellä saatavilla olevat tiedot löytyvät kohdista Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, mutta mitään annostussuositusta ei voida antaa.
Antotapa
MYLOTARG annetaan laskimoon, ja se on saatettava käyttökuntoon ja laimennettava ennen antoa (ks. kohta Käyttö- ja käsittelyohjeet). Kun valmiste on saatettu käyttökuntoon pitoisuuteen 1 mg/ml, injektiopullosta saatava määrä on 4,5 mg (4,5 ml). Käyttökuntoon saatettu ja laimennettu liuos annetaan 2 tuntia kestävänä infuusiona laskimoon tarkassa kliinisessä seurannassa, johon kuuluu sydämen syketiheyden, verenpaineen ja ruumiinlämmön tarkkailu. MYLOTARG-valmistetta ei tule antaa laskimoon nopeana tai hyvin nopeana boluksena (ks. kohta Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Maksatoksisuus, mukaan lukien maksan veno-okklusiivinen tauti (VOD)
MYLOTARG-hoitoa saaneilla potilailla on ilmoitettu maksatoksisuutta, mukaan lukien henkeä uhkaavaa ja toisinaan kuolemaan johtanutta maksan vajaatoimintaa ja maksan veno-okklusiivista tautia (VOD) (ks. kohta Haittavaikutukset).
Mahdollisten riskitekijöiden analyysin perusteella VOD:n riski on suurentunut aikuispotilailla, jotka ovat saaneet MYLOTARG-hoitoa monoterapiana joko ennen kantasolusiirtoa (HSCT) tai sen jälkeen, sekä potilailla, joilla on keskivaikea tai vaikea maksan vajaatoiminta (ks. kohta Haittavaikutukset).
VOD-riskin vuoksi potilasta on tarkkailtava tiiviisti VOD:n merkkien ja oireiden varalta. Näitä voivat olla muun muassa ALAT‑, ASAT‑, bilirubiini‑ ja alkalisen fosfataasin arvojen suureneminen (määritettävä ennen jokaista MYLOTARG-annosta), hepatomegalia (mahdollisesti kivulias), nopea painonnousu ja askites. Pelkkää bilirubiiniarvoa seuraamalla ei ehkä tunnisteta kaikkia potilaita, joilla on VOD-riski. Jos potilaan maksa-arvoissa ilmenee poikkeamia, maksa-arvoja ja maksatoksisuuden kliinisiä merkkejä ja oireita tulisi seurata tiheämmin. Jos potilas siirtyy kantasolusiirtoon, maksa-arvoja tulee seurata tarkoin kantasolusiirron jälkeen asianmukaisella tavalla. Varmaa yhteyttä ei todettu VOD:n ja kantasolusiirron ajankohdan välillä, kun suurempia MYLOTARG-annoksia käytettiin monoterapiana; ALFA-0701-tutkimuksessa suositeltiin kuitenkin pitämään viimeisen MYLOTARG-annoksen jälkeen 2 kuukauden tauko ennen kantasolusiirtoa.
Maksatoksisuuden merkkien ja oireiden hoito voi vaatia annostelun tilapäistä keskeyttämistä tai MYLOTARG-hoidon lopettamista (ks. kohta Annostus ja antotapa). Jos VOD ilmaantuu, MYLOTARG-hoito tulee lopettaa ja potilasta tulee hoitaa tavanomaisen hoitokäytännön mukaan.
Infuusioon liittyvät reaktiot (myös anafylaksia)
Kliinisissä tutkimuksissa ilmoitettiin infuusioon liittyviä reaktioita, myös anafylaksiaa (ks. kohta Haittavaikutukset). Valmisteen markkinoille tulon jälkeen on ilmoitettu kuolemaan johtaneita infuusioreaktioita. Infuusioon liittyvien reaktioiden merkkejä ja oireita voivat olla kuume ja vilunväristykset ja harvemmin hypotensio, takykardia ja hengitysoireet, joita voi ilmetä antoa seuraavien ensimmäisten 24 tunnin aikana. MYLOTARG-infuusio on annettava tarkassa kliinisessä seurannassa, johon kuuluu syketiheyden, verenpaineen ja ruumiinlämmön tarkkailu. Esilääkitystä kortikosteroidilla, antihistamiinilla ja parasetamolilla suositellaan 1 tunti ennen MYLOTARG-valmisteen antoa (ks. kohta Annostus ja antotapa). Infuusio on keskeytettävä heti, jos viitteitä vaikeasta reaktiosta ilmenee, etenkin hengenahdistusta, bronkospasmi tai kliinisesti merkittävä hypotensio. Potilasta on tarkkailtava, kunnes merkit ja oireet ovat hävinneet kokonaan. Hoidon lopettamista on harkittava vakavasti, jos potilaalle kehittyy anafylaksian merkkejä tai oireita, mukaan lukien vaikeita hengitysoireita tai kliinisesti merkittävää hypotensiota (ks. kohta Annostus ja antotapa).
Myelosuppressio
Kliinisissä tutkimuksissa on raportoitu neutropeniaa, trombosytopeniaa, anemiaa, leukopeniaa, kuumeista neutropeniaa, lymfopeniaa ja pansytopeniaa. Osa tapauksista on ollut henkeä uhkaavia tai johtanut kuolemaan (ks. kohta Haittavaikutukset). Neutropenian yhteydessä voi ilmetä komplikaatioina infektioita ja trombosytopenian yhteydessä verenvuotoreaktioita. Infektioita ja verenvuotoreaktioita on raportoitu, ja osa tapauksista oli henkeä uhkaavia tai kuolemaan johtavia.
Ennen jokaista MYLOTARG-annosta tulisi määrittää täydellinen verenkuva. Potilasta on tarkkailtava hoidon aikana infektion ja verenvuodon merkkien ja oireiden tai myelosuppression muiden vaikutusten varalta. Potilasta on tarkkailtava hoidon aikana ja sen jälkeen tavanomaisin kliinisin ja laboratoriotutkimuksin.
MYLOTARG-annostelua voidaan joutua siirtämään tai hoito voidaan joutua lopettamaan pysyvästi vaikean infektion, verenvuodon tai myelosuppression muiden vaikutusten, mukaan lukien vaikean neutropenian tai pitkittyneen trombosytopenian, hoitamiseksi (ks. kohta Annostus ja antotapa).
Tuumorilyysioireyhtymä
Kliinisissä tutkimuksissa on raportoitu tuumorilyysioireyhtymää (ks. kohta Haittavaikutukset). Valmisteen markkinoille tulon jälkeen on ilmoitettu kuolemaan johtaneita tuumorilyysioireyhtymän tapauksia, joita on komplisoinut akuutti munuaisten vajaatoiminta. Tuumorilyysioireyhtymän kehittymisriskin pienentämiseksi hyperleukosyyttistä AML:aa sairastavilla potilailla tulisi ennen MYLOTARG-valmisteen antoa harkita leukoreduktiota hydroksiurealla tai leukafereesillä, jotta perifeerisen veren valkosolumäärä saadaan alle arvon 30 x 109/l (ks. kohta Annostus ja antotapa).
Potilasta on tarkkailtava tuumorilyysioireyhtymän merkkien ja oireiden varalta ja hoidettava tavanomaisen lääketieteellisen käytännön mukaisesti. Tuumorilyysiin liittyvän hyperurikemian kehittyminen on pyrittävä estämään asianmukaisin keinoin, kuten nesteytyksellä ja hyperurikemiaa estävien lääkkeiden (kuten allopurinolin) tai muiden hyperurikemian hoidossa käytettävien aineiden (kuten rasburikaasin) annolla.
Sytogeneettisesti suuren riskin AML
MYLOTARG-valmisteen teho on osoitettu AML-potilailla, joilla on sytogeneettisesti pieni tai keskisuuri riski, mutta tehon määrään suuren riskin potilailla liittyy epävarmuutta. Kun MYLOTARG-valmistetta käytetään yhdessä daunorubisiinin ja sytarabiinin kanssa äskettäin diagnosoidun de novo AML:n hoitoon, sytogeneettisen testauksen tulosten valmistuttua tulee harkita, onko kyseisen potilaan kohdalla MYLOTARG-hoidon jatkamisesta mahdollisesti saatava hyöty riskejä suurempi (ks. kohta Farmakodynamiikka).
Ehkäisy
Naisia, jotka voivat tulla raskaaksi, ja tällaisten naisten kumppaneita on kehotettava käyttämään kahta tehokasta raskaudenehkäisymenetelmää MYLOTARG-hoidon aikana ja vähintään 7 kuukautta (naiset) tai 4 kuukautta (miehet) viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Apuaine
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste voidaan valmistella edelleen antoa varten natriumia sisältäviin liuoksiin (ks. kohdat Annostus ja antotapa ja Käyttö- ja käsittelyohjeet), mikä pitää ottaa huomioon potilaan kaikista lähteistä saamassa natriumin kokonaismäärässä.
Yhteisvaikutukset
MYLOTARG-valmisteella ei ole tehty kliinisiä lääkeyhteisvaikutustutkimuksia. Katso kohdasta Farmakokinetiikka in vitro ‑tutkimusten saatavilla olevat tiedot.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/raskaudenehkäisy miehillä ja naisilla
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskaaksi tuloa MYLOTARG-hoidon aikana.
Naisia, jotka voivat tulla raskaaksi, ja tällaisten naisten kumppaneita on kehotettava käyttämään kahta tehokasta raskaudenehkäisymenetelmää MYLOTARG-hoidon aikana ja vähintään 7 kuukautta (naiset) tai 4 kuukautta (miehet) viimeisen annoksen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja gemtutsumabi-otsogamisiinin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
MYLOTARG-valmistetta ei saa käyttää raskauden aikana, ellei äidin mahdollisesti saama hyöty ole suurempi kuin sikiölle mahdollisesti koituva riski. Sikiölle mahdollisesta aiheutuvasta vaarasta on kerrottava raskaana oleville naisille, gemtutsumabi-otsogamisiini-hoidon aikana raskaaksi tuleville potilaille ja hoitoa saaville miespotilaille, joiden kumppani tulee raskaaksi hoidon aikana.
Imetys
Ei ole tietoa gemtutsumabi-otsogamisiinin tai sen metaboliittien erittymisestä ihmisen rintamaitoon tai vaikutuksista rintaruokittuun lapseen tai maidontuotantoon. Koska MYLOTARG-hoito aiheuttaa rintaruokituille lapsille haittavaikutusten riskin, hoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen saamisesta ei saa imettää (ks. kohta Prekliiniset tiedot turvallisuudesta).
Hedelmällisyys
Tietoja potilaiden hedelmällisyydestä ei ole saatavilla. Prekliinisten löydösten perusteella gemtutsumabi-otsogamisiinihoito voi vaarantaa miehen ja naisen hedelmällisyyden (ks. kohta Prekliiniset tiedot turvallisuudesta). Sekä miesten että naisten tulisi saada ennen hoitoa tietoa toimenpiteistä hedelmällisyyden säilyttämiseksi.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
MYLOTARG-valmisteella on kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita tulisi kehottaa varovaisuuteen autoa ajettaessa ja koneita käytettäessä, sillä MYLOTARG-hoidon aikana voi ilmetä väsymystä, heitehuimausta ja päänsärkyä (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
MYLOTARG-valmisteen yleinen turvallisuusprofiili perustuu tietoihin, jotka on saatu akuuttia myelooista leukemiaa sairastaneista potilaista ALFA‑0701-yhdistelmähoitotutkimuksessa, monoterapiatutkimuksissa ja valmisteen markkinoille tulon jälkeen. Yhdistelmähoitotutkimuksesta saatua turvallisuustietoa valikoiduista hoidon aikana ilmenneistä haittatapahtumista (kaikkien vaikeusasteiden verenvuodot, kaikkien vaikeusasteiden VOD ja vaikeat infektiot) pidetään tärkeimpänä MYLOTARG-valmisteen turvallisuusprofiilin ymmärtämisen kannalta. Kaikki edellä mainitut hoidon aikana ilmenneet haittatapahtumat on arvioitu lääkkeestä aiheutuneiksi haittavaikutuksiksi. Johtuen edellä kuvatusta suppeasta tietojenkeruusta yhdistelmähoitotutkimuksen laboratoriotulokset on sisällytetty taulukkoon 5. Haittavaikutusten täydellistä karakterisointia varten tiedot haittavaikutuksista fraktioimattomalla hoito-ohjelmalla tehdyistä monoterapiatutkimuksista (tutkimukset 201/202/203) ja valmisteen markkinoille tulon jälkeisistä kokemuksista on esitetty taulukossa 6 ja tiedot fraktioidulla hoito-ohjelmalla tehdystä monoterapiatutkimuksesta B1761031 on esitetty jäljempänä olevassa kohdassa.
ALFA‑0701-yhdistelmähoitotutkimuksessa kliinisesti merkittäviä vakavia haittavaikutuksia olivat maksatoksisuus, mukaan lukien VOD (3,8 %), verenvuoto (9,9 %), vaikea infektio (41,2 %) ja tuumorilyysioireyhtymä (1,5 %). Monoterapiatutkimuksissa (tutkimukset 201/202/203) kliinisesti merkittäviä vakavia haittavaikutuksia olivat myös infuusioon liittyvät reaktiot (2,5 %), trombosytopenia (21,7 %) ja neutropenia (34,3 %). Monoterapiatutkimuksessa B1761031 kliinisesti merkittäviin vakaviin haittavaikutuksiin kuuluivat infektio (30,0 %), kuumeinen neutropenia (22,0 %), kuume (6,0 %), verenvuoto (4,0 %), trombosytopenia (4,0 %), anemia (2,0 %) ja takykardia (2,0 %).
Yhdistelmähoitotutkimuksessa yleisimmät (> 30 %) haittavaikutukset olivat verenvuoto ja infektio. Monoterapiatutkimuksissa (tutkimukset 201/202/203) yleisimpiin (> 30 %) haittavaikutuksiin kuuluivat kuume, pahoinvointi, infektio, vilunväristykset, verenvuoto, oksentelu, trombosytopenia, väsymys, päänsärky, stomatiitti, ripuli, vatsakipu ja neutropenia. Monoterapiatutkimuksessa B1761031 yleisimpiin (> 30 %) haittavaikutuksiin kuuluivat infektio (50,0 %), kuumeinen neutropenia (40,0 %) ja verenvuoto (32,0 %).
Yhdistelmähoitotutkimuksessa hoidon pysyvään lopettamiseen yleisimmin (≥ 1 %) johtaneet haittavaikutukset olivat trombosytopenia, VOD, verenvuoto ja infektio. Monoterapiatutkimuksissa (tutkimukset 201/202/203) hoidon pysyvään lopettamiseen yleisimmin (≥ 1 %) johtaneet haittavaikutukset olivat infektio, verenvuoto, monielinvaurio ja VOD. Monoterapiatutkimuksessa B1761031 hoidon pysyvään lopettamiseen johtaneet haittavaikutukset olivat infektio ja kuume.
Haittavaikutustaulukko
Haittavaikutukset on esitetty elinjärjestelmittäin ja esiintymistiheyksittäin seuraavan käytännön mukaan: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 5. Valikoidut** haittavaikutukset potilailla, jotka saivat MYLOTARG-hoitoa yhdistelmähoitotutkimuksessa (ALFA‑0701)
Elinjärjestelmä Esiintymistiheys Suositeltu termi | MYLOTARG + daunorubisiini + sytarabiini (n = 131) | daunorubisiini + sytarabiini (n = 137) | ||
Kaikki vaikeusasteet % | Vaikeusaste 3/4 % | Kaikki vaikeusasteet % | Vaikeusaste 3/4 % | |
| Infektiot | ||||
| Hyvin yleiset | ||||
| Infektio*a | 77,9 | 76,3 | 77,4 | 74,4 |
| Verisuonisto | ||||
| Hyvin yleiset | ||||
| Verenvuoto*b | 90,1 | 20,6 | 78,1 | 8,8 |
| Maksa ja sappi | ||||
| Yleiset | ||||
| Maksan veno-okklusiivinen tauti*c | 4,6 | 2,3 | 1,5 | 1,5 |
| Tutkimukset*** | ||||
| Hyvin yleiset | ||||
| Alentunut hemoglobiini | 100 | 86,2 | 100 | 89,7 |
| Pienentynyt verihiutalemäärä | 100 | 100 | 100 | 100 |
| Pienentynyt valkosolumäärä | 100 | 100 | 99,3 | 99,3 |
| Pienentynyt lymfosyyttien (absoluuttinen) määrä | 98,5 | 90,7 | 97,8 | 89,6 |
| Pienentynyt neutrofiilimäärä | 97,7 | 96,1 | 98,5 | 97,0 |
| Hyperglykemia | 92,0 | 19,2 | 91,1 | 17,8 |
| Suurentunut aspartaattiaminotransferaasi (ASAT) | 89,2 | 14,0 | 73,9 | 9,0 |
| Pidentynyt protrombiiniaika | 84,8 | 3,3 | 89,1 | 0 |
| Aktivoitu partiaalinen tromboplastiiniaika pidentynyt | 80,0 | 6,4 | 57,5 | 5,5 |
| Suurentunut alkalinen fosfataasi | 79,7 | 13,3 | 68,9 | 5,3 |
| Suurentunut alaniiniaminotransferaasi (ALAT) | 78,3 | 10,9 | 81,3 | 15,7 |
| Suurentunut veren bilirubiini | 51,6 | 7,1 | 50,8 | 3,8 |
| Hyperurikemia | 32,5 | 2,6 | 28,5 | 0 |
Lyhenne: n = potilaiden lukumäärä.
* Mukaan lukien kuolemaan johtanut.
** Tässä äskettäin diagnosoitua AML:aa koskeneessa tutkimuksessa kerättiin vain valikoituja turvallisuustietoja.
*** Esiintymistiheys perustuu laboratorioarvoihin (vaikeusaste NCI CTCAE v4.03 mukaan)
a. Infektio kattaa seuraavat: sepsis ja bakteremia (53,4 %), sieni-infektio (15,3 %), alahengitystieinfektio (5,3 %), bakteeri-infektio (9,2 %), maha-suolikanavan infektio (8,4 %), ihoinfektio (2,3 %) ja muut infektiot (28,4 %).
b. Verenvuoto kattaa seuraavat: keskushermoston verenvuoto (3,1 %), maha-suolikanavan yläosan verenvuoto (33,6 %), maha-suolikanavan alaosan verenvuoto (17,6 %), ihonalainen verenvuoto (60,3 %), muu verenvuoto (64,9 %) ja nenäverenvuoto (62,6 %).
c. Maksan veno-okklusiivinen tauti kattaa seuraavat raportoidut suositellut termit: veno-okklusiivinen tauti ja maksan veno-okklusiivinen tauti*.
Taulukko 6. Haittavaikutukset MYLOTARG-monoterapiatutkimuksissa*** ja valmisteen markkinoille tulon jälkeen
Elinjärjestelmä Esiintymistiheys Suositeltu termi | Kaikki vaikeusasteet % | Vaikeusaste 3/4 % |
| Infektiot | ||
| Hyvin yleiset | ||
| Infektio*a | 68,2 | 32,8 |
| Veri ja imukudos | ||
| Hyvin yleiset | ||
| Kuumeinen neutropenia | 19,1 | 11,6 |
| Trombosytopeniab | 48,4 | 48,0 |
| Neutropeniac | 30,3 | 29,2 |
| Anemiad | 27,1 | 24,2 |
| Leukopeniae | 26,7 | 26,7 |
| Yleiset | ||
| Pansytopeniaf | 5,0 | 4,3 |
| Lymfopeniag | 3,6 | 3,2 |
| Immuunijärjestelmä | ||
| Yleiset | ||
| Infuusioon liittyvä reaktioh | 7,6 | 3,6 |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleiset | ||
| Hyperglykemiai | 11,2 | 6,9 |
| Ruokahalun heikkeneminen | 27,1 | 6,1 |
| Yleiset | ||
| Tuumorilyysioireyhtymä** | 2,5 | 1,8 |
| Hermosto | ||
| Hyvin yleiset | ||
| Päänsärky | 38,3 | 12,3 |
| Sydän | ||
| Hyvin yleiset | ||
| Takykardiaj | 13,0 | 4,3 |
| Verisuonisto | ||
| Hyvin yleiset | ||
| Verenvuoto*k | 67,1 | 23,8 |
| Hypotensiol | 20,2 | 14,8 |
| Hypertensiom | 17,3 | 10,5 |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleiset | ||
| Hengenahdistusn | 27,4 | 12,6 |
| Tuntematon | ||
| Interstitiaalinen keuhkokuume* | ||
| Ruoansulatuselimistö | ||
| Hyvin yleiset | ||
| Oksentelu | 60,6 | 33,6 |
| Ripuli | 33,9 | 14,8 |
| Vatsakipuo | 33,2 | 7,2 |
| Pahoinvointi | 71,1 | 39,3 |
| Stomatiittip | 36,1 | 12,3 |
| Ummetus | 25,3 | 5,0 |
| Yleiset | ||
| Askites | 2,9 | 0,4 |
| Dyspepsia | 8,7 | 1,1 |
| Esofagiitti | 1,8 | 0,7 |
| Tuntematon | ||
| Neutropeeninen koliitti* | ||
| Maksa ja sappi | ||
| Hyvin yleiset | ||
| Transaminaasien nousuq | 24,5 | 18,8 |
| Hyperbilirubinemiar | 13,0 | 10,5 |
| Yleiset | ||
| VOD*s | 2,9 | 1,1 |
| Hepatomegalia | 2,5 | 0,7 |
| Keltaisuus | 2,2 | 1,1 |
| Maksan poikkeava toimintat | 2,5 | 1,4 |
| Suurentunut gammaglutamyylitransferaasi | 1,8 | 0,7 |
| Melko harvinaiset | ||
| Maksan vajaatoiminta*# | 0,4 | 0,4 |
| Budd-Chiarin oireyhtymä# | 0,4 | 0,4 |
| Iho ja ihonalainen kudos | ||
| Hyvin yleiset | ||
| Ihottumau | 19,9 | 5,8 |
| Yleiset | ||
| Punoitusv | 9,4 | 2,2 |
| Kutina | 5,4 | 0,4 |
| Munuaiset ja virtsatiet | ||
| Tuntematon | ||
| Vuotava kystiitti* | ||
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleiset | ||
| Kuumew | 82,7 | 52,3 |
| Edeemax | 21,3 | 3,2 |
| Väsymysy | 41,2 | 11,2 |
| Vilunväristykset | 67,9 | 17,3 |
| Yleiset | ||
| Monielinvaurio* | 2,2 | 0,7 |
| Tutkimukset | ||
| Hyvin yleiset | ||
| Suurentunut veren laktaattihydrogenaasi | 16,6 | 7,2 |
| Yleiset | ||
| Suurentunut veren alkalinen fosfataasi | 8,7 | 6,1 |
* Mukaan lukien kuolemaan johtanut.
** Mukaan lukien kuolemaan johtaneet haittavaikutukset valmisteen markkinoille tulon jälkeen.
*** MYLOTARG uusiutuneen AML:n hoidossa (9 mg/m2) (tutkimukset 201/202/203).
# Yksittäistapauksia.
a.Infektio kattaa seuraavat: sepsis ja bakteremia (25,6 %), sieni-infektio (10,5 %), alahengitystieinfektio (13,0 %), ylähengitystieinfektio (4,3 %), bakteeri-infektio (3,6 %), virusinfektio (24,2 %), maha-suolikanavan infektio (3,3 %), ihoinfektio (7,9 %) ja muut infektiot (19,5 %). Valmisteen markkinoille tulon jälkeen on raportoitu myös (esiintymistiheydeltään tuntemattomia) keuhkojen sieni-infektioita, mukaan lukien keuhkojen mykoosi ja Pneumocystis jirovecii ‑keuhkokuume*, ja bakteeri-infektioita, mukaan lukien Stenotrophomonas-infektio.
b. Trombosytopenia kattaa seuraavat raportoidut suositellut termit: pienentynyt verihiutalemäärä ja trombosytopenia*.
c. Neutropenia kattaa seuraavat raportoidut suositellut termit: neutropenia, granulosytopenia ja pienentynyt neutrofiilimäärä.
d. Anemia kattaa seuraavat raportoidut suositellut termit: anemia ja pienentynyt hemoglobiini.
e. Leukopenia kattaa seuraavat raportoidut suositellut termit: leukopenia ja pienentynyt valkosolumäärä.
f. Pansytopenia kattaa seuraavat raportoidut suositellut termit: pansytopenia ja luuytimen vajaatoiminta.
g. Lymfopenia kattaa seuraavat raportoidut suositellut termit: lymfopenia ja pienentynyt lymfosyyttimäärä.
h. Infuusioon liittyvä reaktio kattaa seuraavat raportoidut suositellut termit: infuusioon liittyvä reaktio, urtikaria, yliherkkyys, bronkospasmi, lääkeyliherkkyys ja urtikaria pistokohdassa#.
i. Hyperglykemia kattaa seuraavat raportoidut suositellut termit: hyperglykemia ja suurentunut verensokeri#.
j. Takykardia kattaa seuraavat raportoidut suositellut termit: takykardia, sinustakykardia, nopeutunut syketiheys# ja supraventrikulaarinen takykardia#.
k. Verenvuoto kattaa seuraavat: keskushermoston verenvuoto (5,1 %), maha-suolikanavan yläosan verenvuoto (21,3 %), maha-suolikanavan alaosan verenvuoto (15,2 %), ihonalainen verenvuoto (28,5 %), muu verenvuoto (32,9 %) ja nenäverenvuoto (28,5 %).
l. Hypotensio kattaa seuraavat raportoidut suositellut termit: hypotensio ja alentunut verenpaine.
m. Hypertensio kattaa seuraavat raportoidut suositellut termit: hypertensio ja suurentunut verenpaine.
n. Hengenahdistus kattaa seuraavat raportoidut suositellut termit: hengenahdistus ja rasitushengenahdistus.
o. Vatsakipu kattaa seuraavat raportoidut suositellut termit: vatsakipu, alavatsakipu, ylävatsakipu, epämiellyttävä tunne vatsassa ja vatsan aristus.
p. Stomatiitti kattaa seuraavat raportoidut suositellut termit: limakalvotulehdus, suunielun kipu, stomatiitti, suun haavautuminen, suukipu, suun limakalvojen rakkulointi, aftainen stomatiitti, kielen haavautuminen, kielikipu, suun limakalvojen punoitus, kielitulehdus# ja suunielun rakkulointi#.
q. Transaminaasiarvojen nousu kattaa seuraavat raportoidut suositellut termit: transaminaasiarvojen nousu, maksasoluvaurio, suurentunut alaniiniaminotransferaasi, suurentunut aspartaattiaminotransferaasi ja suurentuneet maksaentsyymit.
r. Hyperbilirubinemia kattaa seuraavat raportoidut suositellut termit: suurentunut veren bilirubiini ja hyperbilirubinemia.
s. Maksan VOD kattaa seuraavat raportoidut suositellut termit: veno-okklusiivinen tauti ja maksan veno-okklusiivinen tauti*#.
t. Maksan epänormaali toiminta kattaa seuraavat raportoidut suositellut termit: maksan toimintakokeiden epänormaalit tulokset ja maksan epänormaali toiminta.
u. Ihottuma kattaa seuraavat raportoidut suositellut termit: ihottuma, ihotulehdus#, allerginen ihottuma#, bulloosinen ihottuma, kosketusihottuma, hilseilevä ihotulehdus#, lääkkeen aiheuttama toistopunoittuma, allerginen kutina# ja punoittava ihottuma#, makulaarinen ihottuma#, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiseva ihottuma, vesikulaarinen ihottuma#.
v. Punoitus kattaa seuraavat raportoidut suositellut termit: katetrointikohdan punoitus, punoitus ja infuusiokohdan punoitus#.
w. Kuume kattaa seuraavat raportoidut suositellut termit: kuume, ruumiinlämmön nousu ja hypertermia.
x. Edeema kattaa seuraavat raportoidut suositellut termit: edeema, kasvojen edeema, perifeerinen edeema, kasvojen turvotus, yleistynyt turvotus ja periorbitaalinen edeema.
y. Väsymys kattaa seuraavat raportoidut suositellut termit: väsymys, voimattomuus, letargia ja huonovointisuus.
Valikoitujen haittavaikutusten kuvaus
Maksatoksisuus, mukaan lukien maksan veno-okklusiivinen tauti (VOD)
Yhdistelmähoitotutkimuksessa kerättiin tietoa VOD:sta ja maksaan liittyvien laboratorioarvojen poikkeamista. Maksatoksisuuteen lukeutuvien haittavaikutusten karakterisoinnissa on hyödynnetty myös monoterapiatutkimuksia.
Yhdistelmähoitotutkimuksessa (n = 131) VODia raportoitiin 6 potilaalla (4,6 %) hoidon aikana tai sen jälkeen; 2 (1,5 %) näistä reaktioista johti kuolemaan (ks. taulukko 5). Näistä VOD-reaktioista 5 (3,8 %) ilmeni 28 päivän kuluessa jostakin gemtutsumabi-otsogamisiiniannoksesta. Yksi VOD-tapahtuma ilmeni yli 28 päivän kuluttua viimeisestä gemtutsumabi-otsogamisiiniannoksesta; tämä tapahtuma ilmeni muutaman päivän kuluttua kantasolusiirron esihoidon aloittamisesta. Mediaaniaika viimeisestä gemtutsumabi-otsogamisiiniannoksesta VOD:n ilmenemiseen oli 9 päivää (vaihteluväli: 2–298 päivää). VOD:ia raportoitiin myös yhdistelmähoitotutkimuksen vertailuhaarassa 2 potilaalla, jotka saivat solunsalpaajahoidon jälkeen uusiutuneeseen AML:aan jatkohoitona MYLOTARG-valmistetta. Näillä molemmilla potilailla VOD ilmeni vasta yli 28 päivän kuluttua gemtutsumabi-otsogamisiinihoidon viimeisestä annoksesta. Toisella näistä potilaista VOD ilmeni 25 päivää hoitoa seuranneen kantasolusiirron jälkeen.
Monoterapiatutkimuksessa B1761031 VOD-tapahtumia ei raportoitu yhdelläkään potilaalla. Yhdellä potilaalla (2,0 %) oli kuitenkin kuolemaan johtanut kapillaarivuoto-oireyhtymä, jonka oireet olivat vastaavia VOD:n kanssa (askites ja hyperbilirubinemia). Vaikeusasteen 3 maksaan liittyviä haittatapahtumia olivat suurentunut gammaglutamyylitransferaasipitoisuus (4,0 %), suurentunut alaniiniaminotransferaasipitoisuus (2,0 %), suurentunut aspartaattiaminotransferaasipitoisuus (2,0 %), hypoalbuminemia (2,0 %) ja suurentunut transaminaasipitoisuus (2,0 %). Yhdelläkään potilaalla ei ollut vaikeusasteen 4 tai 5 maksatoksisuutta.
Mahdollisten riskitekijöiden analyysin mukaan VOD kehittyi 2,6 kertaa todennäköisemmin (95 %:n luottamusväli: 1,448–4,769) fraktioimatonta MYLOTARG-hoitoa monoterapiana saaneille aikuispotilaille ja potilaille, jotka olivat saaneet kantasolusiirron ennen gemtutsumabi-otsogamisiinialtistusta, verrattuna potilaisiin, jotka eivät olleet saaneet kantasolusiirtoa ennen gemtutsumabi-otsogamisiinihoitoa. VOD ilmeni 2,9 kertaa todennäköisemmin (95 %:n luottamusväli: 1,502–5,636) potilailla, jotka olivat saaneet kantasolusiirron gemtutsumabi-otsogamisiinihoidon jälkeen, verrattuna potilaisiin, jotka eivät olleet saaneet kantasolusiirtoa gemtutsumabi-otsogamisiinihoidon jälkeen. VOD kehittyi 8,7 kertaa todennäköisemmin (95 %:n luottamusväli: 1,879–39,862) potilaille, joilla oli keskivaikea/vaikea maksan vajaatoiminta lähtötilanteessa, verrattuna potilaisiin, joilla tällaista ei ollut lähtötilanteessa.
Potilaita tulee seurata maksatoksisuuden varalta kohdassa Varoitukset ja käyttöön liittyvät varotoimet suositellulla tavalla. Maksatoksisuuden merkkien ja oireiden hoito voi vaatia valmisteen annostelun keskeyttämisen tai MYLOTARG-hoidon lopettamisen (ks. kohta Annostus ja antotapa).
Myelosuppressio
Yhdistelmähoitotutkimukseen osallistuneilla potilailla oli aiemmin hoitamaton de novo AML, jota hoidettiin fraktioiduilla gemtutsumabi-otsogamisiiniannoksilla yhdessä solunsalpaajien kanssa. Vaikeusasteen 3/4 leukosyyttien vähenemistä havaittiin 131 potilaalla (100 %), neutrofiilien 124 potilaalla (96,1 %) ja verihiutaleiden 131 potilaalla (100 %).
Induktiovaiheessa verihiutalemäärä korjaantui 109 potilaalla (83,2 %) arvoon 50 x 109/l ja 99 potilaalla (75,6 %) arvoon 100 x 109/l. Verihiutalemäärä korjaantui 34 päivässä (mediaani) arvoon 50 x 109/l ja 35 päivässä (mediaani) arvoon 100 x 109/l. Ensimmäisen konsolidaatiovaiheen aikana verihiutalemäärä korjaantui 92 potilaalla (94,8 %) arvoon 50 x 109/l ja 71 potilaalla (73,2 %) arvoon 100 x 109/l. Verihiutalemäärä korjaantui 32 päivässä (mediaani) arvoon 50 x 109/l ja 35 päivässä (mediaani) arvoon 100 x 109/l. Toisen konsolidaatiovaiheen aikana verihiutalemäärä korjaantui 80 potilaalla (97,6 %) arvoon 50 x 109/l ja 70 potilaalla (85,4 %) arvoon 100 x 109/l. Verihiutalemäärä korjaantui 36,5 päivässä (mediaani) arvoon 50 x 109/l ja 43 päivässä (mediaani) arvoon 100 x 109/l.
Hoitoon vastanneista potilaista (CR, täydellinen remissio ja CRp, täydellinen remissio ilman täydellistä trombosyyttien korjautumista) 22 potilaalla (20,4 %) ilmeni trombosytopeniaa, jossa verihiutalemäärä pysyi arvossa < 50 x 109/l hoidon aloittamisen jälkeen 45 päivää. Pitkittynyttä trombosytopeniaa sairastavien potilaiden lukumäärä pysyi suhteellisen samana eri hoitovaiheissa (induktiovaiheessa 8 potilasta [7,4 %], ensimmäisessä konsolidaatiovaiheessa 8 potilasta [8,5 %] ja toisessa konsolidaatiovaiheessa 10 potilasta [13,2 %]).
Induktiovaiheessa neutrofiilimäärä korjaantui dokumentoidusti 121 potilaalla (92,4 %) B-Neut-arvoon 0,5 x 109/l ja 118 potilaalla (90,1 %) arvoon 1,0 x 109/l. Neutrofiilimäärän korjaantuminen edellä mainittuihin arvoihin kesti 25 päivää (mediaani). Hoidon ensimmäisessä konsolidaatiovaiheessa neutrofiilimäärä korjaantui 94 potilaalla (96,9 %) arvoon 0,5 x 109/l ja 91 potilaalla (94 %) arvoon 1,0 x 109/l. Neutrofiilimäärä korjaantui 21 päivässä (mediaani) arvoon 0,5 x 109/l ja 25 päivässä (mediaani) arvoon 1,0 x 109/l. Hoidon toisen konsolidaatiovaiheen aikana neutrofiilimäärä korjaantui 80 potilaalla (97,6 %) arvoon 0,5 x 109/l ja 79 potilaalla (96,3 %) arvoon 1,0 x 109/l. Neutrofiilimäärä korjaantui 22 päivässä (mediaani) arvoon 0,5 x 109/l ja 27 päivässä (mediaani) arvoon 1,0 x 109/l.
Yhdistelmähoitotutkimukseen osallistuneilla potilailla (n = 131) oli aiemmin hoitamaton de novo AML, jota hoidettiin fraktioiduilla gemtutsumabi-otsogamisiiniannoksilla yhdessä solunsalpaajien kanssa. Tässä tutkimuksessa 102 potilaalla (77,9 %) ilmeni vaikeita (vaikeusasteen ≥ 3) infektioita (syy-yhteydestä riippumatta). Yhden potilaan (0,8 %) raportoitiin kuolleen hoitoon liittyneen septisen sokin vuoksi. Kuolemaan johtanut vaikea infektio raportoitiin 2 potilaalla (1,53 %) MYLOTARG-ryhmässä ja 4 potilaalla (2,92 %) vertailuryhmässä.
Yhdistelmähoitotutkimuksessa (n = 131) raportoitiin kaikkien vaikeusasteiden verenvuotoreaktioita 118 potilaalla (90,1 %) ja vaikeusasteen 3/4 verenvuotoreaktioita 27 potilaalla (20,6 %). Yleisimmät vaikeusasteen 3 verenvuotoreaktiot olivat verioksennus (3,1 %), veriyskä (3,1 %) ja verivirtsaisuus (2,3 %). Vaikeusasteen 4 verenvuotoreaktioita raportoitiin 4 potilaalla (3,1 %) (maha-suolikanavan verenvuoto, verenvuoto ja 2 potilaalla keuhkorakkuloiden verenvuoto). Kuolemaan johtaneita verenvuotoreaktioita raportoitiin 3 potilaalla (2,3 %) (aivojen verenpurkauma, kallonsisäinen verenpurkauma ja kovakalvonalainen verenpurkauma).
Monoterapiatutkimuksessa B1761031 (n = 50) vaikeusasteen 3/4 infektioita raportoitiin 10 potilaalla (20 %). Yleisimmin (≥ 5,0 %) raportoituja vaikeusasteen 3/4 infektioita olivat sepsis ja keuhkokuume, joita kumpaakin todettiin 3 potilaalla (6,0 %). Kuudella (6) potilaalla (12,0 %) oli vaikeusasteen 5 infektio (sepsis 4 potilaalla [8,0 %], epätyypillinen keuhkokuume ja COVID-19-koronavirustautiin liittyvä keuhkokuume, joita kumpaakin 1 potilaalla [2,0 %]). Kaikkien vaikeusasteiden verenvuototapahtumia raportoitiin 16 potilaalla (32,0 %). Vaikeusasteen 3/4 verenvuototapahtumia esiintyi 2 potilaalla (4,0 %) (vaikeusasteen 3 mahaverenvuotoa ja vaikeusasteen 4 traumaattista kallonsisäistä verenvuotoa, joita kumpaakin 1 potilaalla). Kuolemaan johtaneita verenvuototapahtumia ei raportoitu.
Vaikean infektion, verenvuodon tai myelosuppression muiden vaikutusten, mukaan lukien vaikean neutropenian tai pitkittyneen trombosytopenian, hoito voi vaatia valmisteen annostelun siirtämistä tai MYLOTARG-hoidon pysyvää lopettamista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisyys
Kaikkien muiden terapeuttisten proteiinien tavoin tämä valmiste voi olla immunogeeninen.
MYLOTARG-valmisteen lääkevasta-aineita arvioitiin elektrokemiluminesenssimenetelmällä (ECL). Potilaille, joiden näytteistä todettiin lääkevasta-aineita, kehitettiin soluperusteinen määritys MYLOTARG-valmistetta neutraloivien vasta-aineiden mittaamiseksi.
Monoterapiatutkimuksessa B1761031, jossa oli mukana 50 uusiutunutta tai hoitoon vastaamatonta CD33-positiivista AML:aa sairastavaa hoitoa saanutta aikuispotilasta, lääkevasta-aineiden ilmaantuvuus oli 12,0 % (6/50) ja neutraloivien vasta-aineiden ilmaantuvuus oli 2,0 % (1/50). Lääkevasta-aineiden esiintymisellä ei ollut tilastollisesti merkitseviä eikä kliinisesti merkittäviä vaikutuksia hP67.6-kokonaisvasta-aineiden tai konjugoituneen kalikeamisiinin farmakokinetiikkaan. Yhdelläkään potilaalla ei esiintynyt anafylaksiaa, yliherkkyyttä eikä muita lääkevasta-aineisiin liittyviä kliinisiä jälkiseurauksia. Ei todettu näyttöä, että lääkevasta-aineiden esiintymisellä olisi suora yhteys johonkin mahdolliseen turvallisuusongelmaan.
MyeChild 01 ‑tutkimuksen annoshakuosassa, jossa oli mukana 54 äskettäin diagnosoitua AML:aa sairastavaa ≥ 12 kuukauden ikäistä hoitoa saanutta pediatrista potilasta, lääkevasta-aineiden kokonaisilmaantuvuus kaikissa kohorteissa oli 2 % (1/49). Potilailla, joilla oli lääkevasta-aineita, ei raportoitu erityisen kiinnostuksen kohteena olevia infuusioon liittyviä reaktioita.
Lääkevasta-aineiden toteaminen riippuu suuresti käytetyn testin herkkyydestä ja tarkkuudesta. Vasta-ainepositiivisuuden ilmaantuvuuteen testissä voivat vaikuttaa useat tekijät, mukaan lukien testimenetelmä, verenkierrossa olevat gemtutsumabi-otsogamisiinipitoisuudet, näytteen käsittely, näytteenoton ajoitus, samanaikaiset hoidot ja perussairaus. Näistä syistä vasta-aineiden ilmaantuvuuden vertailu gemtutsumabi-otsogamisiinin ja muiden valmisteiden välillä voi olla harhaanjohtavaa.
Pediatriset potilaat
Aiemmin hoitamaton AML
MYLOTARG-valmisteen turvallisuutta ja tehoa lasten ja alle 15-vuotiaiden nuorten aiemmin hoitamattoman AML:n hoidossa ei ole varmistettu (ks. kohta Annostus ja antotapa).
Päättyneessä satunnaistetussa vaiheen 3 pediatrisessa AAML0531-tutkimuksessa (ks. kohta Farmakodynamiikka) selvitettiin gemtutsumabi-otsogamisiinin yhdistämistä intensiiviseen ensilinjan hoitoon 0–29‑vuotiailla lapsilla (93,7 % potilaista < 18 vuotta) ja nuorilla aikuisilla (6,3 % potilaista) (yhteensä n = 1 063), joilla oli vastadiagnosoitu de novo AML. Gemtutsumabi-otsogamisiinin turvallisuusprofiilin todettiin olevan samankaltainen kuin tutkimuksissa, joissa aikuispotilaiden de novo AML:aa hoidettiin gemtutsumabi-otsogamisiinin ja intensiivisen solunsalpaajahoidon yhdistelmällä. Gemtutsumabi-otsogamisiinin optimaalista annosta pediatrisille potilaille ei ole kuitenkaan varmistettu, koska AAML0531-tutkimuksen toisen tehostamisjakson aikana annetun toisen gemtutsumabi-otsogamisiiniannoksen jälkeen neutrofiilimäärän korjaantuminen pitkittyi (> 59 päivää) useammalla gemtutsumabi-otsogamisiiniryhmän potilaalla kuin vertailuryhmän potilaalla (21,0 % vs 11,5 %), ja enemmän potilaita kuoli remission aikana (5,5 % vs 2,8 %).
Pediatrisen MyeChild 01 ‑tutkimuksen annoshakuosassa (ks. kohta Farmakodynamiikka) gemtutsumabi-otsogamisiinin ja induktiohoidon (sytarabiini joko mitoksantronin tai liposomaalisen daunorubisiinin kanssa) yhdistelmän turvallisuusprofiili 54:llä ≥ 12 kuukauden ikäisellä äskettäin diagnosoitua AML:ää sairastavalla lapsella oli samankaltainen kuin turvallisuusprofiili muissa tutkimuksissa, joissa gemtutsumabi-otsogamisiini yhdistettiin intensiiviseen solunsalpaajahoitoon de novo AML:ää sairastavilla aikuisilla ja pediatrisilla potilailla. Kaikkien vaikeusasteiden infektioiden esiintyvyys oli 57,4 %. Kaikissa kohorteissa yleisimmin raportoidut vaikeusasteen ≥ 3 haittavaikutukset olivat kuumeinen neutropenia (92,6 %), trombosytopenia (90,7 %), neutropenia (87,0 %) ja anemia (83,3 %). Kaikissa kohorteissa yleisimmin raportoidut vakavat haittavaikutukset olivat kuumeinen neutropenia (29,6 %) ja infektio (14,8 %). Vakavaa kuumeista neutropeniaa ilmeni 13,3 %:lla kohortin 1 potilaista, 15,0 %:lla kohortin 2 potilaista ja 57,9 %:lla kohortin 3 potilaista. Hoitojakson 1 tai 2 jälkeiseen päivään 45 mennessä 27,8 %:lla potilaista neutrofiilimäärä ei ollut korjautunut arvoon 1,0 × 109/l ja 11,1 %:lla potilaista verensiirrosta riippumaton trombosyyttimäärä ei ollut korjautunut arvoon 80 × 109/l dokumentoidun luuydinaplasian/hypoplasian vuoksi. VOD ilmeni kantasolusiirron jälkeen 13 %:lla potilaista. Kuolemaan johtanut VOD todettiin 1,9 %:lla potilaista.
Uusiutunut tai hoitoon vastaamaton AML
MYLOTARG-valmisteen turvallisuutta ja tehoa pediatristen potilaiden uusiutuneen tai hoitoon vastaamattoman AML:n hoidossa ei ole varmistettu (ks. kohdat Käyttöaiheet ja Annostus ja antotapa).
Taulukossa 7 on esitetty turvallisuutta koskevat tulokset MYLOTARG-hoitoa pediatrisilla potilailla arvioineiden tutkimusten systemaattisesta kirjallisuuskatsauksesta.
Taulukko 7. MYLOTARG-hoitoa saaneiden uusiutunutta tai hoitoon vastaamatonta AML:aa sairastavien pediatristen potilaiden turvallisuutta koskevat tulokset systemaattisesta kirjallisuuskatsauksesta
| Monoterapia | Yhdistelmähoitoa | |||||||||||
| Fraktioitub MYLOTARG-annostus | Fraktioimatonb MYLOTARG-annostus | Fraktioitub MYLOTARG-annostus | Fraktioimatonb MYLOTARG-annostus | |||||||||
| Tutki-musten lkm | N tutki-musta kohden (vaihte-luväli) | Esiin-tyvyysc (%) | Tutki-musten lkm | N tutki-musta kohden (vaihte-luväli) | Esiin-tyvyys (%) | Tutki-musten lkm | N tutki-musta kohden (vaihte-luväli) | Esiin-tyvyys (%) | Tutki-musten lkm | N tutki-musta kohden (vaihte-luväli) | Esiin-tyvyys (%) | |
| VOD | 1 | 6 | 0 | 10 | 5–30 | 6,8 | 2 | 3–17 | 0 | 5 | 5–84 | 4,4 |
| VOD kantasolusiirron jälkeen | Ei raportoitu | 5 | 4–14 | 19,1 | 2 | 3–8 | 0 | 2 | 12–28 | 14,7 | ||
| Kuolemad | 1 | 6 | 0 | 4 | 6–29 | 10,8 | Ei raportoitu | 3 | 5–45 | 6,5 | ||
| Infektio | 5 tutkimusta; N tutkimusta kohden (vaihteluväli) 12–30; 28,4 % | 4 tutkimusta; N tutkimusta kohden (vaihteluväli) 12–84; 42,2 % | ||||||||||
| Myelosuppressioe | Kaikissa tutkimuksissa lähes kaikilla potilailla (> 90 %) ilmeni myelosuppressio | |||||||||||
a. Kun MYLOTARG annettiin yhdistelmähoitona, sytarabiini oli tutkittavan yhdistelmän osana 8 tutkimuksessa 9:stä. b. Fraktioitu annostus viittaa MYLOTARG-annokseen 3 mg/m2 päivinä 1, 4, 7. Fraktioimaton annostus viittaa MYLOTARG-hoitoon (kokonaisannoksen vaihteluväli 1,8 mg/m2 – 9 mg/m2) 2 kertaa hoitosyklissä vähintään 14 päivän välein. c. Esiintyvyydet kaikissa tutkimuksissa arvioitiin käyttäen käänteistä varianssipainotusta kiinteillä vaikutuksilla. Osuudet muunnettiin Freeman–Tukeyn kaksinkertaisella arcsin-muunnoksella ennen tutkimusten yhdistämistä, ja arvioitu yhdistetty esiintyvyys muunnettiin takaisin käyttäen tutkimuksen otoskokojen harmonista keskiarvoa. d. 30 päivän kuluessa viimeisestä MYLOTARG-annoksesta. e. Analysoiduissa tapauksissa mediaaniaika veriarvojen korjaantumiselle (määritelty verihiutaleille arvoksi 20 x 109/l tai 50 x 109/l ja neutrofiileille arvoksi 0,5 x 109/l) oli verihiutaleilla 42–48 päivää ja neutrofiileilla 30–37 päivää. | ||||||||||||
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisessä käytössä ei ole ilmoitettu yhtään MYLOTARG-yliannostustapausta. Yli 9 mg/m2 ‑kerta-annoksia aikuisille ei ole tutkittu. MYLOTARG-yliannostusta tulee hoitaa yleisillä elintoimintoja tukevilla toimilla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX02
Vaikutusmekanismi
Gemtutsumabi-otsogamisiini on CD33-antigeeniin kohdentuva vasta-ainekonjugoitu solunsalpaaja (ADC). Gemtutsumabi on humanisoitu immunoglobuliiniluokan G alatyypin 4 (IgG4) vasta-aine, joka tunnistaa spesifisesti ihmisen CD33‑antigeenin. Valmisteen vasta-aineosa sitoutuu spesifisesti CD33-antigeeniin, joka on siaalihaposta riippuvainen adheesioproteiini. Sitä on myelooisten leukemiablastien ja myelomonosyyttisen solulinjan epäkypsien normaalien solujen pinnalla, mutta ei normaalien hematopoieettisten kantasolujen pinnalla. Valmisteen sisältämä pieni molekyyli, N‑asetyyli-gamma-kalikeamisiini, on sytotoksinen puolisynteettinen luonnontuote. N‑asetyyli-gamma-kalikeamisiini on yhdistetty kovalenttisesti vasta-aineeseen asetyyli-butyyli (4‑(4‑asetyylifenoksi) butaanihappo) ‑linkkerin välityksellä. Prekliiniset tiedot viittaavat siihen, että gemtutsumabi-otsogamisiinin aktiivisuus syöpää vastaan perustuu ADC:n vasta-aine-osan sitoutumiseen CD33‑antigeeniä ilmentäviin syöpäsoluihin, sitä seuraavaan ADC/CD33-kompleksin siirtymiseen solun sisään (internalisaatio) ja N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidin vapautumiseen solun sisällä linkkerin hydrolyyttisen pilkkoutumisen myötä. N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidin aktivoituminen aiheuttaa DNA:n kaksoisjuosteiden katkoksia, mistä seuraa solusyklin pysähtyminen ja ohjelmoitu solukuolema.
Oletettavasti prosentuaalisesti suuren osan CD33‑antigeenikohdista on saturoiduttava, jotta kalikeamisiinin kulkeutuminen leukemiablasteihin olisi maksimaalista. Useissa yhdellä vaikuttavalla aineella tehdyissä tutkimuksissa on mitattu CD33:n saturoitumista MYLOTARG-annoksen jälkeen uusiutuneen ja hoitoon vastaamattoman AML:n hoidossa. Kaikissa tutkimuksissa havaittiin lähes maksimaalinen perifeerisen CD33:n saturaatio MYLOTARG-annoksen jälkeen kaikilla vähintään 2 mg/m² ‑annoksilla. Tämä viittaa siihen, että jo pieni gemtutsumabi-otsogamisiiniannos riittää sitomaan kaikki saatavilla olevat CD33‑kohdat.
Kliininen teho ja turvallisuus
ALFA‑0701-tutkimus aiemmin hoitamatonta de novo AML:aa sairastavilla potilailla
MYLOTARG-valmisteen tehoa ja turvallisuutta arvioitiin satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa, jossa verrattiin MYLOTARG-valmisteella täydennettyä tavanomaista kahden solunsalpaajan daunorubisiinin ja sytarabiinin (DA) induktiohoitoa pelkkään DA‑induktiohoitoon. ALFA‑0701-tutkimukseen mukaan otettujen potilaiden oli oltava 50–70‑vuotiaita ja sairastettava aiemmin hoitamatonta de novo AML:aa. Tutkimuksesta suljettiin pois potilaat, joilla oli akuutti promyelosyyttileukemia (APL, AML3), myelodysplastisesta oireyhtymästä (MDS) johtuva AML tai sekundaarinen AML.
Ensisijainen päätetapahtuma oli tapahtumavapaa elinaika (event-free survival, EFS). Toissijaiset päätetapahtumat olivat täydellisen remission (CR) ja täydellisen remission ilman täydellistä trombosyyttien korjautumista (CRp) saavuttaneiden osuudet, elinaika ilman taudin uusiutumista (relapse-free survival, RFS), kokonaiselinaika (overall survival, OS) ja DA‑yhdistelmähoidon turvallisuus sekä MYLOTARG-valmisteen kanssa että ilman sitä.
Tähän tutkimukseen satunnaistettiin yhteensä 271 potilasta: 135 sai induktiohoitona 3+7 DA ja fraktioidusti 3 × 3 mg/m2 MYLOTARG-annosta, ja 136 sai pelkästään 3+7 DA (ks. kohta Annostus ja antotapa). Tutkimuksessa sai antaa satunnaistamisryhmästä riippumatta toisen induktiohoidon DA‑yhdistelmällä ilman MYLOTARG-valmistetta. Ryhmästä riippumatta niille potilaille, jotka eivät saaneet toista induktiohoitoa eivätkä saavuttaneet CR:ää induktion jälkeen, voitiin antaa idarubisiinista, sytarabiinista ja granulosyyttikasvutekijästä (G‑CSF) koostuva salvage-hoito.
Potilaille, jotka saavuttivat CR:n tai CRp:n, annettiin kaksi jaksoa konsolidaatiohoitoa tutkimuksen alussa tehdyn satunnaistamisen mukaan: daunorubisiinin ja sytarabiinin yhdistelmä joko MYLOTARG-valmisteen kanssa tai ilman sitä. Remission saavuttaneet potilaat soveltuivat myös allogeeniseen kantasolujen siirtoon. Viimeisen MYLOTARG-annoksen ja kantasolusiirron välillä suositeltiin pitämään vähintään 2 kuukauden tauko.
Kaikki potilaat mukaan lukien mediaani-ikä oli 62 vuotta (vaihteluväli 50–70 vuotta), ja useimpien potilaiden (87,8 %) toimintakykyä kuvaava ECOG (Eastern Cooperative Oncology Group) ‑pistemäärä oli lähtötilanteessa 0–1. Hoitoryhmät olivat lähtötilanteen ominaisuuksien suhteen tasapainossa sukupuolta lukuun ottamatta: MYLOTARG-ryhmässä oli enemmän miehiä (54,8 %) kuin pelkkää DA‑hoitoa saaneessa ryhmässä (44,1 %). Kaiken kaikkiaan 59,0 %:lla potilaista oli osoitetusti pienen riskin/keskiriskin tauti NCCN (National Comprehensive Cancer Network) ‑riskiluokituksen mukaan ja 65,3 %:lla pienen riskin/keskiriskin tauti ELN (European LeukaemiaNet) 2010 ‑riskiluokituksen mukaan. CD33-ilmentymä AML-blasteissa määritettiin virtaussytometrialla 194 potilaalta 271:stä (71,6 %). Virtaussytometriatulokset harmonisoitiin paikallisista laboratorioanalyyseistä. Muutamilla potilailla (13,7 %) CD33:n ilmentyminen oli heikkoa (alle 30 %:ssa blasteista).
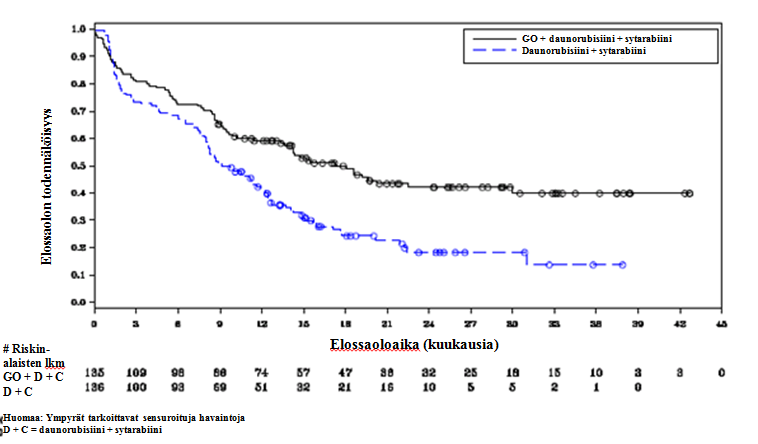
Tutkimuksen ensisijaisen tavoite saavutettiin eli tutkimuksessa osoitettiin, että fraktioitujen MYLOTARG-annosten (3 annosta × 3 mg/m2) lisääminen tavanomaiseen solunsalpaajainduktiohoitoon paransi aiemmin hoitamattomassa de novo AML:ssa EFS:ää tilastollisesti merkitsevästi ja kliinisesti merkittävästi. Mediaani EFS oli 17,3 kuukautta (95 %:n luottamusväli: 13,4–30,0) MYLOTARG-ryhmässä ja 9,5 kuukautta (95 %:n luottamusväli: 8,1–12,0) pelkkää DA‑hoitoa saaneessa ryhmässä; riskitiheyksien suhde (hazard ratio, HR) 0,562 (95 %:n luottamusväli: 0,415–0,762); 2‑suuntainen p‑arvo = 0,0002 log-rank-testillä. Taulukossa 8 on yhteenveto ALFA‑0701-tutkimuksen tehoa koskevista tuloksista, ja kuvassa 1 on Kaplan-Meierin kuvaaja EFS:stä.
Taulukko 8. ALFA‑0701-tutkimuksen tehoa koskevat tulokset (mITT-populaatio)
| MYLOTARG + daunorubisiini + sytarabiini | daunorubisiini + sytarabiini | |
| Tapahtumavapaa elinaika (tutkijan arvio) | n = 135 | n = 136 |
| Tapahtumia, n (%) | 73 (54,1) | 102 (75,0) |
| Mediaani EFS, kuukausia [95 %:n luottamusväli]a | 17,3 [13,4–30,0] | 9,5 [8,1–12,0] |
| 2 vuoden EFS:n todennäköisyys [95 %:n luottamusväli]b | 42,1 [32,9–51,0] | 18,2 [11,1–26,7] |
| 3 vuoden EFS:n todennäköisyys [95 %:n luottamusväli]b | 39,8 [30,2–49,3] | 13,6 [5,8–24,8] |
| Riskitiheyksien suhde [95 %:n luottamusväli]c | 0,562 [0,415–0,762] | |
| p‑arvod | 0,0002 | |
| Elinaika ilman taudin uusiutumista (tutkijan arvio) | n = 110 | n = 100 |
| Tapahtumia, n (%) | 49 (44,5) | 66 (66,0) |
| Mediaani RFS, kuukausia [95 %:n luottamusväli]a | 28,0 [16,3–NE] | 11,4 [10,0–14,4] |
| Riskitiheyksien suhde [95 %:n luottamusväli]c | 0,526 [0,362–0,764] | |
| p‑arvod | 0,0006 | |
| Kokonaiselinaika | n = 135 | n = 136 |
| Kuolemia, n (%) | 80 (59,3) | 88 (64,7) |
| Mediaani OS, kuukausia [95 %:n luottamusväli]a | 27,5 [21,4–45,6] | 21,8 [15,5–27,4] |
| Riskitiheyksien suhde [95 %:n luottamusväli]c | 0,807 [0,596–1,093] | |
| p‑arvod | 0,1646 | |
| Vasteiden määrä (tutkijan arvio) | n = 135 | n = 136 |
| Kokonaisvaste % [95 %:n luottamusväli]e | 81,5 [73,89–87,64] | 73,5 [65,28–80,72] |
| CR | 70,4 | 69,9 |
| CRp | 11,1 | 3,7 |
| Riskiero [95 %:n luottamusväli]f | 7,95 [-3,79–19,85] | |
| p‑arvog | 0,1457 |
EFS:n ensisijaisen määritelmän mukaan: tapahtumien päivämäärät (induktion epäonnistuminen, taudin uusiutuminen tai kuolema) tutkijan arvion mukaan.
mITT-populaatioon kuuluivat kaikki satunnaistetut potilaat, paitsi ennen hoidon aloittamista osallistumissuostumuksensa peruneet. Potilaat analysoitiin alussa osoitetun satunnaistamisryhmän mukaan.
Lyhenteet: CR = täydellinen remissio, CRp = täydellinen remissio ilman täydellistä trombosyyttien korjaantumista, EFS = tapahtumavapaa elinaika, mITT = modifioitu hoitoaikeen mukainen, n = lukumäärä, NE (not estimable) = ei arvioitavissa, OS = kokonaiselinaika, RFS = elinaika ilman taudin uusiutumista.
a. Mediaani Kaplan-Meierin-menetelmällä arvioituna, luottamusväli perustuu Brookmeyerin ja Crowleyn menetelmään, jossa käytetty log-log-muunnosta.
b. Arvioitu Kaplan-Meierin kuvaajan perusteella. Todennäköisyys (%) on laskettu rajatulomenetelmällä. Luottamusväli on laskettu elossaolon todennäköisyyden log-log-muunnoksesta normaalilla likiarviomenetelmällä ja Greenwoodin kaavalla.
c. Coxin verrannollisten riskitiheyksien mallin perusteella vs daunorubisiini + sytarabiini.
d. Log-rank-testin 2‑suuntainen p‑arvo.
e. Vaste = CR + CRp.
f. Kokonaisvasteen ero, luottamusväli perustuu Santnerin ja Snellin menetelmään.
g. Fisherin eksakti testin perusteella.
Kuva 1. Tapahtumavapaan elinajan (tutkijan arvio) Kaplan-Meierin kuvaaja ALFA‑0701-tutkimuksessa (mITT-populaatio)
Huomaa: Ympyrät tarkoittavat sensuroituja havaintoja D + C = daunorubisiini + sytarabiini # Riskin - alaisten lkm GO + D + C D + C Elossaolon todennäköisyys Elossaoloaika (kuukausia)
Lyhenteet: C = sytarabiini, D = daunorubisiini, GO = gemtutsumabi-otsogamisiini, mITT = modifioidun hoitoaikeen mukainen.
