
FASENRA injektioneste, liuos, esitäytetty kynä 30 mg, injektioneste, liuos, esitäytetty ruisku 30 mg
Vaikuttavat aineet ja niiden määrät
Esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 30 mg benralitsumabia* 1 ml:ssa liuosta.
Esitäytetty kynä
Yksi esitäytetty kynä sisältää 30 mg benralitsumabia* 1 ml:ssa liuosta.
*Benralitsumabi on humanisoitu monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste), esitäytetty ruisku
Injektioneste, liuos (injektioneste), esitäytetty kynä (Fasenra Pen)
Kliiniset tiedot
Käyttöaiheet
Astma
Fasenra on tarkoitettu lisälääkkeeksi vaikeaa eosinofiilistä astmaa sairastavien aikuispotilaiden ylläpitohoitoon, kun suuriannoksisen inhaloitavan kortikosteroidin ja pitkävaikutteisen β-agonistin yhdistelmällä ei ole saavutettu riittävää hoitotasapainoa (ks. kohta Farmakodynamiikka).
Eosinofiilinen granulomatoottinen polyangiitti (EGPA)
Fasenra on tarkoitettu lisälääkkeeksi aikuispotilaille, joilla on uusiutunut tai vaikeahoitoinen eosinofiilinen granulomatoottinen polyangiitti (ks. kohta Farmakodynamiikka).
Hypereosinofiilinen oireyhtymä (HES)
Fasenra on tarkoitettu lisälääkkeeksi hypereosinofiilista oireyhtymää sairastaville aikuisille ja vähintään 12‑vuotiaille nuorille, jotka painavat vähintään 35 kg, kun riittävää hoitotasapainoa ei ole saavutettu eikä taudille ole löydetty ei-hematologista sekundaarista syytä (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus ja antotapa
Fasenra-hoidon saa aloittaa vain lääkäri, joka on perehtynyt niiden sairauksien diagnosointiin ja hoitoon, joihin benralitsumabi on tarkoitettu (ks. kohta Käyttöaiheet).
Kun potilaalle tai potilasta hoitavalle henkilölle on neuvottu oikea ihon alle pistämisen tekniikka ja kerrottu yliherkkyysreaktioiden merkeistä ja oireista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), hän tai häntä hoitava henkilö voi antaa Fasenra-valmisteen, jos potilaalla ei tiedetä aiemmin ilmenneen anafylaksiaa ja jos lääkäri katsoo sen olevan asianmukaista, ja potilaan vointia tarvittaessa seurataan. Valmisteen antamista itse harkitaan vain potilailla, jotka ovat jo aiemmin saaneet Fasenra-hoitoa.
Annostus
Fasenra on tarkoitettu pitkäaikaiseen hoitoon. Päätös hoidon jatkamisesta on tehtävä vähintään kerran vuodessa. Päätöksen on perustuttava taudin vaikeusasteeseen, siihen, miten hyvin tauti pysyy hallinnassa, ja veren eosinofiilipitoisuuksiin.
Astma
Benralitsumabin suositeltu annos on 30 mg injektiona ihon alle niin, että 3 ensimmäistä annosta annetaan 4 viikon välein ja sen jälkeen annosväli on 8 viikkoa.
Eosinofiilinen granulomatoottinen polyangiitti
Benralitsumabin suositeltu annos on 30 mg injektiona ihon alle 4 viikon välein.
Jos potilaalle kehittyy henkeä uhkaavia eosinofiilisen granulomatoottisen polyangiitin ilmenemismuotoja, hoidon jatkamisen tarve on arvioitava, koska Fasenra-valmistetta ei ole tutkittu tässä potilasryhmässä.
Hypereosinofiilinen oireyhtymä
Benralitsumabin suositeltu annos aikuisille ja vähintään 12‑vuotiaille nuorille, jotka painavat vähintään 35 kg, on 30 mg injektiona ihon alle 4 viikon välein.
Jos potilaalle kehittyy henkeä uhkaavia hypereosinofiilisen oireyhtymän ilmenemismuotoja, hoidon jatkamisen tarve on arvioitava, koska Fasenra-valmistetta ei ole tutkittu tässä potilasryhmässä.
Antamatta jäänyt annos
Jos injektio jää antamatta suunniteltuna päivänä, annostelua on jatkettava mahdollisimman pian lääkärin määräämällä annostusohjelmalla, kaksinkertaista annosta ei saa antaa.
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäillä potilailla (ks. kohta Farmakokinetiikka).
Munuaisten tai maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on munuaisten tai maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Fasenra-valmisteen turvallisuutta ja tehoa astmaa sairastavien, 6–17 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Saatavissa olevan 6–11‑vuotiaita lapsia koskevan vähäisen tiedon ja 12−17‑vuotiaita nuoria koskevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Fasenra-valmisteen turvallisuutta ja tehoa astmaa sairastavien, alle 6‑vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Fasenra-valmisteen turvallisuutta ja tehoa eosinofiilistä granulomatoottista polyangiittia sairastavien, alle 18‑vuotiaiden lasten ja nuorten hoidossa ei ole varmistettu.
Fasenra-valmisteen turvallisuutta ja tehoa hypereosinofiilista oireyhtymää sairastavien, alle 12‑vuotiaiden lasten hoidossa ei ole varmistettu. Fasenra-valmisteen turvallisuutta alle 35 kg painavien nuorten (12–17‑vuotiaiden) potilaiden hoidossa ei ole varmistettu. Tälle alaryhmälle ei voida antaa suosituksia annostuksesta.
Antotapa
Tämä lääkevalmiste annetaan injektiona ihon alle.
Fasenra annetaan injektiona reiteen tai vatsaan, vähintään 5 cm:n päähän navasta. Jos injektion antaa terveydenhuollon ammattilainen tai potilasta hoitava henkilö, se voidaan antaa myös olkavarteen. Injektiota ei saa antaa aristaville, punoittaville tai kovettuneille ihoalueille eikä alueille, joilla on mustelmia.
Esitäytetyn ruiskun ja esitäytetyn kynän yksityiskohtaiset anto-ohjeet on esitetty kohdassa ”Käyttöohje”.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Astman pahenemisvaiheet
Fasenra-valmistetta ei pidä käyttää astman akuuttien pahenemisvaiheiden hoitoon.
Potilaita on neuvottava ottamaan yhteys lääkäriin, jos astma ei ole hallinnassa tai pahenee hoidon aloittamisen jälkeen.
Kortikosteroidit
Fasenra-hoidon aloittamisen jälkeen kortikosteroidien käytön äkillinen lopettaminen ei ole suositeltavaa. Jos kortikosteroidiannosten pienentäminen katsotaan tarkoituksenmukaiseksi, se on tehtävä asteittain ja lääkärin valvonnassa.
Yliherkkyysreaktiot
Akuutteja systeemisiä reaktioita, mukaan lukien anafylaktisia reaktioita ja yliherkkyysreaktioita (kuten nokkosihottumaa, nokkosjäkälää ja ihottumaa), on ilmennyt benralitsumabin antamisen jälkeen (ks. kohta Haittavaikutukset). Nämä reaktiot saattavat ilmetä tuntien kuluessa antamisesta, mutta joissakin tapauksissa ne ilmaantuvat viiveellä (vuorokausien kuluessa).
Aiempi benralitsumabiin liittymätön anafylaksia saattaa olla Fasenra-valmisteen antamisen jälkeisen anafylaksian riskitekijä (ks. kohta Vasta-aiheet). Kliinisen hoitokäytännön mukaisesti potilaita on tarkkailtava riittävän pitkään Fasenra-valmisteen antamisen jälkeen.
Yliherkkyysreaktion ilmaantuessa Fasenra-hoito on keskeytettävä pysyvästi ja on aloitettava asianmukainen hoito.
Loisinfektiot (matoinfektiot)
Eosinofiilit saattavat osallistua immuunivasteen kehittymiseen joillekin matoinfektioille. Potilaat, joilla oli todettu matoinfektio, suljettiin pois kliinisistä tutkimuksista. Ei tiedetä, vaikuttaako benralitsumabi potilaan vasteeseen matoinfektioita vastaan.
