
XOFIGO injektioneste, liuos 1100 kBq/ml
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksia ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi millilitra liuosta sisältää 1100 kBq radium Ra 223-dikloridia (radium–223-dikloridi), joka vastaa 0,58 ng radium-223:a referenssipäivänä. Radium esiintyy liuoksessa vapaana ionina.
Kukin injektiopullo sisältää 6 ml liuosta (6,6 MBq radium-223-dikloridia referenssipäivänä).
Radium-223 on alfahiukkasia säteilevä aine, jonka puoliintumisaika on 11,4 vuorokautta. Radium-223:n spesifinen aktiivisuus on 1,9 MBq/ng.
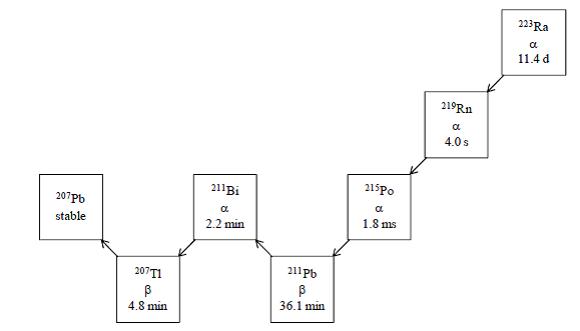
Radium-223:n kuusivaiheinen hajoaminen lyijy-207:ksi tapahtuu lyhytaikaisten tytärnuklidien kautta, ja siihen liittyy useita, energioiltaan ja emissiotodennäköisyyksiltään erilaisia alfa-, beeta- ja gammaemissioita. Radium-223:sta ja sen tytärnuklideista alfahiukkasina säteilevän energian osuus on 95,3 % (energia-alue 5,0-7,5 MeV). Beetahiukkasina säteilevä osuus on 3,6 % (keskimääräiset energiat ovat 0,445 MeV ja 0,492 MeV), ja gammasäteilynä säteilevä osuus on 1,1 % (energia-alue 0,01-1,27 MeV).
Kuva 1:Radium-223:n hajoamisketju, fysikaaliset puoliintumisajat ja hajoamistavat:
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Xofigo on tarkoitettu metastaattisen kastraatioresistentin eturauhassyövän hoitoon aikuispotilaille, joilla on oireilevia luustometastaaseja, mutta ei tiedossa olevia viskeraalisia metastaaseja, joko monoterapiana tai yhdessä luteinisoivan hormonin vapauttajahormonin (LHRH) analogin kanssa taudin progressiossa vähintään kahden aiemman metastaattisen kastraatioresistentin eturauhassyövän systeemisen hoitolinjan (muiden kuin LHRH-analogien) jälkeen, tai joita ei voida hoitaa muilla systeemisillä metastaattisen kastraatioresistentin eturauhassyövän hoidoilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ehto
Valmistetta saavat antaa vain radioaktiivisten aineiden käsittelyyn valtuutetut henkilöt tähän tarkoitukseen varatuissa tiloissa sen jälkeen, kun pätevä lääkäri on tutkinut potilaan.
Annostus ja antotapa
Annostus
Xofigo-valmisteen annostelu: kerta-annos 55 kBq:n aktiivisuus painokiloa kohti annetaan 4 viikon välein, yhteensä 6 injektiota.
Xofigo-valmisteen turvallisuutta ja tehoa 6 injektiota ylittävän hoidon osalta ei ole tutkittu.
Annettavan tilavuuden laskentaa koskevat tarkemmat tiedot, ks. kohta Radiofarmaseuttisten valmisteiden valmistusohjeet.
Erityispotilasryhmät
Iäkkäät
Iäkkäiden (≥ 65-vuotiaat) ja nuorempien potilaiden (< 65-vuotiaat) välillä ei havaittu eroja hoidon yleisessä turvallisuudessa ja tehossa faasin III tutkimuksessa. Annosta ei tarvitse muuttaa iäkkäille potilaille.
Maksan vajaatoiminta
Xofigo-valmisteen turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Koska radium-223 ei metaboloidu maksassa eikä poistu sapen kautta, maksan vajaatoiminnan ei odoteta vaikuttavan radium-223-dikloridin farmakokinetiikkaan. Annosta ei tarvitse muuttaa maksan vajaatoimintaa sairastaville potilaille.
Munuaisten vajaatoiminta
Faasin III kliinisessä tutkimuksessa ei havaittu relevantteja eroja turvallisuudessa ja tehossa potilailla, jotka sairastivat lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma (Krea-Cl): 50-80 ml/min) ja potilailla, joiden munuaisten toiminta oli normaalia. Tietoja potilaista, joilla on kohtalainen (Krea-Cl: 30-50 ml/min) munuaisten vajaatoiminta, on niukasti. Potilaista, joilla on vaikea (Krea-Cl < 30 ml/min) munuaisten vajaatoiminta tai loppuvaiheen munuaistauti, ei ole tietoja. Koska erittyminen virtsaan on erittäin vähäistä ja poistuminen tapahtuu pääasiassa ulosteisiin, munuaisten vajaatoiminnan ei odoteta vaikuttavan radium-223-dikloridin farmakokinetiikkaan. Annosta ei tarvitse muuttaa munuaisten vajaatoimintaa sairastaville potilaille.
Pediatriset potilaat
Käyttöaiheessa eturauhassyöpä ei pediatristen potilaiden hoito Xofigo- valmisteella ole perusteltua.
Antotapa
Xofigo on tarkoitettu laskimonsisäiseen käyttöön. Se tulee antaa hitaana (yleensä 1 minuutin kuluessa) injektiona.
Laskimonsisäinen veritie tai kanyyli on huuhdeltava isotonisella natriumkloridiliuoksella 9 mg/ml (0,9 %) injektiota varten, ennen Xofigo-valmisteen injisointia ja sen jälkeen.
Ks. kohdista Käyttö- ja käsittelyohjeet ja Radiofarmaseuttisten valmisteiden valmistusohjeet lisäohjeita lääkevalmisteen käyttöä koskien.
Vasta-aiheet
Xofigo-valmistetta ei saa käyttää yhdessä abirateroniasetaatin ja prednisonin/prednisolonin kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Samanaikainen käyttö abirateronin ja prednisonin/prednisolonin kanssa tai muiden systeemisten syöpähoitojen kuin LHRH-analogien kanssa
Välianalyysin tiedot kliinisestä tutkimuksesta osoittivat luunmurtumien riskin lisääntyneen sekä mahdollisesti kuolleisuuden riskin lisääntyneen potilailla, jotka saivat Xofigo-valmistetta yhdessä abirateroniasetaatin ja prednisonin/prednisolonin kanssa verrattuna potilaisiin, jotka saivat lumelääkettä yhdessä abirateroniasetaatin ja prednisonin/prednisolonin kanssa (ks. kohta Farmakodynamiikka). Tutkimukseen osallistui oireettomia tai vähäoireisia, luustoon levinnyttä kastraatioresistenttiä eturauhassyöpää sairastavia potilaita, jotka eivät olleet aiemmin saaneet kemoterapiaa.
Tämän vuoksi Xofigo-valmistetta ei saa käyttää yhdessä abirateroniasetaatin ja prednisonin/prednisolonin kanssa (ks. kohta Vasta-aiheet).
Xofigo-valmisteen tehoa ja turvallisuutta yhdistettynä muihin syöpähoitoihin kuin LHRH-analogeihin ei ole vahvistettu; kuolleisuuden ja murtumien lisääntynyt riski on mahdollinen. Siksi Radium-223:n yhdistäminen muiden systeemisten syöpähoitojen kuin LHRH-analogien kanssa ei ole suositeltavaa.
Turvallisesta ajanjaksosta, jonka jälkeen Xofigo-valmistetta voidaan antaa abirateroniasetaatin ja prednisonin/prednisolonin yhdistelmähoidon jälkeen ja päinvastoin, on vähän tietoa. Xofigo-valmisteen ja abirateronin eliminaation puoliintumisajan perusteella on suositeltavaa, ettei Xofigo-hoitoa aloiteta vähintään viiteen päivään abirateroniasetaatin ja prednisonin/prednisolonin yhdistelmähoidon viimeisen annon jälkeen. Systeemistä syöpähoitoa ei saa aloittaa vähintään 30 päivään viimeisen Xofigo-annoksen jälkeen.
Sellaisten potilaiden hoito, joilla on oireettomia tai vähäoireisia luustometastaaseja
Kliinisessä tutkimuksessa, jossa Xofigo lisättiin abirateroniasetaatin ja prednisonin/prednisolonin yhdistelmähoitoon potilaille, joilla oli oireeton tai vähäoireinen kastraatioresistentti eturauhassyöpä, havaittiin lisääntynyt kuoleman ja murtumien riski.
Xofigo-hoidon hyötyä aikuisille, joilla on kastraatioresistentti eturauhassyöpä ja vain oireettomia luustometastaaseja, ei ole vahvistettu. Siksi Xofigo-valmisteen käyttö ei ole suositeltavaa sellaisten aikuisten hoitoon, joilla on kastraatioresistentti eturauhassyöpä ja vain oireettomia luustometastaaseja. Aikuisilla, joilla on kastraatioresistentti eturauhassyöpä ja vähäoireisia luustometastaaseja, hoidon hyöty pitää arvioida tarkoin riskejä suuremmaksi ottaen huomioon, että hyödyn saamiseksi tarvitaan todennäköisesti vilkasta osteoblastien toimintaa (ks. Farmakodynamiikka).
Potilaat, joilla on vain vähäinen määrä osteoblastisia luustometastaaseja
Kliinisissä tutkimuksissa potilailla, joilla oli alle kuusi luustometastaasia, oli lisääntynyt murtumien riski mutta ei tilastollisesti merkitsevää hyötyä elossaoloaikaan. Etukäteen määritetyn alaryhmän analyysi osoitti myös, että kokonaiselossaoloaika ei parantunut merkittävästi potilailla, joiden alkalisen fosfataasin kokonaismäärä oli < 220 U/l. Siksi radium-223:a ei suositella potilaille, joilla on vain vähäinen määrä osteoblastisia luustometastaaseja (ks. kohta Farmakodynamiikka).
Luuydinsupressio
Luuydinsupressiota, etenkin trombosytopeniaa, neutropeniaa, leukopeniaa ja pansytopeniaa, on raportoitu Xofigo-valmisteella hoidetuilla potilailla (ks. kohta Haittavaikutukset).
Sen vuoksi lähtötilanteessa ja ennen jokaista Xofigo-annosta potilaiden veriarvot on tutkittava. Ennen kuin valmistetta annetaan ensimmäisen kerran, neutrofiilien kokonaismäärän (ANC) pitää olla ≥ 1,5 x 109/l, verihiutalemäärän ≥ 100 x 109/l ja hemoglobiinin ≥ 10,0 g/dl. Ennen seuraavan annoksen antamista ANC-arvon pitää olla ≥ 1,0 x 109/l ja verihiutalemäärän ≥ 50 x 109/l. Mikäli näissä arvoissa ei tapahdu paranemista 6 viikon kuluessa viimeisestä Xofigo-annoksesta, vaikka potilasta on hoidettu standardihoidon mukaisesti, Xofigo-hoitoa tulee jatkaa vain huolellisen hyöty-riski-arvioinnin jälkeen.
Potilaita, joilla on todettu luuytimen reservien vähenemistä esimerkiksi aikaisemman sytotoksisen kemoterapian ja/tai sädehoidon (EBRT) jälkeen, tai eturauhassyöpäpotilaita, joilla on edennyt diffuusi infiltraatio luuhun (EOD4; “superscan”), on hoidettava varovaisuutta noudattaen. Faasin III tutkimuksen aikana näillä potilailla havaittiin lisääntyneitä hematologisia haittavaikutuksia, kuten neutropeniaa ja trombosytopeniaa (ks. kohta Haittavaikutukset).
Xofigo-hoidon jälkeisen sytotoksisen kemoterapian tehoa ja turvallisuutta ei ole varmistettu. Rajoitetusti saatavilla olevan tiedon mukaan potilailla, jotka saivat kemoterapiaa Xofigo-valmisteen jälkeen, oli samanlainen hematologinen profiili verrattuna potilaisiin, jotka saivat kemoterapiaa plasebon jälkeen (ks. myös kohta Farmakodynamiikka).
Crohnin tauti ja haavainen koliitti
Xofigo-valmisteen turvallisuutta ja tehoa Crohnin tautia tai haavaista koliittia sairastavilla potilailla ei ole tutkittu. Johtuen Xofigo-valmisteen erittymisestä ulosteeseen, säteily voi johtaa akuutin tulehduksellisen suolistosairauden pahenemiseen. Xofigo-valmistetta saa antaa vain huolellisen hyöty-riskiarvion jälkeen potilaille, joilla on akuutti tulehduksellinen suolistosairaus.
Selkäytimen kompressio
Potilaat, joilla on hoitamaton uhkaava tai jo olemassa oleva selkäytimen kompressio, hoidetaan standardihoidon mukaisesti, jos se on kliinisesti aiheellista, ennen Xofigo-hoidon jatkamista tai aloittamista.
Luunmurtumat
Xofigo lisää luunmurtumien riskiä. Kliinisessä tutkimuksessa Xofigo-valmisteen lisääminen abirateroniasetaattiin ja prednisoniin/prednisoloniin lisäsi murtumien ilmaantuvuutta Xofigo-tutkimushaarassa noin kolminkertaiseksi (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Lisääntynyt murtumien riski havaittiin erityisesti potilailla, joilla oli ollut osteoporoosi, ja potilailla, joilla oli alle kuusi luustometastaasia. Xofigo-valmisteen uskotaan kerääntyvän kohtiin, joissa on nopea luun aineenvaihdunta, kuten kohdat, joissa on rappeuttava luuston sairaus (osteoporoosi) tai äskettäinen (mikro-)murtuma, sillä ne lisäävät murtumien riskiä. Muut tekijät, kuten steroidien samanaikainen käyttö, saattavat lisätä murtumien riskiä edelleen.
Ennen radium-223-hoidon aloittamista luuston tila (esim. gammakuvauksella, luuston mineraalitiheyden mittauksella) ja potilaiden lähtötilanteen murtumariski (esim. osteoporoosi, alle kuusi luustometastaasia, murtumien riskiä lisäävä hoito, matala painoindeksi) on arvioitava huolellisesti, ja niitä on seurattava tarkasti vähintään 24 kuukauden ajan. Ehkäisevien toimenpiteiden, kuten bisfosfonaattien tai denosumabin, käyttöä pitää harkita ennen Xofigo-hoidon aloittamista tai jatkamista (ks. kohta Haittavaikutukset). Jos potilaan lähtötilanteen murtumariski on suuri, hoidon hyöty tulee arvioida tarkoin riskejä suuremmaksi. Potilaiden luunmurtumat on stabiloitava ortopedisesti ennen Xofigo-hoidon aloittamista tai jatkamista.
Leuan osteonekroosi
Bisfosfonaateilla ja Xofigo-valmisteella hoidetuilla potilailla leuan osteonekroosin kehittymisen kasvanutta riskiä ei voida poissulkea. Faasin III tutkimuksessa Xofigo-haarassa on potilailla raportoitu leuan osteonekroositapauksia 0,67 prosentilla (4/600) verrattuna lumelääkehaaran 0,33 prosenttiin (1/301) potilaista. Kaikki potilaat, joilla todettiin leuan osteonekroosi, olivat kuitenkin altistuneet aiemmin tai samanaikaisesti bisfosfonaateille (esim. tsoledronihappo) sekä aiemmin saadulle kemoterapialle (esim. dosetakseli).
Sekundaariset syöpäkasvaimet
Xofigo lisää potilaan pitkäaikaista kumulatiivista kokonaisaltistusta säteilylle. Pitkäaikaiseen kumulatiiviseen säteilyaltistukseen voi liittyä lisääntynyt syövän ja periytyvien vaikutusten riski. Erityisesti osteosarkooman, myelodysplastisen oireyhtymän ja leukemian riski voi olla kasvanut. Xofigo-valmisteen indusoimia syöpätapauksia ei ole raportoitu kliinisissä tutkimuksissa kolmen vuoden seurannassa.
Suolistotoksisuus
Xofigo lisää ripulin, pahoinvoinnin ja oksentelun esiintyvyyttä (ks. kohta Haittavaikutukset), mikä voi johtaa kuivumiseen. Nesteiden saantia suun kautta ja potilaiden nestetilaa on seurattava huolellisesti. Potilaita on kehotettava ottamaan yhteyttä lääkäriin, mikäli heillä ilmenee vaikeaa tai jatkuvaa ripulia, pahoinvointia tai oksentelua. Potilaat, joilla ilmenee kuivumisen tai hypovolemian merkkejä tai oireita, on hoidettava asianmukaisesti.
Ehkäisy miehille
Säteilyyn liittyvien mahdollisten spermatogeneesiin kohdistuvien vaikutusten vuoksi miehiä tulee neuvoa käyttämään tehokkaita ehkäisymenetelmiä Xofigo-hoidon aikana ja 6 kuukautta sen jälkeen (ks. kohta Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Annetusta tilavuudesta riippuen tämä lääke sisältää enintään 54 mg (2,35 mmol) natriumia per annos, joka vastaa 2,7 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
Kliinisiä yhteisvaikutustutkimuksia ei ole tehty.
Kalsium- ja fosfaattilisän ja/tai D-vitamiinin käytön keskeyttämistä muutama päivä ennen Xofigo-hoidon aloittamista on harkittava, koska yhteisvaikutuksia kalsiumin ja fosfaattien kanssa ei voida pois sulkea.
Xofigo-valmisteen kanssa samaan aikaan annetulla kemoterapialla voi olla luuydinsupressiota lisääviä vaikutuksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Xofigo-valmisteen ja samanaikaisen kemoterapian turvallisuutta ja tehoa ei ole osoitettu.
Raskaus ja imetys
Raskaudenehkäisy miehillä
Xofigo-valmisteella ei ole suoritettu eläinten lisääntymistutkimuksia.
Koska säteilyyn on liitetty mahdollisia spermatogeneesiä koskevia vaikutuksia, miehiä on neuvottava käyttämään tehokkaita ehkäisymenetelmiä Xofigo-hoidon aikana ja enintään 6 kuukauden ajan sen jälkeen.
Raskaus ja imetys
Xofigo ei ole tarkoitettu naisille. Raskaana olevien, mahdollisesti raskaana olevien tai imettävien naisten ei pidä käyttää Xofigo-valmistetta.
Hedelmällisyys
Xofigo-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja.
Eläinkokeiden perusteella on olemassa mahdollinen riski, että Xofigo-valmisteen lähettämällä säteilyllä voi olla toksisia vaikutuksia miesten sukupuolirauhasiin tai spermatogeneesiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Miespotilaiden on syytä kysyä neuvoa sperman pakastamisesta ennen hoitoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Xofigo-valmisteella ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuudesta
Xofigo-valmisteen yleinen turvallisuusprofiili perustuu Xofigo-valmisteella hoidettujen faasin III tutkimuksen 600 potilaan tietoihin.
Useimmin havaittuja haittavaikutuksia (≥ 10 %) Xofigo-valmistetta saaneilla potilailla olivat ripuli, pahoinvointi, oksentelu, trombosytopenia ja luunmurtuma.
Vakavimpia haittavaikutuksia olivat trombosytopenia ja neutropenia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja alla esitetty "Valikoitujen haittavaikutusten kuvaus").
Taulukoitu yhteenveto haittavaikutuksista
Xofigo-valmistetta käytettäessä havaitut haittavaikutukset on esitetty alla olevassa taulukossa (ks. taulukko 1). Ne on luokiteltu elinjärjestelmittäin. Sopivinta MedDRA-termiä käytetään kuvaamaan tiettyä reaktiota ja sen synonyymejä sekä siihen liittyviä oireita.
Kliinisissä tutkimuksissa havaitut haittavaikutukset on luokiteltu niiden esiintymistiheyden mukaan. Esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset, joita on raportoitu kliinisissä tutkimuksissa Xofigo-valmisteella hoidetuilla potilailla
| Elinjärjestelmä (MedDRA) | Hyvin yleinen | Yleinen | Melko harvinainen |
| Veri ja imukudos | Trombosytopenia | Neutropenia, pansytopenia, leukopenia | Lymfopenia |
| Ruoansulatuselimistö | Ripuli, oksentelu, pahoinvointi | ||
| Luusto, lihakset ja sidekudos | Luunmurtuma | Ostenekroosi | |
| Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktiot |
Valikoitujen haittavaikutusten kuvaus
Luunmurtumat
Xofigo lisää luunmurtumien riskiä (ks. kohta Farmakodynamiikka). Kliinisissä tutkimuksissa bisfosfonaattien tai denosumabin samanaikainen käyttö vähensi murtumien ilmaantuvuutta potilailla, joita hoidettiin radium-223-monoterapialla. Murtumia on ilmennyt jopa 24 kuukautta ensimmäisen radium-223-annoksen jälkeen.
Trombosytopenia ja neutropenia
Trombosytopeniaa (kaikki asteet) esiintyi 11,5 %:lla Xofigo-valmisteella hoidetuista potilaista ja 5,6 %:lla lumelääkettä saaneista potilaista. 3. ja 4. asteen trombosytopenia havaittiin 6,3 %:lla potilaista, joita oli hoidettu Xofigo-valmisteella, ja 2 %:lla potilaista, jotka olivat saaneet lumelääkettä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kaiken kaikkiaan 3. ja 4. asteen trombosytopenia oli harvinaisempaa potilailla, jotka eivät olleet aikaisemmin saaneet dosetakselia (2,8 % Xofigo-valmisteella hoidetuista potilaista vs. 0,8 % lumelääkettä saaneista potilaista) verrattuna potilaisiin, jotka olivat aikaisemmin saaneet dosetakselia (8,9 % Xofigo-valmisteella hoidetuista potilaista vs. 2,9 % lumelääkettä saaneista potilaista). EOD4 (”superscan”) potilailla, trombosytopeniaa (kaikki asteet) raportoitiin 19,6 % potilaista, joita hoidettiin Xofigo-valmisteella ja 6,7 % potilaista , joita saivat lumelääkettä. Asteen 3. ja 4. trombosytopeniaa havaittiin 5,9 % Xofigo-valmisteella hoidetuista potilaista ja 6,7 % lumelääkettä saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropeniaa (kaikki asteet) ilmoitettiin 5 %:lla Xofigo-valmisteella hoidetuista potilaista ja 1 %:lla lumelääkettä saaneista potilaista. 3. ja 4. asteen neutropenia havaittiin 2,2 %:lla potilaista, joita oli hoidettu Xofigo-valmisteella, ja 0,7 %:lla potilaista, jotka olivat saaneet lumelääkettä. Kaiken kaikkiaan 3. ja 4. asteen neutropenia oli harvinaisempaa potilailla, jotka eivät olleet aikaisemmin saaneet dosetakselia (0,8 % Xofigo-valmisteella hoidetuista potilaista vs. 0,8 % lumelääkettä saaneista potilaista) verrattuna potilaisiin, jotka olivat aikaisemmin saaneet dosetakselia (3,2 % Xofigo-valmisteella hoidetuista potilaista vs. 0,6 % lumelääkettä saaneista potilaista).
Vaiheen I tutkimuksessa neutrofiilien ja verihiutaleiden alimmat arvot esiintyivät 2 3 viikkoa Xofigo-kerta-annoksen laskimoon antamisen jälkeen.
Pistoskohdan reaktiot
1. ja 2. asteen pistospaikan reaktioita, kuten ihottumaa, kipua ja turvotusta, raportoitiin 1,2 %:lla Xofigo-valmisteella hoidetuista potilaista ja 0 %:lla lumelääkettä saaneista potilaista.
Sekundaariset pahanlaatuiset kasvaimet
Xofigo vaikuttaa osaltaan potilaan pitkäaikaiseen kumulatiiviseen kokonaissäteilyaltistukseen. Pitkäaikaiset kumulatiiviset säteilyaltistukset saattavat liittyä syövän sekä perinnöllisten poikkeavuuksien lisääntyneeseen riskiin. Erityisesti osteosarkooman, myelodysplastisen oireyhtymän ja leukemian riski voi kasvaa.
Kolmen vuoden seuranta-aikana Xofigo-valmisteen aiheuttamia syöpätapauksia ei ole raportoitu kliinisissä tutkimuksissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta -tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ei ole raportoitu tapauksia, joissa Xofigo-valmistetta olisi vahingossa annettu yliannostuksena kliinisissä tutkimuksissa.
Spesifistä vasta-ainetta ei ole. Yliannostustapauksessa on aloitettava yleinen elintoimintoja ylläpitävä hoito, mukaan lukien mahdollisen hematologisen ja maha-suolikanavaan liittyvän toksisuuden tarkkailu.
Faasin I kliinisessä tutkimuksessa tutkittiin yksittäisiä aktiivisuudeltaan enintään 276 kBq:n Xofigo-annoksia painokiloa kohti, annosta rajoittavaa toksisuutta ei havaittu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Terapeuttiset radioaktiiviset lääkevalmisteet, muut terapeuttiset radioaktiiviset lääkevalmisteet, muut terapeuttiset radioaktiiviset lääkevalmisteet, ATC-koodi: V10XX03
Vaikutusmekanismi
Xofigo on terapeuttinen alfahiukkasia säteilevä lääkevalmiste.
Sen aktiivinen ainesosa radium-223 (radium-223–dikloridina) jäljittelee kalsiumia ja kohdistaa vaikutuksensa selektiivisesti luuhun, erityisesti luustometastaasialueille, muodostamalla yhdisteitä luumineraalin, hydroksiapatiitin, kanssa. Alfasäteilijöiden suuri lineaarinen energiansiirtokyky (80 keV/µm) johtaa tiheästi tapahtuviin kasvainsolujen kaksisäikeisten DNA-ketjujen katkeamisiin, minkä seurauksena syntyy voimakas sytotoksinen vaikutus. Lisäksi vaikutukset kasvaimen mikroympäristöön mukaan lukien osteoblastit ja osteoklastit, myötävaikuttavat in vivo tehoon. Radium-223:n säteilemien alfahiukkasten kantama on alle 100 µm (vähemmän kuin 10 solun halkaisijaa), jolloin ympäröivän normaalin kudoksen vauriot jäävät mahdollisimman pieniksi.
Farmakodynaamiset vaikutukset
Faasin II satunnaistetussa tutkimuksessa seerumin kaikkien viiden luun aineenvaihdunnan biomerkkiaineen osalta oli Xofigo-valmisteen eduksi merkitsevä ero verrattuna lumelääkkeeseen. (Nämä biomerkkiaineet olivat luun alkalinen fosfataasi [AFOS], kokonais-AFOS ja prokollageeni I:n aminoterminaalinen propeptidi [PINP], luun resorption merkkiaineet: kollageeni I:n poikkisitoutuva karboksiterminaalinen telopeptidi / seerumin kollageeni I:n poikkisitoutuva karboksiterminaalinen telopeptidi [S-CTX-I] ja kollageeni-I:n poikkisidoksinen karboksiterminaalinen telopeptidi [ICTP]).
Sydämen sähköinen toiminta / QT-ajan pidentyminen
Laskimoon annetun Xofigo-injektion jälkeen ei havaittu merkittäviä QTc-ajan pidentymiseen liittyviä vaikutuksia plaseboon verrattaessa 29 potilaan alaryhmässä faasin III tutkimuksessa (ALSYMPCA).
Kliininen teho ja turvallisuus
Xofigo-valmisteen kliinistä turvallisuutta ja tehoa on selvitetty kaksoissokkoutetussa satunnaistetussa faasin III moniannoksisessa monikeskustutkimuksessa (ALSYMPCA; EudraCT 2007-006195-1) kastraatioresistenteilla eturauhassyöpäpotilailla, joilla oli oireilevia luustometastaaseja. Potilaat, joilla oli viskeraalisia metastaaseja tai yli 3 cm suuri pahanlaatuinen lymfadenopatia, suljettiin pois tutkimuksesta.
Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika. Tärkeimpiä päätetapahtumia olivat kliinisesti merkittävien luustotapahtumien ilmaantuminen (symptomatic skeletal events, SSE) sekä aika alkalisen fosfataasin kokonaismäärän progressioon (AFOS), aika prostataspesifisen antigeenin (PSA) progressioon, vasteet alkalisen fosfataasin kokonaismäärässä ja alkalisen fosfataasin kokonaismäärän normalisoituminen.
Etukäteen suunnitellussa välianalyysissä (varmistusanalyysi) tietojenkeräyspäivänä 809 potilasta oli satunnaistettu suhteessa 2:1 saamaan parasta standardihoitoa ja Xofigo-valmistetta 55 kBq/kg laskimoon 4 viikon välein 6 hoitosyklin ajan (N=541) tai vastaavaa lumelääkettä ja parasta standardihoitoa (N=268). Parhaaseen standardihoitoon sisältyivät mm. paikallinen ulkoinen sädehoito, bisfosfonaatit, kortikosteroidit, antiandrogeenit, estrogeenit, estramustiini tai ketokonatsoli.
Päivitetty deskriptiivinen turvallisuus- ja kokonaiselossaoloaika-analyysi tehtiin 921 satunnaistetulla potilaalla ennen vaihtovuoroisuuden toteuttamista (ts. ennen kuin lumelääkeryhmän potilaille tarjottiin mahdollisuutta saada Xofigo-hoitoa).
Lähtötilanteessa Xofigo-valmistetta ja lumelääkettä saaneet potilasryhmät olivat demografisesti ja taudin ominaisuuksien suhteen (välianalyysin potilaat) keskenään samanlaiset. Alla ne esitetään Xofigo-ryhmän potilaiden osalta:
- potilaiden keskimääräinen ikä oli 70 vuotta (ikäjakauma 49-90 vuotta)
- 87 %:lla tutkimukseen osallistuneista potilaista ECOG-suorituskykyluokka oli 0-1
- 41 % sai bisfosfonaatteja
- 42 % potilaista ei ollut saanut aikaisemmin dosetakselia, koska heidät arvioitiin hoitoon soveltumattomiksi tai he kieltäytyivät dosetakselista
- 46 %:lla potilasta ei ollut kipuja tai heidän kipunsa kuului WHO:n asteikon luokkaan 1 (oireeton tai lievästi oireinen) ja 54 %:lla oli WHO:n asteikon luokan 2 3 mukaisia kipuja
- 16 %:lla potilaista oli < 6 luustometastaasia, 44 %:lla potilaista oli 6 20 luustometastaasia, 40 %:lla potilaista oli yli 20 luustometastaasia tai koko luuston voimakas merkkiainekertymä.
Hoitojakson aikana 83 % potilaista sai luteinisoivaa hormonia vapauttavan hormonin (LHRH) agonisteja ja 26 % potilaista sai antiandrogeeneja samanaikaisesti.
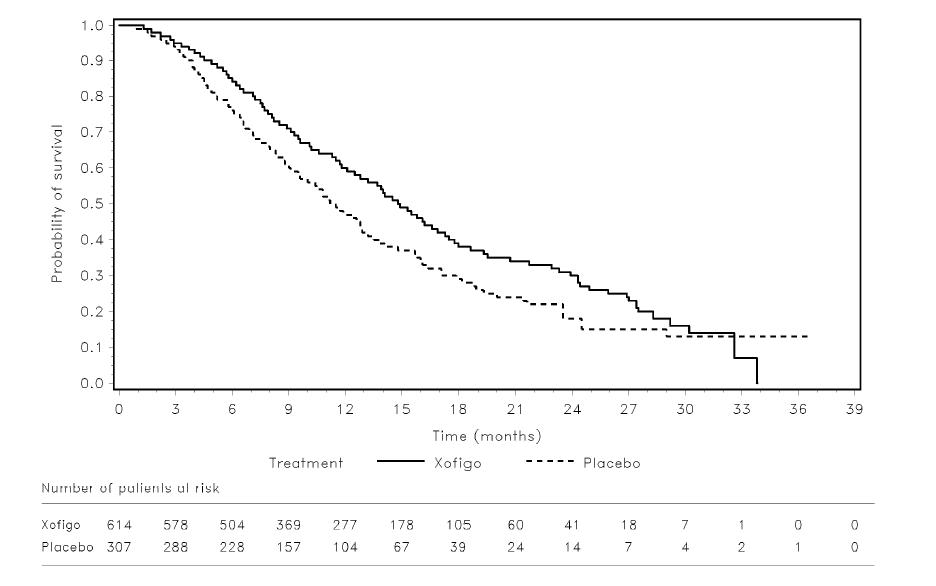
Kokonaiselossaoloaika oli merkitsevästi pitempi Xofigo-valmisteella hoidetuilla potilailla, jotka saivat myös parasta standardihoitoa, kuin lumelääkkeellä hoidetuilla potilailla, jotka saivat myös parasta standardihoitoa (ks. taulukko 2 ja kuva 2) sekä välianalyysin, että päivitetyn analyysin tulosten perusteella. Lumelääkeryhmässä havaittiin enemmän muuhun kuin eturauhassyöpään liittyviä kuolemia kuin Xofigo-ryhmän potilailla (4,8 % (26/541) Xofigo-ryhmässä verrattuna 8,6 % (23/268) lumelääkeryhmässä).
Taulukko 2: Faasin III ALSYMPCA-tutkimuksen elossaolotulokset
| Xofigo | Lumelääke | |
| Välianalyysi | N=541 | N=268 |
| Kuolemantapauksia (%) | 191 (35,5 %) | 123 (45,9 %) |
| Kokonaiselossaoloajan mediaani (kuukautta) (95 % CI) | 14,0 (12,1 - 15,8) | 11,2 (9,0 - 13,2) |
| Riskisuhdeb (95 % CI) | 0,695 (0,552 - 0,875) | |
| p-arvoa (2-suuntainen) | 0,00185 | |
| Päivitetty analyysi | N=614 | N=307 |
| Kuolemantapauksia (%) | 333 (54,2 %) | 195 (63,5 %) |
| Kokonaiselossaoloajan mediaani (kuukautta) (95 % CI) | 14,9 (13,9 - 16,1) | 11,3 (10,4 - 12,8) |
| Riskisuhdeb (95 % CI) | 0,695 (0,581 - 0,832) | |
CI = luottamusväli
a Faasin 3 ALSYMPCA-tutkimus lopetettiin tehon vuoksi välianalyysin jälkeen. Koska päivitetty analyysi esitetään vain deskriptiivisessä tarkoituksessa, p-arvoa ei esitetä.
b Riskisuhde (Xofigo-valmisteen ero lumelääkkeeseen verrattuna) < 1 suosii Xofigo-valmistetta.
Kuva 2: Kaplan-Meier-käyrät kokonaiselossaoloajasta (päivitetty analyysi)
Välianalyysin ja päivitetyn analyysin tuloksista ilmeni merkitsevä paraneminen kaikkien pääasiallisten toissijaisten päätetapahtumien osalta Xofigo-haarassa verrattuna lumelääkehaaraan (ks. taulukko 3). Tapahtumaan kuluneen ajan (time to event) –tietoja AFOS:in progressiosta tuki tilastollisesti merkitsevä etu AFOS:in normalisoitumisesta ja AFOS-vasteista viikolla 12.
Taulukko 3: Tehon toissijaiset päätetapahtumat faasin III ALSYMPCA-tutkimuksesta (välianalyysi)
| Ilmaantuvuus | Time-to-event analyysi (95 % CI) | |||||||
| [potilaista %] | [kuukausien mediaani] | Riskisuhde < 1 suosii Xofigo-valmistetta | p-arvo | |||||
| Xofigo N=541 | Lumelääke N=268 | Xofigo N=541 | Lumelääke N=268 | |||||
| Oireileva luustotapahtuma (SSE) | SSE yhdistetty päätetapahtumata | 132 (24,4 %) | 82 (30,6 %) | 13,5 (12,2-19,6) | 8,4 (7,2-EA)b | 0,610 (0,461-0,807) | 0,00046 | |
| Oireilevien luustotapahtumien (SSE) alaryhmät | Ulkoinen sädehoito kivun lievittämiseksi | 122 (22,6 %) | 72 (26,9 %) | 17,0 (12,9-EA) | 10,8 (7,9-EA) | 0,649 (0,483-0,871) | 0,00375 | |
| Selkäytimen kompressio | 17 (3,1 %) | 16 (6,0 %) | EA | EA | 0,443 (0,223-0,877) | 0,01647 | ||
| Kirurginen toimenpide | 9 (1,7 %) | 5 (1,9 %) | EA | EA | 0,801 (0,267-2,398 | 0,69041 | ||
| Luunmurtumat | 20 (3,7 %) | 18 (6,7 %) | EA | EA | 0,450 (0,236-0,856) | 0,01255 | ||
| Kokonais-AFOS:in pregressioc | 79 (14,6 %) | 116 (43,3 %) | EA | 3,7 (3,5-4,1) | 0,162 (0,120-0,220) | < 0,00001 | ||
| PSA:n progressiod | 288 (53,2 %) | 141 (52,6 %) | 3,6 (3,5-3,7) | 3,4 (3,3-3,5) | 0,671 (0,546-0,826) | 0,00015 | ||
AFOS = alkalinen fosfataasi; CI = luottamusväli; EA = ei arvioitavissa; PSA = prostataspesifinen antigeeni; SSE = oireileva luustotapahtuma (symptomatic skeletal event)
a Määritelty joksikin seuraavista: ulkoinen sädehoito kivun lievittämiseksi, patologinen murtuma, selkäytimen kompressio tai kasvaimeen liittyvä ortopedinen kirurginen toimenpide.
b ei arvioitavissa johtuen riittämättömistä tapahtumista mediaanin jälkeen
c Määritelty ≥ 25 %:n nousuksi verrattuna lähtötilanteeseen/alimpaan arvoon.
d Määritelty ≥ 25 %:n nousuksi ja absoluuttisen arvon ≥ 2 ng/ml:n nousuksi verrattuna lähtötilanteeseen/alimpaan arvoon.
Elossaoloajan alaryhmäanalyysi
Elossaoloajan alaryhmäanalyysissä osoitettiin Xofigo-hoidolla johdonmukainen hyöty, joka ei riippunut bisfosfonaattien käytöstä lähtötilanteessa eikä dosetakselin aikaisemmasta käytöstä.
Hoidolla ei saavutettu tilastollisesti merkitsevää kokonaiselossaoloajan hyötyä niiden potilaiden alaryhmissä, joilla oli alle kuusi metastaasia (hoidon riskisuhde (HR) radium-223:lle lumelääkkeeseen verrattuna oli 0,901; 95 %:n luottamusväli (CI) [0,553 ‑ 1,466], p=0,674) tai lähtötilanteen kokonais-AFOS < 220 U/l (HR 0,823; 95 %:n CI [0,633 ‑ 1,068], p=0,142) faasin III ALSYMPCA-tutkimuksessa. Siksi teho saattaa olla heikentynyt potilailla, joilla osteoblastien toiminta luustometastaaseissa on vähäistä.
Elämänlaatu
Terveyteen liittyvää elämänlaatua (HRQOL) arvioitiin faasin III ALSYMPCA-tutkimuksen aikana käyttäen tätä varten tarkoitettuja kyselylomakkeita: EQ-5D (yleinen) ja FACT-P (eturauhassyöpään erityisesti liittyvä). Molemmat ryhmät kokivat elämänlaadun heikkenemistä. Elämänlaadun heikkeneminen oli hitaampaa Xofigo-hoidon aikana verrattuna lumelääkkeeseen mitattuna EQ-5D utiliteetti-indeksipistemäärällä (-0,040 vs. -0,109; p = 0,001), EQ-5D VAS-pistemäärällä (-2,661 vs. -5,860; p = 0,018) ja FACT-P kokonaispistemäärällä (-3,880 vs. -7,651 p = 0,006), mutta julkaistuja pienimpiä merkittäviä eroja (minimally important differences) ei saavutettu. On niukasti todisteita siitä, että elämänlaadun heikkenemisen viivästyminen jatkuu hoitojakson jälkeen.
Kivunlievitys
Faasin III ALSYPMCA-tutkimuksen tulokset osoittivat positiivisen vaikutuksen luukipuun, mitattuna ajassa, joka kului ennen kivunlievityksenä annettua ulkoista sädehoitoa (EBRT) ja koska harvemmat potilaat ilmoittivat luukivun haittatapahtumana Xofigo-ryhmässä.
Sytotoksisten valmisteiden käyttö hoidon jälkeen
Suhteessa 2:1 satunnaistetun ALSYMPCA-tutkimuksen kuluessa 93 (15,5 %) Xofigo-ryhmän potilaista ja 54 (17,9 %) lumeryhmän potilaista sai sytotoksista kemoterapiaa vaihtelevina aikoina viimeisen hoidon jälkeen. Hematologisissa laboratorioarvoissa ei ollut ilmeisiä eroja näiden kahden ryhmän välillä.
Samanaikainen käyttö abirateronin ja prednisonin/prednisolonin kanssa
Xofigo-valmisteen, abirateroniasetaatin ja prednisonin/prednisolonin samanaikaisen käytön kliinistä tehoa ja turvallisuutta tarkasteltiin satunnaistetussa ja lumekontrolloidussa faasin III monikeskustutkimuksessa (ERA-223-tutkimus), johon osallistui 806 oireetonta tai vähäoireista, luustoon levinnyttä kastraatioresistenttiä eturauhassyöpää sairastavaa potilasta, jotka eivät olleet aiemmin saaneet kemoterapiaa. Tutkimuksen sokkoutus purettiin etuajassa puolueettoman valvontaryhmän suosituksesta. Välianalyysin tiedot osoittivat luunmurtumien ilmaantuvuuden lisääntyneen (28,6 % vs. 11,4 %) ja kokonaiselossaoloajan mediaanin laskeneen (30,7 kuukautta vs. 33,3 kuukautta, HR 1,195, 95 %:n CI [0,950 - 1,505], p=0,13) potilailla, jotka saivat Xofigo-hoitoa yhdessä abirateroniasetaatin ja prednisonin/ prednisolonin kanssa verrattuna potilaisiin, jotka saivat lumelääkettä yhdessä abirateroniasetaatin ja prednisonin/prednisolonin kanssa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Xofigo-valmisteen käytöstä kategorian pahanlaatuiset neoplasmat (lukuun ottamatta keskushermoston kasvaimia, hematopoieettisia ja imukudoksen neoplasmoja) sekä multippelin myelooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa / ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Yleinen johdanto
Farmakokineettiset, biojakautumiseen liittyvät ja dosimetriatiedot on saatu kolmesta faasin I tutkimuksesta. Farmakokineettiset tiedot kerättiin 25 potilaalta, joiden annosten aktiivisuudet vaihtelivat välillä 51-276 kBq/kg. Farmakokineettiset, biojakautumiseen liittyvät ja dosimetriatiedot kerättiin 6 potilaalta, jotka saivat 110 kBq/kg aktiivisuusannoksen kahdesti 6 viikon välein, ja 10 potilaalta, joiden annoksen aktiivisuus oli 55, 110 tai 221 kBq/kg.
Imeytyminen
Xofigo annetaan injektiona laskimoon ja siten sen biologinen hyötyosuus on 100 %
Jakautuminen ja kertyminen elimiin
Laskimoon annetun injektion jälkeen radium-223 eliminoituu verestä nopeasti ja kiinnittyy ensisijaisesti luustoon ja luustometastaaseihin tai erittyy suoleen.
Viidentoista minuutin kuluttua injektiosta 20 % injisoidusta aktiivisuudesta oli jäljellä veressä. 4 tunnin kuluttua injektiosta noin 4 % injisoidusta aktiivisuudesta oli jäljellä veressä. Se laski alle 1 %:iin, kun injektiosta oli kulunut 24 tuntia. Jakautumistilavuus oli suurempi kuin veritilavuus, mikä osoittaa jakautumista perifeerisiin tiloihin.
10 minuuttia injektion jälkeen aktiivisuutta havaittiin luustossa ja suolessa. Kun injektiosta oli kulunut 4 tuntia, radioaktiivisen annoksen keskimääräinen pitoisuus luustossa oli noin 61 % ja suolessa 49 %. .
Merkittävää kertymää ei havaittu muissa elimissä, esimerkiksi sydämessä, maksassa, munuaisissa, virtsarakossa ja pernassa, 4 tunnin kuluttua injektiosta.
Biotransformaatio
Radium-223 on isotooppi, joka hajoaa eikä metaboloidu.
Eliminaatio
Erittyminen ulosteeseen on tärkein poistumistie elimistöstä. Noin 5 % erittyy virtsaan, eikä erittymisestä sappeen ole todisteita.
7 vuorokautta injektion jälkeen tehdyt koko elimistön mittaukset osoittavat, että 76 % annetun annoksen aktiivisuudesta erittyi elimistöstä (hajoaminen huomioitu). Radium-223-dikloridin eliminaation nopeuteen maha-suolikanavasta vaikuttaa ruoansulatuskanavan sisällön läpikulkuajan suuri vaihtelevuus väestössä; suolen normaali tyhjentymisfrekvenssi vaihtelee välillä kerran päivässä - kerran viikossa.
Lineaarisuus/ei-lineaarisuus
Radium-223-dikloridin farmakokinetiikka oli tutkitulla aktiivisuusalueella (51-276 kBq/kg) lineaarinen.
Pediatriset potilaat
Xofigo-valmisteen turvallisuutta ja tehoa ei ole tutkittu alle 18-vuotiailla lapsilla ja nuorilla.
Prekliiniset tiedot turvallisuudesta
Systeeminen toksisuus
Rotilla tehdyissä kerta- ja toistuvan annoksen toksisuustutkimuksissa pääasialliset löydökset olivat vähentynyt painon nousu, hematologiset muutokset, alentunut seerumin alkalinen fosfataasi ja mikroskooppiset löydökset luuytimessä (hematopoieettisten solujen ehtyminen, fibroosi), pernassa (toissijainen ekstramedullaarinen hematopoieesi) ja luustossa (osteosyyttien, osteoblastien, osteoklastien ehtyminen, side- ja luukudoksen leesiot, kasvulevyn/kasvulinjan häiriintyminen/hajoaminen). Nämä löydökset liittyivät säteilystä johtuviin hematopoieesin heikkenemiseen ja osteogeneesin vähenemiseen. Pienin annosaktiivisuus, jolla niitä esiintyi, oli 22 kBq/painokilo (0,4 kertaa kliinisesti suositeltu annos).
Koirilla hematologisia muutoksia havaittiin alkaen pienimmästä 55 kBq/kg:n annosaktiivisuudesta, joka on kliinisesti suositeltu annos. Annosta rajoittavaa myelotoksisuutta havaittiin koirilla yhden antokerran jälkeen, kun annettiin 497 kBq radium-223-dikloridia/painokilo (9 kertaa kliinisesti suositeltu annosaktiivisuus).
Toistuvan kliinisesti suositellun annosaktiivisuuden 55 kBq/painokilo (joka neljäs viikko kuuden kuukauden ajan) jälkeen kahdelle koiralle kehittyi dislokoitumattomia lantion murtumia. Koska hoitoa saaneiden eläinten muilla luuston aluilla osteolyysia esiintyi trabekulaarisessa luussa vaihtelevassa määrin, osteolyysiin liittyvää spontaania murtumaa ei voida sulkea pois. Näiden löydösten kliininen merkitsevyys on tuntematon.
Verkkokalvon irtoamisia havaittiin koirilla sen jälkeen, kun niille oli annettu kerralla injektiona 166 ja 497 kBq:n annosaktiivisuudet painokiloa kohti (3 ja 9 kertaa suositeltu annos), mutta ei sen jälkeen, kun niille annettiin toistuva 55 kBq:n aktiivisuus /painokilo yhden kerran 4 viikossa 6 kuukauden ajan. Verkkokalvon irtautumisen tarkkaa mekanismia ei tunneta, mutta kirjallisuudesta löytyvät tiedot viittaavat siihen, että radium kertyy erityisesti koiran silmän tapetum lucidum -kalvoon. Koska ihmisillä ei ole tapetum lucidum -kalvoa, näiden löydösten kliininen merkitys ihmisten kannalta on epäselvä. Kliinisissä tutkimuksissa ei ole raportoitu yhtään verkkokalvon irtoamistapausta ihmisillä.
Elimissä, jotka osallistuvat radium-223-dikloridin erittämiseen, ei havaittu histologisia muutoksia.
Osteosarkoomia, jotka ovat luustoon etsiytyvien radionuklideiden tunnettu vaikutus, havaittiin kliinisesti relevanteilla annoksilla rotissa 7-12 kuukautta hoidon aloittamisen jälkeen. Osteosarkoomia ei havaittu koirilla tehdyissä tutkimuksissa. Osteosarkoomatapauksia ei ole raportoitu Xofigo-valmisteella tehdyissä kliinisissä tutkimuksissa. Potilaiden riskiä saada osteosarkoomia radium-223 altistuksen myötä ei tällä hetkellä tiedetä. Muiden neoplastisten muutosten kuin osteosarkoomien esiintymistä raportoitiin myös pitemmän jakson (12-15 kuukautta) toksisuustutkimuksissa rotilla (ks. kohta Haittavaikutukset).
Alkiotoksisuus / Lisääntymistoksisuus
Lisääntymis- ja kehitystoksisuutta koskevia tutkimuksia ei ole tehty. Yleisesti ottaen radionuklidit saavat aikaan lisääntymiseen ja kehitykseen liittyviä vaikutuksia.
Hyvin pieni määrä epänormaaleja spermatosyyttejä havaittiin urosrottien kivesten joissakin siementiehyissä sen jälkeen, kun niille oli annettu yhdellä kerralla ≥ 2270 kBq radium-223-dikloridia painokiloa kohti (≥ 41-kertaisesti kliinisesti suositeltu aktiivisuus). Kivekset vaikuttivat muutoin toimivan normaalisti ja lisäkiveksissä havaittiin normaali määrä spermatosyyttejä. Kohdun polyyppejä (endometriaalinen strooma) havaittiin naarasrotilla sen jälkeen, kun niille oli annettu yhdellä kerralla tai toistuvasti ≥ 359 kBq radium-223-dikloridia painokiloa kohti (≥ 6,5-kertaisesti kliinisesti suositeltu aktiivisuus).
Koska radium-223 jakautuu pääasiassa luuhun, mahdollinen miehen sukupuolirauhasiin kohdistuvien toksisten vaikutusten riski kastraatioresistenttiä eturauhassyöpää sairastavilla syöpäpotilailla on hyvin alhainen, mutta sitä ei voida sulkea pois (ks. kohta Raskaus ja imetys).
Genotoksisuus / karsinogeenisuus
Xofigo-valmisteen genotoksisuutta ja karsinogeenisuutta ei ole tutkittu. Yleensä radionuklideita pidetään genotoksisina ja karsinogeenisinä.
Farmakologinen turvallisuus
Elintärkeissä elinjärjestelmissä, ts. sydän ja verisuonet (koira), hengityselimet tai keskushermosto (rotta), ei havaittu merkittäviä vaikutuksia sen jälkeen, kun oli annettu kerta-annos aktiivisuuksia 497-1100 kBq painokiloa kohti (9 [koira] - 20 [rotta] kertaa kliinisesti suositeltu aktiivisuus).
Farmaseuttiset tiedot
Apuaineet
Injektionesteisiin käytettävä vesi, natriumsitraatti, natriumkloridi, vetykloridihappo.
Apuaineet, joiden vaikutus tunnetaan
Yksi ml liuosta sisältää 0,194 mmol (vastaa 4,5 mg) natriumia.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
28 vuorokautta
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita. Radioaktiivisten lääkevalmisteiden säilytyksessä on noudatettava radioaktiivisia aineita koskevia kansallisia määräyksiä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XOFIGO injektioneste, liuos
1100 kBq/ml (L:ei) 6 ml (5449,27 €)
PF-selosteen tieto
Väritön, tyypin I lasista valmistettu injektiopullo, joka on suljettu harmaalla bromobutyyli-kumitulpalla, jossa joko on tai ei ole etyleeni-tetrafluoroetyleeni (EFTA) kelmupäällystettä, Molemmat kumitulpat on päällystetty alumiinitiivisteellä. Injektionpullo sisältää 6 ml injektionestettä, liuosta.
Injektiopullo säilytetään lyijyastiassa.
Valmisteen kuvaus:
Kirkas, väritön isotoninen liuos, jonka pH on 6,0-8,0.
Käyttö- ja käsittelyohjeet
Yleiset varoitukset
Radioaktiivisia aineita saavat ottaa vastaan, käyttää ja antaa vain valtuutetut henkilöt tähän tarkoitukseen varatuissa kliinisissä tiloissa. Radioaktiivisten aineiden vastaanotossa, säilytyksessä, käytössä, kuljettamisessa ja hävittämisessä on noudatettava asianomaisen kansallisen viranomaislaitoksen antamia määräyksiä ja/ tai asianmukaisia lupia.
Radioaktiivisia lääkevalmisteita on käsiteltävä säteilyturvallisuuden ja farmaseuttisten laatuvaatimusten edellyttämällä tavalla. Asianmukaisia aseptisia varotoimia on noudatettava.
Säteilysuojaus
Xofigo-valmisteen radioaktiivisuuden mittaaminen ja kontaminaatioiden havaitseminen tavanomaisilla välineillä on mahdollista radium-223:n ja sen tytärnuklidien hajoamiseen liittyvän gammasäteilyn ansiosta.
Radioaktiivisten aineiden antamisesta aiheutuu mahdollisesti muihin ihmisiin kohdistuvia ulkoisia säteilyriskejä tai elimistön nesteiden (virtsa, ulosteet, oksennus) roiskumisen aiheuttaman kontaminaation riskejä. Säteilyltä on siksi suojauduttava paikallisten ja kansallisten säteilyturvallisuusmääräysten mukaisesti. Huolellisuutta on noudatettava käsiteltäessä materiaaleja, jotka ovat kosketuksissa elimistön nesteiden kanssa (esim. vuodevaatteet). Vaikka radium-223 lähettää ennen kaikkea alfasäteilyä, gamma- ja beetasäteily liitetään radium-223:n ja sen radioaktiivisten tytärisotooppien hajoamiseen. Potilasannosten käsittelyyn liittyvä ulkoinen säteilyaltistus on huomattavasti alhaisempi verrattuna muihin terapeuttisiin radiofarmaseuttisiin lääkevalmisteisiin, sillä annettu radioaktiivisuus on yleensä alle 8 MBq. Jotta säteilyaltistus jäisi mahdollisimman pieneksi, on suositeltavaa - sen lisäksi että noudatetaan ALARA (“As Low As Reasonably Achievable”) –periaatetta eli oleskella mahdollisimman vähän säteilyalueilla, pysytellä mahdollisimman kaukana säteilylähteistä ja käyttää riittävää suojausta.
Käyttämätön radioaktiivinen lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Xofigo-valmisteen valmistelun tai antamisen yhteydessä käytettyjä materiaaleja on käsiteltävä kuten radioaktiivista jätettä.
Korvattavuus
XOFIGO injektioneste, liuos
1100 kBq/ml 6 ml
- Ei korvausta.
ATC-koodi
V10XX03
Valmisteyhteenvedon muuttamispäivämäärä
28.07.2025
Dosimetria
Absorboitunut säteilyannos laskettiin kliinisten biodistribuutiotietojen perusteella. Absorboituneiden annosten laskelmat tehtiin OLINDA/EXM-ohjelmistolla (Organ Level INternal Dose Assessment/EXponential Modeling). Tämä ohjelmisto perustuu Medical Internal Radiation Dose (MIRD) -algoritmiin, jota käytetään laajalti beeta- ja gammasäteilyä lähettäviin radionuklidien laskentaan. Ensisijaisesti alfasäteilyä lähettävän radium-223:a varten lisäoletuksia tehtiin suolen, punaisen luuytimen ja luu-/osteogeenisten solujen osalta, jotta saataisiin parhaat mahdolliset absorboituneen annoksen laskelmat Xofigo-valmisteelle, ottaen huomioon sen havaitun biodistribuution ja erityispiirteet (ks. Taulukko 4).
Taulukko 4. Laskettu elimiin absorboitunut säteilyannos
| Kohde-elin | Alfa-säteily1 (Gy/MBq) | Beeta-säteily1 (Gy/MBq) | Gamma-säteily1 (Gy/MBq) | Kokonaisannos (Gy/MBq) | Variaatio- kerroin
|
| Lisämunuaiset | 0,00000 | 0,00002 | 0,00009 | 0,00012 | 56 |
| Aivot | 0,00000 | 0,00002 | 0,00008 | 0,00010 | 80 |
| Rinnat | 0,00000 | 0,00002 | 0,00003 | 0,00005 | 120 |
| Sappirakon seinämä | 0,00000 | 0,00002 | 0,00021 | 0,00023 | 14 |
| LLI:n2 seinämä | 0,00000 | 0,04561 | 0,00085 | 0,04645 | 83 |
| Ohutsuolen seinämä | 0,00319 | 0,00360 | 0,00047 | 0,00726 | 45 |
| Mahalaukun seinämä | 0,00000 | 0,00002 | 0,00011 | 0,00014 | 22 |
| ULI:n3 seinämä | 0,00000 | 0,03149 | 0,00082 | 0,03232 | 50 |
| Sydämen seinämä | 0,00161 | 0,00007 | 0,00005 | 0,00173 | 42 |
| Munuaiset | 0,00299 | 0,00011 | 0,00011 | 0,00321 | 36 |
| Maksa | 0,00279 | 0,00010 | 0,00008 | 0,00298 | 36 |
| Keuhkot | 0,00109 | 0,00007 | 0,00005 | 0,00121 | --4 |
| Lihas | 0,00000 | 0,00002 | 0,00010 | 0,00012 | 41 |
| Munasarjat | 0,00000 | 0,00002 | 0,00046 | 0,00049 | 40 |
| Haima | 0,00000 | 0,00002 | 0,00009 | 0,00011 | 43 |
| Punainen luuydin | 0,13217 | 0,00642 | 0,00020 | 0,13879 | 41 |
| Osteogeeniset solut | 1,13689 | 0,01487 | 0,00030 | 1,15206 | 41 |
| Iho | 0,00000 | 0,00002 | 0,00005 | 0,00007 | 79 |
Perna | 0,00000 | 0,00002 | 0,00007 | 0,00009 | 54 |
| Kivekset | 0,00000 | 0,00002 | 0,00006 | 0,00008 | 59 |
| Kateenkorva | 0,00000 | 0,00002 | 0,00003 | 0,00006 | 109 |
| Kilpirauhanen | 0,00000 | 0,00002 | 0,00005 | 0,00007 | 96 |
| Virtsarakon seinämä | 0,00371 | 0,00016 | 0,00016 | 0,00403 | 63 |
| Kohtu | 0,00000 | 0,00002 | 0,00023 | 0,00026 | 28 |
| Koko elimistö | 0,02220 | 0,00081 | 0,00012 | 0,02312 | 16 |
1Koska suurimpaan osaan tutkitusta pehmytkudoksesta ei ollut kertynyt radium‑223:a, alfasäteilyn osuus elimen kokonaisannoksesta on merkitty näiden elinten kohdalla nollaksi.
2LLI: paksusuolen alaosa (lower large intestine)
3ULI: paksusuolen yläosa (upper large intestine)
4Keuhkoihin absorboituneen annoksen arvio perustuu malliin, johon on yhdistetty kaikkien tutkittavien verestä saadut aika-aktiivisuustulokset
Xofigo-valmisteella tehdyissä kliinisissä tutkimuksissa havaitut hematologiset haittavaikutukset ovat esiintyvyydeltään ja vaikeusasteeltaan paljon alhaisempia kuin mitä voisi odottaa laskettujen punaiseen luuytimeen absorboituneiden annosten perusteella. Tämä saattaa liittyä alfahiukkassäteilyn spatiaaliseen jakautumiseen, mikä johtaa punaiseen luuytimeen kohdistuvan säteilyannoksen epätasaisuuteen.
Radiofarmaseuttisten valmisteiden valmistusohjeet
Tämä lääkevalmiste pitää tarkastaa silmämääräisesti ennen käyttöä. Xofigo on kirkas, väritön liuos. Älä käytä sitä, jos siinä on värimuutoksia, hiukkasia tai jos pakkaus on vioittunut.
Xofigo on käyttövalmis liuos, eikä sitä saa laimentaa tai sekoittaa mihinkään muihin liuoksiin.
Yksi injektiopullo on tarkoitettu yhtä käyttökertaa varten.
Potilaalle annettavan annoksen tilavuus lasketaan käyttäen seuraavia lukuja:
- potilaan paino (kg)
- annosmäärä (55 kBq/painokilo)
- radioaktiivisen lääkevalmisteen radioaktiivisuuspitoisuus (1100 kBq/ml) referenssipäivänä (ref. pvm). Referenssipäivä on merkitty injektiopulloon ja lyijyastian etikettiin.
- hajoamiskerroin (decay correction, DK- kerroin) radium‑223:n fysikaalisen hajoamisen korjaamiseksi. Jokaisessa injektiopullossa on mukana (pakkausselosteen edellä) DK- kerrointaulukko.
Ruiskuun vedetyn potilasannoksen radioaktiivisuus varmistetaan mittaamalla se oikein kalibroidulla aktiivisuusmittarilla.
Potilaalle annettava kokonaistilavuus lasketaan seuraavasti:
| Annettava tilavuus (ml) | = | Paino (kg) x aktiivisuus (55 kBq/painokilo) |
| DK-kerroin x 1100 kBq/ml |
Käyttämätön radioaktiivinen lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Lisätietoa tästä lääkevalmisteesta on Euroopan lääkeviraston verkkosivuilla http://www.ema.europa.eu/.
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi