
ZYTIGA tabletti, kalvopäällysteinen 500 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 500 mg abirateroniasetaattia, joka vastaa 446 mg:aa abirateronia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 253,2 mg laktoosimonohydraattia ja 13,5 mg natriumia.
Lääkemuoto
Kalvopäällysteinen tabletti
Kliiniset tiedot
Käyttöaiheet
Zytiga on tarkoitettu käytettäväksi yhdistelmänä prednisonin tai prednisolonin kanssa:
- äskettäin diagnosoidun korkean riskin etäpesäkkeisen hormonisensitiivisen eturauhassyövän hoitoon miehille yhdistelmänä androgeenideprivaatiohoidon kanssa (ks. kohta Farmakodynamiikka)
- etäpesäkkeisen kastraatioresistentin eturauhassyövän hoitoon miehille, joiden tauti on oireeton tai oireet ovat lieviä androgeenideprivaatiohoidon epäonnistuttua ja joille solunsalpaajahoito ei ole vielä kliinisesti aiheellista (ks. kohta Farmakodynamiikka)
- etäpesäkkeisen kastraatioresistentin eturauhassyövän hoitoon miehille, joiden tauti on edennyt dosetakseliin pohjautuvan solunsalpaajahoidon aikana tai sen jälkeen.
Annostus ja antotapa
Tätä lääkevalmistetta saa määrätä asianmukainen terveydenhuollon ammattilainen.
Annostus
Suositusannos on 1000 mg (kaksi 500 mg:n tablettia) vuorokaudessa kerta-annoksena, jota ei saa ottaa ruoan kanssa (ks. jäljempänä Antotapa). Zytiga-tablettien ottaminen ruoan kanssa lisää systeemistä altistusta abirateronille (ks. kohdat Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset ja Farmakokinetiikka).
Prednisonin tai prednisolonin annostus
Zytiga-tabletteja käytetään etäpesäkkeisen hormonisensitiivisen eturauhassyövän hoitoon yhdessä prednisonin tai prednisolonin 5 mg:n vuorokausiannoksen kanssa.
Zytiga-tabletteja käytetään etäpesäkkeisen kastraatioresistentin eturauhassyövän hoitoon yhdessä prednisonin tai prednisolonin 10 mg:n vuorokausiannoksen kanssa.
Lääkkeellistä kastraatiota luteinisoivan hormonin vapauttajahormonin (LHRH) analogeilla on jatkettava hoidon aikana, jos potilaalle ei ole tehty kirurgista kastraatiota.
Seurantaa koskevat suositukset
Seerumin transaminaasipitoisuudet on mitattava ennen Zytiga-hoidon aloittamista, kolmen ensimmäisen hoitokuukauden aikana kahden viikon välein ja sen jälkeen kuukausittain. Verenpainetta, seerumin kaliumpitoisuutta ja nesteen kertymistä elimistöön on seurattava kuukausittain. Jos potilaalla on merkittävä kongestiivisen sydämen vajaatoiminnan riski, häntä on kuitenkin seurattava kolmen ensimmäisen hoitokuukauden ajan kahden viikon välein ja sen jälkeen kuukausittain (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos potilaalla on ennestään hypokalemia tai hypokalemia kehittyy Zytiga-hoidon aikana, on harkittava potilaan kaliumpitoisuuden pitämistä tasolla ≥ 4,0 mM.
Jos potilaalle kehittyy ≥ 3. asteen toksisuutta, kuten korkeaa verenpainetta, hypokalemiaa, turvotusta ja muuntyyppistä, mineralokortikoideihin liittymätöntä toksisuutta, hoito on keskeytettävä ja toksisuuden asianmukainen hoito on aloitettava. Zytiga-hoitoa ei saa jatkaa ennen kuin toksisuusoireet ovat lieventyneet 1. asteeseen tai korjautuneet hoitoa edeltäneelle tasolle.
Jos joko Zytiga-, prednisoni- tai prednisolonivuorokausiannos jää ottamatta, hoitoa jatketaan seuraavana päivänä ottamalla tavanomainen vuorokausiannos.
Maksatoksisuus
Jos potilaalle kehittyy hoidon aikana maksatoksisuutta (ALAT- tai ASAT-arvo suurenee yli viisinkertaiseksi normaaliarvojen ylärajaan nähden), hoito on heti keskeytettävä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoito voidaan aloittaa uudelleen pienemmällä annoksella 500 mg (yksi tabletti) kerran vuorokaudessa sen jälkeen, kun maksan toimintakokeiden tulokset ovat palautuneet potilaan hoitoa edeltäneelle tasolle. Jos potilaan hoito aloitetaan uudelleen, seerumin transaminaasiarvoja on seurattava kolmen kuukauden ajan vähintään kahden viikon välein ja sen jälkeen kuukausittain. Jos maksatoksisuus uusiutuu pienemmän 500 mg:n vuorokausiannoksen käytön yhteydessä, hoito on lopetettava.
Jos potilaalle kehittyy vaikea-asteista maksatoksisuutta (ALAT- tai ASAT-arvo 20-kertainen normaaliarvojen ylärajaan [upper limit of normal, ULN] nähden) hoidon missä tahansa vaiheessa, hoito on lopetettava eikä potilasta saa hoitaa uudelleen tällä valmisteella.
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa (Child-Pugh-luokka A) ennestään sairastavan potilaan annosta ei tarvitse muuttaa.
Keskivaikean maksan vajaatoiminnan (Child-Pugh-luokka B) on osoitettu suurentavan abirateronin systeemistä altistusta noin nelinkertaisesti, kun abirateroniasetaattia on annettu 1 000 mg:n kerta-annos suun kautta (ks. kohta Farmakokinetiikka). Toistuvien abirateroniasetaattiannosten kliinisestä turvallisuudesta ja tehosta ei ole tietoa annettaessa valmistetta potilaille, joilla on keskivaikea tai vaikea maksan vajaatoiminta (Child-Pugh-luokka B tai C). Annoksen säätämistä ei voida ennakoida. Jos potilaalla on kohtalainen maksan vajaatoiminta, Zytiga-hoitoa on harkittava tarkoin, ja hoidon hyötyjen on tällaiselle potilaalle oltava selvästi riskejä suuremmat (ks. kohdat Annostus ja antotapa sekä Farmakokinetiikka). Zytiga-hoitoa ei saa antaa, jos potilaalla on vaikea maksan vajaatoiminta (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakokinetiikka).
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden annostusta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Eturauhassyöpää ja vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidosta ei kuitenkaan ole kliinistä kokemusta. Tämän potilasryhmän hoidossa kehotetaan varovaisuuteen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Ei ole asianmukaista käyttää Zytiga-valmistetta pediatristen potilaiden hoitoon.
Antotapa
Zytiga otetaan suun kautta.
Tabletit on otettava kerta-annoksena kerran päivässä tyhjään mahaan. Zytiga on otettava vähintään kaksi tuntia ruokailun jälkeen. Ruokaa ei saa syödä vähintään tuntiin Zytiga-tablettien ottamisen jälkeen. Zytiga-tabletit niellään kokonaisina veden kanssa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Naiset, jotka ovat tai saattavat olla raskaana (ks. kohta Raskaus ja imetys).
- Vaikea-asteinen maksan vajaatoiminta [Child-Pugh-luokka C (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet sekä Farmakokinetiikka)].
- Zytiga-valmisteen ja prednisonin tai prednisolonin käyttö yhdessä radium-223:n (Ra-223:n) kanssa on vasta-aiheista.
Varoitukset ja käyttöön liittyvät varotoimet
Mineralokortikoidiylimäärästä aiheutuva korkea verenpaine, hypokalemia, nesteen kertyminen elimistöön ja sydämen vajaatoiminta
Zytiga saattaa aiheuttaa korkeaa verenpainetta, hypokalemiaa ja nesteen kertymistä elimistöön (ks. kohta Haittavaikutukset) CYP17-entsyymin toiminnan estymisestä aiheutuvan mineralokortikoidipitoisuuden suurenemisen seurauksena (ks. kohta Farmakodynamiikka). Kortikosteroidin samanaikainen anto suppressoi adrenokortikotrooppisen hormonin (ACTH:n) erittymistä, jolloin haittavaikutusten ilmaantuvuus ja vaikeusaste vähenevät. Hoidossa on noudatettava varovaisuutta, jos verenpaineen nousu, hypokalemia (esim. sydänglykosideja käyttävillä potilailla) tai nesteen kertyminen elimistöön (esim. potilaat, joilla on sydämen vajaatoimintaa, vaikea-asteinen tai epästabiili angina pectoris, äskettäin ollut sydäninfarkti, kammioperäisiä rytmihäiriöitä tai vaikea-asteista munuaisten vajaatoimintaa) saattavat pahentaa potilaan perussairautta.
Zytiga-tablettien käytössä on oltava varovainen, jos potilas on aiemmin sairastanut sydän- ja verisuonitautia. Vaiheen 3 Zytiga-tutkimuksiin ei otettu mukaan potilaita, joiden verenpaine ei ollut hallinnassa, joilla oli kliinisesti merkityksellinen sydäntauti, minkä osoitti sydäninfarkti tai tromboottinen valtimotapahtuma edellisten kuuden kuukauden aikana, vaikea-asteinen tai epästabiili angina pectoris tai New York Heart Association (NYHA) -luokan III tai IV (tutkimus 301) tai NYHA-luokan II–IV (tutkimukset 3011 ja 302) sydämen vajaatoiminta tai sydämen ejektiofraktio < 50 %. Tutkimuksiin 3011 ja 302 ei otettu mukaan potilaita, joilla oli sydämen eteisvärinää tai muita hoitoa vaativia sydämen rytmihäiriöitä. Hoidon turvallisuutta potilaille, joiden vasemman kammion ejektiofraktio (LVEF) on < 50 % tai jotka sairastavat NYHA-luokan III tai IV (tutkimus 301) tai NYHA-luokan II–IV (tutkimukset 3011 ja 302) sydämen vajaatoimintaa, ei varmistettu.
Ennen kuin hoitoa annetaan potilaalle, jolla on merkityksellinen kongestiivisen sydämen vajaatoiminnan riski (esim. aiemmin ollut sydämen vajaatoimintaa, huonossa hoitotasapainossa oleva korkea verenpaine tai sydäntapahtumia, kuten iskeeminen sydäntauti), potilaan sydämen toiminnan tutkimista (esim. kaikututkimusta) on harkittava. Sydämen vajaatoiminta on hoidettava ja sydämen toiminta on optimoitava ennen Zytiga-hoitoa. Potilaalla esiintyvä korkea verenpaine, hypokalemia ja nesteen kertyminen elimistöön on korjattava ja saatava hoitotasapainoon. Verenpainetta, seerumin kaliumpitoisuutta, nesteen kertymistä elimistöön (painon nousu, raajojen turvotus) sekä muita kongestiiviseen sydämen vajaatoimintaan viittaavia oireita ja löydöksiä on seurattava kolmen ensimmäisen hoitokuukauden ajan kahden viikon välein ja sen jälkeen kuukausittain ja poikkeavuudet on korjattava. Potilailla, joilla on esiintynyt Zytiga-hoidon yhteydessä hypokalemiaa, on havaittu QT-ajan pitenemistä. Sydämen toiminta on tutkittava kliinisten vaatimusten mukaisesti, asianmukainen hoito on aloitettava ja tämän hoidon lopettamista on harkittava, jos sydämen toiminta heikkenee kliinisesti merkityksellisesti (ks. kohta Annostus ja antotapa).
Maksatoksisuus ja maksan vajaatoiminta
Kontrolloiduissa kliinisissä tutkimuksissa esiintyi huomattavaa maksaentsyymipitoisuuden suurenemista, joka johti hoidon lopettamiseen tai annoksen muuttamiseen (ks. kohta Haittavaikutukset). Seerumin transaminaasipitoisuus on mitattava ennen hoidon aloittamista, kolmen ensimmäisen hoitokuukauden aikana kahden viikon välein ja sen jälkeen kuukausittain. Jos potilaalle kehittyy maksatoksisuuteen viittaavia kliinisiä oireita tai löydöksiä, seerumin transaminaasipitoisuus on mitattava heti. Jos ALAT- tai ASAT-arvo suurenee hoidon missä tahansa vaiheessa yli viisinkertaiseksi normaaliarvojen ylärajaan (ULN) nähden, hoito on keskeytettävä heti ja maksan toimintaa on seurattava tarkoin. Hoitoa voidaan jatkaa vasta, kun maksan toimintakokeiden tulokset ovat palautuneet potilaan hoitoa edeltäneisiin arvoihin, ja annosta on tällöin pienennettävä (ks. kohta Annostus ja antotapa).
Jos potilaalle kehittyy vaikeaa maksatoksisuutta (ALAT- tai ASAT-arvo 20-kertainen normaaliarvojen ylärajaan [ULN] nähden) milloin tahansa hoidon aikana, hoito on lopetettava eikä potilasta saa hoitaa uudelleen.
Aktiivista tai oireista virushepatiittia sairastavia potilaita ei otettu mukaan kliinisiin tutkimuksiin, joten Zytiga-valmisteen käytöstä tämän potilasryhmän hoitoon ei ole tietoja.
Useiden abirateroniasetaattiannosten kliinisestä turvallisuudesta ja tehosta ei ole tietoja, kun niitä annetaan kohtalaista tai vaikeaa maksan vajaatoimintaa sairastaville potilaille (Child–Pugh-luokka B tai C). Jos potilaalla on kohtalainen maksan vajaatoiminta, Zytiga-hoitoa on harkittava tarkoin, ja hoidon hyötyjen on tällaiselle potilaalle oltava selvästi riskejä suuremmat (ks. kohdat Annostus ja antotapa sekä Farmakokinetiikka). Zytiga-hoitoa ei saa antaa, jos potilaalla on vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa, Vasta-aiheet sekä Farmakokinetiikka).
Myyntiluvan saamisen jälkeen on raportoitu harvinaisina tapauksina akuuttia maksan vajaatoimintaa ja fulminanttia hepatiittia; osa näistä tapauksista on johtanut potilaan kuolemaan (ks. kohta Haittavaikutukset).
Kortikosteroidihoidon lopettaminen ja hoito stressitilanteissa
Potilasta on seurattava adrenokortikaalisen vajaatoiminnan varalta ja hoidossa on oltava varovainen, jos prednisoni- tai prednisolonihoito lopetetaan. Jos Zytiga-hoitoa jatketaan kortikosteroidihoidon lopettamisen jälkeen, potilasta on seurattava mineralokortikoidiylimäärän oireiden havaitsemiseksi (ks. edellä).
Prednisoni- tai prednisolonihoitoa saavien potilaiden kortikosteroidiannostusta saattaa olla aiheellista suurentaa epätavallisen stressin yhteydessä ennen stressiä aiheuttavaa tilannetta, sen aikana ja jälkeen.
Luuntiheys
Alentunutta luuntiheyttä voi esiintyä miehillä, joilla on pitkälle edennyt etäpesäkkeinen eturauhassyöpä. Zytiga-valmisteen käyttö yhdessä glukokortikoidin kanssa voi tehostaa tätä vaikutusta.
Aiempi ketokonatsolin käyttö
Hoitovaste saattaa olla heikompi, jos potilas on aiemmin saanut ketokonatsolia eturauhassyövän hoitoon.
Hyperglykemia
Glukokortikoidien käyttö saattaa lisätä hyperglykemiaa, joten diabeetikoiden verensokeripitoisuutta on mitattava tiheästi.
Hypoglykemia
Hypoglykemiaa on raportoitu käytettäessä Zytiga-valmistetta yhdistelmänä prednisonin/prednisolonin kanssa potilaille, jotka sairastavat ennestään diabetesta ja käyttävät pioglitatsonia tai repaglinidia (ks. kohta Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset). Tästä syystä diabetespotilaiden verensokeripitoisuutta pitää seurata.
Käyttö solunsalpaajien kanssa
Zytiga-tablettien ja sytotoksisen solunsalpaajahoidon samanaikaisen käytön turvallisuutta ja tehoa ei ole varmistettu (ks. kohta Farmakodynamiikka).
Apuaineintoleranssi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä. Tämä lääkevalmiste sisältää 27 mg (1,17 mmol) natriumia per kahden tabletin annos, joka vastaa 1,35 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Mahdolliset riskit
Etäpesäkkeistä eturauhassyöpää sairastavilla miehillä, mukaan lukien Zytiga-hoitoa saavat potilaat, saattaa esiintyä anemiaa ja seksuaalisia toimintahäiriöitä.
Vaikutukset luustolihaksiin
Zytiga-hoitoa saaneilla potilailla on raportoitu myopatiaa ja rabdomyolyysia. Useimmat tapaukset kehittyivät kuuden ensimmäisen hoitokuukauden aikana ja potilaat toipuivat Zytiga-hoidon lopettamisen jälkeen. Samanaikaisessa hoidossa lääkevalmisteilla, joihin tiedetään liittyvän myopatiaa/rabdomyolyysiä, suositellaan noudattamaan varovaisuutta.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Voimakkaita CYP3A4:n indusoijia on vältettävä hoidon aikana (paitsi jos käytettävissä ei ole muuta hoitovaihtoehtoa), koska tällöin on olemassa riski, että altistus abirateronille pienenee (ks. kohta Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset).
Abirateroni ja prednisoni/prednisoloni yhdessä Ra-223:n kanssa
Abirateronin ja prednisonin/prednisolonin käyttö yhdessä Ra-223:n kanssa on vasta-aiheista (ks. kohta Vasta-aiheet) oireettomilla tai vähäoireisilla eturauhassyöpää sairastavilla potilailla kliinisissä tutkimuksissa todetun lisääntyneen luunmurtumien riskin ja mahdollisesti lisääntyneen kuolleisuuden riskin vuoksi.
On suositeltavaa, että Ra-223-hoitoa ei aloiteta vähintään viiteen päivään Zytiga-valmisteen ja prednisonin/prednisolonin yhdistelmän viimeisen annon jälkeen.
Yhteisvaikutukset
Ruoan vaikutus abirateroniin
Valmisteen antaminen ruoan kanssa lisää abirateronin imeytymistä huomattavasti. Tämän lääkevalmisteen tehoa ja turvallisuutta ruokailun yhteydessä otettuna ei ole varmistettu, joten sitä ei saa ottaa ruoan kanssa (ks. kohdat Annostus ja antotapa sekä Farmakokinetiikka).
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Muiden lääkevalmisteiden mahdolliset vaikutukset abirateronialtistukseen
Kliinisessä farmakokineettisessä yhteisvaikutustutkimuksessa, jossa terveet koehenkilöt saivat ensin vahvaa CYP3A4:n indusoijaa rifampisiinia 600 mg/vrk 6 vuorokauden ajan ja sen jälkeen 1 000 mg abirateroniasetaattia kerta-annoksena, abirateronin AUC∞-arvon keskiarvo pieneni 55 prosenttia.
Voimakkaita CYP3A4:n indusoijia (esim. fenytoiinia, karbamatsepiinia, rifampisiinia, rifabutiinia, rifapentiinia, fenobarbitaalia, mäkikuismaa [Hypericum perforatum]) on vältettävä hoidon aikana, paitsi jos muuta hoitovaihtoehtoa ei ole käytettävissä.
Erillisessä terveillä koehenkilöillä tehdyssä kliinisessä farmakokineettisessä yhteisvaikutustutkimuksessa vahvan CYP3A4:n estäjän ketokonatsolin samanaikaisella annolla ei ollut kliinisesti merkityksellistä vaikutusta abirateronin farmakokinetiikkaan.
Abirateronin mahdolliset vaikutukset altistukseen muille lääkevalmisteille
Abirateroni on lääkeaineita maksassa metaboloivien entsyymien CYP2D6 ja CYP2C8 estäjä. Abirateroniasetaatin vaikutuksia (yhdistelmänä prednisonin kanssa) CYP2D6-substraatin dekstrometorfaanin kerta-annokseen selvittäneessä tutkimuksessa systeeminen altistus (AUC) dekstrometorfaanille suureni noin 2,9-kertaiseksi. Dekstrometorfaanin aktiivisen metaboliitin, dekstrofaanin, AUC24-arvo suureni noin 33 %.
Varovaisuutta suositellaan käytössä CYP2D6-entsyymin välityksellä aktivoituvien tai metaboloituvien lääkevalmisteiden kanssa, etenkin jos lääkkeen terapeuttinen indeksi on kapea. Jos CYP2D6-entsyymin välityksellä metaboloituvan lääkevalmisteen terapeuttinen indeksi on kapea, kyseisen lääkkeen annoksen pienentämistä on harkittava. Esimerkkejä CYP2D6-entsyymin välityksellä metaboloituvista lääkevalmisteista ovat metoprololi, propranololi, desipramiini, venlafaksiini, haloperidoli, risperidoni, propafenoni, flekainidi, kodeiini, oksikodoni ja tramadoli (näistä kolme viimeisintä tarvitsee CYP2D6-entsyymiä aktiivisen analgeettisen metaboliitin muodostumiseen).
CYP2C8-entsyymin välityksellä vaikuttavien lääkkeiden välisiä yhteisvaikutuksia terveillä tutkittavilla selvittäneessä tutkimuksessa pioglitatsonin AUC suureni 46 % ja pioglitatsonin aktiivisten metaboliittien, M-III:n ja M-IV:n, AUC-arvot pienenivät 10 %, kun pioglitatsonia annettiin yhdessä 1 000 mg:n abirateroniasetaattikerta-annoksen kanssa. Potilaita on seurattava CYP2C8:n substraattiin liittyvien toksisuuden oireiden havaitsemiseksi, jos samanaikaisesti käytettävän CYP2C8:n substraatin terapeuttinen indeksi on kapea. Esimerkkejä CYP2C8:n välityksellä metaboloituvista lääkevalmisteista ovat mm. pioglitatsoni ja repaglinidi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pääasiallisten metaboliittien abirateronisulfaatin ja N-oksidiabirateronisulfaatin osoitettiin in vitro estävän lääkeaineita maksasoluihin kuljettavan OATP1B1:n toimintaa, minkä seurauksena OATP1B1:n välityksellä eliminoituvien lääkevalmisteiden pitoisuudet saattavat suurentua. Kuljettajaproteiineihin perustuvien yhteisvaikutusten varmistamiseksi ei ole kliinisiä tietoja saatavilla.
QT-aikaa tunnetusti pidentävien lääkkeiden samanaikainen käyttö
Androgeenideprivaatiohoito saattaa pidentää QT-aikaa, joten Zytiga-valmisteen käytössä on oltava varovainen, kun samanaikaisesti käytetään QT-aikaa tunnetusti pidentäviä lääkevalmisteita tai lääkevalmisteita, jotka voivat aiheuttaa kääntyvien kärkien takykardiaa (torsades de pointes), kuten ryhmän IA (esim. kinidiini, disopyramidi) tai luokan III (esim. amiodaroni, sotaloli, dofetilidi, ibutilidi) rytmihäiriölääkkeitä, metadonia, moksifloksasiinia, psykoosilääkkeitä, tms.
Spironolaktonin samanaikainen käyttö
Spironolaktoni sitoutuu androgeenireseptoriin ja saattaa suurentaa prostataspesifisen antigeenin (PSA) pitoisuuksia. Spironolaktonin ja Zytiga-valmisteen samanaikaista käyttöä ei suositella (ks. kohta Farmakodynamiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Tämän lääkevalmisteen käytöstä ihmisellä raskauden aikana ei ole tietoja eikä Zytiga-valmiste ole tarkoitettu naisille, jotka voivat tulla raskaaksi.
Ehkäisy miehille ja naisille
Ei tiedetä, esiintyykö abirateronia tai sen metaboliitteja siemennesteessä. Jos potilas on sukupuoliyhteydessä raskaana olevan naisen kanssa, on käytettävä kondomia. Jos potilas on sukupuoliyhteydessä hedelmällisessä iässä olevan naisen kanssa, on käytettävä kondomia ja sen lisäksi toista tehokasta ehkäisymenetelmää. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Raskaus
Zytiga ei ole tarkoitettu naisille ja on vasta-aiheista naisille, jotka ovat tai saattavat olla raskaana (ks. kohdat Vasta-aiheet ja Prekliiniset tiedot turvallisuudesta).
Imetys
Zytiga ei ole tarkoitettu naisille.