Käyttö sytogeneettisesti suuren riskin AML:ssa
ALFA‑0701-tutkimuksen alaryhmäanalyyseissa MYLOTARG-valmisteen lisääminen tavanomaiseen yhdistelmäsolunsalpaajahoitoon ei parantanut EFS:ää sytogenetiikan perusteella suuren riskin potilasalaryhmässä (HR 1,11; 95 %:n luottamusväli: 0,63–1,95). EFS ja OS analysoituna sytogeneettisen riskiluokituksen ja sytogeneettisen/molekulaarisen riskiluokituksen mukaan on esitetty seuraavissa taulukoissa 9 ja 10.
Taulukko 9. Tapahtumavapaa elinaika (tutkijan arvio) AML-riskiluokitusten mukaan ALFA-0701-tutkimuksessa (mITT-populaatio)
| MYLOTARG + daunorubisiini + sytarabiini | daunorubisiini + sytarabiini | |
| Sytogenetiikka (pieni riski/keskiriski), n | 94 | 95 |
| Tapahtumia, n (%) | 44 (46,8) | 68 (71,6) |
| Mediaani EFS, kuukausia [95 %:n luottamusväli]a | 22,5 [15,5–NE] | 11,6 [8,3–13,7] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 0,460 [0,313–0,676] | |
| p-arvoc | < 0,0001 | |
| Sytogenetiikka (suuri riski), n | 27 | 30 |
| Tapahtumia, n (%) | 23 (85,2) | 26 (86,7) |
| Mediaani EFS, kuukausia [95 %:n luottamusväli]a | 4,5 [1,1–7,4] | 2,8 [1,6–8,7] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 1,111 [0,633–1,949] | |
| p-arvoc | 0,7151 | |
| ELN (pieni riski/keskiriski), n | 86 | 91 |
| Tapahtumia, n (%) | 40 (46,5) | 63 (69,2) |
| Mediaani EFS, kuukausia [95 %:n luottamusväli]a | 22,5 [15,5–NE] | 12,2 [8,5–14,3] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 0,485 [0,325–0,724] | |
| p-arvoc | 0,0003 | |
| ELN (suuri riski), n | 37 | 36 |
| Tapahtumia, n (%) | 27 (73,0) | 32 (88,9) |
| Mediaani EFS, kuukausia [95 %:n luottamusväli]a | 7,4 [3,7–14,3] | 4,0 [1,7–8,6] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 0,720 [0,430–1,205] | |
| p-arvoc | 0,2091 | |
ALFA-0701-tutkimusta ei ollut suunniteltu siten, että sen avulla selvitettäisiin prospektiivisesti MYLOTARG-valmisteesta saatavaa hyötyä alaryhmissä; analyysit on esitetty vain kuvailevassa tarkoituksessa. EFS:n ensisijaisen määritelmän mukaan: tapahtumien päivämäärät (induktion epäonnistuminen, taudin uusiutuminen tai kuolema) tutkijan arvion mukaan. mITT-populaatioon kuuluivat kaikki satunnaistetut potilaat, paitsi ennen hoidon aloittamista osallistumissuostumuksensa peruneet. Potilaat analysoitiin alussa osoitetun satunnaistamisryhmän mukaan. Lyhenteet: AML = akuutti myelooinen leukemia, EFS = tapahtumavapaa elinaika, ELN = European LeukaemiaNet, mITT = modifioitu hoitoaikeen mukainen, n = lukumäärä, NE (not estimable) = ei arvioitavissa. a. Mediaani Kaplan-Meierin-menetelmällä arvioituna, luottamusväli perustuu Brookmeyerin ja Crowleyn menetelmään, jossa käytetty log-log-muunnosta. b. Coxin verrannollisten riskitiheyksien mallin perusteella vs daunorubisiini + sytarabiini. c. Log-rank-testin 2‑suuntainen p‑arvo. | ||
Taulukko 10. Kokonaiselinaika AML-riskiluokitusten mukaan ALFA-0701-tutkimuksessa (mITT-populaatio)
| MYLOTARG + daunorubisiini + sytarabiini | daunorubisiini + sytarabiini | |
| Sytogenetiikka (pieni riski/keskiriski), n | 94 | 95 |
| Kuolemia, n (%) | 51 (54,3) | 57 (60,0) |
| Mediaani OS, kuukausia [95 %:n luottamusväli]a | 38,6 [24,4–NE] | 26,0 [18,9–39,7] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 0,747 [0,511–1,091] | |
| p-arvoc | 0,1288 | |
| Sytogenetiikka (suuri riski), n | 27 | 30 |
| Kuolemia, n (%) | 24 (88,9) | 24 (80,0) |
| Mediaani OS, kuukausia [95 %:n luottamusväli]a | 12,0 [4,2–14,2] | 13,5 [9,4–27,3] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 1,553 [0,878–2,748] | |
| p-arvoc | 0,1267 | |
| ELN (pieni riski/keskiriski), n | 86 | 91 |
| Kuolemia, n (%) | 44 (51,2) | 53 (58,2) |
| Mediaani OS, kuukausia [95 %:n luottamusväli]a | 45,6 [25,5–NE] | 26,9 [19,3–46,5] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 0,730 [0,489–1,089] | |
| p-arvoc | 0,1216 | |
| ELN (suuri riski), n | 37 | 36 |
| Kuolemia, n (%) | 31 (83,8) | 29 (80,6) |
| Mediaani OS, kuukausia [95 %:n luottamusväli]a | 13,2 [7,0–18,5] | 13,5 [10,8–19,8] |
| Riskitiheyksien suhde [95 %:n luottamusväli]b | 1,124 [0,677–1,867] | |
| p-arvoc | 0,6487 | |
ALFA-0701-tutkimusta ei ollut suunniteltu siten, että sen avulla selvitettäisiin prospektiivisesti MYLOTARG-valmisteesta saatavaa hyötyä alaryhmissä; analyysit on esitetty vain kuvailevassa tarkoituksessa. mITT-populaatioon kuuluivat kaikki satunnaistetut potilaat, paitsi ennen hoidon aloittamista osallistumissuostumuksensa peruneet. Potilaat analysoitiin alussa osoitetun satunnaistamisryhmän mukaan. AML = akuutti myelooinen leukemia, ELN = European LeukaemiaNet, mITT = modifioitu hoitoaikeen mukainen, n = lukumäärä, NE (not estimable) = ei arvioitavissa, OS = kokonaiselinaika a. Mediaani Kaplan-Meierin-menetelmällä arvioituna, luottamusväli perustuu Brookmeyerin ja Crowleyn menetelmään, jossa käytetty log-log-muunnosta. b. Coxin verrannollisten riskitiheyksien mallin perusteella vs daunorubisiini + sytarabiini. c. Log-rank-testin 2‑suuntainen p‑arvo. | ||
Pediatriset potilaat
Aiemmin hoitamaton AML
COG AAML0531
Satunnaistetussa tutkimuksessa (COG AAML0531) arvioitiin tavanomaista solunsalpaajahoitoa sekä yksinään annettuna että yhdistelmänä MYLOTARG-valmisteen kanssa vastadiagnosoiduilla de novo AML lapsi- (93,7 % potilaista < 18 vuotta) ja nuorilla aikuispotilailla (6,3 % potilaista) (yhteensä n = 1 063). Keskimääräinen ikä oli 8,9 vuotta (vaihteluväli: 0–29 vuotta). Potilaat satunnaistettiin saamaan joko pelkästään tavanomaiset 5 solunsalpaajakuuria tai nämä kuurit ja 2 MYLOTARG-annosta (3 mg/m2/annos), siten että ensimmäinen annos annettiin ensimmäisen induktiohoidon aikana ja toinen annos toisen tehostamishoidon aikana. Tutkimus osoitti, että MYLOTARG-valmisteen lisääminen intensiiviseen solunsalpaajahoitoon paransi EFS:ää (3 vuotta: 50,6 % vs 44,0 %; HR 0,838; 95 %:n luottamusväli: 0,706–0,995; p = 0,0431) de novo AML:ssa uusiutumisriskin pienenemisen ansiosta. MYLOTARG-ryhmässä oli myös viitteitä kokonaiselinajan pidentymiseen, ei kuitenkaan tilastollisesti merkitsevästi (3 vuotta: 72,4 % vs 67,6 %; HR 0,904; 95 %:n luottamusväli: 0,721–1,133; p = 0,3799). Toisaalta huomattiin, että toksisuus (remission jälkeinen toksinen kuolleisuus) lisääntyi pienen riskin AML‑potilailla. Tämän katsottiin johtuvan pitkittyneestä neutropeniasta, joka ilmeni sen jälkeen, kun gemtutsumabi-otsogamisiinia annettiin toisen tehostamishoidon aikana (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Kaiken kaikkiaan 29 potilasta (5,5 %) MYLOTARG-ryhmässä ja 15 potilasta (2,8 %) vertailuryhmässä kuoli remission aikana. Gemtutsumabi-otsogamisiinin optimaalista annosta pediatrisille potilaille ei näin ollen ole varmistettu (ks. kohta Annostus ja antotapa).
MyeChild 01
Pediatrisen MyeChild 01 ‑tutkimuksen pääasiallisessa annoshakuosassa tutkittiin MYLOTARG-annosten 3 mg/m2 lukumäärää (enintään 3 annokseen saakka; kukin annos oli rajoitettu yhteen 5 mg:n injektiopulloon / annos), joka voidaan induktiohoidossa yhdistää turvallisesti sytarabiiniin ja mitoksantroniin tai sytarabiiniin ja liposomaaliseen daunorubisiiniin. Keskeisiä sisäänottokriteerejä olivat potilaan ikä ≥ 12 kuukautta – < 18 vuotta tutkimukseen mukaan otettaessa ja AML:n / suuren riskin MDS:n (luuytimessä > 10 % blasteja) / isoloituneen myeloblastooman diagnoosi ilman aiempaa hoitoa. Tutkimuksessa oli 3 kohorttia, joissa annettiin eri määrä MYLOTARG-infuusioita induktiovaiheen aikana: kohortissa 1 (n = 15) potilaat saivat MYLOTARG-kerta-annoksen (3 mg/m2) solunsalpaajahoidon induktiovaiheen hoitojakson 1 päivänä 4, kohortissa 2 (n = 20) potilaat saivat MYLOTARG-kerta-annoksen (3 mg/m2) solunsalpaajahoidon induktiovaiheen hoitojakson 1 päivinä 4 ja 7 ja kohortissa 3 (n = 19) potilaat saivat MYLOTARG-kerta-annoksen (3 mg/m2) solunsalpaajahoidon induktiovaiheen hoitojakson 1 päivinä 4, 7 ja 10. Mukaan otetuista 55 potilaasta 30 (54,5 %) oli 2 – < 12 vuotiaita, 32 (58,2 %) oli miespuolisia, ja kaikkien potilaiden iän mediaani oli 7,0 (vaihteluväli: 1–17) vuotta. Vaikka teho oli toissijainen päätetapahtuma, hoitoa saaneista potilaista (n = 54) paras kokonaisvaste (CR+CRi) todettiin 49 potilaalla (90,7 %; 95 %:n luottamusväli: 79,7–96,9 %) (80,0 % kohortissa 1, 95,0 % kohortissa 2 ja 94,7 % kohortissa 3). Hoitojakson 2 jälkeen 35 (71,4 %) potilaan raportoitiin olleen MRD-negatiivisia (58,3 % kohortissa 1, 78,9 % kohortissa 2 ja 72,2 % kohortissa 3). MyeChild 01 ‑tutkimus on edelleen käynnissä. Gemtutsumabi-otsogamisiinin optimaalista annosta pediatrisille potilaille ei ole vielä varmistettu (ks. kohta Annostus ja antotapa).
Uusiutunut tai hoitoon vastaamaton AML
MYLOTARG-hoidon arvioimiseksi pediatrisilla potilailla, jotka sairastivat uusiutunutta tai hoitoon vastaamatonta AML:aa, tehtiin systemaattinen kirjallisuuskatsaus näitä potilaita koskevista tutkimuksista. Katsaus käsitti 16 julkaistun artikkelin ja yhdysvaltalaisen tutkimuksen (US Expanded Access Study) 454 potilasta, jotka saivat MYLOTARG-hoitoa joko monoterapiana (kerta-annos tai fraktioitu annostus) tai yhdistelmähoitona (ks. kohta Haittavaikutukset). Tutkimusten mediaanikoko oli 15 potilasta ja vaihteluväli 5–105 potilasta. Potilaiden ikä vaihteli 0 vuodesta (minimi) 22,3 vuoteen (maksimi), ja mediaani-ikä oli 8,7 vuotta hoitoajankohtana.
Suurin osa tutkimuksista toteutettiin erityislupakäyttönä (compassionate use) (70,6 %). MYLOTARG annettiin monoterapiana 47,1 %:ssa tutkimuksista, osana yhdistelmähoitoa 23,5 %:ssa tutkimuksista ja kumpanakin hoitomuotona 29,4 %:ssa tutkimuksista. MYLOTARG-kokonaisannostus vaihteli annoksesta 1,8 mg/m2 annokseen 9 mg/m2. Kun MYLOTARG annettiin yhdistelmähoitona, sytarabiiniin perustuvaa hoito-ohjelmaa käytettiin 8 tutkimuksessa 9:stä. 23,5 %:ssa tutkimuksista suurin osa potilaista sai fraktioituja (3 mg/m2 päivinä 1, 4, 7) MYLOTARG-annoksia, kun taas 35,3 %:ssa tutkimuksista annetut annokset olivat suurempia kuin 3 mg/m2. Useimmissa tutkimuksissa MYLOTARG annettiin induktiohoitona (82,4 %).
MYLOTARG-monoterapiassa vasteiden määrä (CR/CRp/CRi; tutkimusten painotettu keskiarvo) oli fraktioitua annostusta käytettäessä 33,3 % (1 tutkimus) ja fraktioimatonta annostusta käytettäessä 24,3 % (9 tutkimusta). Yhdistelmähoidossa vasteiden määrä oli fraktioimatonta MYLOTARG-annostusta käytettäessä 49,0 % (3 tutkimusta) ja fraktioitua MYLOTARG-annostusta käytettäessä 38,8 % (2 tutkimusta).
MYLOTARG-hoidon tunnettuihin haittatapahtumiin (myelosuppressio, infektiot, kaikki VOD-tapahtumat, VOD kantasolusiirron jälkeen ja kuolema) liittyvät turvallisuutta koskevat tiedot (ks. kohta Haittavaikutukset ja taulukko 7) ovat peräisin kirjallisuudesta.
Analyysia rajoittavia tekijöitä ovat mm. joidenkin tutkimusten pieni otoskoko, tutkimusten heterogeenisuus ja puuttuvat vertailutiedot tässä asetelmassa.
Sydämen elektrofysiologia
MYLOTARG-valmisteen vaikutusta korjattuun QT-aikaan arvioitiin monoterapiatutkimuksessa B1761031 viidelläkymmenellä (50) aikuisella potilaalla, joilla oli uusiutunut tai hoitoon vastaamaton CD33-positiivinen AML. Plasman terapeuttisilla pitoisuuksilla QTcF-ajan suurin keskimääräinen muutos lähtötilanteesta oli 5,10 ms (90 %:n luottamusväli: 2,15–8,06 ms). Yhdelläkään potilaalla maksimi QTcF-aika ei pidentynyt lähtötilanteesta > 60 ms, eikä yhdenkään potilaan QTcF-aika ollut > 480 ms. Yhdessä (1) tapauksessa samalla potilaalla oli sekä eteisvärinää (vaikeusaste 3) että supraventrikulaarista takykardiaa (vaikeusaste 3). Sydämen johtumiseen liittyviä vaikeusasteen 4 tai vaikeusasteen 5 haittatapahtumia ei raportoitu.
Pitoisuus–QTc-aika-analyysin perusteella odotettavissa oleva QTcF-ajan muutos (mediaani) lähtötilanteesta oli hP67.6-kokonaisvasta-aineen osalta 0,842 ms (95 %:n luottamusväli: -1,93–3,51 ms) plasmassa havaitulla keskimääräisellä Cmax-arvolla. Konjugoitumattoman kalikeamisiinin osalta QTcF-ajan odotettavissa oleva muutos (mediaani) lähtötilanteesta oli 0,602 ms (95 %:n luottamusväli: ‑2,17–2,72 ms) plasmassa havaitulla likimääräisellä Cmax-arvolla, kun MYLOTARG-valmistetta annettiin suositellulla hoito-ohjelmalla.
Farmakokinetiikka
Gemtutsumabi-otsogamisiini on vasta-ainekonjugoitu solunsalpaaja. Se koostuu CD33-antigeeniin kohdentuvasta monoklonaalisesta vasta-aineesta (hP67.6), joka on yhdistetty kovalenttisesti sytotoksiseen aineeseen, N‑asetyyli-gamma-kalikeamisiiniin. Gemtutsumabi-otsogamisiinin farmakokinetiikka kuvataan määrittämällä vasta-aineen (hP67.6) farmakokineettiset ominaisuudet ja kalikeamisiinijohdosten konjugoitunut ja konjugoimaton määrä.
Kliiniset farmakokineettiset tiedot kerättiin monoterapiana annetun MYLOTARG-hoito-ohjelman jälkeen (3 mg/m2, enintään yksi 5 mg:n injektiopullo päivinä 1, 4, 7) aikuisista potilaista, joilla oli uusiutunut / hoitoon vastaamaton AML. Altistukset mitattiin useiden annosten jälkeen AUC336- ja Cmax-arvojen geometrisenä keskiarvona, ja mittaustulokset olivat: konjugoitunut kalikeamisiini 461 500 pg∙h/ml ja 11 740 pg/ml; hP67.6-kokonaisvasta-aine 26 820 ng∙h/ml ja 585,6 ng/ml. Konjugoitumatonta kalikeamisiinia koskevia farmakokineettisiä tietoja ei esitetä, koska sen pysyvyyteen plasmassa liittyy epävarmuutta.
Jakautuminen
N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi sitoutuu in vitro noin 97‑prosenttisesti ihmisen plasman proteiineihin. N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi on in vitro P‑glykoproteiinin (P‑gp) substraatti. hP67.6‑vasta-aineen kokonaisjakautumistilavuuden (V1:n [13,0 l] ja V2:n [6,91 l] summa) todettiin olevan potilailla noin 20 litraa.
Biotransformaatio
Gemtutsumabi-otsogamisiinin oletetaan metaboloituvan ensisijaisesti N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidin hydrolyyttisen vapautumisen kautta. In vitro ‑tutkimusten mukaan N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi metaboloituu suuressa määrin, ensisijaisesti disulfidiosan ei-entsymaattisen pelkistymisen kautta. Näin saatavien metaboliittien aktiivisuuden (sytotoksisuuden) odotetaan olevan merkittävästi heikentynyt.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Muiden lääkevalmisteiden vaikutus gemtutsumabi-otsogamisiiniin
N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi metaboloituu in vitro ensisijaisesti ei-entsymaattisen pelkistymisen kautta. Siksi gemtutsumabi-otsogamisiinin anto samanaikaisesti sytokromi P450:n (CYP) tai lääkeaineita metaboloivien uridiinidifosfaattiglukuronosyylitransferaasi (UGT) ‑entsyymien estäjien tai induktorien kanssa ei todennäköisesti muuta altistusta N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidille.
Populaatiofarmakokineettisten analyysien perusteella gemtutsumabi-otsogamisiinin, hydroksiurean, daunorubisiinin ja sytarabiinin yhdistelmän ei ennusteta muuttavan hP67.6:n tai konjugoimattoman kalikeamisiinin farmakokinetiikkaa kliinisesti merkittävästi.
Gemtutsumabi-otsogamisiinin vaikutus muihin lääkevalmisteisiin
Vaikutus CYP-substraatteihin
Kliinisesti merkittävinä pitoisuuksina N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi ja gemtutsumabi-otsogamisiini estivät vain vähän seuraavien CYP-entsyymien aktiivisuutta in vitro: CYP1A2, CYP2A6 (tutkittu vain gemtutsumabi-otsogamisiinilla), CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A4/5.Kliinisesti merkittävinä pitoisuuksina N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi ja gemtutsumabi-otsogamisiini indusoivat vain vähän seuraavien CYP-entsyymien aktiivisuutta in vitro: CYP1A2, CYP2B6 ja CYP3A4.
Vaikutus UGT‑substraatteihin
Kliinisesti merkittävinä pitoisuuksina N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi esti vain vähän seuraavien UGT‑substraattien aktiivisuutta in vitro: UGT1A1, UGT1A4, UGT1A6, UGT1A9 ja UGT2B7.
Vaikutus lääkkeiden kuljetusproteiinien substraatteihin
Kliinisesti merkittävinä pitoisuuksina N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi esti vain vähän seuraavien kuljetusproteiinien aktiivisuutta in vitro: P‑gp, rintasyövän resistenssiproteiini (BCRP), sappisuolapumppu (BSEP), monilääkeresistenssiproteiini MRP2, monilääke- ja toksisten aineiden ekstruusioproteiini MATE1 ja MATE2K, orgaanisten anionien kuljettajat OAT1 ja OAT3, orgaanisten kationien kuljettaja OCT1 ja OCT2 ja orgaanisten anionien kuljettajapolypeptidit OATP1B1 ja OATP1B3.
Vaikutus samanaikaisesti annettuihin solunsalpaajiin
Populaatiofarmakokineettisten analyysien perusteella gemtutsumabi-otsogamisiinin yhdistämisen daunorubisiiniin ja sytarabiiniin ei ennusteta muuttavan näiden aineiden farmakokinetiikkaa kliinisesti merkittävästi.
Eliminaatio
Gemtutsumabi-otsogamisiinin farmakokinetiikkaa kuvasti hyvin kaksitilamalli, jossa on lineaariset ja aikariippuvaiset puhdistumakomponentit. MYLOTARG-valmistetta monoterapiana (3 mg/m2, enintään yksi 5 mg:n injektiopullo päivinä 1, 4, 7) saaneilla 50 potilaalla, joilla oli uusiutunut tai hoitoon vastaamaton AML, hP67.6-kokonaisvasta-aineen puhdistuma oli 0,288 l/h ja terminaalisen eliminaation puoliintumisajaksi (t½) arvioitiin 96,6 h.
Farmakokinetiikka erityisryhmissä
Ikä, etninen tausta ja sukupuoli
Populaatiofarmakokineettisen analyysin perusteella ikä, etninen tausta ja sukupuoli eivät vaikuttaneet merkittävästi gemtutsumabi-otsogamisiinin jakautumiseen elimistössä.
Maksan vajaatoiminta
Gemtutsumabi-otsogamisiinin käyttöä maksan vajaatoimintaa sairastavilla potilailla ei ole selvitetty varsinaisissa farmakokineettisissä tutkimuksissa.
Populaatiofarmakokineettisen analyysin perusteella lievän maksan vajaatoiminnan (National Cancer Institute Organ Dysfunction Working Group [NCI ODWG] ‑työryhmän määritelmän mukaan) ei odoteta vaikuttavan gemtutsumabi-otsogamisiinin (hP67.6‑vasta-aineen ja konjugoimattoman kalikeamisiinin) puhdistumaan. Analyysissa oli mukana 405 potilasta seuraavista maksan vajaatoiminnan NCI ODWG ‑luokista: lievä (B1, n = 58 ja B2, n = 19), keskivaikea (C, n = 6) tai normaali (n = 322) (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Gemtutsumabi-otsogamisiinin käyttöä munuaisten vajaatoimintaa sairastavilla potilailla ei ole selvitetty varsinaisissa farmakokineettisissä tutkimuksissa.
Populaatiofarmakokineettisessä analyysissa, jossa oli mukana 406 potilasta, gemtutsumabi-otsogamisiinin puhdistuma oli lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma (CrCl) 60–89 ml/min, n = 149) tai keskivaikeaa munuaisten vajaatoimintaa (CrCl 30–59 ml/min, n = 47) sairastavilla samaa luokkaa kuin potilailla, joilla oli normaali munuaisten toiminta (CrCl ≥ 90 ml/min, n = 209). Gemtutsumabi-otsogamisiinin farmakokinetiikkaa ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Kliiniset farmakokineettiset tiedot kerättiin induktiovaiheen MYLOTARG-hoito-ohjelman (3 mg/m2, enintään yksi 5 mg:n injektiopullo päivinä 4, 7 ja 10) jälkeen ≥ 12 kuukauden ikäisistä pediatrisista potilaista, joilla oli äskettäin diagnosoitu AML. Altistukset mitattiin kolmannen annoksen jälkeen AUCtau- ja Cmax-arvojen geometrisinä keskiarvoina, ja mittaustulokset olivat seuraavat: konjugoitunut kalikeamisiini 777 300 pg.h/ml ja 24 340 pg/ml; hP67.6-kokonaisvasta-aine 46 500 ng.h/ml ja 1 336 ng/ml.
Prekliiniset tiedot turvallisuudesta
Toistuvan annon toksisuus
Toksisuudet kohdistuivat pääasiassa maksaan, luuytimeen, imukudoselimiin, veriarvoihin (punasolumassan ja valkosolujen, lähinnä lymfosyyttien, määrän pieneneminen), munuaisiin, silmiin ja urosten ja naaraiden lisääntymiselimiin. Vaikutukset rotan maksaan, munuaisiin ja urosten lisääntymiselimiin sekä apinan imukudoksiin (altistuksella, joka oli AUC168‑arvon perusteella rotalla noin 18 kertaa ja apinalla 36 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen) olivat korjaantumattomia. Haitalliset vaikutukset naarasapinoiden lisääntymiselimiin ja silmiin todettiin 12 viikon tutkimuksessa (lisääntymiselimiin altistuksella, joka oli AUC168‑arvon perusteella noin 193 kertaa ja silmiin altistuksella, joka oli AUC168‑arvon perusteella noin 322 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen). Eläimillä todettujen korjaantumattomien löydösten merkitys ihmiselle on epäselvä. Eläimillä ei ole havaittu hermostovaikutuksia MYLOTARG-annostelun jälkeen. Rotalla havaittiin hermostomuutoksia muiden vasta-aine-kalikeamisiinikonjugaattien käytön yhteydessä.
Genotoksisuus
Gemtutsumabi-otsogamisiini on todettu klastogeeniseksi. Tämä on johdonmukaista, koska kalikeamisiini ja muut syövän hoidossa käytettävät enediyni-antibiootit aiheuttavat tunnetusti DNA-katkoksia. N‑asetyyli-gamma-kalikeamisiinidimetyylihydratsidi (vapautunut sytotoksiini) on todettu mutageeniseksi ja klastogeeniseksi.
Karsinogeenisuus
Gemtutsumabi-otsogamisiinilla ei ole tehty varsinaisia karsinogeenisuustutkimuksia. Rotille kehittyi toksisuustutkimuksissa preneoplastisia muutoksia (minimaalinen tai vähäinen soikeiden solujen hyperplasia) maksassa altistuksella, joka oli AUC168‑arvon perusteella noin 54 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen. Apinoilla ei havaittu preneoplastisia eikä neoplastisia muutoksia altistuksella, joka oli AUC168‑arvon perusteella enintään noin 115 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen. Näiden eläimillä todettujen löydösten merkitys ihmiselle on epäselvä.
Lisääntymistoksisuus
Naarasrottien hedelmällisyyttä koskeneessa tutkimuksessa havaittiin lukumääräisesti hieman vähemmän keltarauhasia ja alkiokuolleisuuden lisääntymistä emotoksisuuden yhteydessä (altistus AUC168‑arvon perusteella noin 9,7 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen). Vaikutuksia naarasapinoiden lisääntymiselimiin havaittiin 12 viikon tutkimuksessa (munasarjojen, munanjohtimen, kohdun ja kohdunkaulan surkastuminen; noin 193 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen).
Urosten hedelmällisyyttä koskeneessa tutkimuksessa ilmeni mm. seuraavia vaikutuksia urosten lisääntymiseen: alkusiemensolujen (spermatogonioiden), varhaissiemensolujen (spermatosyyttien), kivesten esisiemensolujen (spermatidien) ja lisäkivesten siemennesteen väheneminen, spermatidien tuman vakuolisaatio ja/tai jättisolujen ilmaantuminen. Muita löydöksiä olivat vaikutukset kiveksiin, lisäkiveksiin ja rintarauhaseen sekä hedelmällisyyteen. Kun urosrottien annettiin jälleen paritella 9 viikon lääketauon jälkeen, vaikutukset siemennesteeseen ja hedelmällisyyteen olivat huonommat, mutta kivesten alkusiemensolujen ja varhaissiemensolujen määrän pieneneminen korjaantui osittain. Vaikutukset urosrotan lisääntymiselimiin olivat joko osittain korjaantuvia tai korjaantumattomia (ks. kohta Raskaus ja imetys). Vaikutuksia urosapinoiden lisääntymiselimiin (kivekset, lisäkivekset, rakkularauhaset) havaittiin altistuksella, joka oli noin 66 kertaa ihmisen kliininen altistus ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen.
Alkio‑/sikiötoksisuutta koskeneen tutkimuksen havaintojen mukaan sikiöpaino pieneni, sikiöiden kylkiluiden aaltoilun ilmaantuvuus suureni ja sikiöiden luiden luutumisen ilmaantuvuus väheni. Alkiokuolleisuuden suurenemiseen ja sikiöiden rakenteellisiin anomalioihin kuului seuraavia: varpaiden epämuodostumat, aortankaaren puuttuminen, eturaajojen putkiluiden anomaliat, lapaluun epämuodostuma, nikamansolmun puuttuminen ja rintalastan yhteensulautuma. Emotoksisuuden yhteydessä havaittiin myös alkiokuolleisuuden suurenemista. Pienin annos, jolla ilmeni alkioihin ja sikiöihin kohdistuneita vaikutuksia, vastasi AUC168‑arvon perusteella 9,7‑kertaista ihmisen kliinistä altistusta ihmiselle annetun kolmannen 3 mg/m2 ‑annoksen jälkeen (ks. kohta Raskaus ja imetys).
Farmaseuttiset tiedot
Apuaineet
Dekstraani 40
Sakkaroosi
Natriumkloridi
Natriumdivetyfosfaattimonohydraatti
Vedetön dinatriumvetyfosfaatti
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
5 vuotta
Käyttökuntoon saatettu ja laimennettu liuos
Suojaa käyttökuntoon saatetut ja laimennetut MYLOTARG-liuokset valolta. Liuokset on käytettävä heti. Käyttökuntoon saatettu tai laimennettu liuos ei saa jäätyä.
Jos valmistetta ei voida käyttää heti:
- Käyttökuntoon saatettua liuosta voidaan säilyttää alkuperäisessä injektiopullossa enintään 16 tuntia jääkaapissa (2–8 °C) tai enintään 3 tuntia huoneenlämmössä (alle 30 °C).
- Laimennettua liuosta voidaan säilyttää enintään 18 tuntia jääkaapissa (2–8 °C) ja enintään 6 tuntia huoneenlämmössä (alle 30 °C). Sallittuun aikaan huoneenlämmössä (alle 30 °C) sisältyy laimennetun liuoksen valmistamiseen tarvittava aika, lämpeneminen tarvittaessa ja anto potilaalle. Enimmäisaika laimennetun liuoksen valmistamisesta sen antoon ei saa ylittää 24 tuntia.
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Säilytä injektiopullo alkuperäiskotelossa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
MYLOTARG kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
5 mg (L:ei) 1 kpl (8380,98 €)
PF-selosteen tieto
Meripihkanvärisestä tyypin 1 lasista valmistettu injektiopullo, jossa on butyylikumitulppa, tiivistekaulus ja repäisykorkki. Injektiopullo sisältää 5 mg gemtutsumabi-otsogamisiinia.
Yksi kotelo sisältää 1 injektiopullon.
Valmisteen kuvaus:
Valkoinen tai lähes valkoinen kakku tai jauhe.
Käyttö- ja käsittelyohjeet
Käytä asianmukaista aseptista tekniikkaa valmisteen käyttökuntoon saattamisessa ja laimentamisessa. Koska MYLOTARG on valolle herkkää, se on suojattava ultraviolettisäteilyltä käyttökuntoon saattamisen, laimentamisen ja annon aikana.
Käyttökuntoon saattaminen
- Laske tarvittava MYLOTARG-annos (mg).
- Anna injektiopullon lämmetä ennen käyttökuntoon saattamista huoneenlämpöiseksi (alle 30 °C) noin 5 minuutin ajan. Saata jokainen 5 mg:n injektiopullo käyttökuntoon 5 ml:lla injektionesteisiin käytettävää vettä, jotta saadaan 1 mg/ml gemtutsumabi-otsogamisiiniliuosta kerta-antoon.
- Pyörittele injektiopulloa varovasti liukenemisen edistämiseksi. Älä ravista.
- Tarkasta käyttökuntoon saatettu liuos hiukkasten ja värjäytymisen varalta. Käyttökuntoon saatetussa liuoksessa voi olla pieniä valkoisia tai lähes valkoisia, läpikuultamattomia tai läpikuultavia ja epämääräisen muotoisia tai säiemäisiä hiukkasia.
- MYLOTARG ei sisällä bakteriostaattisia säilytysaineita.
- Jos käyttökuntoon saatettua liuosta ei voida käyttää heti, sitä voidaan säilyttää alkuperäisessä injektiopullossa enintään 16 tuntia jääkaapissa (2–8 °C) tai enintään 3 tuntia huoneenlämmössä (alle 30 °C). Herkkä valolle. Ei saa jäätyä.
Laimentaminen
- Laske, kuinka paljon käyttökuntoon saatettua liuosta tarvitaan, jotta saadaan potilaan kehon pinta-alaan perustuva asianmukainen annos. Vedä laskemasi liuosmäärä injektiopullosta ruiskuun. MYLOTARG-injektiopullot sisältävät 5 mg lääkevalmistetta ilman ylitäyttöä. Kun valmiste on ohjeen mukaisesti saatettu käyttökuntoon pitoisuuteen 1 mg/ml, injektiopullosta saatava määrä on 4,5 mg (4,5 ml). Herkkä valolle. Hävitä injektiopulloon mahdollisesti käyttämättä jäänyt käyttökuntoon saatettu liuos.
-
Annokset on sekoitettava pitoisuuteen 0,075–0,234 mg/ml seuraavien ohjeiden mukaisesti:
- Alle 3,9 mg:n annokset tulee valmistella ruiskulla annostelua varten. Lisää käyttökuntoon saatettu MYLOTARG-liuos 9 mg/ml (0,9 %) natriumkloridi-injektioliuosta sisältävään ruiskuun siten, että lopulliseksi pitoisuudeksi saadaan 0,075–0,234 mg/ml. Herkkä valolle.
- 3,9 mg:n annos tai tätä suuremmat annokset laimennetaan ruiskussa tai infuusiopussissa sopivaan tilavuuteen 9 mg/ml (0,9 %) natriumkloridi-injektioliuoksella siten, että lopulliseksi pitoisuudeksi saadaan 0,075–0,234 mg/ml. Herkkä valolle.
- Kääntele infuusiosäiliötä varovasti ylösalaisin laimennetun liuoksen sekoittamiseksi. Älä ravista.
- Kun laimennus 9 mg/ml (0,9 %) natriumkloridi-injektioliuoksella on tehty, MYLOTARG-liuos tulee infusoida heti. Jos laimennettua valmistetta ei käytetä heti, sitä voidaan säilyttää enintään 18 tuntia jääkaapissa (2–8 °C) ja enintään 6 tuntia huoneenlämmössä (alle 30 °C). Sallittuun aikaan huoneenlämmössä (alle 30 °C) sisältyy laimennetun liuoksen valmistamiseen tarvittava aika, lämpeneminen tarvittaessa ja anto potilaalle. Enimmäisaika laimennetun liuoksen valmistamisesta sen antoon ei saa ylittää 24 tuntia. Herkkä valolle. Ei saa jäätyä.
- Infuusiosäiliöksi suositellaan polyvinyylikloridista (PVC) (DEHP:iä sisältävä), etyleenivinyyliasetaatista (EVA) tai polyolefiinista (polypropeeni ja/tai polyeteeni) valmistettua säiliötä.
Anto
- Laimennettu liuos on suodatettava. MYLOTARG-infuusiossa on käytettävä niukasti proteiinia sitovaa polyeetterisulfonista (PES) valmistettua 0,2 mikrometrin kiinteää (in-line) suodatinta.
- Ruiskulla annettavissa annoksissa on käytettävä sisähalkaisijaltaan hyvin pientä (microbore) infuusioletkua, jossa on niukasti proteiinia sitova polyeetterisulfonista (PES) valmistettu 0,2 mikrometrin kiinteä (in-line) suodatin.
- Laskimoinfuusioon tarkoitettu pussi tai ruiskut on suojattava infuusion aikana valolta erityisellä suojapeitteellä (myös UV-suoja). Infuusioletkua ei tarvitse suojata valolta.
- Anna laimennettu liuos 2 tunnin infuusiona. Infuusion anto on saatava päätökseen ennen kuin laimennetulle liuokselle sallittu 6 tunnin säilytysaika huoneenlämmössä (alle 30 °C) kuluu umpeen.
- Infuusioletkuiksi suositellaan PVC:stä (joko DEHP:iä sisältävä tai sisältämätön), polyuretaanista tai polyeteenistä valmistettuja letkuja.
Älä sekoita tai infusoi MYLOTARG-valmistetta muiden lääkevalmisteiden kanssa.
Ks. myös kohdan Kestoaika tiedot laimentamisesta, säilytyksestä ja infuusiosta.
Hävittäminen
Syöpälääkkeitä koskevia vaarallisen jätteen hävittämismenetelmiä on noudatettava.
Korvattavuus
MYLOTARG kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
5 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX02
Valmisteyhteenvedon muuttamispäivämäärä
12.10.2023
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com