
SYNFLORIX injektioneste, suspensio, esitäytetty ruisku
Vaikuttavat aineet ja niiden määrät
1 annos (0,5 ml):
Pneumokokkipolysakkaridi serotyyppi 11,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 41,2 3 mikrogrammaa
Pneumokokkipolysakkaridi serotyyppi 51,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 6B1,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 7F1,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 9V1,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 141,2 1 mikrogramma
Pneumokokkipolysakkaridi serotyyppi 18C1,3 3 mikrogrammaa
Pneumokokkipolysakkaridi serotyyppi 19F1,4 3 mikrogrammaa
Pneumokokkipolysakkaridi serotyyppi 23F1,2 1 mikrogramma
1 adsorboitu alumiinifosfaattiin yhteensä 0,5 milligrammaa Al3+
2 konjugoitu proteiini D kantajaproteiiniin
(tuotettu ei-tyypitettävissä olevasta Haemophilus influenzae -kannasta) 9–16 mikrogrammaa
3 konjugoitu tetanustoksoidikantajaproteiiniin 5–10 mikrogrammaa
4 konjugoitu difteriatoksoidikantajaproteiiniin 3–6 mikrogrammaa
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, suspensio (injektio).
Kliiniset tiedot
Käyttöaiheet
Aktiivinen immunisaatio Streptococcus pneumoniae -bakteerin aiheuttamaa invasiivista tautia, keuhkokuumetta ja äkillistä välikorvantulehdusta vastaan vähintään 6 viikon ikäisillä pikkulapsilla ja lapsilla viidenteen syntymäpäivään asti. Tietoa suojasta spesifisiä pneumokokkiserotyyppejä vastaan esitetään kohdissa Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka.
Synflorixin käytön tulee perustua virallisesti hyväksyttyihin suosituksiin ottaen samalla huomioon pneumokokkitautien merkitys eri ikäryhmissä sekä erot epidemiologiassa eri maantieteellisillä alueilla.
Annostus ja antotapa
Annostus
Synflorixin rokotusohjelman tulee perustua virallisiin suosituksiin.
Rokotusohjelma suositellaan annettavaksi loppuun Synflorixilla, jos ensimmäinen annos on ollut Synflorixia.
Vähintään 6 viikon ja enintään 6 kuukauden ikäiset pikkulapset
Kolmen annoksen perusrokotussarja
Optimaalisen suojan saavuttamiseksi suositeltu immunisaatiosarja koostuu neljästä 0,5 ml:n annoksesta. Pikkulasten perusrokotussarja koostuu kolmesta 0,5 ml:n annoksesta, joiden välillä on oltava vähintään 1 kuukausi. Ensimmäinen annos annetaan yleensä kahden kuukauden iässä. Ensimmäinen annos voidaan kuitenkin antaa jo 6 viikon iässä. Tehosteannosta (4. annos) suositellaan vähintään 6 kuukauden kuluttua viimeisimmästä perusrokotusannoksesta ja se voidaan antaa 9 kuukauden iästä lähtien (mieluiten 12–15 kuukauden iässä) (ks kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Kahden annoksen perusrokotussarja
Vaihtoehtoisesti, kun Synflorix annetaan osana lasten yleistä rokotusohjelmaa, sarja voi koostua kolmesta 0,5 ml:n annoksesta. Ensimmäinen annos voidaan antaa jo 6 viikon iässä ja toinen annos annetaan kaksi kuukautta myöhemmin. Tehosteannosta (3. annos) suositellaan aikaisintaan 6 kuukauden kuluttua viimeisimmästä perusrokotusannoksesta ja se voidaan antaa 9 kuukauden iästä lähtien (mieluiten 12–15 kuukauden iässä) (ks. kohta Farmakodynamiikka).
Pikkulapset, jotka ovat syntyneet ennenaikaisesti 27−36 viikkoa kestäneen raskauden jälkeen
Keskosilla, jotka ovat syntyneet vähintään 27 viikkoa kestäneen raskausajan jälkeen, suositeltu rokotusohjelma koostuu neljästä 0,5 ml:n annoksesta. Perusrokotussarja pikkulapsilla käsittää kolme annosta. Ensimmäinen annos annetaan kahden kuukauden iässä. Annosten välillä tulee olla vähintään yksi kuukausi. Tehosteannosta (4. annos) suositellaan aikaisintaan kuuden kuukauden kuluttua viimeisestä perusrokotusannoksesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Rokottamattomat ≥ 7 kuukauden ikäiset pikkulapset ja lapset
- 7–11 kuukauden ikäiset pikkulapset: Rokotussarja koostuu kahdesta 0,5 ml:n perusannoksesta, joiden välillä on oltava vähintään 1 kuukausi. Tehosteannosta (3. annos) suositellaan toisena ikävuotena, jolloin annosvälin on oltava vähintään 2 kuukautta viimeisimmästä perusannoksesta.
- 12 kuukauden – 5 vuoden ikäiset lapset: Rokotussarja koostuu kahdesta 0,5 ml:n annoksesta, joiden välillä on oltava vähintään 2 kuukautta.
Erityispotilaat
Henkilöille, joilla on invasiiviselle pneumokokkitaudille altistavia tiloja (kuten ihmisen immuunikatovirusinfektio (HIV), sirppisoluanemia (SCD) tai pernan toimintahäiriö), Synflorixia voidaan antaa yllämainittujen aikataulujen mukaisesti. Poikkeuksena ovat pikkulapset, joiden rokotussarja aloitetaan 6 viikon − 6 kuukauden iässä; heille tulee ensisijaisesti antaa kolmen annoksen perusrokotussarja (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Pediatriset potilaat
Synflorixin turvallisuutta ja tehoa ei ole osoitettu yli 5 vuoden ikäisillä lapsilla.
Synflorixin ja muiden konjugoitujen pneumokokkirokotteiden käyttö
Sekä Synflorixin että 13‑valenttisen konjugoidun pneumokokkirokotteen (PCV13) käytöstä osana samaa rokotusohjelmaa on saatavilla vain vähän kliinisiä tietoja (ks. kohta Farmakodynamiikka).
Antotapa
Rokote tulee antaa injektiona lihakseen. Suositeltavat antopaikat pikkulapsilla on ulomman reisilihaksen etu-yläosa ja lapsilla olkavarren hartialihas.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai jollekin apuaineelle, jotka on lueteltu kohdassa Apuaineet, tai jollekin kantajaproteiinille.
Kuten muillakin rokotteilla, Synflorixin antaminen vakavaa akuuttia kuumetautia sairastaville on siirrettävä myöhempään ajankohtaan. Vähäinen infektio, kuten nuhakuume, ei ole rokotteen vasta-aihe.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi annetun valmisteen nimi ja eränumero on kirjattava selkeästi ylös.
Ennen rokotusta
Kuten yleensäkin, kun rokotus annetaan pistoksena, potilasta on seurattava harvinaisten anafylaktisten reaktioiden varalta rokotuksen jälkeen ja asianmukaisesta hoitovalmiudesta huolehdittava.
Apnea on potentiaalinen riski annettaessa perusrokotussarja hyvin ennenaikaisesti syntyneille vauvoille (vauvat, jotka ovat syntyneet ≤ 28 viikkoa hedelmöityksestä). Näillä vauvoilla tulee harkita hengitystoiminnan seurantaa 48–72 tunnin ajan, varsinkin, jos vauvalla on esiintynyt hengitystoiminnan kypsymättömyyttä. Tässä lapsiryhmässä rokotuksen tuoma hyöty on korkea. Rokotuksia ei tästä syystä tule jättää antamatta tai lykätä.
Synflorixia ei saa missään olosuhteissa antaa suoneen tai ihonsisäisesti. Synflorixin antamisesta ihon alle ei ole tietoa.
Pyörtymistä voi esiintyä yli 2-vuotiailla lapsilla minkä tahansa rokotuksen jälkeen, tai jopa ennen sitä, psykogeenisena vasteena neulanpistokselle. Pyörtymisestä johtuvien vammojen ehkäisemiseksi on oltava valmiudet.
Kuten yleensäkin, kun rokotus annetaan lihakseen, Synflorixia on annettava varoen henkilöille, joilla on trombosytopenia tai hyytymishäiriö sillä im-rokotus voi heillä johtaa verenvuotoon.
Tietoa rokotteen antamasta suojasta
Kurkkumätä-, jäykkäkouristus- ja Haemophilus influenzae tyyppi b -rokotuksista annettuja virallisia suosituksia on noudatettava.
Tiedot Synflorixin tarjoamasta suojasta muita kuin rokotteen sisältämiä pneumokokki serotyyppejä vastaan, paitsi ristireaktiivista serotyyppiä 19A:ta vastaan (ks. kohta Farmakodynamiikka.), tai ei-tyypitettävissä olevaa Haemophilus influenzae -kantoja vastaan ovat riittämättömät. Synflorix ei suojaa muilta mikro-organismeilta.
Kuten mikä tahansa rokote Synflorix ei ehkä suojaa kaikkia henkilöitä invasiiviselta pneumokokkitaudilta, keuhkokuumeelta eikä välikorvantulehdukselta, vaikka taudin aiheuttaja on rokoteserotyyppiä tai ristireaktiivista serotyyppiä 19A. Lisäksi on huomattava, että rokotteen sisältämien Streptococcus pneumoniae -serotyyppien lisäksi useat muut mikro-organismit aiheuttavat välikorvantulehdusta ja keuhkokuumetta. Tästä syystä kokonaissuoja näitä tauteja vastaan lienee rajoitettu ja huomattavasti heikompi kuin suoja invasiivista tautia vastaan, jonka aiheuttaja on jokin rokoteserotyypistä tai serotyyppi 19A (ks. kohta Farmakodynamiikka).
Kliinisissä tutkimuksissa Synflorix sai aikaan immuunivasteen kaikille rokotteen kymmenelle serotyypille, mutta vasteen suuruudessa oli eroja eri serotyyppien välillä. Toiminnallinen immuunivaste serotyypeille 1 ja 5 oli matalampi kuin vaste kaikille muille rokotteen serotyypeille. Ei tiedetä, johtaako tämä matalampi toiminnallinen immuunivaste serotyypeille 1 ja 5 matalampaan suojatehoon näiden serotyyppien aiheuttamaa invasiivista tautia, keuhkokuumetta tai välikorvantulehdusta vastaan (ks. kohta Farmakodynamiikka)
Synflorix on annettava lapsille rokotusohjelman alkaessa olevan ikäsuosituksen mukainen annostus (ks. kohta Annostus ja antotapa).
Immunosupressiivinen hoito ja immuunikato
Rokotuksen jälkeen vasta-aineiden muodostuminen saattaa jäädä puutteelliseksi lapsilla, joiden immuunivaste on heikentynyt. Syitä immuunivasteen heikentymiseen voivat olla mm. immunosuppressiivinen hoito, geneettinen tekijä, HIV-infektio, raskaudenaikainen altistuminen antirotreviraaliselle lääkitykselle ja/tai HIV:lle tai jokin muu syy.
Turvallisuutta ja immunogeenisuutta koskevat tiedot ovat saatavilla pikkulapsille, joilla on HIV-infektio (oireeton tai lievät oireet WHO:n luokittelun mukaan), HIV-positiivisten äitien synnyttämille HIV-negatiivisille pikkulapsille, sirppisoluanemiaa sairastaville lapsille ja pernan toimintahäiriötä sairastaville lapsille (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Potilaille, joilla on jokin muu erityinen immuunivajaustila, Synflorixin turvallisuutta ja immunogeenisuutta koskevat tiedot eivät ole saatavilla. Tällöin rokotusta on harkittava yksilötasolla (ks. kohta Annostus ja antotapa).
