
ZALTRAP infuusiokonsentraatti, liuosta varten 25 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusiokonsentraattia liuosta varten sisältää 25 mg afliberseptia*.
Yksi 4 ml injektiopullo konsentraattia sisältää 100 mg afliberseptia.
Yksi 8 ml injektiopullo konsentraattia sisältää 200 mg afliberseptia.
* Aflibersepti valmistetaan kiinanhamsterin munasarjasolulinjassa (CHO-K1, nisäkäsperäinen ekspressiojärjestelmä) yhdistelmä-DNA-tekniikalla.
Apuaine(et), joiden vaikutus tunnetaan:
Yksi 4 ml injektiopullo sisältää 0,484 mmol eli 11,118 mg natriumia, ja yksi 8 ml injektiopullo sisältää 0,967 mmol eli 22,236 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Zaltrap on tarkoitettu käytettäväksi yhdessä irinotekaania/5-fluorourasiilia/foliinihappoa sisältävän solunsalpaajahoidon (FOLFIRI) kanssa levinneen kolorektaalisyövän hoitoon aikuisille, joiden tauti on resistentti oksaliplatiinia sisältävälle hoidolle tai on edennyt sen jälkeen.
Ehto
Valmiste annetaan syövän lääkehoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Zaltrap annetaan syövän lääkehoitoon perehtyneen lääkärin valvonnassa.
Annostus
Zaltrap-valmisteen suositusannos on 4 mg/kg infuusiona laskimoon 1 tunnin aikana ennen FOLFIRI-hoitoa. Tämä muodostaa yhden hoitojakson.
Käytettävään FOLFIRI-hoitoon kuuluu irinotekaania (180 mg/m2 infuusiona laskimoon 90 minuutin kuluessa) ja foliinihappoa (d- ja l-isomeerien raseeminen seos; 400 mg/m2 infuusiona laskimoon 2 tunnin kuluessa), jotka annetaan yhtä aikaa päivänä 1 Y-liittimen kautta. Niiden jälkeen annetaan 5-fluorourasiilia (5-FU) 400 mg/m2 boluksena laskimoon, minkä jälkeen annetaan 2400 mg/m2 5-FU-annos jatkuvana laskimoinfuusiona 46 tunnin kuluessa.
Hoitojaksot toistetaan 2 viikon välein.
Zaltrap-hoitoa jatketaan, kunnes tauti etenee tai kehittyy sietämätöntä toksisuutta.
Annosmuutokset
Zaltrap-hoito on lopetettava seuraavissa tilanteissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet):
- Vaikea verenvuoto
- Ruoansulatuskanavan perforaatio
- Fistelimuodostus
- Hypertensio, jota ei saada asianmukaisesti hallintaan verenpainelääkityksellä, tai hypertensiivinen kriisi tai hypertensiivinen enkefalopatia
- Sydämen vajaatoiminta ja pienentynyt ejektiofraktio
- Tromboemboliset valtimotapahtumat
- Asteen 4 tromboemboliset laskimotapahtumat (myös keuhkoemboliat)
- Nefroottinen oireyhtymä tai tromboottinen mikroangiopatia
- Vaikeat yliherkkyysreaktiot (mm. bronkospasmi, hengenahdistus, angioedeema ja anafylaksia) (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet)
- Haavan huono paraneminen, joka edellyttää lääketieteellistä hoitoa
- Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES; tunnetaan myös nimellä reversiibeli posteriorinen leukoenkefalopatiaoireyhtymä, RPLS)
Zaltrap-hoito on tauotettava vähintään 4 viikon ajaksi ennen elektiivistä leikkausta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
| Zaltrap-/FOLFIRI-hoidon siirtäminen tai annosmuutokset | |
| Neutropenia tai trombosytopenia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) | Zaltrap-/FOLFIRI-hoidon antoa siirretään, kunnes neutrofiiliarvot ovat ≥ 1,5 x 109/l tai trombosyyttiarvot ≥ 75 x 109/l. |
| Kuumeinen neutropenia tai neutropeeninen sepsis | Irinotekaaniannosta pienennetään myöhemmissä hoitojaksoissa 15–20 %. Jos tila uusiutuu, myös 5-FU-annoksia (bolus ja infuusio) pienennetään myöhemmissä hoitojaksoissa 20 %. Jos tila uusiutuu, kun irinotekaani- ja 5-FU-annoksia on pienennetty, voidaan harkita Zaltrap-annoksen pienentämistä tasolle 2 mg/kg. Granulosyyttikasvutekijän (G-CSF) käyttöä voidaan harkita. |
Zaltrap-valmisteen aiheuttamat lievät tai keskivaikeat yliherkkyysreaktiot (mm. kuumat aallot, ihottuma, nokkosihottuma ja kutina) | Infuusio keskeytetään, kunnes reaktio korjautuu. Kortikosteroideja ja/tai antihistamiineja voidaan käyttää kliinisen tarpeen mukaan. Myöhempien hoitojaksojen aikana voidaan harkita kortikosteroidien ja/tai antihistamiinien käyttöä esihoitona. |
| Vaikeat yliherkkyysreaktiot (mm. bronkospasmi, hengenahdistus, angioedeema ja anafylaksia) (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet) | Zaltrap-/FOLFIRI-hoito lopetetaan ja potilaalle annetaan asianmukaista lääketieteellistä hoitoa. |
| Zaltrap-hoidon siirtäminen ja annosmuutokset | |
| Hypertensio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Zaltrap-hoito keskeytetään, kunnes hypertensio saadaan hallintaan. Jos lääketieteellisesti merkittävä tai vaikea hypertensio uusiutuu parhaasta mahdollisesta hoidosta huolimatta, Zaltrap-hoito keskeytetään, kunnes hypertensio saadaan hallintaan. Myöhemmissä hoitojaksoissa käytetään pienempää 2 mg/kg annosta. |
| Proteinuria (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Zaltrap-hoito keskeytetään, jos virtsan proteiinimäärä on ≥ 2 g / 24 h, ja aloitetaan uudelleen, kun virtsan proteiinimäärä on < 2 g / 24 h. Jos tila uusiutuu, hoito keskeytetään, kunnes virtsan proteiinimäärä on < 2 g / 24 h. Tämän jälkeen annos pienennetään tasolle 2 mg/kg. |
| FOLFIRI-hoidon annosmuutokset, kun hoitoa käytetäänyhdessä Zaltrap-hoidon kanssa | |
| Vaikea stomatiitti ja palmoplantaarinen erytrodysestesia-oireyhtymä | 5-FU-bolusta pienennetään ja infuusioannosta pienennetään 20 %. |
Vaikea ripuli | Irinotekaaniannosta pienennetään 15–20 %. Jos vaikea ripuli uusiutuu myöhemmän hoitojakson aikana, Jos vaikea ripuli jatkuu molemmista annosleikkauksista Ripulilääkevalmisteita ja nesteytystä voidaan käyttää tarpeen mukaan. |
Irinotekaaniin, 5-fluorourasiiliin tai foliinihappoon liittyvä muu toksisuus, ks. kyseisten valmisteiden voimassa olevat valmisteyhteenvedot.
Erityisryhmät
Iäkkäät
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa 28,2 % potilaista oli ≥ 65-vuotiaita, mutta alle 75-vuotiaita. 5,4 % potilaista oli ≥ 75-vuotiaita. Iäkkäiden ihmisten Zaltrap-annosta ei tarvitse muuttaa.
Maksan vajaatoiminta
Zaltrap-hoidon käyttöä maksan vajaatoimintapotilaille ei ole tutkittu muodollisissa tutkimuksissa (ks. kohta Farmakokinetiikka). Kliiniset tiedot viittaavat siihen, että afliberseptiannosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Afliberseptin käytöstä vaikeaa maksan vajaatoimintaa sairastaville ei ole tietoja.
Munuaisten vajaatoiminta
Zaltrap-hoidon käyttöä munuaisten vajaatoimintapotilaille ei ole tutkittu muodollisissa tutkimuksissa (ks. kohta Farmakokinetiikka). Kliiniset tiedot viittaavat siihen, että aloitusannosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista on hyvin vähän tietoa, joten näiden potilaiden hoidossa on noudatettava varovaisuutta.
Pediatriset potilaat
Ei ole asianmukaista käyttää Zaltrap-valmistetta pediatrisille potilaille etäpesäkkeisen kolorektaalisyövän hoidossa.
Antotapa
Zaltrap-valmisteen saa antaa vain infuusiona laskimoon 1 tunnin aikana. Zaltrap-konsentraatti on hyperosmolaalista (1000 mOsmol/kg), joten laimentamatonta Zaltrap-konsentraattia ei saa antaa nopeana laskimoinjektiona eikä boluksena laskimoon. Zaltrap-valmistetta ei saa antaa injektiona silmän lasiaiseen (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Kukin infuusiokonsentraattia sisältävä injektiopullo on tarkoitettu vain yhtä käyttökertaa (yhtä annosta) varten.
Ennen lääkkeen käsittelyä tai antoa huomioon otettavat varotoimet
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa ja antamiseen sopivat infuusiolaitteet.
Vasta-aiheet
Yliherkkyys afliberseptille tai kohdassa Apuaineet mainituille apuaineille.
Käyttö silmään / silmän lasiaiseen, sillä Zaltrap on hyperosmoottista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
FOLFIRI-hoidon vaikuttavien aineiden (irinotekaani, 5-FU, foliinihappo) vasta-aiheet, ks. kyseisten valmisteiden voimassa olevat valmisteyhteenvedot.
Varoitukset ja käyttöön liittyvät varotoimet
Verenvuoto
Afliberseptihoitoa saaneilla potilailla on ilmoitettu suurentunutta verenvuotoriskiä, mm. vaikeiden ja joskus kuolemaan johtaneiden verenvuototapahtumien riskin suurenemista (ks. kohta Haittavaikutukset).
Potilaiden vointia on seurattava ruoansulatuskanavan verenvuodon ja muun vaikean verenvuodon oireiden ja löydösten varalta. Afliberseptia ei saa antaa potilaille, joilla on vaikeaa verenvuotoa (ks. kohta Annostus ja antotapa).
Zaltrap-/FOLFIRI-hoitoa saaneilla potilailla on ilmoitettu trombosytopeniaa. On suositeltavaa seurata täydellistä verenkuvaa ja trombosyyttiarvoja lähtötilanteessa, ennen kunkin afliberseptihoitojakson alkua ja kliinisen tarpeen mukaan. Zaltrap/FOLFIRI-hoidon antoa siirretään, kunnes trombosyyttiarvo on ≥ 75 x 109/l (ks. kohta Annostus ja antotapa).
