
STIVARGA tabletti, kalvopäällysteinen 40 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 40 mg regorafenibia.
Lääkemuoto
Tabletti, kalvopäällysteinen.
Kliiniset tiedot
Käyttöaiheet
Stivarga on tarkoitettu monoterapiana aikuispotilaiden hoitoon,
- joilla on metastasoitunut kolorektaalisyöpä (CRC) ja joita on aiemmin hoidettu käytettävissä olleilla hoidoilla tai joille näitä hoitoja ei ole pidetty sopivina. Tällaisia hoitoja ovat fluoropyrimidiinipohjainen kemoterapia, anti-VEGF-hoito ja anti-EGFR-hoito (ks. kohta (ks. kohta Farmakodynamiikka).
- joilla on ei-leikattavissa oleva tai metastaattinen ruoansulatuskanavan stroomakasvain (GIST), joka on edennyt edeltävästä imatinibi- ja sunitinibihoidosta huolimatta tai jotka eivät siedä sunitinibia ja imatinibia.
- joilla on hepatosellulaarinen karsinooma (HCC) ja joita on aiemmin hoidettu sorafenibilla.
Ehto
Valmistetta saavat määrätä sellaiset lääkärit, joilla on kokemusta syöpälääkkeiden antamisesta.
Annostus ja antotapa
Annostus
Regorafenibin suositeltu annos on 160 mg (4 kpl 40 mg:n tablettia) otettuna kerran päivässä 3 viikon ajan, jota seuraa 1 viikon tauko. Tämä 4 viikon jakso katsotaan yhdeksi hoitojaksoksi.
Jos potilas unohtaa ottaa annoksen, se on otettava samana päivänä heti potilaan muistaessa sen. Potilas ei saa ottaa kahta annosta samana päivänä unohdetun annoksen korvaamiseksi. Jos potilas oksentaa regorafenibin käytön jälkeen, hänen ei pidä ottaa enempää tabletteja.
Hoitoa jatketaan niin kauan kuin siitä todetaan olevan hyötyä tai kunnes ilmenee kohtuutonta toksisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaat, joiden suorituskykyluokka (performance status, PS) on 2 tai korkeampi, suljettiin pois kliinisistä tutkimuksista. Potilaista, joiden PS ≥ 2, on vain vähäisiä tietoja.
Annoksen säätäminen
Annostelutauko ja/tai annoksen pienentäminen saattaa olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden perusteella. Annosta muutetaan 40 mg (yksi tabletti) kerrallaan. Pienin suositeltu vuorokausiannos on 80 mg. Suurin vuorokausiannos on 160 mg.
Tiedot suositelluista annosmuutoksista ja toimenpiteistä käsi-jalka-ihoreaktion / palmoplantaarisen erytrodysestesian ilmetessä, ks. taulukko 1.
Taulukko 1: Suositellut annosmuutokset ja toimenpiteet käsi-jalka-ihoreaktiossa
| Ihon toksisuusaste | Esiintyminen | Suositellut annosmuutokset ja toimenpiteet |
| 1. aste | Kaikki esiintymiset | Pidä annostaso ennallaan ja aloita välittömästi oireita lievittävät hoitotoimenpiteet. |
| 2. aste | 1. esiintyminen | Pienennä annosta 40 mg (yhden tabletin verran) ja aloita välittömästi hoitotoimenpiteet. Jos oireet eivät parane annoksen pienentämisestä huolimatta, keskeytä hoito vähintään 7 vuorokaudeksi, kunnes toksisuus lieventyy 0.-1. asteelle. Annosta voidaan nostaa uudelleen lääkärin harkinnan mukaan. |
| Oireet eivät parane 7 vrk:n kuluessa tai 2. esiintyminen | Keskeytä hoito, kunnes toksisuus lieventyy 0.-1. asteelle. Hoitoa uudelleen aloitettaessa pienennä annosta 40 mg (yhden tabletin verran). Annosta voidaan nostaa uudelleen lääkärin harkinnan mukaan. | |
| 3. esiintyminen | Keskeytä hoito, kunnes toksisuus lieventyy 0.-1. asteelle. Hoitoa uudelleen aloitettaessa pienennä annosta 40 mg (yhden tabletin verran). Annosta voidaan nostaa uudelleen lääkärin harkinnan mukaan. | |
| 4. esiintyminen | Lopeta Stivarga-hoito pysyvästi. | |
| 3. aste | 1. esiintyminen | Aloita hoitotoimenpiteet välittömästi. Keskeytä hoito vähintään 7 vuorokaudeksi, kunnes toksisuus lieventyy 0.-1. asteelle. Hoitoa uudelleen aloitettaessa pienennä annosta 40 mg (yhden tabletin verran). Annosta voidaan nostaa uudelleen lääkärin harkinnan mukaan. |
| 2. esiintyminen | Aloita hoitotoimenpiteet välittömästi. Keskeytä hoito vähintään 7 vuorokaudeksi, kunnes toksisuus lieventyy 0.-1. asteelle. Hoitoa uudelleen aloitettaessa pienennä annosta 40 mg (yhden tabletin verran). | |
| 3. esiintyminen | Lopeta Stivarga-hoito pysyvästi. |
Katso taulukosta 2 suositellut toimenpiteet ja annosmuutokset, jos maksan toimintakokeiden tulosten huononemisen katsotaan liittyvän Stivarga-hoitoon (ks. myös kohta Varoitukset ja varotoimet).
Taulukko 2: Suositellut toimenpiteet ja annosmuutokset, kun maksan toimintakokeiden tulokset ovat poikkeavia lääkkeestä johtuen
| Todettu ALAT- ja/tai ASAT-arvon kohoaminen | Esiintyminen | Suositellut toimenpiteet ja annosmuutokset |
| ≤ 5 kertaa yli normaalin ylärajan (ULN) (enintään 2. aste) | Kaikki esiintymiset | Jatka Stivarga-hoitoa. Seuraa maksan toimintaa viikoittain, kunnes transaminaasiarvot palaavat tasolle < 3 kertaa ULN* (1. aste) tai lähtötasolle. |
| > 5 kertaa ULN – ≤ 20 kertaa ULN (3. aste) | 1. esiintyminen | Keskeytä Stivarga-hoito. Seuraa transaminaasiarvoja viikoittain, kunnes ne palaavat tasolle < 3 kertaa ULN tai lähtötasolle. Uudelleenaloitus: Jos mahdollinen hyöty on suurempi kuin hepatotoksisuuden riski, aloita Stivarga-hoito uudelleen, pienennä annosta 40 mg (yhden tabletin verran) ja seuraa maksan toimintaa viikoittain vähintään 4 viikon ajan. |
| Uudelleenesiintyminen | Lopeta Stivarga-hoito kokonaan. | |
| > 20 kertaa ULN (4. aste) | Kaikki esiintymiset | Lopeta Stivarga-hoito kokonaan. |
| > 3 kertaa ULN (2. aste tai suurempi) kun samalla bilirubiini > 2 kertaa ULN | Kaikki esiintymiset | Lopeta Stivarga-hoito kokonaan. Seuraa maksan toimintaa viikoittain tilanteen korjaantumiseen tai lähtötasolle palautumiseen saakka. Poikkeus: Gilbertin oireyhtymää sairastavia potilaita, joiden transaminaasiarvot kohoavat, on hoidettava edellä annettujen suositusten mukaisesti ALAT- ja/tai ASAT-arvojen kohoamisen osalta. |
* viitearvojen yläraja
Maksan vajaatoiminta
Regorafenibi eliminoituu pääasiassa maksan kautta.
Kliinisissä tutkimuksissa ei ole todettu olennaisia eroja altistuksessa, turvallisuudessa tai tehossa potilailla, jotka sairastavat lievää maksan vajaatoimintaa (Child Pugh A) ja joiden maksan toiminta on normaalia. Annoksen sovittaminen ei ole tarpeen lievää maksan vajaatoimintaa sairastavilla potilailla. Koska kohtalaista maksan vajaatoimintaa (Child Pugh B) sairastavista potilaista on saatavana vain vähän tietoa, annossuositusta ei voida antaa. Näille potilaille suositellaan kokonaisturvallisuuden tarkkaa seurantaa (ks. kohdat Varoitukset ja varotoimet sekä Farmakokinetiikka).
Stivarga-valmistetta ei suositella käytettäväksi potilailla, joilla on vaikea maksan vajaatoiminta (Child Pugh C), koska Stivarga-valmistetta ei ole tutkittu tässä ryhmässä.
Munuaisten vajaatoiminta
Saatavilla olevat kliiniset tiedot osoittavat samanlaista altistumista regorafenibille ja sen metaboliiteille (M-2 ja M-5) potilailla, joilla on lievä, kohtalainen tai vaikea munuaisten vajaatoiminta verrattuna potilaisiin, joilla munuaisten toiminta on normaalia. Annoksen säätäminen lievää, kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla ei ole tarpeen (ks. myös kohta Farmakokinetiikka).
Iäkkäät potilaat
Kliinisissä tutkimuksissa ei ole todettu olennaisia eroja altistuksessa, turvallisuudessa tai tehossa vanhempien (vähintään 65-vuotiaiden) ja nuorempien potilaiden välillä. (ks. myös kohta Farmakokinetiikka).
Sukupuoli
Kliinisissä tutkimuksissa ei ole todettu olennaisia eroja altistuksessa, turvallisuudessa tai tehossa mies- ja naispotilaiden välillä. Annoksen säätäminen sukupuolen perusteella ei ole tarpeen (ks. myös kohta Farmakokinetiikka).
Etnisten ryhmien väliset erot
Kliinisissä tutkimuksissa ei ole todettu olennaisia eroja altistuksessa tai tehossa eri etnisiin ryhmiin kuuluvien potilaiden välillä. Stivarga-valmisteella hoidetuilla aasialaisilla (erityisesti japanilaisilla) potilailla havaittiin enemmän käsi-jalka-ihoreaktioita / palmoplantaarista erytrodysestesiaa, vaikeita poikkeamia maksan toimintakokeissa ja maksan vajaatoimintaa verrattuna kaukaasialaisiin. Kliinisissä tutkimuksissa Stivarga-hoitoa saaneet aasialaiset potilaat olivat pääasiassa Itä-Aasiasta (∼90 %). Regorafenibin käytöstä mustaihoisilla potilailla on vain vähän tietoa. Annoksen säätäminen etnisen ryhmän perusteella ei ole tarpeen (ks. kohta Farmakokinettiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää Stivarga-valmistetta pediatrisille potilaille levinneen kolorektaalisyövän (CRC), ruoansulatuskanavan stroomakasvainten ja hepatosellulaarisen karsinooman hoidossa.
Stivarga-valmisteen turvallisuutta ja tehoa lapsille ja nuorille (alle 18 vuotta) ei ole varmistettu (ks. kohta Farmakodynamiikka).
Antotapa
Stivarga-valmiste otetaan suun kautta.
Stivarga-valmiste on otettava samaan aikaan joka päivä. Tabletit on nielaistava kokonaisina veden kanssa kevyen aterian (rasvapitoisuus alle 30 %) jälkeen. Kevyt (vähärasvainen) ateria voi sisältää esimerkiksi 1 annoksen muroja (noin 30 g), 1 lasin rasvatonta maitoa, 1 hillolla päällystetyn paahtoleipäviipaleen, 1 lasin omenamehua, ja 1 kupin kahvia tai teetä (520 kaloria, 2 g rasvaa).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Vaikutukset maksaan
Stivarga-valmisteella hoidetuilla potilailla on usein todettu poikkeava tulos maksan toimintakokeessa (alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT] ja bilirubiini). Pienellä osalla potilaista on raportoitu vaikeita poikkeamia maksan toimintakokeissa (3. tai 4. aste) ja maksan vajaatoiminnan kliinisiä oireita (mukaan lukien kuolemaan johtavia) (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa Stivarga-hoitoa saaneilla aasialaisilla (erityisesti japanilaisilla) potilailla havaittiin enemmän vaikeita poikkeamia maksan toimintakokeissa ja maksan vajaatoimintaa kuin kaukaasialaisilla (ks. kohta Annostus ja antotapa).
Maksan toimintakokeiden (ALAT, ASAT ja bilirubiini) suorittamista ennen Stivarga-hoidon aloittamista ja tulosten tarkkaa seurantaa (vähintään kahden viikon välein) suositellaan hoidon kahden ensimmäisen kuukauden ajan. Sen jälkeen säännöllistä seurantaa jatketaan vähintään kuukausittain ja kliinisen tarpeen mukaisesti.
Regorafenibi on uridiinidifosfaatti-glukuronosyylitransferaasi 1A1:n (UGT1A1:n) estäjä (ks. kohta Yhteisvaikutukset). Gilbertin oireyhtymää sairastavilla potilailla saattaa esiintyä lievää, epäsuoraa (konjugoimatonta) hyperbilirubinemiaa.
Potilailla, joilla maksan toimintakokeiden tulosten huononemisen katsotaan johtuvan Stivarga-valmisteesta (ts. mikään muu syy, kuten posthepaattinen kolestaasi tai sairauden eteneminen, ei ole todennäköinen), on noudatettava taulukon 2 mukaisia annosmuutos- ja seurantaohjeita (ks. kohta Annostus ja antotapa).
Regorafenibi eliminoituu pääasiassa maksan kautta.
Kokonaisturvallisuuden tarkkaa seurantaa suositellaan potilailla, joilla on lievä tai kohtalainen maksan vajaatoiminta (ks. myös kohdat Annostus ja antotapa sekä Farmakokinetiikka). Stivarga-valmistetta ei suositella käytettäväksi potilaille, joilla on vaikea maksan vajaatoiminta (Child Pugh C), koska Stivarga-valmistetta ei ole tutkittu tällä väestöryhmällä ja altistus voisi näillä potilailla suurentua.
Infektiot
Stivarga-valmisteeseen on liittynyt lisääntynyttä infektioiden esiintymistä, joista jotkut ovat olleet kuolemaanjohtavia (ks. kohta Haittavaikutukset).
Stivarga-hoidon keskeyttämistä on harkittava tapauksissa, joissa infektio pahenee.
Verenvuoto
Stivarga-valmisteeseen on liittynyt kohonnut verenvuototapahtumien esiintyneisyys. Jotkut niistä johtivat kuolemaan (ks. kohta Haittavaikutukset). Veri- ja hyytymisarvoja on seurattava potilailla, joilla on verenvuodolle altistavia sairauksia, sekä potilailla, joita hoidetaan hyytymisenestolääkkeillä (esim. varfariini ja fenprokumoni) tai samanaikaisesti muilla verenvuotoriskiä lisäävillä lääkevalmisteilla. Maksakirroosipotilaat tulisi hoitosuositusten mukaisesti seuloa ruokatorven laskimolaajentumien varalta ja ne tulisi hoitaa ennen Stivarga-hoidon aloittamista. Stivarga-hoidon lopettamista kokonaan on harkittava, jos potilaalla esiintyy vaikea, kiireellistä hoitoa vaativa verenvuoto.
Maha-suolikanavan perforaatio ja fisteli
Maha-suolikanavan perforaatioita (mukaan lukien kuolemaan johtaneita) ja fisteleitä on raportoitu Stivarga-valmisteella hoidetuilla potilailla (ks. kohta Haittavaikutukset). Näiden tapahtumien tiedetään myös olevan yleisiä sairauteen liittyviä komplikaatioita potilailla, joilla on vatsaontelon sisäisiä malignniteetteja. Stivarga-hoidon keskeyttämistä suositellaan potilaille, joille kehittyy maha-suolikanavan perforaatio tai fisteli.
Sydäniskemia ja -infarkti
Stivarga-valmisteeseen on liitetty lisääntynyt sydänlihasiskemian ja sydäninfarktin ilmaantuvuus (ks. kohta Haittavaikutukset). Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli epästabiili angina pectoris tai joilla angina pectoris oli alkanut uudelleen (3 kuukauden kuluessa Stivarga-hoidon aloittamisesta), joilla oli äskettäin ollut sydäninfarkti (6 kuukauden kuluessa Stivarga-hoidon aloittamisesta) ja joilla oli New York Heart Association (NYHA) luokan 2 tai korkeamman luokan sydämen vajaatoiminta.
Potilaita, joilla on aiemmin ollut iskeeminen sydänsairaus, on seurattava sydänlihasiskemian kliinisten löydösten ja oireiden varalta. Potilaille, joille kehittyy sydäniskemia ja/tai -infarkti, suositellaan Stivarga-valmisteen keskeyttämistä, kunnes oireet ovat hävinneet. Päätös Stivarga-hoidon aloittamisesta uudelleen on tehtävä kyseiselle potilaalle mahdollisesti aiheutuvien hyötyjen ja riskien huolellisen arvioinnin perusteella. Jos oireet eivät häviä, Stivarga-valmisteen käyttö on lopetettava kokonaan.
Hyperammoneeminen enkefalopatia
Hyperammoneemista enkefalopatiaa on havaittu regorafenibin käytön yhteydessä, mukaan lukien kuolemaan johtaneet tapaukset (ks. kohta Haittavaikutukset). Jos potilaalla ilmenee selittämätöntä letargiaa tai psyykkisen tilan muutoksia, on mitattava ammoniakkipitoisuus ja aloitettava asianmukainen kliininen hoito. Jos hyperammoneeminen enkefalopatia on vahvistettu, on harkittava regorafenibihoidon lopettamista pysyvästi.
Posteriorinen reversiibeli leukoenkefalopatia –oireyhtymä (PRES)
Stivarga-hoitoon liittyen on raportoitu PRES-oireyhtymä (ks. kohta Haittavaikutukset). PRES-oireyhtymän löydöksiä ja oireita ovat mm. kouristuskohtaukset, päänsärky, psyykkisen tilan muutos, näköhäiriöt tai kortikaalinen sokeus, johon voi liittyä hypertensio. PRES-diagnoosi on vahvistettava aivojen kuvantamisella. Potilaille, joille kehittyy PRES, suositellaan Stivarga-valmisteen käytön keskeyttämistä sekä hypertension hoitoa ja muiden oireiden hoitamista.
Arteriaalinen hypertensio
Stivarga-valmisteeseen on liitetty lisääntynyt arteriaalisen hypertension ilmaantuvuus (ks. kohta Haittavaikutukset). Verenpaine on mitattava ennen Stivarga-hoidon aloittamista. Verenpaineen seuranta ja hypertension hoito asianmukaisten hoitokäytäntöjen mukaan on suositeltavaa. Jos hypertensio on vaikea tai jatkuva asianmukaisesta hoidosta huolimatta, hoito on lopetettava tilapäisesti ja/tai annosta pienennettävä hoitavan lääkärin harkinnan mukaan (ks. kohta Annostus ja antotapa). Hypertensiivisen kriisin ilmetessä Stivarga-hoito on lopetettava.
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen Stivarga-hoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Tromboottinen mikroangiopatia
Tromboottinen mikroangiopatia, mukaan lukien tromboottinen trombosytopeeninen purppura, on liitetty regorafenibin käyttöön (ks. kohta Haittavaikutukset). Tromboottisen mikroangiopatian mahdollisuutta on harkittava potilailla, joilla on hemolyyttinen anemia, trombosytopenia, väsymystä, vaihtelevia neurologisia oireita ja merkkejä, munuaisten vajaatoimintaa ja kuumetta. Regorafenibihoito on lopetettava potilailta, joille kehittyy tromboottinen mikroangiopatia, ja nopea hoito on tarpeen.
Tromboottisen mikroangiopatian vaikutusten on havaittu häviävän hoidon lopettamisen jälkeen.
Haavan paranemiseen liittyvät komplikaatiot
Koska lääkkeet, joilla on antiangiogeenisia ominaisuuksia, voivat estää haavan paranemisen tai haitata sitä, Stivarga-hoidon keskeyttämistä tilapäisesti suositellaan varotoimenpiteenä potilaille, joille tehdään suuria kirurgisia toimenpiteitä. Päätöksen Stivarga-hoidon jatkamisesta suuren kirurgisen toimenpiteen jälkeen tulee perustua kliiniseen arvioon haavan asianmukaisesta paranemisesta.
Ihotoksisuus
Käsi jalka ihoreaktio eli palmoplantaarinen erytrodysestesia ja ihottuma ovat yleisimmät Stivarga-hoidon yhteydessä todetut dermatologiset haittavaikutukset (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa Stivarga-hoitoa saaneilla aasialaisilla (erityisesti japanilaisilla) potilailla havaittiin enemmän käsijalka-ihoreaktioita kuin kaukaasialaisilla (ks. kohta Annostus ja antotapa). Käsi-jalka-ihoreaktion ehkäisytoimenpiteitä ovat mm. kovettumien tarkastus sekä pehmustettujen kenkien ja käsineiden käyttö jalkapohjiin ja kämmeniin kohdistuvan paineen välttämiseksi. Käsi jalka ihoreaktiota voidaan hoitaa mm. keratolyyttisillä voiteilla (esim. urea, salisyylihappo tai alfahydroksyylihappopohjaiset voiteet, joita levitetään ohut kerros vain kyseisille alueille), sekä kosteuttavilla voiteilla, joita levitetään runsaasti oireiden lievittämiseksi. Annoksen pienentämistä ja/tai Stivarga-hoidon keskeyttämistä tilapäisesti tai, vaikeissa tai jatkuvissa tapauksissa, Stivarga-hoidon lopettamista kokonaan on harkittava (ks. kohta Annostus ja antotapa).
Poikkeavat biokemialliset ja metaboliset laboratoriokokeiden tulokset
Stivarga-hoitoon on liitetty poikkeavien elektrolyyttiarvojen (mukaan lukien hypofosfatemia, hypokalsemia, hyponatremia ja hypokalemia) lisääntynyt esiintyvyys ja poikkeavia aineenvaihduntaan liittyviä arvoja (mukaan lukien kilpirauhasta stimuloivan hormonin, lipaasin ja amylaasin lisääntyminen). Nämä poikkeavuudet ovat yleensä lieviä tai kohtalaisia, niihin ei liity kliinisiä oireita eivätkä ne yleensä vaadi annostelun keskeyttämistä tai annoksen pienentämistä. Stivarga-hoidon aikana suositellaan biokemiallisten ja aineenvaihduntaan liittyvien parametrien seurantaa sekä tarvittaessa asianmukaisen korvaushoidon aloittamista normaalin hoitokäytännön mukaisesti. Annostelun keskeyttämistä tai annoksen pienentämistä tai Stivarga-hoidon lopettamista kokonaan on harkittava, jos merkittävästi poikkeavat arvot jatkuvat tai palaavat uudelleen (ks. kohta Annostus ja antotapa).
Tärkeää tietoa joistakin ainesosista:
Tämä lääkevalmiste sisältää 56,06 mg natriumia per vuorokausiannos (160 mg), joka vastaa 3 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Yksi vuorokausiannos (160 mg) sisältää 1,68 mg lesitiiniä (peräisin soijasta).
Yhteisvaikutukset
CYP3A4:n ja UGT1A9:n estäjät / CYP3A4:n induktorit
In vitro -tiedot viittaavat siihen, että regorafenibi metaboloituu sytokromi CYP3A4:n ja uridiinidifosfaatti-glukuronyylitransferaasin UGT1A9 välityksellä.
Kun voimakasta CYP3A4:n estäjää ketokonatsolia (400 mg 18 vuorokauden ajan) annettiin regorafenibin kerta-annoksen (160 mg 5. päivänä) kanssa, keskimääräinen altistuminen (AUC) regorafenibille nousi noin 33 %, ja keskimääräinen altistuminen aktiivisille aineenvaihduntatuotteille M-2 (N-oksidi) ja M-5 (N-oksidi ja N-desmetyyli) laski noin 90 %. Voimakkaiden CYP3A4:n toiminnan estäjien (esim. klaritromysiini, greippimehu, itrakonatsoli, ketokonatsoli, posakonatsoli, telitromysiini ja vorikonatsoli) samanaikaisen käytön välttäminen on suositeltavaa, sillä niiden vaikutusta regorafenibin ja sen metaboliittien vakaan tilan altistukseen ei ole tutkittu.
Voimakkaan UGT1A9:n estäjän (esim. mefenaamihappo, diflunisaali ja niflumiinihappo) samanaikaista antamista regorafenibihoidon aikana on vältettävä, koska niiden vaikutusta regorafenibin ja sen metaboliittien vakaan tilan altistukseen ei ole tutkittu.
Kun voimakasta CYP3A4:n induktoria rifampisiinia (600 mg 9 vuorokauden ajan) annettiin regorafenibin kerta-annoksen (160 mg 7. päivänä) kanssa, keskimääräinen altistuminen (AUC) regorafenibille laski noin 50 %, keskimääräinen altistuminen aktiiviselle aineenvaihduntatuotteelle M-5 nousi 3-4-kertaisesti ja aktiiviselle aineenvaihduntatuotteelle M-2 altistuminen pysyi muuttumattomana. Muut voimakkaat CYP3A4:n induktorit (esim. fenytoiini, karbamatsepiini, fenobarbitaali ja mäkikuisma) voivat myös lisätä regorafenibin aineenvaihduntaa. Koska regorafenibin plasmapitoisuuden väheneminen voi johtaa tehon heikkenemiseen, voimakkaita CYP3A4:n induktoreita on vältettävä tai on harkittava sellaisen vaihtoehtoisen samanaikaisen lääkevalmisteen valitsemista, jolla ei ole lainkaan tai on vain vähäinen CYP3A4:n indusointikyky.
UGT1A1- ja UGT1A9-substraatit
In vitro -tiedot osoittavat, että sekä regorafenibi että sen aktiivinen aineenvaihduntatuote M-2 estävät glukuronidaatiota uridiinidifosfaatti-glukuronyylitransferaasien UGT1A1 ja UGT1A9 välityksellä, pitoisuuksissa, jotka saavutetaan in vivo vakaassa tilassa. M-5 puolestaan estää vain UGT1A1:aa. Kun regorafenibin annon jälkeen pidettiin 5 päivän tauko ennen irinotekaanin antoa, SN-38:n (UGT1A1:n substraatti ja irinotekaanin aktiivinen aineenvaihduntatuote) AUC nousi noin 44 %. Myös irinotekaanin AUC nousi noin 28 %. Tämä osoittaa, että regorafenibin antaminen samanaikaisesti voi lisätä systeemistä altistumista UGT1A1- ja UGT1A9-substraateille.
Rintasyövän resistenssiproteiinin (BCRP) ja P-glykoproteiinin substraatit
Regorafenibin anto (160 mg 14 vuorokauden ajan) ennen kerta-annoksena annettavaa rosuvastatiinia (5 mg), joka on BRCP:n substraatti, aiheutti 3,8-kertaisen kasvun rosuvastatiinin keskimääräiselle altistumiselle (AUC) sekä ja 4,6-kertaisen kasvun plasman huippupitoisuudessa (Cmax).
Tämä viittaa siihen, että regorafenibin samanaikainen anto muiden BCRP:n substraattien (esim. metotreksaatti, fluvastatiini, atorvastatiini) kanssa voi lisätä näiden plasmapitoisuuksia. Tämän vuoksi on suositeltavaa tarkkailla potilaita huolellisesti BCRP:n substraattien lisääntyneen altistuksen merkkien ja oireiden varalta.
Kliiniset tiedot viittaavat siihen, että regorafenibi ei vaikuta digoksiinin farmakokinetiikkaan. Tämän vuoksi regorafenibia voidaan antaa samanaikaisesti P-glykoproteiinin substraattien, kuten digoksiinin, kanssa, ilman kliinisesti merkittävää lääkeinteraktioita.
P-glykoproteiinin ja BCRP:n estäjät / P-glykoproteiinin ja BCRP:n induktorit ja BCRP
In vitro -tutkimukset osoittavat, että aktiiviset aineenvaihduntatuotteet M‑2 ja M‑5 ovat P-glykoproteiinin ja BCRP:n substraatteja. BCRP:n ja P-glykoproteiinin estäjät ja induktorit voivat vaikuttaa M‑2:n ja M‑5:n altistukseen. Näiden löydösten kliinistä merkitystä ei tunneta (ks. myös kohta Farmakokinetiikka).
CYP-isoformien selektiiviset substraatit
In vitro -tiedot osoittavat, että regorafenibi on sytokromien CYP2C8 (Ki-arvo 0,6 mikromolaaria), CYP2C9 (Ki-arvo 4,7 mikromolaaria) ja CYP2B6 (Ki-arvo 5,2 mikromolaaria) kilpaileva estäjä pitoisuuksissa, jotka saavutetaan in vivo vakaassa tilassa (plasman huippupitoisuus 8,1 mikromolaaria). In vitro CYP3A4:ää (Ki-arvo 11,1 mikromolaaria) ja CYP2C19:ää (Ki-arvo 16,4 mikromolaaria) estävä vaikutus oli vähäisempi.
Kliinisessä testisubstraattitutkimuksessa, jossa regorafenibia annettiin 160 mg 14 vuorokauden ajan, arvioitiin annostelun vaikutusta seuraavien CYP-testisubstraattien farmakokinetiikkaan: CYP2C8 (rosiglitatsoni), CYP2C9 (S-varfariini), CYP2C19 (omepratsoli) ja CYP3A4 (midatsolaami).
Farmakokineettiset tiedot osoittavat, että regorafenibia voidaan antaa samanaikaisesti CYP2C8:n, CYP2C9:n, CYP3A4:n ja CYP2C19:n substraattien kanssa ilman kliinisesti merkittävää interaktiota (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antibiootit
Pitoisuus/aika-profiili viittaa siihen, että regorafenibi ja sen aineenvaihduntatuotteet saattavat käydä läpi enterohepaattisen kierron (ks. kohta Farmakokinetiikka). Huonosti imeytyvän antimikrobisen aineen, neomysiinin, jota käytetään mahasuolikanavan mikroflooran hävittämiseen (mikä saattaa haitata regorafenibin enterohepaattista kiertoa), antaminen samanaikaisesti ei vaikuttanut regorafenibille altistumiseen. Aktiivisten aineenvaihduntatuotteiden M-2 ja M-5, joilla on osoitettu olevan regorafenibia vastaava farmakologinen aktiivisuus in vitro ja in vivo, altistuminen väheni noin 80 %. Tämän neomysiinin yhteisvaikutuksen kliinistä merkitystä ei tiedetä, mutta se saattaa johtaa regorafenibin tehon heikkenemiseen. Muiden antibioottien farmakokineettisiä yhteisvaikutuksia ei ole tutkittu.
Sappihappoja sitovat aineet
Regorafenibi, M-2 ja M-5 käyvät todennäköisesti läpi enterohepaattisen kierron (ks. kohta Farmakokinetiikka). Sappihappoja sitovat aineet, kuten kolestyramiini ja cholestagel, voivat reagoida regorafenibin kanssa muodostaen liukenemattomia komplekseja. Nämä liukenemattomat kompleksit voivat vaikuttaa imeytymiseen (tai takaisinimeytymiseen) ja siten johtaa mahdollisesti alentuneeseen altistumiseen. Näiden mahdollisten yhteisvaikutusten kliininen merkitys on tuntematon, mutta ne saattavat johtaa regorafenibin tehon heikkenemiseen.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / Raskauden ehkäisy naisilla ja miehillä
Hedelmällisessä iässä oleville naisille on kerrottava, että regorafenibi voi aiheuttaa haittaa sikiölle. Hedelmällisessä iässä olevien naisten ja miesten on käytettävä tehokasta raskaudenehkäisyä hoidon aikana ja enintään 8 viikon ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja regorafenibin käytöstä raskaana oleville naisille.
Vaikutusmekanisminsa vuoksi regorafenibin epäillään vahingoittavan sikiötä, kun sitä annetaan raskauden aikana. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Stivarga-valmistetta ei pidä käyttää raskauden aikana, ellei tämä ole selkeästi välttämätöntä vielä senkin jälkeen, kun on tarkoin otettu huomioon äidin tarpeet ja sikiöön kohdistuvat riskit.
Imetys
Ei tiedetä, erittyvätkö regorafenibi tai sen metaboliitit ihmisen rintamaitoon.
Rotilla regorafenibi tai sen metaboliitit erittyvät rintamaitoon. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Regorafenibi voi haitata vastasyntyneen kasvua ja kehitystä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Rintaruokinta on lopetettava Stivarga-hoidon ajaksi.
Hedelmällisyys
Stivarga-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Eläintutkimusten tulokset osoittavat, että regorafenibi voi heikentää uroksen ja naaraan hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia Stivarga-valmisteen vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty. Jos potilailla ilmenee keskittymis- ja reaktiokykyä haittaavia oireita Stivarga-hoidon aikana, on suositeltavaa olla ajamatta ja käyttämättä koneita, kunnes vaikutus häviää.
Haittavaikutukset
hteenveto turvallisuudesta
Stivarga-valmisteen yleinen turvallisuusprofiili perustuu kliinisissä tutkimuksissa yli 800 hoidetusta potilaasta saatuihin tietoihin. Näihin sisältyivät lumelääkkeellä kontrolloidusta vaiheen III tutkimuksesta saadut levinnyttä kolorektaalisyöpää (CRC) sairastaneiden 636 potilaan tiedot, 132 potilaan tiedot, joilla oli ruoansulatuskanavan stroomakasvain (GIST) ja 374 potilaan tiedot, joilla oli hepatosellulaarinen karsinooma (HCC).
Näissä tutkimuksissa regorafenibin turvallisuusprofiili oli yhdenmukainen vaiheen III B-tutkimuksesta saatujen turvallisuustulosten kanssa, joka tehtiin 2872 levinnyttä kolorektaalisyöpää sairastaville potilaille, joiden sairaus oli edennyt standardihoidon jälkeen.
Vakavimmat lääkkeen haittavaikutukset Stivarga-valmistetta saavilla potilailla ovat vaikea maksavaurio, verenvuoto, maha-suolikanavan perforaatio ja infektio.
Useimmiten havaitut lääkkeen haittavaikutukset (≥ 30 %) Stivarga-valmistetta saavilla potilailla ovat kipu, käsi-jalka-ihoreaktio, astenia/väsymys, ripuli, ruokahalun ja syömisen väheneminen, hypertensio ja infektio.
Yhteenveto haittavaikutuksista taulukon muodossa
Stivarga-valmisteella hoidetuilla potilailla kliinisissä tutkimuksissa ilmoitetut haittavaikutukset esitetään taulukossa 3. Ne on luokiteltu elinjärjestelmän mukaan ja sopivinta MedDRA-termiä käytetään kuvaamaan tiettyä reaktiota ja sen synonyymiä sekä siihen liittyviä oireita.
Haittavaikutukset on ryhmitelty niiden esiintyvyyden mukaan. Esiintyvyydet on määritelty seuraavan käytännön mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja tuntematon (saatavissa oleva tieto ei riitä arviointiin).
Haittavaikutukset on esitetty kussakin esiintyvyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Stivarga-valmisteella hoidetuilla potilailla kliinisissä tutkimuksissa ja markkinoille tulon jälkeen raportoidut haittavaikutukset
| Infektiot | |
| Hyvin yleinen | Infektio* |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
| Harvinainen | Keratoakantooma / ihon okasolusyöpä |
| Veri ja imukudos | |
| Hyvin yleinen | Trombosytopenia, anemia |
| Yleinen | Leukopenia |
| Harvinainen | Tromboottinen mikroangiopatia |
| Immuunijärjestelmä | |
| Melko harvinainen | Yliherkkyysreaktiot |
| Umpieritys | |
| Yleinen | Kilpirauhasen vajaatoiminta |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | Vähentynyt ruokahalu ja syöminen |
| Yleinen | Hypokalemia, hypofosfatemia, hypokalsemia, hyponatremia, hypomagnesemia, hyperurikemia, dehydraatio |
| Hermosto | |
| Yleinen | Päänsärky, vapina, perifeeerinen neuropatia |
| Harvinainen | Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES) |
| Tuntematon | Hyperammoneeminen enkefalopatia |
| Sydän | |
| Melko harvinainen | Sydäninfarkti, sydänlihasiskemia |
| Verisuonisto | |
| Hyvin yleinen | Verenvuoto*, kohonnut verenpaine |
| Melko harvinainen | Hypertensiivinen kriisi |
| Tuntematon | Aneurysmat ja valtimon dissekaatiot |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | Dysfonia |
| Ruoansulatuselimistö | |
| Hyvin yleinen | Ripuli, stomatiitti, oksentelu, pahoinvointi |
| Yleinen | Makuaistin häiriöt, suun kuivuminen, ruokatorven refluksitauti, gastroenteriitti |
| Melko harvinainen | Maha-suolikanavan perforaatio*, maha-suolikanavan fisteli, pankreatiitti |
| Maksa ja sappi | |
| Hyvin yleinen | Hyperbilirubinemia, Transaminaasien nouseminen |
| Melko harvinainen | Vaikea maksavaurio*# |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | Käsi-jalka-ihoreaktio**, ihottuma |
| Yleinen | Hiustenlähtö, Kuiva iho, kesivä ihottuma |
| Melko harvinainen | Kynsisairaus, erythema multiforme |
| Harvinainen | Stevens-Johnsonin syndrooma, toksinen epidermaalinen nekrolyysi |
| Luusto, lihakset ja sidekudos | |
| Yleinen | Lihaskouristukset |
| Munuaiset ja virtsatiet | |
| Yleinen | Proteinuria |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | Astenia/uupumus, kipu***, kuume, limakalvotulehdus |
| Tutkimukset | |
| Hyvin yleinen | Painonlasku |
| Yleinen | Amylaasien nouseminen, lipaasien nouseminen, poikkeava INR-arvo (International normalised ratio) |
* Kuolemaan johtavia tapauksia on raportoitu
** Palmoplantaarinen erytrodysestesia MedDRA-terminologiassa
*** Yleisimmin ilmoitetut kiputyypit (≥ 10 %) ovat vatsakipu ja selkäkipu
# Kansainvälisen DILI-asiantuntijatyöryhmän (DILI = drug-induced liver injury eli lääkkeen aiheuttama maksavaurio) DILI-kriteerien mukaan.
Valikoitujen haittavaikutusten kuvaus
Useimmissa tapauksissa maksavaurio, maksan toimintahäiriöt alkoivat hoidon ensimmäisten 2 kuukauden aikana, ja niille oli ominaista hepatosellulaarinen vaurio, jossa transaminaasit kohosivat yli 20-kertaisiksi normaaliarvojen ylärajaan nähden, ja jota seurasi bilirubiinipitoisuuden nousu. Kliinisissä tutkimuksissa suurempi esiintyvyys vaikeita kuolemaan johtaneita maksavaurioita havaittiin japanilaisilla (noin 1,5 %) Stivarga-valmisteella hoidetuilla potilailla verrattuna ei-japanilaisiin potilaisiin (< 0,1 %).
Lumelääkekontrolloiduissa vaiheen III tutkimuksissa Stivarga-hoitoa saaneilla potilailla hemorragian kokonaisesiintyvyys oli 18,2 % potilailla, joita hoidettiin Stivarga-valmisteella ja 9,5 % potilailla, jotka saivat lumelääkettä. Useimmat Stivarga-valmisteella hoidetuilla potilailla ilmenneet verenvuototapahtumat olivat lieviä tai kohtalaisia (1. ja 2. aste: 15,2 %), useimmin nenäverenvuoto (6,1 %). Kuolemaan johtavat tapaukset olivat Stivarga-valmisteella hoidetuilla potilailla melko harvinaisia (0,7 %), ja niihin sisältyi aivoperäisiä hengitysteiden, maha-suolikanavan sekä urogenitaalialueen tapauksia.
Lumelääkekontrolloidussa vaiheen III tutkimuksissa infektioita todettiin useammin Stivarga-valmisteella hoidetuilla potilailla kuin lumelääkettä saavilla potilailla (kaikki asteet: 31,6 % vs. 17,2 %). Useimmat Stivarga-valmisteella hoidetuilla potilailla todetut infektiot olivat vaikeudeltaan lieviä tai kohtalaisia (1. ja 2. aste: 23,0 %), ja niitä olivat mm. virtsatietulehdukset (5,7 %), nasofaryngiitti (4,0 %), mukokutaaniset ja systeemiset sienitulehdukset (3,3 %) sekä keuhkokuume (2,6 %). Infektioihin liittyviä kuolemantapauksia havaittiin useammin potilailla, joita hoidettiin Stivarga-valmisteella (1,0 %) kuin potilailla, jotka saivat lumelääkettä (0,3 %). Tapaukset olivat pääasiassa hengitystietapahtumia.
Lumelääkekontrolloiduissa vaiheen III tutkimuksissa käsi-jalka-ihoreaktion kokonaisesiintyvyys oli Stivarga-hoitoa saaneilla potilailla suurempi kuin lumelääkettä saaneilla potilailla potilailla (kaikki asteet: 51,4 % vs. 6,5 % CRC, 66,7 % vs. 15,2 % GIST ja 51,6 % vs. 7,3 % HCC). Useimmat käsi-jalka-ihoreaktiotapaukset Stivarga-hoitoa saaneilla potilailla ilmenivät ensimmäisen hoitojakson aikana ja olivat vaikeudeltaan lieviä tai kohtalaisia (1. ja 2. aste: 34,3 % CRC, 44,7 % GIST ja 39,3 % HCC). 3. asteen käsi-jalka-ihoreaktion esiintyvyys oli 17,1 % CRC-, 22,0 % GIST- ja 12,3 % HCCpotilailla. Käsi-jalka -ihoreaktion kokonaisesiintyvyys oli suurempi Stivarga-valmisteella hoidetuilla aasialaisilla potilailla verrattuna muihin etnisiin ryhmiin (74,8% CRC-, 88,2% GIST- ja 67,1 % HCC-potilailla). 3. asteen käsi-jalka-ihoreaktion esiintyvyys oli aasialaisilla 20,5% (CRC), 23,5% (GIST) ja 13,5 % (HCC) (ks. kohdat Annostus ja antotapa sekä Varoitukset ja käyttöön liittyvät varotoimet).
Lumelääkekontrolloiduissa vaiheen III tutkimuksissa hypertension kokonaisesiintyvyys oli suurempi potilailla, joita hoidettiin Stivarga-valmisteella verrattuna lumelääkettä saaneisiin potilaisiin (29,6 % vs. 7,5 % CRC, 60,6 % vs. 25,8 % GIST ja 31,0 % vs. 6,2 % HCC). Useimmat hypertensiotapaukset Stivarga-hoitoa saaneilla potilailla ilmenivät ensimmäisen hoitojakson aikana ja olivat vaikeudeltaan lieviä tai kohtalaisia (1. ja 2. aste: 20,9 % CRC-, 31,8 % GIST- ja 15,8 % HCC-potilailla). 3. asteen hypertension esiintyvyys oli 8,7 % CRC-, 28,0 % GIST- ja 15,2 % HCC-potilailla. GIST-tutkimuksessa raportoitiin yksi 4. asteen hypertensiotapaus.
Lumelääkekontrolloiduissa vaiheen III tutkimuksissa hoidosta aiheutuneen proteinurian kokonaisesiintyvyys oli 9,1 % Stivarga-valmisteella hoidetuilla potilailla, kun lumelääkettä saaneilla vastaava luku oli 1,9 %. Stivarga-ryhmässä 35,6 % ja lumelääkeryhmässä 54,5 % potilaista ei parantunut/tilanne ei korjaantunut.
Kaikissa kliinisissä tutkimuksissa sydänsairauteen liittyviä tapahtumia (kaikki asteet) raportoitiin useammin (13,7 % vs. 6,5 %) 75-vuotiailla tai vanhemmilla Stivarga-valmisteella hoidetuilla potilailla (N=410) verrattuna alle 75-vuotiaisiin (N=4108) Stivarga-valmisteella hoidettuihin potilaisiin.
Poikkeavat laboratoriokokeiden tulokset
Taulukossa 4 ja, taulukossa 4a esitetään lumelääkekontrolloiduissa vaiheen III tutkimuksissa todetut hoidosta aiheutuneet poikkeavat laboratoriokokeiden tulokset (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet.)
Taulukko 4: Levinnyttä kolorektaalisyöpää sairastavilla potilailla lumelääkekontrolloidussa vaiheen III tutkimuksessa raportoidut hoidosta aiheutuvat poikkeavat laboratoriokokeiden tulokset (CORRECT), GIST (GRID) ja HCC (RESORCE)
| mCRC (CORRECT) | GIST (GRID) | HCC (RESORCE) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laboratorio- parametri (% tutkituista näytteistä) | Stivarga ja BSC (n= 500) | Plasebo ja BSC (n=253) | Stivarga ja BSC (n= 500) | Plasebo ja BSC (n=253) | Stivarga ja BSC (n= 132) | Plasebo ja BSC (n= 66) | Stivarga ja BSC (n=132) | Plasebo ja BSC (n= 66) | Stivarga ja BSC (n= 374) | Plasebo ja BSC (n=193) | Stivarga ja BSC (n= 374) | Plasebo ja BSC (n=193) |
| Aste a | Aste b | Aste b | ||||||||||
| Kaikki asteet % | Aste 3/4 % | Kaikki asteet % | Aste 3/4 % | Kaikki asteet % | Aste 3/4 % | |||||||
Veri ja imukudos Hemoglobiinin lasku Trombosytopenia Neutropenia Lymfopenia |
78,5 40,5 2,8 54,1 |
66,3 16,8 0 34,8 |
5,3 2,8 0,6 9,3 |
2,8 0,4 0 4,0 |
75,0 12,9 15,9 29,9 |
72,7 1,5 12,1 24,2 |
3,0 0,8 3,1 7,6 |
1,5 1,5 3,0 3,0 |
72,5 63,1 13,6 67,8 |
71,3 50,0 14,9 58,5 |
6,0 5,4 3,0 17,4 |
4,8 0 1,0 11,7 |
Aineenvaihdunta ja ravitsemus Hypokalsemia Hypokalemia Hypofosfatemia |
59,3 25,7 57,4 |
18,3 8,3 11,1 |
1,2 4,3 31,1 |
1,2 0,4 3,6 |
16,7 20,5 54,5 |
4,5 3,0 3,1 |
1,5 3,0 21,2 |
0 0 1,5 |
23,4 30,7 70,4 |
10,1 9,0 31,4 |
0,3 4,3 33,9 |
0 2,1 6,9 |
Maksa ja sappi Hyperbilirubinemia ASAT-arvon kohoaminen ALAT-arvon kohoaminen |
44,6 65,0
45,2 |
17,1 45,6
29,8 |
12,2 5,9
5,5 |
8,4 5,2
3,2 |
33,3 58,3
39,4 |
12,1 47,0
39,4 |
3,8 3,8
4,6 |
1,5 3,0
1,5 |
78,2 92,7
70,4 |
54,5 84,3
58,6 |
15,9 17,8
6,2 |
15,7 19,9
4,7 |
Munuaiset ja virtsatiet Proteinuria |
83,6 |
61,0 |
1,8 |
0,8 |
59,2 |
52,5 |
3,1 |
3,4 |
51,0 |
36,5 |
16,7 |
3,1 |
Tutkimukset INR-arvon kohoaminen * Lipaasien nousu Amylaasin nousu |
23,7
46,0 25,5 |
16,6
18,7 16,7 |
4,2
11,4 2,6 |
1,6
4,4 2,4 |
9,3
14,4 - |
12,5
4,6 - |
1,6
0,8 - |
4,7
0 - |
44,4
40,5 23,0 |
35,4
27,0 19,0 |
0,7
14,2 2,8 |
2,1
8,7 2,7 |
a CTCAE -luokitus (Common Terminology Criteria for Adverse Events (CTCAE)), versio 3.0
b CTCAE -luokitus (Common Terminology Criteria for Adverse Events (CTCAE)), versio 4.0
* International normalized ratio
Paras tukihoito = BCS (Best Supportive Care)
Verrattuna maailmanlaajuiseen vaiheen III kolorektaalisyöpätutkimukseen (CORRECT), johon osallistui pääasiassa (~80 %) kaukaasialaisia potilaita, Stivarga-hoitoa saaneilla potilailla havaittiin enemmän maksaentsyymien pitoisuuksien nousua Aasiassa tehdyssä vaiheen III kolorektaalisyöpätutkimuksessa (CONCUR), johon osallistui pääasiassa (> 90 %) itäaasialaisia potilaita.
Taulukko 4a: Levinnyttä kolorektaalisyöpää sairastavilla potilailla tehdyssä, lumelääkekontrolloidussa vaiheen III tutkimuksessa raportoidut hoidosta aiheutuvat poikkeavat maksan entsyymikokeiden tulokset aasialaisilla potilailla (CONCUR)
| Laboratorioparametri, ( % tutkituista näytteistä) | Stivarga ja BSC§ (N=136) | Lumelääke ja BSC§ (N=68) | ||||
| Kaikki asteet* | 3. aste* | 4. aste* | Kaikki asteet* | 3. aste* | 4. aste* | |
| Bilirubiinin nousu | 66,7 | 7,4 | 4,4 | 32,8 | 4,5 | 0,0 |
| ASAT-arvon kohoaminen | 69,6 | 10,4 | 0,7 | 47,8 | 3,0 | 0,0 |
| ALAT-arvon kohoaminen | 54,1 | 8,9 | 0,0 | 29,9 | 1,5 | 0,0 |
§ Paras tukihoito (Best Supportive Care)
* CTCAE-luokitus (Common Terminology Criteria for Adverse Events), versio 4.0
Näissä lumelääkekontrolloidussa vaiheen III kilpirauhasen toimintaa stimuloivalla hormonilla (TSH) tehdyissä testeissä ilmeni yleisesti ottaen, että TSH oli lähtötason jälkeen normaaliarvojen ylärajaa suurempi (> ULN) 34,6 %:lla Stivarga-valmisteella hoidetuista potilaista ja 17,2 %:lla lumelääkettä saaneista potilaista. Lähtötason jälkeen > 4 kertaa normaaliarvojen ylärajaa suurempi (> ULN) TSH raportoitiin 6,5 %:lla Stivarga-valmisteella hoidetuista potilaista ja 1,3 %:lla lumelääkettä saaneista potilaista. Lähtötason jälkeen normaaliarvojen alarajan alapuolella oleva (< LLN) vapaan trijodityroniinin (FT3) pitoisuus raportoitiin 29,2 %:lla Stivarga-valmisteella hoidetuista potilaista ja 20,4 %:lla lumelääkettä saaneista potilaista. Lähtötason jälkeen normaaliarvojen alarajan alapuolella (< LLN) oleva vapaan tyroksiinin (FT4) pitoisuus ilmoitettiin 8,1 %:lla Stivarga-valmisteella hoidetuista potilaista ja 5,6 %:lla lumelääkettä saaneista potilaista. Kaiken kaikkiaan 4,6 % Stivarga-valmisteella hoidetuista potilaista kehittyi hypotyreoosi, joka edellytti hormonikorvaushoitoa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurin kliinisesti tutkittu Stivarga-annos oli 220 mg vuorokaudessa. Tämän annoksen yhteydessä useimmiten todetut haittavaikutukset olivat dermatologiset tapahtumat, dysfonia, ripuli, limakalvotulehdus, suun kuivuminen, vähentynyt ruokahalu, hypertensio ja uupumus.
Stivarga-valmisteen yliannostukselle ei ole erityistä vastalääkettä. Jos epäillään yliannosta, Stivarga-hoito keskeytetään viipymättä, terveydenhoitoalan ammattilainen aloittaa hoitotoimenpiteet ja potilasta tarkkaillaan, kunnes hänen tilansa vakiintuu kliinisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EX05
(Vaikutusmekanismi ja farmakodynaamiset vaikutukset
Regorafenibi on suun kautta otettava kasvaimen aktiivisuutta vähentävä aine, joka estää voimakkaasti proteiinikinaaseja, mukaan lukien kasvaimen angiogeneesiin (VEGFR1, -2, -3, TIE2), onkogeneesiin (KIT, RET, RAF-1, BRAF, BRAFV600E), metastaasiin (VEGFR3, PDGFR, FGFR) ja kasvaimen immuniteettiin (CSF1R) liittyviä kinaaseja. Regorafenibi inhiboi erityisesti mutatoitunutta KIT:iä, joka on merkittävä onkogeeninen tekijä ruoansulatuskanavan stroomakasvaimissa ja siten se estää kasvainsolujen proliferaatiota. Prekliinisissä tutkimuksissa regorafenibilla todettiin voimakas kasvaimen kasvua hidastava vaikutus lukuisissa kasvaintyypeissä mukaan lukien kolorektaali-, ruoansulatuskanavan stromaaliset ja hepatosellulaariset kasvaintyypit. Vaikutus välittyy todennäköisesti sen antiangiogeenisten ja antiproliferatiivisten vaikutusten kautta. Lisäksi regorafenibi laski kasvaimeen liittyvien makrofagien määrää ja regorafenibilla on todettu antimetastaattisia vaikutuksia in vivo. Tärkeillä ihmisen aineenvaihduntatuotteilla (M-2 ja M-5) havaittiin samanlainen teho regorafenibiin verrattuna in vitro- ja in vivo -malleissa.
Kliininen teho ja turvallisuus
Metastaattinen kolorektaalisyöpä (CRC)
Stivarga-valmisteen kliinistä tehoa ja turvallisuutta on arvioitu kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa vaiheen III monikeskustutkimuksessa (CORRECT) levinnyttä kolorektaalisyöpää sairastavilla potilailla, joiden sairaus on edennyt standardihoidon jälkeen.
Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika (overall survival, OS). Toissijaiset päätetapahtumat olivat taudin etenemisvapaa elossaoloaika (progression-free survival, PFS), objektiivinen kasvaimen hoitovaste (objective tumor response rate, ORR) ja taudin hallinta (disease control rate, DCR).
Yhteensä 760 potilasta satunnaistettiin 2:1 saamaan 160 mg regorafenibia (neljä 40 mg regorafenibia sisältävää Stivarga-tablettia) suun kautta kerran päivässä (n=505) ja parasta tukihoitoa (Best Supportive Care, BSC) tai sopivaa lumelääkettä (n=255) ja parasta tukihoitoa 3 hoitoviikon ajan, jota seurasi yksi hoitovapaa viikko. Keskimääräinen regorafenibin vuorokausiannos oli 147 mg.
Potilaat saivat hoitoa sairauden etenemiseen tai kohtuuttoman toksisuuden ilmenemiseen asti. Ennalta suunniteltu tehon välianalyysi suoritettiin 432 kuolemantapauksen jälkeen. Tutkimus oli sokkouttamaton sen jälkeen, kun tämä suunniteltu kokonaiselossaoloajan välianalyysi oli ylittänyt ennalta määritetyn tehorajan.
760 satunnaistetun potilaan iän mediaani oli 61 vuotta. 61 % oli miehiä, 78 % oli kaukaasialaisia, ja kaikkien potilaiden ECOG-suorituskykyluokka oli lähtötasolla 0 tai 1. Suorituskykyluokka (PS) ≥ 2 ilmoitettiin Stivarga-hoidon aikana 11,4 %:lla potilaista. Hoidon keston mediaani ja vuorokausiannos, samoin kuin annosmuutosten nopeus ja annoksen pienentäminen olivat samanlaisia kuin niillä lumelääkettä saaneilla (8,3 %) potilailla, joiden suorituskykyluokaksi oli ilmoitettu ≥ 2. Suurin osa potilaista, joiden suorituskykyluokka oli ≥ 2, keskeyttivät hoidon taudin edetessä. Tauti sijaitsi ensisijassa paksusuolessa (65 %), peräsuolessa (29 %) tai kummassakin (6 %). KRAS-mutaatio raportoitiin 57 %:lla potilaista tutkimuksen alkaessa.
Useimmat potilaat (52 %) olivat saaneet aiemmin enintään 3 hoitolinjaa levinneeseen tautiin. Aikaisempia hoitoja olivat mm. fluoropyrimidiinipohjainen kemoterapia, anti-VEGF-hoito ja jos potilaalla oli KRAS-villityypin kasvain, anti-EGFR-hoito.
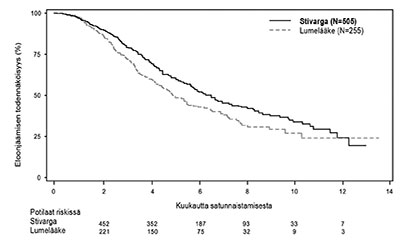
Stivarga-valmisteen ja parhaan tukihoidon yhdistäminen johti huomattavasti pidempään eloonjäämisaikaan verrattuna lumelääkkeen ja parhaan tukihoidon yhdistelmään riskisuhteen ollessa 0,774 (p=0,005178 ositettu log-rank -testi) ja kokonaiselossaoloajan mediaanin ollessa 6,4 kuukautta vs. 5,0 kuukautta [95 % CI 0,636, 0,942] (ks. taulukko 5 ja kuva 1). Taudin etenemisvapaa elossaoloaika oli huomattavasti pidempi Stivarga-valmistetta ja parasta tukihoitoa saaneilla potilailla (riskisuhde: 0,494, p<0,000001, ks. taulukko 5). Vaste (täydellinen vaste tai osittainen vaste) oli Stivarga-valmisteella hoidetuilla potilailla 1 % ja lumelääkkeellä hoidetuilla potilailla 0,4 % % (p=0,188432, 1-puolinen). Taudin hallintaprosentti (disease control rate, DCR) (täydellinen vaste tai osittainen vaste tai stabiili tauti) oli merkitsevästi suurempi Stivarga-valmisteella hoidetuilla potilailla (41,0 % vs. 14,9 %, p<0,000001, 1-puolinen).
Taulukko 5: CORRECT-tutkimuksen tehoa kuvaavat tulokset
| Tehon parametri | Riskisuhde* (95 % CI) | P-arvo (yksipuolinen) | Mediaani (95 % CI) | |
| Stivarga ja BSC§ (N=505) | Lumelääke ja BSC§ (N=255) | |||
| Kokonaiselossaoloaika (OS) | 0,774 (0,636, 0,942) | 0,005178 | 6,4 kuukautta (5,9, 7,3) | 5,0 kuukautta (4,4, 5,8) |
| Taudin etenemisvapaa elossaoloaika (PFS)** | 0,494 (0,419, 0,582) | < 0,000001 | 1,9 kuukautta (1,9, 2,1) | 1,7 kuukautta (1,7, 1,7) |
§ Paras tukihoito (Best Supportive Care)
* Riskisuhde < 1 Stivarga-valmisteen eduksi
** perustuu tutkijan arvioon kasvaimen vasteesta
Kuva 1: Kaplan-Meierin käyrä kokonaiselossaoloajasta (OS)
Kokonaiselossaoloajan ja etenemisvapaan elossaoloajan alaryhmäanalyysit iän (<65; ≥65), sukupuolen, ECOG-suorituskykyluokan (ECOG PS), taudin ensisijaisen sijaintipaikan, ajan ensimmäisestä diagnoosista levinneeseen tautiin, aikaisempien syöpälääkitysten, aikaisempien levinneen taudin hoitolinjojen ja KRAS mutaatiostatuksen mukaan osoittivat hoitovaikutuksen suosivan regorafenibia verrattuna lumelääkkeeseen.
Edeltävän KRAS mutaatiostatuksen alaryhmäanalyysi osoitti kokonaiselossaoloajan olevan pidempi regorafenibilla kuin lumelääkkeellä potilailla, joilla oli KRAS villityypin kasvain. Potilailla, joilla oli KRAS mutaatio kasvaimessa, elossaoloajan pitenemisen raportoitiin olevan numeerisesti vähäisempi. Riippumatta potilaan KRAS mutaatiostatuksesta, etenemisvapaan elossaoloajan hoitovaste oli parempi regorafenibilla kuin lumelääkkeellä. Kokonaiselossaoloajan riskisuhde (95% luottamusväli) oli 0,653 (0,476-0,895) potilailla, joilla oli KRAS villityypin kasvain ja 0,867 (0,670-1,123) potilailla, joiden kasvaimessa oli KRAS mutaatio, ilman että osoitettiin olevan eroa hoitovasteessa (ei merkitsevä yhteisvaikutustesti). Etenemisvapaan elossaoloajan riskisuhde (95% luottamusväli) oli 0,475 (0,362-0,623) potilailla, joilla oli KRAS villityypin kasvain ja 0,525 (0,425-0,649) potilailla, joilla oli KRAS mutaatio kasvaimessa.
Stivarga-valmisteen tehoa ja turvallisuutta arvioitiin toisessa vaiheen III kansainvälisessä satunnaistetussa kaksoissokkoutetussa lumelääkekontrolloidussa monikeskustutkimuksessa 204:lla edeltävää hoitoa saaneella, aasialaisella potilaalla (> 90 % itäaasialaisia), joilla oli levinnyt kolorektaalisyöpä, joka oli edennyt fluoropyrimidiinipohjaisen kemoterapian epäonnistuttua. Vain 59,5 % CONCUR-tutkimukseen osallistuneista potilaista oli saanut aiemmin VEGF-kasvutekijöihin tai EGFR-reseptoreihin kohdistunutta lääkehoitoa. Ensisijainen tehon päätetapahtuma oli kokonaiselossaoloaika (OS). Stivarga-valmisteen ja parhaan tukihoidon (BSC) yhdistäminen johti huomattavasti pidempään eloonjäämisaikaan verrattuna lumelääkkeen ja parhaan tukihoidon yhdistelmään riskisuhteen ollessa 0,550 (p = 0,000159 ositettu log rank -testi) ja kokonaiselossaoloajan mediaanin ollessa 8,8 kuukautta vs. 6,3 kuukautta [95 % CI 0,395; 0,765]. Myös taudin etenemisvapaa elossaoloaika (PFS) oli huomattavasti pidempi Stivarga-valmistetta ja parasta tukihoitoa saaneilla potilailla (riskisuhde: 0,311; p<0,000001), PFS 3,2 kuukautta (Stivarga-hoito) vs. 1,7 kuukautta (lumelääkehoito). Stivargan ja parhaan tukihoidon turvallisuusprofiili oli CONCUR-tutkimuksessa johdonmukainen CORRECT-tutkimuksessa havaitun turvallisuusprofiilin kanssa.
Ruoansulatuskanavan stroomakasvaimet (GIST)
Stivarga-valmisteen kliinistä tehoa ja turvallisuutta on arvioitu kansainvälisessä satunnaistetussa kaksoissokkoutetussa lumelääkekontrolloidussa vaiheen III monikeskustutkimuksessa (GRID) potilailla, joilla on ruoansulatuskanavan stroomakasvain (GIST) ja joita oli aiemmin hoidettu kahdella tyrosiinikinaasin estäjällä (imatinibi ja sunitinibi).
Ensisijaisen päätetapahtuman eli etenemisvapaan elossaoloajan (PFS) analyysi suoritettiin 144 PFS-tapahtuman jälkeen (keskitetty sokkoutettu analyysi). Myös toissijaisia päätetapahtumia, mukaan lukien aika taudin etenemiseen (time to progression, TTP) ja kokonaiselossaoloaika (OS) (välianalyysi), analysoitiin.
Kaiken kaikkiaan 199 GIST-potilasta satunnaistettiin suhteessa 2:1 saamaan 3 viikon hoidon ajan joko 160 mg regorafenibia suun kautta kerran päivässä ja parasta tukihoitoa (BSC; n=133) tai lumelääkettä ja parasta tukihoitoa (n=66). Tämän jälkeen seurasi 1 viikko ilman hoitoa. Potilaan saama keskimääräinen päivittäinen regorafenibiannos oli 140 mg.
Potilaat jatkoivat hoitoa sairauden etenemiseen tai kohtuuttoman toksisuuden ilmenemiseen asti. Lumelääkeryhmän potilaille, joilla todettiin sairauden eteneminen, tarjottiin mahdollisuutta avoimeen regorafenibihoitoon (cross-over -mahdollisuus). Regorafenibiryhmän potilaille, joilla ilmeni taudin eteneminen ja joille tutkijan mielestä regorafenibihoidosta oli kliinistä hyötyä, tarjottiin mahdollisuus jatkaa avointa regorafenibihoitoa.
199 satunnaistetun potilaan keski-ikä oli 58 vuotta, 64 % oli miehiä, 68 % oli kaukaasialaisia ja lähtötilanteessa kaikkien potilaiden ECOG-suorituskykyluokka oli 0 tai 1. Kokonaismediaaniaika taudin viimeisimmästä etenemisestä tai relapsista satunnaistamiseen oli 6 viikkoa.
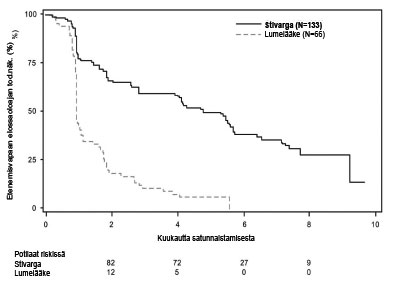
Regorafenibilla ja parhaalla tukihoidolla (BSC) saavutettiin merkitsevästi pitempi etenemisvapaa elossaoloaika (PFS) verrattuna lumelääkkeeseen ja parhaaseen tukihoitoon (BSC) riskisuhteen ollessa 0,268 [95 % CI 0,185; 0,388] ja PFS:n mediaanin ollessa 4,8 kuukautta vs. 0,9 kuukautta (p < 0,000001). Taudin etenemisen tai kuoleman suhteellinen riski väheni noin 73,2 % regorafenibilla hoidetuilla potilailla verrattuna lumelääkkeellä hoidettuihin potilaisiin (ks. taulukko 7, kuva 2). Etenemisvapaan elossaoloajan (PFS) suureneminen oli johdonmukainen riippumatta iästä, sukupuolesta, maantieteellisestä alueesta, aikaisemmista hoitolinjoista tai ECOG-suorituskykyluokasta.
Aika taudin etenemiseen (TTP) oli merkitsevästi pitempi regorafenibia ja parasta tukihoitoa (BSC) saaneilla potilailla kuin potilailla, jotka saivat lumelääkettä ja parasta tukihoitoa (BSC) riskisuhteen ollessa 0,248 [95 % CI 0,170; 0,364] ja mediaanin TTP:n ollessa 5,4 kuukautta vs. 0,9 kuukautta (p < 0,000001) (ks. taulukko 7).
Kokonaiselossaoloaika-analyysin riskisuhde (HR) oli 0,772 (95 % CI, 0,423; 1,408; p = 0,199); mediaania kokonaiselossaoloaikaa ei saavutettu kummassakaan tutkimushaarassa). Taudin etenemisen jälkeen 85 % lumelääkeryhmän potilaista siirtyi regorafenibihoitoon (ks. taulukko 7, kuva 3).
Taulukko 6: GRID-tutkimuksen tehoa kuvaavat tulokset
| Tehon parametri | Riskisuhde* (95 % CI) | P-arvo (yksipuolinen) | Mediaani (95 % CI) | |
| Stivarga ja BSC§ (N=133) | Lumelääke ja BSC§ (N=66) | |||
| Etenemisvapaa elossaoloaika (PFS) | 0,268 (0,185; 0,388) | < 0,000001 | 4,8 kuukautta (4,0; 5,7) | 0,9 kuukautta (0,9; 1,1) |
| Aika taudin etenemiseen (TTP) | 0,248 (0,170;0,364) | < 0,000001 | 5,4 kuukautta (4,1; 5,7) | 0,9 kuukautta (0,9; 1,1) |
| Kokonais-elossaoloaika (OS) | 0,772 (0,423; 1,408) | 0,199 | ES** | ES** |
§ Paras tukihoito (Best Supportive Care)
* Riskisuhde < 1 Stivarga-valmisteen eduksi
** ES: ei saavutettu
Kuva 2: Kaplan-Meierin käyrät etenemisvapaasta elossaoloajasta (PFS)
Kuva 3: Kaplan-Meierin käyrät kokonaiselossaoloajasta (OS)
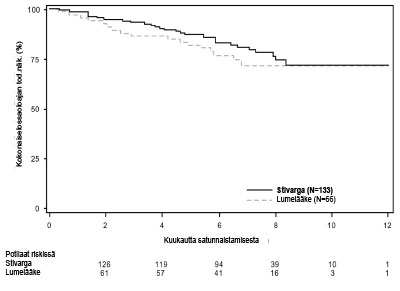
Lisäksi 56 potilasta, joita hoidettiin lumelääkkeellä ja parhaalla tukihoidolla (BSC) sai avointa Stivarga-hoitoa taudin etenemisen jälkeen ja kaikkiaan 41 potilasta, joita hoidettiin Stivarga-valmisteella ja parhaalla tukihoidolla (BSC) jatkoi Stivarga-hoidossa taudin etenemisen jälkeen. Mediaanit toissijaiset etenemisvapaat elossaoloajat (PFS) (mitattuna tutkijan arvioinnin mukaan) olivat 5,0 ja 4,5 kuukautta.
Hepatosellulaarinen karsinooma (HCC)
Stivarga-valmisteen kliinistä tehoa ja turvallisuutta on arvioitu kansainvälisessä, satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa vaiheen III monikeskustutkimuksessa (RESOURCE) hepatosellulaarista karsinoomaa sairastavilla potilailla, joita oli aiemmin hoidettu sorafenibilla.
Tehon ensisijainen päätetapahtuma oli kokonaiselossaoloaika (overall survival, OS). Toissijaiset päätetapahtumat olivat taudin etenemisvapaa elossaoloaika (progression-free survival, PFS), aika taudin etenemiseen (time to progression, TTP), objektiivinen kasvaimen hoitovaste ja taudin hallinta.
Yhteensä 573 hepatosellulaarista karsinoomaa sairastavaa potilasta satunnaistettiin 2:1 saamaan joko 160 mg regorafenibia suun kautta kerran päivässä (n=379) ja parasta tukihoitoa (Best Supportive Care, BSC) tai vastaavaa lumelääkettä (n=194) ja parasta tukihoitoa 3 hoitoviikon ajan, minkä jälkeen seurasi yksi hoitovapaa viikko. Keskimääräinen regorafenibin vuorokausiannos oli 144 mg. Potilaat voivat osallistua tutkimukseen, jos heillä sorfenibihoidon aikana todettiin radiologisesti sairauden etenemistä ja maksantoimintaluokitus oli Child-Pugh A. Potilaat, jotka pysyvästi keskeyttivät sorafenibihoidon siihen liittyvän toksisuuden vuoksi tai jotka sietivät alle 400 mg sorafenibia vuorokaudessa ennen hoidon keskeyttämistä, rajattiin pois. Potilaat satunnaistettiin 10 viikon kuluessa viimeisen sorafenibihoidon jälkeen ja he jatkoivat Stivarga-hoitoa siihen asti kunnes kliinisesti tai radiologisesti todettiin taudin etenemistä tai toksisia vaikutuksia, joita ei voitu hyväksyä. Potilaat saattoivat kuitenkin jatkaa Stivarga-hoitoa taudin etenemisen jälkeen tutkijan harkinnan mukaan
Potilaiden taustatiedot ja taudin lähtötason ominaisuudet olivat vertailukelpoisia Stivarga-valmistetta ja lumelääkettä saavissa ryhmissä. Ne esitetään kaikkien 573 satunnaistetun potilaan osalta alla:
- keskimääräinen ikä: 63 vuotta
- miehiä: 88 %
- kaukasialaisia: 36 %, aasialaisia: 41 %
- ECOG-suorituskykyluokka 0: 66 % tai ECOG-suorituskykyluokka 1: 34 %
- Child-Pugh A: 98 %, Child-Pugh B: 2 %
- HCC:n etiologia: hepatiitti-B (38 %), hepatiitti-C (21 %), alkoholiin liittymätön rasvamaksatulehdus (7 %)
- Potilaista 19 %:lla ei ollut makroskooppista vaskulaarista invaasiota tai kasvainta maksan ulkopuolella
- Barcelona Clinic Liver Cancer (BCLC) aste B: 13 %; BCLC aste C: 87 %
- lokoregionaalinen toimenpide: transarteriaalinen kemoembolisaatio tai kemoinfuusio: 61 %
- sädehoito ennen regorafenibihoitoa: 15 %
- sorafenibihoidon keskimääräinen pituus: 7,8 kuukautta
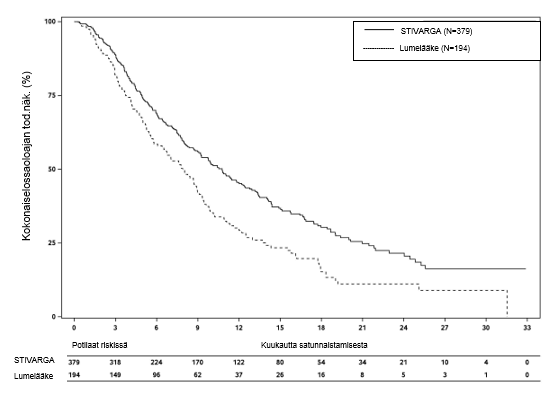
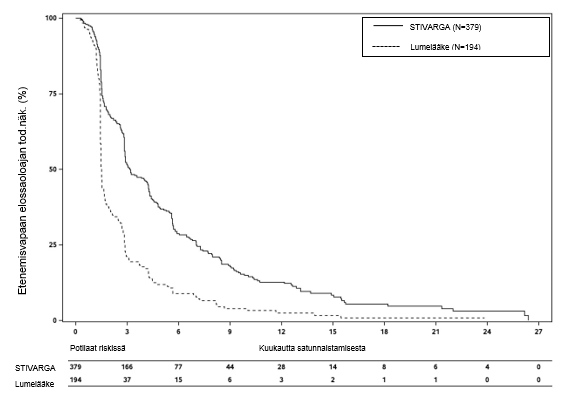
Kokonaiseloaika oli merkitsevästi pitempi Stivarga-valmistetta ja parasta tukihoitoa (BSC) saaneilla potilailla verrattuna potilaisiin, jotka saivat lumelääkettä ja parasta tukihoitoa (BSC). Riskisuhde oli 0,624 [95 % CI 0,498; 0,782], p=0,000017 ositettu log rank -testi ja mediaanin kokonaiseloaika oli 10,6 kuukautta vs.7,8 kuukautta (ks. taulukko 7 ja kuva 4).
Taulukko 7: RESORCE-tutkimuksen tehon tulokset
| Tehon parametri | Riskisuhde* (95% CI) | P‑arvo (yksipuolinen) | Mediaani (95% CI) | |
Stivarga ja BSC§ (N=379) | Lumelääke ja BSC§ (N=194) | |||
Kokonais-elossaoloaika (OS)
| 0,624 (0,498; 0,782) | 0,000017 | 10,6 kuukautta (9.1; 12,1) | 7,8 kuukautta (6,3; 8,8) |
Etenemisvapaa elossaoloaika (PFS) **
| 0,453 (0,369; 0,555) | <0,000001 | 3,1 kuukautta (2,8; 4;2) | 1,5 kuukautta (1,4; 1,6) |
Aika taudin etenemiseen (TTP) ** | 0,439 (0,355, 0,542) | <0,000001 | 3,2 kuukautta (2,9; 4;2) | 1,5 kuukautta (1,4; 1,6) |
Prosenttiosuus
| ||||
| Objektiivinen hoitovaste (ORR)**# | Ei oleellinen | 0,003650 | 11 % | 4 % |
| Taudin hallinta (DCR)**# | Ei oleellinen | <0,000001 | 65 % | 36 % |
§ Paras tukihoito (Best Supportive Care)
* Riskisuhde < 1 Stivarga-valmisteen eduksi
** perustuu tutkijan arvioon kasvaimen hoitovasteeseen RECIST-kriteerein
# Hoitovaste (disease control rate, DCR) (täydellinen tai osittainen vaste), taudin hallinta (täydellinen vaste tai osittainen vaste tai stabiili tauti 6 viikon ajan)
Kuva 4: Kaplan-Meierin käyrät kokonaiselossaoloajasta (OS)
Kuva 5: Kaplan-Meierin käyrät etenemisvapaasta elossaoloajasta (PFS) (mRECIST)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Stivarga-valmisteen käytöstä paksusuolen ja peräsuolen adenokarsinooman,ruoansulatuskanavan stroomakasvainten ja hepatosellulaarisen karsinooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa / ohjeet käytöstä pediatristen potilaiden hoidossa).
Satunnaistettu, kontrolloitu avoin monikeskustutkimus (FaR-RMS-tutkimuksen CT3-osa) arvioi regorafenibin turvallisuutta ja tehoa yhdessä vinkristiinin ja irinotekaanin (VIRR) kanssa verrattuna tavanomaiseen hoitoon vinkristiinillä, irinotekaanilla ja temotsolomidilla (VIRT) potilailla, joilla oli rabdomyosarkooman ensimmäinen tai myöhempi relapsi.
Kun ensimmäisen hyödyttömyysvälianalyysin aikana arvioitiin 103 potilaan tiedot, mukaan lukien 84 pediatrista potilasta, jotka olivat iältään 6 kuukaudesta alle 18 vuoteen, tutkimuksen rekrytointi päätettiin ilman ensisijaisen päätetapahtuman, yhden vuoden tautitapahtumavapaan elossaoloajan (EFS), muodollista arviointia. Hyödyttömyysraja saavutettiin (EFS HR > 1) viitaten alhaiseen todennäköisyyteen sille, että VIRR-yhdistelmä saavuttaisi protokollan määrittämän 15 %:n paremmuuden VIRT-yhdistelmään verrattuna.
Farmakokinetiikka
Imeytyminen
Regorafenibi saavuttaa keskimääräisen plasman huipputason noin 2,5 mg/l noin 3−4 tunnin kuluttua suun kautta otetun 160 mg regorafenibin kerta-annoksen jälkeen, joka annetaan neljänä 40 mg sisältävänä tablettina. 60 mg tai 100 mg kerta-annosten jälkeen tablettien keskimääräinen suhteellinen hyötyosuus oli vastaavasti 69 % ja 83 % suun kautta otettuun liuokseen verrattuna.
Regorafenibin ja sen tärkeimpien farmakologisesti aktiivisten aineenvaihduntatuotteiden (M-2 ja M-5) pitoisuudet olivat suurimmillaan vähärasvaisen (kevyen) aamiaisen jälkeen annettuna verrattaessa joko runsaasti rasvaa sisältävään aamiaiseen tai paastotilanteeseen. Regorafenibin altistuminen lisääntyi 48 %, kun se annettiin runsasrasvaisen aamiaisen yhteydessä ja 36 %, kun se annettiin vähärasvaisen aamiaisen yhteydessä verrattaessa paastotilanteeseen. Aineenvaihduntatuotteiden M-2 (N-oksidi) ja M-5 (N-oksidi ja N-desmetyyli) altistuminen on suurempi, kun regorafenibi annetaan vähärasvaisen aamiaisen yhteydessä verrattuna paastotilanteeseen, ja pienempi, kun se annetaan runsasrasvaisen aterian yhteydessä verrattuna paastotilanteeseen.
Jakautuminen
Regorafenibin ja sen tärkeimpien verenkierrossa esiintyvien aineenvaihduntatuotteiden pitoisuus/aika-profiilit plasmassa osoittivat useita huippuarvoja 24 tunnin annosvälillä, mikä liittyy enterohepaattiseen kiertoon. Regorafenibin sitoutuminen ihmisen plasman proteiineihin in vitro on voimakasta (99,5 %). M-2:n ja M-5:n sitoutuminen proteiineihin in vitro on suurempi (99,8 % ja 99,95 %, tässä järjestyksessä) kuin regorafenibin sitoutuminen. Aineenvaihduntatuotteet M-2 ja M-5 ovat P-gp:n heikkoja substraatteja. Aineenvaihduntatuote M-5 on heikko BCRP-substraatti.
Biotransformaatio
Regorafenibin aineenvaihdunta tapahtuu pääsääntöisesti maksassa. Se läpikäy oksidatiivisen aineenvaihdunnan, jonka välittää CYP3A4, sekä glukuronidaation, jonka välittää UGT1A9. Plasmassa on identifioitu regorafenibille kaksi pääaineenvaihduntatuotetta ja kuusi muuta aineenvaihduntatuotetta. Regorafenibin verenkierrossa ilmenevät pääaineenvaihduntatuotteet ovat M-2 (N-oksidi) ja M-5 (N-oksidi ja N-desmetyyli), jotka ovat farmakologisesti aktiivisia ja joiden pitoisuudet vastaavat regorafenibia vakaassa tilassa. M-2 metaboloituu edelleen CYP3A4:n välittämän oksidatiivisen aineenvaihdunnan kautta sekä UGT1A9:n välittämän glukuronidaation kautta.
Aineenvaihduntatuotteet saattavat vähentyä tai hydrolysoitua maha-suolikanavassa mikrobiflooran vaikutuksesta, mikä mahdollistaa konjugoimattoman vaikuttavan aineen ja aineenvaihduntatuotteiden takaisinimeytymisen (enterohepaattinen kierto).
Eliminaatio
Suun kautta annetun regorafenibin ja sen aineenvaihduntatuotteen M-2 keskimääräinen eliminaation puoliintumisaika plasmassa oli 20−30 tuntia eri tutkimuksissa. Aineenvaihduntatuotteen M-5 keskimääräinen eliminaation puoliintumisaika on noin 60 tuntia (vaihteluväli 40−100 tuntia).
Noin 90 % radioaktiivisesta annoksesta havaittiin 12 vuorokauden kuluessa annostelusta: noin 71 % annoksesta erittyi ulosteisiin (47 % kanta-aineena, 24 % aineenvaihduntatuotteina) ja noin 19 % annoksesta erittyi virtsaan glukuronideina. Glukuronidien erittyminen virtsaan väheni vakaassa tilassa alle 10 %. Ulosteissa löydetty kanta-aine saattaa johtua glukuronidien intestinaalisesta hajoamisesta tai aineenvaihduntatuote M-2:n (N-oksidin) vähenemisestä sekä imeytymättömästä regorafenibista.
Mikrobikasvusto saattaa pelkistää M-5:n M-4:ksi maha-suolikanavassa, jolloin M-4:n takaisinimeytyminen on mahdollista (enterohepaattinen kiertokulku). Lopuksi M-5 erittyy M-4:n kautta M-6:na (karboksyylihappona) ulosteeseen.
Lineaarisuus/ei-lineaarisuus
Regorafenibin systeeminen altistuminen vakaassa tilassa lisääntyy annosriippuvaisesti 60 mg saakka ja vähemmän kuin annosriippuvaisesti annoksen ollessa yli 60 mg. Regorafenibin kertyminen vakaassa tilassa johtaa noin 2 kertaiseen plasmapitoisuuden nousuun, mikä on yhdenmukainen eliminaation puoliintumisajan ja annosvälin kanssa. Vakaassa tilassa regorafenibin saavuttama keskimääräinen plasman huipputaso on noin 3,9 mg/l (8,1 mikromolaaria) suun kautta annetun 160 mg regorafenibiannoksen jälkeen, ja keskimääräisen plasmapitoisuuden huippu-minimisuhde on alle 2.
Molemmilla aineenvaihduntatuotteilla, sekä M-2:lla että M-5:lla, kertyminen on ei-lineaarista, mikä saattaa johtua enterohepaattisesta kierrätyksestä tai UGT1A9-reitin saturoitumisesta. Vaikka M-2:n ja M-5:n pitoisuudet plasmassa regorafenibin kerta-annoksen jälkeen ovat paljon alhaisempia kuin sen emoyhdisteen pitoisuudet, M-2:n ja M-5:n vakaan tilan plasmapitoisuudet ovat verrattavissa regorafenibin vastaaviin pitoisuuksiin.
Maksan vajaatoiminta
Regorafenibille ja sen aineenvaihduntatuotteille M-2 ja M-5 altistuminen on verrattavissa lievää maksan vajaatoimintaa (Child Pugh A) sairastavilla potilailla sekä normaalin maksantoiminnan omaavilla potilailla.
Kohtalaista maksan vajaatoimintaa (Child Pugh B) sairastavista potilaista saadut rajalliset tiedot osoittavat altistumisen olevan samanlainen verrattuna potilaisiin, joiden maksantoiminta on normaalia, 100 mg regorafenibin kerta-annoksen jälkeen. Tietoja potilaista, joilla on Child Pugh -luokan C (vaikea) maksan vajaatoiminta, ei ole. Regorafenibi eliminoituu pääasiassa maksan kautta ja altistuminen voi lisääntyä tässä potilasryhmässä.
Munuaisten vajaatoiminta
Saatavissa olevat kliiniset tiedot ja fysiologiaan perustuva farmakokineettinen mallinnus osoittavat samanlaista altistumista vakaassa tilassa regorafenibille ja sen metaboliiteille M-2 ja M-5 potilailla, jotka sairastavat lievää tai kohtalaista munuaisten vajaatoimintaa verrattuna normaalin munuaistoiminnan omaaviin potilaisiin. Vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla altistuminen regorafenibille on samanlaista verrattuna potilaisiin, joilla munuaisten toiminta on normaalia. Kun taas altistuminen metaboliiteille (M-2 ja M-5) pieneni noin 30 % vakaassa tilassa. Tämän ei katsota olevan kliinisesti merkityksellistä.
Regorafenibin farmakokinetiikkaa ei ole tutkittu loppuvaiheen munuaissairautta sairastavilla potilailla. Fysiologiaan perustuva farmakokineettinen mallinnus ei kuitenkaan ennusta olennaista altistumisen muutosta näillä potilailla.
Iäkkäät potilaat
Iällä ei ollut vaikutusta regorafenibin farmakokinetiikkaan tutkitulla ikävälillä (29−85 vuotta).
Sukupuoli
Sukupuoli ei vaikuta regorafenibin farmakokinetiikkaan.
Etnisten ryhmien väliset erot
Altistuminen regorafenibille eri aasialaisten väestöjen (kiinalaiset, japanilaiset, korealaiset) keskuudessa on samalla vaihteluvälillä kuin kaukaasialaisten keskuudessa.
Sydämen elektrofysiologia / pidentynyt QT-väli
QTc-aikaa pidentäviä vaikutuksia ei todettu 160 mg regorafenibin annon jälkeen vakaassa tilassa erityisessä QT-tutkimuksessa, joka suoritettiin syöpää sairastavilla mies- ja naispotilailla.
Prekliiniset tiedot turvallisuudesta
Systeeminen toksisuus
Kun oli annettu toistuvia annoksia hiirille, rotille ja koirille, haittavaikutuksia todettiin useissa elimissä, ensisijassa munuaisissa, maksassa, ruoansulatuskanavassa, kilpirauhasessa, sydämessä, lymfo-/hemopoieettisessa järjestelmässä, endokriinisessa järjestelmässä, sukupuolielimissä ja ihossa. Rotilla tehdyssä 26 viikon toistetun annoksen toksisuustutkimuksessa sydämen eteiskammioläppien paksuuntumisen esiintyvyyden havaittiin hieman lisääntyneen. Tämä saattaa johtua ikään liittyvän fysiologisen tapahtumaketjun kiihtymisestä. Näitä vaikutuksia ilmeni, kun eläinten altistuminen oli vähemmän tai yhtä paljon kuin otaksuttavissa oleva ihmisen altistuminen (perusteena AUC-vertailu). Muutokset hampaissa ja luissa ja haittavaikutukset sukupuolielimissä olivat selvempiä nuorilla ja kasvuikäisillä eläimillä samoin kuin nuorilla rotilla viitaten mahdolliseen riskiin lapsille ja nuorille.
Lisääntymis- ja kehitystoksisuus
Erityisiä tutkimuksia hedelmällisyydestä ei ole suoritettu. Kuitenkin on huomioitava regorafenibin mahdollinen heikentävä vaikutus miesten ja naisten lisääntymiseen, sillä kiveksissä, munasarjoissa ja kohdussa todettiin morfologisia muutoksia annettaessa rotille ja koirille toistuvia annoksia, kun altistuminen oli vähäisempää kuin otaksuttavissa oleva ihmisen altistuminen (perusteena AUC-vertailu). Todetut muutokset olivat ainoastaan osittain palautuvia.
Kaneilla todettiin regorafenibin vaikutus kohdunsisäiseen kehitykseen, kun altistuminen oli vähemmän kuin otaksuttavissa oleva ihmisen altistuminen (perusteena AUC-vertailu). Suurimpia muutoksia olivat virtsateiden, sydämen ja tärkeimpien verisuonien sekä luuston epämuodostumat.
Genotoksisuus ja karsinogeenisuus
Hiirillä suoritetuissa vakiotesteissä in vitro ja in vivo ei todettu merkkejä regorafenibin genotoksisesta potentiaalista.
Tutkimuksia regorafenibin karsinogeenisesta potentiaalista ei ole tehty.
Ympäristöön kohdistuvien riskien arviointi
Tutkimuksissa regorafenibin ympäristöön kohdistuvista riskeistä on osoitettu, että regorafenibilla on potentiaalia olla pysyvä, bioakkumulatiivinen ja toksinen ympäristölle ja voi aiheuttaa vaaraa pintavesistöille ja sedimenteille (ks. kohta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin: Mikrokiteinen selluloosa, kroskarmelloosinatrium, magnesiumstearaatti, povidoni (K 25), vedetön kolloidinen piidioksidi.
Kalvopäällyste: Punainen rautaoksidi (E172), keltainen rautaoksidi (E172), lesitiini (peräisin soijasta), makrogoli 3350, osittain hydrolysoitu polyvinyylialkoholi, talkki, titaanidioksidi (E171).
Apuaineet, joiden vaikutus tunnetaan
Yksi vuorokausiannos (160 mg) sisältää 2,438 mmol (56,06 mg) natriumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yksi vuorokausiannos (160 mg) sisältää 1,68 mg lesitiiniä (peräisin soijasta) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Avatussa pullossa lääkevalmisteen on osoitettu olevan stabiili 7 viikon ajan. Sen jälkeen lääke on hävitettävä.
Säilytys
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pidä pullo tiiviisti suljettuna.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
STIVARGA tabletti, kalvopäällysteinen
40 mg (L:ei) 3 x 28 kpl (2750,84 €)
PF-selosteen tieto
Valkoinen läpinäkymätön HDPE pullo, joka on suljettu tiivisteellä varustetulla PP/PP (polypropyleeni) -kierrekorkilla, ja jossa on kuivausaine (molekyyliseula).
Yksi pullo sisältää 28 kalvopäällysteistä tablettia.
Pakkauskoot: 84 (3 pulloa à 28 tablettia) kalvopäällysteistä tablettia sisältävä pakkaus.
Valmisteen kuvaus:
Vaaleanpunainen kalvopäällysteinen, soikionmuotoinen tabletti, jonka pituus on 16 mm ja leveys 7 mm ja jonka toisella puolella on merkintä "BAYER" ja toisella puolella "40".
Käyttö- ja käsittelyohjeet
Pidä kuivausaine pullossa.
Tämä lääkevalmiste voi aiheuttaa vaaraa ympäristölle (ks. kohta Prekliiniset tiedot turvallisuudesta). Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
STIVARGA tabletti, kalvopäällysteinen
40 mg 3 x 28 kpl
- Ylempi erityiskorvaus (100 %). Regorafenibi: Hepatosellulaarisen karsinooman hoito, metastasoituneen kolorektaalisyövän hoito, ei-leikattavissa olevan tai metastaattisen ruoansulatuskanavan stroomakasvaimen (GIST) hoito erityisin edellytyksin (176).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Regorafenibi: Maksasolukarsinooman, metastasoituneen kolorektaalisyövän, ei-leikattavissa olevan tai metastaattisen ruoansulatuskanavan stroomakasvaimen (GIST) hoito erityisin edellytyksin (369).
ATC-koodi
L01EX05
Valmisteyhteenvedon muuttamispäivämäärä
27.02.2026
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi