
TASIGNA kapseli, kova 50 mg, 150 mg, 200 mg
Vaikuttavat aineet ja niiden määrät
Tasigna 50 mg kapseli, kova
Yksi kova kapseli sisältää nilotinibihydrokloridimonohydraattia määrän, joka vastaa 50 mg nilotinibia.
Apuaine(et), jonka vaikutus tunnetaan
Yksi kova kapseli sisältää 39,03 mg laktoosimonohydraattia.
Tasigna 150 mg kapseli, kova
Yksi kova kapseli sisältää nilotinibihydrokloridimonohydraattia määrän, joka vastaa 150 mg nilotinibia.
Apuaine(et), jonka vaikutus tunnetaan
Yksi kova kapseli sisältää 117,08 mg laktoosimonohydraattia.
Tasigna 200 mg kapseli, kova
Yksi kova kapseli sisältää nilotinibihydrokloridimonohydraattia määrän, joka vastaa 200 mg nilotinibia.
Apuaine(et), jonka vaikutus tunnetaan
Yksi kova kapseli sisältää 156,11 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli.
Kliiniset tiedot
Käyttöaiheet
Tasigna on tarkoitettu seuraavien sairauksien hoitoon:
- aikuis‑ ja lapsipotilaiden äskettäin todettu kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen krooninen myelooinen leukemia (KML),
- aikuispotilaiden kroonisessa vaiheessa ja akseleraatiovaiheessa oleva Philadelphia‑kromosomipositiivinen KML silloin, kun aiempi hoito, mukaan lukien imatinibihoito, on osoittautunut tehottomaksi tai potilas ei ole sietänyt sitä. Tutkimustuloksia tehosta blastikriisivaiheen Philadelphia‑kromosomipositiivista kroonista myelooista leukemiaa sairastavilla potilailla ei ole,
- lapsipotilaiden kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen KML, kun aiempi hoito, mukaan lukien imatinibihoito, on osoittautunut tehottomaksi tai potilas ei ole sietänyt sitä.
Ehto
Hoidon saa aloittaa kroonisen myelooisen leukemian (KML) diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Tasigna‑hoidon aloittaa aina KML:n diagnostiikkaan ja hoitoon perehtynyt lääkäri.
Annostus
Hoitoa jatketaan niin pitkään kuin siitä on havaittavaa kliinistä hyötyä tai kunnes ilmenee sietämätöntä toksisuutta.
Jos potilaalta unohtuu annos, hänen ei pidä ottaa ylimääräistä annosta vaan seuraava määrätty annos otetaan tavanomaiseen tapaan.
Annostus Philadelphia‑kromosomipositiivisen KML:n hoidossa aikuispotilailla
Suositusannos:
- 300 mg kahdesti vuorokaudessa potilailla, joilla on äskettäin todettu kroonisessa vaiheessa oleva KML
- 400 mg kahdesti vuorokaudessa potilailla, joilla on kroonisessa tai akseleraatiovaiheessa oleva KML, kun aiempi hoito on osoittautunut tehottomaksi tai potilas ei ole sietänyt sitä.
Annostus Philadelphia‑kromosomipositiivisen KML:n hoidossa lapsipotilailla
Lapsipotilaiden annostus on yksilöllinen ja perustuu kehon pinta‑alaan (mg/m2). Suositeltu nilotinibiannos on 230 mg/m2 kahdesti vuorokaudessa pyöristettynä lähimpään 50 mg:n annokseen (kerta‑annos saa olla enintään 400 mg) (ks. taulukko 1). Kovien Tasigna‑kapselien eri vahvuuksia voidaan yhdistellä halutun annoksen saavuttamiseksi.
Alle 2‑vuotiaiden lapsipotilaiden hoidosta ei ole kokemusta. Äskettäin todettua KML:ää sairastavien alle 10‑vuotiaiden lapsipotilaiden hoidosta ei ole tietoja, ja imatinibille resistenttien tai sitä huonosti sietävien alle 6‑vuotiaiden lapsipotilaiden hoidosta on niukasti tietoa.
Taulukko 1 Nilotinibin annostelu lapsille annoksella 230 mg/m2 kahdesti vuorokaudessa
| Kehon pinta‑ala | Annos (mg) (kahdesti vuorokaudessa) |
| Enintään 0,32 m2 | 50 mg |
| 0,33–0,54 m2 | 100 mg |
0,55–0,76 m2 | 150 mg |
| 0,77–0,97 m2 | 200 mg |
0,98–1,19 m2 | 250 mg |
| 1,20–1,41 m2 | 300 mg |
1,42–1,63 m2 | 350 mg |
| ≥ 1,64 m2 | 400 mg |
Aikuisen kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen KML, johon nilotinibia on annettu ensilinjan hoitona ja tällä on saavutettu pitkäkestoinen syvä molekulaarinen vaste (MR4.5)
Hoidon lopettamista voidaan harkita soveltuvilla Philadelphia‑kromosomipositiivisilla KML‑aikuispotilailla, joiden tauti on kroonisessa vaiheessa ja jotka ovat saaneet nilotinibihoitoa annoksella 300 mg kahdesti vuorokaudessa vähintään 3 vuoden ajan, jos syvä molekulaarinen vaste on säilynyt vähintään yhden vuoden ajan juuri ennen hoidon lopettamista. Nilotinibihoidon lopettamisesta päättää lääkäri, jolla on kokemusta KML‑potilaiden hoidosta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Jos soveltuva potilas lopettaa nilotinibihoidon, BCR‑ABL‑transkriptitasoa ja täydellistä verenkuvaa (jossa on mukana erittelylaskenta) on seurattava kerran kuukaudessa ensimmäisen vuoden ajan, sitten 6 viikon välein toisen vuoden ajan ja tämän jälkeen 12 viikon välein. BCR‑ABL‑transkriptimääriä on seurattava kvantitatiivisella diagnostisella testillä, joka on validoitu molekulaarisen vastetason mittaamiseen kansainvälisellä asteikolla (IS) ja jonka herkkyys on vähintään MR4,5 (BCR‑ABL/ABL ≤ 0,0032 % IS).
Jos potilas menettää MR4‑vasteen (MR4 = BCR‑ABL/ABL ≤ 0,01 % IS) mutta ei MMR‑vastetta (MMR = BCR‑ABL/ABL ≤ 0,1 % IS) hoitovapaan vaiheen aikana, BCR‑ABL‑transkriptimääriä on seurattava 2 viikon välein, kunnes BCR‑ABL‑määrät palautuvat tasolle MR4–MR4,5. Jos BCR‑ABL‑määrät pysyvät tasolla MMR–MR4 vähintään 4 peräkkäisellä mittauskerralla, potilas voi palata alkuperäiseen seuranta‑aikatauluun.
Jos potilas menettää MMR‑vasteen, hoito on aloitettava uudelleen 4 viikon kuluessa siitä hetkestä jolloin remission menettämisen tiedetään tapahtuneen. Nilotinibihoito aloitetaan uudelleen annoksella 300 mg kahdesti vuorokaudessa tai alennetulla annoksella 400 mg kerran vuorokaudessa, jos potilaan annosta oli pienennetty ennen hoidon lopettamista. Jos potilas aloittaa nilotinibihoidon uudelleen, BCR‑ABL‑transkriptitasoa on seurattava kuukausittain, kunnes MMR‑vaste saavutetaan uudelleen, ja 12 viikon välein tämän jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aikuisen kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen KML, kun nilotinibihoidolla on saavutettu pitkäkestoinen syvä molekulaarinen vaste (MR4,5) aiemman imatinibihoidon jälkeen
Hoidon lopettamista voidaan harkita soveltuvilla aikuispotilailla, joiden Philadelphia‑kromosomipositiivinen KML on kroonisessa vaiheessa ja jotka ovat saaneet nilotinibihoitoa vähintään 3 vuoden ajan, jos syvä molekulaarinen vaste on säilynyt vähintään yhden vuoden ajan juuri ennen hoidon lopettamista. Nilotinibihoidon lopettamisesta päättää lääkäri, jolla on kokemusta KML‑potilaiden hoidosta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Jos soveltuva potilas lopettaa nilotinibihoidon, BCR‑ABL transkriptitasoa ja täydellistä verenkuvaa (jossa on mukana erittelylaskenta) on seurattava kerran kuukaudessa ensimmäisen vuoden ajan, sitten 6 viikon välein toisen vuoden ajan ja tämän jälkeen 12 viikon välein. BCR‑ABL‑transkriptimääriä on seurattava kvantitatiivisella diagnostisella testillä, joka on validoitu molekulaarisen vastetason mittaamiseen kansainvälisellä asteikolla (IS) ja jonka herkkyys on vähintään MR4,5 (BCR‑ABL/ABL ≤ 0,0032 % IS).
Jos MR4‑vaste (MR4 = BCR‑ABL/ABL ≤ 0,01 % IS) menetetään vahvistetusti hoitovapaan vaiheen aikana (MR4‑vasteen menetys todetaan kahdella perättäisellä mittauskerralla, joiden välillä on vähintään 4 viikon tauko) tai merkittävä molekulaarinen vaste (MMR = BCR‑ABL/ABL ≤ 0,1 % IS) menetetään, hoito on aloitettava uudelleen 4 viikon kuluessa tiedossa olevasta remission menetyshetkestä. Nilotinibihoito aloitetaan uudelleen annoksella 300 mg tai 400 mg kahdesti vuorokaudessa. Jos potilas aloittaa nilotinibihoidon uudelleen, BCR‑ABL‑transkriptitasoa on seurattava kuukausittain, kunnes aiempi huomattava molekulaarinen vaste tai MR4‑vaste saavutetaan uudelleen, ja 12 viikon välein tämän jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen muuttaminen tai sovittaminen
Jos potilaalle kehittyy perussairauteen liittymätöntä hematologista toksisuutta (neutropenia, trombosytopenia), Tasigna‑hoito on mahdollisesti keskeytettävä tilapäisesti ja/tai annosta on pienennettävä (ks. taulukko 2).
Taulukko 2 Annoksen muuttaminen neutropenian ja trombosytopenian takia
Aikuispotilaiden äskettäin todettu kroonisen vaiheen KML annoksella 300 mg x 2 ja imatinibiresistentti tai ‑intolerantti kroonisen vaiheen KML annoksella 400 mg x 2 | ANC* < 1,0 x 109/l ja/tai trombosyyttiarvo < 50 x 109/l |
|
Aikuispotilaiden imatinibiresistentti tai ‑intolerantti akseleraatiovaiheen KML, annos 400 mg x 2 | ANC* < 0,5 x 109/l ja/tai trombosyyttiarvo < 10 x 109/l |
|
Lapsipotilaiden äskettäin todettu kroonisen vaiheen KML annoksella 230 mg/m2 x 2 ja imatinibiresistentti tai ‑intolerantti kroonisen vaiheen KML annoksella 230 mg/m2 x 2 | ANC* < 1,0 x 109/l ja/tai trombosyytit < 50 x 109/l |
|
* ANC = absoluuttinen neutrofiiliarvo.
Lääkitys on keskeytettävä, jos potilaalle kehittyy kliinisesti merkittävää kohtalaista tai vaikeaa ei‑hematologista toksisuutta, ja potilaita on seurattava ja hoidettava asianmukaisesti. Jos äskettäin todettua kroonisen vaiheen KML:ää sairastavan aikuispotilaan aiempi annos oli 300 mg x 2, jos imatinibiresistenttiä tai ‑intoleranttia kroonisen tai akseleraatiovaiheen KML:ää sairastavan aikuispotilaan aiempi annos oli 400 mg x 2 tai jos lapsipotilaan aiempi annos oli 230 mg/m2 x 2, hoitoa voidaan jatkaa annoksella 400 mg kerran vuorokaudessa (aikuiset) tai 230 mg/m2 kerran vuorokaudessa (lapset), kun toksisuus on korjaantunut. Jos aiempi annos oli 400 mg kerran vuorokaudessa (aikuiset) tai 230 mg/m2 kerran vuorokaudessa (lapset), hoito lopetetaan. Annoksen suurentamista takaisin 300 mg:aan kahdesti vuorokaudessa aikuispotilailla, joilla on äskettäin diagnosoitu kroonisen vaiheen KML, tai 400 mg:aan kahdesti vuorokaudessa aikuispotilailla, joilla on imatinibiresistentti tai ‑intolerantti kroonisen tai akseleraatiovaiheen KML, tai tasolle 230 mg/m2 kahdesti vuorokaudessa lapsipotilailla, tulee harkita, jos se on kliinisesti perusteltua.
Seerumin lipaasiarvojen suureneminen: Asteen 3‑4 seerumin lipaasiarvojen suurenemisen yhteydessä aikuispotilaiden annosta pienennetään tasolle 400 mg kerran vuorokaudessa tai hoito keskeytetään. Lapsipotilailla hoito tauotetaan, kunnes tapahtuma korjautuu asteen ≤ 1 tasolle. Jos aiempi annos oli 230 mg/m2 kahdesti vuorokaudessa, hoitoa voidaan tämän jälkeen jatkaa annoksella 230 mg/m2 kerran vuorokaudessa. Jos aiempi annos oli 230 mg/m2 kerran vuorokaudessa, hoito lopetetaan. Seerumin lipaasiarvot tulee tutkia kerran kuukaudessa tai kliinisen tarpeen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Bilirubiiniarvojen ja maksan transaminaasiarvojen suureneminen: Asteen 3‑4 bilirubiini‑ ja maksan transaminaasiarvojen suurenemisen yhteydessä aikuispotilaiden annosta pienennetään tasolle 400 mg kerran vuorokaudessa tai hoito keskeytetään. Jos lapsipotilaalla esiintyy asteen ≥ 2 bilirubiiniarvojen suurenemista tai asteen ≥ 3 maksan transaminaasiarvojen suurenemista, hoito tauotetaan, kunnes arvot palaavat asteen ≤ 1 tasolle. Jos aiempi annos oli 230 mg/m2 kahdesti vuorokaudessa, hoitoa voidaan tämän jälkeen jatkaa annoksella 230 mg/m2 kerran vuorokaudessa. Jos aiempi annos oli 230 mg/m2 kerran vuorokaudessa ja tilanteen korjautuminen asteen ≤ 1 tasolle kestää yli 28 vrk, hoito lopetetaan. Bilirubiiniarvot ja maksan transaminaasiarvot tulee tutkia kerran kuukaudessa tai kliinisen tarpeen mukaan.
Erityisryhmät
Iäkkäät henkilöt
Noin 12 % äskettäin todetun kroonisen vaiheen KML:n vaiheen III tutkimukseen ja noin 30 % imatinibiresistentin tai ‑intolerantin kroonisen ja akseleraatiovaiheen KML:n vaiheen II tutkimukseen osallistuneista potilaista oli vähintään 65‑vuotiaita. 65‑vuotiaiden tai sitä vanhempien potilaiden ja 18‑65‑vuotiaiden aikuisten välillä ei todettu merkittäviä turvallisuus‑ tai tehokkuuseroja.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintapotilailla ei ole tehty kliinisiä tutkimuksia.
Nilotinibi ja sen metaboliitit eivät erity virtsaan, joten munuaisten vajaatoimintapotilailla ei ole odotettavissa kokonaispuhdistuman laskua.
Maksan vajaatoiminta
Maksan vajaatoiminnalla on vähäinen vaikutus nilotinibin farmakokinetiikkaan. Annoksen sovittaminen ei ole tarpeen maksan vajaatoimintaa sairastaville potilaille. Maksan vajaatoimintaa sairastavia potilaita tulee kuitenkin hoitaa varoen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydänhäiriöt
Kliinisiin tutkimuksiin ei otettu potilaita, joilla oli merkittäviä tai huonossa hoitotasapainossa olevia sydänsairauksia (esim. äskettäinen sydäninfarkti, kongestiivinen sydämen vajaatoiminta, epästabiili angina pectoris tai kliinisesti merkittävä bradykardia). Varovaisuutta tulee noudattaa potilailla, joilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.) on merkittäviä sydän‑ ja verisuonisairauksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Nilotinibihoidon yhteydessä on ilmoitettu seerumin kokonaiskolesterolipitoisuuksien suurenemista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lipidipitoisuudet on määritettävä ennen nilotinibihoidon aloittamista ja arvioitava 3 ja 6 kuukauden kuluttua hoidon aloittamisesta sekä vähintään kerran vuodessa pitkäaikaisen hoidon yhteydessä.
Nilotinibihoidon yhteydessä on ilmoitettu veren glukoosipitoisuuden suurenemista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Verensokeri on määritettävä ennen nilotinibihoidon aloittamista ja seurattava hoidon aikana.
Pediatriset potilaat
Tasigna‑valmisteen turvallisuus ja teho kroonisessa vaiheessa olevaa Philadelphia‑kromosomipositiivista KML:ää sairastavien 2 – < 18 vuoden ikäisten lasten hoidossa on varmistettu (ks. kohdat Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka). Alle 2‑vuotiaiden lapsipotilaiden hoidosta tai akseleraatio‑ tai blastikriisivaiheessa olevaa Philadelphia‑kromosomipositiivista KML:ää sairastavien lapsipotilaiden hoidosta ei ole kokemusta. Äskettäin todettua KML:ää sairastavien alle 10‑vuotiaiden pediatristen potilaiden hoidosta ei ole tietoja, ja imatinibille resistenttien tai sitä huonosti sietävien alle 6‑vuotiaiden lapsipotilaiden hoidosta on niukasti tietoa.
Antotapa
Tasigna tulee ottaa tyhjään mahaan kahdesti vuorokaudessa noin 12 tunnin välein. Kovat kapselit nielaistaan kokonaisina veden kera. Potilaan on oltava syömättä 2 tuntia ennen annoksen ottamista ja vähintään tunti annoksen ottamisen jälkeen.
Jos potilas ei pysty nielemään kovia kapseleita, kunkin kovan kapselin sisältö voidaan sekoittaa yhteen teelusikalliseen omenasosetta, joka nautitaan heti. Omenasosetta ei saa käyttää yhtä teelusikallista enempää, eikä muuta ruokaa kuin omenasosetta saa käyttää (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Myelosuppressio
Nilotinibihoito voi aiheuttaa (National Cancer Institute ‑laitoksen Common Toxicity Criteria‑kriteerien [CTC] vaikeusasteen 3 ja 4) trombosytopeniaa, neutropeniaa ja anemiaa. Niitä esiintyy useammin imatinibiresistenttiä tai ‑intoleranttia KML:ää, etenkin akseleraatiovaiheessa olevaa KML:ää sairastavilla potilailla. Täydellinen verenkuva tulee ottaa ensimmäisten 2 kuukauden ajan kahden viikon välein ja sen jälkeen kerran kuukaudessa tai kliinisen tarpeen mukaan. Myelosuppressio oli yleensä korjaantuvaa ja hoidettiin tavallisesti Tasigna‑hoidon tilapäisellä keskeytyksellä tai annosta pienentämällä (ks. kohta Annostus ja antotapa).
QT‑ajan piteneminen
Nilotinibin on osoitettu pidentävän (iholta otetun EKG:n QT‑ajan perusteella mitattua) sydämen kammioiden repolarisaatioaikaa lääkepitoisuuksista riippuvaisella tavalla aikuis‑ ja lapsipotilailla.
Vaiheen III tutkimuksessa äskettäin diagnosoitua kroonisen vaiheen KML:ää sairastavilla potilailla, jotka saivat 300 mg nilotinibia kahdesti vuorokaudessa, QTcF‑ajan keskimääräinen muutos lähtötilanteesta oli vakaassa tilassa 6 msek. QTcF‑aika ei ollut yhdelläkään potilaalla yli 480 msek. Kääntyvien kärkien takykardiaa ei esiintynyt.
Imatinibiresistenteillä ja ‑intoleranteilla 400 mg nilotinibia kahdesti vuorokaudessa saaneilla KML potilailla tehdyssä vaiheen II tutkimuksessa QTcF‑ajan keskimääräinen muutos lähtötilanteesta oli vakaassa tilassa 5 msek kroonisen vaiheen KML:ää sairastavilla potilailla ja 8 msek akseleraatiovaiheen KML:ää sairastavilla potilailla. QTcF‑aika oli > 500 msek alle 1 %:lla näistä potilaista. Kliinisissä tutkimuksissa ei havaittu kääntyvien kärkien takykardiaa.
Terveillä vapaaehtoisilla tehdyssä tutkimuksessa, joissa lääkealtistukset vastasivat potilailla todettuja altistuksia, QTcF‑ajan keskimääräinen lumelääkkeeseen suhteutettu muutos lähtötilanteesta oli 7 msek (luottamusväli ± 4 msek). Yhdenkään tutkimushenkilön QTcF‑aika ei ollut > 450 msek. Tutkimuksen aikana ei todettu lainkaan kliinisesti merkittäviä rytmihäiriöitä. Huomionarvoista on, että yhdelläkään potilaalla ei todettu kääntyvien kärkien takykardiaa (ohimenevää tai pitkäkestoista).
QT‑ajan merkitsevää pitenemistä voi esiintyä, jos nilotinibi otetaan epäasianmukaisella tavalla yhdessä voimakkaiden CYP3A4‑estäjien kanssa ja/tai sellaisten lääkevalmisteiden kanssa, joiden tiedetään voivan pidentää QT‑aikaa, ja/tai ruoan kanssa (ks. kohta Yhteisvaikutukset.). Samanaikainen hypokalemia ja hypomagnesemia voivat voimistaa tätä vaikutusta entisestään. QT‑ajan piteneminen voi aiheuttaa potilaalle kuolemaan johtavien tapahtumien riskin.
Tasignaa on annettava varoen potilaille, joiden QTc‑aika on pidentynyt tai joilla on merkitsevä QTc‑ajan pitenemisen riski, kuten potilailla:
- joilla on synnynnäinen pitkän QT‑ajan oireyhtymä,
- joilla on huonossa hoitotasapainossa oleva tai merkitsevä sydäntauti, esim. äskettäin sairastettu sydäninfarkti, kongestiivinen sydämen vajaatoiminta, epästabiili angina pectoris tai kliinisesti merkitsevä bradykardia.
- jotka käyttävät rytmihäiriölääkitystä tai muita QT‑ajan pitenemistä aiheuttavia aineita.
Potilasta on seurattava tarkoin siltä varalta, että hoito vaikuttaa QTc‑aikaan. Potilaalle on suositeltavaa tehdä EKG‑tutkimus ennen nilotinibihoidon aloittamista sekä kliinisen tarpeen mukaan. Mahdollinen hypokalemia tai hypomagnesemia on korjattava ennen Tasignan antoa, ja potilasta on seurattava säännöllisesti hoidon aikana näiden ilmiöiden varalta.
Äkillinen kuolema
Melko harvinaisia (0,1 – 1 %) äkillisiä kuolemantapauksia on raportoitu imatinibiresistenttiä tai ‑intoleranttia kroonisen tai akseleraatiovaiheen KML:ää sairastavilla potilailla, joilla on sairaushistoriassa sydänsairaus tai merkittäviä sydänsairauksien riskitekijöitä. Taustalla olevan pahanlaatuisen sairauden lisäksi potilailla oli usein myös muita sairauksia ja lääkityksiä. Poikkeamat kammiorepolarisaatiossa saattoivat olla myötävaikuttavia tekijöitä. Äskettäin diagnosoitua kroonisen vaiheen KML:ää sairastavia potilaita koskeneessa vaiheen III tutkimuksessa ei esiintynyt äkkikuolematapauksia.
Nesteretentio ja edeema
Äskettäin diagnosoitua KML:ää sairastavilla potilailla suoritetuissa faasin III tutkimuksissa vaikeita lääkkeeseen liittyviä nesteretentiotapauksia (kuten pleuraeffuusiota, keuhkoedeemaa ja perikardiaalista effuusiota) raportoitiin melko harvoin (0,1–1 %:lla potilaista). Samankaltaisia tapauksia on ilmoitettu myös markkinoille tulon jälkeisissä raporteissa. Nopean ja odottamattoman painonnousun syyt on selvitettävä huolellisesti. Jos vaikea‑asteiseen nesteen kertymiseen viittaavia merkkejä havaitaan nilotinibihoidon aikana, on nesteen kertymisen etiologia arvioitava ja potilas hoidettava tilanteeseen sopivalla tavalla (lisätiedot hoidon sovittamiseksi ei‑hematologisten toksisuuksien yhteydessä, ks. kohta Annostus ja antotapa).
Kardiovaskulaariset tapahtumat
Kardiovaskulaarisia tapahtumia on raportoitu eräässä äskettäin diagnosoitua KML:ää sairastavilla potilailla suoritetussa, satunnaistetussa faasin III tutkimuksessa sekä markkinoille tulon jälkeisessä seurannassa. Em. kliinisessä tutkimuksessa, jossa potilaiden keskimääräinen tutkimuslääkitysaika oli 60,5 kk, asteen 3–4 kardiovaskulaarisina tapahtumina raportoitiin perifeeristä valtimoahtaumatautia (1,4 % annoksella 300 mg nilotinibia kahdesti vuorokaudessa ja 1,1 % annoksella 400 mg nilotinibia kahdesti vuorokaudessa), iskeemistä sydänsairautta (2,2 % annoksella 300 mg nilotinibia kahdesti vuorokaudessa ja 6,1 % annoksella 400 mg nilotinibia kahdesti vuorokaudessa) sekä iskeemisiä aivoverenkiertotapahtumia (1,1 % annoksella 300 mg nilotinibia kahdesti vuorokaudessa ja 2,2 % annoksella 400 mg nilotinibia kahdesti vuorokaudessa). Potilaita on ohjeistettava hakeutumaan välittömästi lääkärin hoitoon, jos heillä ilmenee akuutteja, kardiovaskulaarisiin tapahtumiin viittaavia merkkejä tai oireita. Jokaisen potilaan kardiovaskulaarinen status on arvioitava ja hänen mahdollisia kardiovaskulaarisia riskitekijöitään on seurattava sekä hoidettava aktiivisesti voimassaolevien ohjeistusten mukaisesti koko nilotinibihoidon ajan. Potilaalle on määrättävä asianmukainen hoito kardiovaskulaaristen riskitekijöiden hallinnassa pitämiseen (lisätiedot hoidon sovittamiseksi ei‑hematologisten toksisuuksien yhteydessä, ks. kohta Annostus ja antotapa).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on tapahtunut kyseisen viruksen pysyvillä kantajilla sen jälkeen, kun potilas on saanut BCR‑ABL‑tyrosiinikinaasin estäjiä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan.
Potilaat on testattava hepatiitti B ‑viruksen varalta ennen nilotinibihoidon aloittamista. Maksasairauksien ja hepatiitti B:n hoitoon perehtyneitä asiantuntijoita on kuultava ennen hoidon aloittamista, jos potilaan hepatiitti B ‑serologia on positiivinen (mukaan lukien potilaat, joilla sairaus on aktiivinen) ja jos potilas saa positiivisen hepatiitti B ‑testituloksen hoidon aikana. Hepatiitti B ‑viruksen kantajia, jotka tarvitsevat nilotinibihoitoa, on seurattava tarkasti aktiivisen hepatiitti B ‑virusinfektion oireiden varalta koko hoidon ajan ja useita kuukausia hoidon jälkeen (ks. kohta Haittavaikutukset).
Erityisseuranta kroonisessa vaiheessa olevaa Philadelphia‑kromosomipositiivista KML‑leukemiaa sairastavilla aikuispotilailla, jotka ovat saavuttaneet pitkäkestoisen syvän molekulaarisen vasteen
Soveltuvuus hoidon lopettamiseen
Hoidon lopettamista voidaan harkita soveltuvilla potilailla, jotka ilmentävät vahvistetusti tyypillisiä BCR‑ABL‑transkripteja (e13a2/b2a2 tai e14a2/b3a2). Potilaalla on oltava tyypillisiä BCR‑ABL‑transkripteja, jotta BCR‑ABL‑taso pystytään kvantifioimaan, molekulaarisen vasteen syvyys pystytään arvioimaan ja molekulaarisen remission mahdollinen menetys pystytään määrittämään nilotinibihoidon lopettamisen jälkeen.
Hoidon lopettaneiden potilaiden seuranta
Hoidon lopetukseen soveltuvien potilaiden BCR‑ABL‑transkriptitasoa on seurattava tiheästi kvantitatiivisella diagnostisella testillä, joka on validoitu molekulaarisen vastetason mittaamiseen ja jonka herkkyys on vähintään MR4,5 (BCR‑ABL/ABL ≤ 0,0032 % IS). BCR‑ABL‑transkriptitaso on määritettävä ennen hoidon lopettamista ja sen aikana (kohdat Annostus ja antotapa ja Farmakodynamiikka).
Merkittävä molekulaarisen vasteen (MMR = BCR‑ABL/ABL ≤ 0,1 % IS) menetys nilotinibia ensilinjan tai toisen linjan hoitona saavilla KML-potilailla tai MR4‑vasteen vahvistettu menetys (MR4‑vasteen [MR4 = BCR‑ABL/ABL ≤ 0,01 % IS] menetys todetaan kahdella perättäisellä mittauskerralla, joiden välillä on vähintään 4 viikon tauko) nilotinibia toisen linjan hoitona saavilla KML-potilailla johtaa hoidon aloittamiseen uudelleen 4 viikon kuluessa siitä hetkestä jolloin remission menettämisen tiedetään tapahtuneen. Molekulaarinen relapsi voi ilmetä hoitovapaan vaiheen aikana, eikä pitkän aikavälin tietoja ole vielä saatavilla. BCR‑ABL‑transkriptitason ja täydellisen verenkuvan (jossa on mukana erittelylaskenta) tiheä seuranta on siten erittäin tärkeää, jotta mahdollinen remission menetys pystytään toteamaan (ks. kohta Annostus ja antotapa). Jos potilas ei ole saavuttanut MMR‑vastetta kolmen kuukauden kuluttua hoidon uudelleenaloittamisesta, on tehtävä BCR‑ABL‑kinaasidomeenin mutaatiomääritys.
Laboratoriokokeet ja seuranta
Veren lipidipitoisuudet
Vaiheen III tutkimuksessa, johon osallistuneilla potilailla oli äskettäin todettu KML, todettiin astetta 3–4 edustavaa kokonaiskolesterolipitoisuuksien suurenemista 1,1 %:lla potilaista, jotka saivat 400 mg nilotinibia kahdesti vuorokaudessa. 300 mg nilotinibia kahdesti vuorokaudessa saaneessa ryhmässä ei kuitenkaan havaittu astetta 3–4 edustavaa kolesterolipitoisuuksien suurenemista (ks. kohta Haittavaikutukset). On suositeltavaa määrittää lipidipitoisuudet ennen nilotinibihoidon aloittamista ja arvioida pitoisuudet 3 ja 6 kuukauden kuluttua hoidon aloittamisesta sekä vähintään kerran vuodessa pitkäaikaisen hoidon yhteydessä (ks. kohta Annostus ja antotapa). Jos HMG‑CoA‑reduktaasin estäjän (lipidipitoisuuksia pienentävä lääke) käyttö on tarpeen, lääkärin tulee tutustua kohtaan 4.5 ennen hoidon aloittamista, sillä tietyt HMG‑CoA‑reduktaasin estäjät metaboloituvat CYP3A4‑välitteisesti.
Veren glukoosipitoisuus
Vaiheen III tutkimuksessa, johon osallistuneilla potilailla oli äskettäin todettu KML, todettiin 3. ‑ 4. asteen verensokerin nousua 6,9 %:lla potilaista, jotka saivat 400 mg nilotinibia kahdesti vuorokaudessa, ja 7,2 %:lla potilaista, jotka saivat 300 mg nilotinibia kahdesti vuorokaudessa. On suositeltavaa määrittää verensokeri ennen Tasigna‑hoidon aloittamista ja seurata sitä hoidon aikana kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa). Jos potilas määritysten perusteella on hoidon tarpeessa, tulee lääkärin noudattaa paikallisia käytäntöjä ja hoitosuosituksia.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Tasignan ja voimakkaiden CYP3A4:n estäjien (mm. ketokonatsoli, itrakonatsoli, vorikonatsoli, klaritromysiini, telitromysiini, ritonaviiri) yhteiskäyttöä tulee välttää. Jos potilas tarvitsee jotakin näistä lääkeaineista, nilotinibihoito tulisi mahdollisuuksien mukaan keskeyttää (ks. kohta Yhteisvaikutukset). Jos hoidon tilapäinen keskeyttäminen ei ole mahdollista, on potilasta seurattava tiiviisti QT‑ajan pitenemisen varalta (ks. kohdat Annostus ja antotapa, Yhteisvaikutukset ja Farmakokinetiikka).
Nilotinibin ja voimakkaiden CYP3A4:n indusorien (esim. fenytoiini, rifampisiini, karbamatsepiini, fenobarbitaali ja mäkikuisma) samanaikainen käyttö pienentää todennäköisesti nilotinibialtistusta kliinisesti merkitsevässä määrin. Siksi nilotinibia saavalle potilaalle tulisikin valita jokin toinen, vähemmän CYP3A4:n toimintaa indusoiva lääkevaihtoehto (ks. kohta Yhteisvaikutukset).
Ruoan vaikutus
Ruoka lisää nilotinibin biologista hyötyosuutta. Tasignaa ei saa ottaa aterian yhteydessä (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset) vaan 2 tuntia aterian jälkeen. Potilaan on oltava syömättä vähintään tunti annoksen ottamisen jälkeen. Greippimehua ja muita CYP3A4:n toimintaa tunnetusti estäviä ruoka‑aineita tulee välttää. Jos potilas ei pysty nielemään kovia kapseleita, kunkin kovan kapselin sisältö voidaan sekoittaa yhteen teelusikalliseen omenasosetta, joka nautitaan heti. Omenasosetta ei saa käyttää yhtä teelusikallista enempää, eikä muuta ruokaa kuin omenasosetta saa käyttää (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoiminnalla on vähäinen vaikutus nilotinibin farmakokinetiikkaan. Nilotinibin 200 mg yksittäisannos aiheutti pitoisuus‑aikakäyrän alle jäävässä alueessa (AUC‑arvo) 35 % kasvun lievää, 35 % keskivaikeaa ja 19 % kasvun vaikeaa maksan vajaatoimintaa sairastavilla potilailla verrattuna ryhmään, jolla oli normaali maksan toiminta. Nilotinibin ennakoitu vakaan tilan huippupitoisuus (Cmax) kasvoi em. ryhmissä vastaavasti 29 %, 18 % ja 22 %. Kliinisiin tutkimuksiin ei ole otettu potilaita, joiden alaniinitransaminaasiarvo (ALAT) ja/tai aspartaattiaminotransferaasiarvo (ASAT) on > 2,5 x viitealueen yläraja (tai > 5 x viitealueen yläraja, mikäli tila liittyy potilaan sairauteen) ja/tai joiden kokonaisbilirubiini on > 1,5 x viitealueen yläraja. Nilotinibi metaboloituu pääasiassa maksassa, joten maksan vajaatoiminta saattaa näin ollen suurentaa nilotinibialtistusta. Maksan vajaatoimintapotilaita on hoidettava varoen (ks. kohta Annostus ja antotapa).
Seerumin lipaasi
Seerumin lipaasiarvojen nousua on todettu. Varovaisuuteen on syytä, jos potilaalla on anamneesissa haimatulehdus. Jos lipaasiarvojen nousuun liittyy vatsaoireita, nilotinibihoito on keskeytettävä ja haimatulehduksen mahdollisuus on suljettava pois asianmukaisin diagnostisin toimenpitein.
Täydellinen gastrektomia
Nilotinibin biologinen hyötyosuus saattaa alentua potilailla joille on tehty täydellinen gastrektomia (ks. kohta Farmakokinetiikka). Näiden potilaiden tiheämpää seuraamista on harkittava.
Tuumorilyysisyndrooma
Kliinisesti merkittävän kuivuman korjaaminen ja korkeiden virtsahappotasojen alentaminen ennen nilotinibihoidon aloittamista on suositeltavaa mahdollisen tuumorilyysisyndrooman (TLS) ilmenemisen vuoksi (ks. kohta Haittavaikutukset).
Laktoosi
Tasigna kovat kapselit sisältävät laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi‑intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Pediatriset potilaat
Lapsilla on todettu laboratorioarvojen poikkeavuuksina lievää tai kohtalaista aminotransferaasiarvojen ja kokonaisbilirubiiniarvon suurenemista useammin kuin aikuisilla, osoittaen korkeampaa maksatoksisuuden riskiä pediatrisilla potilailla (ks. kohta Haittavaikutukset). Maksan toimintaa (bilirubiiniarvoja ja maksan transaminaasiarvoja) seurataan kerran kuukaudessa tai kliinisen tarpeen mukaan. Bilirubiiniarvojen ja maksan transaminaasiarvojen suurenemista hoidetaan tauottamalla nilotinibi tilapäisesti, pienentämällä annosta ja/tai lopettamalla nilotinibihoito (ks. kohta Annostus ja antotapa). Pediatrisilla KML-potilailla tehdyssä tutkimuksessa dokumentoitiin kasvun hidastumista nilotinibihoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). On suositeltavaa seurata pediatristen potilaiden kasvua tiiviisti nilotinibihoidon aikana.
Yhteisvaikutukset
Tasignaa saa käyttää yhdessä hematopoieettisten kasvutekijöiden, kuten erytropoietiinin tai granulosyyttikasvutekijän (G‑CSF) kanssa, mikäli se on kliinisesti aiheellista. Sitä saa käyttää yhdessä hydroksiurean tai anagrelidin kanssa, mikäli se on kliinisesti aiheellista.
Nilotinibi metaboloituu enimmäkseen maksassa, ja oksidatiiviseen metaboliaan osallistuu todennäköisesti suurimmassa määrin CYP3A4. Nilotinibi on myös monia lääkeaineita ulos solusta pumppaavan P‑glykoproteiinin (P‑gp) substraatti. CYP3A4:n ja/tai P‑glykoproteiinin toimintaan vaikuttavat aineet saattavat siis vaikuttaa myös systeemisen nilotinibin imeytymiseen ja eliminaatioon.
Aineet, jotka voivat suurentaa nilotinibin pitoisuutta seerumissa
Samanaikainen nilotinibin anto imatinibin (P‑gp:n ja CYP3A4:n substraatti ja moderaattori) kanssa aiheutti lievän CYP3A4:n ja/tai P‑gp:n aktiivisuuden eston. Imatinibin pitoisuus‑aikakäyrän alle jäävä alue (AUC‑arvo) lisääntyi 18 % ‑ 39 % ja nilotinibin AUC‑arvo lisääntyi 18 % ‑ 40 %. Nämä muutokset eivät todennäköisesti ole kliinisesti merkitseviä.
Nilotinibialtistus kolminkertaistui terveillä vapaaehtoisilla, jotka saivat samanaikaisesti voimakasta CYP3A4:n estäjää ketokonatsolia. Siksi voimakkaiden CYP3A4:n estäjien kuten ketokonatsolin, itrakonatsolin, vorikonatsolin, ritonaviirin, klaritromysiinin ja telitromysiinin samanaikaista käyttöä tulee välttää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myös kohtalaisessa määrin CYP3A4‑toimintaa estävät aineet saattavat suurentaa nilotinibialtistusta. Potilaalle tulee harkita muita lääkevaihtoehtoja, jotka eivät estä CYP3A4:n toimintaa joko lainkaan tai juuri lainkaan.
Aineet, jotka voivat pienentää nilotinibin pitoisuutta seerumissa
Voimakas CYP3A4:n indusoija rifampisiini pienentää nilotinibin huippupitoisuutta (Cmax) 64 %:lla ja nilotinibin pitoisuus‑aikakäyrän alle jäävää aluetta (AUC) 80 %:lla. Rifampisiinia ja nilotinibia ei tule käyttää samanaikaisesti.
Nilotinibin ja muiden CYP3A4:ää indusoivien lääkkeiden (esim. fenytoiini, karbamatsepiini, fenobarbitaali ja mäkikuisma) samanaikainen käyttö pienentää todennäköisesti myös nilotinibialtistusta kliinisesti merkitsevässä määrin. CYP3A4:n induktoreja tarvitseville potilaille tulee valita muita lääkevaihtoehtoja, jotka eivät indusoi entsyymien toimintaa niin voimakkaasti.
Nilotinibin liukoisuus riippuu pH:sta, sen liukoisuus heikkenee pH:n noustessa. Terveillä vapaaehtoisilla, jotka saivat esomepratsolia 40 mg kerran päivässä viiden päivän ajan, vatsan pH nousi merkittävästi, mutta nilotinibin imeytyminen vähentyi vain vähän (Cmax väheni 27 % ja AUC0‑∞ väheni 34 %). Nilotinibia voidaan tarpeen mukaan käyttää samanaikaisesti esomepratsolin tai muiden protonipumpun estäjien kanssa.
Terveillä henkilöillä tehdyssä tutkimuksessa nilotinibin farmakokinetiikassa ei havaittu merkittävää muutosta, kun nilotinibia annettiin 400 mg kerta‑annos 2 tuntia ennen ja 10 tuntia famotidiinin jälkeen. Tämän vuoksi H2‑salpaaja voidaan ottaa noin 10 tuntia ennen ja noin 2 tuntia Tasigna‑annoksen jälkeen, jos sen samanaikainen käyttö on välttämätöntä.
Yllä mainitussa tutkimuksessa antasidin (alumiinihydroksidi/magnesiumhydroksidi/simetikoni) anto 2 tuntia ennen tai jälkeen nilotinibin 400 mg kerta‑annoksen ei myöskään muuttanut nilotinibin farmakokinetiikkaa. Tämän vuoksi antasidia voidaan tarvittaessa ottaa noin 2 tuntia ennen tai noin 2 tuntia Tasigna‑annoksen jälkeen.
Aineet, joiden systeemiseen pitoisuuteen nilotinibi voi vaikuttaa
Nilotinibi estää CYP3A4:n, CYP2C8:n, CYP2C9:n, CYP2D6:n ja UGT1A1:n toimintaa kohtalaisen voimakkaasti in vitro, ki‑arvon ollen alhaisimmillaan CYP2C9:n suhteen (ki=0,13 mikroM).
Terveille vapaaehtoisille tehdyssä kerta‑annos lääkeinteraktiotutkimuksessa, jossa annettiin 25 mg varfariinia, joka on herkkä CYP2C9 substraatti, ja 800 mg nilotinibia, ei nähty mitään muutoksia varfariinin farmakokineettisissä parametreissa tai varfariinin farmakodynamiikassa kun mitattiin protrombiiniaikaa (PT) ja INR‑arvoa. Vakaasta tilasta ei ole saatavilla tietoja. Tämä tutkimus viittaa siihen että kliinisesti merkitsevä lääkeinteraktio nilotinibin ja varfariinin välillä on vähemmän todennäköinen varfariini‑annoksilla 25 mg:aan asti. Vakaan tilan tietojen puuttumisesta johtuen, suositellaan varfariinin farmakodynaamisten parametrien kontrollia (PT tai INR) nilotinibin aloittamisen jälkeen (ainakin 2 ensimmäisen viikon aikana).
Kun KML‑potilaille annettiin 400 mg nilotinibia kahdesti päivässä 12 vuorokauden ajan, kasvoi systeeminen altistus suun kautta otetulle midatsolaamille (CYP3A4:n substraatti) 2,6‑ (AUC) ja 2,0‑kertaisesti (Cmax). Nilotinibi on kohtalainen CYP3A4:n estäjä. Tämän seurauksena systeeminen altistus muille lääkevalmisteille, jotka pääasiallisesti metaboloituvat CYP3A4:n kautta (esim. tietyt HMG‑CoA‑reduktaasin estäjät), saattaa kasvaa samanaikaisen nilotinibin annon yhteydessä. Asianmukainen seuranta ja annosmuutokset voivat olla tarpeen, jos samanaikaisesti nilotinibin kanssa käytetään lääkevalmisteita, jotka ovat CYP3A4:n substraatteja ja joilla on kapea terapeuttinen indeksi (koskee mm, muttei ainoastaan, alfentaniilia, siklosporiinia, dihydroergotamiinia, ergotamiinia, fentanyyliä, sirolimuusia ja takrolimuusia).
Nilotinibin käyttö yhdessä pääasiallisesti CYP3A4-välitteisesti eliminoituvien statiinien kanssa voi suurentaa statiinien aiheuttaman myopatian kuten rabdomyolyysin riskiä.
Rytmihäiriölääkkeet ja muut QT‑aikaa mahdollisesti pidentävät aineet
Nilotinibia on annettava varoen potilaille, joiden QT‑aika on pidentynyt tai saattaa pidentyä. Näitä ovat mm. rytmihäiriölääkkeitä kuten amiodaronia, disopyramidia, prokaiiniamidia, kinidiiniä tai sotalolia käyttävät potilaat sekä muita QT‑aikaa mahdollisesti pidentäviä lääkevalmisteita kuten klorokiinia, halofantriinia, klaritromysiiniä, haloperidolia, metadonia ja moksifloksasiinia käyttävät potilaat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääke‑ruokainteraktiot
Ruoka lisää nilotinibin imeytymistä ja hyötyosuutta sekä suurentaa lääkeaineen pitoisuuksia seerumissa (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Greippimehua ja muita CYP3A4:n toimintaa tunnetusti estäviä ruoka‑aineita tulee välttää.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä nilotinibihoidon aikana ja kaksi viikkoa hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja nilotinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Tasignaa ei pitäisi käyttää raskauden aikana, ellei naisen kliininen tila edellytä nilotinibihoitoa. Jos sitä kuitenkin käytetään raskauden aikana, on potilaalle kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Jos nilotinibihoitoa saava nainen harkitsee raskautta, hoidon lopettamista voidaan harkita hoidon lopettamisen soveltuvuuskriteerien perusteella, kuten kohdissa Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet on kuvattu. Potilaista on rajallisesti raskautta koskevaa tietoa hoitovapaan remission tavoittelemisen aikana. Jos raskautta suunnitellaan hoitovapaan remission aikana, potilaalle on kerrottava mahdollisesta nilotinibihoidon uudelleenaloittamisen tarpeesta raskauden aikana (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö nilotinibi ihmisen rintamaitoon. Olemassa olevat toksikologiset tiedot koe‑eläimistä ovat osoittaneet nilotinibin erittyvän rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea, minkä takia käyttäjä ei saa imettää Tasigna-hoidon aikana eikä kahteen viikkoon viimeisen annoksen jälkeen.
Hedelmällisyys
Eläinkokeet eivät viitanneet uros‑ ja naarasrottien hedelmällisyyteen kohdistuviin vaikutuksiin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tasignalla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Jos potilaalla esiintyy huimausta, väsymystä, näköhäiriöitä tai muita sellaisia haittavaikutuksia, jotka saattavat vaikuttaa hänen kykyynsä ajaa autoa tai käyttää koneita turvallisesti, hänen on kuitenkin suositeltavaa välttää näitä toimia kunnes haittavaikutukset ovat hävinneet (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Turvallisuusprofiili perustuu yhdistettyihin tietoihin 3 422 potilaasta, jotka saivat 13 kliinisessä tutkimuksessa Tasigna-hoitoa hyväksyttyihin käyttöaiheisiin: aikuis- ja lapsipotilaiden äskettäin todettu kroonisessa vaiheessa oleva Philadelphia-kromosomipositiivinen krooninen myelooinen leukemia (KML) (5 kliinistä tutkimusta, joihin osallistui 2 414 potilasta), aikuispotilaiden kroonisessa vaiheessa ja akseleraatiovaiheessa oleva Philadelphia-kromosomipositiivinen KML silloin, kun aiempi hoito, mukaan lukien imatinibihoito, on osoittautunut tehottomaksi tai potilas ei ole sietänyt sitä (6 kliinistä tutkimusta, joihin osallistui 939 potilasta), ja lapsipotilaiden kroonisessa vaiheessa oleva Philadelphia-kromosomipositiivinen KML, kun aiempi hoito, mukaan lukien imatinibihoito, on osoittautunut tehottomaksi tai potilas ei ole sietänyt sitä (2 kliinistä tutkimusta, joihin osallistui 69 potilasta). Yhdistetyt tiedot kuvaavat 9 039,34:ää potilasvuotta altistusta.
Nilotinibin turvallisuusprofiili on yhdenmukainen kaikissa käyttöaiheissa.
Yhdistettyjen turvallisuustietojen yleisimmät haittavaikutukset (ilmaantuvuus ≥ 15 %) olivat ihottuma (26,4 %), ylähengitystieinfektio (mukaan lukien nielutulehdus, nenänielutulehdus ja nuha) (24,8 %), päänsärky (21,9 %), hyperbilirubinemia (mukaan lukien veren bilirubiinipitoisuuden suureneminen) (18,6 %), nivelkipu (15,8 %), väsymys (15,4 %), pahoinvointi (16,8 %), kutina (16,7 %) ja trombosytopenia (16,4 %).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa havaitut ja markkinoilletulon jälkeen ilmoitetut haittavaikutukset (taulukko 3) on lueteltu MedDRA-elinjärjestelmä- ja yleisyysluokittain. Yleisyysluokat määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3 Haittavaikutukset
Infektiot | |
Hyvin yleiset | Ylähengitystieinfektio (mukaan lukien nielutulehdus, nenänielutulehdus, nuha) |
Yleiset | Follikuliitti, keuhkoputkitulehdus, kandidiaasi (mukaan lukien suun kandidiaasi), keuhkokuume, gastroenteriitti, virtsatieinfektio |
Melko harvinaiset | Herpesvirusinfektio, peräaukon absessi, kandidiaasi (Candida-infektio), furunkkeli, sepsis, ihonalainen absessi, jalkasilsa |
Harvinaiset | Hepatiitti B:n uudelleenaktivoituminen |
Hyvän‑ ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
Melko harvinaiset | Ihon papillooma |
Harvinaiset | Suun papillooma, paraproteinemia |
Veri ja imukudos | |
Hyvin yleiset | Anemia, trombosytopenia |
Yleiset | Leukopenia, leukosytoosi, neutropenia, trombosytoosi |
Melko harvinaiset | Eosinofilia, kuumeinen neutropenia, lymfopenia, pansytopenia |
Immuunijärjestelmä | |
Melko harvinaiset | Yliherkkyys |
Umpieritys | |
Hyvin yleiset | Kasvun hidastuminen |
Yleiset | Hypotyreoosi |
Melko harvinaiset | Hypertyreoosi |
Harvinaiset | Sekundaarinen hyperparatyreoosi, tyreoidiitti |
Aineenvaihdunta ja ravitsemus | |
Yleiset | Elektrolyyttitasapainon häiriö (mukaan lukien hypomagnesemia, hyperkalemia, hypokalemia, hyponatremia, hypokalsemia, hyperkalsemia, hyperfosfatemia), diabetes, hyperglykemia, hyperkolesterolemia, hyperlipidemia, hypertriglyseridemia, ruokahalun heikkeneminen, kihti, hyperurikemia, hypofosfatemia (mukaan lukien veren fosfaattipitoisuuden pieneneminen) |
Melko harvinaiset | Nestehukka, ruokahalun lisääntyminen, dyslipidemia, hypoglykemia |
Harvinaiset | Ruokahalun häiriöt, tuumorilyysioireyhtymä |
Psyykkiset häiriöt | |
Yleiset | Masennus, unettomuus, ahdistuneisuus |
Melko harvinaiset | Muistinmenetys, sekavuustila, desorientaatio |
Harvinaiset | Dysforia |
Hermosto | |
Hyvin yleiset | Päänsärky |
Yleiset | Huimaus, hypestesia, parestesia, migreeni |
Melko harvinaiset | Aivoverisuonitapahtuma, kallonsisäinen verenvuoto / aivoverenvuoto, iskeeminen aivohalvaus, ohimenevä aivoverenkiertohäiriö (TIA), aivoinfarkti, tajunnanmenetys (mukaan lukien pyörtyminen), vapina, tarkkaavaisuuden häiriöt, hyperestesia, dysestesia, letargia, perifeerinen neuropatia, levottomien jalkojen oireyhtymä, kasvohalvaus |
Harvinaiset | Kallonpohjavaltimon stenoosi, aivoedeema, näköhermotulehdus |
Silmät | |
Yleiset | Sidekalvotulehdus, silmien kuivuminen (mukaan lukien kseroftalmia), silmien ärsytys, (kovakalvon, sidekalvon, silmän) verekkyys, näön hämärtyminen |
Melko harvinaiset | Näön heikkeneminen, sidekalvon verenvuoto, näöntarkkuuden heikkeneminen, silmäluomien turvotus, silmäluomitulehdus, valonvälähdysten näkeminen, allerginen sidekalvotulehdus, kaksoiskuvat, silmän verenvuoto, silmäkipu, silmien kutina, silmien turvotus, silmäpinnan sairaus, silmäkuopan ympäristön turvotus, valonarkuus |
Harvinaiset | Korioretinopatia, papillödeema |
Kuulo ja tasapainoelin | |
Yleiset | Kiertohuimaus, korvakipu, tinnitus |
Melko harvinaiset | Kuulon heikkeneminen (hypoakuusi) |
Sydän | |
Yleiset | Angina pectoris, sydämen rytmihäiriöt (mukaan lukien eteis‑kammiokatkos, sydämen lepatus, sydämen kammiolisälyönnit, takykardia, eteisvärinä, bradykardia), sydämentykytys, EKG:ssä todettava QT‑ajan piteneminen, sepelvaltimotauti |
Melko harvinaiset | Sydäninfarkti, sydämen sivuääni, perikardiumeffuusio, sydämen vajaatoiminta, diastolinen toimintahäiriö, vasen haarakatkos, perikardiitti |
Harvinaiset | Syanoosi, ejektiofraktion pieneneminen |
Yleisyys tuntematon | Kammion toimintahäiriö |
Verisuonisto | |
Yleiset | Hypertensio, punastelu, perifeerinen valtimoahtaumatauti |
Melko harvinaiset | Hypertensiivinen kriisi, katkokävely, perifeerinen valtimostenoosi, verenpurkauma, arterioskleroosi, hypotensio, tromboosi |
Harvinaiset | Verenvuotosokki |
Hengityselimet, rintakehä ja välikarsina | |
Hyvin yleiset | Yskä |
Yleiset | Hengenahdistus, hengenahdistus rasituksen yhteydessä, nenäverenvuoto, suun ja nielun kipu |
Melko harvinaiset | Keuhkopöhö, pleuraeffuusio, interstitiaalinen keuhkosairaus, pleurakipu, keuhkopussitulehdus, nielun ärsytys, dysfonia, pulmonaalinen hypertensio, hengityksen vinkuminen |
Harvinaiset | Nielun ja kurkunpään kipu |
Ruoansulatuselimistö | |
Hyvin yleiset | Pahoinvointi, ylävatsakipu, ummetus, ripuli, oksentelu |
Yleiset | Haimatulehdus, epämukava tunne vatsan alueella, vatsan pullotus, ilmavaivat, vatsakipu, dyspepsia, gastriitti, ruokatorven refluksitauti, peräpukamat, suutulehdus |
Melko harvinaiset | Ruoansulatuskanavan verenvuoto, mustat ulosteet, suun haavaumat, ruokatorvikipu, suun kuivuminen, hampaiden vihlominen (hampaiden hyperestesia), makuaistin häiriöt, enterokoliitti, mahahaava, ientulehdus, palleatyrä, peräsuolen verenvuoto |
Harvinaiset | Ruoansulatuskanavan haavauman perforaatio, verioksennukset, ruokatorven haavaumat, haavainen ruokatorvitulehdus, retroperitoneaalinen verenvuoto, subileus |
Maksa ja sappi | |
Hyvin yleiset | Hyperbilirubinemia (mukaan lukien veren bilirubiinipitoisuuden suureneminen) |
Yleiset | Maksan toimintahäiriö |
Melko harvinaiset | Maksatoksisuus, toksinen hepatiitti, keltaisuus, kolestaasi, hepatomegalia |
Iho ja ihonalainen kudos | |
Hyvin yleiset | Ihottuma, kutina, hiustenlähtö |
Yleiset | Öinen hikoilu, ekseema, nokkosihottuma, voimakas hikoilu, kontuusiot, akne, dermatiitti (mukaan lukien allerginen, eksfoliatiivinen ja aknen kaltainen ihotulehdus), ihon kuivuus, eryteema |
Melko harvinaiset | Kesivä ihottuma, lääkeihottuma, ihon kipu, mustelmanmuodostus, kasvojen turvotus, rakkulat, ihokystat, erythema nodosum, hyperkeratoosi, petekiat, valoherkkyys, psoriaasi, ihon värimuutokset, ihon kesiminen, ihon hyperpigmentaatio, ihon hypertrofia, ihon haavaumat |
Harvinaiset | Erythema multiforme, käsi‑jalkaoireyhtymä, talirauhasen liikakasvu, ihoatrofia |
Luusto, lihakset ja sidekudos | |
Hyvin yleiset | Lihaskipu, nivelkipu, selkäkipu, raajakipu |
Yleiset | Rinnan luiden ja lihasten kipu, luu‑ ja lihaskipu, niskakipu, lihasheikkous, lihaskouristukset, luukipu |
Melko harvinaiset | Tuki‑ ja liikuntaelimistön jäykkyys, nivelten turvotus, niveltulehdus, kylkikipu |
Munuaiset ja virtsatiet | |
Yleiset | Tiheä virtsaamistarve, dysuria |
Melko harvinaiset | Virtsaamispakko, tihentynyt virtsaamistarve yöllä, kromaturia, verivirtsaisuus, munuaisten vajaatoiminta, virtsainkontinenssi |
Sukupuolielimet ja rinnat | |
Yleiset | Erektiohäiriöt, menorragia |
Melko harvinaiset | Rintojen kipu, gynekomastia, nännien turvotus |
Harvinaiset | Rintojen kovettumat |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleiset | Väsymys, kuume |
Yleiset | Rintakipu (mukaan lukien ei‑sydänperäinen rintakipu), kipu, epämukava tunne rinnassa, huonovointisuus, voimattomuus ja ääreisosien turvotus, vilunväreet, influenssan kaltaiset oireet |
Melko harvinaiset | Kasvojen turvotus, nesteen kertyminen alaraajoihin, kehon lämpötilan muuttumisen tunne (mukaan lukien kuumotus ja palelu), paikallinen turvotus |
Harvinaiset | Äkkikuolema |
Tutkimukset | |
Hyvin yleiset | ALAT‑arvon suureneminen, lipaasiarvojen suureneminen |
Yleiset | Hemoglobiinin lasku, suurentunut veren amylaasipitoisuus, suurentunut ASAT‑arvo, suurentunut veren alkalisen fosfataasin pitoisuus, suurentunut gammaglutamyylitransferaasipitoisuus, suurentunut veren kreatiinikinaasipitoisuus, painon lasku, painon nousu, suurentunut kreatiniinipitoisuus, suurentunut kokonaiskolesterolipitoisuus |
Melko harvinaiset | Suurentunut veren laktaattidehydrogenaasipitoisuus, suurentunut veren ureapitoisuus, suurentunut veren konjugoitumattoman bilirubiinin pitoisuus, suurentunut veren parathormonin pitoisuus, suurentunut veren triglyseridipitoisuus, pienentyneet globuliinipitoisuudet, suurentuneet kolesterolipitoisuudet (mukaan lukien LDL‑ ja HDL‑pitoisuudet), suurentunut troponiiniarvo |
Harvinaiset | Pienentynyt veren glukoosipitoisuus, pienentynyt veren insuliinipitoisuus, suurentunut veren insuliinipitoisuus, pienentynyt C‑peptidipitoisuus |
Huom. Kaikkia haittavaikutuksia ei havaittu pediatrisissa tutkimuksissa.
Valikoitujen haittavaikutusten kuvaus
Äkillinen kuolema
Melko harvinaisia (0,1 – 1 %) äkillisiä kuolemantapauksia on raportoitu potilailla, jotka osallistuivat kliinisiin Tasigna‑tutkimuksiin ja/tai erityislupakäyttö‑ohjelmiin ja joilla oli imatinibiresistentti tai ‑intolerantti kroonisen tai akseleraatiovaiheen KML ja taustalla sydänsairaus tai merkittäviä sydänsairauksien riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hepatiitti B:n uudelleenaktivoituminen
Hepatiitti B:n uudelleenaktivoitumista on ilmoitettu BCR‑ABL‑tyrosiinikinaasin estäjien käytön yhteydessä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Nilotinibin turvallisuutta kroonisen vaiheen Philadelphia‑kromosomipositiivista KML:ää sairastavien lapsipotilaiden (2 – < 18 vuoden ikäisten) hoidossa (n = 58) on arvioitu yhdessä päätutkimuksessa 60 kuukauden ajalta (ks. kohta Farmakodynamiikka). Lapsipotilailla haittavaikutusten esiintymistiheys, tyyppi ja vaikeusaste ovat yleensä vastanneet aikuisilla todettuja. Poikkeuksena ovat hyperbilirubinemia/veren bilirubiiniarvon nousu (aste 3/4: 10,3 %) ja transaminaasiarvojen suureneminen (asteen 3/4 ASAT‑arvon suureneminen: 1,7 %, asteen 3/4 ALAT‑arvon suureneminen: 12,1 %), joita ilmoitettiin useammin kuin aikuispotilailla. Bilirubiinipitoisuutta ja maksan transaminaasiarvoja on seurattava hoidon aikana (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kasvun hidastuminen pediatrisilla potilailla
Pediatrisilla KML-potilailla tehdyssä tutkimuksessa havaittiin kasvun hidastumista (vähintään kahden persentiilipääkäyrän ylittymistä kasvukäyrästöllä verrattuna lähtötilanteeseen) 8 potilaalla: viidellä (8,6 %) ylittyi kaksi persentiilipääkäyrää ja kolmella (5,2 %) ylittyi kolme persentiilipääkäyrää verrattuna lähtötilanteeseen, kun altistuksen mediaanikesto oli 51,9 kk potilailla, joilla oli äskettäin todettu kroonisessa vaiheessa oleva Philadelphia-kromosomipositiivinen KML ja 59,9 kk potilailla, joilla oli kroonisessa vaiheessa oleva Philadelphia-kromosomipositiivinen KML ja aiempi imatinibi/dasatinibihoito oli osoittautunut tehottomaksi tai imatinibihoito huonosti siedetyksi. Kasvun hidastumiseen liittyviä tapahtumia ilmoitettiin 3 potilaalla (5,2 %). On suositeltavaa seurata pediatristen potilaiden kasvua tiiviisti nilotinibihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tahallisesta nilotinibin yliannostuksesta on yksittäisiä raportteja, joissa määrittelemätön määrä kovia Tasigna‑kapseleita nieltiin yhdessä alkoholin ja muiden lääkkeiden kanssa. Potilailla esiintyi neutropeniaa, oksentelua ja uneliaisuutta. EKG‑muutoksia tai maksatoksisuutta ei raportoitu. Potilaiden raportoitiin toipuneen tapauksista.
Yliannostustapauksessa seurataan potilaan tilaa ja annetaan tarvittavaa oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, BCR‑ABL‑tyrosiinikinaasin estäjät, ATC‑koodi: L01EA03
Vaikutusmekanismi
Nilotinibi on BCR‑ABL‑onkoproteiinin ABL‑tyrosiinikinaasin potentti estäjä sekä solulinjoissa että primaarisissa Philadelphia‑kromosomipositiivisissa leukemiasoluissa. Se sitoutuu suurella affiniteetilla ATP:n sitoutumiskohtiin ja estää voimakkaasti villin tyypin BCR‑ABL:n toimintaa ja tehoaa 32:een 33:sta imatinibiresistentistä BCR‑ABL‑mutaatiotyypistä. Tämän biokemiallisen vaikutuksen ansiosta nilotinibi estää selektiivisesti leukemiasolujen lisääntymistä ja indusoi apoptoosia sekä solulinjoissa että KML‑potilaiden primaarisissa Philadelphia‑kromosomipositiivisissa leukemiasoluissa. KML‑hiirimalleissa ainoana lääkkeenä suun kautta annettu nilotinibi vähensi kasvaintaakkaa ja pidensi elinaikaa.
Farmakodynaamiset vaikutukset
Nilotinibi ei vaikuta lainkaan tai juuri lainkaan useimpiin muihin tutkittuihin proteiinikinaaseihin mukaan lukien Src. Poikkeuksia tästä ovat PDGF‑, KIT‑ ja efriini‑reseptorikinaasit, joita Tasigna estää pitoisuuksilla, jotka saavutetaan KML:n hoitoon suositelluilla peroraalisilla hoitoannoksilla (ks. taulukko 4).
Taulukko 4 Nilotinibin kinaasiprofiili (fosforylaatio IC50 nM)
BCR‑ABL | PDGFR | KIT |
20 | 69 | 210 |
Kliininen teho
Äskettäin diagnosoidun kroonisen vaiheen KML:n kliiniset tutkimukset
Nilotinibin tehon määrittämiseksi imatinibiin verrattuna toteutettiin avoin, satunnaistettu vaiheen III monikeskustutkimus, johon osallistuneilla 846 aikuispotilaalla oli sytogeneettisesti vahvistettu äskettäin diagnosoitu Philadelphia‑kromosomipositiivinen kroonisen vaiheen KML. Potilaat oli diagnosoitu enimmillään 6 kk tutkimusta ennen, eivätkä he olleet saaneet aiempaa hoitoa (paitsi hydroksiureaa ja/tai anagrelidia). Potilaat satunnaistettiin suhteessa 1:1:1 saamaan joko nilotinibia 300 mg kahdesti vuorokaudessa (n=282), nilotinibia 400 mg kahdesti vuorokaudessa (n=281) tai imatinibia 400 mg kerran vuorokaudessa (n=283). Satunnaistamisen yhteydessä tutkimushenkilöt stratifioitiin diagnoosihetken Sokal‑riskipisteiden mukaisesti.
Lähtötilanteessa hoitoryhmät olivat hyvin vertailukelpoisia keskenään. Mediaani‑ikä oli 47 vuotta molemmissa nilotinibiryhmissä ja 46 vuotta imatinibiryhmässä, ja ≥ 65‑vuotiaiden osuus oli nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä 12,8 %, nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä 10,0 % ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä 12,4 %. Miespotilaita oli hiukan enemmän kuin naisia (miehiä oli nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä 56,0 %, nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä 62,3 % ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä 55,8 %). Yli 60 % kaikista potilaista oli valkoihoisia ja 25 % kaikista potilaista oli aasialaisia.
Ensisijainen analyysi suoritettiin, kun kaikki 846 potilasta oli ollut hoidossa 12 kk:n ajan (tai lopettaneet aiemmin). Myöhempiin analyyseihin otettiin mukaan potilaat, jotka suorittivat 24, 36, 48, 60 ja 72 kk pituisen hoidon loppuun (tai keskeyttivät hoidon tätä aiemmin). Hoidon keston mediaani oli noin 70 kk kaikissa nilotinibihoitoryhmissä ja 64 kk imatinibiryhmässä. Todellisen annoksen mediaani oli nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä 593 mg/vrk, nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä 772 mg/vrk ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä 400 mg/vrk. Tämä tutkimus jatkuu edelleen.
Ensisijainen tehon päätetapahtuma oli huomattava molekulaarinen vaste (MMR) 12 kk:n kohdalla. MMR:n määritelmänä oli ≤ 0,1 % BCR‑ABL/ABL % kansainvälisellä asteikolla RQ‑PCR‑menetelmällä mitattuna, mikä vastaa BCR‑ABL‑transkriptien määrän vähenemistä ≥ 3 logaritmiyksikön verran standardoidusta lähtötilanteesta. Nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmän MMR‑vaste 12 kk:n kohdalla oli tilastollisesti merkitsevästi suurempi kuin imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä (44,3 % vs 22,3 %, p<0,0001). Myös nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmän MMR‑vaste 12 kk:n kohdalla oli tilastollisesti merkitsevästi suurempi kuin imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä (42,7 % vs 22,3 %, p<0,0001).
MMR‑vasteet 3, 6, 9 ja 12 kk:n kohdalla olivat nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä 8,9 %, 33,0 %, 43,3 % ja 44,3 %, nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä 5,0 %, 29,5 %, 38,1 % ja 42,7 % ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä 0,7 %, 12,0 %, 18,0 % ja 22,3 %.
Taulukossa 5 esitetään MMR‑vasteet 12, 24, 36, 48, 60 ja 72 kk kohdalla.
Taulukko 5 MMR‑vasteprosentti
Nilotinibi 300 mg x 2 n = 282 (%) | Nilotinibi 400 mg x 2 n = 281 (%) | Imatinibi 400 mg x 1 n = 283 (%) | |
MMR 12 kk kohdalla | |||
Vaste (95 % lv) | 44,31 (38,4–50,3) | 42,71 (36,8–48,7) | 22,3 (17,6–27,6) |
MMR 24 kk kohdalla | |||
Vaste (95 % lv) | 61,71 (55,8–67,4) | 59,11 (53,1–64,9) | 37,5 (31,8–43,4) |
MMR 36 kk kohdalla2 | |||
Vaste (95 % lv) | 58,51 (52,5–64,3) | 57,31 (51,3–63,2) | 38,5 (32,8–44,5) |
MMR 48 kk kohdalla3 | |||
Vaste (95 % lv) | 59,91 (54,0–65,7) | 55,2 (49,1–61,1) | 43,8 (38,0–49,8) |
MMR 60 kk kohdalla4 | |||
Vaste (95 % lv) | 62,8 (56,8–68,4) | 61,2 (55,2–66,9) | 49,1 (43,2–55,1) |
MMR 72 kk kohdalla5 | |||
Vaste (95 % lv) | 52,5 (46,5–58,4) | 57,7 (51,6–63,5) | 41,7 (35,9–47,7) |
1 Cochran–Mantel–Haenszelin testin p‑arvo vasteisiin (vs imatinibi 400 mg) < 0,0001
2 Kunkin ajankohdan osalta vasteen saavuttaneiksi lasketaan vain kyseisellä hetkellä MMR‑vastealueella olleet potilaat. Yhteensä 199 potilaan (35,2 % kaikista potilaista) MMR‑vastetta ei voitu arvioida 36 kk kohdalla (87 potilasta nilotinibi 300 mg x 2 ‑ryhmästä ja 112 potilasta imatinibiryhmästä); syynä olivat puuttuvat/arviointikelvottomat PCR‑määritystulokset (n = 17), epätyypilliset transkriptit lähtötilanteessa (n = 7) tai tutkimuksen keskeyttäminen ennen 36 kk ajankohtaa (n = 175).
3 Kunkin ajankohdan osalta vasteen saavuttaneiksi lasketaan vain kyseisellä hetkellä MMR‑vastealueella olleet potilaat. Yhteensä 305 potilaan (36,1 % kaikista potilaista) MMR‑vastetta ei voitu arvioida 48 kk kohdalla (98 potilasta nilotinibi 300 mg x 2 ‑ryhmästä, 88 potilasta nilotinibi 400 mg x 2 ‑ryhmästä ja 119 potilasta imatinibiryhmästä); syynä olivat puuttuvat/arviointikelvottomat PCR‑määritystulokset (n = 18), epätyypilliset transkriptit lähtötilanteessa (n = 8) tai tutkimuksen keskeyttäminen ennen 48 kk ajankohtaa (n = 279).
4 Kunkin ajankohdan osalta vasteen saavuttaneiksi lasketaan vain kyseisellä hetkellä MMR‑vastealueella olleet potilaat. Yhteensä 322 potilaan (38,1 % kaikista potilaista) MMR‑vastetta ei voitu arvioida 60 kk kohdalla (99 potilasta nilotinibi 300 mg x 2 ‑ryhmästä, 93 potilasta nilotinibi 400 mg x 2 ‑ryhmästä ja 130 potilasta imatinibiryhmästä); syynä olivat puuttuvat/arviointikelvottomat PCR‑määritystulokset (n = 9), epätyypilliset transkriptit lähtötilanteessa (n = 8) tai tutkimuksen keskeyttäminen ennen 60 kk ajankohtaa (n = 305).
5 Kunkin ajankohdan osalta vasteen saavuttaneiksi lasketaan vain kyseisellä hetkellä MMR‑vastealueella olleet potilaat. Yhteensä 395 potilaan (46,7 % kaikista potilaista) MMR‑vastetta ei voitu arvioida 72 kk kohdalla (130 potilasta nilotinibi 300 mg x 2 ‑ryhmästä, 110 potilasta nilotinibi 400 mg x 2 ‑ryhmästä ja 155 potilasta imatinibiryhmästä); syynä olivat puuttuvat/arviointikelvottomat PCR‑määritystulokset (n = 25), epätyypilliset transkriptit lähtötilanteessa (n = 8) tai tutkimuksen keskeyttäminen ennen 72 kk ajankohtaa (n = 362).
MMR‑vasteet eri ajankohtiin mennessä (vasteen saavuttaneiksi lasketaan potilaat, jotka saavuttivat MMR‑vasteen kyseisenä ajankohtana tai sitä ennen) esitetään MMR‑vasteen kumulatiivisena ilmaantuvuutena (ks. kuva 1).
Kuva 1 MMR‑vasteen kumulatiivinen ilmaantuvuus
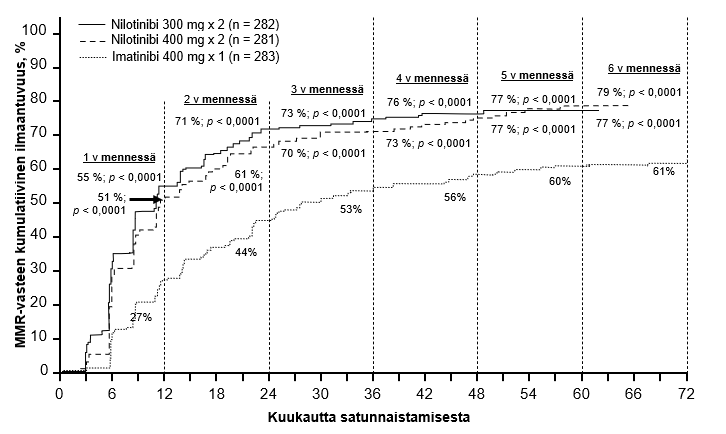
Kaikkia Sokal‑riskiryhmiä tarkasteltaessa kaikkien ajankohtien MMR‑vasteet olivat molemmissa nilotinibiryhmissä johdonmukaisesti suuremmat kuin imatinibiryhmässä.
Retrospektiivisessä analyysissä 91 prosenttia (234/258) 300 mg nilotinibia kahdesti vuorokaudessa saaneista potilaista oli kolmen kuukauden hoidon jälkeen saavuttanut BCR‑ABL‑tason ≤ 10 % verrattuna 67 prosenttiin (176/264) 400 mg imatinibia kerran vuorokaudessa saaneista potilaista. 72 kuukauden kohdalla kokonaiselossaololuvut olivat paremmat niillä potilailla, jotka kolmen kuukauden hoidon jälkeen olivat saavuttaneet BCR‑ABL‑tason ≤ 10 % verrattuna potilaisiin, jotka eivät olleet saavuttaneet tätä molekulaarisen vasteen tasoa [94,5 % vs 77,1 % (p = 0,0005)].
Ensimmäiseen MMR‑vasteeseen kuluneen ajan Kaplan–Meier‑analyysin perusteella MMR‑vaste saavutettiin eri ajankohtina todennäköisemmin sekä nilotinibia 300 mg että 400 mg kahdesti vuorokaudessa saavien ryhmissä kuin imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä (nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmän välinen riskisuhde = 2,17 ja stratifioitu log‑rank p<0,0001; nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia 400 mg kerran vuorokaudessa saavien ryhmän välinen riskisuhde = 1,88 ja stratifioitu log‑rank p<0,0001).
Taulukossa 6 esitetään niiden potilaiden osuus, joiden molekulaarinen vaste oli kansainvälisellä asteikolla ≤ 0,01 % ja ≤ 0,0032 % eri ajankohtien kohdalla. Kuvissa 2 ja 3 puolestaan esitetään niiden potilaiden osuus, joiden molekulaarinen vaste oli kansainvälisellä asteikolla ≤ 0,01 % ja ≤ 0,0032 % eri ajankohtiin mennessä. ≤ 0,01 % molekulaarinen vaste kansainvälisellä asteikolla vastaa BCR‑ABL‑transkriptien ≥ 4 log vähenemistä standardoidusta lähtötilanteesta ja ≤ 0,0032 % molekulaarinen vaste vastaa ≥ 4,5 log vähenemistä.
Taulukko 6 ≤ 0,01 % molekulaarisen vasteen (4 log vähenemisen) ja ≤ 0,0032 % molekulaarisen vasteen (4,5 log vähenemisen) saavuttaneiden potilaiden osuus
Nilotinibi 300 mg x 2 n = 282 (%) | Nilotinibi 400 mg x 2 n = 281 (%) | Imatinibi 400 mg x 1 n = 283 (%) | ||||
≤ 0,01 % | ≤ 0,0032 % | ≤ 0,01 % | ≤ 0,0032 % | ≤ 0,01 % | ≤ 0,0032 % | |
12 kk kohdalla | 11,7 | 4,3 | 8,5 | 4,6 | 3,9 | 0,4 |
24 kk kohdalla | 24,5 | 12,4 | 22,1 | 7,8 | 10,2 | 2,8 |
36 kk kohdalla | 29,4 | 13,8 | 23,8 | 12,1 | 14,1 | 8,1 |
48 kk kohdalla | 33,0 | 16,3 | 29,9 | 17,1 | 19,8 | 10,2 |
60 kk kohdalla | 47,9 | 32,3 | 43,4 | 29,5 | 31,1 | 19,8 |
72 kk kohdalla | 44,3 | 31,2 | 45,2 | 28,8 | 27,2 | 18,0 |
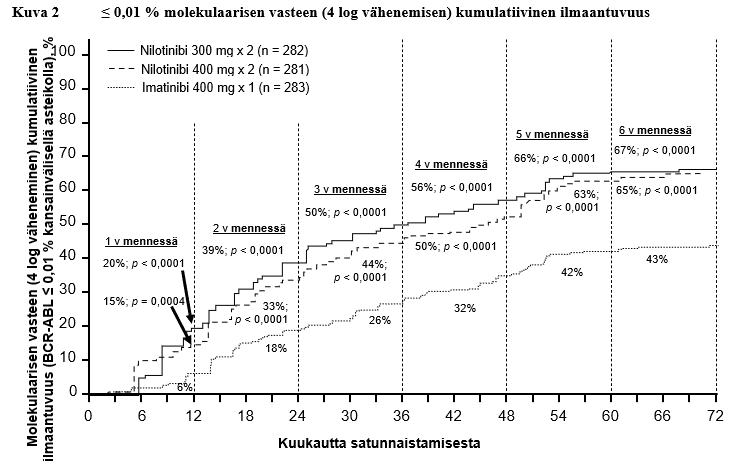
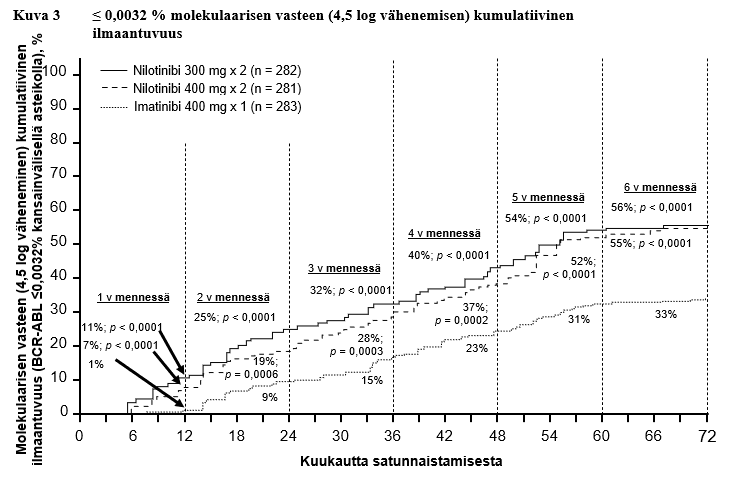
Ensimmäisen MMR‑vasteen kestoa koskevien Kaplan‑Meier ‑laskelmien perusteella MMR‑vasteen säilyttäneiden potilaiden osuudet niistä potilaista, jotka saavuttivat MMR‑vasteen, olivat 72 kk:n kohdalla 92,5 % (luottamusväli 95 %: 88,6‑96,4 %) nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 92,2 % (luottamusväli 95 %: 88,5‑95,9 %) nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 88,0 % (luottamusväli 95 %: 83,0‑93,1 %) imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä.
Täydellisen sytogeneettisen vasteen (CCyR) määritelmänä oli 0 % Ph‑positiivisia metafaaseja luuytimessä perustuen vähintään 20 arvioituun metafaasiin. Paras CCyR‑vaste 12 kk:n kuluessa (ml. potilaat, jotka saavuttivat CCyR‑vasteen 12 kk:n kohdalla tai sitä ennen vastaten hoitoon) oli tilastollisesti suurempi molemmissa nilotinibiryhmissä (300 ja 400 mg kahdesti vuorokaudessa) kuin imatinibiryhmässä (400 mg kerran vuorokaudessa), ks. taulukko 7.
CCyR‑vaste 24 kk:een mennessä (mukaan lukien potilaat, jotka saavuttivat CCyR‑vasteen 24 kk kohdalla tai sitä ennen) oli tilastollisesti merkitsevästi suurempi sekä nilotinibia 300 mg kahdesti vuorokaudessa että 400 mg kahdesti vuorokaudessa saavien ryhmässä verrattuna imatinibia 400 mg kerran vuorokaudessa saavien ryhmään.
Taulukko 7 Paras CCyR‑vasteprosentti
Nilotinibi 300 mg x 2 n = 282 (%) | Nilotinibi 400 mg x 2 n = 281 (%) | Imatinibi 400 mg x 1 n = 283 (%) | |
12 kk:n kohdalla | |||
Vaste (95 % lv) | 80,1 (75,0; 84,6) | 77,9 (72,6; 82,6) | 65,0 (59,2; 70,6) |
Ei vastetta | 19,9 | 22,1 | 35,0 |
CMH‑testin vasteprosentin p‑arvo (vs imatinibi 400 mg kerran vuorokaudessa) | < 0,0001 | 0,0005 | |
24 kk:n kohdalla | |||
Vaste (95 % lv) | 86,9 (82,4; 90,6) | 84,7 (79,9; 88,7) | 77,0 (71,7; 81,8) |
Ei vastetta | 13,1 | 15,3 | 23,0 |
CMH‑testin vasteprosentin p‑arvo (vs imatinibi 400 mg kerran vuorokaudessa) | 0,0018 | 0,0160 |
Kaplan‑Meier ‑laskelmien perusteella CCyR‑vasteen säilyttäneiden potilaiden osuudet niistä potilaista, jotka saavuttivat vasteen, olivat 72 kk:n kohdalla 99,1 % (luottamusväli 95 %: 97,9–100 %) nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 98,7 % (luottamusväli 95 %: 97,1–100 %) nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 97,0 % (luottamusväli 95 %: 94,7–99,4 %) imatinibia kerran vuorokaudessa saavien ryhmässä.
Taudin eteneminen akseleraatiovaiheeseen tai blastikriisivaiheeseen määriteltiin satunnaistamispäivästä ensimmäiseen dokumentoituun akseleraatiovaiheeseen tai blastikriisivaiheeseen etenemiseen tai KML:n aiheuttamaan kuolemaan kuluneeksi ajaksi. Tauti eteni hoidon aikana akseleraatiovaiheeseen tai blastikriisivaiheeseen yhteensä 17 potilaalla: 2 potilaalla nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 3 potilaalla nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 12 potilaalla imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä. Niiden potilaiden arvioidut osuudet, joiden tauti ei ole edennyt akseleraatiovaiheeseen tai blastikriisivaiheeseen 72 kk:n kohdalla, olivat edellä mainituissa ryhmissä 99,3 %, 98,7 % ja 95,2 % (nilotinibi 300 mg x 2 vs imatinibi x 1: riskisuhde = 0,1599 ja p = 0,0059 stratifioidulla log‑rank‑testillä; nilotinibi 400 mg x 2 vs imatinibi x 1: riskisuhde = 0,2457 ja p = 0,0185 stratifioidulla log‑rank‑testillä). Kahden vuoden kohdalla suoritetun analyysin jälkeen ei hoitojen aikana raportoitu yhtään uutta taudin etenemistapausta akseleraatio‑ tai blastikriisivaiheeseen.
Kun taudin etenemisen kriteereihin laskettiin mukaan klonaalinen kehitys, yhteensä 25 hoidetun potilaan tauti eteni akseleraatiovaiheeseen tai blastikriisivaiheeseen raja‑arvona käytettyyn päivään mennessä (3 nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 5 nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 17 imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä). Niiden potilaiden arvioidut osuudet, joiden tauti ei 72 kk:n kohdalla ole edennyt akseleraatiovaiheeseen tai blastikriisivaiheeseen mukaan lukien klonaalinen kehitys, olivat edellä mainituissa ryhmissä 98,7 %, 97,9 % ja 93,2 % (nilotinibi 300 mg x 2 vs imatinibi x 1: riskisuhde = 0,1626 ja p = 0,0009 stratifioidulla log‑rank‑testillä; nilotinibi 400 mg x 2 vs imatinibi x 1: riskisuhde = 0,2848 ja p = 0,0085 stratifioidulla log‑rank‑testillä).
Yhteensä 55 potilasta kuoli hoidon tai hoidon lopettamisen jälkeisen seurantavaiheen aikana (21 potilasta nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 11 potilasta nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 23 potilasta imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä). Näistä 55 potilaasta 26:n kuolema liittyi KML:ään (6 tapausta nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmässä, 4 tapausta nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmässä ja 16 tapausta imatinibia 400 mg kerran vuorokaudessa saavien ryhmässä). Arvioidut elossa olevien potilaiden osuudet 72 kk:n kohdalla olivat näissä ryhmissä 91,6 %, 95,8 % ja 91,4 % (riskisuhde = 0,8934 ja p = 0,7085 stratifioidulla log‑rank‑testillä nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia saavien ryhmän välillä, riskisuhde = 0,4632 ja p = 0,0314 stratifioidulla log‑rank‑testillä nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia saavien ryhmän välillä). Kun ainoastaan KML:ään liittyvät kuolemat lasketaan tapahtumiksi, arvioidut kokonaiselossaololuvut 72 kk:n kohdalla olivat näissä ryhmissä 97,7 %, 98,5 % ja 93,9 % (riskisuhde = 0,3694 ja p = 0,0302 stratifioidulla log‑rank‑testillä nilotinibia 300 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia saavien ryhmän välillä, riskisuhde = 0,2433 ja p = 0,0061 stratifioidulla log‑rank‑testillä nilotinibia 400 mg kahdesti vuorokaudessa saavien ryhmän ja imatinibia saavien ryhmän välillä).
Kroonisen ja akseleraatiovaiheen imatinibiresistentin tai ‑intolerantin KML:n kliiniset tutkimukset
Avoimessa, kontrolloimattomassa vaiheen II monikeskustutkimuksessa selvitettiin nilotinibin tehoa aikuisilla KML‑potilailla, jotka olivat joko resistenttejä imatinibille tai eivät sietäneet sitä. Kroonisen vaiheen KML:lle ja akseleraatiovaiheen KML:lle oli erilliset hoitoryhmät. Tehokkuusmääritykset perustuivat 321 kroonisessa vaiheessa olevaan potilaaseen ja 137 akseleraatiovaiheessa olevaan potilaaseen. Hoidon keston mediaani oli kroonisen vaiheen potilailla 561 vuorokautta ja akseleraatiovaiheen potilailla 264 vuorokautta (ks. taulukko 8). Tasignaa annettiin säännöllisesti (kahdesti vuorokaudessa 2 tuntia aterian jälkeen; potilaat olivat syömättä vähintään tunnin ajan lääkkeenannon jälkeen), ellei potilaalle kehittynyt merkkejä vasteen riittämättömyydestä tai taudin etenemisestä. Annos oli 400 mg kahdesti vuorokaudessa, ja sen sai tarvittaessa suurentaa 600 mg:aan kahdesti vuorokaudessa.
Taulukko 8 Nilotinibialtistuksen kesto
Krooninen vaihe n=321 | Akseleraatiovaihe n=137 | |
Hoidon keston mediaani vuorokausina (25.–75. persentiilit) | 561 (196‑852) | 264 (115‑595) |
Imatinibiresistenteiksi katsottiin potilaat, jotka eivät saavuttaneet täydellistä hematologista vastetta (3 kuukauden kuluessa), sytogeneettistä vastetta (6 kuukauden kuluessa) tai huomattavaa sytogeneettistä vastetta (12 kuukauden kuluessa) tai joiden tauti eteni aiemman sytogeneettisen tai hematologisen vasteen saavuttamisen jälkeen. Imatinibi‑intoleranteiksi katsottiin potilaat, jotka keskeyttivät imatinibihoidon toksisuuden vuoksi eivätkä olleet saavuttaneet huomattavaa sytogeneettistä vastetta tutkimuksen aloitusvaiheessa.
Kaiken kaikkiaan 73 % potilaista oli imatinibiresistenttejä ja 27 % potilaista ei sietänyt imatinibia. Potilaiden KML‑tautihistoria oli yleensä pitkä, ja useimmat potilaat olivat saaneet lukuisia muita syöpälääkkeitä kuten imatinibia, hydroksiureaa ja interferonia. Joillekin oli jopa tehty tulokseton kantasolujen siirto (taulukko 9). Suurimman aiemmin käytetyn imatinibiannoksen mediaani oli 600 mg/vrk. Suurin aiemmin käytetty imatinibiannos oli ≥ 600 mg/vrk 74 %:lla kaikista potilaista, ja 40 % potilaista oli saanut imatinibia annoksena ≥ 800 mg/vrk.
Taulukko 9 KML‑sairaushistorian ominaispiirteet
Krooninen vaihe (n=321) | Akseleraatiovaihe (n=137)* | |
Diagnoosista kuluneen ajan mediaani (kk) (vaihteluväli) | 58 (5‑275) | 71 (2‑298) |
Imatinibi Resistenssi Intoleranssi, ilman huomattavaa sytogeneettistä vastetta | 226 (70 %) 95 (30 %) | 109 (80 %) 27 (20 %) |
Imatinibihoidon keston mediaani (vrk) (25.–75. persentiilit) | 975 (519‑1 488) | 857 (424‑1 497) |
Aiempi hydroksiureahoito | 83 % | 91 % |
Aiempi interferonihoito | 58 % | 50 % |
Aiempi luuydinsiirto | 7 % | 8 % |
*Yhden potilaan tiedot imatinibiresistenssistä/‑intoleranssista puuttuvat.
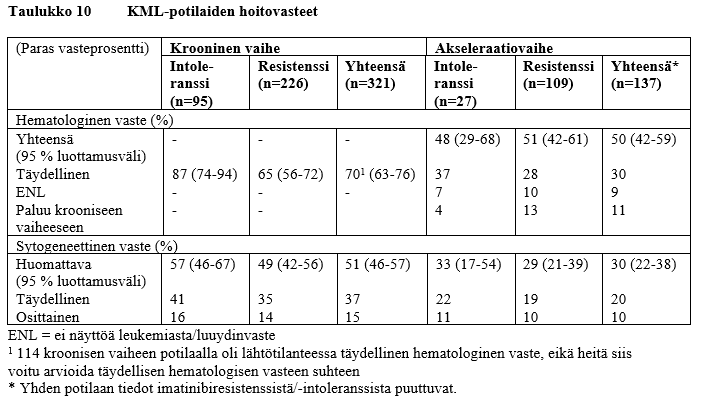
Kroonisen vaiheen KML:ää sairastavilla potilailla ensisijainen päätetapahtuma oli huomattava sytogeneettinen vaste (MCyR), joka määriteltiin Ph+ hematopoieettisten solujen häviämiseksi (CCyR, täydellinen sytogeneettinen vaste) tai merkitseväksi vähenemiseksi tasolle < 35 % Ph+ metafaaseja (osittainen sytogeneettinen vaste). Kroonisen vaiheen potilaiden täydelliset hematologiset vasteet (CHR) arvioitiin toissijaisena päätetapahtumana. Akseleraatiovaiheen KML:ää sairastavilla potilailla ensisijainen päätetapahtuma oli varmistettu hematologinen kokonaisvaste (HR), joka määriteltiin joko täydelliseksi hematologiseksi vasteeksi, leukemian merkkien puuttumiseksi tai krooniseen vaiheeseen palaamiseksi.
Krooninen vaihe
Kroonisen vaiheen KML:ää sairastavista 321 potilaasta 51 % saavutti huomattavan sytogeneettisen vasteen. Suurin osa nilotinibihoitoon vastanneista potilaista saavutti huomattavan sytogeneettisen vasteen nopeasti 3 kuukauden kuluessa (mediaani 2,8 kuukautta) nilotinibihoidon aloittamisesta, ja nämä vasteet ovat olleet pitkäkestoisia. Ajan mediaani täydellisen sytogeneettisen vasteen saavuttamiseen oli hieman yli 3 kuukautta (mediaani 3,4 kuukautta). 77 %:lla (95 % luottamusväli: 70‑84 %) huomattavan sytogeneettisen vasteen saavuttaneista potilaista oli vaste edelleen 24 kuukauden kuluttua. Huomattavan sytogeneettisen vasteen mediaanikestoa ei ole vielä saavutettu. 85 %:lla (95 % luottamusväli: 78‑93 %) täydellisen sytogeneettisen vasteen saavuttaneista potilaista oli vaste edelleen 24 kuukauden kuluttua. Täydellisen sytogeneettisen vasteen mediaanikestoa ei ole vielä saavutettu. Potilaat, joilla oli lähtötilanteessa täydellinen hematologinen vaste, saavuttivat huomattavan sytogeneettisen vasteen muita nopeammin (1,9 kuukautta vs 2,8 kuukautta). Niistä kroonisen vaiheen KML:ää sairastavista potilaista, joilla ei ollut lähtötilanteessa täydellistä hematologista vastetta, 70 % saavutti sen myöhemmin. Täydellisen hematologisen vasteen saavuttamiseen kuluneen ajan mediaani oli 1 kuukausi ja vasteen mediaanikesto 32,8 kuukautta. Kroonisen vaiheen KML:ää sairastavien potilaiden arvioitu kokonaiselossaololuku 24 kuukauden hoidon jälkeen oli 87 %.
Akseleraatiovaihe
Akseleraatiovaiheen KML:ää sairastavista 137 potilaasta 50 %:n varmistettiin saavuttaneen hematologisen vasteen. Suurin osa nilotinibihoitoon vastanneista potilaista saavutti hematologisen vasteen nopeasti (mediaani 1,0 kuukautta), ja nämä vasteet ovat olleet pitkäkestoisia (varmistetun hematologisen vasteen mediaanikesto oli 24,2 kuukautta). 53 %:lla (95 % luottamusväli: 39‑67 %) hematologisen vasteen saavuttaneista potilaista oli vaste edelleen 24 kuukauden kuluttua. 30 % potilaista saavutti huomattavan sytogeneettisen vasteen, ja vasteen saavuttamiseen kuluneen ajan mediaani oli 2,8 kuukautta. 63 %:lla (95 % luottamusväli: 45‑80 %) huomattavan sytogeneettisen vasteen saavuttaneista oli vaste edelleen 24 kuukauden kuluttua. Huomattavan sytogeneettisen vasteen mediaanikesto oli 32,7 kuukautta. Akseleraatiovaiheen KML:ää sairastavien potilaiden arvioitu kokonaiselossaololuku 24 kuukauden hoidon jälkeen oli 70 %.
Kahden hoitoryhmän hoitovaste on esitetty taulukossa 10.
Tutkimustuloksia tehosta blastikriisivaiheessa Philadelphia‑kromosomipositiivista kroonista myelooista leukemiaa sairastavilla potilailla ei ole vielä saatavilla. Vaiheen II tutkimuksessa oli erilliset hoitoryhmät myös Tasignan tutkimiseksi sellaisilla kroonisen vaiheen ja akseleraatiovaiheen KML:ää sairastavilla potilailla, jotka olivat saaneet useita aiempia hoitoja (mm. tyrosiinikinaasin estäjiä) imatinibin lisäksi. Näistä potilaista 30/36 (83 %) oli hoitoresistenttejä, mutta ei intolerantteja. Nilotinibin tehoa arvioitiin 22:lla kroonisen vaiheen KML:ää sairastavalla potilaalla, ja huomattava sytogeneettinen vaste saavutettiin 32 %:lla ja täydellinen hematologinen vaste 50 %:lla potilaista. Nilotinibin tehoa arvioitiin 11:llä akseleraatiovaiheen KML:ää sairastavalla potilaalla, ja hematologinen vaste saavutettiin 36 %:lla potilaista.
42 %:lta kroonisen vaiheen ja 54 %:lta akseleraatiovaiheen KML:ää sairastavista potilaista, jotka otettiin mukaan mutaatiomäärityksiin, löytyi imatinibihoidon epäonnistumisen jälkeen 24 erilaista BCR‑ABL‑mutaatiota. Tasigna osoittautui tehokkaaksi potilailla, joilla oli erilaisia imatinibiresistenssiä aiheuttavia BCR‑ABL‑mutaatioita T315I‑mutaatiota lukuun ottamatta.
Hoidon lopetus kroonisessa vaiheessa olevaa Philadelphia‑kromosomipositiivista KML‑leukemiaa sairastavilla aikuispotilailla, jotka ovat saaneet nilotinibia ensilinjan hoitona ja jotka ovat saavuttaneet pitkäkestoisen syvän molekulaarisen vasteen
Avoimeen, yksiryhmäiseen tutkimukseen otettiin 215 aikuispotilasta, joilla oli kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen KML ja jotka olivat saaneet nilotinibia ensilinjan hoitona ≥ 2 vuoden ajan ja saavuttaneet MolecularMD MRDx BCR‑ABL ‑testillä mitatun MR4,5‑vasteen. Tutkimuksessa he jatkoivat nilotinibihoitoa vielä 52 viikon ajan (nilotinibihoidon vakautushoitovaihe). 190 näistä 215 potilaasta (88,4 %) siirtyi TFR‑vaiheeseen saavutettuaan vakautushoitovaiheessa pitkäkestoisen syvän molekulaarisen vasteen, joka määriteltiin seuraavasti:
- edeltävissä neljässä kvartaalimäärityksessä (jotka tehtiin 12 viikon välein) todettiin vähintään MR4,0‑tason vaste (BCR‑ABL/ABL ≤ 0,01 % IS), joka säilyi yhden vuoden ajan
- viimeisin määritystulos oli MR4,5 (BCR‑ABL/ABL ≤ 0,0032 % IS)
- enintään kaksi määritystulosta välillä MR4,0–MR4,5 (0,0032 % IS < BCR‑ABL/ABL ≤ 0,01 % IS).
Ensisijainen päätetapahtuma oli niiden potilaiden prosenttiosuus, joilla oli MMR‑vaste 48 viikon kuluttua hoitovapaan remissiovaiheen aloittamisesta (kaikki potilaat, joiden hoito jouduttiin aloittamaan uudelleen, katsottiin vastetta saavuttamattomiksi).
Taulukko 11 Hoitovapaa remissiovaihe (TFR) ensilinjan nilotinibihoidon jälkeen
TFR-vaiheeseen siirtyneet potilaat | 190 | |
Viikot TFR-vaiheen aloituksesta | 48 viikkoa | 264 viikkoa |
Vähintään MMR-vasteen säilyttäneet potilaat | 98 (51,6 %, [95 % lv: 44,2–58,9]) | 79[2] (41,6 %, 95 % lv: 34,5–48,9) |
TFR-vaiheen lopettaneet potilaat | 93[1] | 109 |
Syynä MMR-vasteen menettäminen | 88 (46,3 %) | 94 (49,5 %) |
Muu syy | 5 | 15 |
MMR-vasteen menettämisen jälkeen hoidon uudelleen aloittaneet potilaat | 86 | 91 |
MMR-vasteen uudelleen saavuttaneet potilaat | 85 (98,8 %) | 90 (98,9 %) |
MR4,5-vasteen uudelleen saavuttaneet potilaat | 76 (88,4%) | 84 (92,3 %) |
[1] Yksi potilas ei menettänyt MMR-vastetta viikkoon 48 mennessä mutta lopetti TFR-vaiheen.
[2] Kahden potilaan osalta PCR-määritystulokset eivät olleet saatavilla viikolla 264, eikä heidän vastettaan siksi huomioitu tiedonkeruun katkaisun aikaan viikolla 264.
Vähintään 50 % kaikista uudelleenhoidetuista potilaista saavutti uudelleen MMR-vasteen 7 viikossa ja MR4,5-vasteen 12,9 viikossa. Uudelleen saavutetun MMR:n kumulatiivinen vasteprosentti viikolla 24 hoidon uudelleenaloituksen jälkeen oli 97,8 % (89 potilasta 91:stä) ja MR4,5:n osalta viikolla 48 vastaavasti 91,2 % (83 potilasta 91:stä).
Kaplan–Meier‑estimoitu hoitovapaan elossaoloajan mediaani oli 120,1 viikkoa (95 % lv: 36,9–ei arvioitavissa) (kuva 4); 91 potilaalla 190:stä (47,9 %) ei ollut hoitovapaaseen elossaoloon liittyvää tapahtumaa.
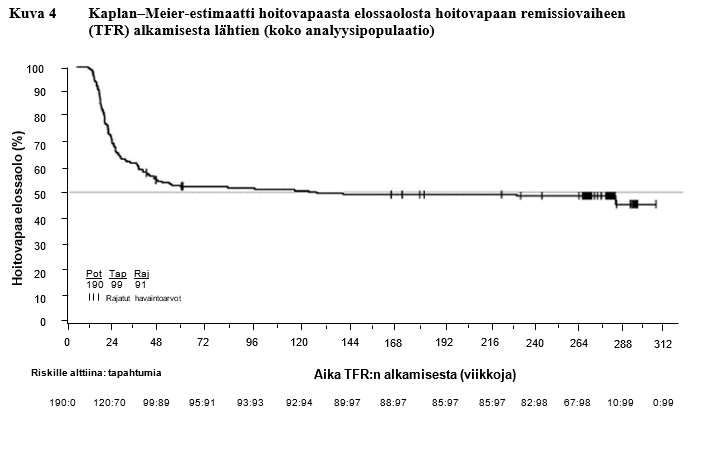
Hoidon lopetus aikuisilla kroonisessa vaiheessa olevilla KML‑potilailla, jotka ovat saavuttaneet nilotinibihoidolla pitkäkestoisen syvän molekulaarisen vasteen (MR4,5) aiemman imatinibihoidon jälkeen
Avoimeen, yksiryhmäiseen tutkimukseen otettiin 163 aikuispotilasta, joilla oli kroonisessa vaiheessa oleva Philadelphia‑kromosomipositiivinen KML, jotka olivat käyttäneet tyrosiinikinaasin estäjiä ≥3 vuoden ajan (imatinibi ensimmäisenä tyrosiinikinaasin estäjähoitona yli 4 viikon ajan ilman imatinibihoidolla saavutettua dokumentoitua MR4,5‑vastetta potilaan siirtyessä nilotinibihoitoon, sitten siirtyminen nilotinibiin vähintään 2 vuodeksi) ja jotka saavuttivat nilotinibihoidolla MolecularMD MRDx BCR‑ABL ‑testillä mitatun MR4,5‑vasteen. Tutkimuksessa potilaiden nilotinibihoitoa jatkettiin vielä 52 viikkoa (nilotinibihoidon vakautushoitovaihe). 126 näistä 163 potilaasta (77,3 %) siirtyi hoitovapaan remission vaiheeseen saavutettuaan vakautushoitovaiheessa pitkäkestoisen syvän molekulaarisen vasteen, joka määriteltiin seuraavasti:
- Edeltävissä neljässä kvartaalimäärityksessä (jotka tehtiin 12 viikon välein) ei todettu vahvistettua MR4,5‑vasteen menetystä (BCR‑ABL/ABL ≤ 0,0032 % IS) yhden vuoden aikana.
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka eivät menettäneet vahvistetusti MR4,0‑vastetta eivätkä menettäneet MMR‑vastetta 48 viikon kuluessa hoidon lopetuksen jälkeen.
Taulukko 12 Hoitovapaa remissiovaihe (TFR) imatinibihoitoa seuranneen nilotinibihoidon jälkeen
TFR-vaiheeseen siirtyneet potilaat | 126 | |
Viikot TFR-vaiheen aloituksesta | 48 viikkoa | 264 viikkoa |
MMR-vasteen säilyttäneet, ei vahvistettua MR4,0-vasteen menettämistä, ei nilotinibihoidon uudelleenaloitusta | 73 (57,9 %, [95% lv: 48,8–66,7]) | 54 (42,9 % [54/126, 95 % lv: 34,1–52,0]) |
TFR-vaiheen lopettaneet potilaat | 53 | 74[1] |
Syynä MR4,0-vasteen vahvistettu menettäminen tai MMR-vasteen menettäminen | 53 (42,1 %) | 61 (82,4 %) |
Muu syy | 0 | 13 |
MMR-vasteen menettämisen tai MR4,0-vasteen vahvistetun menettämisen jälkeen hoidon uudelleen aloittaneet potilaat | 51 | 59 |
MR4,0-vasteen uudelleen saavuttaneet potilaat | 48 (94,1 %) | 56 (94,9 %) |
MR4,5-vasteen uudelleen saavuttaneet potilaat | 47 (92,2 %) | 54 (91,5 %) |
[1] Kahdella potilaalla oli MMR-vaste (PCR-määrityksessä) viikolla 264, mutta he lopettivat hoidon myöhemmin eikä heille enää tehty uusia PCR-määrityksiä.
Kaplan–Meier‑estimaattien perusteella nilotinibihoidon mediaanikesto ennen MR4,0‑vasteen saavuttamista uudelleen oli 11,1 viikkoa (95 % lv: 8,1–12,1) ja sen mediaanikesto ennen MR4,5‑vasteen saavuttamista uudelleen taas 13,1 viikkoa (95 % lv: 12,0–15,9). Uudelleen saavutetun MR4,0:n kumulatiivinen vasteprosentti oli 94,9 % (56 potilasta 59:stä) ja MR4,5:n osalta vastaavasti 91,5 % (54 potilasta 59:stä) 48 viikkoa hoidon uudelleenaloituksen jälkeen.
Kaplan–Meier‑estimoitu hoitovapaan elossaoloajan mediaani oli 224 viikkoa (95 % lv: 39,9–ei arvioitavissa) (kuva 5); 63 potilaalla 126:sta (50,0 %) ei ollut hoitovapaaseen elossaoloon liittyvää tapahtumaa.
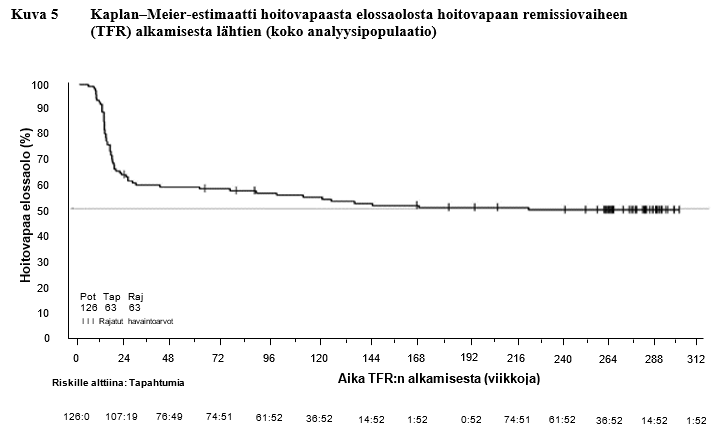
Pediatriset potilaat
Nilotinibilla tehdyssä pediatrisessa päätutkimuksessa yhteensä 58 2 – <18-vuotiasta potilasta (25 potilaalla oli äskettäin todettu kroonisen vaiheen Ph‑positiivinen KML ja 33 potilaalla kroonisen vaiheen Ph‑positiivinen KML ja joko resistenssi imatinibille/dasatinibille tai intoleranssi imatinibille) sai nilotinibihoitoa annoksella 230 mg/m2 kahdesti vuorokaudessa pyöristettynä lähimpään 50 mg annokseen (kerta-annos enintään 400 mg). Tärkeimmät tulokset on esitetty yhteenvetona taulukossa 13.
Taulukko 13 Nilotinibilla tehdyn pediatrisen päätutkimuksen tulokset
Äskettäin diagnosoitu kroonisen vaiheen Ph+ KML (n = 25) | resistentti tai intolerantti kroonisen vaiheen Ph+ KML (n = 33) | |
Hoidon mediaanikesto kuukausina (vaihteluväli) | 51,9 (1,4 - 61,2) | 60,5 (0,7 - 63,5) |
Mediaani (vaihteluväli) todellinen annosintensiteetti (mg/m2/päivä) | 377,0 (149 - 468) | 436,9 (196 - 493) |
Suhteellinen annosintensiteetti (%) verrattuna suunniteltuun annokseen 230 mg/m2 kahdesti päivässä | ||
Mediaani (vaihtelu-väli) | 82,0 (32 - 102) | 95,0 (43 - 107) |
Potilaiden määrä, joilla > 90 % | 12 (48,0 %) | 19 (57,6 %) |
MMR (BCR‑ABL/ABL ≤0, 1 % IS) jaksossa 12, (95 % lv) | 60 % (38,7 - 78,9) | 48,5 % (30,8 - 66,5) |
MMR jaksoon 12 mennessä, (95 % lv) | 64,0 % (42,5 - 82,0) | 57,6 % (39,2 - 74,5) |
MMR jaksoon 66 mennessä, (95 % lv) | 76,0 % (54,9 - 90,6) | 60,6 % (42,1 - 77,1) |
Mediaaniaika MMR-vasteeseen kuukaudessa (95 % lv) | 5,56 (5,52 - 10,84) | 2,79 (0,03 - 5,75) |
Potilaiden lukumäärä (%), jotka saavuttivat MR4,0:n (BCR-ABL/ABL ≤0,01 % IS) jaksoon 66 mennessä | 14 (56,0 %) | 9 (27,3 %) |
Potilaiden lukumäärä (%), jotka saavuttivat MR4,5:n (BCR-ABL/ABL ≤0,01 % IS) jaksoon 66 mennessä | 11 (44,0 %) | 4 (12,1 %) |
Vahvistettu MMR-menetys potilailla, jotka saavuttivat MMR-arvon | 3 19:stä | Ei yhtään 20:stä |
Uusi mutaatio hoidon aikana | Ei yhtään | Ei yhtään |
Taudin eteneminen hoidon aikana | 1 potilas vastasi väliaikaisesti kiihtymisvaiheen/blastikriisivaiheen etenemisen teknistä määritelmää* | 1 potilas eteni kiihtymisvaiheeseen/blastikriisivaiheeseen 10,1 kuukauden hoidon jälkeen |
Kokonaiseloonjäänti | ||
Tapahtumia | 0 | 0 |
Kuolema hoidon aikana | 3 (12 %) | 1 (3 %) |
Kuolema eloonjäännin seuraamisen aikana | Ei arvioitavissa | Ei arvioitavissa |
* yksi potilas vastasi väliaikaisesti kiihtymisvaiheen/blastikriisivaiheen etenemisen teknistä määritelmää (lisääntyneen basofiilisolumäärän takia) kuukausi nilotinibin aloittamisen jälkeen (tilapäinen 13 päivän hoidon keskeyttäminen ensimmäisen jakson aikana). Potilas pysyi tutkimuksessa, palasi krooniseen vaiheeseen ja oli CHR:ssä ja CCyR:ssä kuuden nilotinibihoitosyklin ajan.
Farmakokinetiikka
Imeytyminen
Nilotinibin huippupitoisuudet saavutetaan 3 tuntia peroraalisen annostelun jälkeen. Noin 30 % suun kautta annetusta nilotinibiannoksesta imeytyy. Nilotinibin absoluuttista biologista hyötyosuutta ei ole määritetty. Juotavaan liuokseen (pH 1,2‑1,3) verrattuna nilotinibikapselin suhteellinen biologinen hyötyosuus on noin 50 %. Terveillä vapaaehtoisilla nilotinibin Cmax suurenee 112 % ja pitoisuus‑aikakäyrän alle jäävä alue (AUC) suurenee 82 % paastoarvoista, mikäli Tasigna otetaan aterian yhteydessä. Nilotinibin biologinen hyötyosuus suureni 29 %, kun Tasigna annettiin 30 minuuttia aterian jälkeen, ja 15 %, kun Tasigna annettiin 2 tuntia aterian jälkeen (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Nilotinibin imeytyminen (suhteellinen biologinen hyötyosuus) saattaa alentua 48 % potilailla, joille on tehty täydellinen gastrektomia ja 22 % potilailla, joille on tehty osittainen gastrektomia.
Jakautuminen
Nilotinibin veri/plasma‑suhde on 0,71. Se sitoutuu plasman proteiineihin noin 98‑prosenttisesti in vitro ‑kokeiden perusteella.
Biotransformaatio
Oksidaatio ja hydroksylaatio ovat tärkeimmät terveillä vapaaehtoisilla todetut metaboliareitit. Nilotinibi on tärkein verenkierrossa (seerumissa) oleva ainesosa. Nilotinibin metaboliiteilla ei ole merkitsevää osuutta sen farmakologiseen vaikutukseen. Nilotinibi metaboloituu pääasiassa CYP3A4:n vaikutuksesta, ja CYP2C8 saattaa osallistua sen metaboliaan vähäisessä määrin.
Eliminaatio
Kun terveille vapaaehtoisille annettiin kerta‑annos radioaktiivisesti merkittyä nilotinibia, yli 90 % annoksesta eliminoitui 7 vuorokauden kuluessa pääasiassa ulosteeseen (94 % annoksesta). Muuttumatonta nilotinibia oli 69 % annoksesta.
Kun lääkettä annettiin toistuvasti kerran vuorokaudessa, sen farmakokineettisten ominaisuuksien perusteella arvioitu ilmeinen eliminaation puoliintumisaika oli noin 17 tuntia. Nilotinibin farmakokinetiikassa todettiin kohtalaisia tai suuria potilaskohtaisia vaihteluja.
Lineaarisuus/ei‑lineaarisuus
Vakaassa tilassa nilotinibialtistus riippui annoksesta, mutta annostasolla > 400 mg kerran vuorokaudessa systeemisen altistuksen suureneminen ei kuitenkaan ollut aivan suoraan verrannollinen annokseen. Päivittäinen systeeminen nilotinibialtistus oli vakaassa tilassa 35 % suurempi annoksella 400 mg kahdesti vuorokaudessa kuin annoksella 800 mg kerran vuorokaudessa. Nilotinibin vakaan tilan systeeminen altistus (AUC) oli annostasolla 400 mg kahdesti vuorokaudessa noin 13,4 % suurempi kuin annostasolla 300 mg kahdesti vuorokaudessa. Nilotinibin alimmat (trough) pitoisuudet ja huippupitoisuudet 12 kk:n aikana olivat noin 15,7 % ja 14,8 % suuremmat annostuksella 400 mg kahdesti vuorokaudessa kuin 300 mg kahdesti vuorokaudessa. Nilotinibialtistus ei suurentunut merkittävästi, kun annos suurennettiin tasolta 400 mg kahdesti vuorokaudessa tasolle 600 mg kahdesti vuorokaudessa.
Vakaa tila saavutettiin yleensä 8. hoitopäivään mennessä. Seerumin nilotinibialtistus suureni ensimmäisen annoksen ja vakaan tilan saavuttamisen välillä noin 2‑kertaiseksi, kun lääke otettiin kerran vuorokaudessa, ja 3,8‑kertaiseksi, kun lääke otettiin kahdesti vuorokaudessa.
Hyväksikäytettävyyttä/biologista samanarvoisuutta koskevat tutkimukset
Kahtena 200 mg:n kovana kapselina annetun 400 mg:n nilotinibikerta‑annoksen, joka annettiin sekoittamalla kummankin kovan kapselin sisältö yhteen teelusikalliseen omenasosetta, on osoitettu olevan bioekvivalentti kerta‑annoksena annetun kahden avaamattoman 200 mg kovan kapselin kanssa.
Pediatriset potilaat
Kun nilotinibia annettiin lapsipotilaille annoksella 230 mg/m2 kahdesti vuorokaudessa pyöristettynä lähimpään 50 mg:aan (kerta‑annos enintään 400 mg), nilotinibin vakaan tilan altistus ja puhdistuma todettiin samankaltaisiksi (korkeintaan 2‑kertaisiksi) kuin aikuispotilailla, joiden annos oli 400 mg kahdesti vuorokaudessa. Nilotinibin farmakokineettinen altistus kerta‑annosten ja toistuvien annosten jälkeen vaikutti olevan verrattavissa 2 – < 10 vuoden ikäisten lapsipotilaiden ja ≥ 10 vuoden – < 18 vuoden ikäisten lapsipotilaiden joukossa.
Prekliiniset tiedot turvallisuudesta
Nilotinibia on arvioitu farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, lisääntymistoksisuutta, fototoksisuutta ja karsinogeenisuutta (rotilla ja hiirillä) selvittäneissä tutkimuksissa.
Farmakologiset turvallisuustutkimukset
Nilotinibi ei vaikuttanut keskushermoston toimintaan eikä hengitystoimintoihin. Kaniinin sydämillä suoritetut in vitro prekliiniset sydänturvallisuustutkimukset osoittivat nilotinibin voivan pidentää QT‑aikaa, sillä se salpasi hERG‑kanavia ja pidensi aktiopotentiaalia. Jopa 39 viikon ajan hoidetuilla koirilla ja apinoilla ei todettu mitään vaikutuksia EKG‑tutkimuksissa eikä koirilla tehdyssä erityisessä telemetriatutkimuksessa.
Toistuvan altistuksen toksisuustutkimukset
Toistuvan altistuksen aiheuttamaa toksisuutta selvittäneissä tutkimuksissa koirille annettiin nilotinibia jopa 4 viikon ajan ja makakeille jopa 9 kuukauden ajan. Tutkimuksissa selvisi, että maksa on nilotinibitoksisuuden ensisijainen kohde‑elin. Koe‑‑eläimillä todettiin mm. alaniiniaminotransferaasin ja alkalisen fosfataasin aktiivisuuden tehostumista ja histopatologisia löydöksiä (pääasiassa sinusoidisolujen ja Kupfferin solujen hyperplasiaa/hypertrofiaa, sappitiehyiden hyperplasiaa ja periportaalista fibroosia). Kliiniskemialliset muutokset korjaantuivat yleensä täysin 4 viikon toipumisjakson aikana, ja myös histologiset muutokset korjaantuivat osittain. Pienimmän maksavaikutuksia aiheuttaneen annostason aikaansaama altistus oli ihmisten annoksen 800 mg/vrk aikaansaamaa altistusta pienempi. Jopa 26 viikon ajan hoidetuilla hiirillä ja rotilla havaittiin vain vähäisiä maksamuutoksia. Rotilla, koirilla ja apinoilla havaittiin enimmäkseen korjaantuvaa kolesteroliarvojen nousua.
Genotoksisuustutkimukset
Genotoksisuustutkimuksia tehtiin bakteerijärjestelmissä in vitro ja nisäkässolujärjestelmissä in vitro ja in vivo sekä metabolian aktivoinnin avulla että ilman sitä. Näissä tutkimuksissa nilotinibin ei todettu aiheuttavan mutaatioita.
Karsinogeenisuustutkimukset
2 vuotta kestäneessä rotilla tehdyssä karsinogeenisuustutkimuksessa ei‑uudiskasvuisten leesioiden pääasiallinen kohde‑elin oli kohtu (laajentuminen, verisuonen pullistuma, endoteliaalisolujen liikakasvu, tulehdus ja/tai epiteelin liikakasvu). Karsinogeenisuustutkimuksessa ei havaittu merkkejä karsinogeenisuudesta nilotinibin annoksilla 5, 15 ja 40 mg/kg/vrk. Altistukset (AUC:llä mitattuna) korkeimmalla annostasolla vastasivat noin kaksin‑kolminkertaista ihmisen päivittäistä vakaan tilan nilotinibialtistusta (AUC:hen perustuen) annoksella 800 mg/vrk.
Hiirillä tehdyssä 26 viikkoa kestäneessä Tg.rasH2‑karsinogeenisuustutkimuksessa, jossa nilotinibiannokset olivat 30, 100 ja 300 mg/kg/vrk, havaittiin ihopapilloomia/karsinoomia annoksella 300 mg/kg, joka vastaa noin 30‑40‑kertaista (AUC:hen perustuen) altistusta ihmisellä suurimmalla hyväksytyllä annoksella 800 mg/vrk (400 mg kahdesti vuorokaudessa). Haittavaikutukseton altistustaso ihon neoplastisten leesioiden suhteen oli 100 mg/vrk, joka vastaa noin 10‑20‑kertaista altistusta ihmisellä suurimmalla hyväksytyllä annoksella 800 mg/vrk (400 mg kahdesti vuorokaudessa). Ei‑uudiskasvuisten leesioiden pääasialliset kohde‑elimet olivat iho (epidermaalinen liikakasvu), kasvuvaiheessa olevat hampaat (yläetuhampaiden kiilteen degeneraatio/atrofia ja etuhampaiden ikenien/hammasperäisen epiteelin tulehdus) sekä kateenkorva (lymfosyyttien vähenemisen kasvanut esiintyvyys ja/tai vakavuus).
Lisääntymistoksisuus- ja hedelmällisyystutkimukset
Nilotinibi ei ollut teratogeeninen, mutta alkio‑ ja sikiötoksisuutta todettiin annoksilla, jotka olivat toksisia myös emolle. Munasolun kiinnittymisen jälkeisiä alkiokuolemia todettiin sekä hedelmällisyystutkimuksessa (jossa hoidettiin sekä uroksia että naaraita) että alkiotoksisuustutkimuksessa (jossa hoidettiin vain naaraita). Alkiotoksisuustutkimuksissa rotilla todettiin alkiokuolemia ja sikiöihin kohdistuvia vaikutuksia (pääasiassa sikiöiden painonlaskua, ennenaikainen kasvoluiden fuusioituminen (poskiluun ja yläleukaluun luutumista yhteen) sekä sisäelinten ja luuston poikkeavuuksia) ja kaniineilla sikiöiden resorption lisääntymistä ja luuston poikkeavuuksia. Rotilla tehdyissä pre‑ ja postnataalitutkimuksissa, emon altistus nilotinibille aiheutti painon laskua pennuissa, johon liittyi muutoksia fyysisen kehittymisen parametreissa sekä merkkejä jälkeläisten heikentyneestä parittelusta ja hedelmällisyydestä. Haittavaikutukseton altistustaso oli naarailla yleensä samaa luokkaa tai pienempi kuin ihmisten nilotinibiannoksella 800 mg/vrk aikaansaatu altistus.
Suurimmillakaan tutkituilla annoksilla (noin 5 x ihmisen suositusannokset) ei havaittu vaikutuksia siittiömäärään, siittiöiden liikkuvuuteen eikä uros‑ eikä naarasrottien hedelmällisyyteen.
Tutkimukset nuorilla eläimillä
Nuorten eläinten kehitystä koskeneessa tutkimuksessa nilotinibia annettiin nuorille rotille letkulla suun kautta ensimmäisestä syntymän jälkeisestä viikosta alkaen nuoreen aikuisikään asti (päivä 70 syntymän jälkeen) annoksilla 2, 6 ja 20 mg/kg/vrk. Tavanomaisten tutkimusparametrien lisäksi arvioitiin kehityksen tärkeimpiä vaiheita, keskushermostovaikutuksia, parittelua ja hedelmällisyyttä. Molemmilla sukupuolilla havaitun painon laskun ja urosten esinahan eriytymisen viivästymisen (saattaa liittyä painon laskuun) perusteella annokseksi, jolla ei nuorilla rotilla havaittu mitään haittavaikutuksia, määriteltiin 6 mg/kg/vrk. Nuorilla eläimillä ei esiintynyt aikuisiin verrattuna suurempaa nilotinibiherkkyyttä. Myös toksisuusprofiili oli nuorilla rotilla samaa luokkaa kuin aikuisilla.
Fototoksisuustutkimukset
Nilotinibin osoitettiin absorboivan UV‑B‑ ja UV‑A‑valoa ja jakautuvan ihoon. Sillä todettiin olevan mahdollista fototoksisia vaikutuksia in vitro, mutta vastaavia vaikutuksia ei ole todettu in vivo. Siksi mahdollisuus, että nilotinibi herkistäisi potilaat valolle, on hyvin pieni.
Farmaseuttiset tiedot
Apuaineet
Tasigna 50 mg kapseli, kova
Kapselin sisältö
Laktoosimonohydraatti
Krospovidoni (tyyppi A)
Poloksameeri 188
Piidioksidi, kolloidinen, vedetön
Magnesiumstearaatti
Kapselin kuori
Liivate
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Painomuste
Shellakka
Musta rautaoksidi (E172)
Propyleeniglykoli
Ammoniumhydroksidi
Tasigna 150 mg kapseli, kova
Kapselin sisältö
Laktoosimonohydraatti
Krospovidoni (tyyppi A)
Poloksameeri 188
Piidioksidi, kolloidinen, vedetön
Magnesiumstearaatti
Kapselin kuori
Liivate
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Painomuste
Shellakka
Musta rautaoksidi (E172)
n-butyylialkoholi
Propyleeniglykoli
Dehydratoitu etanoli
Isopropyylialkoholi
Ammoniumhydroksidi
Tasigna 200 mg kapseli, kova
Kapselin sisältö
Laktoosimonohydraatti
Krospovidoni (tyyppi A)
Poloksameeri 188
Piidioksidi, kolloidinen, vedetön
Magnesiumstearaatti
Kapselin kuori
Liivate
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Painomuste
Shellakka (E904)
Dehydratoitu alkoholi
Isopropyylialkoholi
Butyylialkoholi
Propyleeniglykoli
Ammoniakki, väkevä
Kaliumhydroksidi
Punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Säilytä alle 30°C.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TASIGNA kapseli, kova
50 mg (L:kyllä) 120 fol (876,97 €)
150 mg (L:kyllä) 112 fol (901,73 €)
200 mg (L:kyllä) 112 fol (1252,31 €)
PF-selosteen tieto
Tasignasta on saatavilla seuraavat pakkauskoot:
Tasigna 50 mg kapseli, kova
PVC/PVDC/Alu‑läpipainopakkaukset
- Pakkaus, jossa on 120 (kolme 40 kapselin pakkausta) kovaa kapselia.
Tasigna 150 mg kapseli, kova
PVC/PVDC/Alu‑läpipainopakkaukset
- Yksikköpakkaukset, joissa on 28 kovaa kapselia (7 päiväliuskaa, joissa kussakin 4 kovaa kapselia) tai 40 kovaa kapselia (5 läpipainopakkausta, joissa kussakin 8 kovaa kapselia).
- Monipakkaukset, joissa on 112 (neljä 28 kapselin pakkausta) kovaa kapselia, 120 (kolme 40 kapselin pakkausta) kovaa kapselia tai 392 (neljätoista 28 kapselin pakkausta) kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Tasigna 200 mg kapseli, kova
PVC/PVDC/Alu‑läpipainopakkaukset
- Yksikköpakkaukset, joissa on 28 kovaa kapselia taskupakkauksessa.
- Yksikköpakkaukset, joissa on 28 kovaa kapselia (7 päiväliuskaa, joissa kussakin 4 kovaa kapselia) tai 40 kovaa kapselia (5 läpipainopakkausta, joissa kussakin 8 kovaa kapselia).
- Monipakkaukset, joissa on 112 (neljä 28 kapselin taskupakkausta) kovaa kapselia.
- Monipakkaukset, joissa on 112 (neljä 28 kapselin pakkausta) kovaa kapselia, 120 (kolme 40 kapselin pakkausta) kovaa kapselia tai 392 (neljätoista 28 kapselin pakkausta) kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Tasigna 50 mg kapseli, kova
Valkoinen tai kellertävä jauhe kokoa 4 olevassa kovassa liivatekapselissa, jossa on läpinäkymätön punainen yläosa ja vaaleankeltainen läpinäkymätön alaosa; yläosaa kiertää mustalla painettu teksti ”NVR/ABL”.
Tasigna 150 mg kapseli, kova
Valkoinen tai kellertävä jauhe kokoa 1 olevissa punaisissa, läpinäkymättömissä, kovissa liivatekapseleissa, joihin on painettu pituussuunnassa mustalla ”NVR/BCR”.
Tasigna 200 mg kapseli, kova
Valkoinen tai kellertävä jauhe kokoa 0 olevissa vaaleankeltaisissa, läpinäkymättömissä, kovissa liivatekapseleissa, joihin on painettu pituussuunnassa punaisella ”NVR/TKI”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TASIGNA kapseli, kova
150 mg 112 fol
200 mg 112 fol
- Ylempi erityiskorvaus (100 %). Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117).
- Peruskorvaus (40 %).
TASIGNA kapseli, kova
50 mg 120 fol
- Ei korvausta.
ATC-koodi
L01EA03
Valmisteyhteenvedon muuttamispäivämäärä
17.12.2025
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com