Ruoansulatuskanavan perforaatio
Afliberseptihoitoa saaneilla potilailla on ilmoitettu ruoansulatuskanavan perforaatioita, myös kuolemaan johtaneita ruoansulatuskanavan perforaatioita (ks. kohta Haittavaikutukset).
Potilaiden vointia on seurattava ruoansulatuskanavan perforaation oireiden ja löydösten varalta. Afliberseptihoito on lopetettava, jos potilaalle kehittyy ruoansulatuskanavan perforaatio (ks. kohta Annostus ja antotapa).
Fistelimuodostus
Afliberseptihoitoa saaneilla potilailla on ilmennyt ruoansulatuskanavan ja muiden alueiden fistelimuodostusta (ks. kohta Haittavaikutukset).
Afliberseptihoito on lopetettava, jos potilaalle kehittyy fisteli (ks. kohta Annostus ja antotapa).
Hypertensio
Zaltrap-/FOLFIRI-hoitoa saaneilla potilailla on havaittu suurentunut riski saada asteen 3–4 hypertensio (mm. hypertensiota ja yksi essentiaalinen hypertensiotapaus) (ks. kohta Haittavaikutukset).
Jos potilaalla on entuudestaan hypertensio, sen hoitotasapainon on oltava asianmukainen ennen afliberseptihoidon aloittamista. Jos hypertension hoitotasapainoa ei saada riittävän hyväksi, aflibersepti¬hoitoa ei pidä aloittaa. On suositeltavaa seurata verenpainetta kahden viikon välein, mm. ennen kutakin afliberseptin antokertaa sekä kliinisen tarpeen mukaan afliberseptihoidon aikana. Jos afliberseptihoidon aikana ilmenee hypertensiota, verenpaine on saatava hallintaan asianmukaisella verenpainelääkityksellä ja verenpainetta on seurattava säännöllisesti. Jos lääketieteellisesti merkittävä tai vaikea hypertensio uusiutuu parhaasta mahdollisesta hoidosta huolimatta, afliberseptihoito keskeytetään, kunnes hypertensio saadaan hallintaan. Myöhemmissä hoitojaksoissa käytetään pienempää 2 mg/kg afliberseptiannosta. Afliberseptihoito on lopetettava kokonaan, jos hypertension hoitotasapainoa ei saada riittävän hyväksi asianmukaisella verenpainelääkityksellä tai afliberseptiannosta pienentämällä tai potilaalle kehittyy hypertensiivinen kriisi tai hypertensiivinen enkefalopatia (ks. kohta Annostus ja antotapa).
Hypertensio voi pahentaa aiempaa sydän- tai verisuonitautia. Zaltrap-hoidossa on noudatettava varovaisuutta, jos potilaalla on anamneesissa kliinisesti merkittävä sydän- tai verisuonitauti, esim. sepelvaltimotauti tai kongestiivinen sydämen vajaatoiminta. Jos potilaalla on NYHA-luokan III tai IV kongestiivinen sydämen vajaatoiminta, Zaltrap-hoitoa ei pidä käyttää.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen ZALTRAP-hoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Sydämen vajaatoiminta ja pienentynyt ejektiofraktio
Zaltrap‑hoitoa saaneilla potilailla on raportoitu sydämen vajaatoimintaa ja pienentynyttä ejektiofraktiota. Vasemman kammion lähtötason ja sen jälkeisen toiminnan säännöllistä arviointia on harkittava, kun potilas saa Zaltrap-lääkettä. Potilaita on seurattava sydämen vajaatoiminnan ja pienentyneen ejektiofraktion oireiden ja löydösten varalta. Zaltrap‑hoito on lopetettava, jos potilaalle kehittyy sydämen vajaatoiminta tai ejektiofraktio pienenee.
Tromboosi- ja emboliatapahtumat
Tromboemboliset valtimotapahtumat
Afliberseptihoitoa saaneilla potilailla on havaittu tromboembolisia valtimotapahtumia (mm. ohimenevä aivoverenkiertohäiriö, aivoverisuonitapahtuma, angina pectoris, sydämensisäinen trombi, sydäninfarkti, valtimoembolia ja iskeeminen koliitti) (ks. kohta Haittavaikutukset).
Afliberseptihoito on lopetettava, jos potilaalle kehittyy tromboembolinen valtimotapahtuma (ks. kohta Annostus ja antotapa).
Tromboemboliset laskimotapahtumat
Afliberseptihoitoa saaneilla potilailla on ilmoitettu tromboembolisia laskimotapahtumia (mm. syvä laskimotromboosi ja keuhkoembolia, joka on joissakin tapauksissa johtanut kuolemaan) (ks. Haittavaikutukset).
Zaltrap-hoito on lopetettava, jos potilaalle kehittyy henkeä uhkaava (asteen 4) tromboembolinen tapahtuma (kuten keuhkoembolia) (ks. kohta Annostus ja antotapa). Jos potilaalle kehittyy asteen 3 syvä laskimotromboosi, hänelle annetaan antikoagulanttihoitoa kliinisen tarpeen mukaan ja afliberseptihoitoa jatketaan. Jos tila uusiutuu asianmukaisesta antikoagulanttihoidosta huolimatta, afliberseptihoito on lopetettava. Jos potilaalla on asteen 3 tai lievempi tromboemboliatapahtuma, hänen vointiaan on seurattava tarkoin.
Proteinuria
Afliberseptihoitoa saaneilla potilailla on havaittu vaikeaa proteinuriaa, nefroottista oireyhtymää ja tromboottista mikroangiopatiaa (ks. kohta Haittavaikutukset).
Ennen kutakin afliberseptin antokertaa tehdään virtsan liuskatesti ja/tai virtsan proteiini-kreatiniinisuhde arvioidaan proteinurian kehittymisen tai pahenemisen varalta. Jos virtsan liuskatestin mukaan proteiinia on ≥ 2+ tai proteiini-kreatiniinisuhde on > 1 tai > 100 mg/mmol, vuorokausivirtsan keräys on tarpeen.
Afliberseptin käyttö keskeytetään, jos virtsan proteiinimäärä on ≥ 2 g / 24 h. Hoito voidaan aloittaa uudelleen, kun proteiinimäärä on < 2 g / 24 h. Jos tila uusiutuu, hoito keskeytetään, kunnes virtsan proteiinimäärä on < 2 g / 24 h. Tämän jälkeen annos pienennetään tasolle 2 mg/kg. Afliberseptihoito on lopetettava, jos potilaalle kehittyy nefroottinen oireyhtymä tai tromboottinen mikroangiopatia (ks. kohta Annostus ja antotapa).
Neutropenia ja neutropeeniset komplikaatiot
Neutropeenisten komplikaatioiden (kuumeinen neutropenia ja neutropeeniset infektiot) ilmaantuvuuden on havaittu suurentuneen Zaltrap-/FOLFIRI-hoitoa saavilla potilailla (ks. kohta Haittavaikutukset).
On suositeltavaa tarkastaa täydellinen verenkuva ja valkosolujen erittelylaskenta lähtötilanteessa ja ennen kunkin afliberseptihoitojakson alkua. Zaltrap-/FOLFIRI-hoidon antoa siirretään, kunnes neutrofiiliarvo on ≥ 1,5 x 109/l (ks. kohta Annostus ja antotapa). G-CSF-hoitoa voidaan harkita asteen ≥ 3 neutropenian ilmaantuessa ensimmäisen kerran sekä sekundaariprofylaksina, jos potilas saattaa kuulua neutropeenisten komplikaatioiden riskiryhmään.
Ripuli ja nestehukka
Vaikean ripulin ilmaantuvuuden on havaittu suurentuneen Zaltrap-/FOLFIRI-hoitoa saavilla potilailla (ks. kohta Haittavaikutukset).
FOLFIRI-hoidon annosta muutetaan (ks. kohta Annostus ja antotapa), ja ripulilääkkeitä ja nesteytystä annetaan tarpeen mukaan.
Yliherkkyysreaktiot
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa ilmoitettiin vaikeita yliherkkyysreaktioita Zaltrap-/FOLFIRI-hoitoa saaneilla potilailla (ks. kohta Haittavaikutukset).
Jos potilaalle kehittyy vaikea yliherkkyysreaktio (mm. bronkospasmi, hengenahdistus, angioedeema tai anafylaksia), afliberseptihoito lopetetaan ja ryhdytään asianmukaisiin lääketieteellisiin toimiin (ks. kohta Annostus ja antotapa).
Jos Zaltrap aiheuttaa lievän tai keskivaikean yliherkkyysreaktion (esim. kuumat aallot, ihottuma, nokkosihottuma tai kutina), afliberseptihoito keskeytetään, kunnes reaktio korjautuu. Kortikosteroidi- ja/tai antihistamiinihoito voidaan aloittaa kliinisen tarpeen mukaan. Myöhempien hoitojaksojen aikana voidaan harkita kortikosteroidien ja/tai antihistamiinien käyttöä esihoitona (ks. kohta Annostus ja antotapa). Varovaisuus on tarpeen, jos hoito on aiemmin aiheuttanut potilaalle yliherkkyysreaktion, sillä joillakin potilailla on havaittu toistuvia yliherkkyysreaktioita estohoidosta (mm. kortikosteroideista) huolimatta.
Haavan huono paraneminen
Aflibersepti huononsi haavojen paranemista eläinmalleissa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Afliberseptihoidon on ilmoitettu saattavan huonontaa haavojen paranemista (haavan avautuminen, anastomoosin vuotaminen) (ks. kohta Haittavaikutukset).
Afliberseptihoito on tauotettava vähintään 4 viikon ajaksi ennen elektiivistä leikkausta.
On suositeltavaa, että afliberseptihoito aloitetaan aikaisintaan 4 viikon kuluttua suuresta leikkauksesta ja vasta kun leikkaushaava on parantunut täysin. Jos kysessä on pieni kirurginen toimenpide (esim. keskuslaskimoportin asentaminen, biopsia, hampaanpoisto), afliberseptihoito voidaan aloittaa (uudelleen), kun leikkaushaava on parantunut täysin. Afliberseptihoito on lopetettava, jos haavan huono paraneminen edellyttää lääketieteellistä hoitoa (ks. kohta Annostus ja antotapa).