Hedelmällisyys
Abirateroniasetaatti vaikutti uros- ja naarasrottien hedelmällisyyteen, mutta nämä vaikutukset korjautuivat täysin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Zytiga-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuustietojen yhteenveto
Vaiheen 3 Zytiga-tutkimusten yhdistetyssä haittavaikutusanalyysissä todettiin, että ≥ 10 %:lla potilaista havaittuja haittavaikutuksia olivat raajojen turvotus, hypokalemia, korkea verenpaine, virtsatieinfektio sekä suurentunut alaniiniaminotransferaasipitoisuus ja/tai suurentunut aspartaattiaminotransferaasipitoisuus. Muita merkittäviä haittavaikutuksia ovat sydämen toimintahäiriöt, maksatoksisuus, luunmurtumat ja allerginen alveoliitti.
Zytiga saattaa aiheuttaa vaikutusmekanisminsa farmakodynaamisena seurauksena korkeaa verenpainetta, hypokalemiaa ja nesteen kertymistä elimistöön. Vaiheen 3 tutkimuksissa havaittiin ennakoituja mineralokortikoidihaittavaikutuksia yleisemmin abirateroniasetaattia kuin lumelääkettä saaneilla potilailla: hypokalemia 18 % vs 8 %, korkea verenpaine 22 % vs 16 % ja nesteen kertyminen elimistöön (raajojen turvotus) 23 % vs 17 %. Abirateroniasetaattihoitoa ja lumehoitoa saaneiden potilaiden vertailussa CTCAE-luokituksen (versio 4.0) 3. ja 4. asteen hypokalemiaa havaittiin 6 %:lla vs 1 %:lla potilaista, CTCAE-luokituksen (versio 4.0) 3. ja 4. asteen hypertensiota havaittiin 7 %:lla vs 5 %:lla potilaista ja 3. ja 4. asteen nesteen kertymistä elimistöön (raajojen turvotusta) havaittiin 1 %:lla vs 1 %:lla potilaista. Mineralokortikoidivaikutukset saatiin yleensä hoidetuksi onnistuneesti lääkehoidolla. Kortikosteroidin samanaikainen käyttö vähentää näiden haittavaikutusten ilmaantuvuutta ja vaikeusastetta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Tutkimuksissa pitkälle edennyttä etäpesäkkeistä eturauhassyöpää sairastaville potilaille, jotka käyttivät LHRH-analogia tai joille oli aiemmin tehty kivesten poistoleikkaus, annettiin Zytiga-valmistetta 1 000 mg:n vuorokausiannoksina yhdistelmänä pienen prednisoni- tai prednisoloniannoksen (joko 5 mg tai 10 mg vuorokaudessa käyttöaiheesta riippuen) kanssa.
Kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen havaitut haittavaikutukset on lueteltu seuraavassa esiintymistiheyksittäin. Esiintymistiheydet on määritelty seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 1: Kliinisissä lääketutkimuksissa ja valmisteen markkinoilletulon jälkeen havaitut haittavaikutukset | |
| Elinjärjestelmä | Haittavaikutus ja esiintymistiheys |
| Infektiot | hyvin yleinen: virtsatieinfektio yleinen: sepsis |
| Immuunijärjestelmä | tuntematon: anafylaktiset reaktiot |
| Umpieritys | melko harvinainen: lisämunuaisten vajaatoiminta |
| Aineenvaihdunta ja ravitsemus | hyvin yleinen: hypokalemia yleinen: hypertriglyseridemia |
| Sydän | yleinen: sydämen vajaatoiminta*, angina pectoris, eteisvärinä, takykardia melko harvinainen: muut sydämen rytmihäiriöt |
| Verisuonisto | hyvin yleinen: hypertensio |
| Hengityselimet, rintakehä ja välikarsina | harvinainen: allerginen alveoliittia |
| Ruoansulatuselimistö | hyvin yleinen: ripuli yleinen: dyspepsia |
| Maksa ja sappi | hyvin yleinen: suurentunut ALAT-arvo ja/tai suurentunut ASAT-arvob harvinainen: fulminantti hepatiitti, akuutti maksan vajaatoiminta |
| Iho ja ihonalainen kudos | yleinen: ihottuma |
| Luusto, lihakset ja sidekudos | melko harvinainen: myopatia, rabdomyolyysi |
| Munuaiset ja virtsatiet | yleinen: verivirtsaisuus |
| Yleisoireet ja antopaikassa todettavat haitat | hyvin yleinen: perifeerinen edeema |
| Vammat ja myrkytykset | yleinen: luunmurtumat** |
* Sydämen vajaatoiminta sisältää kongestiivisen sydämen vajaatoiminnan, vasemman kammion toimintahäiriön ja pienentyneen ejektiofraktion b Suurentunut alaniiniaminotransferaasipitoisuus ja/tai suurentunut aspartaattiaminotransferaasipitoisuus käsittää suurentuneen ALAT-arvon, suurentuneen ASAT-arvon ja maksan toiminnan poikkeavuudet. | |
Abirateroniasetaattihoitoa saaneilla potilailla esiintyi seuraavia CTCAE-luokituksen (versio 4.0) 3. asteen haittavaikutuksia: hypokalemia 5 %; virtsatieinfektio 2 %, suurentunut alaniiniaminotransferaasipitoisuus ja/tai suurentunut aspartaattiaminotransferaasipitoisuus 4 %, korkea verenpaine 6 %, luunmurtumat 2 %; raajojen turvotus, sydämen vajaatoiminta ja eteisvärinä, kutakin 1 %. CTCAE-luokituksen (versio 4.0) 3. asteen hypertriglyseridemiaa ja angina pectorista esiintyi < 1 %:lla potilaista. CTCAE-luokituksen (versio 4.0) 4. asteen virtsatieinfektioita, suurentuneita alaniiniaminotransferaasipitoisuuksia ja/tai suurentuneita aspartaattiaminotransferaasipitoisuuksia, hypokalemiaa, sydämen vajaatoimintaa, eteisvärinää ja luunmurtumia esiintyi < 1 %:lla potilaista.
Korkean verenpaineen ja hypokalemian ilmaantuvuuden havaittiin olevan suuremmat hormonisensitiivistä eturauhassyöpää sairastavassa potilasryhmässä (tutkimus 3011). Korkeaa verenpainetta havaittiin hormonisensitiivistä eturauhassyöpää sairastavassa potilasjoukossa 36,7 %:lla potilaista (tutkimus 3011) verrattuna 11,8 %:iin potilaista tutkimuksessa 301 ja 20,2 %:iin potilaista tutkimuksessa 302. Hypokalemiaa havaittiin hormonisensitiivistä eturauhassyöpää sairastavassa potilasjoukossa 20,4 %:lla potilaista (tutkimus 3011) verrattuna 19,2 %:iin potilaista tutkimuksessa 301 ja 14,9 %:iin potilaista tutkimuksessa 302.
Haittavaikutusten ilmaantuvuus ja vaikeusaste olivat suuremmat potilaiden alaryhmässä, jossa ECOG-suorituskykypisteet olivat lähtötilanteessa 2, samoin kuin iäkkäillä potilailla (≥ 75-vuotiailla).
Joidenkin haittavaikutusten kuvaus
Kardiovaskulaariset vaikutukset
Kolmeen vaiheen 3 tutkimukseen ei otettu mukaan potilaita, joilla oli huonossa hoitotasapainossa oleva verenpainetauti, kliinisesti merkityksellinen sydäntauti, minkä oli osoittanut sydäninfarkti, tai valtimon tromboottinen tapahtuma kuuden edellisen kuukauden aikana, vaikea-asteinen tai huonossa hoitotasapainossa oleva angina pectoris tai NYHA-luokan III tai IV (tutkimus 301) tai NYHA-luokan II–IV (tutkimukset 3011 ja 302) sydämen vajaatoiminta tai jos sydämen ejektiofraktioksi oli mitattu < 50 %. Kaikki tutkimuksiin mukaan otetut (sekä vaikuttavaa hoitoa että lumehoitoa saaneet potilaat) saivat samanaikaisesti androgeenideprivaatiohoitoa, pääasiassa LHRH-analogeilla, mihin on liittynyt diabetesta, sydäninfarkti, aivoverenkiertohäiriöitä ja sydänperäisiä äkkikuolemia. Abirateroniasetaattihoitoa tai lumelääkettä saaneille potilaille ilmaantui vaiheen 3 tutkimuksissa sydämeen ja verisuonistoon liittyviä haittavaikutuksia seuraavasti: eteisvärinä 2,6 % vs 2,0 %, takykardia 1,9 % vs 1,0 %, angina pectoris 1,7 % vs 0,8 %, sydämen vajaatoiminta 0,7 % vs 0,2 % ja sydämen rytmihäiriöt 0,7 % vs 0,5 %.
Maksatoksisuus
Abirateroniasetaattihoitoa saaneilla potilailla on raportoitu maksatoksisuutta, kuten kohonneita ALAT- ja ASAT-arvoja ja kokonaisbilirubiinipitoisuuksia. Vaiheen 3 kliinisissä tutkimuksissa abirateroniasetaattihoitoa saaneista potilaista noin 6 %:lla on raportoitu 3. ja 4. asteen maksatoksisuutta (esim. ALAT- tai ASAT-arvon suurenemista yli viisinkertaiseksi normaaliarvojen ylärajaan nähden tai bilirubiinipitoisuuden suurenemista yli 1,5-kertaiseksi normaaliarvojen ylärajaan nähden) tyypillisesti kolmen ensimmäisen hoitokuukauden aikana. Tutkimuksessa 3011 havaittiin 3. tai 4. asteen maksatoksisuutta 8,4 %:lla Zytiga-hoitoa saaneista potilaista. Kymmenen Zytiga-hoitoa saaneen potilaan hoito keskeytettiin maksatoksisuuden vuoksi; kahdella heistä oli 2. asteen maksatoksisuutta, kuudella oli 3. asteen maksatoksisuutta ja kahdella oli 4. asteen maksatoksisuutta. Tutkimuksessa 3011 yksikään potilas ei kuollut maksatoksisuuden seurauksena. Vaiheen 3 kliinisissä tutkimuksissa maksan toimintakokeiden tulokset suurenivat todennäköisemmin, jos potilaan ALAT- tai ASAT-arvo oli koholla jo ennen hoitoa kuin jos potilaan arvot olivat hoidon alussa normaalit. Kun joko ALAT- tai ASAT-arvon havaittiin kohonneen yli viisinkertaiseksi normaaliarvojen ylärajaan nähden tai bilirubiinipitoisuuden havaittiin kohonneen yli kolminkertaiseksi normaaliarvojen ylärajaan nähden, abirateroniasetaattihoito keskeytettiin tai lopetettiin. Kahdessa tapauksessa esiintyi maksan toimintakokeiden tulosten huomattavaa suurenemista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Näiden kahden potilaan maksan toiminta oli hoidon alussa normaali, mutta heidän ALAT- tai ASAT-arvonsa suurenivat 15–40-kertaisiksi normaaliarvojen ylärajaan nähden ja bilirubiinipitoisuus suureni 2–6-kertaiseksi normaaliarvojen ylärajaan nähden. Kun hoito lopetettiin, kummankin potilaan maksan toimintakokeiden tulokset normalisoituivat ja toinen potilas sai uudelleen hoitoa eivätkä maksa-arvot enää suurentuneet. Tutkimuksessa 302 havaittiin ALAT- tai ASAT-arvon 3. tai 4. asteen suurenemista 35 (6,5 %) abirateroniasetaattihoitoa saaneella potilaalla. Kohonnut aminotransferaasipitoisuus korjautui 3 potilasta lukuun ottamatta kaikilla potilailla (näistä kolmesta potilaasta kahdelle ilmaantui useita uusia maksametastaaseja ja yhden potilaan ASAT-arvo pysyi suurentuneena noin 3 viikkoa viimeisen abirateroniasetaattiannoksen jälkeen). Vaiheen 3 kliinisissä tutkimuksissa 1,1 %:n abirateroniasetaattihoitoa saaneista potilaista ja 0,6 %:n lumelääkettä saaneista potilaista raportoitiin keskeyttäneen hoidon suurentuneen ALAT- ja ASAT-arvon tai maksan toimintahäiriön vuoksi. Maksatoksisuudesta aiheutuneita kuolemia ei raportoitu.
Maksatoksisuuden riskiä pienennettiin kliinisissä tutkimuksissa siten, että potilasta ei otettu mukaan tutkimukseen, jos hän tutkimuksen alkaessa sairasti hepatiittia tai hänellä oli merkittäviä poikkeavuuksia maksan toimintakokeiden tuloksissa. Tutkimukseen 3011 ei otettu mukaan potilaita, joilla oli lähtötilanteessa ALAT- ja ASAT-arvo > 2,5-kertainen normaaliarvojen ylärajaan nähden, bilirubiinipitoisuus > 1,5 -kertainen normaaliarvojen ylärajaan nähden tai aktiivinen tai oireinen virushepatiitti tai krooninen maksasairaus; askitesta tai maksan toimintahäiriöstä johtuva verenvuotosairaus. Tutkimukseen 301 ei otettu mukaan potilaita, joiden ALAT- ja ASAT-arvo oli ennen hoidon aloittamista ≥ 2,5-kertainen normaaliarvojen ylärajaan nähden, kun potilaan maksassa ei ollut etäpesäkkeitä, ja > 5-kertainen normaaliarvojen ylärajaan nähden, kun potilaalla oli maksassa etäpesäkkeitä. Tutkimukseen 302 osallistumaan soveltuviksi ei katsottu potilaita, joilla oli maksassa etäpesäkkeitä, eikä siihen otettu mukaan potilaita, joiden ALAT- ja ASAT-arvo oli ennen hoidon aloittamista ≥ 2,5-kertainen normaaliarvojen ylärajaan nähden. Jos kliinisiin tutkimuksiin osallistuvalle potilaalle kehittyi maksan toimintakokeiden poikkeavuuksia, nämä hoidettiin tehokkaasti keskeyttämällä hoito ja sallimalla hoidon jatkaminen vasta, kun maksan toimintakokeiden tulokset olivat palautuneet potilaalla hoitoa edeltävälle tasolle (ks. kohta Annostus ja antotapa). Potilaan hoitoa ei enää jatkettu, jos ALAT- tai ASAT-arvo oli suurentunut > 20-kertaiseksi normaaliarvojen ylärajaan nähden. Tämän potilasryhmän hoidon jatkamisen turvallisuutta ei tiedetä. Maksatoksisuuden mekanismia ei tunneta.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Zytigan yliannostuksesta ihmisillä on vähän tietoa.
Spesifistä vasta-ainetta ei ole. Yliannostuksen yhteydessä valmisteen antaminen on lopetettava ja potilaalle on annettava yleisiä elintoimintoja tukevaa hoitoa, johon sisältyy potilaan sydämen toiminnan seuraaminen rytmihäiriöiden varalta sekä hypokalemian ja nesteen elimistöön kertymisen oireiden ja löydösten seuranta. Maksan toimintaa on myös seurattava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Endokrinologiset lääkeaineet, muut hormoniantagonistit ja vastaavat valmisteet, ATC-koodi: L02BX03.
Vaikutusmekanismi
Abirateroniasetaatti (Zytiga) muuntuu in vivo abirateroniksi, joka on androgeenin biosynteesin estäjä. Abirateroni estää selektiivisesti erityisesti entsyymiä 17α-hydroksylaasi/C17,20-lyaasi (CYP17). Tämä entsyymi ilmentyy kiveksissä, lisämunuaisessa ja eturauhaskasvaimen kudoksessa tapahtuvassa androgeenien biosynteesissä ja sitä tarvitaan tähän biosynteesiin. CYP17 katalysoi pregnenolonin ja progesteronin muuntumista testosteronin esiasteiksi, DHEA:ksi ja androsteenidioniksi 17α-hydroksylaation ja C17,20-sidoksen katkeamisen kautta. CYP17:n toiminnan estyminen lisää myös mineralokortikoidituotantoa lisämunuaisissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Androgeeneille herkkä eturauhassyöpä reagoi androgeenipitoisuutta pienentävään hoitoon. Androgeenideprivaatiohoito, kuten LHRH-analogihoito tai kivesten poisto, vähentävät androgeenien muodostumista kiveksissä, mutta eivät vaikuta androgeenien muodostumiseen lisämunuaisissa tai kasvaimessa. Zytiga-hoito yhdessä LHRH-analogihoidon (tai kivesten poistoleikkauksen) kanssa pienentää seerumin testosteronipitoisuuden havaitsemisrajan alapuolelle (kaupallisilla määritysmenetelmillä).
Farmakodynaamiset vaikutukset
Zytiga pienentää seerumin testosteronipitoisuuden ja muiden androgeenien pitoisuudet pienemmiksi kuin LHRH-analogihoito tai kivesten poistoleikkaus yksinään. Tämä johtuu CYP17-entsyymin selektiivisestä estymisestä, koska CYP17-entsyymiä tarvitaan androgeenien biosynteesiin. PSA on eturauhassyöpää sairastavien potilaiden biomarkkeri. Vaiheen 3 tutkimuksessa potilailla, joiden taksaania sisältävä solunsalpaajahoito oli epäonnistunut, 38 %:lla abirateroniasetaattihoitoa saaneista potilaista hoitoa edeltävä PSA-pitoisuus pieneni vähintään 50 % verrattuna 10 %:iin lumelääkkeellä hoidetuista potilaista.
Kliininen teho ja turvallisuus
Teho varmistettiin kolmessa satunnaistetussa, lumelääkekontrolloidussa vaiheen 3 kliinisessä monikeskustutkimuksessa (tutkimukset 3011, 302 ja 301) potilailla, jotka sairastivat etäpesäkkeistä hormonisensitiivistä eturauhassyöpää tai etäpesäkkeistä kastraatioresistenttiä eturauhassyöpää. Tutkimuksessa 3011 mukana olleet potilaat olivat saaneet etäpesäkkeisen hormonisensitiivisen eturauhassyövän diagnoosin äskettäin (enintään 3 kuukautta ennen satunnaistamista) ja heillä oli korkean riskin ennustetekijöitä. Korkean riskin ennusteeksi määriteltiin, että potilaalla oli vähintään kaksi seuraavista kolmesta riskitekijästä: 1) Gleason-pisteet ≥ 8; 2) luuston kuvauksessa todettu kolme tai sitä useampi leesio; 3) mitattavissa oleva viskeraalinen (imusolmukkeita lukuun ottamatta) etäpesäke. Vaikuttavaa hoitoa saaneelle ryhmälle annettiin Zytiga-valmistetta 1 000 mg:n vuorokausiannoksina yhdistettynä kerran päivässä annettavaan pieneen prednisoniannokseen (5 mg), joiden lisäksi annettiin tavanomainen androgeenideprivaatiohoito (LHRH:n agonisti tai kivesten poistoleikkaus). Vertailuryhmän potilaat saivat androgeenideprivaatiohoitoa ja lumehoitoa sekä Zytiga- että prednisonihoidon sijaan. Tutkimuksessa 302 mukana olleet potilaat eivät olleet saaneet aiemmin dosetakselia, kun taas tutkimuksessa 301 mukana olleet potilaat olivat aiemmin saaneet dosetakselia. Potilaat saivat LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus. Vaikuttavaa hoitoa saaneelle ryhmälle annettiin Zytiga-valmistetta 1 000 mg:n vuorokausiannoksina yhdistettynä kaksi kertaa päivässä annettavaan pieneen prednisoni- tai prednisoloniannokseen (5 mg). Verrokkipotilaat saivat lumelääkettä ja pienen prednisoni- tai prednisoloniannoksen (5 mg) kaksi kertaa päivässä.
Seerumin PSA-pitoisuuden riippumattomat muutokset eivät aina ennusta kliinistä hyötyä. Sen vuoksi kaikissa näissä tutkimuksissa suositeltiin, että potilaat jatkavat tutkimushoitoa siihen saakka, kunnes he täyttävät kyseisen tutkimuksen osalta määritellyt tutkimuksen päättymiseen johtavat kriteerit.
Spironolaktonin käyttö ei ollut sallittua missään näistä tutkimuksista, koska spironolaktoni sitoutuu androgeenireseptoriin ja saattaa suurentaa PSA-pitoisuuksia.
Tutkimus 3011 (äskettäin diagnosoitua korkean riskin hormonisensitiivistä eturauhassyöpää sairastavat potilaat)
Tutkimukseen 3011 mukaan otettujen potilaiden (n = 1 199) iän mediaani oli 67 vuotta. Zytiga-hoitoa saaneiden potilaiden lukumäärä etnisen taustan mukaan ryhmiteltynä oli valkoihoisia 832 (69,4 %), aasialaisia 246 (20,5 %), tummaihoisia tai afroamerikkalaisia 25 (2,1 %), muita 80 (6,7 %), ei tiedossa / ei raportoitu 13 (1,1 %) ja Amerikan intiaaneja tai Alaskan alkuperäiskansojen väestöä 3 (0,3 %). Eastern Cooperative Oncology Group (ECOG) -suorituskykypisteet olivat 97 %:lla potilaista 0 tai 1. Potilaita, joilla tiedettiin olevan etäpesäkkeitä aivoissa, huonossa hoitotasapainossa oleva verenpainetauti, merkittävä sydänsairaus tai NYHA-luokan II–IV sydämen vajaatoimintaa, ei otettu mukaan tutkimukseen. Potilaita, joiden etäpesäkkeistä eturauhassyöpää oli aiemmin hoidettu lääkehoidolla, sädehoidolla tai leikkauksella, ei otettu mukaan tutkimukseen, lukuun ottamatta potilaita, jotka olivat saaneet enintään 3 kuukauden ajan androgeenideprivaatiohoitoa tai 1 hoitojakson palliatiivista sädehoitoa tai joille oli tehty leikkaus etäpesäkkeisen sairauden aiheuttamien oireiden hoitamiseksi. Ensisijaisia tehon päätetapahtumia olivat kokonaiselossaoloaika ja radiologinen tautivapaa elossaoloaika (radiographic progression-free survival, rPFS). Kipua kuvaavien Brief Pain Inventory Short Form (BPI-SF) -pisteiden mediaani oli lähtötilanteessa 2,0 sekä aktiivista hoitoa että lumehoitoa saaneessa ryhmässä. Ensisijaisten päätetapahtumien lisäksi hyötyä arvioitiin myös mittaamalla aikaa aikaa luustoon liittyviin tapahtumiin, aikaa seuraavaan eturauhassyöpähoitoon, aikaa solunsalpaajahoidon aloittamiseen, aikaa kivun pahenemiseen ja aikaa PSA-arvon etenemiseen. Hoitoa jatkettiin, kunnes sairaus eteni, potilas vetäytyi tutkimuksesta, ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai potilas kuoli.
Radiologinen tautivapaa elossaoloaika määriteltiin ajaksi satunnaistamisesta sairauden radiologisesti todettuun etenemiseen tai potilaan kuolemaan mistä tahansa syystä. Radiologisesti todettuun etenemiseen kuuluivat sairauden eteneminen luuston kuvauksen perusteella (muokattujen Prostate Cancer Working Group-2 [PCWG2] -kriteerien mukaisesti) tai todettujen pehmytkudosleesioiden eteneminen TT- tai magneettikuvauksen perusteella (RECIST 1.1 -kriteerien mukaisesti).
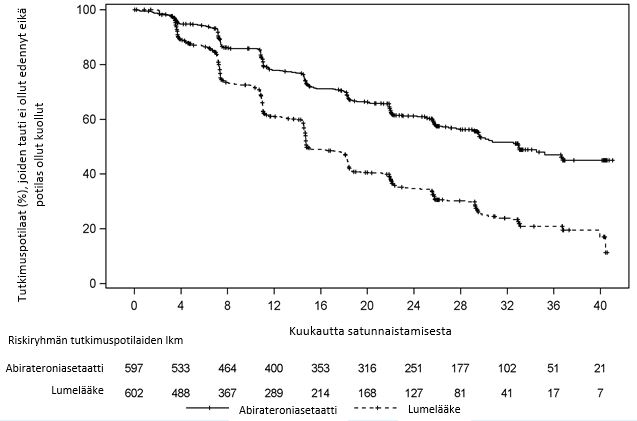
Hoitoryhmien välillä havaittiin merkittävä ero radiologisessa tautivapaassa elossaoloajassa (ks. taulukko 2 ja kuva 1).
Taulukko 2: Radiologinen tautivapaa elossaoloaika, ositettu analyysi; hoitoaikeen mukainen (ITT) potilasjoukko (tutkimus PCR3011) | ||
| AA-P | Lumelääke |
Satunnaistettuja tutkimuspotilaita | 597 | 602 |
Tapahtumia | 239 (40,0 %) | 354 (58,8 %) |
Sensuroituja | 358 (60,0 %) | 248 (41,2 %) |
|
|
|
Aika tapahtumaan (kuukautta) |
|
|
Mediaani (95 %:n CI) | 33,02 (29,57; NE) | 14,78 (14,69; 18,27) |
Vaihteluväli | (0,0+; 41,0+) | (0,0+; 40,6+) |
|
|
|
p-arvoa | < 0,0001 |
|
Riskisuhde (HR) (95 %:n CI)b | 0,466 (0,394; 0,550) |
|
Huom.: + = sensuroitu havainto, NE = ei arvioitavissa (not estimable). rPFS-tapahtuman määrittelyssä on huomioitu radiologisesti todettu eteneminen ja kuolema. AA-P = tutkimuspotilaat, jotka saivat abirateroniasetaattia ja prednisonia. a p-arvo on saatu ECOG-suorituskykypisteillä (0/1 tai 2) ja viskeraalisella leesiolla (ei tai kyllä) ositetulla log-rank-testillä. b Riskisuhde perustuu ositettuun suhteellisen riskin malliin. Riskisuhde < 1 suosii AA-P-ryhmää. | ||
| Kuva 1: Kaplan–Meier-käyrät radiologisesta tautivapaasta elossaoloajasta; hoitoaikeen mukainen (ITT) potilasjoukko (tutkimus PCR3011) |
 |
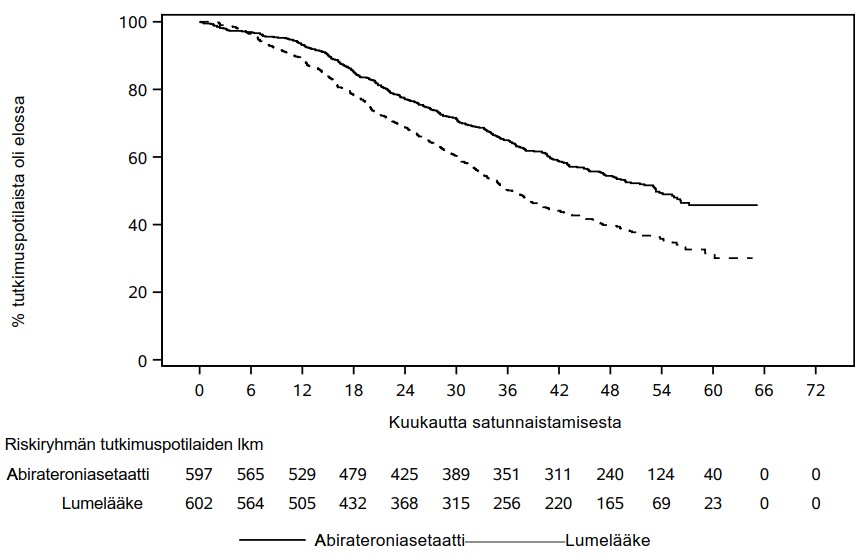
Kokonaiselossaoloaika parani tilastollisesti merkitsevästi, kun abirateroniasetaatti ja prednisoni yhdistettiin androgeenideprivaatiohoitoon ja kuoleman riski pieneni 34 % lumelääkkeen ja androgeenideprivaatiohoidon yhdistelmään verrattuna (HR = 0,66; 95 %:n CI: 0,56; 0,78; p < 0,0001) (ks. taulukko 3 ja kuva 2).
Taulukko 3: Joko Zytiga- tai lumelääkehoitoa saaneiden potilaiden kokonaiselossaoloaika; hoitoaikeen mukainen (ITT) potilasjoukko (tutkimus PCR3011) | ||
Kokonaiselossaoloaika | Zytigan ja prednisonin yhdistelmä (N = 597) | Lumelääke (N = 602) |
Kuolemantapauksia (%) | 275 (46 %) | 343 (57 %) |
Elossaoloajan mediaani (kuukautta) | 53,3 | 36,5 |
(95 %:n CI) | (48,2; NE) | (33,5; 40,0) |
Riskisuhde (HR) (95 %:n CI)1 | 0,66 (0,56; 0,78) | |
NE = ei arvioitavissa (not estimable) | ||
| Kuva 2: Kaplan–Meier-käyrät kokonaiselossaoloajasta; tutkimuksen PCR3011 analyysin hoitoaikeen mukainen (ITT) potilasjoukko |
 |
Potilaiden alaryhmäanalyysit suosivat yhdenmukaisesti Zytiga-hoitoa. Abirateroniasetaatin ja prednisonin yhdistelmän hoitovaikutus radiologiseen tautivapaaseen elossaoloaikaan ja kokonaiselossaoloaikaan oli yhdenmukaisesti suotuisa kaikissa koko tutkimusryhmän ennalta määritellyissä alaryhmissä, lukuun ottamatta niiden potilaiden alaryhmää, joiden ECOG-suorituskykypisteet olivat 2, heillä hyötyä ei havaittu. Otoskoko ryhmässä oli kuitenkin pieni (n = 40), mikä rajoittaa merkityksellisten päätelmien tekemistä.
Kokonaiselossaoloajassa ja radiologisessa tautivapaassa ajassa osoitettujen paranemisten lisäksi Zytiga-hoidosta osoitettiin olevan hyötyä lumehoitoon verrattuna kaikissa prospektiivisesti määritellyissä toissijaisissa päätetapahtumissa.
Tutkimus 302 (solunsalpaajilla aiemmin hoitamattomat potilaat)
Tähän tutkimukseen otettiin mukaan potilaita, jotka eivät olleet aiemmin saaneet solunsalpaajahoitoa ja joiden tauti oli oireeton tai oireet olivat lieviä ja joille solunsalpaajahoito ei ollut vielä kliinisesti aiheellista. Jos pahimmasta kivusta annetut Brief Pain Inventory-Short Form (BPI-SF) -pisteet olivat edellisten 24 tunnin aikana 0–1, potilaan katsottiin olleen oireeton, ja pisteiden 2–3 katsottiin merkitsevän lieviä oireita.
Tutkimukseen 302 (n = 1 088) mukaan otettujen Zytiga-hoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneiden potilaiden iän mediaani oli 71 vuotta ja lumelääkettä yhdistelmänä prednisonin tai prednisolonin kanssa saaneiden potilaiden iän mediaani oli 70 vuotta. Zytiga-hoitoa saaneiden potilaiden määrä rodun mukaan jaettuna oli 520 valkoihoista (95,4 %), 15 mustaihoista (2,8 %), 4 aasialaista (0,7 %) ja 6 muita (1,1 %). 76 % kummankin hoitoryhmän potilaista oli saanut Eastern Cooperative Oncology Group (ECOG) -suorituskykypisteet 0, ja 24 % potilaista oli saanut ECOG-suorituskykypisteet 1. Viidelläkymmenellä prosentilla potilaista oli etäpesäkkeitä yksinomaan luustossa. Lisäksi 31 %:lla potilaista oli etäpesäkkeitä luustossa ja pehmytkudoksessa tai imusolmukkeissa ja 19 %:lla potilaista oli etäpesäkkeitä yksinomaan pehmytkudoksessa tai imusolmukkeissa. Tutkimuksesta suljettiin pois potilaat, joilla oli viskeraalisia etäpesäkkeitä. Muita tehon ensisijaisia päätetapahtumia olivat kokonaiselossaolo ja radiologinen tautivapaa elossaoloaika (radiographic progression-free survival, rPFS). Hoidon hyötyä potilaalle arvioitiin muiden ensisijaisten päätetapahtumien lisäksi mittaamalla aikaa opiaattien käyttöön syöpäkivun hoitoon, aikaa sytotoksisen solunsalpaajahoidon aloittamiseen, aikaa ECOG-suorituskykypisteiden huononemiseen ≥ 1 pistettä ja aikaa taudin etenemiseen PSA-pitoisuuden perusteella, mikä perustui Prostate Cancer Working Group-2 (PCWG2) -kriteereihin. Tutkimushoito lopetettiin, kun todettiin taudin kiistaton kliininen eteneminen. Hoito voitiin lopettaa myös silloin, kun taudin radiologinen eteneminen tutkijan arvion perusteella varmistui.
Radiologista tautivapaata elossaoloaikaa (rPFS) arvioitiin PCWG2-kriteerien (luustomuutokset) ja modifioitujen RECIST-kriteerien (Response Evaluation Criteria In Solid Tumors) (pehmytkudosmuutokset) mukaisella sekvenssikuvauksella. Radiologista tautivapaata elossaoloaikaa analysoitiin arvioimalla taudin radiologista etenemistä keskitetysti.
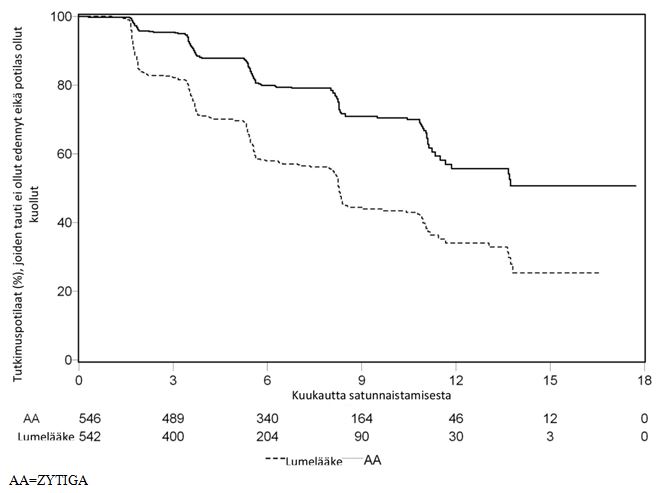
Radiologisen tautivapaan elossaoloajan suunniteltuna analysointiajankohtana todettiin 401 tapahtumaa, jolloin 150 (28 %) Zytiga-hoitoa saaneella potilaalla ja 251 (46 %) lumehoitoa saaneella potilaalla todettiin radiologinen löydös taudin etenemisestä tai potilas oli kuollut. Radiologisessa tautivapaassa elossaoloajassa todettiin merkittävä ero hoitoryhmien välillä (ks. taulukko 4 ja kuva 3).
Taulukko 4: Tutkimus 302: Potilaiden radiologinen tautivapaa elossaoloaika Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneilla, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus | ||
| Zytiga (N = 546) | Lumelääke (N = 542) | |
| Radiologinen tautivapaa elossaoloaika (rPFS) | ||
| Taudin eteneminen tai kuolema | 150 (28 %) | 251 (46 %) |
| rPFS:n mediaani, kuukautta | Ei saavutettu | 8,3 |
| (95 %:n CI) | (11,66; NE) | (8,12; 8,54) |
| p-arvo* | < 0,0001 | |
| Riskisuhde (HR)** | 0,425 | |
| (95 %:n CI) | (0,347; 0,522) | |
| NE= Ei arvioitu (Not estimated) * p-arvo on saatu lähtötilanteen ECOG-suorituskykypisteiden (0 tai 1) ositetusta log-rank-testistä ** Riskisuhde (Hazard ratio, HR) < 1 osoittaa Zytiga-hoidon paremmuuden | ||
| Kuva 3: Kaplan–Meier-käyrät radiologisesta tautivapaasta elossaoloajasta Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneilla potilailla, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus | |
 |
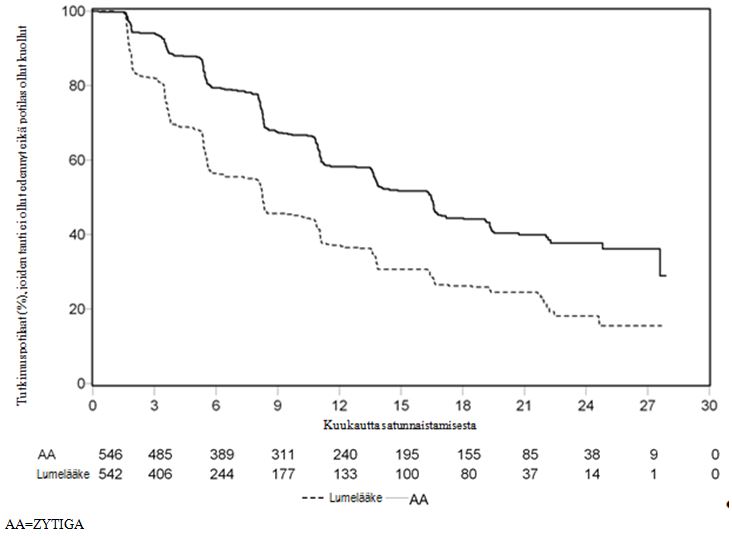
Tutkimuspotilastietojen keräämistä jatkettiin kuitenkin kokonaiselossaolon (overall survival, OS) toiseen välianalyysiin saakka. Tutkija kävi seurantana toteutettuna herkkyysanalyysinä läpi radiologisen tautivapaan elossaoloajan radiologiset tiedot, tiedot on esitetty taulukossa 5 ja kuvassa 4.
Kuudellasadallaseitsemällä (607) potilaalla todettiin radiologinen löydös taudin etenemisestä tai potilas oli kuollut: 271 (50 %) abirateroniasetaattia saaneessa ryhmässä ja 336 (62 %) lumelääkeryhmässä. Abirateroniasetaattihoito pienensi radiologisen löydöksen riskiä taudin etenemisestä tai kuoleman riskiä 47 % lumelääkkeeseen verrattuna (HR = 0,530; 95 %:n CI [0,451; 0,623]; p < 0,0001). rPFS:n mediaani oli abirateroniasetaattiryhmässä 16,5 kuukautta ja lumelääkeryhmässä 8,3 kuukautta.
Taulukko 5: Tutkimus 302: Potilaiden radiologinen tautivapaa elossaoloaika Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneilla, kun potilaat saivat lisäksi LHRH-agonistihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus (toisen välianalyysin eli tutkijan arvioiman kokonaiselossaolon ajankohtana) | ||
| Zytiga (N = 546) | Lumelääke (N = 542) | |
| Radiologinen tautivapaa elossaoloaika (rPFS) | ||
| Taudin eteneminen tai kuolema | 271 (50 %) | 336 (62 %) |
| rPFS:n mediaani, kuukautta | 16,5 | 8,3 |
| (95 %:n CI) | (13,80; 16,79) | (8,05; 9,43) |
| p-arvo* | < 0,0001 | |
| Riskisuhde (HR)** | 0,530 | |
| (95 %:n CI) | (0,451; 0,623) | |
| * p-arvo on saatu lähtötilanteen ECOG-suorituskykypisteiden (0 tai 1) mukaan ositetusta log-rank-testistä ** Riskisuhde (HR) < 1 osoittaa Zytiga-hoidon paremmuuden | ||
Kuva 4: Kaplan–Meier-käyrät radiologisesta tautivapaasta elossaoloajasta Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneista potilaista, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus (toisen välianalyysin eli tutkijan arvioiman kokonaiselossaolon ajankohtana) | |
 |
Suunniteltu välianalyysi kokonaiselossaolosta tehtiin, kun oli todettu 333 kuolemaa. Tutkimuksen sokkoutus purettiin havaitun kliinisen hyödyn laajuuden perusteella, ja lumehoitoryhmän potilaille annettiin mahdollisuus saada Zytiga-hoitoa. Kokonaiselossaolo oli pidempi Zytiga-hoidon yhteydessä lumelääkkeeseen verrattuna, ja kuolemanriski oli 25 % pienempi (HR = 0,752; 95 %:n CI: [0,606, 0,934], p = 0,0097), mutta kokonaiselossaolon tulokset eivät olleet valmiit eikä välianalyysissa saavutettu tilastollisen merkitsevyyden suhteen etukäteen määriteltyä keskeyttämisrajaa (ks. taulukko 6). Elossaolon seurantaa jatketaan tämän välianalyysin jälkeen.
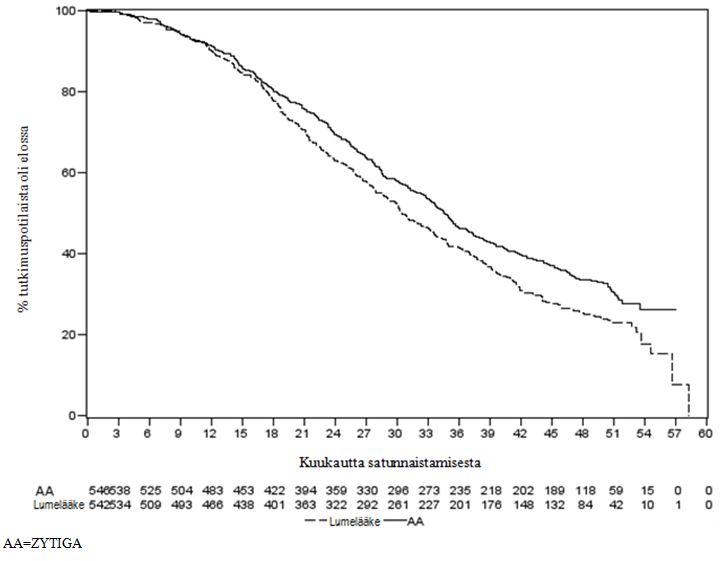
Suunniteltu kokonaiselossaolon loppuanalyysi tehtiin, kun oli todettu 741 kuolemaa (seuranta-ajan mediaani 49 kuukautta). Zytiga-hoitoa saaneista potilaista oli kuollut kuusikymmentäviisi prosenttia (354/546) verrattuna 71 %:iin (387/542) lumehoitoa saaneista potilaista. Zytiga-hoitoa saaneessa ryhmässä osoitettiin tilastollisesti merkitsevä hyöty kokonaiselossaolon suhteen, sillä kuolemanriski oli pienentynyt 19,4 % (HR = 0,806; 95 %:n CI: [0,697; 0,931], p = 0,0033) ja kokonaiselossaolon mediaani oli pidentynyt 4,4 kuukautta (Zytiga-hoidossa 34,7 kuukautta, lumehoidossa 30,3 kuukautta) (ks. taulukko 6 ja kuva 5). Tulosten tällainen paraneminen osoitettiin, vaikka 44 % lumehoitoryhmän potilaista sai myöhemmin Zytiga-hoitoa.
Taulukko 6: Tutkimus 302: Potilaiden kokonaiselossaolo Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneilla, kun potilaat saivat lisäksi LHRH-agonistihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus | ||
| Zytiga (N = 546) | Lumelääke (N = 542) | |
| Elossaolon välianalyysi | ||
| Kuolemantapauksia (%) | 147 (27 %) | 186 (34 %) |
| Elossaoloajan mediaani (kuukautta) | Ei saavutettu | 27,2 |
| (95 %:n CI) | (NE; NE) | (25,95; NE) |
| p-arvo* | 0,0097 | |
| Riskisuhde (HR)** | 0,752 | |
| (95 %:n CI) | (0,606; 0,934) | |
| Elossaolon loppuanalyysi | ||
| Kuolemantapauksia | 354 (65 %) | 387 (71 %) |
| Kokonaiselossaoloajan mediaani (kuukautta) (95 %:n CI) | 34,7 (32,7; 36,8) | 30,3 (28,7; 33,3) |
| p-arvo* | 0,0033 | |
| Riskisuhde (HR)** | 0,806 | |
| (95 %:n CI) | (0,697; 0,931) | |
| NE= Ei arvioitu (Not Estimated) * p-arvo on saatu lähtötilanteen ECOG-suorituskykypisteiden (0 tai 1) mukaan ositetusta log-rank-testistä ** Riskisuhde (HR) < 1 osoittaa Zytiga-hoidon paremmuuden | ||
Kuva 5: Kaplan–Meier-elossaolokäyrät Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneista potilaista, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus, loppuanalyysi | |
 |
Kokonaiselossaoloajassa ja rPFS:ssä havaitun paranemisen lisäksi kaikki toissijaiset päätemuuttujat olivat Zytiga-hoidon yhteydessä lumelääkehoitoa paremmat seuraavasti:
Aika PSA-pitoisuuden etenemiseen PCWG2-kriteerien perusteella: Ajan mediaani PSA-pitoisuuden etenemiseen oli Zytiga-hoitoa saaneilla potilailla 11,1 kuukautta ja lumelääkettä saaneilla potilailla 5,6 kuukautta (HR = 0,488; 95 %:n CI: [0,420; 0,568], p < 0,0001). Aika PSA-pitoisuuden etenemiseen oli Zytiga-hoidon yhteydessä noin kaksinkertainen (HR = 0,488). Niiden tutkimuspotilaiden osuus, joilla PSA-vaste varmistui, oli Zytiga-ryhmässä suurempi kuin lumelääkeryhmässä (62 % vs 24 %; p < 0,0001). Jos potilaalla oli mitattavissa oleva pehmytkudossairaus, huomattavasti useammalla Zytiga-ryhmän potilaalla havaittiin täydellinen tai osittainen vaste.
Aika opiaattien käyttöön syöpäkivun hoitoon: Ajan mediaani opiaattien käyttöön eturauhassyövästä aiheutuvan kivun hoitoon oli Zytiga-hoitoa saaneilla potilailla loppuanalyysin ajankohtana 33,4 kuukautta ja lumelääkettä saaneilla potilailla se oli 23,4 kuukautta (HR = 0,721; 95 %:n CI: [0,614; 0,846], p = < 0,0001).
Aika sytotoksisen solunsalpaajahoidon aloittamiseen: Ajan mediaani sytotoksisen solunsalpaajahoidon aloittamiseen oli Zytiga-hoitoa saaneilla potilailla 25,2 kuukautta ja lumelääkettä saaneilla potilailla 16,8 kuukautta (HR = 0,580; 95 %:n CI: [0,487; 0,691], p < 0,0001).
Aika ECOG-suorituskykypisteiden huononemiseen ≥ 1 pistettä: Ajan mediaani ECOG-suorituskykypisteiden huononemiseen ≥ 1 pistettä oli Zytiga-hoitoa saaneilla potilailla 12,3 kuukautta ja lumelääkettä saaneilla potilailla 10,9 kuukautta (HR = 0,821; 95 %:n CI: [0,714; 0,943], p = 0,0053).
Tutkimuksen seuraavat päätetapahtumat osoittivat Zytiga-hoidon tilastollisesti merkitsevän paremmuuden:
Objektiivinen vaste: Objektiiviseksi vasteeksi määriteltiin niiden tutkimuspotilaiden osuus, joilla oli mitattavissa oleva tauti ja jotka saivat RECIST-kriteerien (imusolmukkeen piti olla hoidon alkaessa kooltaan ≥ 2 cm, jotta se katsottiin kohdemuutokseksi) perusteella täydellisen tai osittaisen vasteen. Niiden tutkimuspotilaiden osuus, joilla oli hoidon alkaessa mitattavissa oleva tauti ja jotka saivat objektiivisen vasteen, oli Zytiga-ryhmässä 36 % ja lumelääkeryhmässä 16 % (p < 0,0001).
Kipu: Zytiga-hoito vähensi huomattavasti (18 %) kivun voimakkuuden keskimääräistä pahenemisriskiä lumelääkkeeseen verrattuna (p = 0,0490). Mediaani aika etenemiseen oli Zytiga-ryhmässä 26,7 kuukautta ja lumelääkeryhmässä 18,4 kuukautta.
Aika FACT-P-pisteiden (kokonaispisteiden) huononemiseen: Zytiga-hoito pienensi FACT-P-pisteiden (kokonaispisteiden) huononemisen riskiä 22 % lumelääkkeeseen verrattuna (p = 0,0028). Mediaani aika FACT-P-pisteiden (kokonaispisteiden) huononemiseen oli Zytiga-ryhmässä 12,7 kuukautta ja lumelääkeryhmässä 8,3 kuukautta.
Tutkimus 301 (potilaat, jotka olivat aiemmin saaneet solunsalpaajahoitoa)
Tutkimukseen 301 mukaan otetut potilaat olivat saaneet aiemmin dosetakselihoitoa. Potilaan taudin etenemistä dosetakselihoidon aikana ei edellytetty, koska tästä solunsalpaajasta aiheutuva toksisuus olisi saattanut johtaa hoidon lopettamiseen. Potilaat jatkoivat tutkimushoitoa, kunnes PSA-pitoisuuden perusteella havaittiin taudin etenevän (varmistettiin 25 %:n suureneminen potilaan lähtötilanteen pitoisuuteen/pienimpään pitoisuuteen nähden) ja samalla todetaan tutkimussuunnitelmassa määritelty taudin radiologinen eteneminen, oireiden eteneminen tai kliininen eteneminen. Ketokonatsolia eturauhassyövän hoitoon aiemmin saaneita potilaita ei otettu tutkimukseen mukaan. Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika.
Tutkimukseen mukaan otettujen potilaiden iän mediaani oli 69 vuotta (vaihteluväli 39−95 vuotta). Zytiga-hoitoa saaneiden potilaiden määrä rodun mukaan jaettuna oli 737 valkoihoista (93,2 %), 28 mustaihoista (3,5 %), 11 aasialaista (1,4 %) ja 14 muita (1,8 %). Tutkimukseen mukaan otetuista potilaista 11 % oli saanut ECOG-suorituskykypisteet 2. Potilaista 70 %:lla oli radiologisia löydöksiä taudin etenemisestä, mihin saattoi liittyä taudin eteneminen PSA-pitoisuuden perusteella. Potilaista 70 % oli saanut yhtä aiempaa sytotoksista solunsalpaajahoitoa ja 30 % oli saanut kahta tällaista hoitoa. 11 %:lla Zytiga-hoitoa saaneista potilaista oli etäpesäkkeitä maksassa.
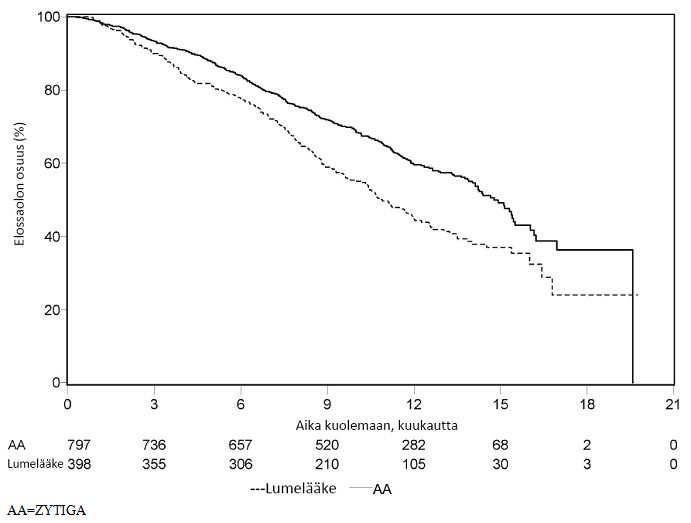
Suunniteltu analyysi tehtiin, kun oli todettu 552 kuolemaa, 42 % (333 potilasta 797 potilaasta) Zytiga-hoitoa saaneista potilaista oli kuollut verrattuna 55 %:iin (219 potilasta 398 potilaasta) lumelääkehoitoa saaneista potilaista. Zytiga-hoitoa saaneilla potilailla havaittiin kokonaiselossaolon mediaanissa tilastollisesti merkitsevä paraneminen (ks. taulukko 7).
| Taulukko 7: Potilaiden kokonaiselossaolo Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneilla, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus | ||
| Zytiga (N = 797) | Lumelääke (N = 398) | |
| Ensisijainen elossaoloajan analyysi | ||
| Kuolemantapauksia (%) | 333 (42 %) | 219 (55 %) |
| Elossaoloajan mediaani (kuukautta) (95 %:n CI) | 14,8 (14,1; 15,4) | 10,9 (10,2; 12,0) |
| p-arvoa | < 0,0001 | |
| HR (95 %:n CI)b | 0,646 (0,543; 0,768) | |
| Päivitetty elossaoloajan analyysi | ||
| Kuolemantapauksia (%) | 501 (63 %) | 274 (69 %) |
| Elossaoloajan mediaani (kuukautta) (95 %:n CI) | 15,8 (14,8; 17,0) | 11,2 (10,4; 13,1) |
| HR (95 %:n CI)b | 0,740 (0,638; 0,859) | |
| ap-arvo on saatu ECOG-suorituskykypisteiden (0–1 vs 2), kipupisteiden (ei kipua vs kipua esiintyy), aiempien solunsalpaajahoitojen lukumäärän (1 vs 2) ja sairauden etenemistyypin (vain PSA vs radiologinen) mukaan ositetusta log-rank-testistä. bHR on saatu ositetusta suhteellisesta riskimallista. HR < 1 osoittaa Zytiga-hoidon paremmuuden. | ||
Muutaman ensimmäisen hoitokuukauden jälkeen kaikkina arviointiajankohtina Zytiga-hoitoa saaneista potilaista oli elossa suurempi osuus verrattuna lumelääkehoitoa saaneisiin potilaisiin (ks. kuva 6).
| Kuva 6: Kaplan-Meier-elossaolokäyrät Zytiga- tai lumelääkehoitoa yhdistelmänä prednisonin tai prednisolonin kanssa saaneista potilaista, kun potilaat saivat lisäksi LHRH-analogihoitoa tai heille oli aiemmin tehty kivesten poistoleikkaus | |
 |
Alaryhmän elossaoloanalyysi osoitti elossaolon olevan johdonmukaisesti suurempi Zytiga-hoidon yhteydessä (ks. kuva 7).
| Kuva 7: Kokonaiselossaoloaika alaryhmittäin: HR ja 95 %:n CI | |
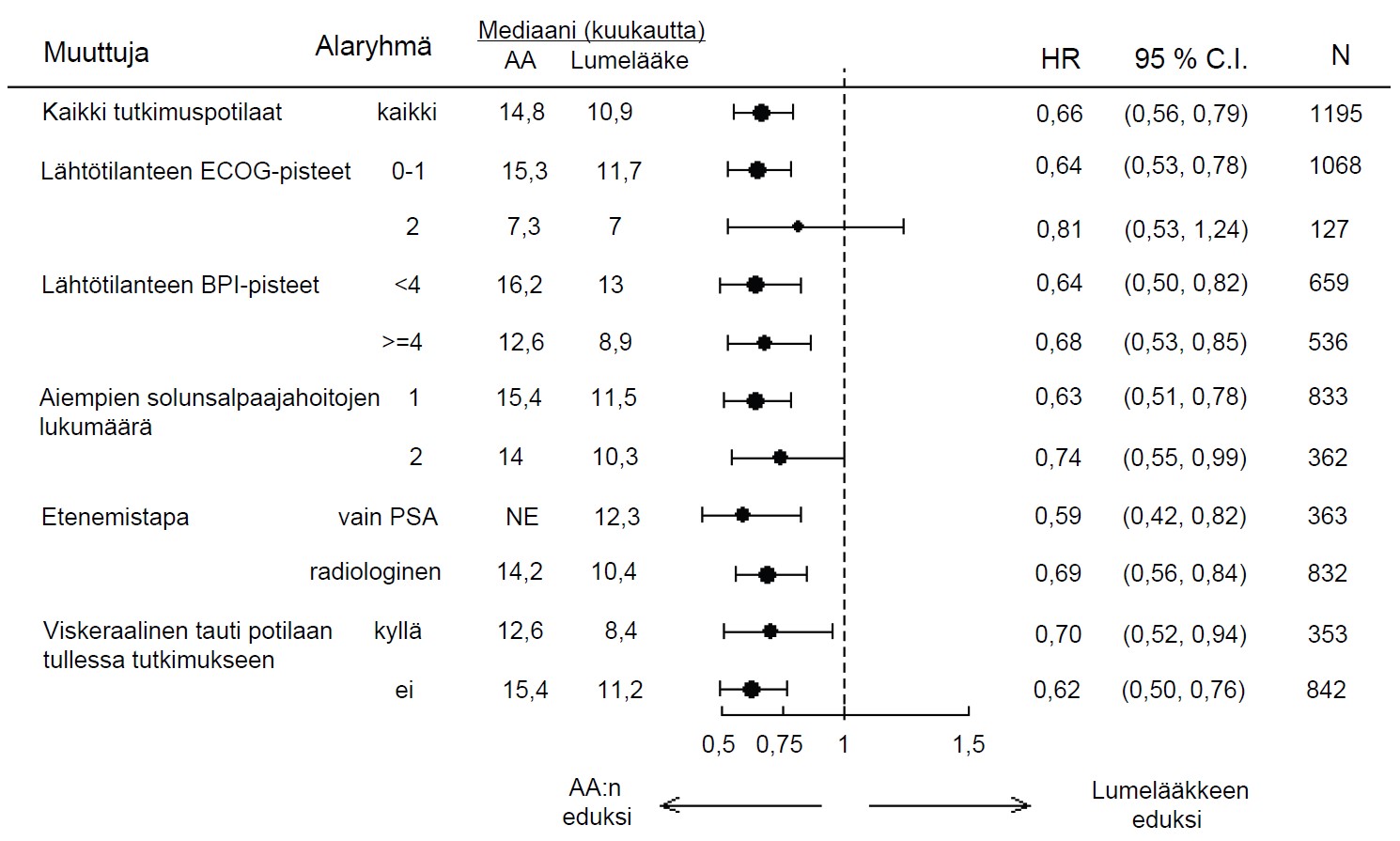 | |
| AA=Zytiga; BPI=suppea kipuarvio (Brief Pain Inventory); C.I.= luottamusväli (confidence interval); ECOG=Eastern Cooperative Oncology Group -toimintakykypisteet; HR=riskisuhde (hazard ratio); NE=ei arvioitavissa (not evaluable) |
Kokonaiselossaoloajassa havaitun paranemisen lisäksi tutkimuksen kaikki toissijaiset päätemuuttujat olivat Zytiga-hoidon yhteydessä paremmat ja tilastollisesti merkitsevät monitestausoikaisun jälkeen seuraavasti:
Zytiga-hoitoa saaneilla potilailla osoitettiin merkittävästi useammin PSA-pitoisuuden kokonaisvaste (määriteltiin ≥ 50 %:n pienenemisenä hoitoa edeltävästä pitoisuudesta) verrattuna lumelääkettä saaneisiin potilaisiin, jolloin vaste esiintyi 38 %:lla Zytiga-hoitoa saaneista verrattuna 10 %:iin lumelääkettä saaneista, p < 0,0001.
Ajan mediaani PSA-pitoisuuden etenemiseen oli Zytiga-hoitoa saaneilla potilailla 10,2 kuukautta ja lumelääkettä saaneilla 6,6 kuukautta (HR = 0,580; 95 %:n CI: [0,462; 0,728], p < 0,0001).
Radiologisen tautivapaan elossaoloajan mediaani oli Zytiga-hoitoa saaneilla potilailla 5,6 kuukautta ja lumelääkettä saaneilla 3,6 kuukautta (HR = 0,673; 95 %:n CI: [0,585; 0,776], p < 0,0001).
Kipu
Niiden potilaiden osuus, joiden kipu lievittyi, oli tilastollisesti merkitsevästi suurempi Zytiga-hoitoa saaneessa ryhmässä kuin lumelääkeryhmässä (44 % vs 27 %, p = 0,0002). Kivun lievitykseen vasteen saanut määriteltiin potilaaksi, joka koki kivun lieventyneen vähintään 30 % hoitoa edeltäneestä tilanteesta kivun pahinta voimakkuutta arvioivan BPI-SF-asteikon pisteiden perusteella edellisten 24 tunnin aikana ilman kipulääkkeiden käytön lisäämistä, kun arviot tehtiin neljän viikon välein ja kivun lieventyminen todettiin kahtena peräkkäisenä arviointikertana. Potilaan kivun lievittyminen analysoitiin vain, jos potilaan kipua arvioivat pisteet olivat lähtötilanteessa ≥ 4 ja lähtötilanteen jälkeen oli vähintään yksi kipua arvioiva pisteytys (N = 512).
Kipu oli pahentunut pienemmällä osalla Zytiga-hoitoa saaneista potilaista lumelääkehoitoa saaneisiin verrattuna 6 kuukauden (22 % vs 28 %), 12 kuukauden (30 % vs 38 %) ja 18 kuukauden (35 % vs 46 %) hoidon jälkeen. Kivun paheneminen määriteltiin pisteiden suurenemisena ≥ 30 % hoitoa edeltäneestä tilanteesta kivun pahinta voimakkuutta arvioivalla BPI-SF-asteikolla edellisten 24 tunnin aikana ilman kipulääkkeiden käytön vähentämistä, mikä havaittiin kahdella peräkkäisellä käynnillä tai kipulääkkeiden käytön lisääntymisellä ≥ 30 %, mikä havaittiin kahdella peräkkäisellä käynnillä. Ajan kivun pahenemiseen 25. persentiili oli Zytiga-ryhmässä 7,4 kuukautta verrattuna 4,7 kuukauteen lumelääkeryhmässä.
Luustoon liittyvät tapahtumat
Luustoon liittyviä tapahtumia esiintyi pienemmällä osalla Zytiga-hoitoa saaneista potilaista lumelääkehoitoa saaneisiin verrattuna 6 kuukauden (18 % vs 28 %), 12 kuukauden (30 % vs 40 %) ja 18 kuukauden (35 % vs 40 %) hoidon jälkeen. Ajan ensimmäiseen luustoon liittyvään tapahtumaan 25. persentiili oli Zytiga-ryhmässä kaksinkertainen verrokkiryhmään verrattuna (9,9 kuukautta Zytiga-ryhmässä verrattuna 4,9 kuukautta lumelääkeryhmässä). Luustoon liittyviksi tapahtumiksi määriteltiin patologiset murtumat, selkäydinkanavan kompressiot, luun palliatiivinen sädehoito tai luukirurgia.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Zytiga-valmisteen käytöstä pitkälle edenneen eturauhassyövän hoidossa kaikissa pediatrisissa potilasryhmissä. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Abirateronin farmakokinetiikkaa on tutkittu abirateroniasetaatin antamisen jälkeen terveillä koehenkilöillä, pitkälle edennyttä etäpesäkkeistä eturauhassyöpää sairastavilla potilailla ja potilailla, joilla ei ollut syöpää, mutta joilla oli maksan tai munuaisten vajaatoimintaa. Abirateroniasetaatti muuntuu in vivo nopeasti abirateroniksi, joka on androgeenien biosynteesin estäjä (ks. kohta Farmakodynamiikka).
Imeytyminen
Kun abirateroniasetaatti annetaan paastotilassa suun kautta, aika abirateronin maksimipitoisuuden saavuttamiseen plasmassa on noin 2 tuntia.
Abirateroniasetaatin antaminen ruoan kanssa suurentaa keskimääräistä systeemistä altistusta abirateronille enintään 10-kertaiseksi (AUC) ja enintään 17-kertaiseksi (Cmax) paastotilaan verrattuna, aterian rasvasisällöstä riippuen. Kun huomioidaan aterian sisällön ja koostumuksen normaali vaihtelu, Zytiga-valmisteen ottaminen aterian yhteydessä saattaa johtaa altistuksen huomattavaan vaihteluun. Zytiga-valmistetta ei siksi saa ottaa ruoan kanssa. Zytiga-tabletit on otettava kerta-annoksena kerran päivässä tyhjään mahaan. Zytiga on otettava vähintään kaksi tuntia ruokailun jälkeen. Ruokaa ei saa syödä vähintään tuntiin Zytiga-tablettien ottamisen jälkeen. Zytiga-tabletit on nieltävä kokonaisina veden kanssa (ks. kohta Annostus ja antotapa).
Jakautuminen
Noin 99,8 % 14C-abirateronista sitoutuu ihmisen plasman proteiineihin. Näennäinen jakaantumistilavuus on noin 5 630 l, mikä viittaa siihen, että abirateroni jakaantuu laajasti ääreiskudoksiin.
Biotransformaatio
Kun 14C-abirateroniasetaatti annetaan kapseleina suun kautta, abirateroniasetaatti hydrolysoituu abirateroniksi, joka metaboloituu tämän jälkeen pääasiassa maksassa sulfaation, hydroksylaation ja oksidaation kautta. Suurin osa verenkierrossa todetusta radioaktiivisuudesta (noin 92 %) on abirateronin metaboliittien muodossa. 15 havaitusta metaboliitista kaksi pääasiallista metaboliittia, abirateronisulfaatti ja N-oksidiabirateronisulfaatti, vastaavat kumpikin noin 43 %:a kokonaisradioaktiivisuudesta.
Eliminaatio
Terveistä koehenkilöistä saatujen tietojen perusteella abirateronin keskimääräinen puoliintumisaika plasmassa on noin 15 tuntia. Kun suun kautta annetaan 1000 mg 14C-abirateroniasetaattia, noin 88 % radioaktiivisesta annoksesta todetaan ulosteessa ja noin 5 % virtsassa. Tärkeimmät ulosteessa esiintyvät yhdisteet ovat muuttumaton abirateroniasetaatti ja abirateroni (abirateroniasetaattia noin 55 % ja abirateronia noin 22 % annetusta annoksesta).
Maksan vajaatoiminta
Abirateroniasetaatin farmakokinetiikkaa tutkittiin potilailla, jotka sairastivat ennestään lievää (Child-Pugh-luokka A) tai keskivaikeaa (Child-Pugh-luokka B) maksan vajaatoimintaa ja terveillä verrokeilla. Systeeminen altistus abirateronille suureni suun kautta annetun 1000 mg:n kerta-annoksen jälkeen noin 11 % lievää maksan vajaatoimintaa ennestään sairastavilla potilailla ja noin 260 % keskivaikeaa maksan vajaatoimintaa ennestään sairastavilla potilailla. Abirateronin keskimääräinen puoliintumisaika piteni noin 18 tuntiin, jos potilaalla oli lievä maksan vajaatoiminta, ja noin 19 tuntiin, jos potilaalla oli keskivaikea maksan vajaatoiminta.
Toisessa tutkimuksessa abirateroniasetaatin farmakokinetiikkaa tutkittiin potilailla, jotka sairastivat ennestään vaikeaa (Child-Pugh-luokka C) maksan vajaatoimintaa (n=8), ja 8 terveellä verrokilla, joiden maksan toiminta oli normaalia. Potilailla, joilla oli vaikea maksan vajaatoiminta, abirateronin AUC-arvo suureni noin 600 % ja vapaan lääkeaineen osuus suureni 80 % verrattuna tutkittaviin, joiden maksan toiminta oli normaalia.
Annosta ei tarvinnut muuttaa, jos potilas sairasti ennestään lievää maksan vajaatoimintaa. Abirateroniasetaattihoitoa on harkittava tarkoin, jos potilaalla on kohtalainen maksan vajaatoiminta ja hoidon hyödyt on tällaiselle potilaalle oltava selvästi riskejä suuremmat (ks. kohdat Annostus ja antotapa sekä Varoitukset ja käyttöön liittyvät varotoimet). Abirateroniasetaattihoitoa ei saa antaa, jos potilaalla on vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa, Vasta-aiheet sekä Varoitukset ja käyttöön liittyvät varotoimet).
Jos potilaalle kehittyy hoidon aikana maksatoksisuutta, hoito voi olla tarpeen keskeyttää ja annosta saattaa olla tarpeen säätää (ks. kohdat Annostus ja antotapa sekä Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Abirateroniasetaatin farmakokineettisiä ominaisuuksia stabiilia hemodialyysihoitoa saavilla loppuvaiheen munuaistautia sairastavilla potilailla verrattiin kaltaistettuihin verrokkeihin, joiden munuaisten toiminta oli normaalia. Systeeminen altistus abirateronille ei suurentunut suun kautta annetun 1000 mg:n kerta-annoksen jälkeen, kun potilas sairasti loppuvaiheen munuaistautia ja sai dialyysihoitoa. Munuaisten vajaatoimintaa sairastavien, myöskään vaikeaa munuaisten vajaatoimintaa sairastavien, potilaiden annosta ei tarvitse pienentää (ks. kohta Annostus ja antotapa). Eturauhassyöpää ja vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidosta ei kuitenkaan ole kliinistä kokemusta. Tämän potilasryhmän hoidossa kehotetaan varovaisuuteen.
Prekliiniset tiedot turvallisuudesta
Kaikissa toksisuutta selvittäneissä eläinkokeissa verenkierrossa olevat testosteronipitoisuudet olivat pienentyneet huomattavasti. Tämän seurauksena havaittiin lisääntymiselinten, lisämunuaisten, aivolisäkkeen ja rintarauhasten painon vähenemistä sekä elinten morfologisia ja/tai histopatologisia muutoksia. Kaikkien muutosten osoitettiin olevan täysin tai osittain korjaantuvia. Lisääntymiselimissä ja androgeenille herkissä elimissä todetut muutokset ovat yhdenmukaisia abirateronin farmakologisten ominaisuuksien kanssa. Kaikki hoitoon liittyneet hormonaaliset muutokset korjautuivat tai niiden osoitettiin olevan häviämässä 4 viikon palautumisjakson aikana.
Uros- ja naarasrotilla tehdyissä hedelmällisyystutkimuksissa abirateroniasetaatti heikensi hedelmällisyyttä, mutta tämä vaikutus korjautui täysin 4–16 viikon kuluessa abirateroniasetaatin annon lopettamisen jälkeen.
Rotalla tehdyssä kehitystoksisuutta selvittäneessä tutkimuksessa abirateroniasetaatti vaikutti tiineyteen mm. alentamalla sikiöiden painoa ja heikentämällä niiden eloonjäämistä. Abirateroniasetaatin ei kuitenkaan todettu ulkoisissa sukuelimissä havaittujen vaikutusten perusteella olevan teratogeeninen.
Kaikki vaikutukset näissä rotalla tehdyissä hedelmällisyys- ja kehitystoksisuustutkimuksissa liittyivät abirateronin farmakologiseen vaikutukseen.
Kaikissa toksisuutta selvittäneissä eläinkokeissa lisääntymiselimissä todettujen muutosten lisäksi farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Abirateroniasetaatti ei ollut karsinogeeninen transgeenisellä (Tg.rasH2) hiirellä tehdyssä kuusi kuukautta kestäneessä tutkimuksessa. Rotalla tehdyssä 24 kuukautta kestäneessä karsinogeenisuustutkimuksessa abirateroniasetaatti lisäsi interstitiaalisolukasvainten ilmaantuvuutta kiveksissä. Löydöksen katsotaan liittyvän abirateronin farmakologiseen vaikutukseen ja olevan rotille ominainen. Abirateroniasetaatti ei ollut karsinogeeninen naarasrotille.
Ympäristöön kohdistuvien riskien arviointi
Vaikuttava aine abirateroni on ympäristöriski vesistöille, etenkin kaloille.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa (silisifioitu)
Kroskarmelloosinatrium
Hypromelloosi 2910 (15 mPa.S)
Laktoosimonohydraatti
Magnesiumstearaatti
Kolloidinen vedetön piidioksidi
Natriumlauryylisulfaatti
Kalvopäällyste
Musta rautaoksidi (E 172)
Punainen rautaoksidi (E 172)
Makrogoli 3350
Polyvinyylialkoholi
Talkki
Titaanidioksidi.
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZYTIGA tabletti, kalvopäällysteinen
500 mg (L:kyllä) 56 fol (1740,33 €)
PF-selosteen tieto
Kartonkisessa taskupakkauksessa oleva PVdC/PE/PVC/alumiiniläpipainopakkaus, jossa 14 kalvopäällysteistä tablettia. Yksi kartonkikotelo sisältää (56 kalvopäällysteistä tablettia) 4 taskupakkausta.
Valmisteen kuvaus:
Purppuranvärinen soikea kalvopäällysteinen tabletti (pituus 20 mm × leveys 10 mm), jonka toisella puolella on merkintä AA ja vastakkaisella puolella 500.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. Tämä lääkevalmiste saattaa aiheuttaa riskin vesistöille (ks. kohta Prekliiniset tiedot turvallisuudesta).
Korvattavuus
ZYTIGA tabletti, kalvopäällysteinen
500 mg 56 fol
- Ylempi erityiskorvaus (100 %). Abirateroni: Eturauhassyövän hoito erityisin edellytyksin (163).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abirateroni: Eturauhassyövän hoito erityisin edellytyksin (352).
ATC-koodi
L02BX03
Valmisteyhteenvedon muuttamispäivämäärä
28.06.2022
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com