
PERJETA infuusiokonsentraatti, liuosta varten 420 mg
Vaikuttavat aineet ja niiden määrät
Yksi 14 ml:n injektiopullo sisältää 420 mg pertutsumabia (pitoisuus 30 mg/ml).
Laimentamisen jälkeen yksi ml liuosta sisältää noin 3,02 mg pertutsumabia aloitusannosta varten ja noin 1,59 mg pertutsumabia ylläpitoannosta varten (ks. kohta Käyttö- ja käsittelyohjeet).
Pertutsumabi on nisäkkään (kiinanhamsterin munasarja) soluissa rekombinantti DNA teknologialla tuotettu humanisoitu monoklonaalinen IgG1-vasta-aine.
Apuaine, jonka vaikutus tunnetaan
Yksi 14 ml:n injektiopullo sisältää 2,8 mg polysorbaattia 20
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Varhaisvaiheen rintasyöpä
Perjeta on tarkoitettu käytettäväksi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
- HER2-positiivista, paikallisesti edennyttä, inflammatorista tai varhaisvaiheen rintasyöpää sairastavien aikuispotilaiden neoadjuvanttihoitoon, kun taudin uusiutumisriski on suuri (ks. kohta Farmakodynamiikka)
- varhaisvaiheen HER2-positiivista rintasyöpää sairastavien aikuispotilaiden adjuvanttihoitoon, kun taudin uusiutumisriski on suuri (ks. kohta Farmakodynamiikka).
Metastasoitunut rintasyöpä
Perjeta on tarkoitettu käytettäväksi yhdistelmänä trastutsumabin ja dosetakselin kanssa HER2-positiivista metastasoitunutta tai paikallisesti uusiutunutta leikkaushoitoon soveltumatonta rintasyöpää sairastavien aikuispotilaiden hoitoon, kun potilas ei ole aiemmin saanut metastasoituneen taudin hoitoon anti-HER2-hoitoa eikä solunsalpaajahoitoa.
Ehto
Tämä valmiste on reseptilääke, jonka määräämiseen liittyy rajoitus, ja hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Valmisteen antajan tulee olla terveydenhuollon ammattilainen, jolla on valmius anafylaksian hoitoon, ja ensiapuvälineiden on oltava saatavilla.
Annostus ja antotapa
Perjeta-hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Perjeta-infuusion antajan pitää olla terveydenhuollon ammattilainen, jolla on valmius anafylaksian hoitoon, ja ensiapuvälineiden on oltava saatavilla.
Annostus
Perjeta-hoitoa saavilla potilailla on oltava HER2 -positiivinen kasvain, mikä määritellään immunohistokemiallisella menetelmällä (IHC3+) ja/tai validoidulla in situ ‑hybridisaatiomenetelmällä (ISH) määritettynä suhdelukuna ≥ 2,0.
Tarkkojen ja toistettavissa olevien tulosten varmistamiseksi testaus on tehtävä erikoistuneessa laboratoriossa, joka voi varmistaa testausmenetelmien validoinnin. Ks. validoidun HER2-testimenetelmän pakkausselosteesta lisätietoja määrityksen toteuttamisesta ja tulkinnasta.
Pertutsumabi-hoidon aloitukseen suositeltu aloitusannos on 840 mg 60 minuutin kestoisena infuusiona laskimoon, jonka jälkeen annetaan kolmen viikon välein ylläpitoannoksena 420 mg 30–60 minuutin kestoisena infuusiona. Jokaisen Perjeta-infuusion päättymisen jälkeen suositellaan potilaan tarkkailua 30–60 minuutin ajan. Myöhemmät trastutsumabi- tai solunsalpaajainfuusiot annetaan vasta tarkkailujakson päätyttyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Perjeta ja trastutsumabi pitää antaa peräkkäin eikä niitä saa sekoittaa samaan infuusiopussiin. Perjeta ja trastutsumabi voidaan antaa kummassa tahansa järjestyksessä. Kun Perjetan kanssa annetaan trastutsumabia, trastutsumabihoidossa suositellaan noudattamaan 3 viikon hoito-ohjelmaa joko
- laskimoon annettavana infuusiona, jolloin hoito aloitetaan suositellulla trastutsumabin aloitusannostuksella 8 mg/painokg, jonka jälkeen annetaan kolmen viikon välein ylläpitoannos 6 mg/painokg
tai
- injektiona ihon alle, jolloin trastutsumabia annetaan vakioannos (600 mg) 3 viikon välein riippumatta siitä, mikä potilaan paino on.
Potilaan saadessa jotakin taksaania Perjeta ja trastutsumabi pitää antaa ennen taksaania.
Perjetan kanssa annettaessa dosetakselihoito voidaan aloittaa annoksella 75 mg/m2, joka voidaan sen jälkeen suurentaa annokseen 100 mg/m2 valitun hoito-ohjelman mukaan ja sen mukaan, miten potilas sietää aloitusannoksen. Dosetakselia voidaan vaihtoehtoisesti antaa alusta lähtien annoksina 100 mg/m2 kolmen viikon välein, tällöinkin valitun hoito-ohjelman mukaan. Jos noudatetaan karboplatiinia sisältävää hoito-ohjelmaa, suositeltu dosetakseliannos on aina 75 mg/m2 (ei annoksen suurentamista). Suositeltu paklitakseliannos on 80 mg/m2 kerran viikossa 12 viikon hoitosykleinä annettaessa Perjetan kanssa adjuvanttihoitona.
Potilaan saadessa jotakin antrasykliiniä sisältävää hoitoa Perjeta ja trastutsumabi pitää antaa koko antrasykliinihoidon päätyttyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Metastasoitunut rintasyöpä
Perjetaa pitää antaa yhdistelmänä trastutsumabin ja dosetakselin kanssa. Perjeta- ja trastutsumabihoitoa voidaan jatkaa niin kauan, kunnes tauti etenee tai ilmaantuu haittavaikutuksia, jotka eivät ole hoidettavissa, vaikka dosetakselihoito lopetettaisiin.
Varhaisvaiheen rintasyöpä
Neoadjuvanttihoidossa Perjetaa annetaan yhdistelmänä trastutsumabin ja solusalpaajahoidon kanssa 3–6 hoitosykliä osana varhaisvaiheen rintasyövän kokonaishoitoa (ks. kohta Farmakodynamiikka).
Adjuvanttihoidossa Perjetaa annetaan yhdistelmänä trastutsumabin kanssa yhteensä yhden vuoden ajan (18 hoitosykliin asti tai kunnes tauti uusiutuu tai ilmaantuu haittavaikutuksia, jotka eivät ole hoidettavissa, riippumatta siitä, mikä näistä tapahtuu ensin) osana varhaisvaiheen rintasyövän kokonaishoitoa ja riippumatta leikkauksen ajankohdasta. Hoitoon pitää kuulua tavanomainen antrasykliiniä ja/tai taksaania sisältävä solunsalpaajahoito. Perjeta- ja trastutsumabihoito pitää aloittaa ensimmäisen taksaania sisältävän hoitosyklin 1. päivänä ja sitä pitää jatkaa, vaikka solunsalpaajahoito lopetettaisiin.
Annosten viivästyminen tai antamatta jääminen
Annosten viivästymistä tai antamatta jäämistä koskevat suositukset, ks. jäljempänä taulukko 1.
Taulukko 1. Annosten viivästymistä tai antamatta jäämistä koskevat suositukset
| Kahden peräkkäisen infuusion välinen aika | Perjeta | trastutsumabi | |
| i.v. | s.c. | ||
| < 6 viikkoa | Pertutsumabiannos 420 mg pitää antaa mahdollisimman pian. Älä odota seuraavaan suunniteltuun antoajankohtaan. Jatka sen jälkeen alkuperäisen hoito-ohjelman noudattamista. | Trastutsumabiannos 6 mg/kg pitää antaa laskimoon mahdollisimman pian. Älä odota seuraavaan suunniteltuun antoajankohtaan. Jatka sen jälkeen alkuperäisen hoito-ohjelman noudattamista. | Trastutsumabin vakioannos 600 mg pitää antaa ihon alle mahdollisimman pian. Älä odota seuraavaan suunniteltuun antoajankohtaan. |
| ≥ 6 viikkoa | Pertutsumabilatausannos 840 mg pitää antaa uudelleen 60 minuutin kestoisena infuusiona laskimoon, jonka jälkeen annetaan 3 viikon välein ylläpitoannos 420 mg laskimoon. | Trastutsumabilatausannos 8 mg/kg pitää antaa uudelleen noin 90 minuutin kestoisena infuusiona laskimoon, jonka jälkeen annetaan 3 viikon välein ylläpitoannos 6 mg/kg laskimoon. | |
Annosmuutokset
Perjeta- tai trastutsumabiannoksen pienentämistä ei suositella. Trastutsumabia koskevat tarkemmat tiedot, ks. valmisteyhteenveto.
Potilaan hoitoa voidaan jatkaa silloinkin, jos hänellä esiintyy solunsalpaajahoidosta aiheutunutta korjautuvaa luuydinlamaa, mutta potilasta on seurattava tällöin tarkoin, jotta neutropenian ilmaantuminen voidaan havaita. Dosetakselin ja muiden solunsalpaajien annosmuutokset, ks. kyseisen valmisteen valmisteyhteenveto.
Jos trastutsumabihoito lopetetaan, Perjeta-hoito on lopetettava.
Sydämen vasemman kammion vajaatoiminta
Perjeta- ja trastutsumabihoito pitää keskeyttää vähintään 3 viikoksi, jos oireet ja löydökset viittaavat kongestiiviseen sydämen vajaatoimintaan (Perjeta-hoito pitää lopettaa mikäli oireinen sydämen vajaatoiminta vahvistetaan, ks. tarkemmat tiedot kohdasta Varoitukset ja käyttöön liittyvät varotoimet).
Metastasoitunutta rintasyöpää sairastavat potilaat
Potilaan vasemman kammion ejektiofraktion (LVEF) pitää olla ennen hoitoa ≥ 50 %. Perjeta- ja trastutsumabihoito pitää keskeyttää vähintään 3 viikon ajaksi, jos
- LVEF pienenee alle 40 %:n
- LVEF on 40–45 % ja se on pienentynyt ≥ 10 prosenttiyksikköä hoitoa edeltäneistä arvoista.
Perjeta- ja trastutsumabihoitoa voidaan jatkaa, jos LVEF on palautunut > 45 %:iin tai 40–45 %:iin, ja ero hoitoa edeltäneisiin arvoihin on < 10 prosenttiyksikköä.
Varhaisvaiheen rintasyöpää sairastavat potilaat
Potilaan vasemman kammion ejektiofraktion (LVEF) pitää olla ennen hoitoa ≥ 55 % (≥ 50 % antrasykliinisolunsalpaajahoidon annon päättymisen jälkeen, jos sitä annetaan). Perjeta- ja trastutsumabihoito pitää keskeyttää vähintään 3 viikon ajaksi, jos
- LVEF pienenee alle 50 %:n ja se on pienentynyt ≥ 10 prosenttiyksikköä hoitoa edeltäneistä arvoista.
Perjeta- ja trastutsumabihoitoa voidaan jatkaa, jos LVEF on palautunut ≥ 50 %:iin tai ero hoitoa edeltäneisiin arvoihin on < 10 prosenttiyksikköä.
Iäkkäät potilaat
Perjeta-hoidon tehossa ei havaittu yleisesti eroja ≥ 65-vuotiaiden ja < 65-vuotiaiden potilaiden välillä. Annosta ei tarvitse muuttaa ≥ 65-vuotiailla potilailla. Yli 75-vuotiaista potilaista on vähän tietoja saatavissa. Ks. kohdasta Haittavaikutukset arviointi Perjeta-hoidon turvallisuudesta iäkkäille potilaille.
Munuaisten vajaatoimintaa sairastavat potilaat
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden pertutsumabiannosta ei tarvitse muuttaa. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoitoon ei voida antaa annossuosituksia, koska farmakokineettisiä tietoja on saatavissa vähän (ks. kohta Farmakokinetiikka).
Maksan vajaatoimintaa sairastavat potilaat
Perjeta-valmisteen tehoa ja turvallisuutta ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Erityisiä annossuosituksia ei voida antaa.
Pediatriset potilaat
Perjeta-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Perjetan käyttö ei ole aiheellista rintasyövän hoidossa tässä ikäryhmässä.
Antotapa
Perjeta annetaan infuusiona laskimoon. Valmistetta ei saa antaa nopeana infuusiona eikä boluksena laskimoon. Ks. kohdista Yhteensopimattomuudet ja Käyttö- ja käsittelyohjeet ohjeet Perjetan laimentamisesta ennen lääkkeen antoa.
Aloitusannoksen yhteydessä suositeltu infuusion kestoaika on 60 minuuttia. Jos potilas sietää ensimmäisen infuusion hyvin, seuraavat infuusiot voidaan antaa 30–60 minuutin kestoisina (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infuusioon liittyvät reaktiot
Jos potilaalle kehittyy infuusioon liittyvä reaktio, saattaa olla tarpeen hidastaa infuusionopeutta tai keskeyttää infuusion anto (ks. kohta Haittavaikutukset). Infuusion antoa voidaan jatkaa kun oireet ovat hävinneet. Oireita voidaan lievittää myös antamalla hoitona happea, beeta-agonisteja, antihistamiineja, nopeaa iv-nesteytystä ja kuumelääkkeitä.
Yliherkkyysreaktiot/anafylaksia
Infuusion anto on keskeytettävä heti pysyvästi, jos potilaalle ilmaantuu NCI-CTCAE-luokituksen gradus 4 reaktio (anafylaksia), bronkospasmi tai akuutti hengitysvaikeusoireyhtymä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi potilaalle annetun valmisteen nimi ja eränumero on tallennettava (tai kirjattava) selkeästi.
Sydämen vasemman kammion toimintahäiriö (mukaan lukien kongestiivinen sydämen vajaatoiminta)
HER2-aktiivisuutta salpaavien lääkevalmisteiden, Perjeta mukaan lukien, käytön yhteydessä on raportoitu vasemman kammion ejektiofraktion (LVEF) pienenemistä.Oireisen vasemman kammion systolisen toimintahäiriön (kongestiivisen sydämen vajaatoiminnan) ilmaantuvuus oli suurempi Perjetaa yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa saaneilla potilailla verrattuna trastutsumabia ja solunsalpaajahoitoa saaneisiin potilaisiin. Potilailla, jotka ovat aikaisemmin saaneet antrasykliinejä tai sädehoitoa rintakehän alueelle, saattaa olla suurempi vasemman kammion ejektiofraktion pienenemisen riski. Oireista sydämen vajaatoimintaa raportoitiin useimmiten adjuvanttihoidon yhteydessä potilailla, jotka saivat antrasykliiniä sisältävää solunsalpaajahoitoa (ks. kohta Haittavaikutukset).
Perjetaa ei ole tutkittu seuraavissa potilasryhmissä: hoitoa edeltävä vasemman kammion ejektiofraktio < 50 %; potilaalla aiemmin esiintynyt kongestiivista sydämen vajaatoimintaa; potilaan vasemman kammion ejektiofraktio pienentynyt < 50 %:iin aiemman trastutsumabista koostuneen liitännäishoidon aikana tai potilaalla on tila, joka saattaa heikentää vasemman kammion toimintaa (esim. huonossa hoitotasapainossa oleva verenpainetauti, äskettäin sairastettu sydäninfarkti, vakavat hoitoa vaativat sydämen rytmihäiriöt) tai aiempi antrasykliinihoito, joka vastaa kumulatiivisesti > 360 mg/m2 doksorubisiinia tai vastaavaa.
Vasemman kammion ejektiofraktio on tutkittava ennen Perjeta-hoidon aloittamista sekä säännöllisesti Perjeta-hoidon aikana (esim. kerran neoadjuvanttihoidon aikana ja 12 viikon välein adjuvanttihoidon aikana tai jos tauti on metastasoitunut), jotta varmistetaan, että vasemman kammion ejektiofraktio on normaaliarvojen puitteissa. Jos vasemman kammion ejektiofraktio on pienentynyt siten kuin kohdassa Annostus ja antotapa on mainittu eikä kohenemista todeta tai jos se on seuraavalla arviointikerralla pienentynyt edelleen, Perjeta- ja trastutsumabihoidon lopettamista on harkittava vakavasti, ellei hyötyjen yksittäiselle potilaalle arvioida olevan riskejä suuremmat.
Sydänriski pitää ottaa tarkoin huomioon ja arvioida yksilöllisesti potilaan hoitotarpeen suhteen ennen kuin Perjetaa käytetään yhdistelmänä jonkin antrasykliinin kanssa. HER2-reseptoriin vaikuttavien lääkeaineiden ja antrasykliinien farmakologisten vaikutusten perusteella sydäntoksisuuden riskin voidaan olettaa olevan suurempi käytettäessä Perjetaa ja antrasykliinejä samanaikaisesti kuin peräkkäin.
Perjetan (yhdistelmänä trastutsumabin ja jonkin taksaanin kanssa) käyttöä peräkkäin epirubisiini- tai doksorubisiinihoidon jälkeen monissa antrasykliinipohjaisissa yhdistelmähoidoissa on tutkittu APHINITY- ja BERENICE-tutkimuksissa. Perjetan ja antrasykliinin samanaikaisen käytön turvallisuudesta on kuitenkin vain suppeita tietoja saatavissa. Perjetaa annettiin TRYPHAENA-tutkimuksessa samanaikaisesti epirubisiinin kanssa, jota käytettiin osana FEC-hoitoa (5-fluorourasiili, epirubisiini, syklofosfamidi) (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Hoitoa saivat vain potilaat, jotka eivät olleet aiemmin saaneet solunsalpaajahoitoa, ja he saivat pieniä kumulatiivisia epirubisiiniannoksia (enintään 300 mg/m2). Tässä tutkimuksessa sydäntä koskeva turvallisuus oli samankaltainen kuin silloin, kun potilaat saivat samaa hoitoa, mutta Perjeta annettiin FEC-solunsalpaajahoidon jälkeen.
Infuusioon liittyvät reaktiot
Perjeta-hoitoon on liittynyt infuusioon liittyviä reaktioita, mukaan lukien kuolemaan johtaneita tapahtumia (ks. kohta Haittavaikutukset). Potilasta pitää tarkkailla ensimmäisen Perjeta-infuusion aikana ja 60 minuutin ajan sen jälkeen, ja 30-60 minuutin ajan seuraavien Perjeta-infuusioiden jälkeen. Jos merkittävä infuusioon liittyvä reaktio ilmaantuu, infuusionopeutta on hidastettava tai infuusion anto keskeytettävä. Potilaalle on tällöin annettava asianmukaista hoitoa. Potilas on tutkittava ja hänen tilaansa on seurattava tarkoin, kunnes oireet ja löydökset häviävät täysin. Jos infuusioon liittyvä reaktio on vaikea-asteinen, hoidon lopettamista pysyvästi pitää harkita. Tätä koskevan kliinisen arvion pitää perustua edellisen reaktion vaikeusasteeseen ja haittavaikutukseen annettuun hoitoon saatuun vasteeseen (ks. kohta Annostus ja antotapa).
Yliherkkyysreaktiot/anafylaksia
Potilaita pitää seurata tarkoin yliherkkyysreaktioiden havaitsemiseksi. Perjeta-hoidon aikana on havaittu vaikea-asteisia yliherkkyysreaktioita, anafylaksiaa ja kuolemaan johtaneita tapahtumia (ks. kohta Haittavaikutukset). Lääkevalmisteet tällaisten reaktioiden hoitamiseen sekä ensiapuvälineet on oltava välittömästi saatavilla. Jos potilaalle ilmaantuu NCI-CTCAE-luokituksen gradus 4 yliherkkyysreaktioita (anafylaksia), bronkospasmi tai akuutti hengitysvaikeusoireyhtymä, Perjeta-hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Kuumeinen neutropenia
Perjetalla, trastutsumabilla ja dosetakselilla hoidetuilla potilailla on suurentunut kuumeisen neutropenian riski verrattuna potilaisiin, joita hoidetaan lumelääkkeellä, trastutsumabilla ja dosetakselilla. Riski on suurentunut erityisesti hoidon ensimmäisten kolmen syklin aikana (ks. kohta Haittavaikutukset). Neutrofiilien määrät olivat metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-tutkimuksessa alhaisimmillaan samanlaiset sekä Perjeta-hoitoa saaneilla potilailla että lumelääkehoitoa saaneilla potilailla. Kuumeisen neutropenian suurempi esiintyvyys Perjeta-hoitoa saaneissa potilaissa liittyi limakalvotulehdusten ja ripulin korkeampaan esiintyvyyteen. Limakalvotulehdusten ja ripulin oireenmukaista hoitoa pitää harkita. Kuumeisen neutropenian haittatapahtumia ei raportoitu dosetakselin annon lopettamisen jälkeen.
Ripuli
Perjeta saattaa aiheuttaa vaikean ripulin. Ripuli on yleisintä taksaanien samanaikaisessa käytössä. Iäkkäillä potilailla (≥ 65-vuotiailla) ripulin riski on suurempi kuin nuoremmilla potilailla (≤ 65-vuotiailla). Ripuli hoidetaan tavanomaisten hoitokäytäntöjen ja ohjeistojen mukaisesti. Varhaista hoitoa loperamidilla, nesteytyksellä ja elektrolyyttien korvaushoidolla pitää harkita, etenkin iäkkäille potilaille sekä silloin, jos ripuli on vaikea-asteista tai pitkittyy. Jos potilaan tila ei lievity yhtään, on harkittava pertutsumabihoidon keskeyttämistä. Kun ripuli on saatu hallintaan, pertutsumabihoidon voi aloittaa uudestaan.
Apuaineet, joiden vaikutus tunnetaan
Perjeta sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Perjeta sisältää polysorbaattia 20. Yksi 14 ml:n injektiopullo sisältää 2,8 mg polysorbaattia 20. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Metastasoitunutta rintasyöpää koskeneen satunnaistetun pivotaalitutkimuksen CLEOPATRA 37 potilaan osatutkimuksessa ei todettu farmakokineettisiä yhteisvaikutuksia pertutsumabin ja trastutsumabin tai pertutsumabin ja dosetakselin välillä. Pertutsumabin ja trastutsumabin tai pertutsumabin ja dosetakselin välillä ei todettu lääkkeiden välisiä yhteisvaikutuksia myöskään farmakokineettisessä populaatioanalyysissä. NEOSPHERE- ja APHINITY-tutkimusten farmakokineettiset tiedot varmistivat, ettei lääkkeiden välisiä yhteisvaikutuksia esiinny.
Pertutsumabin vaikutusta samanaikaisesti annettujen solunsalpaajien (dosetakselin, paklitakselin, gemsitabiinin, kapesitabiinin, karboplatiinin ja erlotinibin) farmakokinetiikkaan selvitettiin viidessä tutkimuksessa. Pertutsumabin ja näiden lääkeaineiden välillä ei todettu viitteitä farmakokineettisistä yhteisvaikutuksista. Pertutsumabin farmakokinetiikka oli näissä tutkimuksissa vastaava kuin tutkimuksissa, joissa sitä tutkittiin ainoana lääkeaineena.
Raskaus ja imetys
Raskauden ehkäisy
Hedelmällisessä iässä olevien naisten pitää käyttää tehokasta raskauden ehkäisyä Perjeta-hoidon aikana ja kuuden kuukauden ajan viimeisen pertutsumabi-annoksen jälkeen.
Raskaus
Pertutsumabin käytöstä raskauden aikana ei ole riittävää tutkimustietoa. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Perjetaa ei suositella annettavaksi raskauden aikana eikä hedelmällisessä iässä oleville naisille ilman tehokasta ehkäisyä.
Imetys
Koska ihmisen IgG erittyy rintamaitoon eikä mahdollisesta imeytymisestä tai lapselle aiheutuvasta haitasta ole tietoja, päätöksen imetyksen tai hoidon lopettamisesta on perustuttava imetyksen hyötyyn imetettävälle lapselle ja naisen Perjeta-hoidosta saamaan hyötyyn (ks. kohta Farmakokinetiikka).
Hedelmällisyys
Eläimillä ei ole tehty erityisiä hedelmällisyystutkimuksia pertutsumabin vaikutusten selvittämiseksi. Cynomolgus-apinoilla tehdyistä toistuvan altistuksen aiheuttamaa toksisuutta koskeneista tutkimuksista ei voida tehdä varmoja päätelmiä miehen lisääntymiselimiin kohdistuvista haittavaikutuksista. Pertutsumabin annon seurauksena sukukypsiin cynomolgus-naarasapinoihin ei havaittu kohdistuvan haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Perjeta-valmisteella on vähäinen vaikutus ajokykyyn tai koneiden käyttökykyyn. Perjeta-hoidon aikana voi esiintyä huimausta (ks. kohta Haittavaikutukset). Jos potilaalle ilmaantuu infuusioon liittyvä reaktio, häntä on kehotettava välttämään auton ajamista ja koneiden käyttämistä, kunnes oireet häviävät.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Perjetan turvallisuutta on tutkittu yli 6000 potilaalla faasin I, II ja III tutkimuksissa, joissa oli mukana eri syöpätyyppejä sairastavia potilaita, joita hoidettiin pääasiassa Perjetalla yhdistelmänä muiden kasvainten kasvua estävien lääkeaineiden kanssa. Tällaisia tutkimuksia olivat pivotaalitutkimukset CLEOPATRA (n = 808), NEOSPHERE (n = 417), TRYPHAENA (n = 225) ja APHINITY (n = 4804) (yhdistetyt tiedot taulukossa 2). Perjetan turvallisuus oli kaikissa tutkimuksissa yleensä yhdenmukainen, mutta haittavaikutusten ilmaantuvuudessa ja yleisimmin esiintyvissä haittavaikutuksissa oli eroja sen mukaan, annettiinko Perjeta monoterapiana vai samanaikaisesti kasvainten kasvua estävien lääkeaineiden kanssa.
Haittavaikutustaulukko
Taulukossa 2 esitetään yhteenveto seuraavien kliinisten pivotaalitutkimusten Perjeta-ryhmien haittavaikutuksista:
- CLEOPATRA, jossa metastasoitunutta rintasyöpää sairastaville potilaille annettiin Perjetaa yhdistelmänä dosetakselin ja trastutsumabin kanssa (n = 453)
- NEOSPHERE (n = 309) ja TRYPHAENA (n = 218), joissa paikallisesti edennyttä, tulehduksellista tai varhaisvaiheen rintasyöpää sairastaville potilaille annettiin neoadjuvanttihoitona Perjetaa yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
- APHINITY, jossa varhaisvaiheen rintasyöpää sairastaville potilaille annetttiin adjuvanttihoitona Perjetaa yhdistelmänä trastutsumabin ja antrasykliiniä sisältävän tai sisältämättömän, taksaania sisältävän solunsalpaajahoidon kanssa (n = 2364).
Taulukossa 2 esitetään lisäksi valmisteen markkinoille tulon jälkeen raportoidut haittavaikutukset. Koska Perjetaa käytettiin näissä tutkimuksissa trastutsumabin ja solunsalpaajien kanssa, on vaikeaa varmistaa haittatapahtuman syy-yhteys tiettyyn lääkevalmisteeseen.
Haittavaikutukset luetellaan seuraavassa MedDRA-elinjärjestelmän ja esiintymistiheysluokan mukaisesti:
Hyvin yleiset ≥ 1/10
Yleiset ≥ 1/100, < 1/10
Melko harvinaiset ≥ 1/1 000, < 1/100
Harvinaiset ≥ 1/10 000, < 1/1 000
Hyvin harvinaiset < 1/10 000
Tuntematon (koska saatavissa oleva tieto ei riitä arviointiin)
Haittavaikutukset on esitetty kussakin yleisyysluokassa ja elinjärjestelmässä haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Näiden yhdistettyjen tietojen perusteella yleisimmät haittavaikutukset (≥ 30 %) olivat ripuli, alopesia, pahoinvointi, väsymys, neutropenia ja oksentelu. Yleisimmät NCI-CTCAE-luokkien 3–4 haittavaikutukset (≥ 10 %) olivat neutropenia ja kuumeinen neutropenia.
Taulukko 2 Yhteenveto haittavaikutuksista Perjetaa saaneilla potilailla kliinisissä tutkimuksissa^ sekä valmisteen markkinoille tulon jälkeen†
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen |
| Infektiot | Nasofaryngiitti | Kynnenvierustulehdus Ylempien hengitysteiden infektio | ||
| Veri ja imukudos | Kuumeinen neutropenia* Neutropenia Leukopenia Anemia | |||
| Immuunijärjestelmä | Infuusioon liittyvä reaktio°°, * | Yliherkkyys°, * Lääkeaineyliherkkyys°, * | Anafylaktinen reaktio°, * | Sytokiinioireyhtymä°° |
| Aineenvaihdunta ja ravitsemus | Heikentynyt ruokahalu | Tuumorilyysioireyhtymä† | ||
| Psyykkiset häiriöt | Unettomuus | |||
| Hermosto | Perifeerinen neuropatia Päänsärky Makuaistin häiriöt Perifeerinen sensorinen neuropatia Heitehuimaus Parestesiat | |||
| Silmät | Lisääntynyt kyynelvuoto | |||
| Sydän | Sydämen vasemman kammion toimintahäiriö** | Kongestiivinen sydämen vajaatoiminta** | ||
| Verisuonisto | Kuumat aallot | |||
| Hengityselimet, rintakehä ja välikarsina | Yskä Nenäverenvuoto Hengenahdistus | Interstitiaalinen keuhkosairaus Pleuraeffuusio | ||
| Ruoansulatuselimistö | Ripuli Oksentelu Stomatiitti Pahoinvointi Ummetus Dyspepsia Vatsakipu | |||
| Iho ja ihonalainen kudos | Alopesia Ihottuma Kynsien häiriöt Kutina Ihon kuivuminen | |||
| Luusto, lihakset ja sidekudos | Lihassärky Nivelsärky Raajakipu | |||
| Yleisoireet ja antopaikassa todettavat haitat | Limakalvojen tulehdus Raajojen turvotus Kuume Väsymys Voimattomuus | Vilunväreet Kipu Turvotus |
^ Taulukossa 2 esitetään yhdistetyt tiedot CLEOPATRA-tutkimuksen koko hoitojaksosta (tietojen keruun katkaisupäivä 11. helmikuuta 2014; Perjeta-hoitosyklien lukumäärän mediaani oli 24) sekä NEOSPHERE-tutkimuksen (Perjeta-hoitosyklien lukumäärän mediaani kaikissa hoitoryhmissä oli 4) ja TRYPHAENA-tutkimuksen (Perjeta-hoitosyklien lukumäärän mediaani kaikissa hoitoryhmissä oli 3–6) neoadjuvanttihoitojaksosta sekä APHINITY-tutkimuksen hoitojaksosta (Perjeta-hoitosyklien lukumäärän mediaani oli 18).
* kuolemaan johtaneita haittavaikutuksia on raportoitu
** kaikkien 4 tutkimuksen koko hoitojakso, vasemman kammion toimintahäiriöiden ja kongestiivisen sydämen vajaatoiminnan ilmaantuvuus kuvastavat yksittäisissä tutkimuksissa raportoituja MedDRA Preferred Terms –termien mukaisia haittavaikutuksia
°Yliherkkyys/anafylaktinen reaktio kuvaa usean haittavaikutuksen ryhmää
°°Infuusioon liittyvä reaktio sisältää useita haittavaikutuksia saman ajanjakson aikana. Ks. ”Valikoitujen haittavaikutusten kuvaukset” alla.
† Valmisteen markkinoille tulon jälkeen raportoidut haittavaikutukset.
Valikoitujen haittavaikutusten kuvaukset
Vasemman kammion toimintahäiriö
Vasemman kammion toimintahäiriöiden ilmaantuvuus oli metastasoitunutta rintasyöpää koskeneen CLEOPATRA-pivotaalitutkimuksen hoitojakson aikana suurempi lumehoitoa saaneessa ryhmässä (8,6 %) kuin Perjeta-hoitoa saaneessa ryhmässä (6,6 %). Myös oireisen vasemman kammion toimintahäiriön ilmaantuvuus oli Perjeta-hoitoa saaneessa ryhmässä pienempi (lumehoitoa saaneessa ryhmässä 1,8 % vs. Perjeta-hoitoa saaneessa ryhmässä 1,5 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaat saivat neoadjuvanttihoitoa koskeneessa NEOSPHERE-tutkimuksessa neoadjuvanttihoitona 4 Perjeta-hoitosykliä. Vasemman kammion toimintahäiriön ilmaantuvuus (koko hoitojakson aikana) oli Perjeta-, trastutsumabi- ja dosetakselihoitoa saaneessa ryhmässä suurempi (7,5 %) verrattuna trastutsumabi- ja dosetakselihoitoa saaneeseen ryhmään (1,9 %). Yhdellä Perjeta- ja trastutsumabihoitoa saaneen ryhmän potilaalla oli oireinen vasemman kammion toimintahäiriö.
Vasemman kammion toimintahäiriön ilmaantuvuus (koko hoitojakson aikana) oli neoadjuvanttihoitoa koskeneessa TRYPHAENA-tutkimuksessa Perjetan ja trastutsumabin yhdistelmää sekä FEC-hoitoa (minkä jälkeen potilaat saivat Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa) saaneessa ryhmässä 8,3 %, Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa ja sen jälkeen FEC-hoitoa (5-fluorourasiili, epirubisiini, syklofosfamidi) saaneessa ryhmässä 9,3 % ja Perjetaa yhdistelmänä TCH-hoidon (dosetakseli, karboplatiini ja trastutsumabi) kanssa saaneessa ryhmässä 6,6 %. Oireisen vasemman kammion toimintahäiriön (kongestiivisen sydämen vajaatoiminnan) ilmaantuvuus oli Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa ja sen jälkeen FEC-hoitoa saaneessa ryhmässä 1,3 % (tässä ei ole mukana potilasta jolla oli oireinen vasemman kammion toimintahäiriö FEC-hoidon aikana ennen kuin potilas sai Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa) ja Perjetaa yhdistelmänä TCH-hoidon kanssa saaneessa ryhmässä myös 1,3 %. Perjetaa yhdistelmänä trastutsumabin ja FEC-hoidon kanssa ja sen jälkeen Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä yhdelläkään potilaalla ei ollut oireista vasemman kammion toimintahäiriötä.
NYHA-luokan III/IV oireisen vasemman kammion systolisen toimintahäiriön (NCI-CTCAE-luokituksen [v.4] mukainen kongestiivinen sydämen vajaatoiminta) ilmaantuvuus oli BERENICE-tutkimuksen neoadjuvanttijaksossa lyhyen antovälin doksorubisiini- ja syklofosfamidihoitoa (adjuvanttisolunsalpaajahoitoa) ja sen jälkeen Perjetaa yhdistelmänä trastutsumabin ja paklitakselin kanssa saaneessa ryhmässä 1,5 %, mutta oireista vasemman kammion systolista toimintahäiriötä ei esiintynyt yhdelläkään potilaalla (0 %) FEC-hoitoa ja sen jälkeen Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä. Oireettoman vasemman kammion systolisen toimintahäiriön ilmaantuvuus (NCI-CTCAE-luokituksen [v.4] mukainen ejektiofraktion pieneneminen) oli lyhyen antovälin adjuvanttisolunsalpaajahoitoa ja sen jälkeen Perjetaa yhdistelmänä trastutsumabin ja paklitakselin kanssa saaneessa ryhmässä 7 % ja FEC-hoitoa ja sen jälkeen Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä 3,5 %.
Oireisen sydämen vajaatoiminnan (NYHA-luokka III tai IV) ja siihen liittyneen vasemman kammion ejektiofraktion pienenemisen vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin ilmaantuvuus oli APHINITY-tutkimuksessa < 1 % (0,9 %:lla Perjeta-hoitoa saaneista potilaista vs 0,5 %:lla lumehoitoa saaneista potilaista). Oireista sydämen vajaatoimintaa kokeneista potilaista tietojen keruun katkaisuajankohtana oli toipunut 55,6 % Perjeta-hoitoa saaneista potilaista ja 71,4 % lumehoitoa saaneista potilaista. (Toipuminen oli määritelty kahdeksi peräkkäiseksi yli 50 %:n vasemman kammion ejektiofraktion mittaustulokseksi.) Valtaosa tapahtumista raportoitiin antrasykliinihoitoa saaneilla potilailla. Oireetonta tai lievästi oireista (NYHA-luokka II) vasemman kammion ejektiofraktion pienenemistä vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin raportoitiin 2,9 %:lla Perjeta-hoitoa saaneista potilaista ja 3,0 %:lla lumehoitoa saaneista potilaista. Näistä potilaista 82,4 % Perjeta-hoitoa saaneista potilaista ja 83,3 % lumehoitoa saaneista potilaista oli tietojen keruun katkaisuajankohtana toipunut.
Infuusioon liittyvät reaktiot
Infuusioon liittyväksi reaktioksi määriteltiin pivotaalitutkimuksissa mikä tahansa tapahtuma, joka raportoitiin yliherkkyydeksi, anafylaktiseksi reaktioksi, akuutiksi infuusioon liittyväksi reaktioksi tai sytokiinioireyhtymäksi, joka esiintyi infuusion aikana tai infuusion antopäivänä. Pivotaalitutkimuksessa CLEOPATRA Perjeta-aloitusannos annettiin päivää ennen trastutsumabia ja dosetakselia, jotta Perjetaan liittyviä vaikutuksia voitiin tutkia. Ensimmäisenä Perjeta-hoidon antopäivänä infuusioon liittyvien reaktioiden kokonaisesiintyvyys oli 9,8 % lumehoitoa saaneessa ryhmässä ja 13,2 % Perjeta-hoitoa saaneessa ryhmässä, ja suurin osa infuusioon liittyvistä reaktioista oli lieviä tai keskivaikeita. Yleisimmät infuusioon liittyvät reaktiot (≥ 1,0 %) Perjeta-hoitoa saaneessa ryhmässä olivat kuume, vilunväristykset, väsymys, päänsärky, voimattomuus, yliherkkyys ja oksentelu.
Toisen hoitosyklin aikana, jolloin kaikkia lääkevalmisteita annettiin samana päivänä, yleisimmät (≥ 1,0 %) infuusioon liittyvät reaktiot Perjeta-hoitoa saaneessa ryhmässä olivat väsymys, makuaistin häiriöt, lääkeyliherkkyys, lihaskipu ja oksentelu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Perjetaa annettiin neoadjuvantti- ja adjuvanttihoitoa koskeneiden tutkimusten kaikissa hoitosykleissä samana päivänä kuin muitakin tutkimuslääkkeitä. Infuusioon liittyviä reaktioita esiintyi ensimmäisenä Perjeta-hoidon (yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa) antopäivänä 18,6–25,0 %:lla potilaista. Infuusioreaktioiden tyyppi ja vaikeusaste olivat yhdenmukaisia niiden CLEOPATRA-tutkimuksessa havaittujen reaktioiden kanssa, jotka esiintyivät hoitosykleissä, joissa Perjetaa annettiin samana päivänä trastutsumabin ja dosetakselin kanssa. Suurin osa reaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita.
Yliherkkyysreaktiot/anafylaksia
Metastasoitunutta rintasyöpää koskeneessa pivotaalitutkimuksessa CLEOPATRA tutkijoiden raportoiman yliherkkyyden/anafylaksian kokonaisesiintyvyys koko hoitojakson aikana oli 9,3 % lumehoitoa saaneessa ryhmässä ja 11,3 % Perjeta-hoitoa saaneessa ryhmässä. Näistä 2,5 % lumehoitoa saaneessa ryhmässä ja 2,0 % Perjeta-hoitoa saaneessa ryhmässä oli vaikeusasteeltaan NCI-CTCAE-luokituksen mukaisia gradus 3–4 haittavaikutuksia. Lumehoitoa saaneessa ryhmässä 2 potilaalla ja Perjeta-hoitoa saaneessa ryhmässä 4 potilaalla esiintyi tapahtumia, jotka tutkija kuvasi anafylaksiaksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suurin osa yliherkkyysreaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita ja ne hävisivät hoidon avulla. Suurimman osan reaktioista arvioitiin tutkimushoitoon tehtyjen muutosten perusteella johtuneen dosetakseli-infuusioista.
Yliherkkyys-/anafylaktiset tapahtumat olivat neoadjuvantti- ja adjuvanttihoitoa koskeneissa tutkimuksissa samankaltaisia kuin CLEOPATRA-tutkimuksessa. NEOSPHERE-tutkimuksessa kahdella Perjetaa ja dosetakselia saaneen ryhmän potilaalla esiintyi anafylaksiaa. Yliherkkyyden/anafylaksian kokonaisesiintyvyys oli sekä TRYPHAENA- että APHINITY-tutkimuksessa suurin Perjetaa ja TCH-hoitoa saaneessa ryhmässä (TRYPHAENA-tutkimuksessa 13,2 % ja APHINITY-tutkimuksessa 7,6 %). Näistä 2,6 % TRYPHAENA-tutkimuksessa ja 1,3 % APHINITY-tutkimuksessa oli vaikeusasteeltaan NCI-CTCAE-luokituksen gradus 3–4.
Kuumeinen neutropenia
Suurimmalla osalla pivotaalitutkimuksen (CLEOPATRA) kummankin hoitoryhmän potilaista esiintyi vähintään yksi leukopeniatapahtuma (63,0 % Perjeta-hoitoryhmän potilaista ja 58,3 % lumehoitoryhmän potilaista), ja suurin osa näistä oli neutropeenisiä tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kuumeista neutropeniaa esiintyi 13,7 %:lla Perjeta-hoitoa saaneista potilaista ja 7,6 %:lla lumehoitoa saaneista potilaista. Kummassakin hoitoryhmässä niiden potilaiden osuus, joilla esiintyi kuumeista neutropeniaa, oli suurin ensimmäisen hoitosyklin aikana ja väheni sen jälkeen tasaisesti. Kuumeisen neutropenian esiintymistiheyden todettiin kummassakin hoitoryhmässä olevan suurentunut aasialaisilla potilailla verrattuna muihin roturyhmiin ja muilta maantieteellisiltä alueilta peräisin oleviin potilaisiin. Aasialaisilla potilailla kuumeisen neutropenian esiintyvyys oli suurempi Perjeta-hoitoa saaneessa ryhmässä (25,8 %) verrattuna lumehoitoa saaneeseen ryhmään (11,3 %).
Kuumeista neutropeniaa esiintyi NEOSPHERE-tutkimuksessa 8,4 %:lla Perjetaa, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 7,5 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa kuumeista neutropeniaa esiintyi 17,1 %:lla Perjetan ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 9,3 %:iin neoadjuvanttihoitona Perjetaa, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Kuumeisen neutropenian ilmaantuvuus oli TRYPHAENA-tutkimuksessa suurempi kuusi hoitosykliä Perjetaa saaneilla potilailla verrattuna kolme hoitosykliä Perjetaa saaneisiin potilaisiin annetusta solunsalpaajahoidosta riippumatta. Neutropenian ja kuumeisen neutropenian ilmaantuvuus oli kummassakin neoadjuvanttitutkimuksessa, samoin kuin CLEOPATRA-tutkimuksessa, suurempi aasialaisilla potilailla verrattuna muihin potilaisiin. Kuumeista neutropeniaa esiintyi NEOSPHERE-tutkimuksessa 8,3 %:lla Perjetaa, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista aasialaisista potilaista verrattuna 4,0 %:iin aasialaisista potilaista, jotka saivat neoadjuvanttihoitona trastutsumabia ja dosetakselia.
Kuumeista neutropeniaa esiintyi APHINITY-tutkimuksessa 12,1 %:lla Perjeta-hoitoa saaneista potilaista ja 11,1 % lumehoitoa saaneista potilaista. Kuumeisen neutropenian ilmaantuvuuden havaittiin CLEOPATRA-, TRYPHAENA- ja NEOSPHERE-tutkimusten tavoin olleen APHINITY-tutkimuksessa suurempi Perjeta-hoitoa saaneilla aasialaisilla potilailla verrattuna muihin etnisiin ryhmiin kuuluviin potilaisiin (Perjeta-hoitoa saaneilla potilailla 15,9 % ja lumehoitoa saaneilla potilailla 9,9 %).
Ripuli
Ripulia esiintyi metastasoitunutta rintasyöpää koskeneessa pivotaalitutkimuksessa CLEOPATRA 68,4 %:lla Perjeta-hoitoa saaneista potilaista ja 48,7 %:lla lumehoitoa saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Useimmat tapahtumat olivat vaikeusasteeltaan lieviä tai kohtalaisia ja niitä esiintyi muutaman ensimmäisen hoitosyklin aikana. NCI-CTCAE-luokituksen mukaisen gradus 3–4 ripulin ilmaantuvuus oli 9,3 % Perjeta-hoitoa saaneilla potilailla verrattuna 5,1 %:iin lumehoitoa saaneilla potilailla. Ripuli kesti pisimmillään (mediaani) 18 vuorokautta Perjeta-hoitoa saaneilla potilailla ja 8 vuorokautta lumehoitoa saaneilla potilailla. Potilaat saivat hyvän vasteen hyvissä ajoin aloitettuun ripulilääkehoitoon.
Ripulia esiintyi NEOSPHERE-tutkimuksessa 45,8 %:lla Perjetaa, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 33,6 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa ripulia esiintyi 72,3 %:lla Perjetan ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 61,4 %:iin neoadjuvanttihoitona Perjetaa, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Useimmat tapahtumat olivat kummassakin tutkimuksessa vaikeusasteeltaan lieviä tai keskivaikeita.
Ripulin ilmaantuvuuden raportoitiin olleen APHINITY-tutkimuksessa suurempi Perjeta-hoitoa saaneilla potilailla (71,2 %) verrattuna lumehoitoa saaneisiin potilaisiin (45,2 %). Graduksen ≥ 3 ripulia raportoitiin Perjeta-haarassa 9,8 %:lla potilaista vs. lumehaarassa 3,7 %:lla potilaista. Valtaosa raportoiduista tapahtumista oli vaikeusasteeltaan gradus 1 tai 2. Ripulin (kaikki gradukset) ilmaantuvuuden raportoitiin olleen suurin kohdennetun hoidon ja taksaanisolunsalpaajahoidon yhdistelmän käytön aikana (Perjeta-hoitohaarassa 61,4 %:lla potilaista vs. lumehoitohaarassa 33,8 %:lla potilaista). Ripulin ilmaantuvuus oli huomattavasti vähäisempää solunsalpaajahoidon lopettamisen jälkeen, sillä solunsalpaajahoidon jälkeisen kohdennetun hoitojakson aikana ripulia esiintyi Perjeta-hoitohaarassa 18,1 %:lla potilaista vs. lumehoitohaarassa 9,2 %:lla potilaista.
Ihottuma
Ihottumaa esiintyi metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-pivotaalitutkimuksessa 51,7 %:lla Perjeta-hoitoa saaneista potilaista verrattuna 38,9 %:iin lumehoitoa saaneista potilaista. Useimpien tapahtumien vaikeusaste oli gradus 1 tai 2, ne esiintyivät kahden ensimmäisen hoitosyklin aikana ja ne vastasivat hyvin hoitosuositusten mukaiseen hoitoon, kuten aknen paikalliseen tai suun kautta otettavaan hoitoon.
Ihottumaa esiintyi NEOSPHERE-tutkimuksessa 40,2 %:lla Perjetaa, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 29,0 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa ihottumaa esiintyi 36,8 %:lla Perjetan ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 20,0 %:iin neoadjuvanttihoitona Perjetaa, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Ihottuman ilmaantuvuus oli annetusta solunsalpaajahoidosta riippumatta suurempi kuusi hoitosykliä Perjetaa saaneilla potilailla verrattuna kolme hoitosykliä Perjetaa saaneisiin potilaisiin.
Ihottumaa esiintyi APHINITY-tutkimuksessa haittavaikutuksena 25,8 %:lla Perjeta-hoitohaaran potilaista vs. 20,3 %:lla lumehoitohaaran potilaista. Valtaosa ihottumista oli vaikeusasteeltaan gradus 1 tai 2.
Laboratorioarvojen poikkeavuudet
NCI-CTCAE-luokituksen (v. 3) mukaisen gradus 3–4 neutropenian ilmaantuvuus oli metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-pivotaalitutkimuksessa vastaavaa kummassakin hoitoryhmässä (86,3 %:lla Perjeta-hoitoa saaneista potilaista ja 86,6 %:lla lumehoitoa saaneista potilaista, mukaan lukien gradus 4 neutropenia 60,7 %:lla Perjeta-hoitoa saaneista potilaista ja 64,8 %:lla lumehoitoa saaneista potilaista).
NCI-CTCAE-luokituksen (v.3) gradus 3–4 neutropenian ilmaantuvuus NEOSPHERE-tutkimuksessa Perjetaa, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneille potilaille oli 74,5 %, mistä graduksen 4 neutropeniaa oli 50,9 %, verrattuna 84,5 %:n ilmaantuvuuteen trastutsumabia ja dosetakselia saaneilla potilailla, mistä graduksen 4 neutropeniaa oli 60,2 %. TRYPHAENA-tutkimuksessa NCI-CTCAE-luokituksen (v.3) graduksen 3–4 neutropenian ilmaantuvuus oli Perjetan ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneilla potilailla 85,3 %, mistä graduksen 4 neutropeniaa oli 66,7 %, ja neoadjuvanttihoitona Perjetaa, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneilla potilailla ilmaantuvuus oli 77,0 %, mistä graduksen 4 neutropeniaa oli 59,5 %.
NCI-CTCAE-luokituksen (v.4) gradus 3–4 neutropenian ilmaantuvuus APHINITY-tutkimuksessa Perjetaa, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille oli 40,6 % verrattuna 39,1 %:n ilmaantuvuuteen lumelääkettä, trastutsumabia ja solunsalpaajahoitoa saaneilla potilailla. Graduksen 4 neutropenian ilmaantuvuus oli 28,3 % Perjetaa, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille ja 26,5 % lumelääkettä, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille.
Iäkkäät potilaat
Seuraavien haittavaikutusten kaikkien vaikeusasteiden ilmaantuvuus oli ≥ 65-vuotiailla potilailla vähintään 5 % suurempi verrattuna < 65-vuotiaisiin potilaisiin: heikentynyt ruokahalu, anemia, painon lasku, voimattomuus, makuaistin häiriöt, perifeerinen neuropatia, hypomagnesemia ja ripuli. Yli 75-vuotiaista potilaista on vähän tietoja saatavilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurinta siedettyä pertutsumabiannosta ei ole määritetty. Kliinisissä tutkimuksissa ei ole tutkittu annosta 25 mg/kg (1727 mg) suurempia kerta-annoksia.
Yliannoksen yhteydessä potilasta on tarkkailtava haittavaikutusten oireiden ja löydösten havaitsemiseksi ja sopiva oireenmukainen hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, monoklonaaliset vasta-aineet, ATC-koodi: L01FD02
Vaikutusmekanismi
Pertutsumabi on humanisoitu, rekombinantti, monoklonaalinen vasta-aine, jonka erityisenä vaikutuskohteena on ihmisen epidermaalisen kasvutekijän reseptorin 2 (HER2) solunulkoinen dimerisaatiodomeeni (aladomeeni II) ja joka siten estää HER2:n, samoin kuin muiden HER-sukuisten reseptorien, kuten EGFR, HER3 ja HER4, ligandiriippuvaista heterodimerisaatiota. Tämän seurauksena pertutsumabi estää ligandin käynnistämää solunsisäistä signaalinvälitystä kahden pääsiallisen signalointireitin kautta, joita ovat mitogeenien aktivoimat proteiinikinaasit (MAP) ja fosfoinositidi-3-kinaasi (PI3K). Näiden signalointireittien estyminen voi johtaa vastaavasti solun kasvun pysähtymiseen ja apoptoosiin. Pertutsumabi toimii lisäksi vasta-aineriippuvaisen soluvälitteisen sytotoksisuuden (antibody-dependent cell-mediated cytotoxicity, ADCC) välittäjänä.
Vaikka pertutsumabi esti yksinään käytettynä ihmisen kasvainsolujen proliferaation, pertutsumabin ja trastutsumabin yhdistelmä lisäsi merkittävästi antituumoriaktiivisuutta HER2-reseptoria yli-ilmentävissä vieraslajisiirremalleissa.
Kliininen teho ja turvallisuus
Metastasoitunutta rintasyöpää koskeva satunnaistettu faasin III tutkimus ja yhden hoitohaaran faasin II tutkimus, kaksi varhaisvaiheen rintasyövän neoadjuvanttihoitoa koskevaa satunnaistettua faasin II tutkimusta (joista toinen kontrolloitu), neoadjuvanttihoitoa koskeva satunnaistamaton faasin II tutkimus ja adjuvanttihoitoa koskeva satunnaistettu faasin III tutkimus tukevat Perjetan tehoa HER2-positiivisen rintasyövän hoidossa.
HER2-reseptorin yli-ilmentyminen määritettiin keskuslaboratoriossa. HER2-reseptorin yli-ilmentymiseksi määriteltiin edellä mainituissa tutkimuksissa immunohistokemiallinen värjäytymistulos 3+ (ICH 3+) tai ISH-testauksen monistumasuhdeluku ≥ 2,0.
Metastasoitunut rintasyöpä
Perjeta yhdistelmänä trastutsumabin ja dosetakselin kanssa
CLEOPATRA (WO20698) on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu faasin III kliininen monikeskustutkimus, joka toteutettiin 808 metastasoitunutta tai paikallisesti uusiutunutta leikkaushoitoon soveltumatonta HER2-positiivista rintasyöpää sairastavalla potilaalla. Tutkimukseen ei otettu mukaan potilaita, joilla oli kliinisesti merkityksellisiä sydämeen liittyviä riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koska tutkimukseen ei otettu mukaan potilaita, joilla oli etäpesäkkeitä aivoissa, Perjetan vaikutuksesta etäpesäkkeisiin aivoissa ei ole tietoja saatavissa. Vain hyvin rajoitettu määrä tietoa on saatavilla potilaista, joilla on leikkaushoitoon soveltumaton, paikallisesti uusiutunut tauti. Potilaat satunnaistettiin suhteessa 1:1 saamaan hoitona lumelääkkeen, trastutsumabin ja dosetakselin yhdistelmää tai Perjetan, trastutsumabin ja dosetakselin yhdistelmää.
Perjeta ja trastutsumabi annettiin vakioannoksina kolmen viikon välein. Potilas sai Perjeta- ja trastutsumabihoitoa, kunnes tauti eteni, potilas perui suostumuksensa tutkimukseen osallistumiseen tai hänelle ilmaantui haittavaikutuksia, jotka eivät olleet hoidettavissa. Dosetakselihoito aloitettiin annoksella 75 mg/m2 infuusiona laskimoon kolmen viikon välein vähintään 6 hoitosyklin ajan. Dosetakseliannos voitiin suurentaa tutkijan harkinnan mukaan annokseen 100 mg/m2, jos potilas sieti alkuannoksen hyvin.
Tutkimuksen ensisijainen päätetapahtuma oli taudin etenemisvapaa aika (PFS), jonka arvioi riippumaton arviointilautakunta (independent review facility, IRF), ja joka määriteltiin ajaksi satunnaistamispäivästä taudin etenemiseen tai (mistä tahansa syystä tapahtuneeseen) kuolemaan, jos potilas kuoli 18 viikon kuluessa kasvaimen viimeisimmästä tutkimuskerrasta. Toissijaisia tehon päätetapahtumia olivat kokonaiselossaoloaika (OS), (tutkijan arvioima) taudin etenemisvapaa aika (PFS), objektiivinen vasteluku (ORR), vasteen kesto ja aika oireiden etenemiseen FACT B Quality of Life ‑elämänlaatukyselyn perusteella.
Kummassakin hoitoryhmässä noin puolella potilaista oli hormonireseptoripositiivinen tauti (määriteltiin estrogeenireseptoripositiiviseksi [ER-positiiviseksi] ja/tai progesteronireseptoripositiiviseksi [PgR-positiiviseksi]) ja kummassakin hoitoryhmässä noin puolet potilaista oli saanut aiemmin adjuvantti- tai neoadjuvanttihoitoa. Suurin osa näistä potilaista oli saanut aiemmin antrasykliiniä ja 11 % kaikista potilaista oli saanut aiemmin trastutsumabia. Yhteensä 43 % kummankin hoitoryhmän potilaista oli saanut aiemmin sädehoitoa. Potilaiden vasemman kammion ejektiofraktion mediaani ennen hoitoa oli 65,0% kummassakin ryhmässä (vaihteluväli 50–88 %).
Hoidon tehon tulokset CLEOPATRA-tutkimuksessa on esitetty yhteenvetona taulukossa 3. Riippumattoman arviointilautakunnan arvioima taudin etenemisvapaa elinaika oli Perjeta-hoitoa saaneessa ryhmässä tilastollisesti merkitsevästi pidempi kuin lumelääkehoitoa saaneessa ryhmässä. Tulokset tutkijan arvioimasta taudin etenemisvapaasta ajasta olivat samankaltaiset kuin riippumattoman arviointilautakunnan arvioimat taudin etenemisvapaan ajan tulokset.
Taulukko 3. Yhteenveto hoidon tehosta CLEOPATRA-tutkimuksessa
| Parametri | Lumelääke + trastu-tsumabi + dosetakseli n = 406 | Perjeta + trastu-tsumabi + dosetakseli n = 402 | Riskisuhde (95 %:n luottamus-väli) | p-arvo |
Taudin etenemisvapaa aika (riippumaton arvio)- ensisijainen päätetapahtuma* Niiden potilaiden lukumäärä, joilla tapahtuma esiintyi Kuukautta (mediaani) | 242 (59 %) 12,4 | 191 (47,5 %) 18,5 | 0,62 [0,51; 0,75] | < 0,0001 |
Kokonaiselinaika - toissijainen päätetapahtuma** Niiden potilaiden lukumäärä, joilla tapahtuma esiintyi Kuukautta (mediaani) | 221 (54,4 %) 40,8 | 168 (41,8 %) 56,5 | 0,68 [0,56; 0,84] | 0,0002 |
Objektiivinen vasteluku (ORR)^ - toissijainen päätetapahtuma Niiden potilaiden lukumäärä, joilla tauti oli mitattavissa Vasteen saaneiden lukumäärä*** Objektiivisen vasteluvun 95 %:n luottamusväli Täydellinen vaste Osittainen vaste Stabiili tauti Etenevä tauti | 336 233 (69,3 %) [64,1; 74,2] 14 (4,2 %) 219 (65,2 %) 70 (20,8 %) 28 (8,3 %) | 343 275 (80,2 %) [75,6; 84,3] 19 (5,5 %) 256 (74,6 %) 50 (14,6 %) 13 (3,8 %) | Objektiivisen vasteluvun ero: 10,8 % [4,2; 17,5] | 0,0011 |
Vasteen kesto †^ n= Viikkoa (mediaani) Mediaanin 95 %:n luottamusväli | 233 54,1 [46; 64] | 275 87,6 [71; 106] |
*Taudin etenemisvapaan ajan ensisijainen analyysi, tiedonkeruun katkaisupäivä 13. toukokuuta 2011.
** Tapahtumaperusteisen kokonaiselinajan loppuanalyysi, tiedonkeruun katkaisupäivä 11. helmikuuta 2014.
*** Potilaan paras kokonaisvaste RECIST-luokituksen mukainen täydellinen vaste tai osittainen vaste.
† Arvioitu potilailla, joiden paras kokonaisvaste on täydellinen vaste tai osittainen vaste.
^ Objektiivinen vasteluku ja vasteen kestoaika perustuvat riippumattoman arviointilautakunnan tekemään arvioon kasvaimesta.
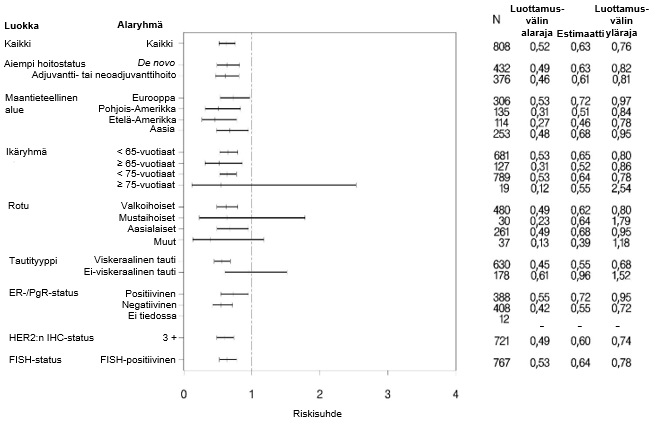
Tulokset olivat yhdenmukaisia ennalta määritellyissä potilaiden alaryhmissä, mukaan lukien alaryhmissä, jotka perustuivat ositustekijöinä käytettyihin maantieteelliseen alueeseen ja aiempaan adjuvantti-/neoadjuvanttihoitoon tai de novo metastasoituneeseen rintasyöpään (ks. kuva 1).
Eksploratiivisessa post hoc ‑analyysissa todettiin, että trastutsumabia aiemmin saaneiden potilaiden (n = 88) riippumattoman arviointilautakunnan arvioima taudin etenemisvapaan ajan riskisuhde oli 0,62 (95 %:n luottamusväli 0,35; 1,07) verrattuna riskisuhteeseen 0,60 (95 %:n luottamusväli 0,43; 0,83) potilailla, jotka olivat saaneet aiempaa hoitoa, mutta joka ei sisältänyt trastutsumabia (n = 288).
Kuva 1. Riippumattoman arviointilautakunnan arvioima taudin etenemisvapaa aika potilasryhmittäin
Tapahtumaperusteisen kokonaiselinajan loppuanalyysi tehtiin, kun 389 potilasta oli kuollut (221 lumehoitoa saaneessa ryhmässä ja 168 Perjeta-hoitoa saaneessa ryhmässä). Perjeta-hoitoa saaneen ryhmän kokonaiselossaolon tilastollisesti merkitsevä hyöty, aikaisemmin havaittu kokonaiselossaolon välianalyysissä tehtynä vuoden kuluttua esisijaisen tehon analyysistä, oli säilynyt (riskisuhde 0,68, p = 0,0002 log-rank-testi). Ajan mediaani kuolemaan oli lumehoitoa saanessa ryhmässä 40,8 kuukautta ja Perjeta-hoitoa saaneessa ryhmässä 56,5 kuukautta (ks. taulukko 3, kuva 2).
Kokonaiselinaikaa kuvaava analyysi tehtiin tutkimuksen lopussa, kun 515 potilasta oli kuollut (280 lumehoitoa saaneessa ryhmässä ja 235 Perjeta-hoitoa saaneessa ryhmässä). Analyysi osoitti, että kokonaiselinajassa todettu tilastollisesti merkitsevä hyöty Perjeta-hoitoa saaneen ryhmän eduksi oli säilynyt 99 kuukauden (mediaani) seurannan ajan (riskisuhde 0,69, p < 0,0001 log-rank-testillä; aika kuolemaan (mediaani) 40,8 kuukautta [lumehoitoa saanut ryhmä] verrattuna 57,1 kuukauteen [Perjeta-hoitoa saanut ryhmä]). Elinajan kiintopiste-estimaatti 8 vuoden kohdalla oli Perjeta-hoitoa saaneessa ryhmässä 37 % ja lumehoitoa saaneessa ryhmässä 23 %.
Kuva 2. Tapahtumaperusteisen kokonaiselinajan Kaplan-Meier-käyrä.
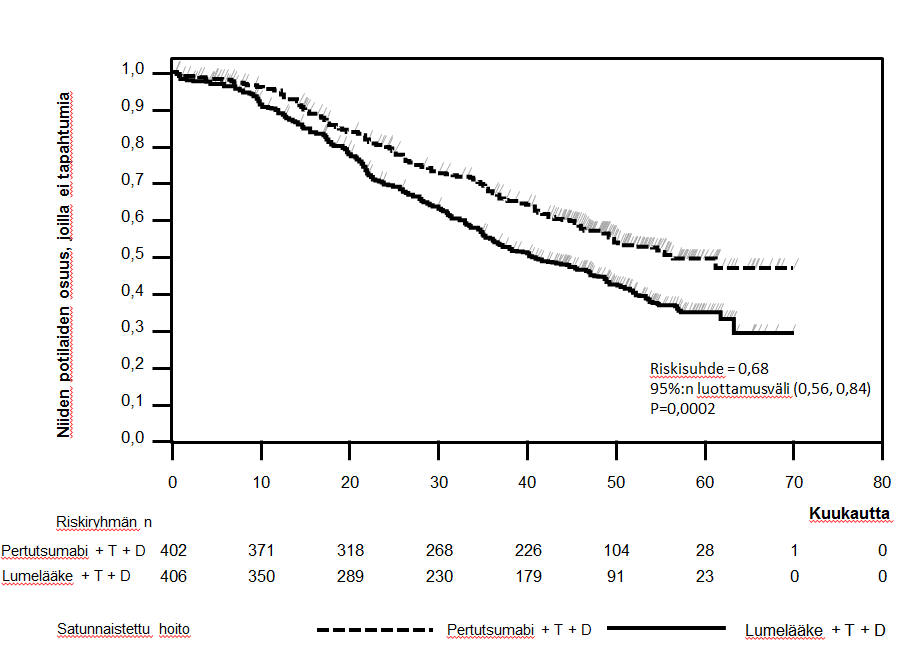
Pertutsumabi (Perjeta); T= trastutsumabi (Herceptin); D= dosetakseli.
Näiden kahden hoitoryhmän välillä ei havaittu tilastollisesti merkitseviä eroja terveyteen liittyvässä elämänlaadussa, jota arvioitiin FACT-B TOI-PFB -pisteytyksellä.
Muut kliinisistä tutkimuksista saadut lisätiedot
BO17929 - yhden hoitoryhmän tutkimus metastasoitunutta rintasyöpää sairastavilla potilailla
BO17929 oli faasin II satunnaistamaton tutkimus, jossa oli mukana metastasoitunutta rintasyöpää sairastavia potilaita, joiden kasvaimet olivat edenneet trastutsumabihoidon aikana. Perjetan ja trastutsumabin yhdistelmän tuloksena saavutettiin vaste 24,2 %:lla potilaista ja lisäksi 25,8 %:lla potilaista havaittiin taudin etenemisen pysähtyneen ainakin kuuden kuukauden ajaksi, mikä viittaa Perjeta-hoidon olevan tehokasta potilailla, joilla tauti on edennyt trastutsumabihoidon aikana.
Varhaisvaiheen rintasyöpä
Neoadjuvanttihoito
Paikallisesti edenneeseen ja inflammatoriseen rintasyöpään katsotaan neoadjuvanttihoidossa liittyvän suuri riski hormonireseptoristatuksesta riippumatta. Varhaisvaiheen rintasyövän riskiarviossa pitää ottaa huomioon kasvaimen koko, hormonireseptoristatus ja etäpesäkkeet imusolmukkeissa.
Käyttö rintasyövän neoadjuvanttihoitoon perustuu patologisella kokonaisvasteella osoitettuun tilan paranemiseen sekä taudin etenemisvapaan ajan pitenemiseen. Nämä eivät kuitenkaan varmista eivätkä mittaa tarkasti hyötyä pitkäaikaisen hoitotuloksen, kuten kokonaiselossaoloajan tai taudin etenemisvapaan ajan, suhteen.
NEOSPHERE (WO20697)
NEOSPHERE on Perjetalla toteutettu faasin II, satunnaistettu, kontrolloitu, monikansallinen monikeskustutkimus, jossa oli mukana 417 äskettäin diagnosoitua varhaisvaiheen, inflammatorista tai paikallisesti edennyttä HER2-positiivista rintasyöpää (T2-4d, primaarikasvaimen läpimitta > 2 cm) sairastavaa aikuista naispotilasta, jotka eivät olleet saaneet aiemmin trastutsumabi-, solunsalpaaja- tai sädehoitoa. Tutkimukseen ei otettu mukaan potilaita, joilla oli metastasoitunut rintasyöpä, rintasyöpä kummassakin rinnassa, kliinisesti merkittäviä sydämen riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) tai vasemman kammion ejektiofraktio < 55 %. Suurin osa potilaista oli alle 65-vuotiaita.
Potilaat satunnaistettiin ennen leikkausta yhteen seuraavista neljästä neoadjuvanttihoitoryhmästä:
- trastutsumabi yhdistelmänä dosetakselin kanssa
- Perjeta yhdistelmänä trastutsumabin ja dosetakselin kanssa
- Perjeta yhdistelmänä trastutsumabin kanssa
- Perjeta yhdistelmänä dosetakselin kanssa.
Satunnaistaminen oli ositettu rintasyövän tyypin (leikattavissa oleva, paikallisesti edennyt tai inflammatorinen) ja ER- tai PgR-positiivisuuden mukaan.
Pertutsumabia annettiin laskimoon aloitusannos 840 mg, minkä jälkeen annettiin 420 mg kolmen viikon välein. Trastutsumabia annettiin laskimoon aloitusannos 8 mg/kg, minkä jälkeen annettiin 6 mg/kg kolmen viikon välein. Dosetakselia annettiin laskimoon aloitusannos 75 mg/m2, minkä jälkeen annettiin 75 mg/m2 tai 100 mg/m2 (jos potilas sieti hoidon) 3 viikon välein. Kaikki potilaat saivat leikkauksen jälkeen 3 hoitosykliä 5-fluorourasiilia (600 mg/m2), epirubisiinia (90 mg/m2) ja syklofosfamidia (600 mg/m2) (FEC-hoitoa) laskimoon kolmen viikon välein sekä trastutsumabia laskimoon kolmen viikon välein, kunnes hoitoa oli annettu vuoden ajan. Pelkästään Perjetan ja trastutsumabin yhdistelmää ennen leikkausta saaneet potilaat saivat leikkauksen jälkeen sekä FEC-hoitoa että dosetakselia.
Tutkimuksen ensisijainen päätetapahtuma oli rinnassa todettu täydellinen patologinen hoitovaste (pathological complete response, pCR) (ypT0/is).Toissijaisia tehon päätetapahtumia olivat kliininen vasteluku, rinnan säästävien leikkausten lukumäärä (vain T2–3 kasvaimet), tauditon elossaoloaika (disease-free survival, DFS) ja taudin etenemisvapaa aika (PFS). Muita eksploratiivisia pCR-lukuja oli mm. taudin leviäminen imusolmukkeisiin (ypT0/isN0 ja ypT0N0).
Demografiset ominaisuudet olivat hyvin tasapainossa (iän mediaani oli 49–50 vuotta, suurin osa [71 %] potilaista oli valkoihoisia), ja kaikki potilaat olivat naisia. Kaikkiaan 7 %:lla potilaista oli inflammatorinen rintasyöpä, 32 %:lla potilaista oli paikallisesti edennyt rintasyöpä ja 61 %:lla potilaista oli leikattavissa oleva rintasyöpä. Kussakin hoitoryhmässä noin puolella potilaista oli hormonireseptoripositiivinen tauti (määriteltiin ER-positiiviseksi ja/tai PgR-positiiviseksi).
Tehon tulokset on esitetty taulukossa 4. Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneilla potilailla havaittiin pCR-luvun (ypT0/is) tilastollisesti merkitsevä paraneminen verrattuna trastutsumabia ja dosetakselia saaneisiin potilaisiin (45,8 % vs 29,0 %, p-arvo = 0,0141). Tulosten havaittiin olevan yhdenmukaisia pCR:n määritelmästä riippumatta. pCR-luvussa todetun eron katsottiin todennäköisesti muodostuvan kliinisesti merkittäväksi eroksi pitkän aikavälin hoitotuloksessa, mitä tuki myös myönteinen kehitys taudin etenemisvapaassa ajassa (PFS) (riskitiheyksien suhde 0,69, 95 %:n luottamusväli 0,34; 1,40) ja taudittomassa elossaoloajassa (DFS) (riskitiheyksien suhde 0,60, 95 %:n luottamusväli 0,28; 1,27).
pCR-luvut sekä Perjeta-hoidosta (Perjeta yhdistelmänä trastutsumabin ja dosetakselin kanssa verrattuna trastutsumabia ja dosetakselia saaneisiin potilaisiin) saatu hyöty olivat pienemmät siinä potilasjoukossa, joilla oli hormonireseptoripositiivisia kasvaimia (ero rintarauhasen pCR-luvussa 6 %), kuin potilasjoukossa, joilla oli hormonireseptorinegatiivisia kasvaimia (ero rintarauhasen pCR-luvussa 26,4 %). pCR-luvut olivat samankaltaiset potilasjoukoissa, joilla oli leikattavissa oleva tai paikallisesti edennyt tauti. Inflammatorista rintasyöpää sairastavia potilaita oli varmojen päätelmien tekemiseksi liian vähän, mutta pCR-luku oli suurempi potilailla, jotka saivat Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa.
TRYPHAENA (BO22280)
TRYPHAENA on faasin II satunnaistettu, kliininen monikeskustutkimus, jossa oli mukana 225 paikallisesti edennyttä, leikattavissa olevaa tai inflammatorista HER2-positiivista rintasyöpää (T2-4d; primaarikasvaimen läpimitta > 2 cm) sairastavaa aikuista naispotilasta, jotka eivät olleet aiemmin saaneet trastutsumabi-, solunsalpaaja- tai sädehoitoa. Tutkimukseen ei otettu mukaan potilaita, joilla oli metastasoitunut rintasyöpä, rintasyöpä kummassakin rinnassa, kliinisesti merkittäviä sydämen riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) tai vasemman kammion ejektiofraktio oli < 55 %. Suurin osa potilaista oli alle 65-vuotiaita. Potilaat satunnaistettiin ennen leikkausta yhteen seuraavista kolmesta neoadjuvanttihoitoryhmästä:
- 3 hoitosykliä FEC-hoitoa, minkä jälkeen 3 hoitosykliä dosetakselia, joista kaikki annettiin samanaikaisesti Perjetan ja trastutsumabin kanssa
- 3 hoitosykliä pelkästään FEC-hoitoa, minkä jälkeen 3 hoitosykliä dosetakselia, joka annettiin samanaikaisesti trastutsumabin ja Perjetan kanssa
- 6 hoitosykliä TCH-hoitoa yhdistelmänä Perjetan kanssa.
Satunnaistaminen oli ositettu rintasyövän tyypin (leikattavissa oleva, paikallisesti edennyt tai inflammatorinen) ja ER- ja /tai PgR-positiivisuuden perusteella.
Pertutsumabia annettiin laskimoon aloitusannos 840 mg, minkä jälkeen annettiin 420 mg kolmen viikon välein. Trastutsumabia annettiin laskimoon aloitusannos 8 mg/kg, minkä jälkeen annettiin 6 mg/kg kolmen viikon välein. FEC-hoitoa (5-fluorourasiili [500 mg/m2], epirubisiini [100 mg/m2], syklofosfamidi [600 mg/m2]) annettiin laskimoon 3 hoitosykliä kolmen viikon välein. Dosetakselia annettiin infuusiona laskimoon aloitusannos 75 mg/m2 kolmen viikon välein, ja annos voitiin suurentaa tutkijalääkärin harkinnan mukaan annokseen 100 mg/m2, jos potilas sieti aloitusannoksen hyvin. Perjetaa yhdistelmänä TCH-hoidon kanssa saaneessa ryhmässä dosetakseli annettiin kuitenkin laskimoon annoksena 75 mg/m2 (annoksen suurentaminen ei ollut sallittua), ja karboplatiinia (AUC 6) annettiin laskimoon kolmen viikon välein. Kaikki potilaat saivat leikkauksen jälkeen trastutsumabia, kunnes hoitoa oli annettu vuoden ajan.
Tämän tutkimuksen ensisijainen päätetapahtuma oli sydämeen liittyvä turvallisuus tutkimuksen neoadjuvanttihoitojakson aikana. Toissijaisia tehon päätetapahtumia olivat rinnan pCR-luku (ypT0/is), tauditon elossaoloaika (DFS), taudin etenemisvapaa aika (PFS) ja kokonaiselossaoloaika.
Demografiset ominaisuudet olivat hoitoryhmien välillä hyvin tasapainossa (iän mediaani oli 49–50 vuotta, suurin osa [77 %] potilaista oli valkoihoisia), ja kaikki potilaat olivat naisia. Kaikkiaan 6 %:lla potilaista oli inflammatorinen rintasyöpä, 25 %:lla potilaista oli paikallisesti edennyt rintasyöpä ja 69 %:lla potilaista oli leikattavissa oleva rintasyöpä. Kussakin hoitoryhmässä noin puolella potilaista oli ER-positiivinen ja/tai PgR-positiivinen tauti.
Kaikissa kolmessa hoitoryhmässä havaittiin suuret pCR-luvut verrattuna sellaisista samankaltaisista hoito-ohjelmista julkaistuihin tietoihin, joihin ei kuulunut pertutsumabia (ks. taulukko 4). Tulosten havaittiin olevan yhdenmukaisia pCR:n määritelmästä riippumatta. pCR-luvut olivat pienemmät potilasjoukossa, jolla oli hormonireseptoripositiivisia kasvaimia (vaihteluväli 46,2–50,0 %), kuin potilasjoukossa, jolla oli hormonireseptorinegatiivisia kasvaimia (vaihteluväli 65,0–83,8 %).
pCR-luvut olivat samankaltaisia potilailla, joilla oli leikattavissa oleva tai paikallisesti edennyt tauti. Inflammatorista rintasyöpää sairastavia potilaita oli varmojen päätelmien tekemiseksi liian vähän.
Taulukko 4. NEOSPHERE (WO20697) ja TRYPHAENA (BO22280): Tehoa koskevat tiedot (Intent-to-Treat-potilaat)
| NEOSPHERE (WO20697) | TRYPHAENA (BO22280) | ||||||
| Parametri | Trastu-tsumabi +dose-takseli N = 107 | Perjeta+ trastu-tsumabi+ dosetakseli N = 107 | Perjeta+ trastu-tsumabi N = 107 | Perjeta +dosetakseli N = 96 | Perjeta+ trastutsumabi+ FEC-hoito◊ Perjeta+ trastutsumabi+ dosetakseli N = 73 | FEC-hoito◊ Perjeta+ trastutsumabi+ dosetakseli N = 75 | Perjeta +TCH-hoito N = 77 |
rinnan pCR-luku (ypT0/is) n (%) [95 %:n luottamus-väli]1 | 31 (29,0 %) [20,6; 38,5] | 49 (45,8 %) [36,1; 55,7] | 18 (16,8 %) [10,3; 25,3] | 23 (24,0 %) [15,8; 33,7] | 45 (61,6 %) [49,5; 72,8] | 43 (57,3 %) [45,4; 68,7] | 51 (66,2 %) [54,6; 76,6] |
pCR-lukujen ero2 [95 %:n luottamus-väli]3 | +16,8 % [3,5; 30,1] | -12,2 % [-23,8; -0,5] | -21,8 % [-35,1; -8,5] | NA | NA | NA | |
| p-arvo (CHH-testin Simesin korjaus)4 | 0,0141 (vs. trastutsumabi+dosetakseli) | 0,0198 (vs. trastutsumabi+dosetakseli) | 0,0030 (vs Perjeta+ trastutsumabi+dosetakseli) | NA | NA | NA | |
rinnan ja imusol-mukkeen pCR-luku (ypT0/is N0) n (%) [95 %:n luottamus-väli] | 23 (21,5 %) [14,1; 30,5] | 42 (39,3 %) [30,3; 49,2] | 12 (11,2 %) [5,9; 18,8] | 17 (17,7 %) [10,7; 26,8] | 41 (56,2 %) [44,1; 67,8] | 41 (54,7 %) [42,7; 66,2] | 49 (63,6 %) [51,9; 74,3] |
ypT0 N0 n (%) [95 %:n luottamus-väli] | 13 (12,1 %) [6,6; 19,9] | 35 (32,7 %) [24,0; 42,5] | 6 (5,6 %) [2,1; 11,8] | 13 (13,2 %) [7,4; 22,0] | 37 (50,7 %) [38,7; 62,6] | 34 (45,3 %) [33,8; 57,3] | 40 (51,9 %) [40,3; 63,5] |
| Kliininen vaste5 | 79 (79,8 %) | 89 (88,1 %) | 69 (67,6 %) | 65 (71,4 %) | 67 (91,8 %) | 71 (94,7 %) | 69 (89,6 %) |
FEC-hoito: 5-fluorourasiili, epirubisiini, syklofosfamidi; TCH-hoito: dosetakseli, karboplatiini ja trastutsumabi, CMH: Cochran–Mantel–Haenszel
1. Yhden näytteen 95 %:n luottamusväli Pearson–Clopperin binomijakaumalla.
2. Perjeta+trastutsumabi+dosetakseli- ja Perjeta+trastutsumabi-hoitoa verrataan trastutsumabi+ dosetakseli-hoitoon, kun taas Perjeta+dosetakseli-hoitoa verrataan Perjeta+trastutsumabi+dosetakseli-hoitoon.
3. Kahden vasteluvun eron likimääräinen 95 %:n luottamusväli Hauck–Andersonin menetelmällä.
4. p-arvo Cochran–Mantel–Haenszelin testillä, johon on tehty Simesin monikerroinkorjaus.
5. Kliininen vaste tarkoittaa potilaita, joilla on neoadjuvanttihoitojakson aikana paras täydellinen tai osittainen kokonaisvaste (rinnan primaarimuutoksessa).
BERENICE (WO29217)
BERENICE on faasin II satunnaistamaton, avoin, monikansallinen monikeskustutkimus, joka tehtiin 401 HER2-positiivista paikallisesti edennyttä tulehduksellista tai varhaisvaiheen rintasyöpää sairastavilla potilailla (joiden kasvainten läpimitta oli > 2 cm tai joiden tauti oli levinnyt imusolmukkeisiin).
Potilaista muodostettiin BERENICE-tutkimuksessa kaksi rinnakkaisryhmää. Potilaat, joille trastutsumabin ja antrasykliinin/taksaanipohjaisen solunsalpaajahoidon yhdistelmästä koostuvan neoadjuvanttihoidon katsottiin sopivan, kohdennettiin saamaan ennen leikkausta toista seuraavista kahdesta hoidosta:
- kohortti A: 4 hoitosykliä, joissa annettiin lyhyen antovälin doksorubisiini- ja syklofosfamidihoitoa kahden viikon välein, jonka jälkeen 4 hoitosykliä Perjetaa yhdistelmänä trastutsumabin ja paklitakselin kanssa
- kohortti B: 4 hoitosykliä FEC-hoitoa, jonka jälkeen 4 hoitosykliä Perjetaa yhdistelmänä trastutsumabin ja dosetakselin kanssa.
Kaikki potilaat saivat leikkauksen jälkeen Perjetaa ja trastutsumabia laskimoon kolmen viikon välein, kunnes hoitoa oli annettu 1 vuoden ajan.
BERENICE-tutkimuksen ensisijainen päätetapahtuma on sydämen turvallisuus tutkimuksen neoadjuvanttijakson aikana. Sydämen turvallisuutta koskeva ensisijainen päätetapahtuma (eli NYHA-luokan III/IV vasemman kammion toimintahäiriöiden ilmaantuvuus ja sydämen vasemman kammion ejektiofraktion pieneneminen) oli yhdenmukainen neoadjuvanttihoidosta aiemmin saatujen tietojen kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Adjuvanttihoito
Adjuvanttihoidossa varhaisvaiheen HER2-positiivista rintasyöpää sairastaviksi potilaiksi, joilla on suuri syövän uusiutumisriski, on APHINITY-tutkimuksen tietojen perusteella määritelty ne potilaat, joiden syöpä on levinnyt imusolmukkeisiin tai joilla on hormonireseptorinegatiivinen tauti.
APHINITY (BO25126)
APHINITY on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu faasin III monikeskustutkimus, jossa oli mukana 4804 varhaisvaiheen HER2-positiivista rintasyöpää sairastavaa potilasta, joiden primaarikasvain oli poistettu leikkauksella ennen satunnaistamista. Potilaat satunnaistettiin sen jälkeen saamaan Perjetaa tai lumelääkettä yhdistelmänä adjuvanttihoitona annetun trastutsumabin ja solunsalpaajan kanssa. Tutkijat valitsivat potilaille yksilöllisesti yhden seuraavista antrasykliiniä sisältävistä tai sisältämättömistä solunsalpaajahoidoista:
- 3 tai 4 hoitosykliä FEC-hoitoa tai 5-fluorourasiilia, doksorubisiinia ja syklofosfamidia (FAC), jonka jälkeen 3 tai 4 hoitosykliä dosetakselia tai 12 hoitosyklin ajan viikoittain paklitakselia
- 4 hoitosykliä AC-hoitoa tai epirubisiinia ja syklofosfamidia (EC-hoitoa), jonka jälkeen 3 tai 4 hoitosykliä dosetakselia tai 12 hoitosyklin ajan viikoittain paklitakselia
- 6 hoitosykliä dosetakselia yhdistelmänä karboplatiinin kanssa.
Pertutsumabi ja trastutsumabi annettiin laskimoon (ks. kohta Annostus ja antotapa) kolmen viikon välein ensimmäisen taksaania sisältävän hoitosyklin 1. hoitopäivästä lähtien yhteensä 52 viikon ajan (enintään 18 hoitosykliä) tai kunnes tauti uusiutui, potilas peruutti suostumuksensa tutkimukseen osallistumisesta tai potilaalle ilmaantui haittavaikutuksia, jotka eivät olleet hoidettavissa. Potilaille annettiin tavanomaisia annoksia 5-fluorourasiilia, epirubisiinia, doksorubisiinia, syklofosfamidia, dosetakselia, paklitakselia ja karboplatiinia.
Solunsalpaajahoidon päätyttyä potilaat saivat sädehoitoa ja/tai hormonihoitoa paikallisen kliinisen hoitokäytännön mukaan.
Tutkimuksen ensisijainen päätetapahtuma oli elossaolo ilman invasiivista tautia (invasive disease-free survival, IDFS), joksi määriteltiin aika satunnaistamisesta invasiivisen rintasyövän paikalliseen tai alueelliseen ensimmäiseen uusiutumiseen samassa rinnassa, uusiutuminen etäpesäkkeenä, invasiivinen rintasyöpä toisessa rinnassa tai kuolema mistä tahansa syystä. Toissijaisia tehon päätetapahtumia olivat elossaolo ilman invasiivista tautia, mukaan lukien uusi muu primaarisyöpä, kokonaiselossaoloaika (OS), tauditon elossaoloaika (DFS), taudin uusiutumisvapaa aika (RFI) ja etäpesäkkeiden uusiutumisvapaa aika (DRFI).
Demografiset tiedot olivat kahden hoitohaaran kesken hyvin tasapainossa. Iän mediaani oli 51 vuotta, ja yli 99 % potilaista oli naisia. Valtaosalla potilaista oli imusolmukkeisiin levinnyt (63 %) ja/tai hormonireseptoripositiivinen tauti (64 %), ja valtaosa oli valkoihoisia (71 %).
45,4 kuukauden (mediaani) seurannan jälkeen APHINITY-tutkimuksessa osoitettiin taudin uusiutumisen tai kuoleman riskin vähentyneen 19 % (riskisuhde [HR] = 0,81; 95 %:n luottamusväli 0,66; 1,00; p-arvo 0,0446) Perjetaa saaneilla potilailla verrattuna potilaisiin, jotka satunnaistettiin saamaan lumehoitoa.
101,2 kuukauden (8,4 vuotta) (mediaani) seurannan jälkeen, kolmannessa kokonaiselossaoloajan välianalyysissä, Perjeta-hoitohaaraan satunnaistetuista potilaista 168 kuoli (7,0 %) ja lumehoitohaaraan satunnaistetuista potilaista 202 kuoli (8,4 %) (riskisuhde [HR] = 0,83; 95 %:n luottamusväli 0,68; 1,02).
Yhteenveto tutkimuksen APHINITY tehoa koskevista tuloksista esitetään taulukossa 5 ja kuvassa 3.
Taulukko 5 Kokonaisteho: hoitoaikeen mukainen (ITT) potilasjoukko
Perjeta + trastutsumabi + solunsalpaajahoito N = 2400 | Lumelääke + trastutsumabi + solunsalpaajahoito N = 2404 | |
| Ensisijainen päätetapahtuma | ||
| Elossaolo ilman invasiivista tautia* | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 171 (7,1 %) | 210 (8,7 %) |
| Riskisuhde [95 %:n luottamusväli] | 0,81 [0,66, 1,00] | |
| p-arvo (Log-Rank-testi, ositettu1) | 0,0446 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 94,1 [93,1; 95,0] | 93,2 [92,2; 94,3] |
| Toissijaiset päätetapahtumat1 | ||
| Elossaolo ilman invasiivista tautia, mukaan lukien uusi muu primaarikasvain* | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 189 (7,9 %) | 230 (9,6 %) |
| Riskisuhde [95 %:n luottamusväli] | 0,82 [0,68; 0,99] | |
| p-arvo (Log-Rank-testi, ositettu1) | 0,0430 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 93,5 [92,5; 94,5] | 92,5 [91,4; 93,6] |
| Tauditon elossaolo* | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 192 (8,0 %) | 236 (9,8 %) |
| Riskisuhde [95 %:n luottamusväli] | 0,81 [0,67; 0,98] | |
| p-arvo (Log-Rank-testi, ositettu1) | 0,0327 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 93,4 [92,4; 94,4] | 92,3 [91,2; 93,4] |
| Kokonaiselossaolo** | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 205 (8,5 %) | 247 (10,3 %) |
| Riskisuhde [95 %:n luottamusväli] | 0,83 [0,69; 1,00] | |
| p‑arvo (Log-Rank-testi, ositettu1) | 0,0441 | |
| 10 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 91,55 [90,38; 92,71] | 89,79 [88,53; 91,06] |
* Elossaolo ilman invasiivista tautia, ensisijainen analyysi, tiedonkeruun katkaisupäivä 19. joulukuuta 2016.
** Tiedot kokonaiselossaolon loppuanalyysistä, tiedonkeruun katkaisupäivä 28. marraskuuta 2024; p‑arvon raja 0,0496.
1. Kaikki analyysit ositettiin imusolmukkeiden statuksen, tutkimussuunnitelman version, keskeisten hormonireseptorien statuksen ja adjuvanttisolunsalpaajahoidon mukaan.
2. 3 vuoden jaksot ilman tapahtumia ja 10 vuoden jaksot ilman tapahtumia saatiin Kaplan-Meierin estimaateista.
Kuva 3 Elossaolo ilman invasiivista tautia: Kaplan-Meier-käyrä
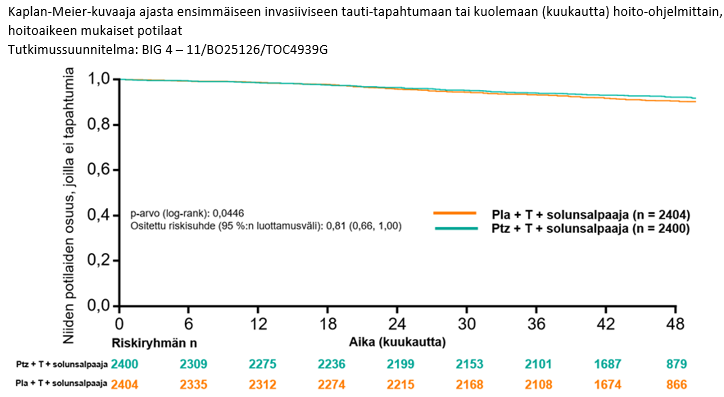
Pla= lumelääke; Ptz= pertutsumabi (Perjeta); T= trastutsumabi.
Elossaolon ilman invasiivista tautia estimaatti 4 vuoden hoidon kohdalla oli Perjeta-hoitoa saaneessa ryhmässä 92,3 % verrattuna 90,6 %:iin lumehoitoa saaneessa ryhmässä. Seurannan mediaani arvion ajankohtana oli 45,4 kuukautta.
Alaryhmäanalyysin tulokset
Perjetan hyödyt olivat primaarianalyysin ajankohtana selkeämmät niiden potilaiden alaryhmissä, joilla uusiutumisriski oli suuri: imusolmukkeisiin levinnyt tai hormonireseptorinegatiivinen tauti (ks. taulukko 6).
Taulukko 6 Alaryhmien tehoa koskevat tulokset imusolmukkeiden statuksen ja hormonireseptorien statuksen mukaan1
| Potilasjoukko | Elossaolon ilman invasiivista tautia koskevien tapahtumien lkm /N yhteensä (%) | Osittamaton riskisuhde (95 %:n luottamusväli) | |
| Perjeta + trastutsumabi + solunsalpaajahoito | Lumelääke + trastutsumabi + solunsalpaajahoito | ||
| Imusolmukkeiden status | |||
| Positiivinen | 139/1503 (9,2 %) | 181/1502 (12,1 %) | 0,77 (0,62; 0,96) |
| Negatiivinen | 32/897 (3,6 %) | 29/902 (3,2 %) | 1,13 (0,68; 1,86) |
| Hormonireseptorien status | |||
| Negatiivinen | 71/864 (8,2 %) | 91/858 (10,6 %) | 0,76 (0,56; 1,04) |
| Positiivinen | 100/1536 (6,5 %) | 119/1546 (7,7 %) | 0,86 (0,66; 1,13) |
1 Ennalta määritellyt alaryhmäanalyysit ilman korjausta useiden vertailujen suhteen, jonka vuoksi tulokset katsotaan deskriptiivisiksi.
Elossaolon ilman invasiivista tautia estimaatit alaryhmässä, jossa potilaiden tauti oli levinnyt imusolmukkeisiin, olivat 3 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 92,0 % verrattuna 90,2 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 89,9 % verrattuna 86,7 %:iin lumehoitoa saaneilla potilailla. Elossaolon ilman invasiivista tautia estimaatit alaryhmässä, jossa potilaiden tauti ei ollut levinnyt imusolmukkeisiin, olivat 3 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 97,5 % verrattuna 98,4 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 96,2 % verrattuna 96,7 %:iin lumehoitoa saaneilla potilailla. Elossaolon ilman invasiivista tautia estimaatit hormonireseptorinegatiivisten potilaiden alaryhmässä olivat 3 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 92,8 % verrattuna 91,2 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 91,0 % verrattuna 88,7 %:iin lumehoitoa saaneilla potilailla.
Elossaolon ilman invasiivista tautia estimaatit hormonireseptoripositiivisten potilaiden alaryhmässä olivat 3 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 94,8 % verrattuna 94,4 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla Perjeta-hoitoa saaneilla potilailla 93,0 % verrattuna 91,6 %:iin lumehoitoa saaneilla potilailla.
Potilaiden raportoimat hoitotulokset
Toissijaisiin päätetapahtumiin kuului potilaiden raportoiman yleisen terveydentilan, rooli- ja fyysisen toimintakyvyn sekä hoidon oireiden arviointi EORTC QLQ-C30- ja EORTC QLQ-BR23 ‑kyselyiden avulla. Potilaiden raportoimien hoitotulosten analyysissä 10 pisteen ero katsottiin kliinisesti merkittäväksi.
Potilaiden fyysistä toimintakykyä, yleistä terveydentilaa ja ripulia koskevissa pisteissä todettiin kummassakin hoitohaarassa kliinisesti merkittävä muutos solunsalpaajahoidon aikana. Fyysistä toimintakykyä kuvaava pisteiden lasku kyseisenä ajankohtana lähtötilanteeseen verrattuna oli Perjeta-hoitohaarassa keskimäärin -10,7 (95 %:n luottamusväli -11,4; -10,0) ja lumehoitohaarassa -10,6 (95 %:n luottamusväli -11,4; -9,9); yleistä terveydentilaa kuvaavien pisteiden lasku oli Perjeta-hoitohaarassa -11,2 (95 %:n luottamusväli -12,2; -10,2) ja lumehoitohaarassa -10,2 (95 %:n luottamusväli -11,1; -9,2). Ripulin oireita kuvaavat pisteet suurenivat Perjeta-hoitohaarassa tasolle +22,3 (95 %:n luottamusväli 21,0; 23,6) ja lumehoitohaarassa tasolle +9,2 (95 %:n luottamusväli 8,2; 10,2).
Kummankin hoitohaaran fyysistä toimintakykyä ja yleistä terveydentilaa kuvaavat pisteet palautuivat tämän jälkeen kohdennetun hoidon aikana lähtötilanteen tasolle. Ripulin oireet palautuivat Perjeta-hoitohaarassa lähtötilanteen tasolle HER2-hoidon jälkeen. Perjetan lisääminen trastutsumabin ja solunsalpaajan yhdistelmään ei vaikuttanut tutkimuksen aikana potilaiden yleiseen roolitoimintakykyyn.
Immunogeenisuus
Potilailta tutkittiin pivotaalitutkimuksessa (CLEOPATRA) useina eri ajankohtina Perjeta-hoitoa neutraloivat lääkevasta-aineet (anti-drug antibodies, ADA). 3,3 %:lla (13/389 potilasta) Perjeta-hoitoa saaneista potilaista ja 6,7 %:lla (25/372 potilasta) lumehoitoa saaneista potilaista todettiin testissä neutraloivia lääkevasta-aineita. BERENICE-tutkimuksessa 4,1 % (16/392) Perjeta-hoitoa saaneista potilaista todettiin neutraloiville lääkevasta-aineille positiivisiksi. Yhdelläkään näistä potilaista ei esiintynyt anafylaktisia/yliherkkyysreaktioita, jotka olisivat selkeästi liittyneet neutraloiviin lääkevasta-aineisiin.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Perjeta-valmisteen käytöstä kaikkien pediatristen potilasryhmien rintasyövän hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Populaatiofarmakokineettinen analyysi tehtiin tiedoista, jotka kattoivat 481 (faasin I, II ja III) kliinisissä tutkimuksissa mukana ollutta potilasta, joilla oli erityyppisiä pitkälle edenneitä syöpäsairauksia ja jotka olivat saaneet Perjeta-hoitoa joko ainoana lääkeaineena tai yhdistelmähoidossa pertutsumabiannoksina 2–25 mg/kg, joita annettiin kolmen viikon välein 30–60 minuutin kestoisina infuusioina laskimoon.
Imeytyminen
Perjeta annetaan infuusiona laskimoon.
Jakautuminen
Kaikissa kliinisissä tutkimuksissa tyypillisen potilaan jakautumistilavuus oli keskitilassa (Vc) 3,11 litraa ja ääreistilassa (Vp) 2,46 litraa.
Biotransformaatio
Pertutsumabin metaboliaa ei ole tutkittu suoranaisesti. Vasta-aineet poistuvat elimistöstä pääasiassa kataboloitumalla.
Eliminaatio
Pertutsumabin puhdistuman (CL) mediaani oli 0,235 l/vrk ja puoliintumisajan mediaani oli 18 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Pertutsumabin farmakokinetiikka on lineaarinen koko suositellulla annosvälillä.
Iäkkäät potilaat
Pertutsumabin farmakokinetiikassa ei havaittu populaatiofarmakokineettisen analyysin perusteella eroa < 65-vuotiaiden (n = 306) ja ≥ 65-vuotiaiden (n = 175) potilaiden välillä.
Munuaisten vajaatoimintaa sairastavat potilaat
Pertutsumabia ei ole tutkittu erityisesti munuaisten vajaatoimintaan keskittyvässä tutkimuksessa. Populaatiofarmakokineettisen analyysin tulosten perusteella altistus pertutsumabille on lievää (kreatiniinipuhdistuma [CLcr] 60–90 ml/min, N = 200) ja keskivaikeaa (CLcr 30–60 ml/min, N = 71) munuaisten vajaatoimintaa sairastavilla potilailla samankaltainen kuin potilailla, joiden munuaisten toiminta on normaali (CLcr yli 90 ml/min, N = 200). Kreatiniinipuhdistuman ja pertutsumabialtistuksen välillä ei havaittu yhteyttä kreatiniinipuhdistuman vaihteluvälialueella (27–244 ml/min).
Muut erityisryhmät
Populaatiofarmakokineettinen analyysi ei viitannut ikään, sukupuoleen tai etniseen taustaan (japanilainen tai muu kuin japanilainen) perustuviin eroihin farmakokinetiikassa. Albumiini ja kehon rasvaton paino lähtötilanteessa olivat tärkeimmät puhdistumaan vaikuttavat kovariaatit. Jos potilaan albumiinipitoisuus oli lähtötilanteessa normaalia suurempi, puhdistuma väheni, ja jos potilaan kehon rasvaton paino oli normaalia suurempi, puhdistuma lisääntyi. Perjeta-hoidon suositelluilla annoksilla ja hoito-ohjelmilla tehdyt herkkyysanalyysit osoittivat kuitenkin, että näiden kahden kovariaatin ääriarvoilla ei ollut merkittävää vaikutusta vakaan tilan tavoitepitoisuuksien saavuttamiseen, mikä todettiin prekliinisissä kasvainten vieraslajisiirremalleissa. Pertutsumabi-annosta ei siksi tarvitse näiden kovariaattien perusteella säätää.
Pertutsumabin farmakokineettiset tulokset NEOSPHERE- ja APHINITY-tutkimuksissa olivat yhdenmukaiset aiemmista populaatiofarmakokineettisistä malleista saatujen ennusteiden kanssa. Pertutsumabin farmakokinetiikassa ei havaittu eroja varhaisvaiheen rintasyöpää ja metastasoitunutta rintasyöpää sairastavien potilaiden välillä.
Prekliiniset tiedot turvallisuudesta
Eläimillä ei ole tehty erityisiä hedelmällisyystutkimuksia pertutsumabin vaikutusten selvittämiseksi.
Cynomolgus-apinoilla tehtyjen toistuvan altistuksen aiheuttamaa toksisuutta selvittäneiden tutkimusten perusteella ei voida vetää täsmällisä johtopäätöksiä urosten lisääntymisjärjestelmään kohdistuvista haittavaikutuksista.
Lisääntymistoksisuustutkimuksia on tehty tiineillä cynomolgus-apinoilla (gestaatiopäivinä 19–50), jolloin aloitusannos oli 30–150 mg/kg, minkä jälkeen annettiin kerran kahdessa viikossa annos 10−100 mg/kg. Näistä annoksista aiheutunut kliinisesti oleellinen altistus oli huippupitoisuuden (Cmax) perusteella 2,5–20 kertaa suurempi kuin ihmiselle suositellulla annoksella. Pertutsumabin anto laskimoon gestaatiopäivinä 19–50 (organogeneesin aikana) aiheutti alkiotoksisuutta, joka lisääntyi annosriippuvasti alkion ja sikiön kuolemaan gestaatiopäivien 25–70 välillä. Alkioista tai sikiöistä kuoli 33 %, kun pertutsumabia annettiin kerran kahdessa viikossa tiineille naarasapinoille annoksilla 10 mg/kg, ja vastaavasti 50 % kuoli annoksilla 30 mg/kg kerran kahdessa viikossa ja 85 % annoksilla 100 mg/kg kerran kahdessa viikossa (annokset olivat huippupitoisuuden (Cmax) perusteella 2,5–20 kertaa suurempia kuin ihmiselle suositeltu annos). Kun gestaatiopäivänä 100 tehtiin keisarileikkaus, kaikissa pertutsumabiannosryhmissä todettiin sikiöveden niukkuutta, vähentynyt keuhkojen ja munuaisten suhteellinen paino sekä mikroskooppista näyttöä munuaisten hypoplasiasta, joka yhdenmukaista munuaisten kehityksen viivästymisen kanssa. Sikiöveden niukkuudesta aiheutuneeksi katsotun sikiön kasvun heikkenemisen lisäksi havaittiin keuhkojen hypoplasiaa (yhdellä kuudesta 30 mg/kg ryhmässä ja yhdellä kahdesta 100 mg/kg ryhmässä), kammioväliseinän vikoja (yhdellä kuudesta 30 mg/kg ryhmässä), ohut kammioväliseinä (yhdellä kahdesta 100 mg/kg ryhmässä) ja vähäisiä luustovikoja (ulkoisia, kolmella kuudesta 30 mg/kg ryhmässä). Kaikkien hoitoryhmien jälkeläisillä raportoitiin pertutsumabialtistukseksi 29–40 % emolla seerumissa gestaatiopäivänä 100 todetusta pitoisuudesta.
Cynomolgus-apinat sietivät viikoittain laskimoon annetun pertutsumabin yleensä hyvin annoksiin 150 mg/kg/annos saakka. Annoksilla 15 mg/kg ja sitä suuremmilla annoksilla havaittiin ajoittaista lievää hoitoon liittynyttä ripulia. Osalla apinoista valmisteen pitkäaikainen (7–26 viikoittaista annosta) anto johti vaikea-asteiseen sekretoriseen ripuliin. Ripuli hoidettiin (lukuun ottamatta yhtä, annoksia 50 mg/kg/annos saanutta eläintä, joka oli lopetettava) tukihoidolla, kuten laskimoon annettavalla nestekorvaushoidolla.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Väkevä etikkahappo
Sakkaroosi
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Perjetan laimentamiseen ei saa käyttää glukoosiliuosta (5 %), koska se ei ole tällaisissa liuoksissa kemiallisesti ja fysikaalisesti stabiili.
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton pakkaus
2 vuotta.
Laimennettu liuos
Käytönaikaiseksi kemialliseksi ja fysikaaliseksi säilyvyydeksi on osoitettu 24 tuntia 30 °C:n lämpötilassa ja 30 vuorokautta 2 °C – 8 ° C:n lämpötilassa suojattuna valolta.
Mikrobiologiselta kannalta valmiste tulisi käyttää heti. Jos valmistetta ei käytetä heti, käyttöä edeltävät säilytysajat ja -olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24 tuntia 2–8 °C:n lämpötilassa, ellei valmistetta ole laimennettu valvotuissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2°C– 8 ºC).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PERJETA infuusiokonsentraatti, liuosta varten
420 mg (L:ei) 1 kpl (14 ml, 30 mg/ml) (3750,18 €)
PF-selosteen tieto
Injektiopullo (tyypin I lasia), jossa (butyylikumi)tulppa ja joka sisältää 14 ml liuosta.
Pakkauksessa 1 injektiopullo.
Valmisteen kuvaus:
Kirkas tai hieman opaalinhohtoinen, väritön tai vaaleankeltainen neste.
Käyttö- ja käsittelyohjeet
Perjeta ei sisällä antimikrobista säilytysainetta. Tämän vuoksi käyttökuntoon saatetun infuusioliuoksen steriiliys on varmistettava huolellisesti, ja terveydenhuollon ammattilaisen pitää saattaa valmiste käyttökuntoon kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa (ks. kohta Kestoaika).
Perjeta on yhtä käyttökertaa varten.
Ei saa ravistaa. 14 ml Perjeta-konsentraattia on vedettävä injektiopullosta käyttäen steriiliä neulaa ja ruiskua ja laimennettava 250 ml:aan 9 mg/ml (0,9 %), tai vaihtoehtoisesti 4,5 mg/ml (0,45 %) natriumkloridi-infuusioliuosta PVC-infuusiopussissa tai PVC:tä sisältämättömässä polyolefiini-infuusiopussissa. Laimentamisen jälkeen yksi ml liuosta sisältää noin 3,02 mg pertutsumabia (840 mg/278 ml) aloitusannosta varten (tarvitaan kaksi injektiopulloa) ja noin 1,59 mg pertutsumabia (420 mg/264 ml) ylläpitoannosta varten (tarvitaan yksi injektiopullo).
Pussia on käänneltävä varovasti ylösalaisin liuoksen sekoittamiseksi, jotta liuokseen ei muodostu vaahtoa.
Parenteraaliset lääkevalmisteet on tarkistettava silmämääräisesti ennen antoa, ettei niissä ole hiukkasia ja värimuutoksia havaittavissa. Mikäli liuoksessa on havaittavissa hiukkasia tai värimuutoksia ei sitä saa käyttää. Infuusio on annettava heti valmisteen käyttökuntoon saattamisen jälkeen (ks. kohta Kestoaika).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Perjeta on yhteensopiva polyvinyylikloridista (PVC) valmistettujen ja PVC:tä sisältämättömien polyolefiinista, mukaan lukien polyeteenista, valmistettujen infuusiopussien kanssa.
Korvattavuus
PERJETA infuusiokonsentraatti, liuosta varten
420 mg 1 kpl
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L01FD02
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com