Leuan osteonekroosi (ONJ)
Leuan osteonekroositapauksia on ilmoitettu Zaltrap-hoitoa saaneilla potilailla, joista useat olivat saaneet aiemmin tai samanaikaisesti laskimonsisäistä bisfosfonaattia, joka on tunnettu leuan osteonekroosin riskitekijä. On noudatettava varovaisuutta, kun Zaltrap-valmistetta ja laskimonsisäistä bisfosfonaattia annetaan samanaikaisesti tai peräkkäin. Invasiivinen hammastoimenpide on myös tunnettu riskitekijä. Hammastarkastusta ja asianmukaisia ennaltaehkäiseviä hammastoimenpiteitä on harkittava ennen Zaltrap-hoidon aloitusta. Jos potilas on aiemmin saanut tai saa parasta aikaa laskimonsisäistä bisfosfonaattihoitoa, invasiivisiä hammastoimenpiteitä on vältettävä, jos mahdollista (ks. kohta Haittavaikutukset).
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)
PRES-oireyhtymää ei ilmoitettu vaiheen III avaintutkimuksessa etäpesäkkeistä kolorektaalisyöpää sairastavilla. Muissa tutkimuksissa PRES-oireyhtymää ilmoitettiin potilailla, jotka saivat afliberseptia joko monoterapiana tai yhdessä muiden syöpälääkkeiden kanssa (ks. kohta Haittavaikutukset).
PRES-oireyhtymän oireita voivat olla psyykkisen tilan muutos, kouristuskohtaukset, pahoinvointi, oksentelu, päänsärky tai näköhäiriöt. PRES-diagnoosi vahvistetaan aivojen magneettikuvauksella.
Afliberseptihoito on lopetettava, jos potilaalle kehittyy PRES (ks. kohta Annostus ja antotapa).
Iäkkäät
Iäkkäillä, ≥ 65-vuotiailla ihmisillä on suurentunut ripulin, huimauksen, voimattomuuden, painon laskun ja nestehukan riski. On suositeltavaa seurata potilaan vointia huolellisesti, jotta ripulin ja nestehukan oireet ja löydökset pystytään toteamaan ja hoitamaan nopeasti ja mahdolliset riskit pystytään minimoimaan (ks. kohta Haittavaikutukset).
Munuaisten vajaatoiminta
Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden afliberseptihoidosta on hyvin vähän tietoa. Afliberseptiannosta ei tarvitse muuttaa (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka).
Toimintakyky ja muut samanaikaiset sairaudet
Jos potilaan ECOG-toimintakykyluokka on ≥ 2 tai hänellä on merkittäviä samanaikaisia sairauksia, huonojen kliinisten tulosten riski voi olla suurentunut ja potilasta on seurattava tarkoin kliinisen tilan huononemisen varhaismerkkien varalta.
Hyväksytyistä käyttöaiheista poikkeava käyttö silmän lasiaiseen
Zaltrap on hyperosmoottinen liuos, joka ei sovi annettavaksi silmän sisään. Zaltrap-valmistetta ei saa antaa injektiona silmän lasiaiseen (ks. kohta Vasta-aiheet).
ZALTRAP sisältää natriumia
Tämä lääkevalmiste sisältää enintään 22 mg natriumia per injektiopullo, mikä vastaa 1,1 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yhteisvaikutukset
Populaatiofarmakokinetiikan analyysissä ja eri tutkimusten vertailuissa ei havaittu mitään farmakokineettisiä yhteisvaikutuksia afliberseptin ja FOLFIRI-hoidon välillä.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / Ehkäisy naisille
Hedelmällisessä iässä olevia naisia on kehotettava välttämään raskautta Zaltrap-hoidon aikana, ja heille on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä. Hedelmällisessä iässä olevien naisten, jotka ovat saaneet ZALTRAP-hoitoa, on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukauden ajan viimeisen hoitoannoksen jälkeen.
Raskaus
Ei ole olemassa tietoja afliberseptin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Angiogeneesi on kriittisen tärkeää sikiönkehityksen kannalta, joten Zaltrap-valmisteen annon jälkeinen angiogeneesin estyminen voi olla haitaksi raskaudelle. Zaltrap-valmistetta saa käyttää raskauden aikana vain, jos hoidon mahdolliset hyödyt ylittävät siihen mahdollisesti liittyvät riskit. Jos potilas tulee raskaaksi Zaltrap-hoidon aikana, hänelle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Imetys
Zaltrap-valmisteen vaikutusta maidoneritykseen sekä lääkkeen erittymistä rintamaitoon ja sen vaikutuksia imetettävään lapseen ei ole tutkittu.
Ei tiedetä, erittyykö aflibersepti ihmisen rintamaitoon. Imetettävään lapseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Zaltrap-hoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Apinatutkimusten perusteella on todennäköistä, että sekä miesten että naisten hedelmällisyys heikkenee afliberseptihoidon aikana (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Zaltrap-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn. Jos potilaalla on näköön, keskittymiskykyyn tai reaktiokykyyn vaikuttavia oireita, häntä on kehotettava välttämään ajamista ja koneiden käyttöä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Zaltrap-valmisteen ja FOLFIRI-hoidon yhdistelmän turvallisuutta arvioitiin vaiheen III tutkimuksessa, johon osallistuneet 1216 potilasta olivat saaneet aiempaa hoitoa etäpesäkkeiseen kolorektaalisyöpään. 611 potilasta sai Zaltrap-valmistetta 4 mg/kg annoksina joka toinen viikko (yksi hoitojakso), ja 605 potilasta sai lume-/FOLFIRI-hoitoa. Potilaiden saamien Zaltrap-/FOLFIRI-hoitojaksojen mediaanimäärä oli 9.
Yleisimpiä haittavaikutuksia (kaikki vaikeusasteet, ilmaantuvuus ≥ 20 %), joiden ilmoitettu ilmaantuvuus oli Zaltrap-/FOLFIRI-ryhmässä vähintään 2 % suurempi kuin lume-/FOLFIRI-ryhmässä, olivat yleisyysjärjestyksessä yleisimmästä alkaen leukopenia, ripuli, neutropenia, proteinuria, aspartaattiaminotransferaasiarvojen (ASAT) suureneminen, stomatiitti, uupumus, trombosytopenia, alaniiniaminotransferaasiarvojen (ALAT) suureneminen, hypertensio, painon lasku, ruokahalun heikkeneminen, nenäverenvuodot, vatsakipu, dysfonia, seerumin kreatiniinipitoisuuden suureneminen ja päänsärky (ks. taulukko 1).
Yleisimmin ilmoitettuja asteen 3–4 reaktioita (ilmaantuvuus ≥ 5 %), joiden ilmoitettu ilmaantuvuus oli Zaltrap-/FOLFIRI-ryhmässä vähintään 2 % suurempi kuin lume-/FOLFIRI-ryhmässä, olivat yleisyysjärjestyksessä yleisimmästä alkaen neutropenia, ripuli, hypertensio, leukopenia, stomatiitti, uupumus, proteinuria ja voimattomuus (ks. taulukko 1).
Yleisimpiä hoidon pysyvään lopettamiseen johtaneita haittavaikutuksia, joita ilmeni ≥ 1 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista, olivat verisuonistoon kohdistuneet haittavaikutukset (3,8 %) kuten hypertensio (2,3 %), infektiot (3,4 %), voimattomuus/uupumus (1,6 % ja 2,1 %), ripuli (2,3 %), nestehukka (1 %), stomatiitti (1,1 %), neutropenia (1,1 %), proteinuria (1,5 %) ja keuhkoembolia (1,1 %).
Haittavaikutusten yhteenvetotaulukko
Zaltrap-/FOLFIRI-hoitoa saaneilla potilailla ilmoitetut haittavaikutukset ja laboratorioarvojen poikkeavuudet verrattuna lume-/FOLFIRI-ryhmän ilmoituksiin esitetään Taulukossa 1 MedDRA-elinjärjestelmäluokittain ja yleisyysluokittain. Taulukon 1 haittavaikutukset on määritelty seuraavasti: mikä tahansa kliininen haittavaikutus tai laboratorioarvojen poikkeavuus, jonka ilmaantuvuus (kaikki asteet) oli etäpesäkkeisen kolorektaalisyövän tutkimuksen afliberseptiryhmässä ≥ 2 % suurempi kuin lumeryhmässä; mukana ovat myös missä tahansa afliberseptitutkimuksessa havaitut haittavaikutukset, joiden kohdalla tämä ilmaantuvuuskynnys ei ylittynyt, mutta jotka vastasivat VEGF:n estäjien ryhmän haittavaikutuksia. Haittavaikutusten voimakkuus esitetään Yhdysvaltain National Cancer Institute -instituutin (NCI) Common Toxicity Criteria -asiakirjan (CTC) version 3.0 mukaisesti. Jokaisessa yleisyysryhmässä haittavaikutukset esitetään vakavuuden mukaan alenevassa järjestyksessä. Yleisyydet perustuvat kyseisen haittavaikutuksen kaikkiin vaikeusasteisiin, ja ne määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1000, < 1/100), harvinaiset (≥ 1/10000, < 1/1000), hyvin harvinaiset (< 1/10000), yleisyys tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 1 – Etäpesäkkeisen kolorektaalisyövän tutkimuksen Zaltrap-/FOLFIRI-ryhmän potilailla ilmoitetut haittavaikutukset
| Elinjärjestelmäluokka Yleisyysluokka | Haittavaikutus | |
| Kaikki asteet | Aste ≥ 3 | |
| Infektiot | ||
| Hyvin yleiset | Infektio (1) | Infektio (1) |
| Yleiset | Neutropeeninen infektio/sepsis (1) Virtsatietulehdus Nenänielutulehdus | Neutropeeninen infektio/sepsis (1) |
| Melko harvinaiset | Virtsatietulehdus | |
| Veri ja imukudos | ||
| Hyvin yleiset | Leukopenia (2) Neutropenia (1), (2) Trombosytopenia (2) | Leukopenia (2) Neutropenia (2) |
| Yleiset | Kuumeinen neutropenia | Kuumeinen neutropenia Trombosytopenia (2) |
| Immuunijärjestelmä | ||
| Yleiset | Yliherkkyys (1) | |
| Melko harvinaiset | Yliherkkyys (1) | |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleiset | Ruokahalun heikkeneminen Painon lasku | |
| Yleiset | Nestehukka (1) | Nestehukka (1) Ruokahalun heikkeneminen Painon lasku |