Konjugoidun pneumokokkirokotteen käyttö ei korvaa 23-valenttisen pneumokokkipolysakkaridirokotteen käyttöä ≥ 2 vuoden ikäisillä lapsilla, joilla on korkea riski saada invasiivinen Streptococcus pneumoniaen aiheuttama tauti (kuten lapset, joilla on sirppisoluanemia, asplenia, HIV-infektio, krooninen sairaus tai muu immuunivajaustila). Riskiryhmiin kuuluville ≥ 2 vuoden ikäisille lapsille, jotka ovat saaneet Synflorix-rokotetta, tulee aina kun se katsotaan suositeltavaksi antaa 23-valenttinen pneumokokkipolysakkaridirokote.
Konjugoidun pneumokokkirokotteen (Synflorix) ja 23-valenttisen pneumokokkipolysakkaridirokotteen välillä tulee olla vähintään 8 viikkoa. Tällä hetkellä ei tiedetä, johtaako pneumokokkipolysakkaridirokotteen antaminen Synflorix-rokotuksen saaneille lapsille myöhemmin heikompaan vasteeseen jatkossa annettaville pneumokokkipolysakkaridi- tai konjugoidulle pneumokokkirokotteille.
Kuumetta alentavien lääkkeiden ennalta ehkäisevä käyttö
Kuumetta alentavien lääkkeiden antaminen ennaltaehkäisevästi tai välittömästi rokotuksen jälkeen voi alentaa rokotuksen jälkeisen kuumereaktion ilmaantuvuutta ja voimakkuutta. Kliiniset tiedot parasetamolilla ja ibuprofeenilla viittaavat siihen, että parasetamolin profylaktinen käyttö saattaa laskea kuumeen ilmaantuvuutta, kun ibuprofeenin profylaktisella käytöllä puolestaan oli rajoitettu vaikutus kuumeen ilmaantuvuuteen. Kliiniset tiedot viittaavat siihen, että parasetamoli saattaa heikentää Synflorixin aikaansaamaa immuunivastetta. Havainnon kliininen merkitys on kuitenkin tuntematon.
Kuumetta alentavien lääkkeiden käyttö on perusteltua:
- kaikilla lapsilla, jotka saavat Synflorixia samanaikaisesti kokosoluhinkuyskärokotteen kanssa, sillä kuumereaktioita esiintyy enemmän (ks. kohta Haittavaikutukset).
- kaikilla lapsilla, joilla on jokin kouristuksia aiheuttava sairaus tai joilla on aikaisemmin esiintynyt kuumekouristuksia.
Kuumetta alentava hoito tulee aloittaa paikallisten hoitosuositusten mukaan.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ˮnatriumitonˮ.
Yhteisvaikutukset
Käyttö muiden rokotteiden kanssa
Synflorix voidaan antaa samanaikaisesti minkä tahansa seuraavan monovalentin rokotteen tai yhdistelmärokotteen kanssa [mukaan lukien DTaP-HBV-IPV/Hib ja DTwP-HBV/Hib]: kurkkumätä-jäykkäkouristus-soluton hinkuyskärokote (DTaP), hepatiitti B ‑rokote (HBV), inaktivoitu poliorokote (IPV), Haemophilus influenzae tyyppi b ‑rokote (Hib), kurkkumätä-jäykkäkouristus-kokosoluhinkuyskärokote (DTwP), tuhkarokko-sikotauti-vihurirokkorokote (MPR), vesirokkorokote (V), konjugoitu meningokokki seroryhmä C- rokote (CRM197 ja TT-konjugaatit), konjugoitu meningokokki A, C, W-135 ja Y -rokote (TT-konjugaatti), oraalinen poliorokote (OPV) ja oraalinen rotavirusrokote. Eri injisoitavat rokotteet tulee aina antaa eri injektiokohtiin.
Kliinisissä tutkimuksissa on osoitettu, että samanaikaisesti annettujen muiden rokotteiden turvallisuusprofiili ja niiden aikaansaama immuunivaste eivät muuttuneet. Poikkeuksen muodostaa vaste inaktivoidulle poliovirus tyyppi 2:lle, joka on vaihdellut eri tutkimuksissa (suojaava vasta-ainetaso saavutettu 78 % - 100 %:lla rokotetuista). Kun konjugoitua meningokokki A, C, W-135 ja Y (TT-konjugaatti) rokotetta annettiin samanaikaisesti Synflorix-tehosteannoksen kanssa toisena ikävuotena lapsille, jotka aikaisemmin olivat saaneet kolmen annoksen perusrokotesarjan Synflorixia, vasta-aineiden geometrisissä keskiarvopitoisuuksissa (GMC) ja opsonofagosyyttisellä menetelmällä mitatuissa geometrisissä keskiarvotittereissä (OPA GMT) havaittiin yhden pneumokokkiserotyypin (18C) kohdalla matalampia tasoja. Samanaikaisella annolla ei ollut vaikutusta rokotteen yhdeksään muuhun pneumokokkiserotyyppiin. Immuunivaste Hib-TT-konjugaatille, difteria-antigeenille ja tetanusantigeenille tehostui. Havainnon kliinistä merkitystä ei tiedetä.
Käyttö systeemisten immunosuppressiivisten lääkkeiden kanssa
Immunosuppressiivista hoitoa saavilla riittävää vastetta ei ehkä saavuteta, kuten ei muidenkaan rokotteiden annon yhteydessä.
Käyttö ennalta ehkäisevästi annettujen kuumetta alentavien lääkkeiden kanssa
Kliiniset tiedot viittaavat siihen, että parasetamolin ennalta ehkäisevä käyttö mahdollisten rokotusten jälkeisten kuumereaktioiden vähentämiseksi saattaa heikentää Synflorixin aikaansaamaa immuunivastetta. Havainnon kliininen merkitys on kuitenkin tuntematon. Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Raskaus ja imetys
Synflorix ei ole tarkoitettu aikuisille. Sen vuoksi valmisteen käytöstä ihmisellä raskauden tai imetyksen aikana ei ole tietoja, eikä eläimillä ole suoritettu lisääntymistutkimuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ei merkityksellinen.
Haittavaikutukset
Haittavaikutusten yhteenveto
Synflorixin turvallisuutta koskeva arviointi perustuu kliinisiin tutkimuksiin, joissa Synflorixia on annettu perusrokotussarjana 63 905 annosta 22 429 terveelle pikkulapselle ja 137 keskoselle. Tehoasteannos on annettu 19 466 lapselle ja 116 keskoselle toisena ikävuotena. Turvallisuutta on myös arvioitu 435 aikaisemmin rokottamattomalla 2–5-vuotiaalla lapsella, joista 285 sai kaksi annosta Synflorixia. Kaikissa tutkimuksissa Synflorix annettiin samanaikaisesti muiden suositeltujen lapsuusiän rokotusten kanssa.
Useimmin perusrokotussarjan annosten jälkeen raportoidut haittavaikutukset pikkulapsilla olivat pistoskohdassa ilmenevä punoitus (41 %) ja ärtyneisyys (55 %). Tehosteannoksen jälkeen yleisimmät haittavaikutukset olivat pistoskohdassa ilmenevä kipu (51 %) ja ärtyneisyys (53 %). Useimmat reaktiot olivat lieviä tai kohtalaisia eivätkä ne kestäneet kauan.
Haittavaikutusten ilmaantuvuudessa tai vaikeusasteessa ei havaittu lisääntymistä perusrokotussarjan edetessä.
Paikallinen reaktogeenisuus perusrokotussarjan jälkeen oli samanlainen < 12 kuukauden ikäisillä pikkulapsilla ja > 12 kuukauden ikäisillä lapsilla. Poikkeuksen muodosti injektiokohdan kipu, jonka insidenssi nousi iän mukana. Kipua raportoitiin yli 39 % pikkulapsista, jotka olivat < 12 kuukauden ikäisiä ja yli 58 % lapsista, jotka olivat > 12 kuukauden ikäisiä.
Tehosteannoksen jälkeen > 12 kuukauden ikäiset lapset saavat todennäköisemmin reaktioita injektiokohdassa verrattuna Synflorixin perusrokotussarjassa havaittuihin frekvensseihin pikkulapsilla.
Urtikariaa raportoitiin 12−23-kuukauden ikäisillä lapsilla useammin (melko harvoin) täydennysrokotuksen jälkeen kuin perusrokotuksen ja tehosterokotuksen jälkeen.
Reaktogeenisuus oli korkeampi lapsilla, jotka saivat kokosoluhinkuyskärokotetta samanaikaisesti. Eräässä kliinisessä tutkimuksessa Synflorixia (N=603) tai 7-valenttista Prevenaria (N=203) annettiin lapsille samanaikaisesti DTPw -rokotteen kanssa. Perusrokotussarjan jälkeen kuumetta raportoitiin Synflorix-ryhmässä ≥ 38 ºC-asteisena 86,1 %:lla ja > 39 ºC-asteisena 14,7 %:lla lapsista ja 7‑valenttisen Prevenar-ryhmässä 82,9 %:lla ja 11,6 %:lla vastaavasti.
Vertailevissa kliinisissä tutkimuksissa paikallisten ja yleisten haittavaikutusten ilmaantuvuus 4 päivän kuluessa jokaisen rokoteannoksen jälkeen oli Synflorix-rokoteannosten jälkeen samaa suuruusluokkaa kuin 7-valenttisen Prevenar-rokoteannosten jälkeen.
Luettelo haittavaikutuksista
Haittavaikutukset (kaikissa ikäryhmissä) on luokiteltu frekvenssin mukaan.
Frekvenssit ilmaistaan seuraavasti:
Hyvin yleiset (≥1/10)
Yleiset (≥1/100, <1/10)
Melko harvinaiset (≥1/1000, <1/100)
Harvinaiset (≥1/10 000, <1/1000)
Hyvin harvinaiset (<1/10 000)
Jokaisen yleisyysluokan sisällä haittavaikutukset on esitetty vakavuudeltaan laskevassa järjestyksessä.
Elinjärjestelmä | Yleisyys | Haittavaikutukset |
Kliiniset lääketutkimukset | ||
Immuunijärjestelmä | Harvinaiset | Allergiset reaktiot (kuten ekseema, allerginen dermatiitti, atooppinen dermatiitti) |
Hyvin harvinaiset | Angioedeema | |
Aineenvaihdunta ja ravitsemus | Hyvin yleiset | Ruokahaluttomuus |
Psyykkiset häiriöt | Hyvin yleiset | Ärtyneisyys |
Melko harvinaiset | Epätavallinen itku | |
Hermosto | Hyvin yleiset | Uneliaisuus |
Harvinaiset | Kouristukset (joihin voi liittyä kuumetta) | |
Verisuonisto | Hyvin harvinaiset | Kawasakin tauti |
Hengityselimet, rintakehä ja välikarsina | Melko harvinaiset | Apnea hyvin ennenaikaisesti syntyneillä vauvoilla (vauvat, jotka ovat syntyneet ≤ 28 viikkoa hedelmöityksestä) (Ks kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Ruoansulatuselimistö | Melko harvinaiset | Ripuli, oksentelu |
Iho ja ihonalainen kudos | Melko harvinaiset | Ihottuma |
Harvinaiset | Urtikaria | |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleiset | Kuume ≥ 38 ºC peräaukosta mitattuna (ikä < 2 vuotta), kipu, punoitus, turvotus injektiokohdassa, |
Yleiset | Kuume > 39 ºC peräaukosta mitattuna (ikä < 2 vuotta), kovettuma injektiokohdassa, | |
Melko harvinaiset | Mustelma, verenvuoto tai kyhmy injektiokohdassa | |
Haittavaikutuksia on lisäksi raportoitu perusrokotussarjan tehosteannoksen jälkeen ja/tai täydennysrokotuksen jälkeen | ||
Hermosto | Melko harvinaiset | Päänsärky (2−5-vuotiaat) |
Ruoansulatuselimistö | Melko harvinaiset | Pahoinvointi (2−5-vuotiaat) |
Yleisoireet ja antopaikassa todettavat haitat | Yleiset | Kuume ≥ 38 °C peräaukosta mitattuna (ikä 2–5 vuotta) |
Melko harvinaiset | Kuume > 40 ºC peräaukosta mitattuna (ikä < 2 vuotta), kuume > 39 °C peräaukosta mitattuna (ikä 2–5 vuotta), pistetyn raajan epämääräinen turvotus, joka joskus on ulottunut läheiseen niveleen, kutina injektiokohdassa | |
Markkinoille tulon jälkeiset tiedot | ||
Immuunijärjestelmä | Hyvin harvinaiset | Anafylaksia |
Hermosto | Harvinaiset | Hypotonis-hyporesponsiivinen episodi |
Erityispotilaat
Synflorixin turvallisuutta arvioitiin perusrokotussarjan saaneilla potilailla; 83 HIV-positiivisella (HIV+/+) pikkulapsella (oireeton tai lievät oireet WHO:n luokittelun mukaan), 101 HIV-positiivisten äitien (HIV +/-) synnyttämällä HIV-negatiivisella pikkulapsella ja 50 sirppisoluanemiaa (SCD) sairastavalla pikkulapsella. Näistä vastaavassa järjestyksessä 76, 96 ja 49 pikkulasta saivat tehosteannoksen. Synflorixin turvallisuutta arvioitiin myös 50 sirppisoluanemiaa sairastavalla lapsella, joiden rokotukset aloitettiin 7–11 kuukauden iässä ja joista kaikki saivat tehosterokotteen ja 50 sirppisoluanemiaa sairastavalla lapsella, joiden rokotukset aloitettiin 12–23 kuukauden iässä. Tulosten mukaan Synflorixin reaktogeenisuus ja turvallisuusprofiili ovat vastaavia näissä suuren riskin ryhmissä ja terveillä lapsilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Tämä mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksia ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: rokotteet, pneumokokkirokotteet ATC-koodi: J07AL52
Epidemiologiset tiedot
Synflorixin 10 pneumokokkiserotyyppiä edustavat Euroopan yleisimpiä sairauksia aiheuttavia pneumokokkiserotyyppejä. Ne aiheuttavat noin 56–90 % kaikista invasiivista pneumokokkisairauksista alle 5-vuotiailla lapsilla. Tässä ikäryhmässä serotyypit 1, 5 ja 7F aiheuttavat 3,3–24,1 % invasiivisista sairauksista maasta ja tutkitusta aikavälistä riippuen.
Etiologiasta riippumatta keuhkokuume on maalimanlaajuisesti johtava syy lasten sairastavuuteen ja kuolleisuuteen. Prospektiivisissa tutkimuksissa Streptococcus pneumonia aiheutti arviolta 30−50 % keuhkokuumetapauksista.
Äkillinen välikorvatulehdus on yleinen, etiologialtaan vaihteleva lapsuusiän tauti. Bakteerit voivat aiheuttaa 60–70 % kaikista äkillisen välikorvatulehduksen episodeista. Maailmanlaajuisesti Streptococcus pneumoniae ja ei-tyypitettävissä oleva Haemophilus influenzae (NTHi) ovat tavallisimmat välikorvatulehdusta aiheuttavat bakteerit.
Teho ja vaikuttavuus kliinisissä tutkimuksissa
Laaja III/IV vaiheen kaksoissokkoutettu, aluesatunnaistettu, kontrolloitu kllininen lääketutkimus suoritettiin Suomessa (FinIP). Lapset satunnaistettiin 4 ryhmään kahden lapsille annettavan perusrokotusohjelman mukaan (3 ja 5 kuukauden iässä annettava 2 annoksen perusrokotusohjelma tai 3, 4 ja 5 kuukauden iässä annettava perusrokotusohjelma, joita molempia seurasi tehosteannos 11 kuukauden iässä). Lapset saivat joko Synflorixia (2/3 alueista) tai kontrollina hepatiittirokotetta (1/3 alueista). Täydennysrokotusryhmissä lapset, jotka olivat 7−11 kuukauden ikäisiä ensimmäistä rokoteannosta annettaessa, saivat Synflorixia tai hepatiitti B -kontrollirokotetta 2 annoksen perusrokotusohjelman mukaan, jota seurasi tehosteannos. Lapset, jotka olivat 12−18 kuukauden ikäisiä ensimmäistä rokoteannosta annettaessa, saivat 2 annosta joko Synflorixia tai hepatiitti A kontrollirokotetta. Keskimääräinen seuranta-aika (alkaen ensimmäisestä rokotuksesta) invasiivisen taudin ja sairaalassa todetun keuhkokuumeen osalta oli 24−28 kuukautta. Upotetussa tutkimuksessa (nested study), jossa lapsia seurattiin noin 21 kuukauden ikään asti, arvioitiin vaikutusta nenänielun kantajuuteen sekä lääkärin toteamaan vanhempien raportoimaan akuuttiin välikorvatulehdukseen.
Argentiinassa, Panamassa ja Kolumbiassa toteutetussa laajassa, satunnaistetussa, kaksoissokkotetussa III-vaiheen tutkimuksessa (Clinical Otitis media and Pneumonia study – COMPAS) terveille 6−16 viikon ikäisille pikkulapsille annettiin joko Synflorixia tai kontrollina hepatiitti B -rokotetta 2, 4 ja 6 kuukauden iässä ja joko Synflorixia tai kontrollina hepatiitti A -rokotetta 15−18 kuukauden iässä.
Invasiivinen pneumokokkitauti (IPD) (näitä ovat sepsis, meningiitti, bakteerien aiheuttama keuhkokuume ja bakteerien esiintyminen veressä)
Vaikuttavuus/teho lapsiryhmässä, jotka olivat tutkimuksen alkaessa alle 7 kuukauden ikäisiä
Rokotteen vaikuttavuus tai teho (VE) IPD:n ehkäisyssä osoitettiin rokotteen sisältämille, viljelyllä varmennetuille pneumokokkiserotyypeille, kun Synflorixia annettiin lapsille joko 2+1 tai 3+1 rokotusohjelman mukaan FinIP-tutkimuksessa tai 3+1 rokotusohjelman mukaan COMPAS-tutkimuksessa (ks. taulukko 1).
Taulukko 1: Rokoteserotyyppien aiheuttamat IPD-tapaukset ja rokotteen vaikuttavuus (FinIP) tai teho (COMPAS) lapsilla, joiden ikä tutkimuksen alkaessa oli alle 7 kuukautta ja jotka olivat saaneet ainakin yhden rokoteannoksen (ryhmä, joka koostui kaikista rokotetuista lapsista)
| IPD:n serotyyppi | FinIP | COMPAS | ||||||
| IPD tapausten määrä | VE (95 % CI) | IPD tapausten määrä | VE (95 % CI) | |||||
| Synflorix 3+1 rokotusohjelma | Synflorix 2+1 rokotusohjelma | Kontrollil(2) | 3+1 rokotusohjelma | 2+1 rokotusohjelma | Synflorix 3+1 rokotusohjelma | Kontrolli | 3+1 rokotusohjelma | |
N 10 273 | N 10 054 | N 10 200 | N 11 798 | N 11 799 | ||||
| Rokoteserotyyppi IPD(1) | 0 | 1 | 12 | 100 %(3) (82,8; 100) | 91,8%(4) (58,3; 99,6) | 0 | 18 | 100 %(5) (77,3;100) |
| Serotyyppi 6B IPD | 0 | 0 | 5 | 100 % (54,9; 100) | 100 % (54,5; 100) | 0 | 2 | - |
| Serotyyppi 14 IPD | 0 | 0 | 4 | 100 % (39,6; 100) | 100 % (43,3; 100) | 0 | 9 | 100 % (49,5;100) |
IPD Invasiivinen pneumokokkitauti
VE Rokotteen vaikuttavuus (FinIP) tai teho (COMPAS)
N Henkilöiden lukumäärä ryhmässä
CI Luottamusväli
(1) FinIP:ssä serotyyppien 6B ja 14 lisäksi, viljelyllä varmennettujen rokoteserotyyppien aiheuttamat IPD-tapaukset olivat serotyyppiä 7F (1 tapaus Synflorix 2+1 klusterissa), ja 18C, 19F ja 23F (1 tapaus jokaisessa kontrolliklusterissa). COMPAS:issa serotyypit 5 (2 tapausta), 18C (4 tapausta) ja 23F (1 tapaus) olivat kontrolliryhmässä, serotyyppien 6B ja 14 lisäksi.
(2) 2 lapsikontrolliklusteriä yhdistettynä
(3) p-arvo<0.0001
(4) p-arvo=0.0009
(5) ATP-ryhmässä VE oli 100 % (95 % CI: 74,3; 100; 0 vastaan 16 tapausta)
FinIP-tutkimuksessa rokotteen kokonaisvaikuttavuus viljelyllä varmennettuja IPD-tapauksia vastaan oli 100 % (95 % CI: 85,6; 100; 0 tapausta vastaan 14 tapausta) 3+1 rokotusohjelmassa, 85,8 % (95 % CI: 49,1; 97,8; 2 tapausta vastaan 14 tapausta) 2+1 rokotusohjelmassa ja 93,0 % (95 % CI: 74,9; 98,9; 2 tapausta vastaan 14 tapausta) perusrokotusohjelmasta riippumatta. COMPAS-tutkimuksessa kokonaisvaikuttavuus oli 66,7 % (95 % CI: 21,8; 85,9; 7 vastaan 21 tapausta).
Vaikuttavuus täydennysrokotuksen jälkeen
Täydennysrokotusohjelmilla rokotettiin 15 447 lasta, joista kukaan Synflorix-ryhmissä ei saanut viljelyllä varmennettua IPD:tä, kun taas kontrolliryhmissä oli 5 rokoteserotyypin aiheuttamaa IPD-tapausta (serotyypit 4, 6B, 7F, 14 ja 19F).
Keuhkokuume
Tehoa keuhkokuumetta vastaan arvioitiin COMPAS-tutkimuksessa. Seuranta-aika ATP-ryhmässä alkoi 2 viikkoa 3. annoksesta ja oli keskimäärin 23 kuukautta (vaihteluväli 0−34 kuukautta) välianalyysissä ja 30 kuukautta (vaihteluväli 0−44 kuukautta) loppuanalyysissä. ATP- seurantajakson välianalyysissä keskimääräinen ikä oli 29 kuukautta (vaihteluväli 4−41 kuukautta) ja loppuanalyysissä 36 kuukautta (vaihteluväli 4−50 kuukautta). Tehosteannoksen saaneiden osuus ATP-ryhmässä oli 92,3 % molemmissa analyyseissä.
Synflorixin teho ensimmäistä todennäköisesti bakteerin aiheuttamaa avosyntyistä keuhkokuume-episodia vastaan (Community Acquired Pneumonia CAP), joka ilmaantui milloin tahansa 2 viikon kuluttua 3. annoksesta, osoitettiin ATP-ryhmän (P-arvo ≤ 0,002) välianalyysissä (tapahtuma-vetoinen, ensisijainen päätetapahtuma). Todennäköisesti bakteerin aiheuttama CAP (B-CAP) määritellään radiologisesti vahvistetuksi CAP:ksi, jonka löydöksiin kuuluu joko rintakehän röntgenkuvassa näkyvä alveolaarinen konsolidaatio/pleuraeffuusio tai ei-alveolaarinen infiltraatio CRP:n ollessa ≥ 40 mg/l.
Rokotteen teho B-CAP:ia vastaan välianalyysissä on esitetty alla (taulukko 2).
Taulukko 2: Niiden henkilöiden lukumäärä ja prosentuaalinen osuus, joilla oli ensimmäinen B-CAP-episodi, joka ilmaantui milloin tahansa 2 viikon kuluttua 3. Synflorix-annoksesta tai kontrollirokoteannoksesta, sekä rokotteen teho (ATP-ryhmä)
Synflorix N=10 295 | Kontrollirokote N=10 201 | Rokotteen teho | ||
| n | % (n/N) | n | % (n/N) | |
| 240 | 2,3 % | 304 | 3,0 % | 22.0 % (95% CI: 7,7; 34,2) |
N Ryhmän henkilöiden lukumäärä
n/% Niiden henkilöiden lukumäärä/prosentuaalinen osuus, joilla raportoitiin ensimmäinen B-CAP-episodi milloin tahansa 2 viikon kuluttua 3. annoksesta.
CI Luottamusväli
Välianalyysissä (ATP-ryhmä) rokotteen teho ensimmäistä CAP-episodia vastaan, joissa löydöksinä oli alveolaarinen konsolidaatio tai pleuraeffuusio (C-CAP, WHO:n määritelmä), oli 25,7 % (95 % CI: 8,4; 39,6). Teho ensimmäistä kliinisesti epäiltyä ja röntgentutkimukseen johtanutta CAP-episodia vastaan oli 6,7 % (95 % CI: 0,7; 12,3).
Lopullisessa analyysissä (ATP-ryhmä) rokotteen teho ensimmäistä B-CAP-episodia vastaan oli 18,2 % (95 % CI:4,1; 30,3), C-CAP:issa 22,4 % (95 % CI: 5,7; 36,1) ja kliinisesti epäillyssä CAP:issa, josta lähete röntgenkuvaan 7,3 % (95 % CI: 1,6; 12,6). Teho baktereemista pneumokokkikeuhkokuumetta tai rokoteserotyyppien aiheuttamaa empyeemaa vastaan oli 100 % CI: 41,9;100). Suoja B-CAP-episodeja vastaan ennen tehosteannosta oli 13,6 % (95 % CI: -11,3; 33,0) ja tehosteannoksen aikaan tai sen jälkeen 21,7 % (95 % CI: 3,4; 36,5). Suoja C-CAP-episodeja vastaan ennen tehosteannosta oli 15,1 % (95 % CI: -15,5; 37,6) ja tehosteannoksen aikaan tai sen jälkeen 26,3 % (95 % CI: 4,4; 43,2)
B-CAP- ja C-CAP-episodien määrän lasku oli suurin < 36 kuukauden ikäisillä lapsilla (rokotteen teho 20,6 % (95 % CI: 6,5; 32,6) ja 24,2 % (95 % CI: 7,4; 38,0, vastaavasti). Tulokset rokotteen tehosta > 36 kuukauden ikäisillä lapsilla viittaavat suojan heikkenemiseen. Suojan säilymistä 36 kuukauden iän jälkeen B-CAP- ja C-CAP-episodeja vastaan ei ole vahvistettu.
Latinalaisessa Amerikassa suoritetun COMPAS-tutkimuksen tuloksia tulee soveltaa varoen, sillä keuhkokuumeen epidemiologia vaihtelee maantieteellisesti.
FinIP tutkimuksessa rokotteen vaikuttavuus sairaalassa todettujen keuhkokuumetapausten (keuhkokuume todettu ICD 10 tautiluokituksen mukaan) vähenemiseen oli 26,7 % (95 % CI: 4,9; 43,5) 3+1 lasten rokotusohjelmassa ja 29,3 % (95 % CI: 7,5; 46,3) 2+1 lasten rokotusohjelmassa. Täydennysrokotuksen vaikuttavuus oli 33,2 % (95 % CI:3,0;53,4) 7−11 kuukauden ryhmässä ja 22,4 % (95% CI:-8,7; 44,8) 12−18 kuukauden ryhmässä.
Äkillinen välikorvatulehdus
COMPAS- ja POET-tutkimuksissa (Pneumococcal Otitis Media Efficacy Trial) tehoa mitattiin pneumokokkikonjugaattirokotteilla, jotka sisälsivät proteiini D:tä. COMPAS-tutkimuksessa rokote oli Synflorix ja POET-tutkimuksessa rokote oli tutkimusvaiheessa oleva 11-valenttinen konjugaattirokote (joka lisäksi sisälsi serotyyppiä 3).
COMPAS-tutkimuksessa 7 214 henkilöä (rokotettu kokonaisryhmä TVC) otettiin mukaan analyysiin, jossa tehoa arvioitiin akuuttia välikorvan tulehdusta vastaan. Näistä 5 989 henkilöä oli ATP-ryhmässä.
Taulukko 3: Rokotteen teho akuuttia välikorvantulehdusta1) vastaan COMPAS-tutkimuksessa
| Äkillisen välikorvatulehduksen tyyppi tai aiheuttaja | Rokotteen teho (95 % CI) |
| ATP(2) | |
| Kliininen välikorvantulehdus | 16,1 % (-1,1;30,4)(3) |
| Mikä tahansa pneumokokkiserotyyppi | 56,1 % (13,4;77,8) |
| 10 rokotteen sisältämää pneumokokkiserotyyppiä | 67,1 % (17,0; 86,9) |
| Ei-tyypitettävissä oleva Haemophilus influenzae (NTHi) | 15,0 %(4) (-83,8;60,7) |
CI Luottamusväli
(1) Ensimmäinen episodi
(2) Korkeintaan 40 kuukauden seuranta-aika alkaen 2 viikon jälkeen kolmannesta perusannoksesta
(3) Ei tilastollisesti merkittävä ennalta määritelty kriteeri (Yksisuuntainen p=0.032). TVC-ryhmässä rokotteen teho ensimmäistä kliinistä äkillistä välikorvantulehdusepisodia vastaan oli kuitenkin 19 % (95 % CI: 4.4;31.4).
(4) Ei tilastollisesti merkittävä.
Tsekissä ja Slovakiassa on suoritettu toinen laaja, 4 907 pikkulasta (ATP-ryhmä) käsittävä, satunnaistettu, kaksoissokkoutettu tutkimus (Pneumococcal Otitis Media Efficacy (POET)). Tutkimuksessa pikkulapsille annettiin joko 11-valenttinen tutkimusrokote (11Pn-PD), joka sisälsi Synflorixin 10 serotyyppiä (sekä serotyypin 3, jolle tehoa ei voitu osoittaa) 3, 4, 5 ja 12–15 kuukauden rokotusohjelmalla tai kontrollirokotetta (hepatiitti A-rokote).
11 Pn-PD-rokotteen teho ensimmäistä äkillisen välikorvatulehduksen episodia vastaan oli 52,6 % (95 % CI: 35,0;65,5), kun episodin aiheutti rokoteserotyyppi. Serotyyppi-spesifinen teho ensimmäistä äkillisen välikorvatulehduksen episodia vastaan osoitettiin serotyypeille 6B (86,5 %, 95 % CI: 54,9; 96,0), 14 (94,8 %, 95 % CI 61,0; 99,3), 19 F (43,3 %, 95 % CI: 6,3; 65,4) ja 23F (70,8 %, 95 % CI: 20,8; 89,2). Muiden serotyyppien kohdalla äkillisen välikorvatulehduksen episodeja esiintyi tutkimuksen aikana liian vähän, jotta tehon suhteen voitaisiin tehdä johtopäätöksiä. Rokotteen teho oli 51,5 % (95 % CI: 36,8; 62,9) mitä tahansa äkillistä välikorvatulehduksen episodia vastaan, jonka aiheuttaja oli mikä tahansa pneumokokkiserotyyppi. Rokotteen teho ensimmäistä äkillistä NTHi-välikorvatulehdusepisodia oli 31,1 % (CI: -3,7; 54,2, ei tilastollisesti merkitsevä). Teho mitä tahansa äkillistä NTHi-välikorvan tulehdusepisodia vastaan oli 35,3 % (95 % CI: 1,8:57,4). Rokotteen arvioitu teho mitä tahansa äkillisen välikorvatulehduksen kliinistä episodia vastaan etiologiasta riippumatta oli 33,6 % (95 % CI: 20,8; 44,3).
Synflorixin suojateho pneumokokkien aiheuttamaa äkillistä välikorvatulehdusta vastaan arvioidaan Synflorixilla olevan samanlainen kuin POET-tutkimuksessa käytetyllä rokotteella, joka oli 11‑valenttinen. Tämä perustuu molempien rokotteiden aikaansaamaan toiminnallisen vasta-ainevasteen verrattavuuteen (OPA).
Muiden bakteeripatogeenien aiheuttamien, tai ei-rokoteserotyyppien/ei-rokotteeseen liittyvien serotyyppien aiheuttamien äkillisten välikorvatulehdusten ilmaantuvuudessa ei havaittu nousua COMPAS-tutkimuksessa (perustuu muutamaan raportoituun tapaukseen) eikä POET-tutkimuksessa.
Vaikuttavuus vanhempien raportoimaan lääkärin toteamaan äkilliseen välikorvantulehdukseen arvioitiin FinIP-tutkimuksen upotetussa tutkimuksessa (nested study). Rokotettujen pikkulasten ryhmässä rokotteen vaikuttavuus tälle äkillisen välikorvatulehduksen päätetapahtumalle oli 6,1 % (95: -2,7; 14,1) 3+1rokotusohjelmassa ja 7,4 % (CI: -2,8; 16,6) 2+1 rokotusohjelmassa.
Vaikutus nenänielun kantajuuteen
Synflorixin teho nenänielun kantajuuteen arvioitiin kahdessa kaksoissokkoutetussa, satunnaistetussa tutkimuksessa käyttäen inaktiivista kontrollia: FinIP-tutkimuksen upotetussa tutkimuksessa (nested study) (5 023 henkilöä) ja COMPAS-tutkimuksessa (1 700 henkilöä).
Sekä COMPAS-tutkimuksessa että upotetussa (nested study) FinIP-tutkimuksessa Synflorix vähensi rokoteserotyyppien kantajuutta. Tehosteannoksen jälkeen havaittiin selvä nousu ei-rokoteserotyypeissä (poislukien rokotteeseen liittyvät). Tulokset eivät olleet tilastollisesti merkitseviä kaikissa COMPAS-analyyseissä. Kokonaisuutena tarkasteltuna oli kuitenkin havaittavissa suuntaus, jossa pneumokokin (nielu)kantajuus väheni.
Molemmissa tutkimuksissa yksittäiset serotyypit 6B ja 19F vähenivät merkittävästi. Upotetussa (nested study) FinIP-tutkimuksessa huomattava väheneminen havaittiin myös yksittäisten serotyyppien 14 ja 23F kohdalla, sekä kolmen annoksen perusrokotussarjassa ristireaktiivisen serotyypin 19A kohdalla.
Kliinisessä tutkimuksessa arvioitiin nenänielun kantajuutta HIV-positiivisilla pikkulapsilla (N = 83) ja HIV-positiivisten äitien synnyttämillä HIV-negatiivisilla pikkulapsilla (N = 101), ja sitä verrattiin HIV-negatiivisten äitien synnyttämiin HIV-negatiivisiin pikkulapsiin (N = 100). Altistuminen HI-virukselle tai HIV-infektio eivät näyttäneet muuttavan Synflorixin vaikutusta pneumokokin kantajuuteen 24–27 kuukauden ikään asti, eli 15 kuukautta tehosteannoksen antamisesta.
Vaikuttavuus markkinoille tulon jälkeisessä seurannassa
Brasiliassa Synflorix otettiin lasten yleiseen rokotusohjelmaan käyttäen 3+1 rokotusohjelmaa pikkulapsilla (2, 4 ja 6 kuukauden iässä ja tehosteannos 12 kuukauden iässä) ja täydennysrokotusta alle 2-vuotiailla. Kaltaistettu tapaus-verrokkitutkimus osoitti, että viljelyllä tai PRC-menetelmällä varmennettujen rokoteserotyyppien aiheuttamat IPD-tapaukset, sekä yksittäisten serotyyppi 6B:n, 14:n tai 19A:n aiheuttamat IPD-tapaukset, vähenivät merkitsevästi. Tutkimus perustui seurantatietoihin, joita kerättiin lähes kolmen vuoden ajan Synflorix-rokotusten aloituksesta.
Taulukko 4: Yhteenveto Synflorixin vaikuttavuudesta IPD-tapauksiin Brasiliassa
| IPD tauti(1) | Sovitettu vaikuttavuus(2) % (95% CI) |
Mikä tahansa rokoteserotyypin aiheuttama IPD(3)
| 83.8% (65.9; 92.3) 81.3% (46.9; 93.4) 87.7% (61.4; 96.1) |
Yksittäisen serotyypin aiheuttama IPD(4)
| 82.8% (23.8; 96.1) 87.7% (60.8; 96.1) 82.2% (10.7; 96.4) |
(1) Viljelyllä tai PCR-menetelmällä varmennettu IPD.
(2) Sovitettu vaikuttavuus edustaa IPD-tapausten prosentuaalista alenemaa Synflorixilla rokotetussa ryhmässä verrattuna ei-rokotettuun ryhmään, sekoittavien tekijöiden vaikutusta kontrolloiden.
(3) Viljelyllä tai PCR-menetelmällä varmennetut serotyyppien 4, 6B, 7F, 9V, 14, 18C, 19F ja 23F tapaukset olivat mukana analyysissä.
(4) Yksittäiset serotyypit, joille saatiin tilastollisesti merkitsevä tulos vaikuttavuusanalyysissä, sekoittavien tekijöiden vaikutusta kontrolloiden (monivertailuisovitusta ei tehty).
Suomessa Synflorix otettiin lasten yleiseen rokotusohjelmaan käyttäen rokotusohjelmaa 2+1 pikkulapsilla (3 ja 5 kuukauden iässä ja tehosteannos 12 kuukauden iässä) ilman täydennysrokotusta. Kun verrattiin IPD-tapausten ilmaantuvuutta ennen ja jälkeen rokotusohjelman, saatiin viitteitä minkä tahansa viljelyllä varmennetun, minkä tahansa rokoteserotyypin ja serotyypin 19A aiheuttaman IPD:n ilmaantuvuuden merkitsevästä alenemisesta.
Taulukko 5: IPD yleisyys ja vastaava yleisyyden alenema Suomessa
| IPD | Ilmaantuvuus 100 000 henkilövuotta kohden | Suhteellinen yleisyyden alenema(1) % (95% CI) | |
| Ennen yleistä rokotusohjelmaa | Yleisen rokotusohjelman jälkeen | ||
| Mikä tahansa viljelyllä varmennettu | 62.9 | 12.9 | 80% (72; 85) |
| Mikä tahansa rokoteserotyyppi(2) | 49.1 | 4.2 | 92% (86; 95) |
| Serotyyppi 19A | 5.5 | 2.1 | 62% (20; 85) |
(1) Suhteellinen yleisyyden aleneminen viittaa siihen, kuinka paljon IPD-tapausten ilmaantuvuus ≤5 ikäisillä lapsilla aleni Synflorix-ryhmässä (seuranta 3 vuotta rokotusohjelman alkamisesta) verrattuna historiallisiin ei-rokotettuihin ryhmiin, jotka olivat ikään ja vuodenajan suhteen sovitettuja (jokaista ryhmää seurattiin 3 vuoden ajan ennen kuin Synflorix otettiin yleiseen rokotusohjelmaan).
(2) Viljelyllä varmennetut serotyyppien 1, 4, 6B, 7F, 9V, 14, 18C, 19F ja 23 tapaukset olivat mukana analyysissä
Quebecissä Kanadassa Synflorix otettiin lasten rokotusohjelmaan (2 perusrokotusannosta alle 6 kuukauden ikäisille lapsille ja tehosteannos 12 kuukauden iässä) sen jälkeen, kun 7-valenttista Prevenar-pneumokokkikonjugaattirokotetta oli käytetty 4,5 vuotta. Perustuen 1,5 vuoden seurantaan, joka alkoi Synflorixin rokotusohjelman käyttöönoton jälkeen ja kun rokotekattavuus oli yli 90 % rokotettavan ikäryhmän lapsissa, havaittiin pienenemistä rokoteserotyyppien aiheuttaman IPD:n ilmaantuvuudessa (valtaosin muutoksia serotyypin 7F aiheuttamassa taudissa). Ei-rokoteserotyyppien aiheuttaman IPD:n ilmaantuvuudessa ei havaittu samanaikaista nousua. Kaiken kaikkiaan IPD-tapausten ilmaantuvuus oli 35/100 000 henkilövuotta Synflorixia saaneissa ryhmissä ja 64/100000 henkilövuotta 7-valenttista Prevenaria saaneissa ryhmissä. Ero on tilastollisesti merkitsevä (p = 0,03). Syy-seurausvaikutusten päätelmiä ei voida tehdä tämäntyyppisistä havainnoivista tutkimuksista.
Immunogeenisuus
Immunologinen vertailukelpoisuus (non-inferiority) 7-valenttisen Prevenarin kanssa
WHO:n suositusten mukaan potentiaalisen tehon osoittaminen invasiivisessa pneumokokkitaudissa ennen myyntiluvan myöntämistä perustui immuunivasteiden vertailuun niiden 7 serotyypin osalta, jotka sisältyvät Synflorixiin ja vertailurokotteeseen. Vertailurokotteena on ollut konjugoitu pneumokokkirokote, jonka suojateho on aikaisemmin osoitettu (7-valenttinen Prevenar). Vertailurokotteeseen kuulumattomien, mutta Synflorixin sisältämien kolmen muun serotyypin immuunivasteet on myös tutkittu.
Head-to-head tutkimuksessa, jossa Synflorixia verrattiin 7-valenttiseen Prevenariin, Synflorixin immuunivasteen samanarvoisuus osoitettiin ELISA-menetelmällä kaikille serotyypeille paitsi 6B:lle ja 23F:lle (yläraja-arvo 96,5 % Cl, ryhmien välinen ero > 10 %) (taulukko 6). Yhden kuukauden kuluttua kolmannesta Synflorix-perusannoksesta rokotetuista 65,9 % saavutti vasta-aineiden raja-arvon (0,20 mikrog/ml) 6B:lle ja 81,4 % 23F:lle, kun rokotus annettiin 2, 3 ja 4 kuukauden perusrokotusohjelmalla. 7-valenttisella Prevenarilla vastaavat prosenttiosuudet kolmen annoksen jälkeen olivat 79,0 % ja 94,1 %. Vasteiden erojen kliininen merkitys on tuntematon, sillä Synflorixin on havaittu olevan tehokas 6B serotyypin aiheuttamassa invasiivisessa pneumokokkitaudissa (IPD) kaksoissokkoutetussa, aluesatunnaistetussa kliinisessä tutkimuksessa (ks. taulukko 1).
Rokotetuista 97,3 %, 99,0 % ja 99,5 % saavutti raja-arvon Synflorixin lisäserotyypeille 1, 5 ja 7F. Vaste oli vähintään yhtä hyvä kuin 7-valenttisen Prevenarin yhdistetty keskimääräinen vaste molempien rokotteiden 7 yhteiselle serotyyppille (95,8 %).
Taulukko 6: Synflorixin ja 7-valenttisen Prevenarin vertaileva analyysi: Niiden henkilöiden prosentuaalinen osuus, joiden vasta-aine-pitoisuus oli > 0,20 mikrog/ml yhden kuukauden kuluttua kolmannesta annoksesta.
| Vasta-aine | Synflorix | 7-valenttinen Prevenar | Raja-arvon ≥ 0.20 mikrog/ml saavuttaneiden ero prosentteina (7-valenttinen Prevenar miinus Synflorix) | ||||
| N | % | N | % | % | 96,5 % CI | ||
| Anti-4 | 1106 | 97,1 | 373 | 100 | 2,89 | 1,71 | 4,16 |
| Anti-6B | 1100 | 65,9 | 372 | 79,0 | 13,12 | 7,53 | 18,28 |
| Anti-9V | 1103 | 98,1 | 374 | 99,5 | 1,37 | -0,28 | 2,56 |
| Anti-14 | 1100 | 99,5 | 374 | 99,5 | -0,08 | -1,66 | 0,71 |
| Anti-18C | 1102 | 96,0 | 374 | 98,9 | 2,92 | 0,88 | 4,57 |
| Anti-19F | 1104 | 95,4 | 375 | 99,2 | 3,83 | 1,87 | 5,50 |
| Anti-23F | 1102 | 81,4 | 374 | 94,1 | 12,72 | 8,89 | 16,13 |
Synflorixin aikaansaamat vasta-aineiden geometriset keskiarvopitoisuudet seitsemää yhteistä serotyyppiä vastaan olivat perusrokotussarjan jälkeen matalammat kuin 7-valenttisen Prevenarin aikaansaamat vastaavat pitoisuudet. Ennen tehosteannosta geometriset keskiarvopitoisuudet (8–12 kuukautta viimeisestä perusannoksesta) olivat yleensä molemmilla rokotteilla samanlaiset. Tehosteannoksen jälkeen geometriset keskiarvopitoisuudet olivat Synflorix-ryhmässä matalammat kuin 7-valenttisen Prevenar-ryhmässä useimpien yhteisten serotyyppien kohdalla.
Samassa tutkimuksessa Synflorixin osoitettiin aikaansaavan toiminnallisia vasta-aineita kaikille serotyypeille. Synflorix-rokotetuista 87,7–100 % ja 7-valenttisen Prevenar-rokotetuista 92,1–100% saavutti OPA-titterin ≥ 8 yhden kuukauden kuluttua kolmannesta annoksesta jokaista molempien rokotteen sisältämää seitsemää serotyyppiä vastaan. Rokotteiden välinen ero oli alle 5 % kaikille yhteisille serotyypeille, 6B ja 23F mukaan lukien, kun verrattiin niiden henkilöiden prosentuaalista osuutta, jotka saavuttivat OPA-titterin ≥ 8. Perus- ja tehosterokotuksen jälkeiset OPA vasta-aineiden keskimääräiset geometriset titterit olivat Synflorix-ryhmässä matalammat kuin 7-valenttisen Prevenar-ryhmässä kaikkien yhteisten seitsemän serotyypin kohdalla paitsi serotyypillä 19F.
Synflorixilla serotyypeille 1, 5 ja 7F vastaava prosentuaalinen osuus rokotetuista, joilla OPA-titteri oli ≥ 8, oli 65,7 %, 90,9 % ja 99,6 % perusrokotussarjan jälkeen ja 91,0 %, 96,3 % ja 100 % tehosteannoksen jälkeen. OPA-vasteet serotyypeille 1 ja 5 olivat suuruudeltaan matalampia kuin kaikille muille serotyypeille. Näiden havaintojen merkitystä suojatehoon ei tiedetä. Vaste serotyypille 7F oli samaa suuruusluokkaa kuin vaste seitsemälle yhteiselle serotyypille.
On osoitettu, että Synflorix saa aikaan immuunivasteen ristireaktiiviselle serotyyppi 19A:lle, jossa rokotetuista 48,8 % (95 % CI: 42,9;54,7) saavutti OPA-titterin ≥ 8 yhden kuukauden kuluttua tehosteannoksesta.
Neljännen annoksen antaminen (tehosteannos) toisena ikävuotena aikaansai anamnestisen vasta-ainevasteen, joka oli mitattavissa ELISA- ja OPA-menetelmällä. Vaste oli havaittavissa rokoteserotyypeille ja ristireaktiiviselle 19A serotyypille, mikä osoittaa immunologisen muistin muodostuneen kolmen annoksen perusrokotussarjan jälkeen.
Immunogeenisuuteen liittyviä lisätietoja
Vähintään 6 viikon ja enintään 6 kuukauden ikäiset pikkulapset
Kolmen annoksen perusrokotussarja
Kliinisissä tutkimuksissa Synflorixin immunogeenisuutta arvioitiin 3 perusrokotusannoksen jälkeen (6 941 koehenkilöä) eri aikatauluilla annettuna (mm. 6-10-14 viikon, 2-3-4 , 3-4-5 tai 2-4-6 kuukauden iässä), sekä neljännen annoksen (tehoste) jälkeen (5 645 koehenkilöä) , joka annettiin vähintään 6 kuukautta viimeisen perusrokotussarjaan kuuluvan annoksen jälkeen 9 kuukauden iästä lähtien. Yleisesti ottaen rokotevasteet olivat eri rokotusohjelmissa keskenään verrannollisia, vaikka 2-4-6 kuukauden rokotusohjelmassa havaittiin jonkin verran korkeampia immuunivasteita.
Kahden annoksen perusrokotussarja
Synflorixin immunogeenisuutta on arvioitu kliinisissä tutkimuksissa kahden annoksen perusrokotusohjelman jälkeen (470 koehenkilöä) annettuna eri aikataulujen mukaan (mukaan lukien 6–14 viikon, 2–4 tai 3–5 kuukauden iässä). Kolmas annos (tehoste) (470 koehenkilöä) annettiin vähintään 6 kuukautta viimeisen perusrokotussarjaan kuuluvan annoksen jälkeen 9 kuukauden iästä lähtien.
Kliinisessä tutkimuksessa arvioitiin Synflorixin immunogeenisuutta neljässä Euroopan maassa henkilöillä, jotka olivat saaneet kaksi tai kolme perusrokotusannosta. Niiden henkilöiden prosentuaalisessa osuudessa, jotka saavuttivat vasta-aineiden pitoisuuden ≥ 0,20 mikrog/ml (ELISA) ei havaittu merkittävää eroa ryhmien välillä. Serotyypeillä 6B ja 23F henkilöiden prosentuaalinen osuus oli matalampi kuin muilla rokoteserotyypeillä (taulukot 7 ja 8). Verrattaessa kahden annoksen perusrokotussarjaa saaneita kolmen annoksen perusrokotussarjan saaneisiin, niiden henkilöiden prosentuaalinen osuus, joilla OPA-titterit olivat ≥ 8, oli matalampi serotyypeille 6B, 18C ja 23F (74,4 %, 82,8 %, 86,3 % kahden annoksen rokotusohjelmassa ja 88,9 %, 96,2 % ja 97,7 % vastaavasti kolmen annoksen rokotusohjelmassa). Kaiken kaikkiaan immuunivaste säilyi heikommin 11 kuukauden iässä annettuun tehosteannokseen saakka kahden annoksen perusrokotusryhmässä. Molemmissa rokotusohjelmissa tehosteannoksen aikaansaama vaste osoitti immunologisen muistin olemassa oloa jokaiselle rokoteserotyypille (taulukot 7 ja 8). Tehosteannoksen jälkeen 2 annoksen perusrokotusohjelmassa havaittiin pienempi prosentuaalinen osuus henkilöitä, joiden OPA-titterit olivat ≥ 8 serotyyppi 5:llä (87,2 % 2 annoksen perusrokotusohjelmassa ja 97,5 % 3 annoksen perusrokotusohjelmassa) ja serotyyppi 6B:llä (81,1 % verrattuna 90,3 %). Kaikki muut vasteet olivat verrattavissa.
Taulukko 7: Kahden annoksen perusrokotussarjan saaneiden henkilöiden prosentuaalinen osuus, joilla vasta-aineiden pitoisuudet olivat ≥ 0,20 mikrog/ml yhden kuukauden kuluttua perusrokotussarjasta ja yhden kuukauden kuluttua tehosteannoksesta
| Vasta-aine | ≥ 0,20 mikrog/ml (ELISA) | |||||
| Perusrokotussarjan jälkeen | Tehosteannoksen jälkeen | |||||
| % | 95 % CI | % | 95 %CI | |||
| Anti-1 | 97,4 | 93,4 | 99,3 | 99,4 | 96,5 | 100 |
| Anti-4 | 98,0 | 94,4 | 99,6 | 100 | 97,6 | 100 |
| Anti-5 | 96,1 | 91,6 | 98,5 | 100 | 97,6 | 100 |
| Anti-6B | 55,7 | 47,3 | 63,8 | 88,5 | 82,4 | 93,0 |
| Anti-7F | 96,7 | 92,5 | 98,9 | 100 | 97,7 | 100 |
| Anti-9V | 93,4 | 88,2 | 96,8 | 99,4 | 96,5 | 100 |
| Anti-14 | 96,1 | 91,6 | 98,5 | 99,4 | 96,5 | 100 |
| Anti-18C | 96,1 | 91,6 | 98,5 | 100 | 97,7 | 100 |
| Anti-19F | 92,8 | 87,4 | 96,3 | 96,2 | 91,8 | 98,6 |
| Anti-23F | 69,3 | 61,3 | 76,5 | 96,1 | 91,7 | 98,6 |
Taulukko 8: Kolmen annoksen perusrokotussarjan saaneiden henkilöiden prosentuaalinen osuus, joilla vasta-aineiden pitoisuudet olivat ≥ 0,20 mikrog/ml yhden kuukauden kuluttua perusrokotussarjasta ja yhden kuukauden kuluttua tehosteannoksesta
| Vasta-aine | ≥ 0,20 mikrog/ml (ELISA) | |||||
| Perusrokotussarjan jälkeen | Tehosteannoksen jälkeen | |||||
| % | 95 % CI | % | 95 %CI | |||
| Anti-1 | 98,7 | 95,3 | 99,8 | 100 | 97,5 | 100 |
| Anti-4 | 99,3 | 96,4 | 100 | 100 | 97,5 | 100 |
| Anti-5 | 100 | 97,6 | 100 | 100 | 97,5 | 100 |
| Anti-6B | 63,1 | 54,8 | 70,8 | 96,6 | 92,2 | 98,9 |
| Anti-7F | 99,3 | 96,4 | 100 | 100 | 97,5 | 100 |
| Anti-9V | 99,3 | 96,4 | 100 | 100 | 97,5 | 100 |
| Anti-14 | 100 | 97,6 | 100 | 98,6 | 95,2 | 99,8 |
| Anti-18C | 99,3 | 96,4 | 100 | 99,3 | 96,3 | 100 |
| Anti-19F | 96,1 | 91,6 | 98,5 | 98,0 | 94,2 | 99,6 |
| Anti-23F | 77,6 | 70,2 | 84,0 | 95,9 | 91,3 | 98,5 |
Samanlaisia vasta-aineiden GMC-arvoja (ELISA) ristireaktiiviselle serotyypille 19A perusrokotussarjan ja tehosteannoksen jälkeen havaittiin kahden annoksen rokotusohjelmassa [0,14 mikrog/ml (95 % CI: 0,12;0,17) ja 0,73 mikrog/ml (95 % CI: 0,58;0,92)] sekä kolmen annoksen rokotusohjelmassa [0,19 mikrog/ml (95 % CI: 0,16;0,24) ja 0,87 mikrog/ml (95 % CI: 0,69;1,11)]. Kahden annoksen rokotusohjelmassa niiden henkilöiden osuus, joilla OPA titterit olivat ≥ 8, sekä geometriset keskiarvotiitterit (GMT), jäivät matalammiksi perusrokotussarjan ja tehosteannoksen jälkeen kuin kolmen annoksen rokotusohjelmassa. Molemmissa rokotusohjelmissa havaittiin anamnestiseen immuunivasteeseen viittaava vaste tehosteannokselle.
Kahden annoksen perusrokotusohjelmassa perusannosten ja tehosteannoksen jälkeen havaittujen matalampien immuunivasteiden kliinistä merkitystä ei tiedetä.
Etelä-Afrikassa tehdyssä tutkimuksessa arvioitiin Synflorixin immunogeenisuutta, kun se annettiin tehosteannoksena 9–10 kuukauden iässä kolmen (6, 10 ja 14 viikon iässä) tai kahden (6 ja 14 viikon iässä) annoksen perusrokotussarjan jälkeen. Perusrokotussarjan jälkeen havaittiin, että niiden koehenkilöiden prosenttiosuus, jotka saavuttivat vasta-ainetasot ja joiden OPA-titterit olivat ≥ 8, oli samanlainen kaikille rokoteserotyypeille sekä kahden että kolmen annoksen jälkeen lukuun ottamatta alempia OPA-prosenttiosuuksia serotyypille 14. Vasta-aineiden GMC:t ja OPA GMT:t olivat matalampia kahden annoksen jälkeen useimmille rokoteserotyypeille.
Perusrokotussarjan jälkeen havaittiin koskien ristireaktiivista serotyyppiä 19A, että vastaava prosenttiosuus koehenkilöistä saavutti vasta-ainekynnyksen ja OPA-titterit ≥ 8 sekä samanlaiset vasta-aineiden GMC:t ja OPA GMT:t molemmissa ryhmissä.
Kaiken kaikkiaan tehosteannosta edeltävä immuunivasteen pysyvyys oli matalampi kahden annoksen kuin kolmen annoksen perusrokotussarjaryhmässä useimmille rokoteserotyypeille ja oli samanlainen serotyypille 19A.
Tehosteannos 9–10 kuukauden iässä
Etelä-Afrikassa tehdyssä tutkimuksessa 9–10 kuukauden iässä annettu tehosteannos aiheutti merkittävää nousua vasta-aineiden GMC:ssä ja OPA GMT:ssä jokaiselle rokoteserotyypille ja serotyypille 19A sekä kahden annoksen että kolmen annoksen perusrokotussarjaryhmissä. Tämä osoittaa, että tapahtuu immuunivasteen primaarista kehittymistä (priming).
Tehosteannos 9–12 kuukauden iässä vs. 15–18 kuukauden iässä
Intiassa tehdyssä tutkimuksessa arvioitiin tehosteannosta, joka annettiin 6, 10 ja 14 viikon iässä annetun perusrokotussarjan jälkeen 9–12 kuukauden iässä (66 lasta) tai 15–18 kuukauden iässä (71 lasta). Tutkimuksessa ei havaittu eroja ryhmien vasta-aineiden GMC:ssä. Ryhmässä, joka sai tehosteannoksen 15–18 kuukauden iässä, havaittiin korkeampia OPA GMT:itä useimmille rokoteserotyypeille ja serotyypille 19A. Tämän havainnon kliinistä merkitystä ei kuitenkaan tiedetä.
Immunologinen muisti
Eurooppalaisen kahden ja kolmen annoksen perusrokotusohjelmia arvioivan tutkimuksen seurannassa osoitettiin vasta-aineiden säilyminen 36−46 kuukauden iässä tutkittavilla, jotka olivat saaneet kahden annoksen perusrokotussarjan jälkeen tehosteannoksen. Tutkittavista vähintään 83,7 % oli edelleen seropositiivisia rokoteserotyypeille ja ristireatiiviselle serotyypille 19A. Tutkittavista, jotka olivat saaneet kolmen annoksen perusrokotussarjan ja tehosteannoksen, vähintään 96,5 % oli edelleen seropositiivisia rokoteserotyypeille ja 86,4 % oli seropositiivisia serotyypille 19A. Yhden 4. ikävuotena annetun Synflorix-täydennysrokotusannoksen jälkeen ELISA vasta-aineiden geometristen keskiarvopitoisuuksien ja OPA geometristen keskiarvotitterien nousun määrä (fold-increase) ennen rokotusta olevalta tasolta rokotuksen jälkeiseen tasoon oli samanlainen kaksi perusannosta saaneilla henkilöillä verrattuna kolme perusannosta saaneisiin henkilöihin. Nämä tulokset osoittavat immunologisen muistin olemassaolon perusrokoteannoksia saaneilla henkilöillä kaikille rokoteserotyypeille ja ristireaktiiviselle serotyypille 19A.
Rokottamattomat ≥ 7 kuukauden ikäiset pikkulapset ja lapset:
Synflorixin aikaansaamaa immuunivastetta rokoteserotyypille ja ristireaktiiviselle serotyypille 19A tutkittiin aikaisemmin rokottamattomilla vanhemmilla lapsilla kolmessa kliinisessä tutkimuksessa.
Ensimmäisessä kliinisessä tutkimuksessa immuunivastetta rokoteserotyypeille ja ristireaktiiviselle serotyypille 19A tutkittiin 7–11 kuukauden ikäisillä, 12–23 kuukauden ikäisillä ja 2–5 vuoden ikäisillä pikkulapsilla ja lapsilla.
- 7–11 kuukauden ikäisille pikkulapsille annettiin kaksi perusannosta ja tehosteannos toisena ikävuotena. Tässä ikäryhmässä immuunivaste oli tehosteannoksen jälkeen yleensä samanlainen kuin lapsilla, jotka olivat saaneet kolme perusannosta ennen kuuden kuukauden ikää.
- 12–23 kuukauden ikäisillä lapsilla immuunivasteet kahden annoksen jälkeen olivat verrattavissa niihin vasteisiin, jotka saatiin < 6 kuukauden ikäisillä lapsilla, jotka olivat saaneet kolme annosta. Poikkeuksen muodostivat rokoteserotyypit 18C ja 19F sekä serotyyppi 19A, joille vaste oli korkeampi 12–23 kuukauden ikäisillä lapsilla.
- 2–5 vuoden ikäisillä lapsilla, joille oli annettu yksi annos, ELISA vasta-aineiden geometriset keskiarvopitoisuudet olivat samanlaiset 6:lle rokoteserotyypille sekä serotyypille 19A, kuin annettaessa < 6 kuukauden ikäisille pikkulapsille kolmen annoksen rokotussarja. Pitoisuudet olivat matalammat 4:lle rokoteserotyypille (serotyypit 1, 5, 14 ja 23F). OPA geometriset keskiarvotitterit olivat samanlaiset tai korkeammat kerta-annoksen jälkeen kuin kolmen annoksen perusrokotussarjan jälkeen < 6 kuukauden ikäisillä pikkulapsilla, paitsi serotyypin 5 osalta.
Toisessa kliinisessä tutkimuksessa yksi annos annettuna neljän kuukauden kuluttua kahden täydennysrokotusannoksen jälkeen, jotka oli annettu 12−20 kuukauden iässä, sai aikaan huomattavan nousun vasta-aineiden ELISA geometrisissä keskiarvopitoisuuksissa ja OPA geometrisissä keskiarvotittereissä (kun vasteita verrattiin ennen viimeistä annosta ja sen jälkeen). Tämä osoittaa, että kaksi täydennysannosta saa aikaan riittävän immunisaation.
Kolmas kliininen tutkimus osoitti, että kahden annoksen rokotussarja annettuna kahden kuukauden välein 36–46 kuukauden ikäisille lapsille aikaansai jokaiselle rokoteserotyypille ja ristireaktiiviselle serotyypille 19A korkeammat ELISA vasta-aineiden geometriset keskiarvopitoisuudet ja OPA geometriset keskiarvotitterit kuin kuukauden kuluttua mitatut arvot kolmen annoksen perusrokotussarjan jälkeen. Niiden koehenkilöiden osuus, joilla ELISA vasta-aine pitoisuudet olivat ≥ 0,20 mikrog/ml tai OPA titterit olivat ≥ 8 jokaiselle rokotteen serotyypille, oli täydennysrokotusryhmässä verrannollinen tai korkeampi kuin kolmen annoksen perusrokotussarjan saaneilla.
Vasta-aineiden pitkäaikaista säilymistä ei ole tutkittu pikkulapsilla, joille on annettu perusrokotussarja ja tehosteannos eikä vanhemmilla lapsilla, joille on annettu kahden annoksen perusrokotussarja.
Kliinisessä tutkimuksessa on osoitettu, että Synflorix voidaan turvallisesti antaa tehosteena toisena ikävuotena lapsille, jotka ovat saaneet kolmen 7-valenttisen Prevenar-annoksen perusrokotussarjan. Tutkimus osoitti, että immuunivaste 7 yhteiselle serotyypille oli verrattavissa siihen vasteeseen, joka saadaan 7-valenttisella Prevenar-tehosteannoksella. On kuitenkin huomattava, että 7-valenttista Prevenaria perusrokotussarjana saaneet lapset eivät muodosta suojaa Synflorixin kolmelle lisäserotyypille (1, 5 ja 7F). Tästä syystä tässä ikäryhmässä yhden Synflorix-annoksen antamaa suojaa ja kestoa ei näiden kolmen serotyypin kohdalla voida arvioida invasiivisessa pneumokokkien aiheuttamassa taudissa eikä välikorvantulehduksessa.
Immunogeenisuustiedot keskosilla
Synflorixin immunogeenisuutta arvioitiin hyvin ennenaikaisesti syntyneillä keskosilla (raskausaika 27−30 viikkoa) (N=42), keskosilla (raskausaika 31−36 viikkoa) (N=82) ja täysiaikaisilla pikkulapsilla (raskausaika > 36 viikkoa) (N=132) tutkimuksessa, jossa koehenkilöille annettiin kolmen annoksen perusrokotussarja 2, 4 ja 6 kuukauden iässä. Immunogeenisuutta arvioitiin neljännen (tehoste) annoksen jälkeen 15−18 kuukauden iässä hyvin ennenaikaisesti syntyneillä keskosilla (N=44), keskosilla (N=69) ja täysiaikaisilla pikkulapsilla (N=127).
Kuukauden kuluttua perusrokotussarjasta (toisin sanoen kolmannen annoksen jälkeen) vähintään 92,7 % koehenkilöistä saavutti ≥ 0,20 mikrog/ml ELISA vasta-ainepitoisuudet kaikille rokoteserotyypeille ja vähintään 81,7 % saavutti ≥ 8 OPA titterit kaikille rokoteserotyypeille, paitsi serotyypille 1 (vähintään 58,8 %:lla OPA titterit ≥ 8). Toisiaan vastaavat vasta-aineiden geometriset keskiarvopitoisuudet ja OPA geometriset keskiarvotitterit havaittiin kaikilla pikkulapsilla. Poikkeuksen muodostivat serotyypit 4, 5,9V ja ristireaktiivinen serotyyppi 19A, joilla geometriset keskiarvopitoisuudet olivat matalammat hyvin ennenaikaisesti syntyneillä, ja serotyyppi 9V, jolla geometriset keskiarvopitoisuudet olivat matalammat keskosilla ja serotyyppi 5, jolla OPA geometriset keskiarvotitterit olivat matalammat hyvin ennenaikaisesti syntyneillä keskosilla. Näiden erojen kliinistä merkitystä ei tunneta.
Kuukauden kuluttua tehosteannoksesta havaittiin nousua vasta-aineiden ELISA geometrisissä keskiarvopitoisuuksissa ja OPA geometrisissä keskiarvotittereissä jokaiselle rokoteserotyypille ja ristireaktiiviselle serotyypille 19A, mikä on osoitus immunologisesta muistista. Vastaavat vasta-aineiden geometriset keskiarvopitoisuudet ja OPA geometriset keskiarvotitterit havaittiin kaikissa lapsiryhmissä. Poikkeuksen muodosti serotyyppi 5, jolla havaittiin matalammat OPA geometriset keskiarvotitterit hyvin ennenaikaisesti syntyneillä. Kaiken kaikkiaan vähintään 97,6 % koehenkilöistä saavutti vasta-aineiden ELISA-pitoisuudet, jotka olivat ≥ 0,20 mikrog/ml ja vähintään 91,9 % saavutti OPA-titterit, jotka olivat ≥ 8.
Immunogeenisuus erityispotilailla
HIV-positiiviset (HIV+/+) pikkulapset ja HIV-positiivisten äitien synnyttämät HIV-negatiiviset (HIV+/-) pikkulapset
Etelä-Afrikassa suoritetussa kliinisessä tutkimuksessa arvioitiin Synflorixin immunogeenisuutta. Tutkimuksessa Synflorix annosteltiin kolmen annoksen perusrokotussarjana (6, 10 ja 14 viikon iässä), jonka jälkeen annettiin tehosteannos (9−10 kuukauden iässä). Tutkimukseen osallistui 70 HIV-positiivista pikkulasta (HIV+/+), 91 HIV-positiivisten äitien synnyttämää HIV-negatiivista (HIV+/-) pikkulasta ja 93 HIV-negatiivisten äitien synnyttämää HIV-negatiivista (HIV -/-) pikkulasta. Tutkimukseen otettiin mukaan vain HIV+/+ -pikkulapsia, joiden taudin WHO-luokitus oli tasolla 1 (oireeton) tai tasolla 2 (lievät oireet).
Suurimmalle osalle rokoteserotyypeistä ryhmien välinen vertailu ei osoittanut eroavaisuuksia immuunivasteissa perusrokotussarjan jälkeen HIV+/+ ja HIV -/- ryhmien tai HIV+/- ja HIV-/- ryhmien välillä. Poikkeuksena HIV +/+ ryhmässä pienempi prosenttiosuus potilaista saavutti OPA titterin ≥ 8 ja OPA GMT oli matalampi. Tämän matalamman perusrokotussarjan jälkeisen OPA vasteen kliininen merkitys ei ole tiedossa. Tulokset eivät osoittaneet ryhmien välillä olevan eroja vasta-aineiden ELISA GMC:ssa ja OPA GMT:ssa ristireaktiiviselle serotyypille 19A.
Synflorixin tehosteannos HIV+/+ ja HIV +/- pikkulapsilla sai aikaan vahvan nousun vasta-aineiden ELISA GMC:ssa ja OPA GMT:ssa jokaiselle rokoteserotyypille ja serotyyppi 19A:lle, mikä osoittaa immunologista kehittymistä (priming). Suurimmalle osalle rokoteserotyypeistä ja serotyyppi 19A:lle ryhmien välinen vertailu ei osoittanut eroja tehosteannoksen jälkeen vasta-aineiden ELISA GMC:ssa ja OPA GMT:ssa ryhmien HIV+/+ ja HIV-/- tai HIV+/- ja HIV-/- välillä.
Proteiini D:tä koskevien tuloksien mukaan perusrokotussarjan ja tehosteannoksen jälkeiset immuunivasteet olivat verrannollisia ryhmien välillä.
Jokaisessa ryhmässä immuunivasteen havaittiin jatkuvan 24−27 kuukauden iässä, eli 15 kuukauteen asti tehosteannoksen jälkeen.
Sirppisoluanemiaa (SCD) sairastavat lapset
Burkina Fasossa tehdyssä kliinisessä tutkimuksessa arvioitiin Synflorixin immunogeenisuutta, kun sitä annettiin 146 lapselle, jotka sairastivat SCD:tä (HbSS-tautia, HbSC-tautia tai beetatalassemiaa), ja verrokkiryhmälle, jossa oli 143 ikävertaistettua lasta, joilla ei ollut SCD:tä. Lapsista, joilla oli SCD, 48 alle 6 kuukauden ikäistä lasta sai primaarirokotuksen 8, 12 ja 16 viikon iässä, jonka jälkeen he saivat tehosteannoksen 9–10 kuukauden iässä, 50 7–11 kuukauden ikäistä ja 48 12–23 kuukauden ikäistä lasta aloittivat kiinniottorokotukset iänmukaisesti. SCD:n ei havaittu vaikuttavan Synflorixin immuunivasteeseen yhdellekään rokoteserotyypeille, serotyyppi 19A:lle tai proteiini D:lle.
Pernan toimintahäiriötä sairastavat lapset
Synflorix-valmisteen immunogeenisuutta ja turvallisuutta arvioitiin pneumokokkirokotetta saamattomilla ja aiemmin sitä saaneilla lapsilla, joilla oli synnynnäinen tai hankinnainen pernattomuus, pernan toimintahäiriö tai komplementtipuutoksia. Tutkimukseen osallistui vain rajallinen määrä lapsia. Tutkittavista kuusi oli 2–5-vuotiaita ja neljäkymmentä oli 6–17-vuotiaita (Synflorix-valmisteen käyttöaihe on lapsilla viidenteen syntymäpäivään asti). Tässä tutkimuksessa Synflorix-valmisteen immunogeenisuus osoitettiin eikä uutta turvallisuuteen liittyvää tietoa todettu.
Säilöntäaineena 2-fenoksietanolia (2-PE) sisältävän Synflorix-valmisteen immunogeenisuus
Synflorix-valmisteen, joka sisältää säilöntäaineena 2-PE:tä (mukana 4 annoksen pakkauksessa), immunogeenisuutta arvioitiin 6, 10 ja 18 viikon iässä rokotetuilla terveillä pikkulapsilla, ja verrattiin Synflorixia ilman lisättyä säilöntäainetta saaneeseen ryhmään (160 tutkittavaa per ryhmä).
Immuunivasteita vertailtiin käyttäen samanarvoisuus(non-inferiority)-kriteereitä vasta-aineiden GMC-suhteen osalta (GMC tutkimusryhmästä, joka sai Synflorixia ilman 2-PE:tä, suhteessa GMC:hen tutkimusryhmästä, joka sai 2-PE:tä sisältävää Synflorixia) jokaiselle kymmenelle rokotteen serotyypille ja ristireaktiiviselle serotyypille 19A.
Samanarvoisuus osoitettiin, sillä vasta-aineiden GMC-suhteiden kaksipuolisen 95 % CI:n yläraja oli alle kaksi jokaiselle kymmenelle rokoteserotyypille ja ja serotyypille 19A. Lisäksi OPA GMT:t olivat samansuuruisia molemmissa ryhmissä.
Synflorix-valmisteen ja 13‑valenttisen konjugoidun pneumokokkirokotteen käyttö samassa rokotusohjelmassa
Synflorix‑valmisteen ja 13‑valenttisen konjugoidun pneumokokkirokotteen (PCV13) käyttöä (vaihdettavuutta) samassa rokotusohjelmassa arvioitiin Meksikossa toteutetussa kliinisessä tutkimuksessa. Imeväisikäisiä, jotka saivat ensin perusrokotussarjana kaksi annosta PCV13‑rokotetta (86 tutkittavaa) tai yhden annoksen PCV13‑rokotetta ja yhden annoksen Synflorix-valmistetta (89 tutkittavaa) ja myöhemmin tehosteannoksena Synflorix-valmistetta 12–15 kuukauden iässä, vertailtiin Synflorix-valmistetta 2+1-rokotusohjelman mukaisesti saaneisiin imeväisikäisiin.
Yhden kuukauden kuluttua perusrokotussarjan ja tehosteannoksen saamisesta niiden imeväisikäisten prosenttiosuudet, joilla vasta-aineet useimmille 10 yleisestä serotyypistä saavuttivat arvon ≥ 0,2 µg/ml ja OPA-titterit nousivat raja-arvoja suuremmiksi, olivat suuret sekä Synflorix-valmistetta että PCV13-rokotetta saaneilla: ≥ 97,7 %:lla vasta-ainepitoisuudet kahdeksassa 10 serotyypistä olivat ≥ 0,2 µg/ml, ja ≥ 92,0 %:lla seitsemässä 10 serotyypistä OPA-titterit olivat raja-arvoja suuremmat. Ristireaktiivisen serotyypin 19A kohdalla vastaavat prosenttiosuudet olivat vähintään 86,5 % ja 88,0 %.
Turvallisuuteen liittyviä huolenaiheita ei tullut ilmi, kun rokote vaihdettiin PCV13-rokotteesta Synflorix-valmisteeseen perusrokotussarjan tai tehosterokotuksen aikana.
Farmakokinetiikka
Ei oleellinen.
Prekliiniset tiedot turvallisuudesta
Tutkimukset 11-valenttisella rokotteella, joka vastaa Synflorixia, eivät tuoneet ilmi mitään erityistä riskiä ihmisille. Päätelmät perustuvat tavanomaisiin farmakologisiin turvallisuustutkimuksiin sekä akuutin ja toistuvan annostuksen toksisuuskokeisiin.
Farmaseuttiset tiedot
Apuaineet
Yhden annoksen säiliö
Natriumkloridi
Injektionesteisiin käytettävä vesi
Neljän annoksen säiliö
Natriumkloridi
2-fenoksietanoli
Injektionesteisiin käytettävä vesi
Adsorbentit, ks. kohta Vaikuttavat aineet ja niiden määrät.
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Yhden annoksen säiliö
4 vuotta
Neljän annoksen säiliö
3 vuotta
Kun neljän annoksen injektiopullo on avattu, rokotetta voidaan säilyttää jääkaapissa (2 °C–8 °C) korkeintaan 28 päivän ajan. Rokote on hävitettävä, jos sitä ei käytetä 28 päivän kuluessa avaamisesta.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Neljän annoksen säiliö
Säilytysolosuhteet injektiopullon ensimmäisen avaamisen jälkeen, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SYNFLORIX injektioneste, suspensio, esitäytetty ruisku
10 x 0,5 ml (ilman neulaa) (-)
PF-selosteen tieto
Esitäytetty ruisku
0,5 ml:aa suspensiota esitäytetyssä ruiskussa (tyypin I lasia), jossa männän tulppa (butyylikumia) ja kuminen kärkikorkki. Pakkauskoot 1, 10 ja 50, neuloilla tai ilman.
Injektiopullo
0,5 ml:aa suspensiota yhden annoksen injektiopullossa (tyypin I lasia), jossa korkki (butyylikumia). Pakkauskoot 1, 10 ja 100.
Neljän annoksen säiliö
2 ml:aa suspensiota neljän annoksen injektiopullossa (tyypin I lasia), jossa korkki (butyylikumia). Pakkauskoot 10 ja 100.
Esitäytetyn ruiskun kärkikorkki ja kuminen männän tulppa sekä injektiopullon korkki ovat valmistettu synteettisestä kumista.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Rokote on samea, valkoinen suspensio.
Käyttö- ja käsittelyohjeet
Esitäytetty ruisku
Esitäytettyä ruiskua varastoitaessa siihen voi muodostua hienojakoinen valkoinen sakka ja kirkas valkoinen pintakerros. Tämä ei ole laadun huonontumisen merkki.
Ennen kuin ruisku otetaan käyttöön sen sisältöä tarkistetaan silmämääräisesti ennen ravistamista ja sen jälkeen vierashiukkasten ja/tai fysikaalisten muutosten varalta. Hävitettävä, jos ulkonäkö on muuttunut.
Rokotteen on annettava lämmetä huoneenlämpöiseksi ennen antoa.
Rokotetta on ravistettava hyvin ennen käyttöä.
Esitäytetyn ruiskun käyttöohje
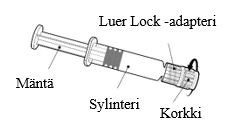 | Pidä kiinni ruiskun sylinteristä, ei männästä. Poista ruiskun korkki kiertämällä vastapäivään. |
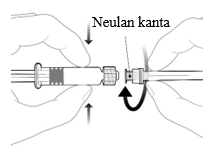 | Neula kiinnitetään ruiskuun yhdistämällä neulan kanta Luer Lock -adapteriin ja kiertämällä neulaa neljänneskierros myötäpäivään, kunnes neula tuntuu kiinnittyvän ruiskuun. Älä vedä ruiskun mäntää ulos sylinteristä. Jos mäntä irtoaa sylinteristä, älä anna rokotetta. |
Injektiopullo
Injektiopulloa varastoitaessa siihen voi muodostua hienojakoinen valkoinen sakka ja kirkas valkoinen pintakerros. Tämä ei ole laadun huonontumisen merkki.
Ennen kuin injektiopullo otetaan käyttöön sen sisältöä tarkistetaan silmämääräisesti ennen ravistamista ja sen jälkeen vierashiukkasten ja/tai fysikaalisten muutosten varalta. Hävitettävä, jos ulkonäkö on muuttunut.
Rokotteen on annettava lämmetä huoneenlämpöiseksi ennen antoa.
Rokotetta on ravistettava hyvin ennen käyttöä.
Injektiopulloa käytettäessä 0,5 ml:n annos vedetään ruiskuun sopivaa steriiliä neulaa käyttäen (21G–25G); varotoimiin on ryhdyttävä kontaminaation estämiseksi.
Ennen annostelua vaihda neula niin, että käytät uutta steriiliä neulaa.
Neljän annoksen säiliö
Injektiopulloa varastoitaessa siihen voi muodostua hienojakoinen valkoinen sakka ja kirkas valkoinen pintakerros. Tämä ei ole laadun huonontumisen merkki.
Ennen kuin injektiopullo otetaan käyttöön sen sisältöä tarkistetaan silmämääräisesti ennen ravistamista ja sen jälkeen vierashiukkasten ja/tai fysikaalisten muutosten varalta. Hävitettävä, jos ulkonäkö on muuttunut.
Rokotteen on annettava lämmetä huoneenlämpöiseksi ennen antoa.
Rokotetta on ravistettava hyvin ennen käyttöä.
Moniannossäiliötä käytettäessä jokainen 0,5 ml annos vedetään ruiskuun sopivaa steriiliä neulaa käyttäen (21G–25G); varotoimiin on ryhdyttävä kontaminaation estämiseksi.
Ennen annostelua vaihda neula niin, että käytät uutta steriiliä neulaa.
Hävitys
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SYNFLORIX injektioneste, suspensio, esitäytetty ruisku
10 x 0,5 ml
- Ei korvausta.
ATC-koodi
J07AL52
Valmisteyhteenvedon muuttamispäivämäärä
19.05.2026
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi