
SOLIRIS infuusiokonsentraatti, liuosta varten 300 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Lääkärin opas
Potilas
Vaikuttavat aineet ja niiden määrät
Ekulitsumabi on humanisoitu monoklonaalinen (IgG2/4κ)-vasta-aine. Sitä tuotetaan NS0-solulinjassa yhdistelmä-DNA-tekniikalla.
Yksi 30 ml:n injektiopullo sisältää 300 mg ekulitsumabia (10 mg/ml).
Laimennuksen jälkeen infuusioliuoksen lopullinen pitoisuus on 5 mg/ml.
Apuaine(et), joiden vaikutus tunnetaan:
natrium (5 mmol/injektiopullo), polysorbaatti 80 (6,6 mg/injektiopullo)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Soliris on tarkoitettu sellaisten aikuisten ja lasten hoitoon, joilla on:
- kohtauksittainen yöllinen hemoglobinuria (paroksysmaalinen nokturnaalinen hemoglobinuria, PNH). Kliinisestä hyödystä on näyttöä potilailla, joilla on hemolyysia ja suureen tautiaktiivisuuteen viittaava kliininen oire (oireita), riippumatta siitä, onko potilas saanut verensiirtoja (ks. kohta Farmakodynamiikka).
- atyyppinen hemolyyttis-ureeminen oireyhtymä (aHUS) (ks. kohta Farmakodynamiikka).
- refraktorinen yleistynyt myasthenia gravis (gMG) vähintään 6-vuotiailla potilailla, joilla on todettu asetyylikoliinireseptorin (AChR) vasta-aineita (ks. kohta Farmakodynamiikka).
Soliris on tarkoitettu aikuisilla:
- neuromyelitis optica -kirjon häiriö (NMOSD) hoitoon potilailla, joilla on todettu akvaporiini 4:n (AQP4) vasta-aineita ja joiden taudinkulku on relapsoiva (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa antaa vain terveydenhoitoalan ammattilainen hyväksytyn käyttöaiheen hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Soliris-hoitoa saa antaa vain terveydenhoitoalan ammattilainen hematologisia häiriöitä, munuaissairautta, neuromuskulaarista tai neuroinflammatorista sairautta sairastavien potilaiden hoitoon perehtyneen lääkärin valvonnassa.
Kotona annettavaa infuusiota voidaan harkita potilaille, jotka ovat sietäneet terveysasemalla/sairaalassa annettuja infuusioita hyvin. Päätös potilaan kotona annettavasta infuusiosta tulee tehdä hoitavan lääkärin arvioinnin ja suosituksen jälkeen. Toimenpiteen suorittaa pätevä terveydenhuollon ammattilainen.
Annostus
Kohtauksittainen yöllinen hemoglobinuria (PNH)aikuisilla
PNH:n annostusohjelma aikuisille potilaille (≥ 18-vuotiaille) koostuu 4 viikon alkuvaiheesta ja sen jälkeisestä ylläpitovaiheesta:
- Alkuvaihe: 600 mg Soliris-valmistetta annetaan 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona joka viikko 4 ensimmäisen viikon ajan
- Ylläpitovaihe: 900 mg Soliris-valmistetta annetaan 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona viidennen viikon ajan, minkä jälkeen 900 mg Soliris-valmistetta annetaan 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona 14 ± 2 päivän välein (ks. kohta Farmakodynamiikka).
Atyyppinen hemolyyttis-ureeminen oireyhtymä (aHUS), refraktorinen yleistynyt myasthenia gravis (gMG) ja neuromyelitis optica -kirjon häiriö (NMOSD)aikuisilla
aHUS:n, refraktorisen gMG:n ja NMOSD:n annostusohjelma aikuisille potilaille (≥ 18-vuotiaille) koostuu 4 viikon alkuvaiheesta ja sen jälkeisestä ylläpitovaiheesta:
- Alkuvaihe: 900 mg Soliris-valmistetta annetaan 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona joka viikko 4 ensimmäisen viikon ajan
- Ylläpitovaihe: 1 200 mg Soliris-valmistetta annetaan 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona viidennen viikon ajan, minkä jälkeen 1 200 mg Soliris-valmistetta annetaan 25−45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona 14 ± 2 päivän välein (ks. kohta Farmakodynamiikka).
Refraktorinen gMG
Käytettävissä olevien tietojen mukaan kliininen vaste saavutetaan Soliris-hoidossa yleensä viikkoon 12 mennessä. Hoidon keskeyttämistä on harkittava sellaisten potilaiden kohdalla, joilla ei todeta näyttöä hoidon hyödystä viikkoon 12 mennessä.
Pediatriset potilaat, joilla on PNH, aHUS tai refraktorinen gMG
Pediatrisia PNH-, aHUS- tai refraktorista gMG:tä sairastavia potilaita, joiden paino on ≥ 40 kg, hoidetaan aikuisten annostussuositusten mukaan.
Alle 40 kg painavien pediatristen PNH- ja aHUS-potilaiden sekä refraktorista gMG:tä sairastavien potilaiden Soliris-annostusohjelma on:
| Potilaan paino | Alkuvaihe | Ylläpitovaihe |
| 30 – < 40 kg | 600 mg viikossa ensimmäisten 2 viikon ajan | 900 mg viikolla 3, minkä jälkeen 900 mg kahden viikon välein |
| 20 – < 30 kg | 600 mg viikossa ensimmäisten 2 viikon ajan | 600 mg viikolla 3, minkä jälkeen 600 mg kahden viikon välein |
| 10 – < 20 kg | 600 mg:n kerta-annos viikolla 1 | 300 mg viikolla 2, minkä jälkeen 300 mg kahden viikon välein |
| 5 – < 10 kg | 300 mg:n kerta-annos viikolla 1 | 300 mg viikolla 2, minkä jälkeen 300 mg kolmen viikon välein |
On vain vähän tietoa pediatrisista potilaista, joilla on PNH tai refraktorinen gMG ja jotka painavat alle 40 kg. Solirisin annostus alle 40 kg painaville pediatrisille potilaille, joilla on PNH tai refraktorinen gMG, on sama kuin suositeltu painon mukainen annos pediatrisille aHUS-potilaille. Perustuen saatavilla oleviin farmakokineettisiin/farmakodynaamisiin tietoihin potilaista, joilla on aHUS ja PNH ja jotka ovat saaneet Soliris-hoitoa, tällä pediatrisille potilaille tarkoitetulla painon mukaisella annosohjelmalla saavutettavan teho- ja turvallisuusprofiilin odotetaan olevan samankaltainen kuin aikuisilla. Alle 40 kg painaville potilaille, joilla on refraktorinen gMG, tällä painonmukaisella annosohjelmalla saavutettavan teho- ja turvallisuusprofiilin odotetaan myös olevan samankaltainen kuin aikuisilla.
Soliris-lisäannos on tarpeen samanaikaisen plasmafereesin, plasmanvaihdon tai tuoreen jääplasman infusoinnin yhteydessä, kuten alla on kuvattu:
| Plasman antotapa | Viimeisin Soliris-annos | Soliris-lisäannos jokaisen plasmafereesin/plasmanvaihdon/plasmainfuusion yhteydessä | Soliris-lisäannoksen antoajankohta |
| Plasmafereesi tai plasmanvaihto | 300 mg | 300 mg jokaisen plasmafereesi- tai plasmanvaihtokerran yhteydessä | 60 minuutin kuluessa jokaisen plasmafereesin tai plasmanvaihdon jälkeen |
| ≥ 600 mg | 600 mg jokaisen plasmafereesi- tai plasmanvaihtokerran yhteydessä | ||
| Infuusiona annettu tuore jääplasma | ≥ 300 mg | 300 mg jokaisen tuoreen jääplasmainfuusion yhteydessä | 60 minuuttia ennen kutakin tuoretta jääplasmainfuusiota |
Soliris-lisäannos on tarpeen samanaikaisen laskimonsisäisen immunoglobuliinihoidon (IVIg) yhteydessä, kuten alla on kuvattu (ks. myös kohta Yhteisvaikutukset):
| Viimeisin Soliris-annos | Soliris-lisäannos | Soliris-lisäannoksen antoajankohta |
| ≥ 900 mg | 600 mg / IVIg-sykli | Mahdollisimman pian IVIg-syklin jälkeen |
| ≤ 600 mg | 300 mg / IVIg-sykli |
Lyhenne: IVIg = laskimonsisäinen immunoglobuliinihoito
Hoidon seuranta
aHUS-potilailla on tarkkailtava tromboottisen mikroangiopatian (TMA) oireita ja löydöksiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet aHUS-potilaiden laboratorioseuranta).
Soliris-hoitoa suositellaan jatkamaan potilaan koko eliniän, ellei Soliris-hoidon lopettamisen ole kliinisesti aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät
Soliris-hoitoa voidaan antaa 65-vuotiaille ja sitä vanhemmille potilaille. Näyttöä erityisten varotoimien tarpeellisuudesta iäkkäitä ihmisiä hoidettaessa ei ole. Kokemukset Soliris-hoidosta tällä potilaspopulaatiolla ovat kuitenkin vielä vähäiset.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse säätää (ks. kohta Farmakodynamiikka).
Maksan vajaatoiminta
Solirisin turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Soliris‑valmisteen turvallisuutta ja tehoa alle 6 vuoden ikäisten lasten refraktorisen gMG:n hoidossa ei ole varmistettu.
Soliris‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten NMOSD:n hoidossa ei ole varmistettu.
Antotapa
Ei saa antaa nopeana injektiona (bolusinjektiona) laskimoon. Solirisin saa antaa vain laskimonsisäisenä infuusiona jäljempänä esitettyjen ohjeiden mukaan.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Laimennettu Soliris-liuos tulee antaa 25–45 minuuttia (35 minuuttia ± 10 minuuttia) kestävänä laskimonsisäisenä infuusiona aikuisille ja 1–4 tuntia kestävänä infuusiona alle 18-vuotiaille pediatrisille potilaille painovoimalla toimivalla nesteensiirtolaitteella, ruiskutyyppisellä pumpulla tai infuusiopumpulla. Laimennettua Soliris-liuosta ei tarvitse suojata valolta infuusion aikana.
Potilaita tulee seurata yhden tunnin ajan infuusion jälkeen. Jos haittavaikutuksia ilmenee Soliris-hoidon antamisen aikana, infuusionopeutta voidaan hidastaa tai infuusio voidaan keskeyttää lääkärin harkinnan mukaan. Jos infuusionopeutta hidastetaan, infuusion kokonaisaika ei saa ylittää aikuisilla kahta tuntia eikä alle 18-vuotiailla pediatrisilla potilailla neljää tuntia.
Kotona annettavien infuusioiden turvallisuudesta on rajallisesti tietoa, joten lisävarotoimenpiteitä kuten valmiutta infuusioreaktioiden tai anafylaksian kiireelliseen hoitoon potilaan kotona suositellaan. Infuusioreaktiot on kuvattu valmisteyhteenvedon kohdissa Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
Vasta-aiheet
Yliherkkyys ekulitsumabille, hiiren proteiineille tai kohdassa Apuaineet mainituille apuaineille.
Soliris-hoitoa ei saa aloittaa potilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet):
- joilla on hoitamaton Neisseria meningitidis -infektio
-
joilla ei ole voimassa olevaa Neisseria meningitidis -rokotusta, paitsi jos estohoitoa sopivilla antibiooteilla käytetään siihen saakka, kunnes rokotuksesta on kulunut kaksi viikkoa.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Solirisin ei odoteta vaikuttavan anemian aplastiseen komponenttiin potilailla, joilla on PNH.
Meningokokki-infektio
Vaikutusmekanisminsa vuoksi Solirisin käyttö lisää potilaan alttiutta saada meningokokki-infektio (Neisseria meningitidis). Minkä tahansa seroryhmän aiheuttamaa meningokokkia voi esiintyä. Infektiovaaran vähentämiseksi kaikki potilaat tulee rokottaa vähintään 2 viikkoa ennen Soliris-hoidon antamista, ellei Soliris-hoidon viivästymisestä aiheutuva riski ole suurempi kuin meningokokki-infektion riskit. Potilaille, jotka aloittavat Soliris-hoidon ennen kuin tetravalentista meningokokkirokotuksesta on kulunut kaksi viikkoa, on annettava estohoitoa sopivilla antibiooteilla niin kauan, kunnes rokotuksesta on kulunut kaksi viikkoa. Rokotteita kaikkia seroryhmiä vastaan, joille rokote on saatavilla, mukaan lukien A, C, Y, W 135 ja B, suositellaan yleisesti patogeenisten meningokokkiseroryhmien estoon. Potilaat on rokotettava ja uudelleen rokotettava voimassa olevien kansallisten rokotussuositusten mukaisesti.
Rokotus voi aktivoida komplementtia entistä enemmän. Sen seurauksena komplementtivälitteisiä tauteja kuten PNH:ta, aHUS:aa, refraktorista gMG:tä ja NMOSD:tä, sairastavilla potilailla perussairauden merkit ja oireet kuten hemolyysi (PNH:ssa) tai tromboottinen mikroangiopatia (aHUS:ssa), voivat pahentua, tai potilas voi saada pahenemisvaiheen (refraktorinen gMG) tai relapsin (NMOSD). Potilaita on siis suositellun rokotuksen jälkeen seurattava tarkoin taudin oireiden varalta.
Rokotus ei ehkä ole riittävä meningokokki-infektion estämiseksi. Antibakteeristen aineiden asianmukaista käyttöä koskevia virallisia ohjeita tulee noudattaa. Soliris-hoitoa saaneilla potilailla on raportoitu vakavia ja kuolemaan johtaneita meningokokki-infektioita. Sepsis on yleinen meningokokki-infektioiden ilmentymä Soliris-hoitoa saavilla potilailla (ks. kohta Haittavaikutukset). Kaikkia potilaita tulee seurata meningokokki-infektion varhaisten oireiden varalta. Jos infektiota epäillään, tulee oireet arvioida välittömästi ja hoitaa sopivilla antibiooteilla tarpeen mukaan. Potilaalle tulee kertoa näistä oireista ja toimenpiteistä sen varalta, että hänen on välittömästi haettava lääkärinhoitoa. Lääkärin on kerrottava potilaalle Soliris-hoidon hyödyistä ja riskeistä ja annettava potilaalle potilasopas ja potilaskortti (katso kuvaus pakkausselosteesta).
Muut systeemiset infektiot
Soliris-hoidon antamisessa tulee sen vaikutusmekanismin vuoksi noudattaa varovaisuutta systeemisiä infektioita sairastavilla potilailla. Potilaiden infektioalttius varsinkin Neisseria-bakteerien ja kapselillisten bakteerien aiheuttamille infektioille saattaa kasvaa. Neisseria-lajien (muiden kuin Neisseria meningitidisin) aiheuttamia vakavia infektioita, mukaan lukien disseminoituneita gonokokki-infektioita, on raportoitu. Potilaiden tietämystä mahdollisista vakavista infektioista ja niiden oireista tulee lisätä antamalla heille tietoja pakkausselosteesta. Lääkärien tulee neuvoa potilaille, miten ehkäistä tippuri-infektioita.
Infuusioon liittyvät reaktiot
Solirisin antaminen voi aiheuttaa infuusioon liittyviä reaktioita tai immunogeenisuutta, joka voi aiheuttaa allergisia tai yliherkkyysreaktioita (myös anafylaksiaa). Kliinisissä tutkimuksissa yksi refraktorista gMG:tä sairastava potilas (0,9 %) sai infuusioon liittyvän reaktion, joka vaati Soliris-hoidon keskeyttämisen. Kukaan pediatrisista potilaista, joilla oli PNH, aHUS, refraktorinen gMG tai NMOSD, ei saanut Soliris-hoidon keskeyttämisen vaativaa infuusioon liittyvää reaktiota. Solirisin antaminen tulee keskeyttää kaikilla potilailla, jotka saavat vaikeita infuusioon liittyviä reaktioita. Näille potilaille tulee antaa asianmukaista hoitoa.
Immunogeenisyys
Soliris-hoitoa saaneilla potilailla havaittiin harvoin vasta-ainevaste kaikissa kliinisissä tutkimuksissa. Alhaisen vasta-ainevasteen esiintyvyyden (3,4 %) raportoitiin olleen lumelääkekontrolloiduissa PNH-tutkimuksissa vastaava kuin lumelääkettä saaneilla potilailla (4,8 %). Soliris-hoitoa saaneista aHUS-potilaista 3:lla 100:sta (3 %:lla) todettiin vasta-aineita Solirisille yhdistelmä-ECL-tutkimuksessa. aHUS-potilaista yhdellä 100:sta (1 %:lla) neutraloivien vasta-aineiden arvot olivat matalia positiivisia.
Lumelääkekontrolloidussa refraktorista gMG:tä koskeneessa tutkimuksessa yhdellekään (0/62) Soliris-hoitoa saaneista potilaista ei kehittynyt lääkevasta-aineita 26 viikon aktiivisen hoidon aikana, kun taas refraktorista gMG:tä sairastavilla potilailla tehdyssä jatkotutkimuksessa yhteensä kolme 117 potilaasta (2,6 %) oli ADA-positiivisia jollakin lähtötilanteen jälkeisellä käynnillä. Positiiviset ADA-tulokset vaikuttivat ohimeneviltä, koska positiivisia tittereitä ei saatu myöhemmillä käynneillä, eikä näillä potilailla havaittu positiivisten ADA-tittereiden vaikutuksiin viittaavia kliinisiä löydöksiä.
NMOSD-potilailla tehdyssä lumelääkekontrolloidussa tutkimuksessa 2:lla 95:stä (2,1 %:lla) Soliris-hoitoa saaneesta potilaasta todettiin lääkevasta-ainevaste lähtötilanteen jälkeen. Kummallakaan potilaalla ei todettu neutraloivia vasta-aineita. Positiiviset ADA-näytteet olivat heikosti positiivisia ja positiivisuus oli ohimenevää.
Vasta-aineiden kehittymisen ja kliinisen vasteen tai haittavaikutusten välillä ei ole todettu korrelaatiota.
Immunisaatio
PNH- ja aHUS-potilaiden ja potilaiden, joilla on refraktorinen gMG tai NMOSD, immunisaatio on suositeltavaa aloittaa voimassa olevien rokotusohjeiden mukaisesti ennen Soliris-hoidon aloittamista. Lisäksi kaikki potilaat tulee rokottaa meningokokki-infektioita vastaan vähintään 2 viikkoa ennen Solirisin antamista, ellei Soliris-hoidon viivästymisestä aiheutuva riski ole suurempi kuin meningokokki-infektion riskit. Jos potilas aloittaa Soliris-hoidon ennen kuin tetravalentista meningokokkirokotuksesta on kulunut kaksi viikkoa, hänelle on annettava estohoitona sopivia antibiootteja, kunnes rokotuksesta on kulunut kaksi viikkoa. Rokotteita kaikkia seroryhmiä vastaan, joilla rokote on saatavilla, mukaan lukien A, C, Y, W 135 ja B, suositellaan yleisesti patogeenisten meningokokkiseroryhmien estoon. Potilaat on rokotettava ja uudelleen rokotettava voimassa olevien kansallisten rokotussuositusten mukaisesti (ks. Meningokokki-infektio).
Alle 18-vuotiaat potilaat on rokotettava Haemophilus influenzae- ja pneumokokki-infektioita vastaan, ja rokotuksissa on noudatettava tarkoin kutakin ikäryhmää koskevia kansallisia rokotussuosituksia.
Rokotus voi aktivoida komplementtia entistä enemmän. Sen seurauksena komplementtivälitteisiä tauteja, kuten PNH:ta, aHUS:aa, refraktorista gMG:tä tai NMOSD:tä, sairastavilla potilailla perussairauden merkit ja oireet kuten hemolyysi (PNH:ssa) tai tromboottinen mikroangiopatia (aHUS:ssa), voivat pahentua, tai potilas voi saada pahenemisvaiheen (refraktorinen gMG) tai relapsin (NMOSD). Potilaita on siis suositellun rokotuksen jälkeen seurattava tarkoin taudin oireiden varalta.
Antikoagulanttihoito
Soliris-hoidon ei pitäisi aiheuttaa muutoksia antikoagulanttihoitoon.
Immunosuppressiohoidot
Refraktorinen gMG
Kun immunosuppressiohoitoja ja koliiniesteraasin estäjähoitoja vähennetään tai lopetetaan, potilaita on seurattava tarkoin sairauden pahenemisesta kertovien merkkien varalta.
Neuromyelitis optica -kirjon häiriö
Kun immunosuppressiohoitoa vähennetään tai hoito lopetetaan, potilaita on seurattava tarkoin NMOSD:n mahdollisesta relapsista kertovien merkkien ja oireiden varalta.
PNH-potilaiden laboratorioseuranta
PNH-potilaita tulee seurata intravaskulaarisen hemolyysin merkkien ja oireiden varalta, mukaan lukien seerumin laktaattidehydrogenaasin (LDH) määrän seuranta. Soliris-hoitoa saavia potilaita tulee seurata vastaavalla tavalla intravaskulaarisen hemolyysin varalta mittaamalla LDH-määriä. Potilaiden annosta täytyy ehkä muuttaa suositellun 14 ± 2 päivän annosteluohjelman puitteissa ylläpitovaiheen aikana (korkeintaan joka 12. päivä).
aHUS-potilaiden laboratorioseuranta
Soliris-hoitoa saavilla aHUS-potilailla on seurattava tromboottisen mikroangiopatian ilmaantumista määrittämällä trombosyyttimäärä, seerumin LDH ja seerumin kreatiniini, minkä perusteella potilaiden annosta saattaa olla tarpeen säätää suositellun 14 ± 2 päivän annosteluohjelman puitteissa ylläpitovaiheen aikana (korkeintaan joka 12. päivä).
PNH-potilaan hoidon keskeyttäminen
Jos PNH-potilas keskeyttää Soliris-hoidon, potilasta tulee seurata tarkoin vakavan intravaskulaarisen hemolyysin merkkien ja oireiden varalta. Vakava hemolyysi voidaan havaita seerumin LDH-määrästä, joka nousee hoitoa edeltänyttä tasoa suuremmaksi, ja mistä tahansa seuraavista tekijöistä: yli 25 prosentin absoluuttinen PNH-kloonin koon pienentyminen (verensiirrosta johtuvan laimentumisen puuttuessa) yhden viikon kuluessa tai nopeammin, hemoglobiinin määrä < 5 g/dl tai vähentyminen > 4 g/dl yhden viikon kuluessa tai nopeammin, angina pectoris, psyykkisen tilan muutos, 50 prosentin nousu seerumin kreatiniinin määrässä tai tromboosi. Soliris-hoidon keskeyttävää potilasta tulee seurata vakavan hemolyysin ja muiden reaktioiden varalta vähintään 8 viikon ajan.
Jos vakava hemolyysi ilmenee Soliris-hoidon keskeyttämisen jälkeen, seuraavia menettelyjä tai hoitoja tulee harkita: verensiirto (punasoluja) tai verenvaihto, jos PNH-punasolujen osuus on > 50 % punasolujen kokonaismäärästä virtaussytometrialla mitattuna, antikoagulaatio, kortikosteroidit tai Soliris-hoidon uudelleen aloittaminen. Kliinisissä PNH-tutkimuksissa Soliris-hoito-ohjelman keskeytti 16 potilasta. Vakavaa hemolyysiä ei ilmennyt.
aHUS-potilaan hoidon keskeyttäminen
Tromboottisia mikroangiopatiakomplikaatioita on havaittu joillakin potilailla jo 4 viikon kuluttua ja enintään 127 viikon kuluttua Soliris-hoidon keskeyttämisestä. Hoidon keskeyttämistä pitää harkita vain, jos se on lääketieteellisesti aiheellista.
Kliinisissä aHUS-tutkimuksissa 61 potilasta (21 pediatrista potilasta) keskeytti Soliris-hoidon. Seuranta-ajan mediaani oli 24 viikkoa. Hoidon keskeyttämisen jälkeen 12 potilaalla havaittiin 15 vaikeaa tromboottista mikroangiopatiakomplikaatiota, ja 2 vaikeaa tromboottista mikroangiopatiakomplikaatiota ilmeni lisäksi 2 potilaalla, jotka käyttivät hyväksyttyä Soliris-annostusta pienempää annostusta (ks. Annostus ja antotapa). Vaikeita tromboottisia mikroangiopatiakomplikaatioita esiintyi potilailla riippumatta siitä, oliko heillä tunnistettu geenimutaatio, suuren riskin polymorfismi tai autovasta-aine. Näillä potilailla esiintyi vakavia lääketieteellisiä lisäkomplikaatioita, joita olivat mm. vaikea munuaistoiminnan heikentyminen, sairauteen liittyvä sairaalahoito ja eteneminen dialyysia vaativaan loppuvaiheen munuaissairauteen. Vaikka Soliris-hoito aloitettiin uudelleen hoidon keskeyttämisen jälkeen, munuaissairaus eteni yhdellä potilaalla loppuvaiheeseen.
Jos aHUS-potilas keskeyttää Soliris-hoidon, potilasta tulee seurata tarkoin komplikaationa ilmaantuvan vaikea-asteisen tromboottisen mikroangiopatian merkkien ja oireiden varalta. Seuranta ei välttämättä riitä vaikeiden mikroangiopatiakomplikaatioiden ennustamiseen eikä ehkäisyyn aHUS-potilailla Soliris-hoidon keskeyttämisen jälkeen.
Hoidon lopettamisen jälkeen komplikaationa ilmaantuva vaikea-asteinen tromboottinen mikroangiopatia voidaan tunnistaa (i) toteamalla mitkä tahansa kaksi seuraavista veriarvoista tai määrittämällä jokin seuraavista veriarvoista toistuvasti: trombosyyttimäärän väheneminen vähintään 25 % hoitoa edeltävästä määrästä tai Soliris-hoidon aikana todetusta suurimmasta trombosyyttimäärästä, seerumin kreatiniinipitoisuuden suureneminen vähintään 25 % hoitoa edeltävään pitoisuuteen tai Soliris-hoidon aikana todettuun pienimpään pitoisuuteen verrattuna tai seerumin laktaattidehydrogenaasipitoisuuden suureneminen vähintään 25 % hoitoa edeltävään pitoisuuteen tai Soliris-hoidon aikana todettuun pienimpään pitoisuuteen verrattuna, tai (ii) toteamalla jokin seuraavista: mielentilan muutos tai kouristuskohtaus, angina pectoris tai hengenahdistus tai verisuonitukos.
Jos vaikea-asteinen tromboottinen mikroangiopatia ilmaantuu komplikaationa Soliris-hoidon lopettamisen jälkeen, on harkittava Soliris-hoidon aloittamista uudelleen, elintoimintoja tukevaa plasmanvaihtoa/plasmainfuusiota tai asianmukaisia elinkohtaisia tukitoimenpiteitä, kuten dialyysihoitoa tukemaan munuaisten toimintaa, mekaanista ventilaatiota tukemaan hengitystoimintoja tai antikoagulantteja.
Refraktorisen gMG:n hoidon lopettaminen:
Soliris-valmisteen käyttöä refraktorisen gMG:n hoitoon on tutkittu vain asetelmassa, jossa lääkkeenanto on ollut jatkuvaa. Soliris-hoidon lopettaneita potilaita on seurattava tarkoin sairauden pahenemisen merkkien ja oireiden varalta.
NMOSD:n hoidon lopettaminen:
Soliris-valmisteen käyttöä NMOSD:n hoitoon on tutkittu vain asetelmassa, jossa lääkkeenanto on ollut jatkuvaa, eikä Soliris-hoidon lopettamisen vaikutuksia ole selvitetty. Soliris-hoidon lopettaneita potilaita on seurattava tarkoin NMOSD:n mahdollisen relapsin merkkien ja oireiden varalta.
Hoito-oppaat
Jokaisen lääkärin, joka aikoo määrätä Soliris-hoitoa, on varmistettava, että hän on tutustunut valmisteen määräämistä koskevaan oppaaseen terveydenhuollon ammattilaisille. Lääkärin on keskusteltava Soliris-hoidon riskeistä ja hyödyistä potilaan kanssa ja annettava potilaalle potilasopas ja potilaskortti.
Potilaalle on kerrottava, että jos hänelle ilmaantuu kuumetta, päänsärkyä, johon liittyy kuumetta ja/tai niskan jäykkyyttä tai herkkyyttä valolle, hänen on hakeuduttava heti lääkäriin, koska nämä oireet saattavat viitata meningokokki-infektioon.
Apuaine(et), joiden vaikutus tunnetaan
Natrium
9 mg/ml:n vahvuisella natriumkloridi-injektionesteellä (0,9 %) laimentamisen jälkeen tämän lääkevalmisteen suurin annos sisältää 0,88 g natriumia per 240 ml, mikä vastaa 44,0 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
4,5 mg/ml:n vahvuisella natriumkloridi-injektionesteellä (0,45 %) laimentamisen jälkeen tämän lääkevalmisteen suurin annos sisältää 0,67 g natriumia per 240 ml, mikä vastaa 33,5 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Polysorbaattia 80
Tämä lääkevalmiste sisältää 6,6 mg polysorbaatti 80:aa per injektiopullo (30 ml:n injektiopullo), joka vastaa enintään 0,66 mg:aa/painokilo aikuispotilaiden ja yli 10 kg painavien lapsipotilaiden suurimmalla annoksella ja enintään 1,32 mg:aa/painokilo 5 – alle 10 kg painavien lapsipotilaiden suurimmalla annoksella. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Ekulitsumabi saattaa heikentää rituksimabin odotettuja farmakodynaamisia vaikutuksia, koska se saattaa estää rituksimabin aiheuttamaa komplementtiriippuvaista sytotoksisuutta.
Plasmanvaihdon, plasmafereesin, infuusiona annetun tuoreen jääplasman ja laskimonsisäisen immunoglobuliinihoidon (IVIg) on osoitettu pienentävän ekulitsumabin pitoisuutta seerumissa. Ekulitsumabin lisäannos on tarpeen näiden yhteydessä. Ks. kohdasta Annostus ja antotapa ohjeet samanaikaisesta plasmanvaihdosta, plasmafereesista, tuoreen jääplasman infusoinnista ja IVIg-hoidosta.
Ekulitsumabin samanaikainen käyttö laskimonsisäisen immunoglobuliinihoidon (IVIg) yhteydessä saattaa vähentää ekulitsumabin tehoa. Potilasta on seurattava huolellisesti ekulitsumabin tehon heikentymisen varalta.
Ekulitsumabin samanaikainen käyttö vastasyntyneen Fc-reseptorin (FcRn) estäjien kanssa saattaa vähentää systeemistä ekulitsumabialtistusta ja vähentää ekulitsumabin tehoa. Potilasta on seurattava huolellisesti ekulitsumabin tehon heikentymisen varalta.
Raskaus ja imetys
Naisille, jotka voivat tulla raskaaksi, tulee harkita riittävän ehkäisyn käyttöä ekulitsumabihoidon ajaksi ja vähintään 5 kuukaudeksi viimeisen annoksen jälkeen.
Raskaus
Ekulitsumabihoitoa saaville raskaana oleville naisille ei ole tehty hyvin kontrolloituja tutkimuksia. Tiedot rajallisesta määrästä ekulitsumabille altistuneita raskauksia (alle 300 raskaudesta) eivät viittaa lisääntyneeseen sikiön epämuodostumien tai sikiöön tai vastasyntyneeseen kohdistuvan toksisuuden riskiin. Hyvin kontrolloitujen tutkimusten puutteesta johtuen asiasta ei kuitenkaan ole täyttä varmuutta. Raskaana olevien naisten kohdalla yksilöllinen hyöty-riskiarviointi on siksi suositeltavaa ennen ekulitsumabihoidon aloittamista ja hoidon aikana. Jos raskaudenaikaisen hoidon katsotaan olevan tarpeen, äidin ja sikiön huolellista, paikallisten suositusten mukaista seurantaa suositellaan.
Ekulitsumabin vaikutusta eläinten lisääntymiseen ei ole tutkittu (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ihmisen IgG-vasta-aineiden tiedetään läpäisevän ihmisen istukan, ja näin ollen on mahdollista, että ekulitsumabilla on komplementin terminaalista osaa estävä vaikutus sikiön verenkierrossa. Soliris-valmistetta tulee siksi antaa raskaana oleville naisille vain, jos se on selvästi tarpeen.
Imetys
Ei ole odotettavissa vaikutuksia vastasyntyneisiin tai imeväisiin, sillä niukat saatavissa olevat tiedot viittaavat siihen, ettei ekulitsumabi erity ihmisen rintamaitoon. Saatavissa olevien tietojen niukkuuden vuoksi rintaruokinnan hyödyt kehitykselle ja terveydelle tulee kuitenkin huomioida yhdessä äidin kliinisen ekulitsumabin tarpeen ja rintaruokittavaan lapseen mahdollisesti kohdistuvien, ekulitsumabista tai äidin perussairaudesta johtuvien haittavaikutusten kanssa.
Hedelmällisyys
Erityisiä tutkimuksia ekulitsumabin vaikutuksesta hedelmällisyyteen ei ole tehty.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Soliris-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Supportiivista turvallisuustietoa saatiin 33:sta kliinisestä tutkimuksesta, joiden puitteissa 1 555 potilasta komplementtivälitteisiä tauteja sairastavista populaatioista (mukaan lukien PNH, aHUS, refraktorinen gMG ja NMOSD) altistui ekulitsumabille. Yleisin haittavaikutus oli päänsärky (ilmeni yleisimmin annostelun alkuvaiheessa), ja vakavin haittavaikutus oli meningokokki-infektio.
Haittavaikutustaulukko
Taulukossa 1 on esitetty spontaaniraportoinnissa ja loppuun suoritetuissa kliinisissä ekulitsumabitutkimuksissa (mukaan lukien PNH- ja aHUS-tutkimuksissa sekä refraktorista gMG:tä ja NMOSD:tä koskevissa tutkimuksissa) raportoidut haittavaikutukset. Ekulitsumabin käyttöön liittyvät haittavaikutukset on lueteltu elinjärjestelmän ja suositettavan termin mukaan jaettuina hyvin yleisiin (≥1/10), yleisiin (≥1/100, <1/10), melko harvinaisiin (≥1/1 000, <1/100), harvinaisiin (≥1/10 000, <1/1 000) ja tuntemattomiin (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Kliinisissä ekulitsumabitutkimuksissa raportoidut sekä valmisteen markkinoille tulon jälkeen todetut haittavaikutukset PNH- ja aHUS- potilailla sekä potilailla, joilla oli refraktorinen gMG tai NMOSD
| MedDRA elinluokat | Hyvin yleinen (≥1/10) | Yleinen (≥1/100, < 1/10) | Melko harvinainen (≥1/1 000, <1/100) | Harvinainen (≥1/10 000, <1/1 000) | Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin) |
| Infektiot | Keuhkokuume, ylähengitysteiden infektio, keuhkoputkitulehdus, nasofaryngiitti, virtsatieinfektio, huuliherpes | Meningokokki-infektiob, sepsis, septinen sokki, peritoniitti, alahengitystieinfektio, sieni-infektio, virusinfektio, paisea, selluliitti, influenssa, gastrointestinaalinen infektio, kystiitti, infektio, sinuiitti, ieninfektio |
Aspergillus-infektioc, bakteeriperäinen niveltulehdusc, urogenitaalinen gonokokki-infektio, | ||
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Pahanlaatuinen melanooma, myelodysplastinen oireyhtymä | ||||
| Veri ja imukudos | Leukopenia, anemia | Trombosytopenia, lymfopenia | Hemolyysi*, hyytymistekijöiden poikkeavuudet, punasolujen sakkautuminen, koagulopatia | ||
| Immuunijärjestelmä | Anafylaktinen reaktio, yliherkkyys | ||||
| Umpieritys | Gravesin tauti | ||||
| Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu | ||||
| Psyykkiset häiriöt | Unettomuus | Masennus, ahdistus, mielialan vaihtelut, unihäiriöt | Poikkeavat unet | ||
| Hermosto | Päänsärky | Heitehuimaus | Parestesia, vapina, makuhäiriö, pyörtyminen | ||
| Silmät | Hämärtynyt näkökyky | Sidekalvon ärsytys | |||
| Kuulo ja tasapainoelin | Tinnitus, kiertohuimaus | ||||
| Sydän | Sydämentykytys | ||||
| Verisuonisto | Hypertensio | Pahentunut hypertensio, hypotensio, kuumat aallot, laskimoiden häiriöt | Hematooma | ||
| Hengityselimet, rintakehä ja välikarsina | Yskä, kurkku- ja nielukipu | Hengenahdistus, nenäverenvuoto, kurkun ärsytys, nenän tukkoisuus, voimakas nuha | |||
| Ruoansulatuselimistö | Ripuli, oksentelu, pahoinvointi, vatsakipu | Ummetus, dyspepsia, vatsan pingottuminen | Gastroesofageaalinen refluksitauti, ienkipu | ||
| Maksa ja sappi | ALAT-arvon nousu, ASAT-arvon nousu, GGT-arvon nousu | Ikterus | Maksavauriod | ||
| Iho ja ihonalainen kudos | Ihottuma, kutina, alopesia | Urtikaria, eryteema, petekiat, liikahikoilu, kuiva iho, ihotulehdus | Ihon pigmenttimuutokset | ||
| Luusto, lihakset ja sidekudos | Nivelkipu, lihaskipu, kipu raajoissa | Lihaskouristukset, luukipu, selkäkipu, niskakipu | Leukalukko, nivelten turvotus | ||
| Munuaiset ja virtsatiet | Munuaisten vajaatoiminta, virtsaamisvaikeudet, verivirtsaisuus | ||||
| Sukupuolielimet ja rinnat | Spontaani erektio | Kuukautishäiriöt | |||
| Yleisoireet ja antopaikassa todettavat haitat | Kuume, väsymys, influenssan tapainen sairaus | Edeema, epämukava tunne rinnassa, astenia, rintakipu, infuusiokohdan kipu, vilunväreet | Ekstravasaatio, infuusiokohdan parestesiat, kuumuuden tunne | ||
| Tutkimukset | Hematokriitin pieneneminen, hemoglobiiniarvon pieneneminen | Coombsin kokeen positiivinen tulosc | |||
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvä reaktio |
Mukana olleet tutkimukset: astma (C07-002), aHUS(C08-002, C08-003, C10-003, C10-004), dermatomyosiitti (C99-006), refraktorinen gMG (C08-001, ECU-MG-301, ECU-MG-302, ECU-MG-303), neuromyelitis optica -kirjon häiriö (ECU-NMO-301, ECU-NMO-302), IMG (C99-004, E99-004), PNH (C02-001, C04-001, C04-002, C06-002, C07-001, E02-001, E05-001, E07-001, M07-005, X03-001, X03-001A), psoriaasi (C99-007), RA (C01-004, C97-001, C99-001, E01-004, E99-001), STEC-HUS (C11-001), SLE (C97-002). MedDRA-versio 26.1.
*Ks. kohta Joidenkin haittavaikutusten kuvaus.
a Paiseisiin sisältyvät seuraavat MedDRA termit: paise raajassa, paise paksusuolessa, paise munuaisessa, ihonalainen paise, hammaspaise, maksapaise, perirektaalinen paise, rektaalinen paise.
b Meningokokki-infektioon sisältyvät seuraavat MedDRA-termit: meningokokki-infektio, meningokokkisepsis, meningokokkiaivokalvotulehdus.
c Valmisteen markkinoilletulon jälkeisistä raporteista tunnistetut lääkehaittavaikutukset
d Saatavissa oleva markkinoilletulon jälkeinen tieto ei riitä esiintyvyyden arviointiin.
Joidenkin haittavaikutusten kuvaus
Kaikissa kliinisissä tutkimuksissa vakavin haittavaikutus oli meningokokin aiheuttama sepsis, joka on yleinen meningokokki-infektioiden ilmentymä Soliris-hoitoa saavilla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Myös muiden Neisseria-lajien aiheuttamia sepsistapauksia on raportoitu, mukaan lukien Neisseria gonorrhoeaen, Neisseria siccan/subflavan sekä määrittämättömien Neisseria-lajien.
Vasta-aineita Solirisille havaittiin 2 prosentilla PNH-potilaista käyttämällä ELISA-koetta sekä 3 prosentilla aHUS-potilaista ja 2 prosentilla NMOSD-potilaista käyttämällä yhdistelmä-ECL-tutkimusta. Refraktorista gMG:tä koskevissa lumelääkekontrolloiduissa tutkimuksissa ei todettu lääkevasta-aineita. Muiden proteiinien tapaan käyttöön liittyy immunogeenisuuden mahdollisuus.
Hemolyysitapauksia on raportoitu, kun Soliris-annos on kliinisissä PNH-tutkimuksissa jäänyt saamatta tai sen saaminen on viivästynyt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tromboottisia mikroangiopatiakomplikaatioita on raportoitu, kun Soliris-annos on kliinisissä aHUS-tutkimuksissa jäänyt saamatta tai sen saaminen on viivästynyt (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Pediatriseen PNH-tutkimukseen M07-005 otettujen pediatristen ja nuorten (11 – < 18-vuotiaiden) potilaiden turvallisuusprofiili vaikutti samankaltaiselta kuin aikuisilla PNH-potilailla todettu. Yleisin pediatrisilla potilailla raportoitu haittavaikutus oli päänsärky.
aHUS-tutkimuksissa C08-002, C08-003, C09-001r ja C10-003 mukana olleiden pediatristen aHUS-potilaiden (iältään 2 kuukautta – < 18-vuotta) turvallisuusprofiili vaikutti samankaltaiselta kuin aikuisilla aHUS-potilailla todettu. Turvallisuusprofiilit eri-ikäisten pediatristen potilaiden alaryhmissä vaikuttavat samankaltaisilta.
Pediatrisilla potilailla (iältään 12 – < 18 vuotta), joilla oli refraktorinen gMG ja jotka olivat mukana tutkimuksessa ECU‑MG‑303, turvallisuusprofiili vaikutti samanlaiselta kuin refraktorista gMG:tä sairastavilla aikuisilla todettu.
Iäkkäät potilaat
Iäkkäiden (≥ 65 vuotta) ja tätä nuorempien (< 65 vuotta) refraktorista gMG:tä sairastavien potilaiden välillä ei ole raportoitu yleisiä eroja turvallisuuden suhteen (ks. kohta Farmakodynamiikka).
Muita sairauksia sairastavat potilaat
Turvallisuutta koskevat tiedot muista kliinisistä tutkimuksista
Samansuuntaisia tietoja turvallisuudesta saatiin 12:sta loppuun suoritetusta kliinisestä tutkimuksesta, joihin osallistui 934 ekulitsumabille altistettua potilasta muista sairauspopulaatioista kuin PNH, aHUS, refraktorinen gMG tai NMOSD. Yksi rokottamaton potilas, jolla diagnosoitiin idiopaattinen membranoottinen glomerulonefropatia, sai meningokokin aiheuttaman meningiitin eli aivokalvontulehduksen. Muita sairauksia kuin PNH, aHUS, refraktorinen gMG tai NMOSD sairastavilla potilailla ilmoitetut haittavaikutukset olivat samankaltaisia kuin PNH- ja aHUS-potilailla sekä potilailla, joilla oli refraktorinen gMG tai NMOSD (ks. taulukko 1 edellä). Näissä kliinisissä tutkimuksissa ei ole tullut ilmi erityisiä haittavaikutuksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen
ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Kliinisissä tutkimuksissa ei ole ilmoitettu yliannostustapauksia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: komplementin estäjät, ATC-koodi: L04AJ01.
Soliris on humanisoitu monoklonaalinen IgG2/4k-yhdistelmävasta-aine. Se sitoutuu ihmisen komplementtiproteiiniin C5 ja estää komplementin terminaalisen osan aktivoitumisen. Soliris-vasta-aine sisältää ihmisen geenien muuttumattomia alueita ja hiiren komplementaarisuuden määrittäviä alueita, jotka on siirretty ihmisen kevyet ja raskas ketjun vaiheteleviin osiin. Soliris koostuu kahdesta 448 aminohapon raskas ketjusta ja kahdesta 214 aminohapon kevyt ketjusta. Sen molekyylipaino on noin 148 kDa.
Solirisia tuotetaan hiiren myelooman ilmentymässä (NS0-solulinja) ja puhdistetaan affiniteetti- ja ionivaihtokromatografialla. Lääkeainemassan valmistusprosessi sisältää myös erityiset virusten inaktivaatio- ja poistovaiheet.
Vaikutusmekanismi
Solirisin vaikuttava aine ekulitsumabi on komplementin terminaalisen osan estäjä, joka sitoutuu suurella affiniteetilla spesifisesti komplementtiproteiiniin C5. Se estää C5:n pilkkoutumisen C5a:ksi ja C5b:ksi ja terminaalisen komplementtikompleksin C5b-9 muodostumisen. Ekulitsumabi säilyttää komplementin aktivaation varhaiset komponentit, jotka ovat tärkeitä mikro-organismien opsonisaatiolle ja immuunikompleksien poistamiselle.
Soliris-hoito estää PNH-potilailla terminaalisen komplementin hallitsemattoman aktivaation ja siitä aiheutuvan komplementtivälitteisen intravaskulaarisen hemolyysin. Noin 35 mikrogrammaa/ml on useimmilla PNH-potilailla riittävä ekulitsumabin pitoisuus seerumissa komplementin terminaalisen osan aiheuttaman intravaskulaarisen hemolyysin olennaisesti täydelliseen estämiseen.
Solirisin antaminen pitkäaikaisesti johti PNH-potilailla nopeasti ja pysyvästi komplementin välittämän hemolyyttisen aktiivisuuden vähenemiseen.
Soliris-hoito estää aHUS-potilailla terminaalisen komplementin hallitsemattoman aktivaation ja siitä aiheutuvan komplementtivälitteisen tromboottisen mikroangiopatian.
Suositusannoksina annettu Soliris vähensi kaikilla potilailla terminaalisen komplementin aktivaatiota nopeasti ja pitkäkestoisesti. Seerumin ekulitsumabipitoisuus noin 50–100 mikrogrammaa/ml on kaikilla aHUS-potilailla riittävä estämään terminaalisen komplementin aktivaation jokseenkin täydellisesti.
Solirisin antaminen pitkäaikaisesti johti aHUS-potilailla nopeasti ja pysyvästi komplementin välittämän tromboottisen mikroangiopatian vähenemiseen.
Potilailla, joilla on refraktorinen gMG, terminaalisen komplementin hallitsematon aktivaatio aiheuttaa solukalvoa tuhoavasta kompleksista (MAC) riippuvaista liukenemista ja C5a:sta riippuvaista tulehdusta hermo-lihasliitoksessa, mikä johtaa neuromuskulaarisen viestinvälityksen häiriöön. Soliris-valmisteen jatkuva anto johtaa välittömään, täydelliseen ja pitkäkestoiseen terminaalisen komplementin aktivaation estoon (ekulitsumabin pitoisuus seerumissa ≥ 116 mikrogrammaa/ml).
NMOSD-potilailla AQP4-autovasta-aineiden aiheuttama terminaalisen komplementin hallitsematon aktivaatio aiheuttaa MAC:sta ja C5a:sta riippuvaista tulehdusta, joka johtaa astrosyyttien nekroosiin ja veri-aivoesteen läpäisevyyden lisääntymiseen sekä ympäröivien oligodendrosyyttien ja neuronien tuhoutumiseen. Soliris-valmisteen jatkuva anto johtaa välittömään, täydelliseen ja pitkäkestoiseen terminaalisen komplementin aktivaation estoon (ekulitsumabin pitoisuus seerumissa ≥ 116 mikrogrammaa/ml).
Kliininen teho ja turvallisuus
Paroksysmaalinen nokturnaalinen hemoglobinuria
Solirisin turvallisuutta ja tehoa PNH-potilailla, joilla oli hemolyysi, arvioitiin satunnaistetussa, lumelääkekontrolloidussa 26 viikkoa kestäneessä kaksoissokkotutkimuksessa (C04-001). PNH-potilaita hoidettiin Solirisilla myös 52 viikkoa kestäneessä yhden haaran tutkimuksessa (C04-002) ja pitkäaikaisessa jatkotutkimuksessa (E05-001). Potilaat saivat meningokokkirokotuksen ennen kuin he saivat Solirista. Kaikissa tutkimuksissa ekulitsumabiannos oli aluksi 600 mg 7 ± 2 päivän välein 4 viikon ajan. Tämän jälkeen annettiin 900 mg 7 ± 2 päivää myöhemmin ja sitten 900 mg 14 ± 2 päivän välein tutkimuksen loppuun asti. Solirista annettiin 25–45 minuutin (35 minuuttia ± 10 minuuttia) laskimonsisäisenä infuusiona. PNH-potilailla käynnistettiin myös havainnoiva noninterventiotyyppinen rekisteritutkimus (M07-001), jossa pyrittiin selvittämään PNH:n luonnollista kulkua hoitamattomilla potilailla ja Soliris-hoidon aikana saavutettavia kliinisiä hoitotuloksia.
Tutkimuksessa C04-001 (TRIUMPH) PNH-potilaat, jotka olivat saaneet vähintään 4 verensiirtoa edellisten 12 kuukauden aikana ja joilla oli virtaussytometrisesti vahvistettuna vähintään 10 % PNH-soluja ja verihiutaleiden määränä vähintään 100 000/mikrolitra, satunnaistettiin joko Soliris-hoitoon (n = 43) tai lumelääkehoitoon (n = 44). Ennen satunnaistamista kaikkia potilaita seurattiin alustavan seurantajakson ajan, jolloin vahvistettiin punasolusiirron tarve ja määritettiin hemoglobiinipitoisuus (asetusarvo), joka määrittäisi kunkin potilaan hemoglobiinin vakiintumisen ja verensiirtotulokset. Hemoglobiinin asetusarvo oli pienempi tai yhtä suuri kuin 9 g/dl oireilevilla potilailla ja pienempi tai yhtä suuri kuin 7 g/dl oireettomilla potilailla. Ensisijaisia lopputapahtumia olivat hemoglobiinin vakiintuminen (potilaat, joilla hemoglobiinipitoisuus pysyi hemoglobiinin asetusarvon yläpuolella ja joilla punasolusiirto pystyttiin välttämään koko 26 viikon jakson ajan) ja verensiirron tarve. Väsymys ja terveyteen liittyvä elämänlaatu olivat merkityksellisiä toissijaisia lopputapahtumia. Hemolyysiä seurattiin lähinnä seerumin LDH-mittauksilla. Lisäksi PNH-punasolujen osuutta seurattiin virtaussytometrialla. Lähtötilanteessa antikoagulantteja ja systeemisiä kortikosteroideja saaneilla potilailla jatkettiin näitä hoitoja. Merkittävät lähtötilanteen ominaisuudet tasapainotettiin (ks. taulukko 2).
Kontrolloimattomassa tutkimuksessa C04-002 (SHEPHERD) PNH-potilaat, joille oli tehty vähintään yksi verensiirto edellisten 24 kuukauden aikana ja joiden verihiutalearvo oli vähintään 30 000 verihiutaletta/mikrolitra, saivat Soliris-hoitoa 52 viikon jakson ajan. Samanaikaisia hoitoja olivat antitromboottiset aineet 63 prosentilla potilaista ja systeemiset kortikosteroidit 40 prosentilla potilaista. Lähtötilanteen ominaisuudet esitetään taulukossa 2.
Taulukko 2: Potilastiedot ja ominaisuudet tutkimuksissa C04-001 ja C04-002
| C04-001 | C04-002 | ||
| Parametri | Lumelääke n = 44 | Soliris n = 43 | Soliris n = 97 |
| Keskimääräinen-ikä (keskihajonta) | 38,4 (13,4) | 42,1 (15,5) | 41,1 (14,4) |
| Sukupuoli - nainen (%) | 29 (65,9) | 23 (53,5) | 49 (50,5) |
| Aiempi aplastinen anemia tai MDS (%) | 12 (27,3) | 8 (18,7) | 29 (29,9) |
| Samanaikaiset antikoagulantit (%) | 20 (45,5) | 24 (55,8) | 59 (61) |
| Samanaikaiset steroidit /immunosuppressiohoidot (%) | 16 (36,4) | 14 (32,6) | 46 (47,4) |
| Hoidon keskeytys | 10 | 2 | 1 |
| Punasolusiirto 12 edellisen kuukauden aikana (mediaani (Q1,Q3)) | 17,0 (13,5, 25,0) | 18,0 (12,0, 24,0) | 8,0 (4,0, 24,0) |
| Keskim. Hb-arvo (g/dl) asetuspisteessä (keskihajonta) | 7,7 (0,75) | 7,8 (0,79) | N/A |
| Hoitoa edeltävä LDH (mediaani, U/l) | 2 234,5 | 2 032,0 | 2 051,0 |
| Vapaa hemoglobiini alussa (mediaani, mg/dl) | 46,2 | 40,5 | 34,9 |
TRIUMPH-tutkimuksessa Soliris-hoitoa saaneilla potilailla hemolyysi väheni merkitsevästi (p < 0,001), mikä lisääntyneen hemoglobiinin stabilisaation osoittamana paransi anemiaa, ja punasolusiirtojen tarve väheni verrattuna lumelääkkeellä hoidettuihin potilaisiin (ks. taulukko 3). Tällaisia vaikutuksia ilmeni kaikkien kolmen tutkimusta edeltävän verensiirtoryhmän (4–14 yksikköä, 15–25 yksikköä, > 25 yksikköä) potilailla. 3 viikon Soliris-hoidon jälkeen potilaat ilmoittivat vähemmän väsymystä ja heidän terveyteen liittyvä elämänlaatunsa parani. Tutkimuksen otoskoon ja keston vuoksi Solirisin vaikutusta tromboottisiin tapahtumiin ei voitu määrittää. SHEPHERD-tutkimuksessa 96 potilasta 97 mukaan otetusta potilaasta oli mukana tutkimuksen loppuun asti (yksi potilas kuoli tromboottisen tapahtuman seurauksena). Seerumin LDH-määrinä mitattu intravaskulaarisen hemolyysin väheneminen kesti koko hoitojakson ajan ja lisäsi verensiirron välttämistä, vähensi punasolusiirron tarvetta ja vähensi väsymystä. Katso taulukko 3.
Taulukko 3: Tehoa koskevat tulokset tutkimuksissa C04-001 ja C04-002
| C04-001 | C04-002* | ||||
| Lumelääke n = 44 | Soliris n = 43 | P-arvo | Soliris n = 97 | P-arvo | |
| Niiden potilaiden osuus, joilla hemoglobiinitaso stabilisoitui 26 viikon hoidon aikana | 0 | 49 | < 0,001 | N/A | |
| Punasolusiirrot hoidon aikana (mediaani) | 10 | 0 | < 0,001 | 0 | < 0,001 |
| Verensiirto vältetty hoidon aikana (%) | 0 | 51 | < 0,001 | 51 | < 0,001 |
| LDH tutkimuksen lopussa (mediaani, U/l) | 2 167 | 239 | < 0,001 | 269 | < 0,001 |
| LDH AUC tutkimuksen lopussa (mediaani, U/l x päivä) | 411 822 | 58 587 | < 0,001 | –632 264 | < 0,001 |
| Vapaa hemoglobiini tutkimuksen lopussa (mediaani, mg/dl) | 62 | 5 | < 0,001 | 5 | < 0,001 |
| FACIT-väsymys (vaikutuksen suuruus) | 1,12 | < 0,001 | 1,14 | < 0,001 | |
* Tutkimuksen C04-002 tulokset viittaavat vertailuun hoitoa edeltävän ja hoidon jälkeisen tilanteen välillä.
Alun perin tutkimuksiin C04-001 ja C04-002 sekä muihin ensimmäisiin tutkimuksiin osallistuneista 195 potilaasta Soliris-hoitoa saaneet PNH-potilaat otettiin mukaan pitkäaikaiseen lisätutkimukseen (E05-001). Intravaskulaarinen hemolyysi väheni kaikilla potilailla koko Solirisille altistamisen ajan. Altistamisaika oli 10–54 kuukautta. Tromboottisia tapahtumia oli vähemmän Soliris-hoidon aikana kuin yhtä pitkän ajanjakson aikana ennen hoitoa. Tämä havainto osoitettiin kuitenkin kontrolloimattomissa kliinisissä tutkimuksissa.
PNH-rekisterin (M07-001) perusteella arvioitiin Soliris-hoidon tehoa PNH-potilailla, jotka eivät olleet saaneet punasolusiirtoja. Potilailla oli suuri tautiaktiivisuus, joka määriteltiin tilanteeksi, jossa hemolyysi oli lisääntynyt (LDH ≥ 1,5 × viitearvojen yläraja) ja potilaalla oli tähän liittyvä kliininen oire (oireita): väsymys, hemoglobinuria, vatsakipu, hengenahdistus, anemia (hemoglobiini < 100 g/l), merkittävä haitallinen verisuonitapahtuma (mukaan lukien tromboosi), nielemisvaikeus tai erektiohäiriö.
PNH-rekisteristä todettiin, että Soliris-hoitoa saaneilla potilailla oli vähemmän hemolyysia ja siihen liittyviä oireita. 6 kuukauden kohdalla Soliris-hoitoa saaneilla potilailla, joilla ei ollut anamneesissa punasolusiirtoja, oli merkitsevästi (p < 0,001) pienemmät LDH-pitoisuudet (LDH-pitoisuuden mediaani 305 U/l; taulukko 4). Lisäksi 74 %:lla Soliris-hoitoa saaneista potilaista, joilla ei ollut anamneesissa siirtoja, todettiin FACIT-väsymyspisteiden parantuneen kliinisesti merkittävästi (ts. suurentuneen vähintään 4 pisteen verran) ja 84 %:lla EORTC-väsymyspisteiden parantuneen kliinisesti merkittävästi (ts. pienentyneen vähintään 10 pisteen verran).
Taulukko 4: Tehotulokset (LDH-pitoisuus ja FACIT-väsymyspisteet) PNH-potilailla, joilla ei ollut anamneesissa verensiirtoja, tutkimus M07-001
| M07-001 | |
| Parametri | Soliris Ei verensiirtoja |
| LDH-pitoisuus lähtötilanteessa (mediaani, U/l) | N = 43 1 447 |
| LDH-pitoisuus 6 kk:n kohdalla (mediaani, U/l) | N = 36 305 |
| FACIT-väsymyspisteet lähtötilanteessa (mediaani) | N = 25 32 |
| FACIT-väsymyspisteet viimeisellä saatavilla olleella arviointikerralla (mediaani) | N = 31 44 |
| FACIT-väsymyspistemäärä mitataan asteikolla 0–52. Mitä suurempi arvo, sitä vähemmän väsymystä. | |
Atyyppinen hemolyyttis-ureeminen oireyhtymä
Solirisin tehoa atyyppisen hemolyyttis-ureemisen oireyhtymän (aHUS) hoidossa arvioitiin 100 potilaan tietojen perusteella, jotka oli saatu neljästä prospektiivisesta kontrolloidusta tutkimuksesta, kolmesta aikuisille ja nuorille potilaille tehdystä tutkimuksesta (C08-002A/B, C08-003A/B ja C10-004) ja yhdestä lapsille ja nuorille potilaille tehdystä tutkimuksesta (C10-003) ja yhdestä 30 potilaalle tehdystä retrospektiivisestä tutkimuksesta (C09-001r).
Tutkimus C08-002A/B oli prospektiivinen, kontrolloitu, avoin tutkimus, jossa oli mukana varhaisvaiheen aHUS-potilaita, joilla oli todettu tromboottisen mikroangiopatian kliinisiä oireita, joita olivat trombosyyttimäärä ≤ 150 × 109/l plasmanvaihdosta/plasmainfuusiosta huolimatta sekä viitearvojen ylärajaa suuremmat LDH- ja seerumin kreatiniinipitoisuudet. Tutkimus C08-003A/B oli prospektiivinen, kontrolloitu, avoin tutkimus, johon osallistui pitempään sairastaneita aHUS-potilaita, joilla ei ollut selkeästi todettavia tromboottisen mikroangiopatian oireita ja jotka saivat pitkäaikaisesti plasmanvaihtoja/plasmainfuusioita (≥ 1 plasmanvaihto/plasmainfuusio kahden viikon välein ja enintään 3 plasmanvaihtoa/plasmainfuusiota viikossa vähintään 8 viikon ajan ennen ensimmäistä annosta). Potilaat saivat kummassakin prospektiivisessa tutkimuksessa Soliris-hoitoa 26 viikon ajan, ja useimmat potilaat osallistuivat pitkäkestoiseen avoimeen jatkotutkimukseen. Kaikkien kumpaankin prospektiiviseen tutkimukseen mukaan otettujen potilaiden ADAMTS-13-aktiivisuus oli yli 5 %.
Potilaat saivat meningokokkirokotuksen ennen Soliris-hoidon aloittamista tai estohoitoa sopivilla antibiooteilla siihen saakka, kunnes rokotuksesta oli kulunut kaksi viikkoa. Aikuisille ja nuorille aHUS-potilaille annettu Soliris-annos oli kaikissa tutkimuksissa 900 mg 7 ± 2 vuorokauden välein neljän viikon ajan, minkä jälkeen annos oli 1 200 mg 7 ± 2 vuorokautta myöhemmin, ja tämän jälkeen 1 200 mg 14 ± 2 vuorokauden välein tutkimuksen kestoajan. Soliris annettiin 35 minuuttia kestävänä infuusiona laskimoon. Pediatristen potilaiden ja alle 40 kg painavien nuorten potilaiden annostus määräytyi farmakokineettisen simulaation perusteella, jossa suositusannos ja hoito-ohjelma määriteltiin painon perusteella (ks. kohta Annostus ja antotapa).
Tutkimuksessa C08-002A/B ensisijainen päätetapahtuma oli trombosyyttimäärän muutos lähtötilanteeseen verrattuna, ja tutkimuksessa C08-003A/B ensisijainen päätetapahtuma oli tromboottisen mikroangiopatian (TMA) tapahtumista vapaa tila. Muita päätetapahtumia olivat TMA:han liittyvien toimenpiteiden lukumäärä, hematologisten arvojen normaalistuminen, täydellinen TMA-vaste, LDH-pitoisuuden muutokset, munuaisten toiminta ja elämänlaatu. TMA-tapahtumista vapaaksi tilaksi määriteltiin seuraavien tapahtumien puuttuminen vähintään 12 viikon ajan: trombosyyttimäärän väheneminen > 25 % lähtötilanteeseen verrattuna, plasmanvaihto/plasmainfuusio ja uusi dialyysihoitokerta. TMA:han liittyviksi toimenpiteiksi määriteltiin plasmanvaihto/plasmainfuusio tai uusi dialyysihoitokerta. Hematologisten arvojen normaalistumiseksi määriteltiin trombosyyttimäärän palautuminen normaaliksi ja LDH-pitoisuuksien pysyminen samalla tasolla pitkäkestoisesti ≥ 2 peräkkäisellä määrityskerralla ≥ 4 viikon aikana. Täydelliseksi TMA-vasteeksi määriteltiin hematologisten arvojen normaalistuminen ja seerumin kreatiniinipitoisuuksien pieneneminen ≥ 25 % pitkäkestoisesti ≥ 2 peräkkäisellä määrityskerralla ≥ 4 viikon aikana.
Potilaiden ominaisuudet lähtötilanteessa esitetään taulukossa 5.
Taulukko 5: Potilastiedot ja ominaisuudet tutkimuksissa C08-002A/B ja C08-003A/B
| Parametri | C08-002A/B | C08-003A/B |
| Soliris n = 17 | Soliris n = 20 | |
| Aika ensimmäisestä diagnoosista seulontaan kuukausina, mediaani (min., maks.) | 10 (0,26, 236) | 48 (0,66, 286) |
| Aika tällä hetkellä esiintyvistä kliinisistä TMA-oireista seulontaan kuukausina, mediaani (min., maks.) | < 1 (< 1, 4) | 9 (1, 45) |
| Tällä hetkellä esiintyviin kliinisiin TMA-oireisiin annettujen plasmanvaihtojen/plasmainfuusioiden lukumäärä, mediaani (min., maks.) | 17 (2, 37) | 62 (20, 230) |
| Plasmanvaihtojen/plasmainfuusioiden lukumäärä ensimmäistä ekulitsumabiannosta edeltävien 7 päivän aikana, mediaani (min., maks.) | 6 (0, 7) | 2 (1, 3) |
| Trombosyyttimäärä lähtötilanteessa (× 109/l), keskiarvo (keskihajonta) | 109 (32) | 228 (78) |
| LDH-pitoisuus lähtötilanteessa (U/l), keskiarvo (keskihajonta) | 323 (138) | 223 (70) |
| Potilaat, joilla ei ole todettu mutaatiota, n (%) | 4 (24) | 6 (30) |
aHUS-tutkimukseen C08-002 A/B osallistuneet potilaat saivat Soliris-hoitoa vähintään 26 viikon ajan. Kun alkuvaiheen 26 viikon hoitojakso päättyi, suurin osa potilaista sai edelleen Soliris-hoitoa osallistumalla jatkotutkimukseen. aHUS-tutkimuksessa C08-002A/B Soliris-hoidon keston mediaani oli noin 100 viikkoa (vaihteluväli: 2 viikosta 145 viikkoon).
Soliris-hoidon aloittamisen jälkeen todettiin terminaalisen komplementin aktiivisuuden vähentyneen ja trombosyyttimäärän lisääntyneen lähtötilanteeseen verrattuna. Terminaalisen komplementin aktiivisuuden vähenemistä todettiin kaikilla potilailla Soliris-hoidon aloittamisen jälkeen. Taulukossa 6 esitetään yhteenveto aHUS-tutkimuksen C08-002A/B tehon tuloksista. Kaikki tehon päätetapahtumavasteet paranivat tai pysyivät ennallaan 2 vuoden hoidon ajan. Täydellinen TMA-vaste säilyi ennallaan kaikilla vasteen saaneilla. Kun hoitoa jatkettiin yli 26 viikon ajan, kahdella potilaalla tämän lisäksi saavutettiin täydellinen TMA-vaste, joka pysyi ennallaan LDH:n normalisoitumisen (1 potilaalla) ja seerumin kreatiniinin vähenemisen (2 potilaalla) vuoksi.
Munuaisten toiminta parani ja pysyi hyvänä Soliris-hoidon aikana glomerulusten laskennallisen suodatusnopeuden (eGFR) perusteella. Neljällä viidestä potilaasta, jotka tarvitsivat dialyysihoitoa tutkimukseen tullessaan, dialyysihoito voitiin lopettaa Soliris-hoidon ajaksi. Yhdelle potilaalle kehittyi uusi dialyysihoidon tarve. Potilaat raportoivat terveyteen liittyvän elämänlaadun parantuneen.
aHUS-tutkimukseen C08-002A/B osallistuneiden potilaiden vaste Soliris-hoitoon oli samanlainen riippumatta siitä, oliko heillä todettu komplementtijärjestelmää sääteleviä proteiineja koodittavien geenien mutaatioita.
aHUS-tutkimukseen C08-003A/B osallistuneet potilaat saivat Soliris-hoitoa vähintään 26 viikon ajan. Kun alkuvaiheen 26 viikon hoitojakso päättyi, suurin osa potilaista sai edelleen Soliris-hoitoa osallistumalla jatkotutkimukseen. aHUS-tutkimuksessa C08-003A/B Soliris-hoidon keston mediaani oli noin 114 viikkoa (vaihteluväli: 26–129 viikkoa). Taulukossa 6 on esitetty yhteenveto aHUS-tutkimuksen C08-003A/B tehon tuloksista.
aHUS-tutkimukseen C08-003A/B osallistuneiden potilaiden vaste Soliris-hoitoon oli samanlainen riippumatta siitä, oliko heillä todettu komplementtijärjestelmää sääteleviä proteiineja koodittavien geenien mutaatioita. Soliris-hoidon aloittamisen jälkeen todettiin terminaalisen komplementin aktiivisuuden vähentyneen kaikilla potilailla. Kaikki tehon päätetapahtumavasteet paranivat tai pysyivät ennallaan 2 vuoden hoidon ajan. Täydellinen TMA-vaste säilyi ennallaan kaikilla vasteen saaneilla. Kun hoitoa jatkettiin yli 26 viikon ajan, kuudella potilaalla tämän lisäksi saavutettiin täydellinen TMA-vaste, joka pysyi ennallaan seerumin kreatiniinin vähenemisen vuoksi.
Yksikään potilaista ei tarvinnut dialyysihoitoa Soliris-hoidon aikana. Munuaisten toiminta parani Soliris-hoidon aikana glomerulusten laskennallisen suodatusnopeuden (eGFR) mediaanin perusteella.
Taulukko 6: Tehoa koskevat tulokset prospektiivisissa aHUS-tutkimuksissa C08-002A/B ja C08-003A/B
| C08-002A/B n = 17 | C08-003A/B n = 20 | |||
| Viikolla 26 | 2 vuoden kuluttua1 | Viikolla 26 | 2 vuoden kuluttua1 | |
| Trombosyyttimäärän normaalistuminen | 14 (82) | 15 (88) | 18 (90) | 18 (90) |
| Kaikki potilaat, n (%) | (57–96) | (64–99) | (68–99) | (68–99) |
| (95 %:n luottamusväli) Potilaat, joiden arvo poikkeava lähtötilanteessa, n/n (%) | 13/15 (87) | 13/15 (87) | 1/3 (33) | 1/3 (33) |
| TMA-tapahtumista vapaa tila, n (%) (95 %:n luottamusväli) | 15 (88) (64–99) | 15 (88) (64–99) | 16 (80) (56–94) | 19 (95) (75–99) |
| TMA-toimenpiteiden lukumäärä | ||||
| Päivittäisen lukumäärän mediaani ennen ekulitsumabihoitoa, (min., maks.) | 0,88 (0,04, 1,59) | 0,88 (0,04, 1,59) | 0,23 (0,05, 1,09) | 0,23 (0,05, 1,09) |
| Päivittäisen lukumäärän mediaani ekulitsumabihoidon aikana, (min., maks.) | 0 (0, 0,31) | 0 (0, 0,31) | 0 | 0 |
p-arvo | p < 0,0001 | p < 0,0001 | P<0,0001 | p < 0,0001 |
| Kroonisen munuaistaudin paheneminen ≥ 1 vaikeusastetta, n (%) (95 %:n luottamusväli) | 10 (59) (33–82) | 12 (71) (44–90) | 7 (35) (15–59) | 12 (60) (36–81) |
| eGFR-arvon muutos ml/min/1,73 m2: mediaani (vaihteluväli) viikolla 26 | 20 (-1, 98) | 28 (3, 82) | 5 (–1, 20) | 11 (–42, 30) |
| eGFR-arvon paraneminen ≥ 15 ml/min/1,73 m2, n (%) (95 %:n luottamusväli) | 8 (47) (23–72) | 10 (59) (33–82) | 1 (5) (0–25) | 8 (40) (19–64) |
| Hemoglobiiniarvon muutos > 20 g/l, n (%) (95 %:n luottamusväli) | 11 (65) (38–86)2 | 13 (76) (50–93) | 9 (45) (23–68)3 | 13 (65) (41–85) |
| Hematologinen normaalistuminen, n (%) (95 %:n luottamusväli) | 13 (76) (50–93) | 15 (88) (64–99) | 18 (90) (68–99) | 18 (90) (68–99) |
| Täydellinen TMA-vaste, n (%) (95 %:n luottamusväli) | 11(65) (38–86) | 13(76) (50–93) | 5 (25) (9–49) | 11 (55) (32–77) |
1 Tietojen koontihetkellä (data cut-off) (20. huhtikuuta 2012)
2 Tutkimus C08-002: kolme potilasta sai punasolutuotantoa stimuloivia lääkevalmisteita (ESA-valmisteita), joiden käyttö lopetettiin ekulitsumabihoidon aloittamisen jälkeen
3 Tutkimus C08-003: 8 potilasta sai punasolutuotantoa stimuloivia lääkevalmisteita (ESA-valmisteita), ja kolme heistä lopetti ESA-valmisteiden käytön ekulitsumabihoidon aikana
aHUS-tutkimukseen C10-004 osallistui 41 potilasta, joilla esiintyi tromboottisen mikroangiopatian (TMA) oireita. Tutkimukseen mukaanoton kriteerit täyttyivät niillä potilailla, joiden verihiutalearvo oli alle normaaliarvon alemman rajan (LLN), joilla oli todisteita hemolyysista kuten seerumin LDH-tason nousu, ja joilla oli seerumin kreatiniinin taso yli normaalitasojen ylärajan ilman jatkuvan dialyysin tarvetta. Potilaiden mediaani-ikä oli 35 vuotta (vaihteluväli: 18–80 vuotta). Kaikilla aHUS-tutkimukseen C10-004 osallistuneilla oli ADAMTS-13-taso yli 5 %. Potilaista 51 %:lla oli tunnistettavissa oleva komplementtia säätelevän tekijän mutaatio tai autovasta-aine. Yhteensä 35 potilasta sai plasmanvaihdon/plasmainfuusion ennen ekulitsumabia. Taulukossa 7 on yhteenveto aHUS-tutkimukseen C10-004 osallistuneiden potilaiden tärkeimmistä lähtötilanteen kliinisistä ja sairauteen liittyvistä ominaisuuksista.
Taulukko 7: aHUS-tutkimukseen C10-004 osallistuneiden potilaiden lähtötilanteen ominaisuudet
| Parametri | aHUS-tutkimusC10-004 N = 41 |
| Aika aHUS-diagnoosista ensimmäiseen tutkimuksessa annettuun annokseen (kuukautta), mediaani (min., maks.) | 0,79 (0,03, 311) |
| Aika nykyisestä kliinisestä TMA-esiintymisestä ensimmäiseen tutkimuksessa annettuun annokseen (kuukautta), mediaani (min., maks.) | 0,52 (0,03, 19) |
| Lähtötilanteen verihiutalemäärä (× 109/l), mediaani (min., maks.) | 125 (16, 332) |
| Lähtötilanteen LDH (U/l), mediaani (minimi–maksimi) | 375 (131, 3 318) |
| Lähtötilanteen eGFR (ml/min/1,73 m2), mediaani (min., maks.) | 10 (6, 53) |
aHUS-tutkimukseen C10-004 osallistuneet potilaat saivat Solirisia vähintään 26 viikon ajan. Ensimmäisten 26 hoitoviikon jälkeen useimmat potilaat valikoituivat saamaan pitkäaikaista hoitoa.
Soliris-hoidon aloittamisen jälkeen todettiin terminaalisen komplementin aktiivisuuden vähentyneen ja trombosyyttimäärän lisääntyneen lähtötilanteeseen verrattuna. Soliris vähensi komplementin välittämää TMA-aktiviteettia, mikä näkyy trombosyyttien mediaanitason nousuna lähtötasosta viikkoon 26. aHUS-tutkimuksessa C10-004 keskimääräinen (± keskihajonta) trombosyyttimäärä nousi 119 ± 66 x109/l:n lähtötilanteesta yhden viikon kuluessa 200 ± 84 x 109/l:een. Tämä vaikutus pysyi ennallaan 26 viikon ajan (keskimääräinen trombosyyttimäärä [± keskihajonta] viikolla 26: 252 ± 70 x 109/l). Munuaistoiminta mitattuna eGFR:llä parani Soliris-hoidon aikana. Niistä 24 potilaasta, jotka tarvitsivat dialyysia lähtötilanteessa, 20 pystyi lopettamaan dialyysin Soliris-tutkimuksen aikana. Taulukossa 8 on yhteenveto aHUS-tutkimuksen C10-004 tehon tuloksista.
Taulukko 8: Prospektiivisen aHUS-tutkimuksen C10-004 tehon tulokset
| Tehon parametri | aHUS-tutkimus C10-004 (N = 41) 26 viikon kohdalla |
| Trombosyyttien määrän muutos viikkoon 26 mennessä (109/l) | 111 (–122, 362) |
| Hematologinen normaalistuminen, n (%) Hematologisen normaalistumisen mediaani kestoaika, viikkoina (vaihteluväli)1 | 36 (88) 46 (10, 74) |
| Täydellinen TMA-vaste, n (%) TMA-vasteen mediaani kestoaika, viikkoina (vaihteluväli)1 | 23 (56) 42 (6, 74) |
| TMA-tapahtumista vapaa tila, n (%) 95 %:n luottamusväli | 37 (90) 77; 97 |
| Päivittäisten TMA-toimenpiteiden lukumäärä, mediaani (vaihteluväli) | |
| Ennen ekulitsumabihoitoa Ekulitsumabihoidon aikana | 0,63 (0, 1,38) 0 (0, 0,58) |
1 Tietojen koontihetkellä (4. syyskuuta 2012), Soliris-hoidon keston mediaani 50 viikkoa (vaihteluväli: 13–86 viikkoa).
Pitkäaikaiseen Soliris-hoitoon (mediaani 52 viikkoa, vaihteluväli 15–126 viikkoa) liittyi kliinisesti merkittävien parannusten lisääntymistä aHUS-aikuispotilailla. Kun Soliris-hoitoa jatkettiin yli 26 viikon ajan, lisäksi kolme potilasta (yhteensä 63 % potilaista) saavutti täydellisen TMA-vasteen ja lisäksi neljä potilasta (yhteensä 98 % potilaista) saavutti hematologisen normalisoitumisen. Viimeisessä arvioinnissa eGFR parani lähtötilanteesta ≥ 15 ml/min/1,73 m2 25 potilaalla 41:stä (61 %).
Refraktorinen yleistynyt myasthenia gravis
Solirisin tehoa refraktorisen gMG:n hoidossa arvioitiin 139 potilaan tietojen perusteella, jotka olivat peräisin kahdesta prospektiivisesta kontrolloidusta tutkimuksesta (tutkimukset C08-001 ja ECU-MG-301) ja yhdestä avoimesta jatkotutkimuksesta (tutkimus ECU-MG-302).
Tutkimus ECU-MG-301 (REGAIN) oli 26 viikkoa kestävä kaksoissokkoutettu, satunnaistettu, lumelääkekontrolloitu vaiheen 3 monikeskustutkimus, jossa tutkittiin Solirista potilailla, joiden aiempi hoito oli epäonnistunut ja joilla oli edelleen oireita. Satakahdeksantoista (118) potilasta 125:stä (94 %) suoritti 26 viikon hoitojakson loppuun ja 117 (94 %) potilasta osallistui sen jälkeen tutkimukseen ECU-MG-302, joka oli avoin, pitkäaikaistehoa ja -turvallisuutta tutkiva monikeskus- ja jatkotutkimus, jossa kaikki potilaat saivat Soliris-hoitoa.
Tutkimuksessa ECU-MG-301 gMG-potilaat, jotka olivat saaneet positiivisen serologisen testituloksen AChR-vasta-aineille, jotka kuuluivat kliinisiin MGFA (Myasthenia Gravis Foundation of America) luokitusluokkiin II–IV ja joiden MG-ADL:n kokonaispistemäärä oli ≥ 6, satunnaistettiin joko Soliris-ryhmään (n = 62) tai lumelääkeryhmään (n = 63). Kaikilla tutkimukseen osallistuneilla potilailla oli refraktorinen gMG, ja he täyttivät seuraavat ennalta määritellyt kriteerit:
1) Vähintään vuoden kestänyt, epäonnistunut hoito kahdella tai useammalla immunosuppressiivisella lääkkeellä (joko yhdistelmähoitona tai monoterapiana), ts. potilailla oli immunosuppressiohoidoista huolimatta häiriöitä päivittäisissä toiminnoissaan.
TAI
2) Vähintään yksi epäonnistunut hoito immunosuppressiivisella lääkkeellä, ja oireiden hallinta oli vaatinut pitkäkestoista plasmanvaihtoa tai IVIg-hoitoa, ts. potilaat olivat tarvinneet säännöllistä plasmanvaihtoa tai IVIg-hoitoa lihasheikkouden hoitoon vähintään joka kolmas kuukausi viimeisten 12 kuukauden aikana.
Potilaat saivat meningokokkirokotuksen ennen Soliris-hoidon aloittamista tai ennalta ehkäisevää lääkitystä sopivilla antibiooteilla 2 viikon ajan rokotuksen jälkeen. Tutkimuksissa ECU-MG-301 ja ECU-MG-302 Soliris-annostus aikuisilla refraktorista gMG:tä sairastavilla potilailla oli 900 mg 7 (± 2) päivän välein 4 viikon ajan, jonka jälkeen annostus oli 1 200 mg viikolla 5 (± 2 päivää) ja sitten 1 200 mg 14 (± 2) päivän välein tutkimuksen keston ajan. Soliris annettiin 35 minuuttia kestävänä laskimoinfuusiona.
Taulukossa 9 on esitetty tutkimukseen ECU-MG-301 osallistuneiden refraktorista gMG:tä sairastavien potilaiden lähtötilanteen ominaisuudet.
Taulukko 9: Potilaiden demografiset tiedot ja ominaisuudet tutkimuksessa ECU-MG-301
| Soliris (n = 62) | Lumelääke (n = 63) | |
Ikä MG-diagnoosin aikaan (vuotta), Keskiarvo (min, maks) | 38,0 (5,9, 70,8) | 38,1 (7,7, 78,0) |
| Naisia, n (%) | 41 (66,1) | 41 (65,1) |
MG:n kesto (vuotta), Keskiarvo (min, maks) | 9,9 (1,3, 29,7) | 9,2 (1,0, 33,8) |
| Lähtötilanteen MG-ADL-pisteet | ||
| Keskiarvo (keskihajonta) | 10,5 (3,06) | 9,9 (2,58) |
| Mediaani | 10,0 | 9,0 |
| Lähtötilanteen QMG-pisteet | ||
| Keskiarvo (keskihajonta) | 17,3 (5,10) | 16,9 (5,56) |
| Mediaani | 17,0 | 16,0 |
| ≥ 3 aikaisempaa immunosuppressiohoitoa* diagnoosin jälkeen, n (%) | 31 (50,0) | 34 (54,0) |
| Potilaat, joilla on ollut aikaisempia pahenemisvaiheita diagnoosin jälkeen,n (%) | 46 (74,2) | 52 (82,5) |
| Potilaat, joilla on ollut aikaisempi MG-kriisi diagnoosin jälkeen, n (%) | 13 (21,0) | 10 (15,9) |
| Aiempi ventilaattorihoito diagnoosin jälkeen, n (%) | 15 (24,2) | 14 (22,2) |
| Aiempi intubaatio diagnoosin jälkeen (MGFA-luokka V), n (%) | 11 (17,7) | 9 (14,3) |
* Immunosuppressiiviset lääkkeet, kuten kortikosteroidit, atsatiopriini, mykofenolaatti, metotreksaatti, siklosporiini, takrolimuusi tai syklofosfamidi.
Tutkimuksessa ECU-MG-301 ensisijainen päätetapahtuma oli muutos lähtötilanteesta päivittäisten toimintojen profiilin (MG-ADL; potilaan raportoima, gMG:n suhteen validoitu tulosmittari) kokonaispisteissä viikolla 26. MG-ADL:n ensisijainen analyysi oli heikoimpaan arvoon perustuva kovarianssianalyysi (Worst-Rank ANCOVA), jossa Solirisin keskiarvo oli 56,6 ja lumelääkkeen 68,3. Arvot perustuivat 125 tutkittavaan (p = 0,0698).
Tärkein toissijainen päätetapahtuma oli muutos lähtötilanteesta QMG:n eli kvantitatiivisen MG-pisteytysjärjestelmän (lääkärin raportoima gMG:n suhteen validoitu tulosmittari) kokonaispisteissä viikolla 26. QMG:n ensisijainen analyysi oli heikoimpaan arvoon perustuva kovarianssianalyysi (Worst-Rank ANCOVA), jossa Solirisin keskiarvo oli 54,7 ja lumelääkkeen 70,7. Arvot perustuivat 125 tutkittavaan (p = 0,0129).
Ensi- ja toissijaisten päätetapahtumien ennalta määrättyjen toistomittausanalyysien tehotulokset on esitetty taulukossa 10.
Taulukko 10: ECU-MG-301-tutkimuksen tehotulosten muutos lähtötilanteesta viikolle 26
| Tehon päätetapahtumat: kokonaispistemäärän muutos lähtötilanteesta viikolla 26 | Soliris (SEM) | Lumelääke (SEM) | Soliris-muutos verrattuna lumelääkkeeseen– pienimmän neliösumman keskiarvojen ero (95 %:n CI) | p-arvo (käytössä toistomittaus-analyysi) |
| MG-ADL | –4,2 (0,49) | –2,3 (0,48) | –1,9 (-3,3, -0,6) | 0,0058 |
| QMG | –4,6 (0,60) | –1,6 (0,59) | –3,0 (-4,6, -1,3) | 0,0006 |
| MGC | –8,1 (0,96) | –4,8 (0,94) | –3,4 (-6,0, -0,7) | 0,0134 |
| MG-QoL-15 | –12,6 (1,52) | –5,4 (1,49) |
–7,2 (–11,5, –3,0) | 0,0010 |
SEM = keskiarvon keskivirhe CI = luottamusväli, MGC = myasthenia gravis -yhteispisteet, MG-QoL-15 = myasthenia gravis -elämänlaatupisteet (15 kohtaa)
Tutkimuksessa ECU-MG-301 MG-ADL-kokonaispisteiden osalta kliinisen vasteen määritelmänä oli vähintään 3 pisteen koheneminen. Niiden potilaiden osuus, jotka olivat saavuttaneet kliinisen vasteen viikolla 26 ilman varalääkettä, oli Soliris-ryhmässä 59,7 % ja lumelääkeryhmässä 39,7 % (p = 0,0229).
Tutkimuksessa ECU-MG-301 QMG-kokonaispisteiden osalta kliinisen vasteen määritelmänä oli vähintään 5 pisteen koheneminen. Niiden potilaiden osuus, jotka olivat saavuttaneet kliinisen vasteen viikolla 26 ilman varalääkettä, oli Soliris-ryhmässä 45,2 % ja lumelääkeryhmässä 19 % (p = 0,0018).
Taulukossa 11 on yhteenveto potilaista, jotka ilmoittivat kliinisestä huononemisesta, ja potilaista, jotka tarvitsivat varalääkettä 26 viikon aikana.
Taulukko 11: Kliininen huononeminen ja varalääkkeen käyttö tutkimuksessa ECU-MG-301
| Muuttuja | Tilastotieto | Lumelääke (N = 63) | Soliris (N = 62) |
| Kliinistä huononemista raportoineiden potilaiden kokonaismäärä | n (%) | 15 (23,8) | 6 (9,7) |
| Varalääkettä tarvinneiden potilaiden kokonaismäärä | n (%) | 12 (19,0) | 6 (9,7) |
Tutkimukseen ECU-MG-301 osallistuneista 125 potilaasta 117 osallistui pitkäkestoiseen jatkotutkimukseen (tutkimus ECU-MG-302), jossa kaikki saivat Solirista. Potilailla, joita oli aiemmin hoidettu Soliris-valmisteella tutkimuksessa ECU-MG-301, havaittiin edelleen Solirisin jatkuva vaikutus kaikilla mittareilla (MG-ADL, QMG, MGC ja MG-QoL15) 130 viikon jatkoajanjaksolla, jolloin ekulitsumabihoitoa jatkettiin tutkimuksessa ECU-MG-302. Potilailla, jotka saivat lumelääkettä tutkimuksessa ECU-MG-301 (tutkimuksen ECU-MG-302 lumelääke/ekulitsumabi-ryhmä), parannusta havaittiin ekulitsumabihoidon aloittamisen jälkeen, ja se säilyi yli 130 viikon ajan tutkimuksessa ECU-MG-302. Kaaviossa 1 esitetään lähtötilanteen jälkeen tapahtuneet muutokset sekä MG-ADL-pisteissä (A) että QMG-pisteissä (B) 26 viikon hoidon jälkeen tutkimuksessa ECU-MG-301 ja 130 viikon hoidon jälkeen (n = 80 potilasta) tutkimuksessa ECU-MG-302.
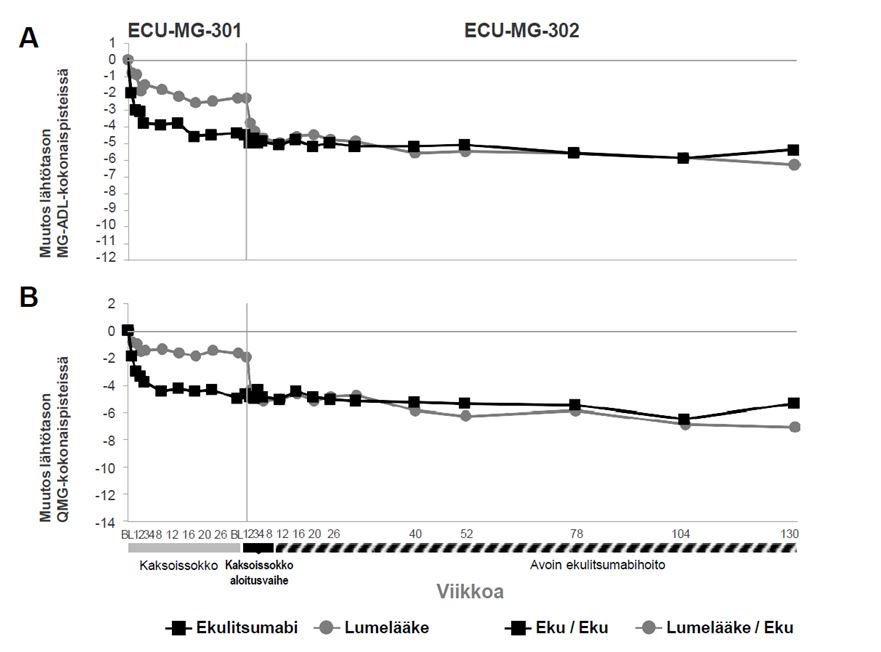
Kaavio 1: Keskimuutokset lähtötasosta MG-ADL-pisteissä (1A) ja QMG-pisteissä (1B) tutkimuksissa ECU-MG-301 ja ECU-MG-302
Tutkimuksessa ECU-MG-302 lääkäreillä oli mahdollisuus säätää taustalla olevia immunosuppressiohoitoja. Tässä asetelmassa vähintään yhden immunosuppressiohoidon vuorokausiannosta pienennettiin 65,0 %:lla potilaista ja olemassa oleva immunosuppressiohoito lopetettiin 43,6 %:lla potilaista. Yleisin syy immunosuppressiohoidon muutokseen oli myasthenia gravis -oireiden lievittyminen.
Kliinisissä tutkimuksissa Soliris-hoitoa annettiin kahdellekymmenellekahdelle (22) (17,6 %) iäkkäälle (> 65 vuotta) refraktorista gMG:tä sairastavalle potilaalle. Ikään liittyviä merkittäviä eroja turvallisuudessa ja tehossa ei havaittu.
Neuromyelitis optica -kirjon häiriö
Solirisin tehoa ja turvallisuutta NMOSD-potilaiden hoidossa arvioitiin sellaisten 143 potilaan tietojen perusteella, jotka olivat peräisin yhdestä kontrolloidusta tutkimuksesta (tutkimus ECU-NMO-301), ja sellaisten 119 potilaan tietojen perusteella, jotka jatkoivat hoitoa yhdessä avoimessa jatkotutkimuksessa (tutkimus ECU-NMO-302).
Tutkimus ECU-NMO-301 oli kaksoissokkoutettu, satunnaistettu, lumelääkekontrolloitu vaiheen 3 monikeskustutkimus, jossa Solirista tutkittiin NMOSD-potilaiden hoidossa.
Tutkimuksessa ECU-NMO-301 NMOSD-potilailla tuli olla positiivinen serologinen testitulos AQP4-vasta-aineille, vähintään kaksi relapsia viimeisten 12 kuukauden aikana tai viimeisten 24 kuukauden aikana kolme relapsia , joista vähintään yksi seulontaa edeltävien 12 kuukauden aikana, sekä pistemäärä ≤ 7 laajennetulla toimintakyvyn rajoittuneisuusasteikolla (EDSS). Potilaat satunnaistettiin suhteessa 2:1 joko Soliris-ryhmään (n = 96) tai lumelääkeryhmään (n = 47).
Taustalla olevat vakaa-annoksiset immunosuppressiohoidot sallittiin tutkimuksen aikana rituksimabia ja mitoksantronia lukuun ottamatta.
Potilaat saivat joko meningokokkirokotuksen vähintään kaksi viikkoa ennen Soliris-hoidon aloittamista tai estohoitoa sopivilla antibiooteilla siihen saakka, kunnes rokotuksesta oli kulunut kaksi viikkoa. NMOSD:n hoitoa koskevassa ekulitsumabin kliinisessä kehitysohjelmassa aikuisille NMOSD-potilaille annettu Soliris-annos oli 900 mg 7 ± 2 vuorokauden välein neljän viikon ajan, minkä jälkeen annos oli 1 200 mg viikolla 5 ± 2 vrk ja tämän jälkeen 1 200 mg 14 ± 2 vuorokauden välein tutkimuksen kestoajan. Soliris annettiin 35 minuuttia kestävänä infuusiona laskimoon.
Suurin osa (90,9 %) potilaista oli naisia. Noin puolet potilaista oli valkoihoisia (49,0 %). Mediaani-ikä ensimmäisen tutkimuslääkeannoksen antohetkellä oli 45 vuotta.
Taulukko 12: Potilaiden sairaushistoria ja lähtötilanteen ominaisuudet tutkimuksessa ECU-NMO-301
| Muuttuja | Tunnusluku | Lumelääke (N = 47) | Ekulitsumabi (N = 96) | Yhteensä (N = 143) |
|---|---|---|---|---|
| NMOSD-historia | ||||
| Ikä NMOSD:n ensimmäisen kliinisen ilmenemisen ajankohtana (vuosina) | Keskiarvo (SD) | 38,5 (14,98) | 35,8 (14,03) | 36,6 (14,35) |
| Mediaani | 38,0 | 35,5 | 36,0 | |
| Min., maks. | 12, 73 | 5, 66 | 5, 73 | |
| Aika NMOSD:n ensimmäisestä kliinisestä ilmenemisestä ensimmäiseen tutkimuslääkeannokseen (vuosina) | Keskiarvo (SD) | 6,601 (6,5863) | 8,156 (8,5792) | 7,645 (7,9894) |
| Mediaani | 3,760 | 5,030 | 4,800 | |
| Min., maks. | 0,51; 29,10 | 0,41; 44,85 | 0,41; 44,85 | |
| Vuotuisten relapsien määrä seulontaa edeltävien 24 kuukauden aikana | Keskiarvo (SD) | 2,07 (1,037) | 1,94 (0,896) | 1,99 (0,943) |
| Mediaani | 1,92 | 1,85 | 1,92 | |
| Min., maks. | 1,0; 6,4 | 1,0; 5,7 | 1,0; 6,4 | |
| Lähtötilanteen ominaisuudet | ||||
| EDSS-pisteet lähtötilanteessa | Keskiarvo (SD) | 4,26 (1,510) | 4,15 (1,646) | 4,18 (1,598) |
| Mediaani | 4,00 | 4,00 | 4,00 | |
| Min., maks. | 1,0; 6,5 | 1,0; 7,0 | 1,0; 7,0 | |
| Ei IST-hoitoja lähtötilanteessa | n (%) | 13 (27,7) | 21 (21,9) | 34 (23,8) |
Lyhenteet: ARR = vahvistettu relapsien määrä; EDSS = laajennettu toimintakyvyn rajoittuneisuusasteikko; IST = immunosuppressiohoito; maks. = maksimi; min. = minimi; NMOSD = neuromyelitis optica -kirjon häiriö; SD = keskihajonta.
Tutkimuksessa ECU-NMO-301 ensisijainen päätetapahtuma oli aika ensimmäiseen tutkimuksen aikana ilmenneeseen relapsiin; ajat vahvisti hoidon suhteen sokkoutettu riippumaton arviointitoimikunta. Lumelääkkeeseen verrattuna ekulitsumabilla todettiin merkitsevä vaikutus ensimmäiseen tutkimuksen aikana ilmenneeseen relapsiin kuluvaan aikaan (suhteellinen riskin vähenemä 94 %; riskisuhde 0,058; p < 0,0001) (kaavio 2). Soliris-hoitoa saavilla potilailla todettu parannus ajassa ensimmäiseen tutkimuksen aikana ilmenneeseen relapsiin oli samankaltainen niillä, jotka saivat samanaikaista immunosuppressiohoitoa, ja niillä, jotka eivät saaneet sitä.
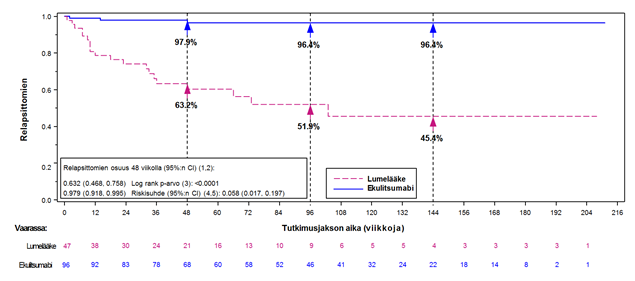
Kaavio 2: Kaplan-Meier -el inaika-arvio ensimmäiseen tutkimuksen aikana vahvistettuun relapsiin kuluneelle ajalle tutkimuksessa ECU-NMO-301 – koko analysoitava populaatio
Huomautus: potilailla, joilla ei vahvistettu tutkimuksen aikana relapsia, aika relapsiin sensuroitiin tutkimusvaiheen lopussa.
Ositetut analyysit perustuvat neljään satunnaistamisjoukkoon:
(i) pieni EDSS-pistearvo satunnaistamisvaiheessa (<=2,0), (ii) suuri EDSS-pistearvo (>=2,5 – <=7) eikä aiempia hoitoja satunnaistamisvaiheessa, (iii) suuri EDSS-pistearvo (>=2,5 – <=7) ja satunnaistamisvaiheessa jatkettiin samaa IST-hoitoa tai samoja IST-hoitoja, jota/joita potilas oli käyttänyt edellisen relapsin jälkeen, (iv) suuri EDSS-pistearvo (>=2,5 – <=7) ja IST-hoitoon tai -hoitoihin oli satunnaistamisvaiheessa tehty muutoksia edellisen relapsin jälkeen.
1 Perustuu Kaplan-Meierin tulorajamenetelmään.
2 Perustuu komplementaariseen log-log-transformaatioon.
3 Perustuu ositettuun log rank -testiin.
4 Perustuu ositettuun Coxin suhteellisen vaaran malliin.
5 Waldin luottamusväli.
Lyhenteet: CI = luottamusväli; EDSS = laajennettu toimintakyvyn rajoittuneisuusasteikko; IST = immunosuppressiohoito
Tutkimuksen aikana ekulitsumabilla vahvistettujen vuotuisten relapsien määrän (ARR) suhde (95 % CI) lumelääkkeeseen nähden oli 0,045 (0,013; 0,151), joka vastaa 95,5 %:n suhteellista laskua tutkimuksen aikana vahvistettujen vuotuisten relapsien määrässä ekulitsumabihoitoa saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin (p < 0,0001) (taulukko 13).
Taulukko 13: Tutkimuksen aikana vahvistettujen vuotuisten relapsien määrä tutkimuksessa ECU-NMO-301 – koko analysoitava populaatio
| Muuttuja | Tunnusluku | Lumelääke (N = 47) | Ekulitsumabi (N = 96) |
|---|---|---|---|
| Relapsien kokonaismäärä | Summa | 21 | 3 |
| Potilasvuosien kokonaismäärä tutkimusvaiheen aikana | n | 52,41 | 171,32 |
| Mukautettu, vahvistettu ARRa | Määrä | 0,350 | 0,016 |
| 95 % CI:n | 0,199; 0,616 | 0,005; 0,050 | |
| Hoidon vaikutusa | Määrien suhde (ekulitsumabi/ lumelääke) | … | 0,045 |
| 95 %:n CI | … | 0,013; 0,151 | |
| p-arvo | … | < 0,0001 |
a Perustuu Poissonin regressioon, jota on mukautettu satunnaistamisjoukkojen ja seulontaa edeltävien 24 kuukauden ARR-arvon mukaan.
Lyhenteet: ARR = vuotuisten relapsien määrä; CI = luottamusväli.
Lumelääkkeeseen verrattuna ekulitsumabihoito vähensi vuotuista sairaalahoitojen määrää (0,04 ekulitsumabia saaneilla vs. 0,31 lumelääkettä saaneilla potilailla), vuotuista kortikosteroidien laskimonsisäisten antojen määrää akuuttien relapsien hoitoon (0,07 ekulitsumabia saaneilla vs. 0,42 lumelääkettä saaneilla potilailla) ja vuotuista plasmanvaihtojen määrää (0,02 ekulitsumabia saaneilla vs. 0,19 lumelääkettä saaneilla potilailla).
Muissa toissijaisissa päätetapahtumissa lähtötilanteen ja tutkimuksen päättymisen välisenä aikana tapahtuneiden muutosten jakauma suosi ekulitsumabihoitoa lumelääkkeeseen nähden kaikilla neurologista haittaa (EDSS-pisteet [p = 0,0597] ja mRS [nimellinen p = 0,0154]), toiminnallista haittaa (HAI [nimellinen p = 0,0002]) ja elämänlaatua (EQ-5D VAS [nimellinen p = 0,0309] ja EQ-5D-indeksi [nimellinen p = 0,0077]) arvioivilla mittareilla.
Tutkimuksen ECU-NMO-302 loppuanalyysi osoitti, että tutkimuksen aikana todettujen vuotuisten relapsien määrä (hoitavan lääkärin määrittämänä) pieneni tilastollisesti merkitsevästi ja kliinisesti merkittävästi ekulitsumabihoitoa saaneilla potilailla aiemmin (tutkimuksessa ECU-NMO-301 seulontaa edeltävien 24 kuukauden aikana) todetuista vuotuisten relapsien määristä muutoksen mediaanin (min., maks.) perusteella (–1,825 [–6,38; 1,02], p < 0,0001).
Tutkimuksessa ECU-NMO-302 lääkäreillä oli mahdollisuus säätää taustalla olevia immunosuppressiohoitoja. Tässä asetelmassa yleisin muutos immunosuppressiohoitoon oli hoitoannoksen pienentäminen, joka tehtiin 21,0 %:lla potilaista. Lisäksi 15,1 %:lla potilaista olemassa oleva immunosuppressiohoito lopetettiin.
Solirista (ekulitsumabi) ei ole tutkittu NMOSD-potilaiden akuuttien relapsien hoidossa.
Pediatriset potilaat
Paroksysmaalinen nokturnaalinen hemoglobinuria
Yhteensä 7 pediatrista PNH-potilasta, joiden painon mediaani oli 57,2 kg (vaihteluväli 48,6–69,8 kg) ja ikä 11–17 vuotta (iän mediaani 15,6 vuotta), sai Solirista tutkimuksessa M07-005.
Ekulitsumabihoito suositellulla annostuksella yhdistettiin pediatrisilla potilailla intravaskulaarisen hemolyysin vähenemiseen seerumin LDH-tasoa mitattaessa. Se johti myös verensiirtojen tarpeen huomattavaan vähenemiseen tai häviämiseen sekä yleisen toimintakyvyn paranemisen trendiin. Ekulitsumabihoidon teho pediatrisilla PNH-potilailla vaikuttaa vastaavan keskeisiin PNH-tutkimuksiin (C04-001 ja C04-002) otetuilla aikuisilla PNH-potilailla todettua tehoa (taulukot 3 ja 14).
Taulukko 14: Tehoa koskevat tulokset pediatrisessa PNH-tutkimuksessa M07-005
| P-arvo | |||
| Keskiarvo (keskihajonta) | Wilcoxonin signed rank -testi | Pariutettu t-testi | |
| LDH-arvon muutos lähtövaiheesta 12 viikon kohdalla (U/l) | –771 (914) | 0,0156 | 0,0336 |
| LDH AUC (U/l x vuorokausi) | –60 634 (72 916) | 0,0156 | 0,0350 |
| Plasman vapaan hemoglobiinin muutos lähtövaiheesta 12 viikon kohdalla (mg/dl) | –10,3 (21,13) | 0,2188 | 0,1232 |
| Tyypin III RBC:n kloonin koon muutos lähtövaiheesta (prosenttia poikkeavista soluista) | 1,80 (358,1) | ||
| Muutos lähtötasolta PedsQLTM4.0 Generic Core -asteikolla 12 viikon kohdalla (potilaat) | 10,5 (6,66) | 0,1250 | 0,0256 |
| Muutos lähtötasolta PedsQLTM4.0 Generic Core -asteikolla 12 viikon kohdalla (vanhemmat) | 11,3 (8,5) | 0,2500 | 0,0737 |
| Muutos lähtötasolta PedsQLTM Multidimensional Fatigue -asteikolla 12 viikon kohdalla (potilaat) | 0,8 (21,39) | 0,6250 | 0,4687 |
| Muutos lähtötasolta PedsQLTM Multidimensional Fatigue -asteikolla 12 viikon kohdalla (vanhemmat) | 5,5 (0,71) | 0,5000 | 0,0289 |
Atyyppinen hemolyyttis-ureeminen oireyhtymä
Yhteensä 15 pediatrista potilasta (ikä 2 kuukaudesta 12 vuoteen) sai Soliris-hoitoa aHUS-tutkimuksessa C09-001r. Potilaista 47 %:lla oli todettu komplementtia säätelevän tekijän mutaatio tai autovasta-aineita. Ajan mediaani aHUS-diagnoosista ensimmäiseen Soliris-annokseen oli 14 kuukautta (vaihteluväli: < 1–110 kuukautta). Ajan mediaani senhetkisistä tromboottisen mikroangiopatian oireista ensimmäiseen Soliris-annokseen oli 1 kuukausi (vaihteluväli: < 1–16 kuukautta). Soliris-hoidon kestoajan mediaani oli iältään < 2-vuotiailla lapsilla (n = 5) 16 viikkoa (vaihteluväli 4–70 viikkoa) ja ikäryhmään 2-vuotiaista < 12-vuotiaisiin kuuluvilla lapsilla (n = 10) 31 viikkoa (vaihteluväli 19–63 viikkoa).
Nämä pediatristen potilaiden tehon tulokset olivat yleisesti ottaen yhdenmukaiset pivotaalitutkimuksiin C08-002 ja C08-003 osallistuneilla aHUS-potilailla todettujen tulosten kanssa (taulukko 6). Yksikään pediatrinen potilas ei tarvinnut uutta dialyysihoitokertaa Soliris-hoidon aikana.
Taulukko 15: aHUS-tutkimukseen C09-001r osallistuneiden pediatristen potilaiden tehon tulokset
| Tehon parametri | < 2-vuotiaat (n = 5) | 2 – < 12-vuotiaat (n = 10) | < 12-vuotiaat (n = 15) |
| Potilaat, joiden trombosyyttimäärä normaalistui, n (%) | 4 (80) | 10 (100) | 14 (93) |
| Täydellinen TMA-vaste, n (%) | 2 (40) | 5 (50) | 7 (50) |
| Päivittäisten TMA-toimenpiteiden lukumäärä, mediaani (vaihteluväli) | |||
| Ennen ekulitsumabihoitoa | 1 (0, 2) | < 1 (0,07, 1,46) | < 1 (0, 2) |
| Ekulitsumabihoidon aikana | < 1 (0, < 1) | 0 (0, < 1) | 0 (0, < 1) |
| Potilaat, joiden glomerulusten laskennallinen suodatusnopeus (eGFR) parani ≥ 15 ml/min/1,73 m2, n (%) | 2 (40) | 6 (60) | 8 (53) |
Kun pediatristen potilaiden tromboottisen mikroangiopatian (TMA) senhetkiset vaikea-asteiset kliiniset oireet olivat kestäneet lyhyemmän aikaa ennen ekulitsumabihoitoa, ekulitsumabihoidon avulla saatiin tromboottinen mikroangiopatia hallintaan ja munuaisten toiminta parani (taulukko 15).
Kun pediatristen potilaiden tromboottisen mikroangiopatian (TMA) senhetkiset vaikea-asteiset kliiniset oireet olivat kestäneet pidemmän aikaa ennen ekulitsumabihoitoa, ekulitsumabihoidon avulla saatiin tromboottinen mikroangiopatia hallintaan. Munuaisten toiminta ei kuitenkaan muuttunut korjautumattoman munuaisvaurion vuoksi (taulukko 16).
Taulukko 16: Tutkimukseen C09-001r osallistuneiden pediatristen potilaiden tehon tulokset tromboottisen mikroangiopatian (TMA) senhetkisten vaikea-asteisten kliinisten oireiden kestoajan mukaan
| TMA:n vaikea-asteisten kliinisten oireiden kestoaika | ||
| < 2 kuukautta N = 10 (%) | > 2 kuukautta N = 5 (%) | |
| Trombosyyttimäärän normaalistuminen | 9 (90) | 5 (100) |
| TMA-tapahtumista vapaa tila | 8 (80) | 3 (60) |
| Täydellinen TMA-vaste | 7 (70) | 0 |
| Glomerulusten laskennallisen suodatusnopeuden (eGFR) paraneminen ≥ 15 ml/min/1,73 m2 | 7 (70) | 0* |
* Yhden potilaan eGFR parani munuaisen siirron jälkeen
Yhteensä 22 pediatrista ja nuorta potilasta (iältään 5 kuukautta – 17 vuotta) sai Solirisia aHUS-tutkimuksessa C10-003.
C10-003-tutkimukseen oton kriteerit täyttyivät niillä potilailla, joiden verihiutalearvo oli alle normaaliarvon alemman rajan (LLN), joilla oli todisteita hemolyysista kuten seerumin LDH:n nousu yli normaaliarvon ylärajan, ja joilla oli seerumin kreatiniinitaso ≥97 persentiilia ikään nähden ilman jatkuvan dialyysin tarvetta. Potilaiden mediaani-ikä oli 6,5 vuotta (vaihteluväli: 5 kuukautta – 17 vuotta). aHUS-tutkimukseen C10-003 osallistuneilla potilailla oli ADAMTS-13-taso yli 5 %. Potilaista 50 %:lla oli tunnistettavissa oleva komplementtia säätelevän tekijän mutaatio tai autovasta-aine. Yhteensä 10 potilasta sai plasmanvaihdon/plasmainfuusion ennen ekulitsumabihoitoa. Taulukossa 17 on yhteenveto aHUS-tutkimukseen C10-003 osallistuneiden potilaiden tärkeimmistä lähtötilanteen kliinisistä ja sairauteen liittyvistä ominaisuuksista.
Taulukko 17: aHUS-tutkimukseen C10-003 osallistuneiden pediatristen ja nuorten potilaiden lähtötilanteen ominaisuudet
| Parametri | 1 kuukausi – <12 vuotta (N = 18) | Kaikki potilaat (N = 22) |
| Aika aHUS-diagnoosista ensimmäiseen tutkimuksessa annettuun annokseen (kuukautta), mediaani (min., maks.) | 0,51 (0,03, 58) | 0,56 (0,03, 191) |
| Aika nykyisestä kliinisestä TMA-esiintymisestä ensimmäiseen tutkimuksessa annettuun annokseen, (kuukautta), mediaani (min., maks.) | 0,23 (0,03, 4) | 0,20 (0,03, 4) |
| Lähtötilanteen verihiutalemäärä (× 109/l), mediaani (min., maks.) | 110 (19, 146) | 91 (19, 146) |
| Lähtötilanteen LDH (U/l), mediaani (min., maks.) | 1 510 (282, 7 164) | 1 244 (282, 7 164) |
| Lähtötilanteen eGFR (ml/min/1,73 m2 ), mediaani (min., maks.) | 22 (10, 105) | 22 (10, 105) |
aHUS-tutkimukseen C10-003 osallistuneet potilaat saivat Solirisia vähintään 26 viikon ajan. Ensimmäisten 26 hoitoviikon jälkeen useimmat potilaat valitsivat pitkäaikaisen hoidon saannin.
Soliris-hoidon aloittamisen jälkeen todettiin terminaalisen komplementin aktiivisuuden vähentyneen kaikilla potilailla. Soliris vähensi komplementin välittämää TMA-aktiviteettia, mikä näkyy trombosyyttien mediaanitason nousuna lähtötasosta viikkoon 26. Keskimääräinen (± keskihajonta) trombosyyttimäärä nousi 88 ± 42 x 109/l:n lähtötilanteesta yhden viikon kuluessa 281 ± 123 x 109/l:een. Tämä vaikutus pysyi ennallaan 26 viikon ajan (keskimääräinen trombosyyttimäärä [± keskihajonta] viikolla 26: 293 ± 106 x 109/l). Munuaistoiminta mitattuna eGFR:llä parani Soliris-hoidon aikana. Niistä 11 potilaasta, jotka tarvitsivat dialyysia lähtötilanteessa, 9 ei tarvinnut dialyysia enää ekulitsumabihoitoa koskevan tutkimuspäivän 15 jälkeen. Vasteet olivat samankaltaisia kaikissa ikäryhmissä 5 kuukaudesta 17 vuoteen. aHUS-tutkimuksessa C10-003 Solirisilla saadut vasteet olivat potilailla samanlaiset huolimatta siitä, oliko heillä vai ei ollut tunnistettavissa olevia komplementtia säätelevän tekijän proteiinien geenimutaatioita tai tekijän H autovasta-aineita.
Taulukossa 18 on yhteenveto aHUS-tutkimuksen C10-003 tehon tuloksista.
Taulukko 18: Prospektiivisen aHUS-tutkimuksen C10-003 tehon tulokset
| Tehon parametri | 1 kuukausi – <12 vuotta (N = 18) 26 viikon kohdalla | Kaikki potilaat (N = 22) 26 viikon kohdalla |
| Täydellinen hematologinen normaalistuminen, n (%) | 14 (78) | 18 (82) |
| Täydellisen hematologisen normaalistumisen kestoajan mediaani, viikkoina (vaihteluväli)1 | 35 (13, 78) | 35 (13, 78) |
| Täydellinen TMA-vaste, n (%) Täydellisen TMA-vasteen kestoajan mediaani, viikkoina (vaihteluväli)1 | 11 (61) 40 (13, 78) | 14 (64) 37 (13, 78) |
| TMA-tapahtumista vapaa tila, n (%) 95 %:n luottamusväli | 17 (94) Ei sovellu | 21 (96) 77; 99 |
| Päivittäisten TMA-toimenpiteiden lukumäärä, mediaani (vaihteluväli) Ennen ekulitsumabihoitoa, mediaani Ekulitsumabihoidon aikana, mediaani | Ei sovellu Ei sovellu | 0,4 (0, 1,7) 0 (0, 1,01) |
| eGFR-parannus ≥15 ml/min/ 1,73 m2, n (%) | 16 (89) | 19 (86) |
| eGFR:n muutos (≥15 ml/min/1,73 m2) viikolla 26, mediaani (vaihteluväli) | 64 (0,146) | 58 (0, 146) |
| CKD-parannus ≥1 asteella, n (%) | 14/16 (88) | 17/20 (85) |
| Plasmanvaihto-/plasmainfuusio tapahtumista vapaa tila, n (%) Uusista dialyysitapahtumista vapaa tila, n (%) 95 %:n luottamusväli | 16 (89) 18 (100) Ei sovellu | 20 (91) 22 (100) 85;100 |
1 tietojen koontihetkellä (12. lokakuuta 2012) Soliris-hoidon keston mediaani 44 viikkoa (vaihteluväli: 1 annos–88 viikkoa).
Pitkäaikaiseen Soliris-hoitoon (mediaani 55 viikkoa, vaihteluväli 1 päivä – 107 viikkoa) liittyi kliinisesti merkittävien parannusten lisääntymistä pediatrisilla ja nuorilla aHUS-potilailla. Kun Soliris-hoitoa-jatkettiin yli 26 viikon ajan, lisäksi yksi potilas (yhteensä 68 % potilaista) saavutti täydellisen TMA-vasteen ja lisäksi kaksi potilasta (yhteensä 91 % potilaista) saavutti hematologisen normalisoitumisen. Viimeisessä arvioinnissa eGFR parani lähtötilanteesta ≥ 15 ml/min/1,73 m2 19 potilaalla 22:sta (86 %). Yksikään potilaista ei tarvinnut uutta dialyysihoitoa Soliris-hoidon aikana.
Refraktorinen yleistynyt myasthenia gravis
Yhteensä 11 pediatrista potilasta, joilla oli refraktorinen gMG, sai Soliris-valmistetta tutkimuksessa ECU‑MG‑303. Hoitoa saaneiden potilaiden kehonpainon mediaani lähtötilanteessa oli 59,7 kg (vaihteluväli 37,2–91,2 kg) ja mediaani-ikä seulonnassa 15 vuotta (vaihteluväli 12–17 vuotta). Kaikilla tutkimukseen mukaan otetuilla potilailla oli refraktorinen gMG ja vähintään yksi seuraavista:
- Vähintään yksi vähintään yhden vuoden kestänyt epäonnistunut immunosuppressiohoito, joka määriteltiin seuraavasti: (i) pitkään jatkunut heikkous, joka hankaloitti päivittäisiä toimintoja, tai (ii) myasthenia graviksen paheneminen ja/tai hoidon aikainen kriisi taikka (iii) sietokyvyttömyys immunosuppressiohoidoille haittavaikutuksen tai oheissairauden (‑sairauksien) vuoksi.
- Ylläpitohoito plasmanvaihdoilla tai IVIg-hoidolla tarpeen oireiden hallitsemiseksi (ts. potilaat, jotka olivat tarvinneet plasmanvaihtoa tai IVIg-hoitoa lihasheikkouden hoitoon säännöllisesti vähintään 3 kuukauden välein seulontaa edeltävien vähintään 12 kuukauden ajan).
Tutkimukseen ECU-MG-303 mukaan otettujen, refraktorista gMG:tä sairastavien pediatristen potilaiden ominaisuudet lähtötilanteessa on kuvattu taulukossa 19.
Taulukko 19: Potilaiden demografiset tiedot ja ominaisuudet tutkimuksessa ECU-MG-303
Ekulitsumabi (n = 11) | ||
|---|---|---|
| Naisia | n (%) | 9 (81,8 %) |
| MG:n kesto (MG:n diagnoosista ensimmäisen tutkimuslääkkeen antopäivämäärään [vuotta]) | Keskiarvo (SD) Mediaani (minimi, maksimi) | 3,99 (2,909) 2,90 (0,1, 8,8) |
| Lähtötilanteen MG-ADL-kokonaispisteet | Keskiarvo (SD) Mediaani (minimi, maksimi) | 5,0 (5,25) 4,0 (0, 19) |
| Lähtötilanteen QMG-kokonaispisteet | Keskiarvo (SD) Mediaani (minimi, maksimi) | 16,7 (5,64) 15,0 (10, 28) |
MGFA-luokka seulonnassa IIb IIIa IIIb IVa IVb | n (%) |
2 (18,2) 3 (27,3) 3 (27,3) 0 3 (27,3) 0 |
Potilaat, joilla on ollut aikaisempia MG:n pahenemisvaiheita, ml. MG-kriisi diagnoosin jälkeen Ei Kyllä Paheneminen MG-kriisi | n (%) |
4 (36,4) 7 (63,6) 6 (54,5) 3 (27,3) |
Pitkäaikainen IVIg-hoito tutkimukseen otettaessa Kyllä Ei | n (%) |
6 (54,5) 5 (45,5) |
Immunosuppressiohoitojen lukumäärä lähtötilanteessa 0 | n (%) |
2 (18,2) 4 (36,4) 5 (45,5) |
Potilaat, jotka saivat immunosuppressiohoitojaa lähtötilanteessa, n (%) Kortikosteroidit Atsatiopriini Mykofenolaattimofetiili Takrolimuusi | n (%) |
8 (72,7) 1 (9,1) 2 (18,2) 3 (27,3) |
aImmunosuppressiohoitoja olivat kortikosteroidit, atsatiopriini, syklofosfamidi, siklosporiini, metotreksaatti, mykofenolaattimofetiili ja takrolimuusi. Kukaan potilaista ei saanut siklosporiinia, syklofosfamidia tai metotreksaattia lähtötilanteessa.
- Lyhenteet: IVIg = laskimonsisäinen immunoglobuliinihoito; MG = myasthenia gravis; MG‑ADL = Myasthenia Gravis Activities of Daily Living profile; MGFA = Myasthenia Gravis Foundation of America; QMG = Quantitative Myasthenia Gravis score for disease severity (kvantitatiivinen pisteytysjärjestelmä myasthenia graviksen vaikeusasteen mukaan); SD = keskihajonta
Tutkimuksen ECU-MG-303 ensisijainen päätetapahtuma oli QMG-kokonaispistemäärän muutos lähtötilanteesta ajan kuluessa varalääkkeestä huolimatta. Soliris-hoitoa saaneilla pediatrisilla potilailla osoitettiin QMG-kokonaispistemäärän tilastollisesti merkitsevää paranemista koko 26 viikon pituisen ensisijaisen arvioitavan hoitovaiheen ajan. Tutkimuksen ECU-MG-303 ensisijaisten ja toissijaisten päätetapahtumien tulokset on esitetty taulukossa 20.
Soliris-hoidon teho refraktorista gMG:tä sairastavilla pediatrisilla potilailla oli yhdenmukainen pivotaalitutkimukseen ECU-MG-301 mukaan otetuilla refraktorista gMG:tä sairastavilla aikuispotilailla havaitun tehon kanssa (taulukko 10).
Taulukko 20: Tehoa koskevat tulokset tutkimuksessa ECU-MG-303
| Tehon päätetapahtumat: Kokonaispistemäärän muutos lähtötilanteesta viikkoon 26 mennessä | Pienimmän neliösumman keskiarvo (SEM) 95 %:n CI |
| QMG | –5,8 (1,2) (–8,40, –3,13) |
| MG-ADL-kokonaispistemäärä | –2,3 (0,6) (–3,63, –1,03) |
| MGC-kokonaispistemäärä | –8,8 (1,9) (–12,92, –4,70) |
an on potilaiden lukumäärä viikkoon 26 mennessä
Lyhenteet: CI = luottamusväli; LS = least squares (pienimmän neliösumman menetelmä); MG-ADL = Myasthenia Gravis Activities of Daily Living profile; MGC = Myasthenia Gravis Composite; ; QMG = Quantitative Myasthenia Gravis score for disease severity (kvantitatiivinen pisteytysjärjestelmä myasthenia graviksen vaikeusasteen mukaan); SEM = keskiarvon keskivirhe; VAS = visuaalinen analoginen asteikko
Tutkimuksessa ECU-MG-303 kliininen vaste QMG- ja MG-ADL-kokonaispistemäärissä määriteltiin vähintään 5 pisteen (QMG) ja vähintään 3 pisteen (MG-ADL) parannukseksi lähtötilanteesta. Kliinisen vasteen saavuttaneiden osuus viikolla 26 varahoidosta riippumatta oli QMG-ryhmässä 70 % ja MG-ADL-ryhmässä 50 %. Viikon 26 tutkimuskäynnille osallistuneet 10 potilasta saavuttivat MGFA‑PIS-statuksen (MGFA Post‑Interventional Status) parannuksen viikolla 26. Seitsemän potilasta (70 %) saavutti refraktorisen gMG:n minimaalisen ilmentymän viikkoon 26 mennessä.
Yhdellä potilaalla (9,1 %) havaittiin kliininen paheneminen (MG-kriisi) ensisijaisen arvioitavan hoitovaiheen aikana. Paheneminen vaati varahoitoa (plasmanvaihto), jota annettiin viikon 22 ja viikon 24 tutkimuskäyntien välillä. Tämän ja hoitavan lääkärin päätöksen takia tälle potilaalle ei tehty QMG-, MG-ADL- tai muita tehon arviointeja viikon 20 jälkeen eikä potilas ollut mukana jatkovaiheessa. Jatkovaiheen aikana kahdella potilaalla esiintyi kliininen paheneminen (MG‑kriisi), joka vaati varahoitoa (toisessa tapauksessa plasmanvaihto ja IVIg‑hoito kliiniseen pahenemiseen ja toisessa tapauksessa IVIg‑hoito ja kaksi lisähoitoa ekulitsumabilla).
Koko refraktorista gMG:tä sairastavia pediatrisia potilaita koskevan tutkimuksen aikana (tutkimus ECU-MG-303) neljän potilaan (4/11) (36,4 %) päivittäistä IST- tai antikolinesteraasiannosta pienennettiin parantuneiden MG-oireiden ansiosta. Lisäksi yhden potilaan (9,1 %) päivittäistä annosta pienennettiin parantuneiden MG‑oireiden ansiosta ja suurennettiin sen jälkeen pahentuneiden MG‑oireiden takia, ja yhdelle potilaalle aloitettiin uusi kortikosteroidihoito pahentuneiden MG‑oireiden takia.
Pitkäaikainen teho
Kaikki potilaat, jotka olivat mukana ensisijaisen hoitovaiheen loppuun asti (N = 10), siirtyivät jatkovaiheeseen, jossa hoitoa annettiin enintään 208 viikon ajan. Vain kaksi potilasta oli mukana jatkovaiheen loppuun asti. Kahdeksan potilasta lopetti tutkimukseen osallistumisen jatkovaiheen aikana. Näistä potilaista neljä siirtyi joko markkinoilla olevalla Soliris- tai Ultomiris-valmisteella annettavaan hoitoon tai toiseen käynnissä olevaan, Ultomiris-valmistetta koskevaan pediatriseen tutkimukseen.
Potilaiden vaste säilyi yhdenmukaisena koko tutkimuksen ajan, ja sen voimakkuus oli samankaltainen kuin alkuperäisen hoitojakson aikana raportoitu vasteen voimakkuus.
Kaavio 3: Toistomittausmalliin perustuva QMG-kokonaispisteiden muutos lähtötilanteesta (pienimmän neliösumman keskiarvo ja 95 %:n luottamusväli) varahoidosta riippumatta viikoilla 1–52
Huomautus: Lähtötilanteen määritelmänä oli viimeinen saatavilla oleva arviointi ennen ensimmäistä tutkimuslääkeinfuusiota.
Huomautus: Arviot perustuvat toistomittaussekamalliin (MMRM), joka käsitti käyntien ja lähtötilanteen arvot.
Keskiarvo = 0. Kovarianssirakenteena käytettiin CS-kovarianssirakennetta.
Neuromyelitis optica -kirjon häiriö
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Soliris-valmisteen käytöstä NMOSD:n hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Farmakokinetiikka ja lääkeaineenvaihdunta
Biotransformaatio
Ihmisen retikuloendoteliaalisen systeemin solut poistavat ja hajottavat endosytoosilla vasta-aineita. Ekulitsumabi sisältää vain luonnollisia aminohappoja eikä sillä ole tunnettuja aktiivisia aineenvaihduntatuotteita. Ihmisen vasta-aineita hajottavat pieniksi peptideiksi ja aminohapoiksi pääasiassa lysosomaaliset entsyymit.
Eliminaatio
Erityisiä tutkimuksia Solirisin erittymis- tai eliminoitumisreiteistä maksan, munuaisten, keuhkojen tai ruoansulatuskanavan kautta ei ole tehty. Normaaleissa munuaisissa vasta-aineet eivät erity eikä niitä voi suodattaa niiden koon vuoksi.
Farmakokineettiset/farmakodynaamiset suhteet
40 PNH-potilaalla käytettiin yksiosaista mallia farmakokineettisten parametrien arviointiin usean annoksen jälkeen. Keskimääräinen puhdistuma oli 0,31 ± 0,12 ml/t/kg, keskimääräinen jakautumistilavuus oli 110,3 ± 17,9 ml/kg ja keskimääräinen eliminaation puoliintumisaika oli 11,3 ± 3,4 päivää. Vakaa tila saavutetaan 4 viikon kuluessa aikuisten PNH-potilaiden annostusohjelmaa käytettäessä.
Farmakodynaaminen aktiivisuus on PNH-potilailla suoraan verrannollinen seerumin ekulitsumabipitoisuuden kanssa. Kun alin pitoisuus on yli ≥ 35 mikrogrammaa/ml, hemolyyttinen toiminta estyy käytännöllisesti katsoen täydellisesti suurimmalla osalla PNH-potilaista.
Tutkimuksissa C08-002A/B ja C08-003A/B Soliris-hoitoa suositellun hoito-ohjelman mukaisesti saaneista 37 aHUS-potilaasta kerättyjen toistuvien annosten farmakokineettisistä tiedoista tehtiin toinen populaatiofarmakokineettinen analyysi tavanomaisella yksitilamallilla. Tämän mallin mukaan Soliris-valmisteen puhdistuma oli tyypillisellä 70 kg:n painoisella aHUS-potilaalla 0,0139 l/h ja jakautumistilavuus oli 5,6 l. Eliminaation puoliintumisaika oli 297 tuntia (noin 12,4 vuorokautta).
Toista väestön farmakokineettista mallia sovellettiin aHUS-tutkimuksessa suositettua Soliris-annosta saavien 22 aHUS-potilaan moniannos-farmakokineettisiin tietoihin. Solirisin puhdistuma ja jakaantumistilavuus ovat painosta riippuvaisia, mikä muodostaa pediatristen potilaiden painoon perustuvan annosohjelman perustan (ks. Annostus ja antotapa). Pediatristen aHUS-potilaiden Solirisin puhdistuma-arvot olivat 10,4 ml/h 70 kg:n painoisilla, 5,3 ml/h 30 kg:n painoisilla ja 2,2 ml/h 10 kg:n painoisilla potilailla. Jakaantumistilavuudet olivat vastaavasti 5,23, 2,76 ja 1,21 litraa. Vastaavat eliminaation puoliintumisajat pysyivät lähes muuttumattomina vaihteluvälillä 349–378 h (noin 14,5–15,8 vuorokautta).
Ekulitsumabin puhdistumaa ja puoliintumisaikaa tutkittiin myös plasmanvaihtotoimenpiteiden aikana. Plasmanvaihto johti ekulitsumabipitoisuuden pienenemiseen noin puoleen 1 tunnin kuluttua toimenpiteestä, ja ekulitsumabin eliminaation puoliintumisaika lyheni 52,4 tuntiin. Lisäannoksen antamista suositellaan, kun Solirista annetaan aHUS-potilaalle, jolle annetaan plasmainfuusioita tai jolle tehdään plasmanvaihto (ks. kohta Annostus ja antotapa).
Kun Solirista annetaan suositusannoksina, terminaalisen komplementin aktiivisuus vähenee kaikilla aHUS-potilailla nopeasti ja pitkäkestoisesti. Farmakodynaaminen aktiivisuus korreloi aHUS-potilailla suoraan seerumin ekulitsumabipitoisuuden kanssa sekä alimman pitoisuuden säilymisen kanssa tasolla noin 50–100 mikrogrammaa/ml, mikä johtaa kaikilla aHUS-potilailla terminaalisen komplementin aktiivisuuden estymiseen jokseenkin täydellisesti.
Farmakokineettiset parametrit ovat yhdenmukaisia PNH:ta, aHUS:aa, refraktorista gMG:tä ja NMOSD:tä sairastavissa potilasryhmissä.
Farmakodynaaminen aktiivisuus, mitattuna < 0,5 mikrog/ml vapaina C5-pitoisuuksina, vastaa jokseenkin täydellistä terminaalisen komplementin aktiivisuuden estoa PNH- ja aHUS-potilailla sekä potilailla, joilla on refraktorinen gMG tai NMOSD.
Erityispopulaatiot
Solirisin farmakokinetiikkaa ei ole arvioitu nimenomaisissa tutkimuksissa erityispopulaatioilla sukupuolen, etnisen taustan, iän (vanhukset) tai munuaisen tai maksan vajaatoiminnan perusteella. Populaatiofarmakokineettiset analyysit tiedoista, jotka on saatu PNH:ta, aHUS:aa, gMG:tä ja NMOSD:tä sairastavilla potilailla tehdyistä tutkimuksista, ovat osoittaneet, ettei sukupuolella, etnisellä taustalla, iällä (vanhukset) tai munuaisten tai maksan vajaatoiminnalla ole vaikutusta ekulitsumabin farmakokinetiikkaan.
Pediatriset potilaat
Ekulitsumabin farmakokinetiikkaa arvioitiin tutkimuksessa M07-005 pediatrisilla PNH-potilailla (iältään 11 – < 18 vuotta), tutkimuksissa C08-002, C08-003, C09-001r ja C10-003 pediatrisilla aHUS-potilailla (iältään 2 kuukautta – < 18 vuotta) sekä tutkimuksessa ECU-MG-303 refraktorista gMG:tä sairastavilla pediatrisilla potilailla (iältään 12 – < 18 vuotta). Populaatiofarmakokineettinen analyysi osoitti painon olevan merkittävä kovariaatti PNH:ssa, aHUS:issa, refraktorisessa gMG:ssä ja NMOSD:ssä ja että pediatristen potilaiden annostus on määritettävä painon perusteella.
Prekliiniset tiedot turvallisuudesta
Ekulitsumabin spesifisyyttä C5:lle ihmisen seerumissa arvioitiin kahdessa in vitro -tutkimuksessa.
Ekulitsumabin ristireaktiivisuutta kudoksessa arvioitiin määrittämällä sitoutuminen 38 ihmiskudoksen valikoimaan. Tässä tutkimuksessa tutkittu C5-ekspressio ihmiskudosvalikoimassa vastaa C5-ekspressiosta julkaistuja raportteja. C5:tä on ilmoitettu esiintyvän sileässä lihaksessa, poikkijuovaisessa lihaksessa ja proksimaalisen munuaistubuluksen epiteelissä. Odottamatonta kudoksen ristireagointia ei havaittu.
Ekulitsumabin vaikutusta eläinten lisääntymiseen ei ole tutkittu, koska valmisteella ei ole farmakologista vaikutusta muilla lajeilla kuin ihmisellä.
26 viikkoa kestäneessä hiirillä tehdyssä toksisuustutkimuksessa, jossa käytettiin keinotekoista rottaeläimen C5:n vastaista vasta-ainetta, hoito ei vaikuttanut tutkittuihin toksisuusparametreihin. Hemolyyttinen aktiivisuus estyi tutkimuksen aikana tehokkaasti sekä naaras- että uroshiirillä.
Hiirillä keinotekoisella terminaalista komplementtia estävällä vasta-aineella tehdyissä lisääntymistoksikologiatutkimuksissa ei havaittu selviä hoitoon liittyviä vaikutuksia tai haittavaikutuksia. Vasta-ainetta käytettiin C5-salpauksen lisääntymisturvallisuuden arviointiin. Näissä tutkimuksissa arvioitiin hedelmällisyyttä ja varhaista alkion kehitystä, kehitystoksisuutta sekä pre- ja postnataalista kehitystä.
Kun emän altistuminen vasta-aineelle tapahtui organogeneesin aikana, suuremmalle vasta-aineannokselle (noin nelinkertainen määrä suositeltuun ihmisen Soliris-annokseen verrattuna painon vertailun perusteella) altistuneiden emojen 230 jälkeläisen joukossa ilmeni kaksi tapausta verkkokalvon dysplasiaa ja yksi napatyrätapaus. Altistuminen ei kuitenkaan lisännyt keskenmenoja tai vastasyntyneiden kuolemia.
Ekulitsumabin genotoksisuutta ja karsinogeenisuutta ei ole tutkittu eläinkokein.
Farmaseuttiset tiedot
Apuaineet
Natriumdivetyfosfaatti (E339), dinatriumvetyfosfaatti (E339), natriumkloridi, polysorbaatti 80 (E433), injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
30 kuukautta.
Lääkevalmiste on käytettävä välittömästi laimentamisen jälkeen. Kemiallisen ja fysikaalisen stabiiliuden on kuitenkin todistettu kestävän 24 tuntia 2-8 °C:n lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C)
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Alkuperäispakkauksessa olevat Soliris-injektiopullot voidaan poistaa jääkaapista vain yhden enintään 3 päivän kestoisen jakson ajaksi. Valmiste on tämän jälkeen säilytettävä jälleen jääkaapissa.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SOLIRIS infuusiokonsentraatti, liuosta varten
300 mg (L:ei) 1 kpl (30 ml (10 mg/ml)) (5401,60 €)
PF-selosteen tieto
30 ml tiivistettä injektiopullossa (tyypin I lasipullo), jossa on tulppa (silikonoitua butyylia) ja tiiviste (alumiinia) sekä irti napsautettava korkki (polypropyleenia).
Pakkauskoko on yksi injektiopullo.
Valmisteen kuvaus:
Kirkas ja väritön liuos, jonka pH on 7,0 ja osmolaliteetti noin 290–310 mOsm/kg.
Käyttö- ja käsittelyohjeet
Ennen kuin Soliris-liuos annetaan, se tulee tarkistaa silmämääräisesti hiukkasten ja värimuutoksen varalta. Älä käytä liuosta, jos siinä näkyy hiukkasia tai värimuutoksia.
Ohjeet:
Käyttövalmiiksi saattaminen ja laimentaminen on tehtävä hyvän käytännön mukaisesti etenkin aseptiikan kannalta.
Vedä Soliris-valmiste kokonaan injektiopullosta steriilillä ruiskulla.
Siirrä suositeltu annos infuusiopussiin.
Laimenna Soliris siten, että sen lopullinen pitoisuus on 5 mg/ml, lisäämällä infuusiopussiin 9 mg/ml natriumkloridi-injektionestettä (0,9 %), 4,5 mg/ml natriumkloridi-injektionestettä (0,45 %) tai 5 % dekstroosia vedessä.
Pitoisuuteen 5 mg/ml laimennetun liuoksen lopullinen määrä on 60 ml 300 mg annoksissa, 120 ml 600 mg annoksissa, 180 ml 900 mg annoksissa ja 240 ml 1 200 mg annoksissa. Liuoksen tulee olla kirkasta ja väritöntä.
Ravista laimennetun liuoksen sisältävää infuusiopussia varovasti, jotta lääkevalmiste ja laimenne sekoittuvat hyvin.
Laimennetun liuoksen tulee antaa lämmetä huoneenlämpöiseksi altistamalla se ympäristön ilmalle, ennen kuin liuos annetaan potilaalle.
Hävitä käyttämätön injektiopulloon jäänyt valmiste.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SOLIRIS infuusiokonsentraatti, liuosta varten
300 mg 1 kpl
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L04AJ01
Valmisteyhteenvedon muuttamispäivämäärä
17.07.2025
Yhteystiedot
Forskaren, Hagaplan 4
SE-113 68 Tukholma/Stockholm
Ruotsi/Sverige
+46 (0)8 557 727 50
alexion.nordics@alexion.com