| Sydän | ||
| Melko harvinaiset | Sydämen vajaatoiminta | |
| Harvinaiset | Pienentynyt ejektiofraktio | |
| Hermosto | ||
| Hyvin yleiset | Päänsärky | |
| Yleiset | Päänsärky | |
| Melko harvinaiset | PRES (1), (4) | PRES (1), (4) |
| Verisuonisto | ||
| Hyvin yleiset | Hypertensio (1) Verenvuoto (1) | Hypertensio |
| Yleiset | Tromboemboliset valtimotapahtumat (1) Tromboemboliset laskimotapahtumat (1) | Tromboemboliset valtimotapahtumat (1) Tromboemboliset laskimotapahtumat (1) Verenvuoto (1) |
| Tuntematon | Aneurysmat ja valtimon dissekaatiot | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleiset | Hengenahdistus Nenäverenvuoto Dysfonia | |
| Yleiset | Suu-nielukipu Voimakas nuha | |
| Melko harvinaiset | Hengenahdistus Nenäverenvuoto Dysfonia Suu-nielukipu | |
| Ruoansulatuselimistö | ||
| Hyvin yleiset | Ripuli (1) Stomatiitti Vatsakipu Ylävatsakipu | Ripuli (1) Stomatiitti |
| Yleiset | Verenvuoto peräaukosta Fisteli (1) Haavainen stomatiitti Peräpukamat Peräaukon kipu Hammassärky | Vatsakipu Ylävatsakipu |
| Melko harvinaiset | Ruoansulatuskanavan perforaatio (1) | Ruoansulatuskanavan perforaatio (1) Verenvuoto peräaukosta Fisteli (1) Haavainen stomatiitti Peräaukon kipu |
| Maksa ja sappi | ||
| Hyvin yleiset | ASAT-arvon suureneminen (2) ALAT-arvon suureneminen (2) | |
| Yleiset | ASAT-arvon suureneminen (2) ALAT-arvon suureneminen (2) | |
| Iho ja ihonalainen kudos | ||
| Hyvin yleiset | Palmoplantaarinen erytrodysestesiaoireyhtymä | |
| Yleiset | Ihon hyperpigmentaatio | Palmoplantaarinen erytrodysestesiaoireyhtymä |
| Melko harvinaiset | Haavan huono paraneminen (1) | Haavan huono paraneminen (1) |
| Luusto, lihakset ja sidekudos | ||
| Melko harvinaiset | Leuan osteonekroosi (ONJ) | |
| Munuaiset ja virtsatiet | ||
| Hyvin yleiset | Proteinuria (1), (3) Suurentunut seerumin kreatiniinipitoisuus | |
| Yleiset | Proteinuria (1), (3) | |
| Melko harvinaiset | Nefroottinen oireyhtymä (1) Tromboottinen mikroangiopatia (1) | Nefroottinen oireyhtymä (1) Tromboottinen mikroangiopatia (1) |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleiset | Voimattomuustilat | Voimattomuustilat |
| Huom. Haittavaikutukset on ilmoitettu MedDRA-version MEDDRA13.1 mukaisesti, ja niiden vaikeusaste on luokiteltu NCI:n CTC-asiakirjan version 3.0 mukaisesti. (1) Ks. ”Tiettyjen haittavaikutusten kuvaus” tässä kohdassa. (2) Perustuu laboratorioarvoihin (% potilaista, joille tehtiin laboratorioarviointeja) (3) Kliinisten ja laboratoriotietojen yhdistelmä (4) Ei ilmoitettu etäpesäkkeisen kolorektaalisyövän tutkimuksessa. PRES-oireyhtymää ilmoitettiin kuitenkin muissa tutkimuksissa potilailla, jotka käyttivät afliberseptia monoterapiana tai yhdessä muiden syöpälääkkeiden kuin FOLFIRI-hoidon kanssa. | ||
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa ≥ 20 %:lla potilaista ilmeni anemiaa, pahoinvointia, oksentelua, ummetusta, hiustenlähtöä, AFOS-pitoisuuksien suurenemista ja hyperbilirubinemiaa. Näiden määrä oli samaa luokkaa molemmissa ryhmissä, eikä Zaltrap-/FOLFIRI-hoitoryhmässä havaittu ilmaantuvuus ollut ≥ 2 % toisen ryhmän ilmaantuvuutta suurempi.
Tiettyjen haittavaikutusten kuvaus
Verenvuoto
Zaltrap-hoitoa saavien potilaiden verenvuotoriski suurenee (koskee myös vaikeita verenvuotoja ja joskus kuolemaan johtavia verenvuototapahtumia). Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa verenvuotoa (kaikki asteet) ilmoitettiin 37,8 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 19,0 %:lla lume-/FOLFIRI-ryhmän potilaista. Yleisin ilmoitettu verenvuototyyppi oli vähäinen (asteen 1–2) nenäverenvuoto, jota ilmeni 27,7 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista potilaista. Asteen 3–4 verenvuotoa, mm. ruoansulatuskanavan verenvuotoa, hematuriaa ja toimenpiteenjälkeistä verenvuotoa, ilmoitettiin 2,9 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 1,7 %:lla lume-/FOLFIRI-ryhmän potilaista. Muissa tutkimuksissa Zaltrap-hoitoa saaneille potilaille on ilmaantunut vaikeaa kallonsisäistä verenvuotoa ja keuhkoverenvuotoa/veriyskää, myös kuolemaan johtaneita tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ruoansulatuskanavan perforaatio
Zaltrap-hoitoa saaneilla potilailla on ilmoitettu ruoansulatuskanavan perforaatioita, myös kuolemaan johtaneita ruoansulatuskanavan perforaatioita. Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa ruoansulatuskanavan perforaatioita (kaikki asteet) ilmoitettiin Zaltrap-/FOLFIRI-ryhmässä 3 potilaalla 611:sta (0,5 %) ja lume-/FOLFIRI-ryhmässä 3 potilaalla 605:stä (0,5 %). Ruoansulatuskanavan perforaatiotapahtuman vaikeusaste oli 3–4 Zaltrap-/FOLFIRI-ryhmän kaikilla 3 potilaalla (0,5 %) ja lume-/FOLFIRI-ryhmässä 2 potilaalla (0,3 %). Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) ruoansulatuskanavan perforaatioiden (kaikki asteet) ilmaantuvuus oli Zaltrap-ryhmässä 0,8 % ja lumeryhmässä 0,3 %. Asteen 3–4 ruoansulatuskanavan perforaatiotapahtumia ilmeni 0,8 %:lla Zaltrap-ryhmän potilaista ja 0,2 %:lla lumeryhmän potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Fistelimuodostus
Zaltrap-hoitoa saaneille potilaille on ilmaantunut ruoansulatuskanavan ja muiden alueiden fistelimuodostusta. Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa fisteleitä (sijainti anaalinen, enterovesikaalinen, enterokutaaninen, kolovaginaalinen tai intestinaalinen) ilmoitettiin Zaltrap-/FOLFIRI-ryhmässä 9 potilaalla 611:sta (1,5 %) ja lume-/FOLFIRI-ryhmässä 3 potilaalla 605:stä (0,5 %). Asteen 3 ruoansulatuskanavan fistelimuodostusta ilmaantui Zaltrap-ryhmässä 2 potilaalle (0,3 %) ja lumeryhmässä 1 potilaalle (0,2 %). Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) fistelien (kaikki asteet) ilmaantuvuus oli Zaltrap-potilailla 1,1 % ja lumehoitoa saaneilla 0,2 %. Asteen 3–4 fisteleitä ilmeni 0,2 %:lla Zaltrap-hoitoa saaneista ja 0,1 %:lla lumehoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hypertensio
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa hypertensiota (kaikki asteet) ilmoitettiin 41,2 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 10,7 %:lla lume-/FOLFIRI-ryhmän potilaista. Zaltrap-/FOLFIRI-hoitoa saaneilla potilailla on todettu asteen 3–4 hypertension riskin suurenemista (mm. hypertensiota ja yksi essentiaalinen hypertensiotapaus). Asteen 3 hypertensiota (joka edellytti aiemman verenpainelääkityksen muuttamista tai hoitoa useammalla kuin yhdellä lääkevalmisteella) ilmoitettiin 1,5 %:lla lume-/FOLFIRI-ryhmän potilaista ja 19,1 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista. Asteen 4 hypertensiota (hypertensiivinen kriisi) ilmoitettiin yhdellä Zaltrap-/FOLFIRI-hoitoa saaneella potilaalla (0,2 %). Zaltrap-/FOLFIRI-hoitoa saaneille potilaille kehittynyt asteen 3–4 hypertensio alkoi 54 %:ssa tapauksista kahden ensimmäisen hoitojakson aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tromboosi- ja emboliatapahtumat
Tromboemboliset valtimotapahtumat
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa tromboembolisia valtimotapahtumia (mm. ohimenevä aivoverenkiertohäiriö, aivoverisuonitapahtuma, angina pectoris, sydämensisäinen trombi, sydäninfarkti, valtimoembolia ja iskeeminen koliitti) ilmoitettiin 2,6 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 1,5 %:lla lume-/FOLFIRI-hoitoa saaneista. Asteen 3–4 tapahtumia ilmeni Zaltrap-/FOLFIRI-ryhmässä 11 potilaalla (1,8 %) ja lume-/FOLFIRI-ryhmässä 3 potilaalla (0,5 %). Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) tromboembolisten valtimotapahtumien (kaikki asteet) ilmaantuvuus oli Zaltrap-potilailla 2,3 % ja lumehoitoa saaneilla 1,7 %. Asteen 3–4 tromboembolisia valtimotapahtumia ilmeni 1,7 %:lla Zaltrap-hoitoa saaneista ja 1,0 %:lla lumehoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tromboemboliset laskimotapahtumat
Tromboembolisten laskimotapahtumien ryhmä kattaa syvät laskimotromboosit ja keuhkoemboliat. Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa tromboembolisia laskimotapahtumia (kaikki asteet) ilmeni 9,3 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 7,3 %:lla lume-/FOLFIRI-ryhmän potilaista. Asteen 3–4 tromboembolisia laskimotapahtumia ilmeni 7,9 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 6,3 %:lla lume-/FOLFIRI-ryhmän potilaista. Keuhkoemboliaa ilmeni 4,6 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 3,5 %:lla lume-/FOLFIRI-ryhmän potilaista. Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) tromboembolisten laskimotapahtumien (kaikki asteet) ilmaantuvuus oli Zaltrap-potilailla 7,1 % ja lumepotilailla 7,1 %.
Proteinuria
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa ilmoitettiin proteinuriaa (jonka tiedot perustuvat sekä kliinisiin että laboratoriotietoihin) 62,2 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 40,7 %:lla lume-/FOLFIRI-ryhmän potilaista. Asteen 3–4 proteinuriaa ilmeni 7,9 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 1,2 %:lla lume-/FOLFIRI-ryhmän potilaista. Nefroottista oireyhtymää ilmeni 2:lla Zaltrap-/FOLFIRI-ryhmän potilaalla (0,5 %), mutta ei yhdelläkään lume-/FOLFIRI-ryhmän potilaalla. Yhdellä Zaltrap-/FOLFIRI-hoitoa saaneella potilaalla oli proteinuriaa ja hypertensiota, ja hänellä todettiin tromboottinen mikroangiopatia. Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) nefroottisen oireyhtymän ilmaantuvuus oli Zaltrap-potilailla 0,5 % ja lumehoitoa saaneilla 0,1 % (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia ja neutropeeniset komplikaatiot
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa 67,8 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ilmoitettiin neutropeniaa (kaikki asteet), kun taas lume-/FOLFIRI-ryhmässä vastaava osuus oli 56,3 %. Asteen 3–4 neutropeniaa ilmeni 36,7 %:lla Zaltrap-/FOLFIRI-ryhmän potilaista ja 29,5 %:lla lume-/FOLFIRI-ryhmän potilaista. Yleisin asteen 3–4 neutropeeninen komplikaatio oli kuumeinen neutropenia, jota ilmeni 4,3 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 1,7 %:lla lume-/FOLFIRI-hoitoa saaneista. Asteen 3–4 neutropeenisiä infektioita / sepsistä ilmaantui 1,5 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 1,2 %:lla lume-/FOLFIRI-hoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektiot
Infektioiden yleisyys oli Zaltrap-/FOLFIRI-hoitoa saaneilla suurempi (kaikki asteet, 46,2 %; asteet 3–4, 12,3 %) kuin lume-/FOLFIRI-hoitoa saaneilla (kaikki asteet, 32,7 %; asteet 3–4, 6,9 %). Infektioita olivat mm. virtsatieinfektio, nenänielutulehdus, ylähengitystieinfektio, keuhkokuume, katetrikohdan infektio ja hammasinfektio.
Ripuli ja nestehukka
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa ripulia (kaikki asteet) todettiin 69,2 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 56,5 %:lla lume-/FOLFIRI-hoitoa saaneista. Nestehukkaa (kaikki asteet) todettiin 9,0 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 3,0 %:lla lume-/FOLFIRI-hoitoa saaneista. Asteen 3–4 ripulia ilmoitettiin 19,3 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 7,8 %:lla lume-/FOLFIRI-hoitoa saaneista. Asteen 3–4 nestehukkaa ilmoitettiin 4,3 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 1,3 %:lla lume-/FOLFIRI-hoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yliherkkyysreaktiot
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa vaikeita yliherkkyysreaktioita ilmoitettiin 0,3 %:lla Zaltrap-/FOLFIRI-hoitoa saaneista ja 0,5 %:lla lume-/FOLFIRI-hoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haavan huono paraneminen
Zaltrap-hoitoon voi liittyä haavan huonoa paranemista (haavan avautuminen, anastomoosin vuoto). Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa haavan huonoa paranemista ilmoitettiin Zaltrap-/FOLFIRI-ryhmässä 3 potilaalla (0,5 %) ja lume-FOLFIRI-ryhmässä 5 potilaalla (0,8 %). Asteen 3 haavan huonoa paranemista ilmoitettiin Zaltrap-/FOLFIRI-ryhmässä 2 potilaalla (0,3 %) mutta lume-/FOLFIRI-ryhmässä ei yhdelläkään potilaalla. Kaikissa kolmessa vaiheen III lumekontrolloidussa kliinisessä tutkimuksessa (kolorektaali-, haima- ja keuhkosyöpäpotilailla) haavan huonon paranemisen (kaikki asteet) ilmaantuvuus oli Zaltrap-potilailla 0,5 % ja lumehoitoa saaneilla 0,4 %. Asteen 3–4 haavan huonoa paranemista ilmeni 0,2 %:lla Zaltrap-hoitoa saaneista mutta ei kenelläkään lumehoitoa saaneista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)
PRES-oireyhtymää ei ilmoitettu vaiheen III avaintutkimuksessa etäpesäkkeistä kolorektaalisyöpää sairastavilla. Muissa tutkimuksissa PRES-oireyhtymää ilmoitettiin potilailla, jotka saivat Zaltrap-valmistetta joko monoterapiana (0,5 %) tai yhdessä muiden syöpälääkkeiden kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muita haittavaikutuksia ja laboratorioarvojen poikkeavuuksia, joiden ilmoitettu yleisyys (kaikki asteet) oli Zaltrap-/FOLFIRI-ryhmässä ≥ 5 % suurempi kuin lume-/FOLFIRI-ryhmässä
Seuraavien haittavaikutusten ja laboratorioarvojen poikkeavuuksien ilmoitettu yleisyys (kaikki asteet) oli Zaltrap-/FOLFIRI-ryhmässä ≥ 5 % suurempi kuin lume-/FOLFIRI-ryhmässä (yleisyysjärjestyksessä yleisimmästä alkaen): leukopenia (78,3 % Zaltrap-/FOLFIRI-ryhmässä ja 72,4 % lume-/FOLFIRI-ryhmässä, kaikki asteet; 15,6 % ja 12,2 %, asteet 3–4), ASAT-arvojen suureneminen (57,5 % ja 50,2 %, kaikki asteet; 3,1 % ja 1,7 %, asteet 3–4), stomatiitti (50,1 % ja 32,9 %, kaikki asteet; 12,8 % ja 4,6 %, asteet 3–4), uupumus (47,8 % ja 39,0 %, kaikki asteet; 12,6 % ja 7,8 %, asteet 3–4), trombosytopenia (47,4 % ja 33,8 %, kaikki asteet; 3,3 % ja 1,7 %, asteet 3–4), ALAT-arvojen suureneminen (47,3 % ja 37,1 %, kaikki asteet; 2,7 % ja 2,2 %, asteet 3–4), ruokahalun heikkeneminen (31,9 % ja 23,8 %, kaikki asteet; 3,4 % ja 1,8 %, asteet 3–4), painon lasku (31,9 % ja 14,4 %, kaikki asteet; 2,6 % ja 0,8 %, asteet 3–4), dysfonia (25,4 % ja 3,3 %, kaikki asteet; 0,5 % ja 0 %, asteet 3–4), päänsärky (22,3 % ja 8,8 %, kaikki asteet; 1,6 % ja 0,3 %, asteet 3–4), voimattomuus (18,3 % ja 13,2 %, kaikki asteet; 5,1 % ja 3,0 %, asteet 3–4), palmoplantaarinen erytrodysestesiaoireyhtymä (11,0 % ja 4,3 %, kaikki asteet; 2,8 % ja 0,5 %, asteet 3–4) ja ihon hyperpigmentaatio (8,2 % ja 2,8 %, kaikki asteet; 0 % ja 0 %, asteet 3–4).
Pediatriset potilaat
Valmisteen turvallisuutta lasten hoidossa ei ole varmistettu.
Muut erityisryhmät
Iäkkäät
Etäpesäkkeisen kolorektaalisyövän avaintutkimuksessa 611 potilasta sai Zaltrap-/FOLFIRI-hoitoa. Heistä 172 (28,2 %) oli ≥ 65-vuotiaita mutta alle 75-vuotiaita, ja 33 (5,4 %) oli ≥ 75-vuotiaita. Iäkkäillä (≥ 65-vuotiailla) haittavaikutusten todennäköisyys voi olla suurempi. Ripulin, huimauksen, voimattomuuden, painon laskun ja nestehukan ilmaantuvuus oli iäkkäillä ihmisillä ≥ 5 % suurempi kuin nuoremmilla. Iäkkäiden ihmisten vointia on seurattava tarkoin ripulin ja mahdollisen nestehukan varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Kolmessa lumekontrolloidussa vaiheen III kliinisessä tutkimuksessa Zaltrap-hoitoa saaneilla potilailla, joilla oli lähtötilanteessa lievä munuaisten vajaatoiminta (N = 352), havaittiin samankaltaisia haittavaikutuksia kuin munuaistoiminnaltaan normaaleilla potilailla (N = 642). Zaltrap-ryhmään kuului pieni määrä potilaita, joilla oli lähtötilanteessa keskivaikea/vaikea munuaisten vajaatoiminta (N = 49). Näillä potilailla muut kuin munuaisiin liittyvät tapahtumat olivat yleisesti ottaen samaa luokkaa kuin munuaistoiminnaltaan normaaleilla potilailla, mutta nestehukan (kaikki asteet) ilmaantuvuus oli > 10 % suurempi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Kuten kaikkien proteiinilääkkeiden käytön yhteydessä, myös Zaltrap-valmistetta käytettäessä saattaa ilmetä immunogeenisuutta.
Kaikissa kliinisissä syöpätautitutkimuksissa todettiin, että pienten lääkevasta-ainetittereiden ilmaantuvuus lähtötilanteen jälkeisissä lääkevasta-ainekokeissa oli samaa luokkaa sekä lumehoitoa että Zaltrap-hoitoa saaneilla (3,3 % ja 3,8 %). Yhdelläkään potilaalla ei todettu suuria afliberseptivasta-ainetittereitä. Zaltrap-ryhmässä 17 potilasta (1,6 %) ja lumeryhmässä 2 potilasta (0,2 %) sai myös positiivisen tuloksen neutraloivien vasta-aineiden määrityksessä. Etäpesäkkeisen kolorektaalisyövän avaintutkimuksen lume-/FOLFIRI-ryhmässä todettiin positiivisia tuloksia lääkevasta-ainekokeessa [18/526 (3,4 %)] useammin kuin Zaltrap-/FOLFIRI-ryhmässä [8/521 (1,5 %)]. Etäpesäkkeisen kolorektaalisyövän avaintutkimuksen lume-/FOLFIRI-ryhmässä todettiin myös positiivisia tuloksia neutraloivien vasta-aineiden määrityksessä [2/526 (0,38 %)] useammin kuin Zaltrap-/FOLFIRI-ryhmässä [1/521 (0,19 %)]. Positiivinen tulos immunogeenisuustutkimuksessa ei vaikuttanut millään havaittavalla tavalla afliberseptin farmakokinetiikkaan kyseisillä potilailla.
Kun otetaan huomioon, että lääkevasta-ainekokeen tulokset olivat samaa luokkaa sekä lume- että Zaltrap-ryhmissä, Zaltrap-hoitoon liittyvän immunogeenisuuden todellinen ilmaantuvuus on todennäköisesti yliarvioitu näissä tutkimuksissa.
Immunogeenisuustiedot riippuvat suuresti kokeen herkkyydestä ja spesifisyydestä. Vasta-ainepositiivisuuden havaittuun ilmaantuvuuteen tietyssä tutkimuksessa vaikuttavat myös monet muut tekijät, mm. näytteen käsittely, näytteenottoajankohta, muut samanaikaiset lääkevalmisteet ja perussairaus. Zaltrap-potilailla todetun vasta-aineilmaantuvuuden vertailu muilla valmisteilla todettuun vasta-aineilmaantuvuuteen voi siis johtaa harhaan.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Afliberseptin turvallisuudesta ei ole tietoa, jos annos on yli 7 mg/kg kahden viikon välein tai yli 9 mg/kg kolmen viikon välein. Yleisimmin havaitut haittavaikutukset olivat näillä annoksilla samankaltaisia kuin hoitoannoksillakin.
Spesifistä vastalääkettä Zaltrap-yliannostukselle ei tunneta. Yliannostustapaukset hoidetaan ryhtymällä asianmukaisiin tukitoimiin, joihin kuuluu etenkin hypertension ja proteinurian seurantaa ja hoitoa. Lääkärin on valvottava potilaan vointia tarkoin mahdollisten haittavaikutusten varalta (ks. kohta Haittavaikutukset).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet, muut syöpälääkkeet, ATC-koodi: L01XX44
Vaikutusmekanismi
Endoteelikasvutekijät A ja B (VEGF-A, VEGF-B) ja istukkakasvutekijä (PlGF) ovat angiogeenisten VEGF-kasvutekijöiden perheeseen kuuluvia kasvutekijöitä, joilla voi olla voimakas mitogeeninen, kemotaktinen ja verisuonten läpäisevyyteen kohdistuva vaikutus endoteelisoluihin. VEGF-A vaikuttaa kahden endoteelisolujen solukalvolla esiintyvän reseptorityrosiinikinaasin (VEGFR-1 ja VEGFR-2) kautta. PlGF ja VEGF-B sitoutuvat vain VEGFR-1:een, jota esiintyy myös valkosolujen solukalvolla. Jos VEGF-A aktivoi näitä reseptoreja liian voimakkaasti, seurauksena voi olla patologinen uudissuonimuodostus ja verisuonten liiallinen läpäisevyys. Myös PlGF-kasvutekijä on yhteydessä patologiseen uudissuonimuodostukseen ja tulehdussolujen hakeutumiseen kasvaimiin.
Aflibersepti tunnetaan tieteellisessä kirjallisuudessa myös nimellä VEGF TRAP. Kyseessä on rekombinantti fuusioproteiini, jossa ihmisen VEGF-reseptorien 1 ja 2 solunulkoisten domeenien VEGF-kasvutekijää sitovat osat on yhdistetty ihmisen IgG1:n Fc-osaan. Aflibersepti valmistetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasolulinjassa (CHO-K-1, nisäkäsperäinen ekspressiojärjestelmä). Aflibersepti on dimeerinen glykoproteiini, jonka proteiinimolekyylipaino on 97 kilodaltonia (kDa). Se on glykosyloitunut, mikä suurentaa molekyylin kokonaismassaa 15 %, joten molekyylin kokonaispaino on 115 kDa.
Aflibersepti toimii VEGF-A:han sitoutuvana liukoisena valereseptorina, jolla on suurempi affiniteetti kuin vastaavilla luontaisilla reseptoreilla. Se vaikuttaa samalla tavalla myös rakenteellisesti samankaltaisiin PlGF- ja VEGF-B-ligandeihin. Aflibersepti sieppaa endogeenisen ligandin ja estää sitä sitoutumasta vastaavaan reseptoriin, jolloin reseptorivälitteinen signaalin muodostus estyy.
Aflibersepti salpaa VEGF-reseptorien aktivoitumisen ja estää endoteelisolujen lisääntymisen, jolloin kasvaimen hapen- ja ravinteidensaannista huolehtivien uudissuonien kasvu estyy.
Aflibersepti sitoutuu ihmisen VEGF-A:han (tasapainotilan dissosiaatiovakio, KD, on 0,5 pM VEGF-A165:n suhteen ja 0,36 pM VEGF-A121:n suhteen), ihmisen PlGF-tekijään (KD 39 pM PlGF-2:n suhteen) ja ihmisen VEGF-B:hen (KD 1,92 pM) ja muodostaa stabiilin, inertin kompleksin, jolla ei ole havaittavaa biologista aktiivisuutta.
Farmakodynaamiset vaikutukset
Afliberseptin anto hiirille, joilla oli ksenotransplantoitu tai allotransplantoitu kasvain, esti eri syöpätyyppien kasvua.
Kliininen teho ja turvallisuus
Zaltrap-valmisteen tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa potilailla, joilla oli etäpesäkkeinen kolorektaalisyöpä ja jotka olivat aiemmin saaneet oksaliplatiinipohjaista hoitoa, johon oli mahdollisesti kuulunut myös bevasitsumabi. Yhteensä 1226 potilasta satunnaistettiin (suhteessa 1:1) saamaan joko Zaltrap-valmistetta (N = 612; 4 mg/kg annos infuusiona laskimoon 1 tunnin kuluessa päivänä 1) tai lumelääkettä (N = 614) yhdessä 5 fluourasiilin ja irinotekaanin kanssa [FOLFIRI: irinotekaani, 180 mg/m2 infuusiona laskimoon 90 minuutin kuluessa, ja foliinihappo (d- ja l-isomeerien raseeminen seos), 400 mg/m2 infuusiona laskimoon 2 tunnin kuluessa; molemmat annettiin yhtä aikaa päivänä 1 Y-liittimen kautta, minkä jälkeen annettiin 400 mg/m2 suuruinen 5-FU-bolus laskimoon ja 2400 mg/m2 5-FU-annos jatkuvana infuusiona laskimoon 46 tunnin kuluessa]. Hoitojaksot toistettiin molemmissa ryhmissä 2 viikon välein. Hoitoa jatkettiin, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta. Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika. Hoitovalinta stratifioitiin ECOG-toimintakykyluokan (0, 1 tai 2) ja aiemman bevasitsumabihoidon (kyllä/ei) mukaan.
Hoitoryhmät olivat hyvässä tasapainossa demografisten tietojen suhteen (ikä, rotu, ECOG-toimintakykyluokka ja aiempi bevasitsumabihoito). Tutkimukseen satunnaistettujen 1226 potilaan mediaani-ikä oli 61 v, ja heistä 58,6 % oli miehiä. 97,8 %:n ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1, ja 2,2 %:n toimintakykyluokka oli lähtötilanteessa 2. Satunnaistetuista 1226 potilaasta 89,4 % lume-/FOLFIRI-ryhmän potilaista ja 90,2 % Zaltrap-/FOLFIRI-ryhmän potilaista oli saanut aiempaa oksaliplatiinipohjaista yhdistelmäsolunsalpaajahoitoa etäpesäkkeisen tai pitkälle edenneen syövän hoitoon. Noin 10 % potilaista (10,4 % lume-/FOLFIRI-ryhmän potilaista ja 9,8 % Zaltrap-/FOLFIRI-ryhmän potilaista) oli saanut aiemmin oksaliplatiinipohjaista liitännäissolunsalpaajahoitoa, ja tauti oli edennyt joko liitännäissolunsalpaajahoidon aikana tai 6 kuukauden kuluessa sen päättymisestä. 373 potilasta (30,4 %) oli saanut oksaliplatiinipohjaisen hoidon yhteydessä bevasitsumabia.
Zaltrap-/FOLFIRI-hoidon yleiset tehoa kuvaavat tulokset verrattuna lume-/FOLFIRI-hoitoon esitetään kuvassa 1 ja taulukossa 2.
Kuva 1 – Kokonaiselossaoloaika (kk) – hoitoryhmittäiset Kaplan–Meier-käyrät – hoitoaikomuspopulaatio (ITT)
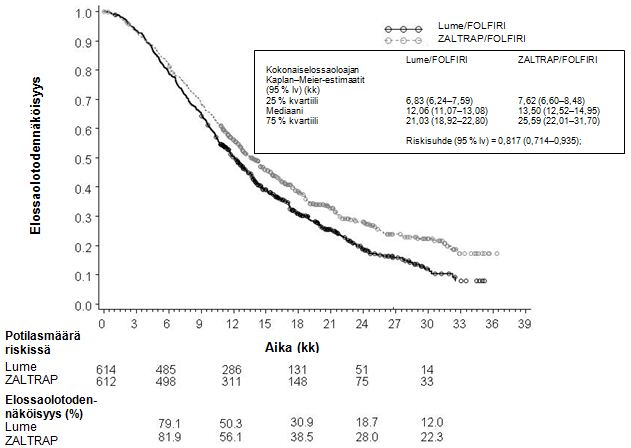
Taulukko 2 – Tärkeimmät tehoa mittaavat päätetapahtumata – ITT-populaatio
| Lume/FOLFIRI (N = 614) | Zaltrap/FOLFIRI (N = 612) | |
| Kokonaiselossaoloaika | ||
| Kuolematapahtumia, n (%) | 460 (74,9 %) | 403 (65,8 %) |
| Kokonaiselossaoloajan mediaani (95 % lv) (kk) | 12,06 (11,07–13,08) | 13,50 (12,52–14,95) |
| Stratifioitu riskisuhde (95 % lv) | 0,817 (0,714–0,935) | |
| Stratifioitu log-rank-testin p-arvo | 0,0032 | |
| Etenemisvapaa elinaikab | ||
| Tapahtumia, n (%) | 454 (73,9 %) | 393 (64,2 %) |
| Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 4,67 (4,21–5,36) | 6,90 (6,51–7,20) |
| Stratifioitu riskisuhde (95 % lv) | 0,758 (0,661–0,869) | |
| Stratifioitu log-rank-testin p-arvo | 0,00007 | |
| Kokonaisvasteprosentti (täydelliset + osittaiset vasteet) (95 % lv) (%)c | 11,1 (8,5–13,8) | 19,8 (16,4–23,2) |
| Stratifioitu Cochran–Mantel–Haenszel-testin p-arvo | 0,0001 | |
a Stratifioitu ECOG-toimintakykyluokan (0, 1, tai 2) ja aiemman bevasitsumabihoidon (kyllä/ei) perusteella.
b Etenemisvapaa elinaika (perustuu riippumattoman arviointitoimikunnan arvioon kasvaimesta): merkitsevyyskynnys 0,0001
c Objektiivinen kokonaisvasteprosentti riippumattoman arviointitoimikunnan mukaan
Kokonaiselossaoloaika ja etenemisvapaa elinaika arvioitiin eri stratifiointitekijöiden mukaan. Zaltrap-/FOLFIRI-hoidon ilmoitettu hoitovaikutus kokonaiselossaoloaikaan oli aiemmin bevasitsumabia käyttäneillä potilailla numeroiden mukaan pienempi kuin potilailla, jotka eivät olleet aiemmin altistuneet bevasitsumabille. Merkkejä hoitovaikutuksen heterogeenisyydestä ei havaittu (interaktiotesti ei merkitsevä). Aiemman bevasitsumabialtistuksen mukaan jaoteltujen tulosten yhteenveto esitetään taulukossa 3.
Taulukko 3 – kokonaiselossaoloaika ja etenemisvapaa elinaika aiemman bevasitsumabialtistuksen mukaan jaoteltuinaa – ITT-populaatio
| Lume/FOLFIRI (N = 614) | Zaltrap/FOLFIRI (N = 612) | |
| Kokonaiselossaoloaika | ||
| Aiempaa bevasitsumabihoitoa saaneet potilaat (n (%)) | 187 (30,5 %) | 186 (30,4 %) |
| Kokonaiselossaoloajan mediaani (95 % lv) (kk) | 11,7 (9,96–13,77) | 12,5 (10,78–15,47) |
| Riskisuhde (95 % lv) | 0,862 (0,676–1,100) | |
| Potilaat, joilla ei aiempaa bevasitsumabihoitoa (n (%)) | 427 (69,5 %) | 426 (69,6 %) |
| Kokonaiselossaoloajan mediaani (95 % lv) (kk) | 12,4 (11,17–13,54) | 13,9 (12,72–15,64) |
| Riskisuhde (95 % lv) | 0,788 (0,671–0,925) | |
| Etenemisvapaa elinaika | ||
| Aiempaa bevasitsumabihoitoa saaneet potilaat (n (%)) | 187 (30,5 %) | 186 (30,4 %) |
| Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 3,9 (3,02–4,30) | 6,7 (5,75–8,21) |
| Riskisuhde (95 % lv) | 0,661 (0,512–0,852) | |
| Potilaat, joilla ei aiempaa bevasitsumabihoitoa (n (%)) | 427 (69,5 %) | 426 (69,6 %) |
| Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 5,4 (4,53–5,68) | 6,9 (6,37–7,20) |
| Riskisuhde (95 % lv) | 0,797 (0,679–0,936) | |
a IVRS-järjestelmän tietojen mukaan
Kokonaiselossaoloajan ja etenemisvapaan elinajan tiedot analysoitiin myös ECOG-toimintakykyluokan mukaan. ECOG-toimintakykyluokassa 0 kokonaiselossaoloajan riskisuhde (95 % lv) oli 0,77 (0,64–0,93); ECOG-toimintakykyluokassa 1 se oli 0,87 (0,71–1,06). ECOG-toimintakykyluokassa 0 etenemisvapaan elinajan riskisuhde (95 % lv) oli 0,76 (0,761–0,91); ECOG-toimintakykyluokassa 1 se oli 0,75 (0,61–0,92).
Jälkianalyysien tietojen yhteenveto esitetään taulukossa 4. Analyyseistä suljettiin pois potilaat, joiden tauti eteni liitännäishoidon aikana tai 6 kuukauden kuluessa siitä. Potilaat on jaoteltu sen mukaan, olivatko he saaneet aiemmin bevasitsumabihoitoa.
Taulukko 4 – Jälkianalyysit, joista vain liitännäishoitoa saaneet potilaat suljettiin poisa, b
| Lume/FOLFIRI (N = 550) | Zaltrap/FOLFIRI (N = 552) | ||
| Aiempaa bevasitsumabihoitoa saaneet potilaat; vain liitännäishoitoa saaneet suljettiin pois (n (%)) | 179 (32,5 %) | 177 (32,1 %) | |
| Kokonaiselossaoloajan mediaani (95 % lv) (kk) | 11,7 (9,66–13,27) | 13,8 (11,01–15,87) | |
| Riskisuhde (95 % lv) | 0,812 (0,634–1,042) | ||
| Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 3,9 (3,02–4,30) | 6,7 (5,72–8,21) | |
| Riskisuhde (95 % lv) | 0,645 (0,498–0,835) | ||
| Potilaat, joilla ei aiempaa bevasitsumabihoitoa; vain liitännäishoitoa saaneet suljettiin pois (n (%)) | 371 (67,5 %) | 375 (67,9 %) | |
| Kokonaiselossaoloajan mediaani (95 % lv) (kk) | 12,4 (11,17–13,54) | 13,7 (12,71–16,03) | |
| Riskisuhde (95 % lv) | 0,766 (0,645–0,908) | ||
| Etenemisvapaan elinajan mediaani (95 % lv) (kk) | 5,3 (4,50–5,55) | 6,9 (6,24–7,20) | |
| Riskisuhde (95 % lv) | 0,777 (0,655–0,921) | ||
a IVRS-järjestelmän tietojen mukaan
b Kun ITT-populaatiosta suljettiin pois potilaat, joiden tauti eteni liitännäishoidon aikana tai 6 kuukauden kuluessa siitä, kokonaiselossaoloajan riskisuhde (95 % lv) oli 0,78 (0,68–0,90) [kokonaiselossaoloajan mediaani (95 % lv) lume-/FOLFIRI-ryhmässä 11,9 kk (10,88–13,01) ja Zaltrap/FOLFIRI-ryhmässä 13,8 kk (12,68–15,44)].
Kokonaiselossaoloajan ja etenemisvapaan elinajan muut alaryhmäanalyysit osoittivat, että Zaltrap-/FOLFIRI-hoidolla saavutettiin parempi hoitovaikutus kuin lume-/FOLFIRI-hoidolla. Alaryhmien muodostusperusteet olivat ikä (< 65; ≥ 65), sukupuoli, etäpesäkkeiden sijainti vain maksassa, hypertensioanamneesi ja affisioituneiden elinten määrä.
Kokonaiselossaoloajan alaryhmäanalyysissä Zaltrap-/FOLFIRI-hoitoa saaneilla < 65-vuotiailla ja ≥ 65-vuotiailla potilailla todetut hyödyt vastasivat johdonmukaisesti koko populaation tuloksia.
VELOUR-tutkimuksessa tehtiin eksploratiiviset biomerkkiaineanalyysit, joihin kuului RAS-mutaatiostatuksen analysointi 482 potilaalla 1 226 potilaasta (n = 240 afliberseptiryhmässä; 242 lumeryhmässä). Potilailla, joiden kasvaimessa oli villin tyypin RAS, kokonaiselossaoloajan (OS) riskisuhde (95 %:n luottamusväli) oli 0,7 (0,5−1,0). Kokonaiselossaoloajan mediaani oli 16,0 kuukautta afliberseptia saaneilla potilailla ja 11,7 kuukautta lumelääkettä saaneilla potilailla. Vastaavasti potilailla, joiden kasvaimessa oli RAS-mutaatioita, kokonaiselossaoloajan riskisuhde oli 0,9 (0,7–1,2) ja mediaani oli 12,6 kuukautta afliberseptia ja 11,2 kuukautta lumelääkettä saaneilla potilailla. Tämä data on eksploratiivista ja tilastollinen vuorovaikutustesti oli ei-merkitsevä (näytön puute hoidon vaikutuksen heterogeenisuudesta villin tyypin RAS- ja RAS-mutaatioalaryhmien välillä).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta tutkia Zaltrap-valmisteen käyttöä kaikkien pediatristen potilasryhmien hoidossa paksusuolen ja peräsuolen adenokarsinooman hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Seuraavat farmakokineettiset tiedot perustuvat laajalti populaatiofarmakokinetiikan analyysiin, johon otettiin tiedot 1507:stä erilaisia pitkälle edenneitä syöpätauteja sairastaneesta potilaasta.
Imeytyminen
Prekliinisissä kasvainmalleissa biologisesti aktiiviset afliberseptiannokset vastasivat annoksia, joilla verenkierron vapaan afliberseptin pitoisuudet ylittävät VEGF-tekijään sitoutuneen afliberseptin pitoisuudet. VEGF-tekijään sitoutuneen afliberseptin pitoisuudet verenkierrossa suurenevat afliberseptiannoksen myötä, kunnes valtaosa saatavilla olevasta VEGF-tekijästä on sitoutunut. Kun afliberseptiannosta suurennetaan edelleen, verenkierron vapaan afliberseptin pitoisuudet suurenivat annosriippuvaisesti, mutta VEGF-tekijään sitoutuneen afliberseptin pitoisuudet suurenivat enää vain vähän.
Potilaille annetaan 4 mg/kg Zaltrap-annos laskimoon kahden viikon välein niin, että verenkierrossa on enemmän vapaata afliberseptia kuin VEGF-tekijään sitoutunutta afliberseptia.
Suositusannoksilla (4 mg/kg kahden viikon välein) vapaan afliberseptin pitoisuudet olivat lähes vakaan tilan luokkaa toiseen hoitojaksoon mennessä, eikä kumuloitumista tapahdu juurikaan (kumulaatiosuhde vakaassa tilassa 1,2 verrattuna ensimmäiseen antokertaan).
Jakautuminen
Vapaan afliberseptin jakautumistilavuus on vakaassa tilassa noin 8 l.
Biotransformaatio
Afliberseptilla ei ole tehty metaboliatutkimuksia, sillä kyseessä on proteiini. Aflibersepti hajoaa oletettavasti pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Vapaa aflibersepti eliminoituu lähinnä sitoutumalla endogeeniseen VEGF-tekijään ja muodostamalla sen kanssa stabiilin, inaktiivisen kompleksin. Kuten muutkin suuret proteiinit, sekä vapaa että sitoutunut aflibersepti eliminoituvat todennäköisesti hitaammin muiden biologisten mekanismien kuten proteolyyttisen katabolian kautta.
Yli 2 mg/kg annoksilla vapaan afliberseptin puhdistuma oli noin 1,0 l/vrk ja terminaalinen puoliintumisaika 6 vrk.
Suurimolekyyliset proteiinit eivät puhdistu munuaisteitse, joten munuaisteitse tapahtuva eliminaatio on todennäköisesti hyvin vähäistä.
Lineaarisuus/ei-lineaarisuus
Lääkkeen jakautuminen riippuu kohdemolekyylistä, joten vapaan afliberseptin puhdistuma on nopeampi (ei-lineaarinen) alle 2 mg/kg annoksilla, mikä johtunee afliberseptin sitoutumisesta suuremmalla affiniteetilla endogeeniseen VEGF-tekijään. Annosvälillä 2–9 mg/kg havaittava lineaarinen puhdistuma johtuu todennäköisesti saturoitumattomista biologisista eliminaatiomekanismeista kuten proteiinikataboliasta.
Muut erityisryhmät
Iäkkäät
Ikä ei vaikuttanut vapaan afliberseptin farmakokinetiikkaan.
Rotu
Populaatioanalyysissä ei havaittu rotuun liittyvää vaikutusta.
Sukupuoli
Sukupuoli oli merkittävin vapaan afliberseptin puhdistuman ja tilavuuden potilaskohtaista vaihtelua selittävä kovariaatti. Miehillä vapaan afliberseptin puhdistuma oli 15,5 % suurempi ja jakautumistilavuus 20,6 % suurempi kuin naisilla. Nämä erot eivät vaikuta altistukseen, sillä annos perustuu painoon. Annosta ei tarvitse muuttaa sukupuolen perusteella.
Paino
Paino vaikutti vapaan afliberseptin puhdistumaan ja jakautumistilavuuteen, ja afliberseptialtistus suureni 29 % potilailla, joiden paino oli ≥ 100 kg.
Maksan vajaatoiminta
Zaltrap-hoidon käyttöä maksan vajaatoimintapotilaille ei ole tutkittu muodollisissa tutkimuksissa. Populaatiofarmakokinetiikan analyysiin otettiin tiedot 1507:stä erilaisia pitkälle edenneitä syöpätauteja sairastaneesta potilaasta, jotka saivat Zaltrap-hoitoa joko solunsalpaajahoidon kanssa tai ilman. Zaltrap-hoitoa sai 63 potilasta, joilla oli lievä maksan vajaatoiminta (kokonaisbilirubiinipitoisuus > 1,0 x – 1,5 x viitearvojen yläraja [ULN] ja ASAT mikä tahansa), ja 5 potilasta, joilla oli keskivaikea maksan vajaatoiminta (kokonaisbilirubiinipitoisuus > 1,5 x – 3 x ULN ja ASAT mikä tahansa). Afliberseptin puhdistuma ei muuttunut näillä lievää ja keskivaikeaa maksan vajaatoimintaa sairastaneilla potilailla. Vaikeaa maksan vajaatoimintaa sairastavista (kokonaisbilirubiinipitoisuus > 3 x ULN ja ASAT mikä tahansa) ei ole tietoa.
Munuaisten vajaatoiminta
Zaltrap-hoidon käyttöä munuaisten vajaatoimintapotilaille ei ole tutkittu muodollisissa tutkimuksissa. Populaatiofarmakokinetiikan analyysiin otettiin tiedot 1507:stä erilaisia pitkälle edenneitä syöpätauteja sairastaneesta potilaasta, jotka saivat Zaltrap-hoitoa joko solunsalpaajahoidon kanssa tai ilman. Tähän populaatioon kuului 549 potilasta, joilla oli lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma 50–80 ml/min), 96 potilasta, joilla oli keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma 30–50 ml/min) ja 5 potilasta, joilla oli vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma < 30 ml/min). Populaatiofarmakokinetiikan analyysissä ei havaittu kliinisesti merkittäviä eroja vapaan afliberseptin puhdistumassa eikä systeemisessä altistuksessa (AUC), kun lievää ja keskivaikeaa munuaisten vajaatoimintaa sairastavia potilaita verrattiin koko tutkittuun populaatioon 4 mg/kg Zaltrap-annoksia käytettäessä. Vaikeaa munuaisten vajaatoimintaa sairastaneiden kohdalla johtopäätöksiä ei voida tehdä, sillä tietoja on hyvin vähän. Vaikeaa munuaisten vajaatoimintaa sairastaneita potilaita oli vähän, ja altistus oli heillä samaa luokkaa kuin munuaistoiminnaltaan normaaleilla potilailla.
Prekliiniset tiedot turvallisuudesta
Toksikologia ja farmakologia eläimillä
Kun afliberseptia annettiin yhden tai kahden viikon välein makakiapinoiden laskimoon enintään 6 kuukauden ajan, seurauksena oli luumuutoksia (kasvulevyn, tukirangan ja muun luuston muutoksia) sekä nenäontelon, munuaisten, munasarjojen ja lisämunuaisten muutoksia. Afliberseptin aiheuttamia muutoksia havaittiin jo pienimmillä tutkituilla annoksilla, joiden tuottama altistus plasmassa oli lähes samaa luokkaa kuin hoitoannoksia käyttävien potilaiden altistus. Useimmat afliberseptin aiheuttamat muutokset korjautuivat 5 kuukauden lääkkeettömän vaiheen jälkeen luusto- ja nenäontelomuutoksia lukuun ottamatta. Useimpien löydösten katsottiin liittyvän afliberseptin farmakologiseen vaikutukseen.
Afliberseptin anto hidasti haavojen paranemista kanilla. Koko ihon paksuutta koskeneissa - ja ihoviiltomalleissa afliberseptin anto heikensi fibroosivastetta, uudissuonimuodostusta, epidermiksen hyperplasiaa/epiteelin uudiskasvua sekä vetolujuutta. Aflibersepti nosti normotensiivisten jyrsijöiden verenpainetta.
Karsinogeenisuus ja mutageenisuus
Afliberseptin karsinogeenisuutta ja mutageenisuutta ei ole arvioitu tutkimuksissa.
Hedelmällisyyden heikentyminen
Afliberseptilla ei ole tehty nimenomaan hedelmällisyyteen kohdistuvaa vaikutusta arvioivia eläintutkimuksia.
Toistuvien annosten toksisuutta arvioineen tutkimuksen tulokset viittaavat siihen, että aflibersepti saattaa heikentää lisääntymistoimintoja ja hedelmällisyyttä. Sukukypsillä naarasmakakeilla todettiin munasarjatoiminnan ja munarakkuloiden kehityksen estymistä. Myös eläinten normaali kiimakierto häiriintyi. Sukukypsillä urosmakakeilla havaittiin siittiöiden liikkuvuuden heikkenemistä ja siittiöiden morfologisten poikkeavuuksien ilmaantuvuuden suurenemista. Näitä vaikutuksia aiheuttava altistus ei eronnut potilaiden altistuksesta. Vaikutukset korjautuivat täysin 8–18 viikon kuluttua viimeisestä injektiosta.
Lisääntymis- ja kehitystoksisuus
Afliberseptin on todettu olevan alkiotoksinen ja teratogeeninen, kun sitä annettiin tiineiden kanien laskimoon 3 päivän välein organogeneesin aikana (tiineyspäivät 6–18) annoksilla, jotka olivat noin 1–15-kertaisia suhteessa ihmisen annokseen (4 mg/kg kahden viikon välein). Havaittuja vaikutuksia olivat emon painon lasku, sikiöiden resorptioiden määrän suureneminen sekä ulkoisten ja sisäelimiin ja luustoon kohdistuvien epämuodostumien ilmaantuvuuden suureneminen sikiöillä.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi, natriumkloridi, natriumsitraattidihydraatti, sitruunahappomonohydraatti, polysorbaatti 20, dinatriumfosfaatti, heptahydraatti, mononatriumfosfaatti, monohydraatti, natriumhydroksidi ja/tai kloorivetyhappo (pH:n säätelyyn), injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden eikä liuottimien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Laimennettuna infuusiopussissa
Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan 2–8 °C lämpötilassa ja 8 tunnin ajan 25 °C lämpötilassa.
Mikrobiologiselta kannalta infuusioneste on käytettävä välittömästi.
Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja säilytysolosuhteet ovat käyttäjän vastuulla. Ne ovat normaalisti kuitenkin enintään 24 tuntia 2–8 °C lämpötilassa, ellei laimennus tapahdu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZALTRAP infuusiokonsentraatti, liuosta varten
25 mg/ml (L:ei) 8 ml (1062,60 €)
PF-selosteen tieto
4 ml konsentraattia 5 ml injektiopullossa, joka on valmistettu kirkkaasta (tyypin I) borosilikaattilasista ja sinetöity siivekkeellisellä tulpalla, jossa on repäisykorkki ja jonka sisällä on päällystetty sinettilevy. Pakkauskoko: 1 tai 3 injektiopulloa.
8 ml konsentraattia 10 ml injektiopullossa, joka on valmistettu kirkkaasta (tyypin I) borosilikaattilasista ja sinetöity siivekkeellisellä tulpalla, jossa on repäisykorkki ja jonka sisällä on päällystetty sinettilevy. Pakkauskoko: 1 injektiopullo.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Konsentraatti on kirkas, väritön tai vaaleankeltainen liuos.
Käyttö- ja käsittelyohjeet
Zaltrap on steriili, säilöntäaineeton ja pyrogeeniton konsentraatti, joten infuusionesteen (liuos) valmistelusta huolehtii terveydenhuoltohenkilöstö turvallisuusohjeita noudattaen ja aseptista tekniikkaa käyttäen.
Zaltrap-valmisteen käsittelyssä on noudatettava varovaisuutta, ja eristyslaitteiden, henkilösuojainten (esim. hansikkaat) ja valmisteluohjeiden käyttöön on kiinnitettävä huomiota.
Infuusionesteen valmistelu
- Zaltrap-injektiopullo tarkastetaan silmämääräisesti ennen käyttöä. Konsentraattiliuoksen on oltava kirkasta, eikä siinä saa olla hiukkasia.
- Vedä potilaan tarvitseman annoksen mukainen tilavuus Zaltrap-konsentraattia injektiopullosta. Infuusionesteen valmisteluun saatetaan tarvita useampia kuin yksi injektiopullo.
- Laimenna konsentraatti haluttuun antotilavuuteen NaCl-liuoksella 9 mg/ml (0,9 %) tai 5 % glukoosi-infuusionesteellä. Lopullisen Zaltrap-infuusionesteen afliberseptipitoisuuden on oltava 0,6–8 mg/ml.
- Käytä DEHP-ftalaattia sisältävästä PVC-muovista tai polyolefiinista valmistettuja infuusiopusseja.
- Laimennettu liuos tarkastetaan silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. Jos siinä on värimuutoksia tai hiukkasia, käyttövalmis liuos on hävitettävä.
- Zaltrap-injektiopullo on tarkoitettu vain yhtä käyttökertaa varten. Injektiopullon tulppaa ei saa läpäistä uudelleen ensimmäisen läpäisykerran jälkeen. Käyttämätön konsentraatti tulee hävittää.
Infuusioliuoksen antaminen
Laimennettu Zaltrap-liuos annetaan infuusiolaitteistolla, jossa on 0,2 mikronin polyeetterisulfonisuodatin.
Infuusiolaitteiston on oltava jotakin seuraavista materiaaleista:
- polyvinyylikloridi (PVC), joka sisältää bis(2-etyyliheksyyli)ftalaattia (DEHP)
- DEHP-vapaa PVC-muovi, joka sisältää trioktyylitrimellitaattia (TOTM)
- polypropeeni
- polyeteenipinnoitettu PVC
- polyuretaani
Polyvinyylideenifluoridista (PVDF) tai nailonista tehtyjä suodattimia ei saa käyttää.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZALTRAP infuusiokonsentraatti, liuosta varten
25 mg/ml 8 ml
- Ei korvausta.
ATC-koodi
L01XX44
Valmisteyhteenvedon muuttamispäivämäärä
05.10.2023
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi