
XEOMIN injektiokuiva-aine, liuosta varten 50 U, 100 U, 200 U
Vaikuttavat aineet ja niiden määrät
XEOMIN 50 yksikköä injektiokuiva-aine, liuosta varten
Yksi injektiopullo sisältää 50 yksikköä Clostridium botulinum A-tyypin neurotoksiinia (150 kD), joka ei sisällä kompleksoivia proteiineja*.
XEOMIN 100 yksikköä injektiokuiva-aine, liuosta varten
Yksi injektiopullo sisältää 100 yksikköä Clostridium botulinum A-tyypin neurotoksiinia (150 kD), joka ei sisällä kompleksoivia proteiineja*.
XEOMIN 200 yksikköä injektiokuiva-aine, liuosta varten
Yksi injektiopullo sisältää 200 yksikköä Clostridium botulinum A-tyypin neurotoksiinia (150 kD), joka ei sisällä kompleksoivia proteiineja*.
* Clostridium botulinum A-tyypin neurotoksiinia, puhdistettu Clostridium Botulinum ‑viljelmästä (Hallin kanta)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine, liuosta varten
Kliiniset tiedot
Käyttöaiheet
XEOMIN on tarkoitettu aikuisille oireenmukaiseen hoitoon seuraavissa tapauksissa:
• blefarospasmi ja hemifasiaalispasmi
• servikaalinen dystonia eli spastinen tortikollis
• yläraajan jäykkyys
• neurologisista sairauksista johtuva krooninen sialorrea.
XEOMIN on tarkoitettu ≥ 12 kg painaville 2–17-vuotiaille lapsille ja nuorille oireenmukaiseen hoitoon seuraavassa tapauksessa:
• neurologisista/hermoston kehitykseen liittyvistä sairauksista johtuva krooninen sialorrea.
Annostus ja antotapa
Koska biologisen aktiivisuuden määrityksissä on yksikköeroja, XEOMIN-valmisteen yksiköihin perustuvat suositusannokset eivät ole keskenään vaihdettavissa muiden A-tyypin botuliinitoksiinia sisältävien valmisteiden annosten kanssa.
Ks. kohdasta Farmakodynamiikka yksityiskohtaiset tiedot kliinisistä tutkimuksista, joissa verrattiin XEOMIN-valmistetta ja tavanomaista A-tyypin botuliinitoksiinikompleksia (900 kD).
XEOMIN-valmistetta saavat antaa vain asianmukaisen pätevyyden saaneet lääkärit, joilla on kokemusta tällaisesta hoidosta ja vaadittavien välineiden käytöstä.
Lääkäri päättää kunkin potilaan osalta sopivasta annoksesta, antotiheydestä ja injektiokohtien määrän. Annoksen suuruus pitää määrittää titraamalla.
Suositeltua XEOMIN-kerta-annosta ei saa ylittää.
Annostus
Blefarospasmi (luomikouristus) ja hemifasiaalispasmi
Suositeltava alkuannos on 1,25–2,5 yksikköä per injektiokohta. Alkuannos ei saa olla yli 25 yksikköä silmää kohti. Kokonaisannos ei saa ylittää yhdellä hoitokerralla 50 yksikköä silmää kohden. Hoitoa ei yleensä saa uusia useammin kuin joka 12. viikko. Hoitokertojen väli pitää määrittää yksilöllisesti potilaan todellisen kliinisen tarpeen mukaan.
Vaikutus alkaa näkyä neljän vuorokauden kuluessa (mediaaniaika) injektion annosta. XEOMIN-käsittelyn vaikutus kestää yleensä noin 3–5 kuukautta. Se voi kuitenkin kestää huomattavasti pidemmän tai lyhyemmän ajan.
Hoitoa toistettaessa annos voidaan suurentaa enintään kaksinkertaiseksi, jos ensimmäisen hoidon vaste on riittämätön. On kuitenkin ilmeistä, että yli 5,0 yksikön suuruisen annoksen injisoiminen kuhunkin kohtaan ei lisää vaikutusta.
Potilaan hemifasiaalispasmi pitää hoitaa samalla tavoin kuin toispuolinen blerafospasmi hoidetaan.
Spastinen tortikollis
Kun spastista tortikollista hoidetaan XEOMIN-valmisteella, annostuksen pitää olla yksilöllinen ja perustua potilaan pään ja kaulan asentoon, mahdollisen kivun sijaintiin, lihashypertrofiaan, potilaan painoon ja hoitovasteeseen.
Ensimmäisellä hoitokerralla saa injisoida enintään 200 yksikköä, ja annosta säädetään seuraavilla hoitokerroilla vasteen mukaan. Kokonaisannos ei saa millään hoitokerralla ylittää 300 yksikköä. Yhteenkään injektion antokohtaan ei saa antaa yli 50 yksikköä.
Vaikutus alkaa ilmetä seitsemän vuorokauden kuluessa (mediaani) injektion annosta. XEOMIN-käsittelyn vaikutus kestää yleensä noin 3–4 kuukautta, mutta vaikutus voi kuitenkin kestää myös huomattavasti pidempään tai olla lyhyempi. Hoitokertojen väliksi ei suositella alle 10 viikkoa. Hoitokertojen väli pitää määrittää yksilöllisesti potilaan todellisen kliinisen tarpeen mukaan.
Käsivarsien jäykkyys
Tarkka annos ja injektointikohtien määrä on sopeutettava lihasten kokoon, määrään ja sijaintiin sekä spastisuuden vakavuusasteeseen sekä mahdolliseen paikalliseen lihasheikkouteen.
Hoitoon suositellut annokset lihasta kohti:
Kliininen tila Lihas | Yksikköä (vaihteluväli) | Injektiokohtien määrä lihasta kohti |
Ranteen fleksio | ||
Flexor carpi radialis | 25–100 | 1–2 |
Flexor carpi ulnaris | 20–100 | 1–2 |
Nyrkkiin puristunut käsi | ||
Flexor digitorum superficialis | 25–100 | 2 |
Flexor digitorum profundus | 25–100 | 2 |
Kyynärpään fleksio | ||
Brachioradialis | 25–100 | 1–3 |
Biceps | 50–200 | 1–4 |
Brachialis | 25–100 | 1–2 |
Kyynärvarren pronataatio | ||
Pronator quadratus | 10–50 | 1 |
Pronator teres | 25–75 | 1–2 |
Kämmeneen suuntautunut peukalo | ||
Flexor pollicis longus | 10–50 | 1 |
Adductor pollicis | 5–30 | 1 |
Flexor pollicis brevis/ Opponens pollicis | 5–30 | 1 |
Olkapään rotaatio/ekstensio/adduktio | ||
Deltoideus, pars clavicularis | 20–150 | 1–3 |
Latissimus dorsi | 25–150 | 1–4 |
Pectoralis major | 20–200 | 1–6 |
Subscapularis | 15–100 | 1–4 |
Teres major | 20–100 | 1–2 |
Kokonaisannos yläraajan jäykkyyden hoitoon saa olla enintään 500 yksikköä hoitokertaa kohti, ja olkapään lihaksiin saa antaa enintään 250 yksikköä.
Potilasraporttien mukaan vaikutus alkoi 4 vuorokauden kuluttua hoidosta. Paras teho lihastonuksen paranemisessa todettiin 4 viikon kuluessa. Hoidon teho kesti yleensä 12 viikkoa, mutta vaikutus voi kuitenkin kestää huomattavasti pidempään tai olla lyhyempi.
Injektioita ei saa yleensä toistaa useammin kuin joka 12. viikko. Hoitokertojen väli pitää määrittää yksilöllisesti potilaan todellisen kliinisen tarpeen mukaan.
Krooninen sialorrea (aikuisilla)
Injektioon käytetään käyttökuntoon saatettua injektionestettä, jonka pitoisuus on 5 yksikköä/0,1 ml.
XEOMIN injisoidaan kummankin puolen korvasylkirauhasiin ja leuanalussylkirauhasiin (yhteensä neljä injektiota yhdellä hoitokerralla). Annos jaetaan korvasylkirauhasten ja leuanalussylkirauhasten kesken suhteessa 3:2 seuraavasti:
Rauhaset | Yksiköt | Tilavuus |
Korvasylkirauhaset | 30/puoli | 0,6 ml / injektio |
Leuanalussylkirauhaset | 20/puoli | 0,4 ml / injektio |
Injektiokohdan on oltava lähellä rauhasen keskikohtaa.
Suositeltu annos hoitokertaa kohden on 100 yksikköä. Enimmäisannosta ei saa ylittää.
Hoitokertojen väli pitää määrittää yksilöllisesti potilaan todellisen kliinisen tarpeen mukaan. Hoidon toistamista alle 16 viikon välein ei suositella.
Krooninen sialorrea (lapsilla/nuorilla)
Käyttökuntoon saatettu liuos, jonka pitoisuus on 2,5 yksikköä/0,1 ml.
XEOMIN injisoidaan kummankin puolen korvasylkirauhasiin ja leuanalussylkirauhasiin (yhteensä neljä injektiota yhdellä hoitokerralla). Painonmukainen annos jaetaan korvasylkirauhasten ja leuanalussylkirauhasten kesken suhteessa 3:2 seuraavassa taulukossa esitetyllä tavalla.
Annostuksesta ei voida antaa suosituksia alle 12 kg:aa painaville lapsille.
Paino | Korvasylkirauhanen, kumpikin puoli | Leuanalussylkirauhanen, kumpikin puoli | Kokonaisannos, molemmat sylkirauhaset, molemmat puolet | ||
Annos rauhasta kohti | Tilavuus injektiota kohti | Annos rauhasta kohti | Tilavuus injektiota kohti | ||
[kg] | [yksikköä] | [ml] | [yksikköä] | [ml] | [yksikköä] |
≥ 12 ja < 15 | 6 | 0,24 | 4 | 0,16 | 20 |
≥ 15 ja < 19 | 9 | 0,36 | 6 | 0,24 | 30 |
≥ 19 ja < 23 | 12 | 0,48 | 8 | 0,32 | 40 |
≥ 23 ja < 27 | 15 | 0,60 | 10 | 0,40 | 50 |
≥ 27 ja < 30 | 18 | 0,72 | 12 | 0,48 | 60 |
≥ 30 | 22,5 | 0,90 | 15 | 0,60 | 75 |
Injektiokohdan on oltava lähellä rauhasen keskikohtaa.
Hoitokertojen väli pitää määrittää yksilöllisesti potilaan todellisen kliinisen tarpeen mukaan. Hoidon toistamista alle 16 viikon välein ei suositella.
Kaikki käyttöaiheet
Jos hoitovastetta ei ilmene kuukauden kuluessa ensimmäisestä injektiosta, on ryhdyttävä seuraaviin toimenpiteisiin:
- lihakseen injisoidun neurotoksiinin kliinisen vaikutuksen varmistaminen, esim. EMG-tutkimuksella erikoisyksikössä
- vasteen puuttumisen selvittäminen; esim. injisoitavien lihasten huono erottaminen, liian pieni annos, puutteellinen injektiotekniikka, fiksoitunut kontraktuura, liian heikko antagonistilihas, vasta-aineiden mahdollinen muodostuminen
- on harkittava, onko A-tyypin botuliinitoksiinihoito sopiva hoitomuoto kyseiselle potilaalle
- jos haittavaikutuksia ei ole ilmennyt ensimmäisen hoitokerran yhteydessä, voidaan antaa toinen käsittely seuraavilla ehdoilla: 1) annosta säädetään viimeisimmän hoidon epäonnistumista koskevan analyysin perusteella, 2) kohdelihakset paikallistetaan käyttäen injektiota ohjaavaa tekniikkaa, kuten elektromyografiaa, 3) ensimmäisen ja seuraavan hoitokerran suositettua vähimmäishoitoväliä noudatetaan.
Pediatriset potilaat
XEOMIN-valmisteen turvallisuutta ja tehoa muuhun kuin kohdassa Käyttöaiheet mainittuun käyttöaiheeseen ei ole selvitetty pediatrisille potilaille. Annostuksesta ei voida antaa suosituksia muuhun käyttöaiheeseen kuin krooniseen sialorreaan ≥ 12 kg painaville 2–17-vuotiaille lapsille ja nuorille .
Pediatristen potilaiden XEOMIN-hoidosta tällä hetkellä saatavissa olevat kliiniset tiedot esitetään kohdassa Farmakodynamiikka.
Antotapa
Kaikki käyttöaiheet
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa. Käyttökuntoon saattamisen jälkeen XEOMIN pitää käyttää yhdellä antokerralla ja vain yhdelle potilaalle.
XEOMIN-liuos on tarkoitettu annettavaksi lihakseen ja rauhaseen (sylkirauhaseen).
Blefarospasmi ja hemifasiaalispasmi
Käyttövalmis XEOMIN-liuos injisoidaan lihakseen sopivalla steriilillä neulalla (esim. 27–30 G / halkaisija 0,30–0,40 mm, pituus 12,5 mm). EMG-ohjauksen käyttö ei ole välttämätöntä. Suositeltava injektiotilavuus on 0,05–0,1 ml.
XEOMIN injisoidaan yläluomessa silmän kehälihaksen (orbicularis oculi) mediaali- ja lateraaliosaan ja alaluomessa kehälihaksen lateraaliosaan. Injektioita voidaan antaa lisäksi kulmakaarien seudussa kehälihaksen lateraaliosaan ja kasvojen yläosaan, jos niissä olevat lihaskouristukset haittaavat näkökykyä.
Potilaille, joilla on toispuolinen blefarospasmi, injektioita annetaan vain kyseiseen silmään.
Potilaan hemifasiaalispasmi pitää hoitaa samalla tavoin kuin toispuolinen blerafospasmi hoidetaan.
Kliinisistä XEOMIN-tutkimuksista ei ole kokemusta kasvojen alaosaan annetuista injektioista. Injektiota ei pidä antaa kasvojen alaosan lihaksiin, koska siihen liittyy merkittävä paikallisen heikkouden riski. Kirjallisuudessa paikallista heikkoutta on raportoitu potilaille hemifasiaalispasmin hoitoon tälle alueelle annettujen botuliinitoksiini-injektioiden jälkeen.
Spastinen tortikollis
Injektiot annetaan sopivalla steriilillä neulalla pintalihaksiin (esim. 25–30 G / halkaisija 0,30–0,50 mm, pituus 37 mm) ja syvempiin lihaksiin (esim. 22 G / halkaisija 0,70 mm, pituus 75 mm). Suositeltava injektiotilavuus on 0,1–0,5 ml per injektiokohta.
Spastisen tortikolliksen hoidossa XEOMIN-valmistetta injisoidaan seuraaviin lihaksiin: sternocleidomastoideus, levator scapulae, scalenus, splenius capitis ja/tai yhteen trapezius-lihakseen tai molempiin trapezius-lihaksiin. Tämä luettelo ei ole täydellinen, sillä mikä tahansa pään asentoa ylläpitävä lihas voi olla osallisena tortikolliksen synnyssä ja voi sen vuoksi olla hoidon tarpeessa. Jos yksittäisten lihasten tunnistaminen on vaikeaa, injektiot pitää antaa esim. EMG- tai ultraääniohjauksessa. Lihasmassa ja hypertrofian tai atrofian aste on otettava huomioon sopivaa annosta määritettäessä.
Useisiin kohtiin annetut XEOMIN-injektiot kattavat dystonisen lihaksen hermotusalueet tasaisemmin, mikä on erityisen edullista suurissa lihaksissa. Optimaalisin injektioiden määrä riippuu kohteena (denervaatio) olevan lihaksen koosta.
Sternocleidomastoideus-lihaksia ei saa injisoida bilateraalisesti, sillä haittavaikutusten riski kasvaa (erityisesti nielemisvaikeudet), jos injektio annetaan molempiin kaulan lihaksiin tai lihakseen annettavat annokset ovat suurempia kuin 100 yksikköä.
Käsivarsien jäykkyys
Käyttökuntoon saatettu XEOMIN-liuos injisoidaan sopivalla steriilillä neulalla (esim. 26 G / läpimitta 0,45 mm ja pituus 37 mm injektoitaessa pintalihaksiin). Syvempiin lihaksiin käytetään pidempää neulaa (esim. 22 G / läpimitta 0,7 mm ja pituus 75 mm).
Injektiota ohjaavan tekniikan, kuten elektromyografian tai ultraäänen, käyttöä suositellaan kohdelihasten paikallistamiseen, jos yksittäisten lihasten tunnistaminen on vaikeaa. Monen injektointikohdan käyttäminen mahdollistaa sen, että XEOMIN pääsee tasaisemmin kosketuksiin hermotusalueen kanssa. Monen injektointikohdan käyttämisestä on hyötyä etenkin suurempien lihasten yhteydessä.
Krooninen sialorrea (aikuisilla/lapsilla/nuorilla)
Käyttövalmis XEOMIN-liuos injisoidaan rauhaseen sopivalla steriilillä neulalla (esim. 27–30 G / halkaisija 0,30–0,40 mm, pituus 12,5 mm).
Aikuisille sylkirauhasten paikallistamiseen voidaan käyttää anatomisia maamerkkejä tai ultraääniohjausta, joskin ultraääniohjaus on suositeltavampi, koska hoitotulos voi siten olla parempi (ks. kohta Farmakodynamiikka).
Lasten ja nuorten hoitoon on käytettävä ultraääniohjausta. Lapsille ja nuorille voidaan ennen injektiota antaa paikallispuudutus (kuten puudutusvoide), sedaatio tai sekä puudutus että sedaatio huolellisen hyöty-haitta-arvioinnin jälkeen ja paikallista käytäntöä noudattaen.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Lihastoimintaan vaikuttava yleistynyt sairaus (esim. myasthenia gravis, Lambert-Eatonin oireyhtymä).
- Infektio tai tulehdus suunnitellussa injektiokohdassa.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yleistä:
Lääkärin on ennen XEOMIN-valmisteen pistämistä tutkittava potilaan anatomia ja aiemmista kirurgisista toimenpiteistä anatomiaan mahdollisesti aiheutuneet muutokset.
XEOMIN-valmisteen antamista verisuoneen on varottava tarkoin.
XEOMIN-valmisteen käytössä pitää olla varovainen:
• jos esiintyy millaisia verenvuotohäiriöitä tahansa
• potilailla, jotka saavat antikoagulanttihoitoa tai muita aineita, joilla saattaa olla antikoagulanttivaikutus.
A-tyypin botuliinitoksiinin kliiniset vaikutukset saattavat joko voimistua tai heiketä toistuvien injektioiden seurauksena. Mahdollisia syitä kliinisen tehon muutoksiin voivat olla liuoksen käyttökuntoon saattamisessa käytettyjen menetelmien erot, erot hoitovälien pituudessa; injektiokohdat tai toksiinin tehon vähäiset vaihtelut käytetystä biologisen testauksen menetelmästä riippuen tai vasteen sekundaarisesta puuttumisesta hoidon aikana.
Paikallinen ja kauas antopaikasta levinnyt toksiinivaikutus
Väärin kohdistetusta A-tyypin botuliinitoksiini-injektiosta aiheutuvana haittavaikutuksena saattaa esiintyä läheisten lihasryhmien tilapäinen paralyysi. Suuret annokset saattavat aiheuttaa kaukana injektion antopaikasta sijaitsevien lihasten paralyysin.
Haittavaikutuksia, jotka saattavat liittyä A-tyypin botuliinitoksiinin leviämiseen kauas antopaikasta, on raportoitu (ks. kohta Haittavaikutukset). Osa näistä voi olla hengenvaarallisia ja joissakin tapauksissa näiden on raportoitu johtaneen kuolemaan, jolloin osaan tapauksista liittyi nielemishäiriöitä, keuhkokuumetta ja/tai huomattavaa heikkokuntoisuutta.
Hoitoannoksia saavilla potilailla voi ilmetä voimakasta lihasheikkoutta. Botuliinitoksiinivalmisteiden injisoinnin jälkeen on raportoitu iatrogeenista botulismia. Potilaita ja heidän omaishoitajiaan on neuvottava, että jos potilaalle ilmaantuu mitä tahansa levinneeseen botuliinitoksiinivaikutukseen sopivia oireita tai löydöksiä tai jos potilaalle ilmaantuu nielemis-, puhe- tai hengityshäiriöitä, on hakeuduttava heti lääkärinhoitoon (ks. kohta Yliannostus).
Nielemisvaikeuksia on raportoitu tapauksissa, joissa injektio on annettu muualle kuin kaulan lihaksiin.
Aiemmat neuromuskulaariset sairaudet
Liiallisen lihasheikkouden riski saattaa olla suurempi potilailla, joilla on jokin neuromuskulaarinen perussairaus, etenkin annettaessa valmistetta lihakseen. A-tyypin botuliinitoksiinivalmistetta tulee käyttää näillä potilailla erikoislääkärin valvonnassa ja vain tilanteissa, joissa hoidon hyötyjen katsotaan olevan sen riskejä suuremmat.
Jos potilaalla on aiemmin esiintynyt aspiraatiota tai nielemisvaikeuksia, hoidon yhteydessä on yleisesti noudatettava varovaisuutta. Äärimmäistä varovaisuutta on noudatettava hoidettaessa näitä potilaita servikaalisen dystonian vuoksi.
XEOMIN-valmisteen käytössä on oltava varovainen:
- potilailla, joilla on amyotrofinen lateraaliskleroosi
- potilailla, joilla on muita sairauksia, jotka aiheuttavat perifeerisiä neuromuskulaarisia toimintahäiriöitä
- jos kohdelihaksissa on huomattavaa heikkoutta tai surkastumista.
Yliherkkyysreaktiot
A-tyypin botuliinitoksiinivalmisteiden käytön yhteydessä on raportoitu yliherkkyysreaktioita. Jos potilaalle ilmaantuu vakava (esim. anafylaktinen reaktio) ja/tai välitön yliherkkyysreaktio, hänelle on aloitettava tarkoituksenmukainen hoito.
Vasta-aineiden muodostuminen
Annosten antaminen liian tihein väliajoin saattaa lisätä vasta-aineiden muodostumisen riskiä. Vasta-aineiden muodostuminen voi aiheuttaa hoidon epäonnistumisen (ks. kohta Annostus ja antotapa).
Vasta-aineiden muodostumisen todennäköisyyttä voidaan minimoida injisoimalla pienin tehokas annos ja pitämällä injektioiden välillä pisin kliinisesti tarkoituksenmukainen antoväli.
Pediatriset potilaat
Pediatrisilla potilailla, joilla oli muita samanaikaisesti esiintyviä häiriöitä tai sairauksia, pääasiassa CP-vamma, on spontaaniraportoinnissa raportoitu muita A-tyypin botuliinitoksiinivalmisteita koskien hyvin harvoin toksiinin mahdollista leviämistä kauas antopaikasta. Annettu annos on tällaisissa tapauksissa ollut yleensä näiden valmisteiden suositeltua annosta suurempi.
Spontaaniraporteissa koskien lapsia, joilla oli vaikea CP-vamma ja joita oli hoidettu botuliinitoksiinivalmisteilla, mukaan lukien lääkkeen myyntiluvasta poikkeava käyttö (esim. niskan alue), raportoidut kuolemantapaukset, joihin on toisinaan liittynyt aspiraatiokeuhkokuume, ovat olleet harvinaisia. Riskin katsotaan olevan erityisen suuri, jos pediatrisen potilaan perusterveydentila on huono tai jos potilaan neurologinen toimintakyky on heikentynyt merkittävästi, jos potilaalla on nielemisvaikeuksia tai jos potilaalla on äskettäin ollut aspiraatiokeuhkokuume tai keuhkosairaus.
Käyttöaihekohtaiset varoitukset
Blefarospasmi ja hemifasiaalispasmi
Injektion antamista yläluomen kohottajalihaksen (m. levator palpebrae superioris) lähelle tulee välttää ptoosin välttämiseksi. Kaksoiskuvia voi ilmetä, jos A-tyypin botuliinitoksiinia pääsee leviämään alempaan vinoon silmälihakseen (m. obliquus oculi inferior ). Tämä haittavaikutus voitaneen välttää, jos vältetään injektioiden antoa alaluomen mediaaliosaan.
A-tyypin botuliinitoksiinin antikolinergisen vaikutuksen takia XEOMIN-valmistette on määrättävä varoen potilaille, joille saattaa kehittyä ahdaskulmaglaukooma.
Luomenreunan uloskääntymisen estämiseksi ei injektiota pidä antaa alaluomen alueelle, ja mahdolliset epiteelivauriot on hoidettava tehokkaasti. Tähän saatetaan tarvita suojaavia tippoja, voiteita, pehmeitä hoitavia piilolaseja tai silmäluomien sulkemista lapulla jne.
Silmän räpyttelyn väheneminen sen jälkeen, kun silmän kehälihakseen (orbicularis oculi) on injisoitu XEOMIN-valmistetta, voi aiheuttaa sarveiskalvon kuivumista, sarveiskalvon päällyskerroksen pitkäaikaisen vaurion ja haavautumisen varsinkin potilailla, joilla on aivohermojen (kasvohermo) häiriöitä. Sarveiskalvon tuntoa on testattava potilailla, joille on aikaisemmin tehty silmäleikkauksia.
Silmäluomien pehmytosiin tulee helposti pieniä verenpurkaumia. Verenpurkaumien riskiä voidaan vähentää, jos injektion antokohtaa painetaan kevyesti välittömästi injektion annon jälkeen.
Spastinen tortikollis
XEOMIN-valmisteen injisoinnissa herkille alueille, kuten kaulavaltimon, keuhkon kärkien ja ruokatorven läheisyyteen, on oltava varovainen.
Aikaisemmin liikkumatta olleita tai liikuntaa harrastamattomia potilaita on huomautettava lisäämään liikuntaa vain vähitellen XEOMIN-injektion jälkeen.
Potilasta tulisi informoida, että spastisen tortikolliksen hoitoon annetut XEOMIN-injektiot voivat aiheuttaa lievää tai vaikeaa dysfagiaa, johon voi liittyä aspiraation ja hengenahdistuksen vaara. Tällöin saatetaan tarvita erityisiä hoitotoimenpiteitä (esim. ruokintaletkun asennus) (ks. myös kohta Haittavaikutukset). Nielemisvaikeuksia voitaneen vähentää, jos sternocleidomastoideukseen injisoitu annos on vähemmän kuin 100 yksikköä. Nielemisvaikeuksien riski on suurempi potilailla, joiden kaulalihakset ovat pienemmät tai joille joudutaan antamaan injektio kumpaankin sternocleidomastoideus-lihakseen. Nielemisvaikeudet johtuvat XEOMIN-valmisteen farmakologisen vaikutuksen leviämisestä ruokatorven lihaksiin.
Käsivarsien jäykkyys
XEOMIN-valmisteen injisoinnissa herkille alueille, kuten kaulavaltimon, keuhkon kärkien ja ruokatorven läheisyyteen, on oltava varovainen.
Aikaisemmin liikkumatta olleita tai liikuntaa harrastamattomia potilaita on huomautettava lisäämään liikuntaa vain vähitellen XEOMIN-injektion jälkeen.
Fokaalisen spastisuuden hoidossa XEOMIN-valmistetta on tutkittu vain tavanomaisten hoito-ohjelmien yhteydessä eikä sen ole tarkoitus korvata näitä hoitomuotoja. On epätodennäköistä, että XEOMIN parantaisi liikkuvuutta tehokkaasti nivelissä, joissa on pysyvä lihaskontraktuura.
Kouristuskohtausten ilmaantumista tai uusiutumista on ilmoitettu tavallisesti potilailla, joilla on taipumus tällaisiin haittoihin. Näiden haittojen yhteyttä botuliinitoksiini-injektioihin ei ole varmistettu.
Krooninen sialorrea (aikuisilla/lapsilla/nuorilla)
Lääkityksen (esimerkiksi aripipratsolin, klotsapiinin, pyridostigmiinin) aiheuttaman sialorrean yhteydessä harkitaan ensimmäiseksi sialorreaa aiheuttavan lääkityksen vaihtamista, annoksen pienentämistä tai jopa lääkityksen lopettamista ennen XEOMIN-valmisteen käyttöä sialorrean hoitoon.
XEOMIN-valmisteen turvallisuutta ja tehoa lääkityksen aiheuttaman sialorrean hoidossa ei ole tutkittu.
Jos XEOMIN-valmisteen käytön yhteydessä ilmenee suun kuivumista, on harkittava annoksen pienentämistä.
Hoidon alussa suositellaan hammastarkastusta. Hammaslääkärille on kerrottava XEOMIN-valmisteen käytöstä sialorrean hoitoon, jotta hän voi päättää asianmukaisista toimista karieksen ehkäisemiseksi.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Aminoglykosidiantibiootit tai muut lääkevalmisteet, jotka vaikuttavat neurotransmissioon hermo-lihasliitoksessa, esim. tubokurariinin kaltaiset lihasrelaksantit, voivat teoriassa voimistaa A-tyypin botuliinitoksiinin vaikutuksia.
Sen vuoksi on oltava erittäin varovainen, jos XEOMIN-valmistetta annetaan aminoglykosidien tai spektinomysiinin käytön aikana. Perifeerisiä lihasrelaksantteja pitää käyttää varoen ja tarvittaessa on pienennettävä lihasrelaksantin aloitusannosta tai käytettävä keskipitkävaikutteisia valmisteita (vekuronium tai atrakurium) pitkävaikutteisten valmisteiden sijasta.
Lisäksi käytettäessä valmistetta kroonisen sialorrean hoitoon pään ja kaulan (sylkirauhaset mukaan lukien) sädehoito ja/tai samanaikainen hoito antikolinergisilla aineilla (esim. atropiinilla, glykopyrroniumilla, skopolamiinilla) saattaa voimistaa toksiinin vaikutusta. XEOMIN-valmisteen käyttö sialorrean hoitoon sädehoidon aikana ei ole suositeltavaa.
4-aminokinoliinit voivat heikentää XEOMIN-valmisteen tehoa.
Raskaus ja imetys
Raskaus
Ei ole olemassa riittäviä tietoja A-tyypin botuliinitoksiinin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmisille ei tunneta. Sen vuoksi XEOMIN-valmistetta ei pitäisi käyttää raskauden aikana, mikäli käyttö ei ole selvästi välttämätöntä tai jos mahdollinen hyöty ei ole suurempi kuin riski.
Imetys
Ei tiedetä, erittyykö A-tyypin botuliinitoksiini rintamaitoon. Sen vuoksi XEOMIN-valmistetta ei saa käyttää imetyksen aikana.
Hedelmällisyys
A-tyypin botuliinitoksiinin käytöstä ei ole kliinisiä tietoja. Kaniineilla ei havaittu haitallisia vaikutuksia urosten tai naaraiden hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
XEOMIN-valmisteella on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilasta on neuvottava, että jos hänellä on voimattomuutta, lihasheikkoutta, huimausta, näköhäiriöitä tai riippuluomet, hänen on vältettävä ajamista tai ryhtymästä muuhun mahdolliseen toimintaan, josta voi aiheutua vaaraa.
Haittavaikutukset
Haittavaikutukset havaitaan tavallisesti käsittelyn jälkeisen viikon aikana, ja ne ovat luonteeltaan tilapäisiä. Haittavaikutukset saattavat liittyä vaikuttavaan aineeseen, injektion antamiseen tai kumpaankin.
Käyttöaiheesta riippumattomat haittavaikutukset
Antoon liittyvät haittavaikutukset
Injektion yhteydessä voi esiintyä paikallista kipua, tulehdusta, tuntoharhoja, hypestesiaa, aristusta, turvotusta, edeemaa, punoitusta, kutinaa, paikallisia infektioita, verenpurkaumia, verenvuotoa ja/tai mustelmia.
Neulan aiheuttama kipu ja/tai ahdistus voi johtaa vasovagaalisiin reaktioihin, mukaan lukien ohimenevä oireinen hypotonia, pahoinvointi, tinnitus ja pyörtyminen.
A-tyypin botuliinitoksiinin lääkeryhmään liittyvät haittavaikutukset
Paikallinen lihasheikkous on yksi A-tyypin botuliinitoksiinin odotettavissa oleva farmakologinen vaikutus.
Toksiinin leviäminen
Kauas antopaikasta levinneeseen toksiiniin liittyviä haittavaikutuksia, joissa oireet vastaavat A-tyypin botuliinitoksiinin vaikutuksia (liiallista lihasheikkoutta, nielemisvaikeuksia ja aspiraatiokeuhkokuumetta, joka on joissakin tapauksissa johtanut potilaan kuolemaan) on raportoitu hyvin harvoin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yliherkkyysreaktiot
Vakavia ja/tai välittömiä yliherkkyysreaktioita, kuten anafylaksiaa, seerumitautia, urtikariaa, pehmytkudosturvotusta ja hengenahdistusta, on raportoitu harvoin. Osa näistä reaktioista on ilmoitettu joko yksinään annetun tavanomaista A-tyypin botuliinitoksiinikompleksia sisältävän valmisteen jälkeen tai käytettynä yhdessä muiden lääkeaineiden kanssa, joiden tiedetään aiheuttavan samankaltaisia reaktioita.
Kliinisessä käytössä todetut haittavaikutukset
Seuraavia haittavaikutuksia on raportoitu XEOMIN-hoidon yhteydessä. Haittavaikutusten yleisyys on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Blefarospasmi
Elinjärjestelmäluokka | Haittavaikutus | Yleisyys |
Hermosto | Päänsärky, kasvohalvaus | Melko harvinainen |
Silmät | Silmäluomien ptoosi | Hyvin yleinen |
Silmien kuivuminen, näön sumeneminen, näkökyvyn heikentyminen | Yleinen | |
Kahtena näkeminen, lisääntynyt kyynelvuoto | Melko harvinainen | |
Ruoansulatuselimistö | Suun kuivuminen | Yleinen |
Nielemisvaikeudet | Melko harvinainen | |
Iho ja ihonalainen kudos | Ihottuma | Melko harvinainen |
Luusto, lihakset ja sidekudos | Lihasheikkous | Melko harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan kipu | Yleinen |
Väsymys | Melko harvinainen |
Hemifasiaalispasmi
Hemifasiaalispasmin yhteydessä esiintyvät haittavaikutukset ovat oletettavasti samankaltaisia kuin blefarospasmin yhteydessä.
Spastinen tortikollis
Elinjärjestelmäluokka | Haittavaikutus | Yleisyys |
Infektiot | Ylempien hengitysteiden infektio | Yleinen |
Hermosto | Päänsärky, presynkopee, huimaus | Yleinen |
Puheen häiriöt | Melko harvinainen | |
Hengityselimet, rintakehä ja välikarsina | Dysfonia, hengenahdistus | Melko harvinainen |
Ruoansulatuselimistö | Nielemisvaikeudet | Hyvin yleinen |
Suun kuivuminen, pahoinvointi | Yleinen | |
Iho ja ihonalainen kudos | Liikahikoilu | Yleinen |
Ihottuma | Melko harvinainen | |
Luusto, lihakset ja sidekudos | Niskakipu, lihasheikkous, lihaskipu, lihasspasmit, muskuloskeletaalinen jäykkyys | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan kipu, voimattomuus | Yleinen |
Spastisen tortikolliksen hoito voi aiheuttaa eriasteisia nielemisvaikeuksia, mikä voi aiheuttaa aspiraation riskin, joka saattaa vaatia hoitotoimenpiteitä. Nielemisvaikeudet voivat kestää kahdesta kolmeen viikkoon injektion jälkeen, mutta yhdessä tapauksessa nielemisvaikeuksien on raportoitu kestäneen viisi kuukautta.
Käsivarsien jäykkyys
Elinjärjestelmäluokka | Haittavaikutus | Yleisyys |
Hermosto | Päänsärky, hypestesia | Melko harvinainen |
Ruoansulatuselimistö | Suun kuivuminen | Yleinen |
Dysfagia, pahoinvointi | Melko harvinainen | |
Luusto, lihakset ja sidekudos | Lihasheikkous, raajakipu, lihaskipu | Melko harvinainen |
Yleisoireet ja antopaikassa todettavat haitat | Voimattomuus | Melko harvinainen |
Injektiokohdan kipu | Tuntematon |
Krooninen sialorrea (aikuisilla)
Elinjärjestelmäluokka | Haittavaikutus | Yleisyys |
Hermosto | Parestesiat | Yleinen |
Puheen häiriöt | Melko harvinainen | |
Ruoansulatuselimistö | Suun kuivuminen, dysfagia | Yleinen |
Syljen muutokset (sakeutuminen), dysgeusia | Melko harvinainen |
Joissakin tapauksissa on ilmoitettu pitkäaikaista (> 110 vuorokautta), vaikea-asteista suun kuivumista, mistä voi aiheutua lisäkomplikaatioita, kuten ientulehdusta, dysfagiaa ja kariesta.
Krooninen sialorrea (lapsilla/nuorilla)
Elinjärjestelmäluokka | Haittavaikutus | Yleisyys |
Ruoansulatuselimistö | Dysfagia | Melko harvinainen |
Syljen muutokset (sakeutuminen), suun kuivuminen, suukipu, karies | Tuntematon |
Markkinoille tulon jälkeinen käyttökokemus
Seuraavia käyttöaiheesta riippumattomia haittavaikutuksia, joiden esiintyvyys on tuntematon, on raportoitu XEOMIN-hoidon yhteydessä valmisteen markkinoille tulon jälkeen:
Elinjärjestelmäluokka | Haittavaikutus |
Immuunijärjestelmä | Yliherkkyysreaktiot, kuten turvotus, edeema (myös kaukana injektiokohdasta), punoitus, kutina, ihottuma (paikallinen ja yleistynyt) ja hengästyneisyys |
Luusto, lihakset ja sidekudos | Lihasten surkastuminen |
Yleisoireet ja antopaikassa todettavat haitat | Flunssan kaltaiset oireet |
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Katso kohdasta Varoitukset ja käyttöön liittyvät varotoimet. tiedot paikalliseen ja kauas antopaikasta levinneeseen toksiinivaikutukseen liittyvistä riskeistä.
Yliannostuksen oireet
A-tyypin botuliinitoksiinin suuret annokset voivat aiheuttaa huomattavan neuromuskulaarisen paralyysin ja monenlaisia siihen liittyviä oireita etäällä injektion antokohdasta. Oireita voivat olla yleinen heikotus, ptoosi, kaksoiskuvat, hengitysvaikeudet, puhevaikeudet, hengityslihasten halvaus tai nielemisvaikeudet, jotka voivat johtaa aspiraatiokeuhkokuumeeseen.
Toimenpiteet yliannostustapauksissa
Yliannostuksen tai toksiinin leviämisen yhteydessä potilaan on oltava lääkärin seurannassa voimakkaan lihasheikkouden tai lihasten paralyysin oireiden havaitsemiseksi. Oireenmukainen hoito saattaa olla tarpeen. Jos hengityslihakset halvaantuvat, hengityksen tukeminen saattaa olla tarpeen.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut lihasrelaksantit, perifeerisesti vaikuttavat lihasrelaksantit,
ATC-koodi: M03AX01
A-tyypin botuliinitoksiini salpaa kolinergisen neurotransmission hermo-lihasliitoksessa estämällä asetyylikoliinin vapautumista. Hermo-lihasliitoksen hermopäätteet eivät enää reagoi hermoimpulsseihin, ja neurotransmitterin eritys motorisissa päätelevyissä estyy (kemiallinen denervaatio). Impulssien transmissio palautuu ennalleen uusien hermopäätteiden muodostumisen myötä ja niiden kytkeytyessä uudelleen motorisiin päätelevyihin.
Vaikutusmekanismi
A-tyypin botuliinitoksiinin vaikutusmekanismia kolinergisissä hermopäätteissä voidaan kuvata nelivaiheisesti etenevänä prosessina:
- sitoutuminen: A-tyypin botuliinitoksiinin raskas ketju sitoutuu poikkeuksellisen selektiivisesti ja suurella affiniteetilla reseptoreihin, joita esiintyy vain kolinergisissä päätteissä.
- soluun siirtyminen: hermopäätteen kalvo supistuu ja toksiini imeytyy hermopäätteeseen (endosytoosi).
- translokaatio: neurotoksiinin raskaan ketjun aminopäätesegmentti muodostaa huokoisen vesikkelin kalvoon, disulfidisidos hajoaa ja neurotoksiinin kevyt ketju siirtyy huokosen kautta sytosoliin.
- vaikutus: kun kevyt ketju on vapautunut, se pilkkoo erittäin spesifisesti kohdeproteiinin (SNAP 25), joka on välttämätön asetyylikoliinin vapautumiselle.
Päätelevyjen toiminnan/ärsykkeiden välittymisen täydellinen palautuminen tapahtuu tavallisesti 3−4 kuukauden kuluessa lihakseen annetun injektion jälkeen, kun hermopäätteet versovat ja kytkeytyvät uudelleen motorisiin päätelevyihin.
Tulokset kliinisistä tutkimuksista
XEOMIN-valmisteen hoidollinen ekvivalenssi A-tyypin botuliinitoksiinikompleksia (onabotulinumtoksiini A, 900 kD) sisältävään vertailuvalmisteeseen, Botoxiin, nähden osoitettiin kahdessa vertailevassa vaiheen III kerta-annostutkimuksessa; yhdessä tutkimuksessa, jossa potilailla oli blefarospasmi (tutkimus MRZ 60201-0003, n = 300), ja toisessa tutkimuksessa, jossa potilailla oli servikaalinen dystonia (tutkimus MRZ 60201-0013, n = 463). Tutkimustulokset viittaavat myös siihen, että XEOMIN-valmisteen ja tämän vertailuvalmisteen teho ja turvallisuusprofiili ovat samankaltaiset potilailla, joilla on blefarospasmi tai servikaalinen dystonia, kun annos muunnetaan suhteessa 1:1 (ks. kohta Annostus ja antotapa).
Blefarospasmi
XEOMIN-valmistetta on tutkittu vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa yhteensä 109 blefarospasmipotilaalla. Potilailla oli hyvänlaatuisen essentiaalin blefarospasmin diagnoosi, ja lähtötilanteen Jankovic Rating Scale (JRS) -pisteytyksen sairauden vaikeusastetta kuvaavan osion pisteet olivat ≥ 2, ja he olivat saaneet vakaan tyydyttävän hoitovasteen aiemmin annettuun vertailuvalmisteeseen (onabotulinumtoksiini A).
Potilaat satunnaistettiin (2:1) saamaan XEOMIN-kerta-annoksen (n = 75) tai lumelääkettä (n = 34) annoksen, joka oli samankaltainen (+/- 10 %) kuin kahdella viimeisimmällä Botox‑injektiohoidon antokerralla ennen heidän tutkimukseen mukaan tuloaan. Suurin tässä tutkimuksessa sallittu annos oli 50 yksikköä silmää kohden, ja keskimääräinen XEOMIN-annos oli 32 yksikköä silmää kohden.
Tehon ensisijainen päätetapahtuma oli JRS-pisteytyksen sairauden vaikeusastetta kuvaavan osion muutos hoitoaikeen mukaisessa (intent-to-treat, ITT) potilasjoukossa lähtötilanteesta viikkoon 6 injektion antamisen jälkeen. Potilaiden puuttuvat arvot korvattiin potilaan viimeisimmällä arvolla (last observation carried forward). Hoitoaikeen mukaisen potilasjoukon JRS-pisteytyksen sairauden vaikeusastetta kuvaavan osion pisteiden muutos XEOMIN-ryhmän ja lumeryhmän välillä lähtötilanteesta viikkoon 6 oli -1,0 (95 %:n luottamusväli -1,4; -0,5) pistettä, joka oli tilastollisesti merkitsevä (p < 0,001).
Potilaiden oli mahdollista jatkaa tutkimuksen jatko-osassa, jos uusi injektio oli tarpeen. Potilaat saivat enimmillään viisi XEOMIN-injektiota, ja lyhyin kahden injektion välinen aika oli vähintään kuusi viikkoa (tutkimuksen kokonaiskesto 48–69 viikkoa, ja maksimiannos 50 yksikköä silmää kohden). XEOMIN‑hoitoa saaneilla tutkittavilla injektioiden välinen aika (mediaani) oli koko tutkimuksen ajan 10,14 (1. väli) – 12,00 viikkoa (2.−5. väli).
Toisen kaksoissokkoutetun, lumekontrolloidun vaiheen III kliinisen tutkimuksen avoimessa jatkovaiheessa selvitettiin XEOMIN-valmisteen tehoa yhteensä 61 potilaalla. Potilailla oli hyvänlaatuisen essentiaalin blefarospasmin diagnoosi, ja lähtötilanteen Jankovic Rating Scale (JRS) ‑pisteytyksen sairauden vaikeusastetta kuvaavan osion pisteet olivat ≥ 2. Potilaat eivät olleet aiemmin saaneet botuliinitoksiinihoitoa eli he eivät olleet saaneet botuliinitoksiinia blefarospasmin hoitoon vähintään 12 kuukauteen ennen XEOMIN-valmisteen antoa. Potilaat satunnaistettiin tutkimuksen päävaiheessa (6−20 viikkoa) saamaan XEOMIN-valmistetta kerta-annoksena 12,5 yksikköä silmää kohden (n = 22), 25 yksikköä silmää kohden (n = 19) tai lumelääkettä (n = 20). Uuden injektion tarvitsevilla potilailla oli mahdollisuus jatkaa mukana tutkimuksessa sen jatkovaiheessa ja saada vielä yksi XEOMIN-injektio.
Tutkimuksen päävaiheessa hoitokertojen välisen ajan mediaani oli lumelääkeryhmässä 6 viikkoa, 12,5 yksikköä silmää kohden saaneessa ryhmässä 11 viikkoa ja 25 yksikköä silmää kohden saaneessa ryhmässä 20 viikkoa. JRS-pisteytyksen sairauden vaikeusastetta kuvaavan osion pisteiden muutoksesta lähtötilanteesta viikkoon 6 tehtiin kovarianssianalyysi (ANCOVA). Tässä analyysissa pienimmän neliösumman keskiarvon ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) oli 25 yksikköä XEOMIN-valmistetta silmää kohden saaneessa ryhmässä -1,2 (-1,9; -0,6), joka oli tilastollisesti merkitsevä, kun taas 12,5 yksikköä XEOMIN-valmistetta saaneessa ryhmässä vastaava ero lumelääkkeeseen verrattuna oli -0,5 (-1,1; 0,2), joka ei ollut tilastollisesti merkitsevä.
Potilaat saivat tutkimuksen jatkovaiheen aikana XEOMIN-injektion (n = 39) annostuksella, jonka keskiarvo oli lähes 25 yksikköä (vaihteluväli: 15–30 yksikköä) silmää kohden, ja hoitokertojen välisen ajan mediaani oli 19,9 viikkoa.
Spastinen tortikollis
XEOMIN-valmistetta on tutkittu vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, monikeskustutkimuksessa yhteensä 233 servikaalista dystoniaa sairastavalla potilaalla. Potilaiden kliininen diagnoosi oli pääasiassa spastinen tortikollis, ja heidän lähtötilanteen Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) ‑kokonaispisteensä olivat ≥ 20. Potilaat satunnaistettiin (1:1:1) saamaan kerta-annoksena 240 yksikköä XEOMIN-valmistetta (n = 81), 120 yksikköä XEOMIN-valmistetta (n = 78) tai lumelääkettä (n = 74). Injektioiden lukumäärän ja antokohdan päätti tutkijalääkäri.
Ensisijainen tehon muuttuja oli hoitoaikeen mukaisen (intent-to-treat, ITT) potilasjoukon TWSTRS-kokonaispisteiden pienimmän neliön keskimuutos lähtötilanteesta viikkoon 4 injektion jälkeen, ja puuttuvat arvot korvattiin potilaan lähtötilanteen arvolla (kattava tilastomalli). TWSTRS-kokonaispisteiden muutos lähtötilanteesta viikkoon 4 oli huomattavasti suurempi XEOMIN‑ryhmissä verrattuna muutokseen lumelääkeryhmässä (kaikkien tilastomallien p < 0,001). Näillä eroilla oli myös kliinistä merkitystä: esim. ‑9,0 pistettä verrattaessa 240 yksikköä lumelääkkeeseen ja ‑7,5 pistettä verrattaessa 120 yksikköä lumelääkkeeseen kattavassa tilastomallissa.
Potilaiden oli mahdollista jatkaa tutkimuksen jatko-osassa, jos uusi injektio oli tarpeen. Potilaat saivat enimmillään viisi XEOMIN-injektiota (120 yksikköä tai 240 yksikköä), ja lyhyin kahden injektion välinen aika oli vähintään kuusi viikkoa (tutkimuksen kokonaiskesto 48–69 viikkoa). Potilaiden uusintahoitoa koskevien pyyntöjen perusteella XEOMIN-hoitoon saadun vasteen kesto (mediaani) tässä tutkimuksessa (sekä kaksoissokkoutettu että avoin jatkotutkimusjakso) oli 12 viikkoa (kvartiilivälit: 9–15 viikkoa). Useimpien hoitokertojen (96,3 %) jälkeen aika uusintahoitoon oli 6–22 viikkoa ja yksittäisissä tapauksissa enimmillään 28 viikkoa.
Käsivarsien jäykkyys (aikuisilla)
Pivotaalitutkimuksessa (kaksoissokkoutettu, lumelääkekontrolloitu monikeskustutkimus) 148 aivohalvauksen jälkeisestä yläraajan jäykkyydestä kärsivää potilasta satunnaistettiin saamaan XEOMIN-valmistetta (n = 73) tai lumelääkettä (n = 75). Kliinisessä tutkimuksessa keskimääräinen kumulatiivinen annos enintään kuuden hoitokerran jälkeen oli 1333 yksikköä (suurin annos 2395 yksikköä) korkeintaan 89 viikon kuluessa.
Ensisijaisella tehoa kuvanneella muuttujalla mitattuna (ranteen fleksoreiden vaste viikolla 4, kun vasteeksi katsottiin vähintään yhden pykälän paraneminen viisiportaisella Ashworth-asteikolla) vasteen saavuttamisen mahdollisuus oli 3,97 kertaa suurempi XEOMIN-hoitoa saaneilla potilailla (hoitovaste: 68,5 %) kuin lumelääkettä saaneilla (hoitovaste: 37,3 %; 95 %:n luottamusväli: 1,90–8,30; p < 0,001, ITT-populaatio).
Vakioannoksella toteutetussa tutkimuksessa ei ollut tarkoitus erotella nais- ja miespotilaita, mutta post-hoc-analyysissä hoitovaste oli parempi naisilla (89,3 %) kuin miehillä (55,6 %), ja hoitoryhmien välinen ero oli tilastollisesti merkitsevä vain naisilla. Miespotilaiden hoitovaste Ashworth-asteikolla mitattuna 4 viikon jälkeen oli XEOMIN-hoitoa saaneilla potilailla kuitenkin jatkuvasti suurempi kaikissa hoidetuissa lihasryhmissä lumelääkkeeseen verrattuna. Potilaiden uusintahoitoa koskevien pyyntöjen perusteella tehon kesto tässä pivotaalitutkimuksessa ja sen jälkeisellä avoimella jatkotutkimusjaksolla oli 14 viikkoa (kvartiilivälit: 13–17 viikkoa), ja useimpien hoitokertojen (95,9 %) jälkeen aika uusintahoitoon oli 12–28 viikkoa.
Hoitovasteessa ei todettu eroja miesten ja naisten välillä pivotaalitutkimuksen avoimessa jatkotutkimuksessa (jossa annostus voitiin määrittää joustavasti), jossa oli mukana 145 potilasta ja hoitokertoja enintään 5. Näin oli myös havainnoitsijan suhteen sokkoutetussa tutkimuksessa (EudraCT-numero 2006-003036-30), jossa arvioitiin kahden eri XEOMIN-laimennoksen tehoa ja turvallisuutta 192 potilaalla, joilla oli eri syistä johtuvaa käsivarren jäykkyyttä.
Toiseen kaksoissokkoutettuun, lumelääkekontrolloituun vaiheen III kliiniseen tutkimukseen osallistui yhteensä 317 aiemmin hoitoa saamatonta, yläraajan jäykkyydestä kärsivää potilasta, joilla aivohalvauksesta oli kulunut vähintään kolme kuukautta. Tutkimuksen päävaiheen aikana annettiin kiinteä kokonaisannos XEOMIN-valmistetta (400 yksikköä) kliinisen kuvan (joko kyynärpään fleksio, ranteen fleksio tai nyrkkiin puristunut käsi) mukaisesti valittuun kohdelihakseen ja muihin jännittyneisiin lihasryhmiin (n=210). Viikolla 4 injektion annon jälkeen nähtiin ensisijaisissa tehoa kuvaavissa muuttujissa tilastollisesti merkitsevä parannus vasteen saaneiden määrässä Ashworth-asteikolla mitattuna tai tilastollisesti merkitsevä muutos lähtötasosta Ashworth-asteikolla mitattuna ja tutkijan arvioimana (Investigator's Global Impression of Change).
296 potilasta sai hoitoa päävaiheen loppuun asti ja osallistui ensimmäiseen avoimen jatkotutkimuksen jaksoon. Jatkotutkimuksessa potilaille annettiin enintään kolme injektiota. Jokainen avoin jatkotutkimusjakso koostui yhdestä hoitokerrasta (kokonaisannos 400 yksikköä XEOMIN-valmistetta, annos jaettiin joustavasti hoidettavien lihasten kesken), minkä jälkeen seurasi 12 viikon mittainen havainnointijakso. Tutkimus kesti kokonaisuudessaan 48 viikkoa.
Olkapään lihasten hoitoa tutkittiin avoimessa vaiheen III tutkimuksessa 155 potilaalla, joilla oli kliinistä hoitoa vaativaa jäykkyyttä sekä ylä- että alaraajoissa. Tutkimusprotokollan mukaisesti yläraajaan sai antaa enintään 600 yksikköä XEOMIN-valmistetta.
Tutkimuksessa todettiin positiivinen yhteys XEOMIN-annoksen suurentamisen ja potilaan tilan kohenemisen välillä Ashworth-asteikolla mitattuna ja muissa tehoa kuvaavissa muuttujissa ilman, että tämä heikensi potilaan turvallisuutta tai XEOMIN-valmisteen siedettävyyttä.
CP-vammasta johtuva ala- ja yläraajojen jäykkyys (lapsilla/nuorilla)
Alaraajojen tutkiminen
Erääseen kaksoissokkoutettuun, rinnakkaisryhmillä tehtyyn vaiheen III kliiniseen annos-vastetutkimukseen otettiin mukaan 311 lasta ja nuorta (2–17-vuotiasta), joilla oli CP-vammasta johtuvaa toispuolista tai molemminpuolista alaraajojen jäykkyyttä. XEOMIN-valmistetta annettiin alaraajojen jäykkyyden hoitoon kolmessa hoitoryhmässä (4 yksikköä/kg [enintään 100 yksikköä], 12 yksikköä/kg [enintään 300 yksikköä] tai 16 yksikköä/kg [enintään 400 yksikköä]) kahden valitun alaraajojen kliinisen mallin (pystyjalka, polven fleksio, reiden adduktio) hoitoon.
Pientä annosta saaneen ryhmän oli tarkoitus toimia tässä tutkimuksessa vertailuryhmänä. Suuren ja pienen annoksen välisessä vertailussa ei osoitettu tilastollisesti merkitseviä eroja primaarisessa eikä yhteisprimaarisessa tehokkuuden päätetapahtumassa. Plantaarifleksoreissa viikolla 4 injektion jälkeen pienimmän neliösumman keskiarvon muutos lähtötilanteesta (keskivirhe, 95 %:n luottamusväli) oli Ashworth-asteikolla suuren annoksen osalta -0,70 (0,061, 95 %:n luottamusväli: ‑0,82; ‑0,58) ja pienen annoksen osalta -0,66 (0,084, 95 %:n luottamusväli: -0,82; -0,50) p-arvon ollessa 0,650. Lihastonuksen väheneminen ei kuvastunut vaikutuksessa toimintakykyyn tai tutkijan arvioon (Investigator’s Global Impression of Change). Riittävää XEOMIN-annostusta lasten ja nuorten alaraajojen jäykkyyden hoitoon ei voida määritellä. Kaksoissokkoutetussa hoidossa ja avoimessa pitkäkestoisessa hoidossa, jossa XEOMIN-valmistetta annettiin neljällä injektiojaksolla, ei havaittu odottamattomia haittavaikutuksia.
Yläraajojen tutkiminen
Toisessa kaksoissokkoutetussa, rinnakkaisryhmillä tehdyssä vaiheen III annos-vastetutkimuksessa XEOMIN-hoitoa sai yhteensä 350 lasta ja nuorta (2–17-vuotiasta), joilla oli CP-vammasta johtuvaa yläraajojen tai sekä ylä- että alaraajojen jäykkyyttä. Tutkimuksen päävaiheessa XEOMIN-valmistetta annettiin yläraajojen jäykkyyden hoitoon (kyynärpään fleksio, ranteen fleksio, nyrkkiin puristunut käsi, kyynärvarren pronaatio, kämmeneen suuntautunut peukalo) tai sekä ylä- että alaraajojen jäykkyyden hoitoon (pystyjalka, polven fleksio, reiden adduktio) yhdellä injektiojaksolla kolmessa hoitoryhmässä: 2–5 yksikköä/kg (enintään 50–125 yksikköä), 6–15 yksikköä/kg (enintään 150−375 yksikköä) ja 8–20 yksikköä/kg (enintään 200–500 yksikköä). Potilaiden hoitoa jatkettiin suurimmalla annoksella tutkimuksen avoimessa jatkovaiheessa, jossa hoito annettiin kolmella injektiojaksolla.
Kyynärpään fleksiossa tai ranteen fleksiossa havaittiin Ashworth-asteikolla lähtötilanteesta viikkoon 4 injektion jälkeen tilastollisesti merkitsevä ero pienen ja suuren annoksen välillä (-0,22 [95 %:n luottamusväli -0,4; -0,04] p = 0,017). Lihastonuksen väheneminen ei kuvastunut vaikutuksessa toimintakykyyn tai tutkijan arvioon (Investigator’s Global Impression of Change). Tämän tutkimuksen perusteella ei siten voitu määritellä riittävää XEOMIN-annostusta pediatristen potilaiden yläraajojen jäykkyyden hoitoon.
Ylä- ja alaraajojen jäykkyyden XEOMIN-hoidossa ei raportoitu odottamattomia turvallisuutta koskevia huolenaiheita, kun hoitoa annettiin enintään neljä injektiojaksoa (14 ± 2 viikkoa kukin).
Krooninen sialorrea (aikuisilla)
Kaksoissokkoutetussa lumekontrolloidussa vaiheen III pivotaalitutkimuksessa oli mukana yhteensä 184 potilasta, joilla oli Parkinsonin tautiin, epätyypilliseen parkinsonismiin, aivohalvaukseen tai traumaattiseen aivovaurioon liittyvää, vähintään kolme kuukautta kestänyttä sialorreaa. Tutkimuksen päävaiheen aikana annettiin kiinteä kokonaisannos XEOMIN-valmistetta (100 tai 75 yksikköä) tai lumelääkettä rauhasiin siten, että korvasylkirauhasiin ja leuanalussylkirauhasiin annettujen annosten suhde oli 3:2.
uSFR (g/min) | GICS (pistemäärä) | ||||
Hoito | Aikapiste | n obs | Pienimmän neliösumman keskiarvo (keskivirhe) | n obs | Pienimmän neliösumman keskiarvo (keskivirhe) |
Lumelääke | 4. viikko | 36 | -0,04 (0,033) | 36 | 0,67 (0,186) |
100 yksikköä | 4. viikko | 73 | -0,13 (0,026) | 74 | 1,25 (0,144) |
100 yksikköä | 8. viikko | 73 | -0,13 (0,026) | 74 | 1,30 (0,148) |
100 yksikköä | 12. viikko | 73 | -0,12 (0,026) | 74 | 1,21 (0,152) |
100 yksikköä | 16. viikko | 73 | -0,11 (0,027) | 74 | 0,93 (0,152) |
uSFR: stimuloimaton syljeneritysnopeus (Unstimulated Salivary Flow Rate); GICS: Global Impression of Change Scale ‑asteikko n obs: todettu lukumäärä | |||||
GICS-asteikolla (Global Impression of Change Scale) havaittiin 4. viikolla vähintään 1 pisteen paraneminen (ensisijainen päätetapahtuma) 73 %:lla 100 yksikköä XEOMIN-valmistetta saaneista potilaista verrattuna 44 %:iin lumeryhmän potilaista. Kummankin ensisijaisen tehoa kuvaavan muuttujan (stimuloimaton syljeneritysnopeus ja GICS-asteikko 4. viikolla injektion jälkeen) varmistava analyysi osoitti 100 yksikköä hoitoa saaneessa ryhmässä tilastollisesti merkitsevää tilan paranemista lumeryhmään verrattuna. Tehon parametrien paraneminen 8. ja 12. viikolla injektion jälkeen oli osoitettavissa ja säilyi viimeiseen havainnointiajankohtaan saakka, joka oli tutkimuksen päävaiheen 16. viikolla. Muut ensisijaiset tehoa kuvaavat muuttujat osoittivat viikolla 4, että tulokset valmisteen annosta ultraääniohjauksessa ovat paremmat verrattuna anatomisten maamerkkien käyttöön (uSFR:n p-arvo 0,019 vs 0,099 ja GICS:n p-arvo 0,003 vs 0,171).
Päävaiheessa oli mukana 173 hoitoa saanutta potilasta sen päättymiseen saakka, minkä jälkeen potilaat siirtyivät jatkovaiheeseen. Jatkovaihe koostui kolmesta annossokkoutetusta hoitosyklistä, joista jokaisessa oli yksi hoitokerta (XEOMIN-kokonaisannos 100 tai 75 yksikköä samassa suhteessa kuin päävaiheessa) ja joita seurasi 16 viikon havainnointivaihe. 151 potilasta oli mukana jatkovaiheessa sen päättymiseen saakka. Jatkovaiheen tulokset vahvistivat päävaiheen havainnot ja osoittivat 100 yksikön annoksina annetun XEOMIN-hoidon jatkamisen hyödyt.
Krooninen sialorrea (lapsilla/nuorilla)
Kaksoissokkoutetussa lumekontrolloidussa vaiheen III kliinisessä tutkimuksessa hoidettiin yhteensä 255 lasta ja nuorta (2−17-vuotiasta), joiden paino oli vähintään 12 kg ja joilla oli neurologisiin sairauksiin tai älylliseen kehitysvammaisuuteen liittyvää kroonista sialorreaa. Tutkimuksen päävaiheen aikana 220 potilaalle (6−17-vuotiaalle) annettiin XEOMIN-valmistetta painoluokan mukaan ja korkeintaan 75 yksikköä tai lumelääkettä. Hoito annettiin rauhasiin ultraääniohjauksessa siten, että korvasylkirauhasiin ja leuanalussylkirauhasiin annettujen annosten suhde oli vastaavasti 3:2.
uSFR (g/min) | GICS (pistemäärä) | ||||
Hoito | Aikapiste | n obs | Pienimmän neliösumman keskiarvo (keskivirhe) | n obs | Pienimmän neliösumman keskiarvo (keskivirhe) |
Lumelääke | 4. viikko | 72 | -0,07 (0,015) | 72 | 0,63 (0,104) |
XEOMIN painon mukaan | 4. viikko | 148 | -0,14 (0,012) | 148 | 0,91 (0,075) |
8. viikko | 146 | -0,16 (0,012) | 146 | 0,94 (0,068) | |
12. viikko | 147 | -0,16 (0,013) | 147 | 0,87 (0,073) | |
16. viikko | 145 | -0,15 (0,013) | 146 | 0,77 (0,070) | |
uSFR: stimuloimaton syljeneritysnopeus (Unstimulated Salivary Flow Rate); GICS: Global Impression of Change Scale ‑asteikko n obs: todettu lukumäärä | |||||
Yhteisprimaaristen tehoa kuvaavien muuttujien (uSFR ja GICS viikolla 4 injektion annon jälkeen) varmistusanalyysi osoitti tilastollisesti merkitsevää ja kliinisesti relevanttia XEOMIN-ryhmän paranemista lumelääkeryhmään verrattuna. Hoitoryhmien välillä havaittiin tilastollisesti merkitseviä eroja kummankin tehoa kuvaavan muuttujan suhteen päävaiheen päättymiseen saakka viikolla 16.
Kaikkia 35:tä lasta (2–5 vuotta) hoidettiin XEOMIN-valmisteella painoluokan mukaisella annostuksella, lumelääkehaaraa ei käytetty kontrollina, ja hoito osoitti tutkittujen tehoa kuvaavien muuttujien paranemisen olevan samanlaista kuin 6–17-vuotiaiden XEOMIN-hoitoryhmässä.
247 potilasta osallistui seuraavaan avoimen jatkojakson ensimmäiseen hoitojaksoon. Avoin jatkojakso koostui vielä kolmesta muusta hoitojaksosta, joista jokainen käsitti yhden hoitokerran ja sitä seuraavan 16 viikon havainnointijakson. Kaikki potilaat saivat XEOMIN-valmistetta saman ennalta määrätyn annostusohjelman mukaan ja samalla annosten suhteella kuin tutkimuksen päävaiheessa. Yhteensä 222 potilasta kävi avoimen jatkojakson loppuun asti. Avoimen jatkojakson tulokset vahvistivat päävaiheen löydökset, jotka osoittivat hoidosta saatavan hyödyn olevan jatkuvaa. Uusia tai odottamattomia turvallisuushuolia ei havaittu.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset XEOMIN-valmisteen käytöstä
- kaikkien pediatristen potilasryhmien dystonian hoidossa
- 0–24 kuukauden ikäisten imeväisten ja taaperoiden lihasspastisuuden ja kroonisen sialorrean hoidossa.
Ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Vaikuttavan aineen yleiset ominaisuudet
Klassisia kinetiikkaa ja jakautumista koskevia tutkimuksia ei voida tehdä A-tyypin botuliinitoksiinilla, koska vaikuttavaa ainetta käytetään erittäin pieniä määriä (pikogramma injektiota kohti) ja se sitoutuu hermopäätteisiin nopeasti ja palautumattomasti.
Natiivi A-tyypin botuliinitoksiini on suurimolekyylipainoinen kompleksi, joka neurotoksiinin lisäksi (150 kD) sisältää muita, ei-toksisia proteiineja, kuten hemagglutiniineja ja nonhemagglutiniineja. Toisin kuin tavanomaiset A-tyypin botuliinitoksiinikompleksia sisältävät valmisteet XEOMIN sisältää puhdasta (150 kD) neurotoksiinia, koska siinä ei ole kompleksoivia proteiineja ja vierasproteiinisisältö on siksi pieni. Annetun vierasproteiinisisällön katsotaan olevan yksi hoidon sekundaarisen epäonnistumisen syy.
A-tyypin botuliinitoksiinin on osoitettu siirtyvän aksonissa retrogradisesti lihakseen annetun injektion jälkeen. Aktiivisen A-tyypin botuliinitoksiinin retrogradista transsynaptista kulkua keskushermostoon ei kuitenkaan ole todettu hoitoannoksia käytettäessä.
Reseptoriin sitoutunut A-tyypin botuliinitoksiini siirtyy hermopäätteeseen endosytoosin välityksellä ennen kohteensa (SNAP-25) saavuttamista ja hajoaa sen jälkeen solussa. Verenkierrossa olevat vapaat A-tyypin botuliinitoksiinimolekyylit, jotka eivät ole sitoutuneet presynaptisiin kolinergisten hermopäätteiden reseptoreihin, fagosytoituvat tai pinosytoituvat ja hajoavat samalla tavoin kuin muutkin verenkierrossa kiertävät vapaat proteiinit.
Vaikuttavan aineen jakautuminen potilaissa
XEOMIN-valmisteella ei ole tehty farmakokineettisiä tutkimuksia ihmisillä edellä mainittujen syiden vuoksi.
Prekliiniset tiedot turvallisuudesta
Kardiovaskulaarista ja suolistoon liittyvää turvallisuusfarmakologiaa koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Lihakseen injisoidun XEOMIN-valmisteen toistuvan annoksen systeemistä toksisuutta koskevissa eläinkokeissa tehdyt löydökset liittyivät lähinnä XEOMIN-valmisteen farmakodynaamisiin vaikutuksiin eli atoniaan, pareesiin ja atrofiaan lihaksessa, johon injektio annetaan.
Vastaavasti leuanalussylkirauhaseen annettu injektio vähensi rauhasen painoa kaikilla annostasoilla, ja suurimmalla annoksella (40 yksikköä/kg) havaittiin sylkirauhasen rauhasrakkulan atrofiaa, kun rotille annettiin XEOMIN-valmistetta neljä injektiota 8 viikon välein.
Paikalliseen siedettävyyteen liittyviä ongelmia ei havaittu. Lisääntymistoksisuustutkimuksissa XEOMIN-valmisteella ei ollut haitallisia vaikutuksia uros- tai naaraskaniinien hedelmällisyyteen, eikä suoria haitallisia vaikutuksia rotan ja/tai kanin alkion tai sikiön kehitykseen tai pre- ja postnataaliseen kehitykseen. Kun XEOMIN-valmistetta annettiin alkiotoksisuutta selvittävissä tutkimuksissa päivittäin tai yhden tai kahden viikon välein annoksina, jotka aiheuttivat emojen painon laskua, keskenmenojen määrä kuitenkin lisääntyi kaniineilla ja rotilla havaittiin sikiöiden painon lievää laskua. Näiden tutkimusten perusteella ei voida olettaa, että emojen jatkuva systeeminen altistus organogeneesin (tuntemattoman) herkän vaiheen aikana aiheuttaisi välttämättä teratogeenisia vaikutuksia.
Nuorilla rotilla vieroituksen jälkeen tehdyssä toksisuustutkimuksessa suurimmilla tutkituilla annoksilla (30 yksikköä/kg/antokerta) todettiin kivesten ituepiteelin atrofiaa ja hypospermiaa ilman vaikutuksia urosten hedelmällisyyteen. Urosten ja naaraiden paritellessa 14 viikon iässä suurta annosta saaneiden urosten parittelusuoritus oli heikompi, mikä saattoi johtua raajojen heikkoudesta tai huomattavasti pienemmästä ruumiinpainosta. Annoksista 10 yksikköä/kg/antokerta ja sitä suuremmista annoksista ei aiheutunut vaikutuksia keltarauhasten keskimääräiseen lukumäärään, mutta alkiokuolemat ennen implantaatiota lisääntyivät. Sitä ei pystytty selvittämään varmasti, johtuiko tämä havainto uroksista vai naaraista.
Kliinisen hoidon turvallisuusmarginaalit olivat vastaavasti suurien kliinisten annosten osalta yleensä pienet.
XEOMIN-valmisteen genotoksisuutta ja karsinogeenisuutta ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Ihmisen albumiini
Sakkaroosi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
XEOMIN 50 yksikköä injektiokuiva-aine, liuosta varten: 3 vuotta
XEOMIN 100 yksikköä injektiokuiva-aine, liuosta varten: 4 vuotta
XEOMIN 200 yksikköä injektiokuiva-aine, liuosta varten: 3 vuotta
Käyttökuntoon saatettu liuos
Valmisteen on osoitettu olevan kemiallisesti ja fysikaalisesti stabiili 24 tuntia 2 °C – 8 °C:ssa.
Mikrobiologiselta kannalta valmiste tulee käyttää välittömästi. Jos sitä ei käytetä heti, säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla eivätkä tavallisesti saa ylittää 24 tuntia 2 ºC − 8 ºC:n lämpötilassa, ellei valmistetta ole saatettu käyttökuntoon kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä alle 25 ºC.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XEOMIN injektiokuiva-aine, liuosta varten
50 U (L:ei) 1 kpl (133,75 €)
100 U (L:ei) 1 kpl (237,22 €)
200 U (L:ei) 1 kpl (404,43 €)
PF-selosteen tieto
Injektiopullo (tyypin 1 lasia), jossa on tulppa (bromobutyylikumia) ja avaamattomuuden osoittava alumiinisuojustiiviste.
XEOMIN 50 yksikköä injektiokuiva-aine, liuosta varten: Pakkauskoot ovat 1, 2, 3 tai 6 injektiopulloa, joista kukin sisältää 50 yksikköä.
XEOMIN 100 yksikköä injektiokuiva-aine, liuosta varten: Pakkauskoot ovat 1, 2, 3, 4 tai 6 injektiopulloa, joista kukin sisältää 100 yksikköä.
XEOMIN 200 yksikköä injektiokuiva-aine, liuosta varten: Pakkauskoot ovat 1, 2, 3, 4 tai 6 injektiopulloa, joista kukin sisältää 200 yksikköä.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen jauhe.
Käyttö- ja käsittelyohjeet
Käyttökuntoon saattaminen
XEOMIN saatetaan käyttökuntoon injektiota varten liuottamalla injektiokuiva-aine 9 mg/ml (0,9-prosenttiseen) natriumkloridi-injektioliuokseen. Käyttökuntoon saattaminen ja laimennus on tehtävä hyvien laboratoriokäytäntöjen mukaisesti, etenkin aseptiset seikat huomioiden.
Injektiopullon sisällön käyttökuntoon saattaminen ja ruiskun valmistelu on hyvä tehdä muovitettujen paperipyyhkeiden päällä kaikkien roiskeiden saamiseksi talteen. Vedä ruiskuun sopiva määrä natriumkloridiliuosta (ks. laimennustaulukkoa). Valmisteen käyttökuntoon saattamiseen suositellaan lyhyttä 20–27 G:n viistokärkistä neulaa. Työnnä neula kohtisuoraan kumitulpan läpi ja ruiskuta liuotin varovasti injektiopulloon, jotta vältät vaahdon muodostumisen. Jos alipaine ei vedä liuotinta injektiopulloon, injektiopullo pitää hävittää. Ruisku irrotetaan injektiopullosta ja XEOMIN sekoitetaan liuottimeen pyörittelemällä ja kääntelemällä injektiopulloa, mutta sitä ei saa ravistaa voimakkaasti. Valmisteen käyttökuntoon saattamiseen käytetty neula pitää tarvittaessa jättää kiinni injektiopulloon, jotta tarvittava määrä liuosta voidaan vetää uuteen, injektion antamiseen soveltuvaan steriiliin ruiskuun.
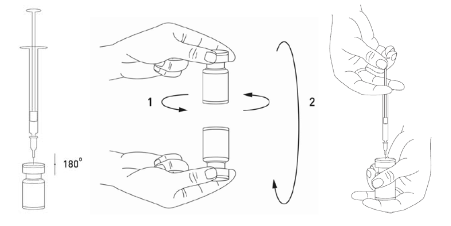
Käyttökuntoon saatettu XEOMIN on kirkas ja väritön liuos.
XEOMIN-valmistetta ei saa käyttää, jos liuos on sameaa tai sisältää haituvia tai hiukkasia.
Tahattoman yliannoksen välttämiseksi ole huolellinen, että käytät valitsemaasi pakkauskokoon nähden oikean määrän liuotinta. Jos samassa toimenpiteessä injektion annossa käytetään erikokoisia XEOMIN-injektiopulloja, on oikea liuotinmäärä varmistettava huolellisesti saatettaessa käyttövalmiiksi tietty yksikkömäärä 0,1 ml:aa kohden. Käytettävä liuotinmäärä on erilainen, kun käytetään 50 yksikköä, 100 yksikköä ja 200 yksikköä sisältävää XEOMIN‑valmistetta. Jokainen ruisku on merkittävä vastaavasti.
Seuraavassa taulukossa on kuvattu 50 yksikköä, 100 yksikköä ja 200 yksikköä sisältävän XEOMIN-valmisteen mahdolliset pitoisuudet:
Saatu annos (yksikköinä/0,1 ml) | Lisätty liuotin (9 mg/ml [0,9-prosenttinen] natriumkloridi-injektioneste, liuos) | ||
50 yksikköä sisältävä injektiopullo | 100 yksikköä sisältävä injektiopullo | 200 yksikköä sisältävä injektiopullo | |
20 yksikköä | 0,25 ml | 0,5 ml | 1 ml |
10 yksikköä | 0,5 ml | 1 ml | 2 ml |
8 yksikköä | 0,625 ml | 1,25 ml | 2,5 ml |
5 yksikköä | 1 ml | 2 ml | 4 ml |
4 yksikköä | 1,25 ml | 2,5 ml | 5 ml |
2,5 yksikköä | 2 ml | 4 ml | Ei mahdollinen. |
2 yksikköä | 2,5 ml | 5 ml | Ei mahdollinen. |
1,25 yksikköä | 4 ml | Ei mahdollinen. | Ei mahdollinen. |
Injektioliuokset, joita on säilytetty kauemmin kuin 24 tuntia, ja käyttämätön injektioliuos on hävitettävä.
Injektiopullojen, ruiskujen ja käytettyjen materiaalien hävittäminen turvallisesti
Käyttämättömät injektiopullot sekä injektiopulloon ja/tai ruiskuun jäljelle jäävä liuos on autoklavoitava. Jäljelle jäänyt XEOMIN voidaan vaihtoehtoisesti inaktivoida lisäämällä siihen jotakin seuraavista liuoksista: etanoli (70 %), isopropanoli (50 %), SDS (0,1 %, anioninen detergentti), laimennettu natriumhydroksidiliuos (0,1 N NaOH) tai laimennettu natriumhypokloriittiliuos (vähintään 0,1 % NaOCl).
Käytettyjä injektiopulloja, ruiskuja ja muita materiaaleja ei saa tyhjentää inaktivoinnin jälkeen, vaan ne on laitettava asianmukaiseen astiaan ja hävitettävä paikallisten vaatimusten mukaisesti.
Suositukset mahdollisiin vaaratilanteisiin A-tyypin botuliinitoksiinin käsittelyssä
- Roiskeet on puhdistettava pyyhkimällä: tähän käytetään kuiva-ainetta siivottaessa jollakin edellä mainitulla liuoksella kyllästettyä imukykyistä pyyhettä, ja käyttövalmiiksi sekoitettua valmistetta siivottaessa kuivaa imukykyistä pyyhettä.
- Kontaminoituneet pinnat on puhdistettava jollakin edellä mainitulla liuoksella kyllästetyllä imukykyisellä pyyhkeellä, minkä jälkeen pinta kuivataan.
- Jos injektiopullo rikkoutuu, toimitaan edellä annettujen ohjeiden mukaisesti, kerätään lasinsirut ja pyyhitään valmiste pois varoen samalla, etteivät lasinsirut aiheuta viiltohaavoja.
- Jos valmistetta pääsee kosketuksiin ihon kanssa, altistunut ihoalue huuhdellaan runsaalla vesimäärällä.
- Jos valmistetta pääsee silmiin, ne huuhdellaan huolellisesti runsaalla vesimäärällä tai silmähuuhdeliuoksella.
- Jos valmistetta pääsee haavaan, viiltohaavaan tai rikkoutuneelle iholle, altistunut alue huuhdellaan huolellisesti runsaalla vesimäärällä. Asianmukaisiin hoitotoimenpiteisiin ryhdytään pistetyn annoksen mukaan.
Näitä valmisteen käytöstä, käsittelystä ja hävittämisestä annettuja ohjeita on noudatettava tarkasti.
Korvattavuus
XEOMIN injektiokuiva-aine, liuosta varten
50 U 1 kpl
100 U 1 kpl
200 U 1 kpl
- Peruskorvaus (40 %).
- Ei korvausta hammaslääkärin määräämästä lääkkeestä.
ATC-koodi
M03AX01
Valmisteyhteenvedon muuttamispäivämäärä
30.10.2025
Yhteystiedot
Gustav III:s Boulevard 32
169 73 Solna
Sweden
+46 (0) 8-36 80 00
www.merztherapeutics.com
nordics.office@merz.com