Potilaan matoinfektio on hoidettava ennen benralitsumabihoidon aloittamista. Jos potilas saa infektion hoidon aikana eikä matolääkkeillä saada hoitovastetta, benralitsumabihoito tulisi keskeyttää, kunnes infektio on hävinnyt.
Elinten toimintaa tai henkeä uhkaava eosinofiilinen granulomatoottinen polyangiitti
Fasenra-valmistetta ei ole tutkittu potilailla, joilla eosinofiilisen granulomatoottisen polyangiitin aktiivinen ilmenemismuoto uhkaa potilaan elintoimintoja tai henkeä (ks. kohta Annostus ja antotapa).
Henkeä uhkaava hypereosinofiilinen oireyhtymä
Fasenra-valmistetta ei ole tutkittu potilailla, joilla hypereosinofiilisen oireyhtymän aktiivinen ilmenemismuoto uhkaa potilaan henkeä (ks. kohta Annostus ja antotapa).
Apuaine, jonka vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,06 mg polysorbaatti 20:tä per 30 mg:n (1 ml:n) annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Satunnaistetussa kaksoissokkoutetussa rinnakkaisryhmillä tehdyssä tutkimuksessa, johon osallistui 103 potilasta, joilla oli vaikea astma ja joiden ikä oli 12−21 vuotta, benralitsumabihoidolla ei näyttänyt olevan vaikutusta kausi-influenssarokotuksen aikaansaamaan humoraaliseen vasta-aineiden tuotantoon. Benralitsumabin ei odoteta vaikuttavan muiden samanaikaisesti annettujen lääkevalmisteiden farmakokinetiikkaan (ks. kohta Farmakokinetiikka).
Sytokromi P450 ‑entsyymit, effluksipumput ja proteiineja sitovat mekanismit eivät vaikuta benralitsumabin puhdistumaan. IL5Rα:n ilmentymisestä hepatosyyteissä ei ole näyttöä. Eosinofiilien häviäminen ei johda proinflammatoristen sytokiinien pitkäaikaisiin systeemisiin muutoksiin.
Raskaus ja imetys
Raskaus
Benralitsumabin käytöstä raskaana oleville naisille on vain vähän tietoja (alle 300 raskaudesta).
Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Benralitsumabin kaltaiset monoklonaaliset vasta-aineet siirtyvät raskauden edetessä istukan läpi lineaarisesti, joten sikiön mahdollinen altistuminen on todennäköisesti suurempi toisen ja kolmannen raskauskolmanneksen aikana.
Varmuuden vuoksi Fasenra-valmisteen käyttöä on suositeltavaa välttää raskauden aikana. Sen antamista raskaana oleville naisille voidaan harkita vain, jos hoidosta odotettavissa oleva hyöty äidille on suurempi kuin sikiölle mahdollisesti koituva riski.
Imetys
Ei tiedetä, erittyvätkö benralitsumabi tai sen metaboliitit ihmisillä äidinmaitoon tai koe-eläimillä maitoon. Imetettävään lapseen kohdistuvia riskejä ei voida sulkea pois.
On päätettävä, lopetetaanko imetys vai pidättäydytäänkö Fasenra-hoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ihmisten hedelmällisyyttä koskevia tietoja ei ole. Eläimillä tehdyissä tutkimuksissa ei ole todettu haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Fasenra-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Benralitsumabilla on samankaltainen turvallisuusprofiili astman, eosinofiilisen granulomatoottisen polyangiitin ja hypereosinofiilisen oireyhtymän hoidossa.
Yleisimmin ilmoitettuja haittavaikutuksia astman hoidossa olivat päänsärky (8 %) ja nielutulehdus (3 %). Päänsärky oli yleisimmin ilmoitettu haittavaikutus eosinofiilisen granulomatoottisen polyantiigin (17 %) ja hypereosinofiilisen oireyhtymän (16 %) hoidossa. Vaikeusasteeltaan erilaisia anafylaktisia reaktioita on ilmoitettu benralitsumabin käytön yhteydessä.
Haittavaikutustaulukko
Seuraavia haittavaikutuksia on raportoitu benralitsumabin käytön yhteydessä astmaa, eosinofiilistä granulomatoottista polyangiittia ja hypereosinofiilista oireyhtymää koskevien kliinisten tutkimusten aikana tai valmisteen markkinoilletulon jälkeen.
Haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä yleisyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutustaulukko
MedDRA:n elinjärjestelmä | Haittavaikutus | Esiintymistiheys |
Infektiot | Nielutulehdusa | Yleinen |
Immuunijärjestelmä | Yliherkkyysreaktiotb Anafylaktinen reaktio | Yleinen Tuntematon |
Hermosto | Päänsärkyc | Yleinen |
Yleisoireet ja antopaikassa | Kuume Pistoskohdan reaktiod | Yleinen |
a. Nielutulehdus, mukaan lukien: ”Nielutulehdus”, ”Bakteeriperäinen nielutulehdus”, ”Virusperäinen nielutulehdus” ja ”Streptokokin aiheuttama nielutulehdus”.
b. Yliherkkyysreaktiot, mukaan lukien: ”Nokkosihottuma”, ”Nokkosjäkälä” ja ”Ihottuma”. Esimerkkejä näihin liittyvistä ilmoitetuista oireista ja tietoja reaktion ilmaantumiseen kuluneesta ajasta on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
c. Hyvin yleinen eosinofiilistä granulomatoottista polyangiittia ja hypereosinofiilista oireyhtymää koskevissa kliinisissä tutkimuksissa.
d. Ks. kohta ”Valikoitujen haittavaikutusten kuvaus”.
Valikoitujen haittavaikutusten kuvaus
Pistoskohdan reaktiot
Lumekontrolloiduissa astmaa koskevissa tutkimuksissa pistoskohdan reaktioita (kuten kipua, punoitusta, kutinaa tai näppylöitä) ilmeni 2,2 %:lla suositelluilla annoksilla benralitsumabia saaneista potilaista ja 1,9 %:lla lumelääkettä saaneista potilaista. Nämä tapahtumat olivat luonteeltaan ohimeneviä.
Pitkän aikavälin turvallisuus
Astmaa sairastavilla tutkimusten 1, 2 ja 3 potilailla tehdyssä 56 viikon mittaisessa jatkotutkimuksessa (tutkimus 4) 842 potilasta sai Fasenra-valmistetta suositellulla annoksella ja jatkoi tutkimuksessa. Turvallisuusprofiili oli kokonaisuutena samanlainen kuin edellä kuvatuissa astmatutkimuksissa. Lisäksi astmaa sairastavilla edellisten tutkimusten potilailla tehdyssä avoimessa, turvallisuutta koskevassa jatkotutkimuksessa (tutkimus 5) 226 potilasta sai Fasenra-valmistetta suositellulla annoksella enintään 43 kuukauden ajan. Edellisten tutkimusten hoitojakson kanssa yhdistettynä seuranta-ajan mediaani on 3,4 vuotta (vaihteluväli 8,5 kuukautta – 5,3 vuotta). Turvallisuusprofiili tämän seurantajakson aikana vastasi Fasenra-valmisteen tunnettua turvallisuusprofiilia.
Pediatriset potilaat
Astma
Pediatrisista potilaista on vain vähän tietoja. Vaiheen 3 tutkimuksiin osallistui 108 iältään 12–17-vuotiasta, astmaa sairastavaa nuorta (tutkimus 1: n = 53, tutkimus 2: n = 55). Näistä potilaista 46 sai lumelääkettä ja 40 sai benralitsumabia 3 ensimmäistä annosta 4 viikon välein ja sen jälkeen 8 viikon välein ja 22 potilasta sai benralitsumabia 4 viikon välein. Tutkimuksiin 1 ja 2 osallistuneet nuoret 12–17-vuotiaat potilaat (n = 86) jatkoivat benralitsumabihoitoa tutkimuksessa 4 enintään 108 viikon ajan. Nuorilla potilailla havaittujen haittavaikutusten esiintymistiheys, tyyppi ja vaikeusaste olivat samanlaisia kuin aikuisilla.
Farmakokinetiikkaa ja farmakodynamiikkaa arvioitiin avoimessa, kontrolloimattomassa, 48 viikon mittaisessa tutkimuksessa pienellä joukolla pediatrisia potilaita (n = 28). Potilailla oli vaikea astma, joka ei ollut hoitotasapainossa. Tutkimuksessa 6-11-vuotiailla potilailla havaittu turvallisuusprofiili oli samanlainen kuin aikuisilla ja nuorilla potilailla (ks. kohta Annostus ja antotapa).
Hypereosinofiilinen oireyhtymä
Vaiheen 3 tutkimukseen osallistui 4 iältään 12–17-vuotiasta, hypereosinofiilista oireyhtymää sairastavaa nuorta. Näistä potilaista 3 sai benralitsumabia ja 1 sai lumelääkettä 4 viikon välein. Hypereosinofiilista oireyhtymää koskeneessa tutkimuksessa benralitsumabia saaneilla 3 nuorella ei tunnistettu uusia haittavaikutuksia verrattuna vahvistettuun turvallisuusprofiiliin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa eosinofilista astmaa sairastaville potilaille annettiin enintään 200 mg:n annoksia injektiona ihon alle. Näissä tutkimuksissa ei todettu näyttöä annosriippuvaisesta toksisuudesta.
Benralitsumabin yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksessa potilaalle on annettava asianmukaista tukihoitoa ja hänen tilaansa on seurattava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: obstruktiivisten hengitystiesairauksien lääkkeet, muut systeemisesti käytettävät obstruktiivisten hengitystiesairauksien lääkkeet, ATC-koodi: R03DX10
Vaikutusmekanismi
Benralitsumabi on eosinofiilien vastainen, humanisoitu, afukosyloitu, monoklonaalinen vasta-aine (IgG1, kappa). Se sitoutuu spesifisesti ihmisen interleukiini 5 -reseptorin (IL5Rα) alfa-alayksikköön. IL-5-reseptori ilmentyy erityisesti eosinofiilien ja basofiilien pinnalla. Koska benralitsumabin Fc-osassa ei ole fukoosia, benralitsumabilla on suuri affiniteetti immuunijärjestelmän efektorisolujen, kuten luonnollisten tappajasolujen, FcɣRIII-reseptoreja kohtaan. Tämä johtaa eosinofiilien ja basofiilien apoptoosiin, koska vasta-aineesta riippuvainen soluvälitteinen sytotoksisuus (ADCC) tehostuu, mikä vähentää eosinofiilistä tulehdusta.
Farmakodynaamiset vaikutukset
Vaikutus veren eosinofiileihin
Astmaa sairastavilla potilailla benralitsumabihoito aiheuttaa veren eosinofiilien lähes täydellisen häviämisen 24 tunnin kuluessa ensimmäisen lääkeannoksen antamisesta. Tämä vaikutus säilyy koko hoidon ajan. Eosinofiilien häviämiseen verestä liittyy seerumin eosinofiilien jyväsproteiinien (eosinofiiliperäisen neurotoksiinin ja eosinofiilisen kationisen proteiinin) väheneminen sekä veren basofiilien väheneminen.
Eosinofiilista granulomatoottista polyangiittia sairastavilla potilailla veren eosinofiilien häviäminen vastasi astmatutkimuksissa havaittua vaikutusta. Veren eosinofiilien häviäminen todettiin ensimmäisenä havaintoajankohtana eli 1 hoitoviikon jälkeen, ja vaikutus säilyi koko 52 viikon hoitojakson ajan.
Hypereosinofiilista oireyhtymää sairastavilla potilailla veren eosinofiilien väheneminen vastasi astmaa ja eosinofiilista granulomatoottista polyangiittia koskeneissa tutkimuksissa havaittua vaikutusta. Veren eosinofiilien väheneminen todettiin ensimmäisenä havaintoajankohtana (viikko 4), ja vaikutus säilyi koko 24 viikon hoitojakson ajan.
Vaikutus hengitysteiden limakalvojen eosinofiileihin
Benralitsumabin vaikutusta hengitysteiden limakalvojen eosinofiileihin arvioitiin astmapotilailla, joiden yskösnäytteessä todettiin suurentuneita eosinofiilipitoisuuksia (vähintään 2,5 %). Tässä 12 viikon mittaisessa vaiheen 1 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa potilaat saivat joko 100 tai 200 mg benralitsumabia ihon alle. Tässä tutkimuksessa hengitysteiden limakalvojen eosinofiilien vähenemän mediaani lähtötilanteeseen nähden oli 96 % benralitsumabiryhmässä ja 47 % lumelääkeryhmässä (p = 0,039).
Kliininen teho
Astma
Benralitsumabin tehoa arvioitiin kolmessa satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa, lumekontrolloidussa 28–56 viikon mittaisessa kliinisessä tutkimuksessa 12–75‑vuotiailla potilailla.
Näissä tutkimuksissa benralitsumabia annettiin 30 mg:n annoksella niin, että 3 ensimmäistä annosta annettiin 4 viikon välein ja sen jälkeen annosväli oli 4 tai 8 viikkoa. Tutkimuksissa valmistetta annettiin taustahoidon lisälääkkeenä ja verrattiin lumelääkkeeseen.
Pahenemisvaiheita arvioineisiin kahteen tutkimukseen SIROCCO (tutkimus 1) ja CALIMA (tutkimus 2) osallistui yhteensä 2 510 potilasta, joilla oli vaikea astma, joka ei ollut hoitotasapainossa. Potilaista 64 % oli naisia. Potilaiden keskimääräinen ikä oli 49 vuotta. Potilailla oli ollut viimeksi kuluneiden 12 kuukauden aikana vähintään kaksi (keskimäärin kolme) astman pahenemisvaihetta, joiden hoitoon oli tarvittu suun kautta otettavaa tai systeemistä kortikosteroidia, ja seulonnassa todetun Asthma Control Questionnaire‑6 ‑pistemäärän (ACQ6) edellytettiin olevan vähintään 1,5 ja keuhkojen toiminnan edellytettiin olleen lähtötilanteessa heikentynyt (ennen bronkodilataattorin antamista mitattu keskimääräinen uloshengityksen sekuntikapasiteetti [ FEV1] 57,5 % viitearvosta) siitä huolimatta, että potilaat saivat suuriannoksista inhaloitavaa kortikosteroidihoitoa (ICS) (tutkimus 1) tai keskisuurilla tai suurilla annoksilla inhaloitavaa kortikosteroidihoitoa (tutkimus 2) ja pitkävaikutteista β‑agonistihoitoa (LABA). Ainakin yhtä lisälääkettä annettiin 51 %:lle tutkimuksen 1 potilaista ja 41 %:lle tutkimuksen 2 potilaista.
Suun kautta annettavan kortikosteroidin käytön vähentämistä arvioineeseen ZONDA‑tutkimukseen (tutkimus 3) osallistui yhteensä 220 astmapotilasta (61 % naisia, keskimääräinen ikä 51 vuotta), jotka saivat tavanomaisena hoitona annetun suuriannoksisen inhaloitavan kortikosteroidin ja pitkävaikutteisen β‑agonistin lisäksi astman hoitotasapainon ylläpitämiseen päivittäin suun kautta kortikosteroidia (OCS) (8–40 mg vuorokaudessa, mediaani 10 mg) sekä 53 %:ssa tapauksista ainakin yhtä lisälääkettä. Tutkimuksessa oli 8 viikon mittainen aloitusjakso, jonka aikana suun kautta annettavan kortikosteroidin määrä titrattiin pienimpään tehokkaaseen annokseen, jolla astman hoitotasapaino säilyi. Potilaiden veren eosinofiilipitoisuudet olivat vähintään 150 solua/μl ja heillä oli ollut vähintään yksi pahenemisvaihe viimeksi kuluneiden 12 kuukauden aikana.
Vaikka tutkimuksissa 1, 2 ja 3 tutkittiin kahta eri annostusohjelmaa, benralitsumabin kolme ensimmäistä annosta on suositeltavaa antaa 4 viikon välein ja sen jälkeen suositeltu annosväli on 8 viikkoa (ks. kohta Annostus ja antotapa), koska tiheämmästä annostelusta ei ole havaittu olevan lisähyötyä. Jäljempänä esitetty yhteenveto tuloksista koskee suositeltua annostusohjelmaa käyttäen saatuja tuloksia.
Pahenemisvaiheita koskevat tutkimukset
Ensisijainen päätemuuttuja oli kliinisesti merkittävien astman pahenemisvaiheiden vuosittainen määrä potilailla, joiden veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/μl ja jotka käyttivät suuriannoksista inhaloitavaa kortikosteroidia ja pitkävaikutteista β-agonistia. Kliinisesti merkittäväksi astman pahenemisvaiheeksi määriteltiin vaikeutunut astma, joka edellytti vähintään 3 vuorokauden mittaista hoitoa suun kautta otettavilla tai systeemisillä kortikosteroideilla ja/tai käyntiä päivystyspoliklinikalla, jolloin tarvittiin suun kautta otettavaa tai systeemistä kortikosteroidia ja/tai sairaalahoitoa. Potilailla, jotka saivat kortikosteroideja suun kautta ylläpitohoitona, kliinisesti merkittäväksi astman pahenemisvaiheeksi määriteltiin suun kautta otettavan tai systeemisen kortikosteroidin vakaan annoksen tilapäinen suurentaminen vähintään 3 vuorokauden ajaksi tai kerta-annos pistoksena annettavaa, kortikosteroidia sisältävää depotvalmistetta.
Molemmissa tutkimuksissa benralitsumabia saaneiden potilaiden vuosittaisten pahenemisvaiheiden määrä väheni merkittävästi verrattuna lumelääkkeeseen potilailla, joiden veren eosinofiilipitoisuus oli ≥ 300 solua/μl. Lisäksi FEV1‑arvon keskimääräisessä muutoksessa lähtötilanteesta nähtiin hyötyä jo viikosta 4 alkaen, ja tämä vaikutus säilyi koko hoidon ajan hoidon lopettamiseen asti (Taulukko 2).
Pahenemisvaiheiden määrän havaittiin vähentyneen lähtötilanteen eosinofiilipitoisuudesta riippumatta, mutta lähtötilanteen suurempien eosinofiilipitoisuuksien todettiin mahdollisesti ennustavan parempaa hoitovastetta erityisesti FEV1:n suhteen.
Taulukko 2. Tutkimuksissa 1 ja 2 hoidon jälkeen todetut, vuosittaista pahenemisvaiheiden määrää ja keuhkojen toimintaa koskevat tulokset eosinofiilipitoisuuksien mukaan
| Tutkimus 1 | Tutkimus 2 | ||
Benralitsumabi | Lumelääke | Benralitsumabi | Lumelääke | |
Veren eosinofiilipitoisuus | n = 267 | n = 267 | n = 239 | n = 248 |
Kliinisesti merkittävät pahenemisvaiheet | ||||
Esiintymistiheys | 0,74 | 1,52 | 0,73 | 1,01 |
Ero | -0,78 | -0,29 | ||
Esiintymistiheyksien suhde | 0,49 (0,37, 0,64) | 0,72 (0,54, 0,95) | ||
p-arvo | < 0,001 | 0,019 | ||
FEV1 (l) ennen bronkodilataattoria | ||||
Keskiarvo lähtötilanteessa | 1,660 | 1,654 | 1,758 | 1,815 |
Parannus lähtötilanteeseen | 0,398 | 0,239 | 0,330 | 0,215 |
Ero (95 %:n luottamusväli) | 0,159 (0,068, 0,249) | 0,116 (0,028, 0,204) | ||
p-arvo | 0,001 | 0,010 | ||
Veren eosinofiilipitoisuus | n = 131 | n = 140 | n = 125 | n = 122 |
Kliinisesti merkittävät pahenemisvaiheet | ||||
Esiintymistiheys | 1,11 | 1,34 | 0,83 | 1,38 |
Ero | -0,23 | -0,55 | ||
Esiintymistiheyksien suhde | 0,83 (0,59, 1,16) | 0,60 (0,42, 0,86) | ||
FEV1 (l) ennen bronkodilataattoria | ||||
Keskimääräinen muutos | 0,248 | 0,145 | 0,140 | 0,156 |
Ero (95 %:n luottamusväli) | 0,102 (-0,003, 0,208) | -0,015 (-0,127, 0,096) | ||
a Hoitoaiepopulaatio (potilaat, jotka saivat suuriannoksista inhaloitavaa kortikosteroidia ja joiden veren eosinofiilipitoisuus oli ≥ 300 solua/μl).
b Ei ollut suunniteltu havaitsemaan eroja hoidon vaikutuksessa potilailla, joiden veren eosinofiiliipitoisuus oli < 300 solua/μl.
Tutkimusten 1 ja 2 yhdistettyjen tulosten mukaan pahenemisvaiheet vähenivät lukumääräisesti enemmän ja FEV1‑arvot paranivat enemmän potilailla, joiden veren eosinofiilipitoisuudet olivat lähtötilanteessa suuremmat.
Tutkimuksessa 1 sairaalahoitoa tai päivystyskäyntiä edellyttäneiden pahenemisvaiheiden esiintymistiheys oli benralitsumabia saaneilla potilailla 0,09 ja lumelääkettä saaneilla potilailla 0,25 (esiintymistiheyksien suhde 0,37, 95 %:n luottamusväli 0,20, 0,67; p ≤ 0,001) ja tutkimuksessa 2 benralitsumabia saaneilla potilailla 0,12 ja lumelääkettä saaneilla 0,10 (esiintymistiheyksien suhde 1,23, 95 %:n luottamusväli 0,64, 2,35; p = 0,538). Tutkimuksen 2 lumehoitohaarassa oli liian vähän tapahtumia, jotta olisi voitu tehdä johtopäätöksiä sairaalahoitoa tai päivystyskäyntejä edellyttäneistä pahenemisvaiheista.
Sekä tutkimuksessa 1 että tutkimuksessa 2 benralitsumabia saaneiden potilaiden astmaoireet (astmaan liittyvä kokonaispistemäärä, Total Asthma Score) vähenivät tilastollisesti merkitsevästi verrattuna lumelääkettä saaneisiin potilaisiin. Samanlaista paranemista benralitsumabin eduksi havaittiin ACQ6- ja AQLQ(S)+12-mittareilla (Standardised Asthma Quality of Life Questionnaire for 12 Years and Older) arvioituna (Taulukko 3).
Taulukko 3. Hoitojen väliset erot arvioituna astmaoireita kuvaavien kokonaispistemäärien keskimääräisellä muutoksella lähtötilanteesta, ACQ6- ja AQLQ(s)+12-pistemäärät hoidon päättyessä – potilaat, jotka saivat suuriannoksista inhaloitavaa kortikosteroidia ja joiden veren eosinofiilipitoisuus oli ≥ 300 solua/μl
| Tutkimus 1 | Tutkimus 2 | ||
Benralitsumabi | Lumelääke | Benralitsumabi | Lumelääke | |
Astmaoireita kuvaava kokonaispistemääräb | ||||
Keskiarvo | 2,68 | 2,74 | 2,76 | 2,71 |
Parannus | -1,30 | -1,04 | -1,40 | -1,16 |
Ero (95 %:n | -0,25 (-0,45, -0,06) | -0,23 (-0,43, -0,04) | ||
p-arvo | 0,012 | 0,019 | ||
ACQ-6 | ||||
Keskiarvo | 2,81 | 2,90 | 2,80 | 2,75 |
Parannus | -1,46 | -1,17 | -1,44 | -1,19 |
Ero (95 %:n | -0,29 (-0,48, -0,10) | -0,25 (-0,44, -0,07) | ||
AQLQ(S)+12 | ||||
Keskiarvo | 3,93 | 3,87 | 3,87 | 3,93 |
Parannus | 1,56 | 1,26 | 1,56 | 1,31 |
Ero (95 %:n | 0,30 (0,10, 0,50) | 0,24 (0,04, 0,45) | ||
a Potilaiden määrä (n) vaihtelee hieman, mikä johtuu niiden potilaiden määristä, joista oli saatavilla kuhunkin muuttujaan liittyvää tietoa. Esitetyt tulokset perustuvat kustakin muuttujasta saatavilla olevaan viimeisimpään tietoon.
b Astman oireita kuvaava asteikko: kokonaispistemäärä nollasta (vähiten) kuuteen (eniten); päivä‑ ja yöaikaan ilmenevien astmaoireiden pistemäärät nollasta (vähiten) kolmeen (eniten). Yksittäiset päivä‑ ja yöaikaan mitatut pistemäärät olivat samanlaiset.
Alaryhmäanalyysit aiemmin ilmenneiden pahenemisvaiheiden mukaan
Tutkimusten 1 ja 2 alaryhmäanalyyseissä todettiin, että ennen tutkimukseen osallistumista ilmenneiden pahenemisvaiheiden suurempi määrä oli paremman hoitovasteen mahdollinen ennustetekijä. Kun tätä tulosta tarkastellaan itsenäisesti tai yhdistettynä veren eosinofiilipitoisuuteen lähtötilanteessa, näiden tekijöiden avulla saatetaan tunnistaa potilaat, jotka voivat saada paremman vasteen benralitsumabihoitoon (Taulukko 4).
Taulukko 4. Pahenemisvaiheiden esiintymistiheys ja keuhkojen toiminta (FEV1) hoidon päättyessä tutkimusta edeltäneen vuoden pahenemisvaiheiden määrän mukaan– potilaat, jotka saivat suuriannoksista inhaloitavaa kortikosteroidia ja joiden veren eosinofiilipitoisuus oli ≥ 300 solua/μl
| Tutkimus 1 | Tutkimus 2 | ||
Benralitsumabi | Lumelääke | Benralitsumabi | Lumelääke | |
Lähtötilanteessa 2 pahenemisvaihetta | ||||
n | 164 | 149 | 144 | 151 |
Pahenemisvaiheiden | 0,57 | 1,04 | 0,63 | 0,62 |
Ero | -0,47 | 0,01 | ||
Esiintymistiheyksien | 0,55 (0,37, 0,80) | 1,01 (0,70, 1,46) | ||
Ennen | 0,343 | 0,230 | 0,266 | 0,236 |
Ero (95 %:n | 0,113 (-0,002, 0,228) | 0,029 (-0,079, 0,137) | ||
Lähtötilanteessa vähintään 3 pahenemisvaihetta | ||||
n | 103 | 118 | 95 | 97 |
Pahenemisvaiheiden | 0,95 | 2,23 | 0,82 | 1,65 |
Ero | -1,28 | -0,84 | ||
Esiintymistiheyksien | 0,43 (0,29, 0,63) | 0,49 (0,33, 0,74) | ||
Ennen | 0,486 | 0,251 | 0,440 | 0,174 |
Ero (95 %:n | 0,235 (0,088, 0,382) | 0,265 (0,115, 0,415) | ||
Suun kautta otettavan kortikosteroidin annoksen pienentämistä arvioineet tutkimukset
Lumekontrolloidussa ZONDA-tutkimuksessa (tutkimus 3) ja yksihaaraisessa, avoimessa PONENTE-tutkimuksessa (tutkimus 6) arvioitiin benralitsumabin vaikutusta suun kautta otettujen kortikosteroidien käytön vähentämiseen ylläpitohoidossa.
Tutkimuksessa 3 ensisijainen päätemuuttuja oli suun kautta otetun kortikosteroidin lopullisen annoksen prosentuaalinen pieneneminen lähtötilanteeseen verrattuna viikoilla 24–28 siten, että astman hoitotasapaino säilyy. Tutkimuksen 3 tuloksista on esitetty yhteenveto taulukossa 5.
Taulukko 5. Benralitsumabin vaikutus suun kautta otettavan kortikosteroidiannoksen pienentämiseen, tutkimus 3
| Benralitsumabi | Lumelääke |
Wilcoxonin järjestyslukutesti (primaarianalyysin menetelmä) | ||
Päivittäin suun kautta otettavan kortikosteroidiannoksen | 75 (60, 88) | 25 (0, 33) |
Wilcoxonin järjestyslukutesti, p-arvo | < 0,001 |
|
Suhteellisten vastamittojen malli (herkkyysanalyysi) | ||
Suun kautta otettavan kortikosteroidin käytön prosentuaalinen väheneminen viikolla 28 | ||
≥ 90 %:n väheneminen | 27 (37 %) | 9 (12 %) |
≥ 75 %:n väheneminen | 37 (51 %) | 15 (20 %) |
≥ 50 %:n väheneminen | 48 (66 %) | 28 (37 %) |
> 0 %:n väheneminen | 58 (79 %) | 40 (53 %) |
Ei muutosta tai suun kautta otettavan kortikosteroidin käyttö ei | 15 (21 %) | 35 (47 %) |
Kerroinsuhde (95 %:n luottamusväli) | 4,12 (2,22, 7,63) |
|
Päivittäisen suun kautta otettavan kortikosteroidiannoksen | 22 (52 %) | 8 (19 %) |
Kerroinsuhde (95 %:n luottamusväli) | 4,19 (1,58, 11,12) |
|
Päivittäisen suun kautta otettavan kortikosteroidiannoksen | 43 (59 %) | 25 (33 %) |
Kerroinsuhde (95 %:n luottamusväli) | 2,74 (1,41, 5,31) |
|
Pahenemisvaiheiden esiintymistiheys | 0,54 | 1,83 |
Esiintymistiheyksien suhde (95 %:n luottamusväli) | 0,30 (0,17, 0,53) |
|
Sairaalahoitoa tai päivystyskäyntiä edellyttäneiden | 0,02 | 0,32 |
Esiintymistiheyksien suhde (95 %:n luottamusväli) | 0,07 (0,01, 0,63) |
|
a Vain potilailla, joiden lähtötilanteen optimaalinen suun kautta otettava kortikosteroidiannos oli enintään 12,5 mg, saatiin suun kautta otettavan kortikosteroidin annosta pienentää 100 % tutkimuksen aikana.
Tutkimuksessa 3 arvioitiin myös keuhkojen toimintaa, astmaoireita kuvaavaa pistemäärää ja ACQ6- ja AQLQ(S)+12-pisteitä. Tulokset olivat samanlaisia kuin tutkimuksissa 1 ja 2.
Tutkimukseen 6 osallistui 598 aikuispotilasta, joilla oli vaikea astma (veren eosinofiilipitoisuus tutkimukseen ottamisen yhteydessä ≥ 150 solua/μl tai viimeksi kuluneiden 12 kuukauden aikana ≥ 300 solua/μl, jos pitoisuus tutkimukseen ottamisen yhteydessä oli < 150 solua/μl) ja jotka tarvitsivat suun kautta otettavaa kortikosteroidia. Ensisijaiset päätemuuttujat olivat niiden potilaiden osuus, jotka lopettivat suun kautta otetun kortikosteroidin käytön siten, että astman hoitotasapaino säilyi, ja niiden potilaiden osuus, joilla suun kautta otetun kortikosteroidin lopullinen annos oli enintään 5 mg siten, että astman hoitotasapaino säilyi ja lisämunuaisen toiminta oli huomioitu. Niiden potilaiden osuus, jotka lopettivat suun kautta otetun kortikosteroidin käytön ylläpitohoitona oli 62,9 %. Niiden potilaiden osuus, joilla suun kautta otetun kortikosteroidin annos oli enintään 5 mg (astman hoitotasapaino säilyi eikä lisämunuaisten toiminta rajoittanut annosta) oli 81,9 %. Vaikutukset suun kautta otettujen kortikosteroidien käytön vähentämiseen olivat samanlaisia riippumatta veren eosinofiilipitoisuudesta tutkimukseen ottamisen yhteydessä (mukaan lukien potilailla, joiden veren eosinofiilipitoisuus oli < 150 solua/μl), ja vaikutukset säilyivät 24–32 viikon mittaisen seurantavaiheen ajan. Vuosittaisten pahenemisvaiheiden määrä tutkimuksessa 6 oli vastaava kuin mitä aiemmissa tutkimuksissa on ilmoitettu.
Pitkäaikaiset jatkotutkimukset
Benralitsumabin pitkän aikavälin tehoa ja turvallisuutta arvioitiin 56 viikon mittaisessa vaiheen 3 BORA-jatkotutkimuksessa (tutkimus 4). Tutkimukseen osallistui tutkimuksista 1, 2 ja 3 yhteensä 2 123 potilasta, joista 2 037 oli aikuisia ja 86 nuoria (vähintään 12‑vuotiaita). Tutkimuksessa 4 arvioitiin benralitsumabin pitkäaikaisia vaikutuksia vuosittaisten pahenemisvaiheiden määrään, keuhkojen toimintaan, ACQ6- ja AQLQ(S)+12-pistemääriin sekä suun kautta annettavan kortikosteroidin vähentyneen käytön säilymiseen kahdella annostusohjelmalla, joita oli tutkittu edeltävissä tutkimuksissa.
Kun käytettiin suositeltua annostusohjelmaa, havaittu pahenemisvaiheiden vuosittaisen määrän väheneminen lumekontrolloiduissa edeltävissä tutkimuksissa 1 ja 2 (potilailla, joiden veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/μl ja jotka käyttivät suuriannoksista inhaloitavaa kortikosteroidia) säilyi toisena hoitovuonna (Taulukko 6). Edeltävissä tutkimuksissa 1 ja 2 benralitsumabia saaneilla potilailla 73 %:lla ei ilmennyt pahenemisvaiheita jatkotutkimuksessa 4.
Taulukko 6. Pahenemisvaiheet pitkäaikaisen hoidon aikanaa
| Lumelääkeb (N = 338) | Benralitsumabi (N = 318) | ||
Tutkimukset 1 ja 2 | Tutkimukset 1 ja 2 | Tutkimus 4 | Tutkimukset 1, 2 ja 4c | |
Esiintymistiheys | 1,23 | 0,65 | 0,48 | 0,56 |
a Potilaat, jotka siirtyivät tutkimukseen 4 edeltävistä tutkimuksista 1 ja 2 ja joiden veren eosinofiilipitoisuus lähtötilanteessa oli ≥ 300 solua/μl ja jotka käyttivät suuriannoksista inhaloitavaa kortikosteroidia.
b Sisältää tutkimuksissa 1 ja 2 lumelääkettä saaneet potilaat enintään edeltävän tutkimuksen päättymiseen saakka (viikkoon 48 tutkimuksessa 1 ja viikkoon 56 tutkimuksessa 2).
c Hoidon kokonaiskesto: 104–112 viikkoa.
Vaikutusten havaittiin säilyneen samalla tavalla koko tutkimuksen 4 ajan keuhkojen toiminnan sekä ACQ6- ja AQLQ(S)+12-pistemäärien osalta (Taulukko 7).
Taulukko 7. Keuhkojen toiminnan sekä ACQ6- ja AQLQ(S)+12-pistemäärien muutokset lähtötilanteestaa
| Tutkimukset 1 ja 2 Lähtötilanneb | Tutkimukset 1 ja 2 Hoidon päättyminenc | Tutkimus 4 Hoidon päättyminend
|
FEV1 (l) ennen bronkodilataattoria | |||
n | 318 | 305 | 290 |
Keskiarvo lähtötilanteessa (keskihajonta) | 1,741 (0,621) | -- | -- |
Muutos lähtötilanteeseen nähden (keskihajonta) e | -- | 0,343 (0,507) | 0,404 (0,555) |
ACQ6 | |||
n | 318 | 315 | 296 |
Keskiarvo lähtötilanteessa (keskihajonta) | 2,74 (0,90) | -- | -- |
Muutos lähtötilanteeseen nähden (keskihajonta) e | -- | -1,44 (1,13) | -1,47 (1,05) |
AQLQ(S)+12 |
| ||
n | 307 | 306 | 287 |
Keskiarvo lähtötilanteessa (keskihajonta) | 3,90 (0,99) | -- | -- |
Muutos lähtötilanteeseen nähden (keskihajonta) e | -- | 1,58 (1,23) | 1,61 (1,21) |
n = niiden potilaiden määrä, joista oli saatavilla tietoja kyseisenä ajankohtana.
a. Veren eosinofiilipitoisuus lähtötilanteessa ≥ 300 solua/μl ja suuriannoksisen inhaloitavan kortikosteroidin käyttö: benralitsumabi annettiin suositellulla annostusohjelmalla
b. Tutkimusten 1 ja 2 lähtötilanteen integroitu analyysi sisältää aikuiset ja nuoret.
c. Integroitu analyysi hoidon päätyttyä tutkimuksessa 1 (viikko 48) ja tutkimuksessa 2 (viikko 56)
d. Hoidon päättymisajankohta tutkimuksessa 4 oli viikko 48 (viimeinen ajankohta aikuisia ja nuoria koskevien tietojen osalta).
e. Lähtötilanne on ennen benralitsumabihoitoa tutkimuksissa 1 ja 2.
Tutkimuksessa 4 arvioitiin tehoa myös potilailla, joiden veren eosinofiilipitoisuus lähtötilanteessa oli < 300 solua/μl, ja se vastasi tutkimuksissa 1 ja 2 todettua tehoa.
Päivittäin suun kautta otettavan kortikosteroidin vähentyneen käytön säilyminen havaittiin myös jatkotutkimuksen aikana tutkimuksesta 3 mukaan otetuilla potilailla (Kuva 1).
Kuva 1. Päivittäin suun kautta otettavan kortikosteroidin prosentuaalinen väheneminen ajan myötä, mediaani (tutkimukset 3 ja 4)a
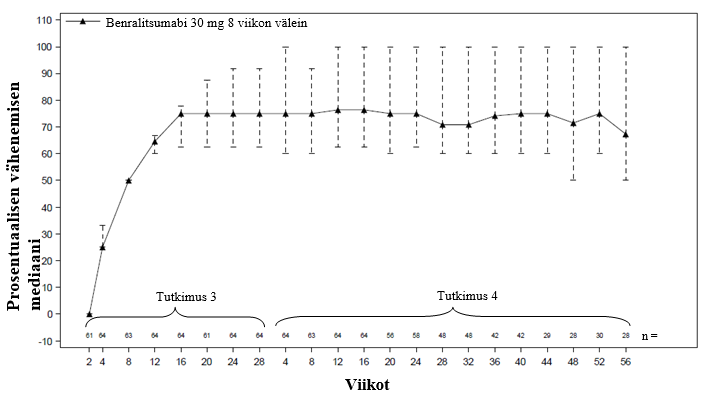
a Edeltävän tutkimuksen 3 potilaat, jotka jatkoivat benralitsumabihoitoa tutkimuksessa 4. Potilaat saivat siirtyä toiseen jatkotutkimukseen osallistuttuaan vähintään 8 viikon ajan tutkimukseen 4 jatkamatta 56 viikon mittaisen jatkovaiheen loppuun asti.
Tutkimuksessa 5, joka oli toinen pitkäaikainen turvallisuutta koskeva jatkotutkimus (ks. kohta Haittavaikutukset), pahenemisvaiheiden vuosittainen määrä (0,47) oli hyväksyttyjä annoksia saaneilla potilailla vastaava kuin mitä ilmoitettiin edeltävissä tutkimuksissa 1, 2 (0,65) ja 4 (0,48).
Eosinofiilinen granulomatoottinen polyangiitti
Benralitsumabin tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa kliinisessä non-inferioriteettitutkimuksessa, jossa hoidon kesto oli 52 viikkoa ja johon otettiin vähintään 18‑vuotiaita potilaita, joilla oli eosinofiilinen granulomatoottinen polyangiitti. Yhteensä 140 potilasta satunnaistettiin saamaan joko 30 mg benralitsumabia tai 300 mg mepolitsumabia ihon alle 4 viikon välein. Tutkimukseen osallistuneilla potilailla oli anamneesissa uusiutunut tai vaikeahoitoinen sairaus, ja he saivat suun kautta otettavaa vakioannoksista kortikosteroidihoitoa (≥ 7,5 mg – ≤ 50 mg prednisolonia/prednisonia vuorokaudessa) joko vakioannoksisen immunosuppressanttihoidon kanssa tai ilman sitä (syklofosfamidin käyttö oli suljettu pois). Lähtötilanteessa suun kautta otettavien kortikosteroidien vuorokausiannoksen mediaani oli 10 mg, ja 36 % potilaista sai immunosuppressanttihoitoa. Suun kautta otettavien kortikosteroidien annosta pienennettiin tutkijalääkärin harkinnan mukaan. Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen, elinten toimintaa tai henkeä uhkaava eosinofiilinen granulomatoottinen polyangiitti.
Remissio
Ensisijainen päätemuuttuja oli niiden potilaiden osuus, jotka olivat remissiossa (määritelmä: Birmingham Vasculitis Activity Score ‑pistemäärä [BVAS] 0 [ei aktiivista vaskuliittia] ja prednisoloni-/prednisoniannos ≤ 4 mg/vrk) sekä viikolla 36 että viikolla 48. Kuten taulukossa 8 esitetään, benralitsumabi osoittautui vähintään samanveroiseksi (non-inferior) mepolitsumabin kanssa ensisijaisen päätetapahtuman suhteen. Taulukossa 8 esitetään myös remission kumulatiivista kestoa ja remission komponentteja koskevat tulokset.
Taulukko 8. Eosinofiilisen granulomatoottisen polyangiitin remissio ja remission komponentit
| Remissio (Suun kautta otettavat kortikosteroidit ≤ 4 mg/vrk + | Suun kautta otettavat kortikosteroidit ≤ 4 mg/vrk | BVAS-pistemäärä = 0 | |||
Benraa N = 70 | Mepob N = 70 | Benraa N = 70 | Mepob N = 70 | Benraa N = 70 | Mepob N = 70 | |
Potilaat, jotka olivat remissiossa sekä viikolla 36 että viikolla 48 | ||||||
Potilaita, n (%)c | 40 (58) | 40 (57) | 42 (61) | 41 (58) | 58 (83) | 59 (84) |
Remissio-osuuksien erot (%)c (95 %:n luottamusväli) (p-arvo) | 1,21
(-14,12, 16,53) (0,88)d | 2,64
(-12,67, 17,95) (0,74)d, e | -1,17
(-13,27, 10,94) (0,85)d, e | |||
Kumulatiivinen kesto 52 viikon aikana, n (%) | ||||||
0 viikkoaf > 0 – < 12 viikkoa 12 – < 24 viikkoa 24 – < 36 viikkoa ≥ 36 viikkoa | 9 (13) 13 (19) 8 (11) 20 (29) 20 (29) | 15 (21) 10 (14) 8 (11) 19 (27) 18 (26) | 9 (13) 11 (16) 9 (13) 19 (27) 22 (31) | 12 (17) 12 (17) 8 (11) 18 (26) 20 (29) | 0 0 2 (3) 6 (9) 62 (89) | 0 2 (3) 2 (3) 7 (10) 59 (84) |
N = potilaiden määrä analyysissä.
a. Benralitsumabi (Benra), 30 mg 4 viikon välein.
b. Mepolitsumabi (Mepo), 300 mg 4 viikon välein.
c. Mallin mukaan korjatut prosenttiosuudet.
d. Käytetään paremmuuden testaamiseen.
e. Ei testattu muodollisesti ennalta määritellyssä monivertailutestausmenettelyssä.
f. Ei saavuttanut remissiota missään vaiheessa.
Niiden potilaiden osuus, jotka saavuttivat remission ensimmäisten 24 hoitoviikon aikana ja pysyivät remissiossa viikolle 52 asti, oli 42 % benralitsumabia saaneilla ja 37 % mepolitsumabia saaneilla (vasteen saavuttaneiden potilaiden osuuksien ero 5,54 %, 95 %:n luottamusväli ‑9,30, 20,37, nimellinen p-arvo 0,46).
Kun käytössä oli vaihtoehtoinen remission määritelmä eli BVAS-pistemäärä 0 ja prednisoloni-/prednisoniannos ≤ 7,5 mg/vrk, ryhmien välillä todettiin vastaava teho näiden päätemuuttujien suhteen.
Ensisijaisen remissiopäätemuuttujan saavuttaneita potilaita oli kaikissa ennalta määritellyissä demografisiin tietoihin ja lähtötilanteen ominaisuuksiin perustuvissa alaryhmissä.
Uusiutuminen
Riskitiheyksien suhde ensimmäiseen uusiutumiseen (vaskuliitti, astma tai sinonasaalinen) kuluneen ajan suhteen oli 0,98 (95 %:n luottamusväli 0,53, 1,82, nimellinen p-arvo 0,95). Uusiutuminen todettiin 30 %:lla benralitsumabia saaneista potilaista ja 30 %:lla mepolitsumabia saaneista potilaista. Uusiutumisten vuosittainen määrä oli 0,50 benralitsumabia saaneilla potilailla ja 0,49 mepolitsumabia saaneilla potilailla (esiintymistiheyksien suhde 1,03, 95 %:n luottamusväli 0,56, 1,90, nimellinen p-arvo 0,93). Uusiutumisen tyypit olivat benralitsumabia saaneilla potilailla vastaavanlaiset kuin mepolitsumabia saaneilla.
Suun kautta otettavat kortikosteroidit
Suun kautta otettavien kortikosteroidien vuorokausiannoksen keskiarvo viikoilla 48–52 on esitetty taulukossa 9. Suun kautta otettavien kortikosteroidien annoksen pienentyminen 100 %:lla todettiin 41 %:lla benralitsumabia saaneista potilaista ja 26 %:lla mepolitsumabia saaneista (ero 15,69 %, 95 %:n luottamusväli 0,67, 30,71, nimellinen p-arvo 0,04).
Taulukko 9.Suun kautta otettavien kortikosteroidien vuorokausiannoksen keskiarvo viikoilla 48–52 EGPA-potilailla
| Potilaiden määrä (%) | |
Benralitsumabia (N = 70) | Mepolitsumabib (N = 70) | |
0 mg > 0 – ≤ 4,0 mg > 4,0 – ≤ 7,5 mg > 7,5 mg | 29 (41) 19 (27) 15 (21) 7 (10) | 19 (27) 30 (43) 13 (19) 8 (11) |
N = potilaiden määrä analyysissä.
a. Benralitsumabi, 30 mg 4 viikon välein.
b. Mepolitsumabi, 300 mg 4 viikon välein.
Astman hallintapisteet (Asthma Control Questionnaire-6, ACQ-6)
ACQ‑6-pistemäärän keskimääräinen muutos lähtötilanteesta oli ‑0,57 benralitsumabia saaneilla potilailla ja ‑0,61 mepolitsumabia saaneilla potilailla (ero 0,05, 95 %:n luottamusväli ‑0,18, 0,27, nimellinen p‑arvo 0,67).
Hypereosinofiilinen oireyhtymä (HES)
Benralitsumabin tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa, lumekontrolloidussa kliinisessä tutkimuksessa, jossa hoidon kesto oli 24 viikkoa ja johon otettiin vähintään 12‑vuotiaita potilaita, joilla oli hypereosinofiilinen oireyhtymä (jolle ei ollut löydetty ei-hematologista sekundaarista syytä). Yhteensä 133 potilasta (129 aikuista ja 4 nuorta), joilla oli hypereosinofiilisen oireyhtymän pahenemisvaiheen oireita tai löydöksiä tai joilla oli ilmennyt vähintään kaksi hypereosinofiilisen oireyhtymän pahenemisvaihetta edeltävien 12 kuukauden aikana, sai satunnaistettua hoitoa. Seulontavaiheessa potilaiden veren eosinofiilipitoisuus oli ≥ 1 000 solua/µl ja potilaat olivat saaneet hypereosinofiiliseen oireyhtymään vakioannoksista hoitoa vähintään 4 viikon ajan. Hypereosinofiilisen oireyhtymän hoitoon kuuluivat suun kautta otettava kortikosteroidihoito, immunosuppressiivinen/sytotoksinen hoito tai muut hypereosinofiiliseen oireyhtymään liittyvät oireenmukaiset hoidot. Tutkimuksesta suljettiin pois potilaat, joilla oli hypereosinofiilisen oireyhtymän aktiivinen henkeä uhkaava ilmenemismuoto tai jotka olivat positiivisia FIP1L1-PDGFRA-kinaasin suhteen. Potilaat satunnaistettiin saamaan 30 mg benralitsumabia tai lumelääkettä ihon alle 4 viikon välein, ja he jatkoivat samanaikaisesti vakioannoksista hypereosinofiilisen oireyhtymän hoitoa. Kaikilla satunnaistetuilla potilailla todettiin seulontavaiheessa vaste 2 päivää kestävälle suun kautta annetulle kortikosteroidihoidolle (veren eosinofiilipitoisuus < 1 000 solua/μl). Tutkimukseen otettiin mukaan kolme potilasta, joilla katsottiin olevan samanaikaisesti eosinofiilinen granulomatoottinen polyangiitti ja hypereosinofiilinen oireyhtymä.
Ensisijainen päätemuuttuja oli aika ensimmäiseen hypereosinofiilisen oireyhtymän pahenemisvaiheeseen. Hypereosinofiilisen oireyhtymän pahenemisvaiheeksi määriteltiin hypereosinofiilisen oireyhtymän kliininen ilmeneminen tai laboratorioarvon poikkeavuus, jonka seurauksena oli suun kautta otettavan kortikosteroidin annostuksen suurentaminen vähintään 10 mg:lla vuorokaudessa vähintään 2 päivän ajaksi tai tällaisen hoidon aloittaminen tai uuden sytotoksisen ja/tai immunosuppressiivisen hoidon annostuksen suurentaminen tai tällaisen hoidon aloittaminen tai sairaalahoito. Benralitsumabihoito pidensi aikaa ensimmäiseen hypereosinofiilisen oireyhtymän pahenemisvaiheeseen (kuva 2) ja pienensi ensimmäisen pahenemisvaiheen riskiä merkitsevästi, 65 %, hoitojakson aikana lumelääkkeeseen verrattuna (riskitiheyksien suhde [HR]: 0,35, 95 %:n luottamusväli 0,18, 0,69, p = 0,0024). Lisäksi suuremmalla osuudella benralitsumabia saaneista potilaista ei ilmennyt pahenemisvaiheita (81 % vs. 58 % lumelääkettä saaneista) ja pienemmällä osuudella benralitsumabia saaneista potilaista ilmeni hypereosinofiilisen oireyhtymän pahenemisvaiheita, joiden seurauksena suun kautta otettavan kortikosteroidin annostusta suurennettiin (12 [18 %]), lumelääkettä saaneisiin verrattuna (28 [42 %]) 24 viikon pituisen hoitojakson aikana.
Kuva 2. Kaplan–Meier-kuvaaja ajasta ensimmäiseen hypereosinofiilisen oireyhtymän pahenemisvaiheeseen verrattuna lumelääkkeeseen
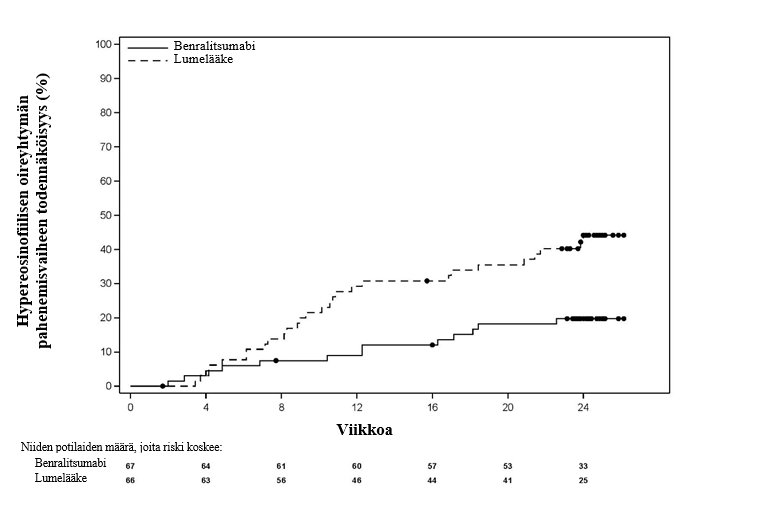
Keskeiset toissijaiset päätemuuttujat olivat niiden potilaiden osuus, joilla ilmeni hypereosinofiilisen oireyhtymän pahenemisvaihe hoitojakson aikana, hypereosinofiilisen oireyhtymän pahenemisvaiheiden esiintymistiheys, aika ensimmäiseen hematologiseen uusiutumiseen ja uupumuksen vaikeusasteen muutos lähtötilanteesta (taulukko 10). Kaikki keskeiset toissijaiset päätemuuttujat olivat tilastollisesti merkitseviä.
Taulukko 10.Keskeiset toissijaiset tehoa koskevat päätetapahtumat, hypereosinofiilista oireyhtymää koskeva tutkimus
| Benralitsumabia | Lumelääke | |
Niiden potilaiden osuus, joilla ilmeni hypereosinofiilisen oireyhtymän pahenemisvaihe | |||
Potilaat, joilla ilmeni vähintään 1 hypereosinofiilisen oireyhtymän pahenemisvaihe tai jotka keskeyttivät tutkimukseen osallistumisenb, n (%) | 15 (22) | 30 (45) | |
Kerroinsuhdec (95 %:n luottamusväli) | 0,31 (0,14, 0,69) | ||
p-arvo | 0,003 | ||
Suhteellinen vähenemä, % (suhteellinen riski [95 %:n luottamusväli]) | 52 % (0,48 [0,29, 0,80]) | ||
Hypereosinofiilisen oireyhtymän pahenemisvaiheiden esiintymistiheys | |||
Esiintymistiheys/vuosi | 0,41 | 1,23 | |
Esiintymistiheyksien suhdec (95 %:n luottamusväli) | 0,34 (0,18, 0,63) | ||
p-arvo | 0,0008 | ||
Aika ensimmäiseen hematologiseen uusiutumiseend | |||
Potilaat, joilla todettiin hematologinen uusiutuminen, n (%) | 5 (7) | 39 (59) | |
Riskitiheyksien suhde (HR)c (95 %:n luottamusväli) | 0,08 (0,03, 0,20) | ||
p-arvo | < 0,0001 | ||
Uupumuksen vaikeusasteen muutos lähtötilanteesta PROMIS Fatigue -mittarin perusteellae | |||
Standardoidun T-arvon absoluuttinen muutos lähtötilanteesta viikolla 24, pienimmän neliösumman keskiarvo (95 %:n luottamusväli) | ‑8,6 (‑10,6, ‑6,6) | ‑3,9 (‑6,0, ‑1,8) | |
Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | ‑4,7 (‑7,6, ‑1,8) | ||
p-arvo | 0,0017 | ||
a. Benralitsumabi 30 mg annettuna 4 viikon välein.
b. Kaikki potilaat, jotka keskeyttivät tutkimukseen osallistumisen kokematta pahenemisvaihetta
(lumelääkettä saaneilla n = 2, benralitsumabia saaneilla n = 2), otettiin mukaan analyysiin pahenemisvaiheen kokeneina.
c. Kerroinsuhde, esiintymistiheyksien suhde tai riskitiheyksien suhde < 1 suosii benralitsumabia.
d. Hematologiseksi uusiutumiseksi määritellään eosinofiilipitoisuus ≥ 1 000 solua/μl.
e. Patient Reported Outcomes Measurement Information System (PROMIS) Fatigue Short Form 7a ‑mittari: standardoitu T‑arvo (vaihteluväli 29,4–83,2). Arvon pieneneminen viittaa lievittymiseen. Benralitsumabi- ja lumelääkehoitojen välillä todettiin nimellisesti merkitsevä ero uupumuksessa viikolla 4, ja ero säilyi koko tutkimuksen ajan.
Immunogeenisuus
Kaiken kaikkiaan hoidon tuottama lääkevasta-ainevaste kehittyi 48–56 viikon hoitojakson aikana pahenemisvaiheita koskevissa vaiheen 3 lumekontrolloiduissa tutkimuksissa 107 potilaalle 809 potilaasta (13 %), jotka sairastivat astmaa ja saivat benralitsumabia suositelluilla annoksilla. Suurin osa vasta-aineista oli neutraloivia ja pysyviä. Benralitsumabin vasta-aineisiin liittyi benralitsumabin lisääntynyt puhdistuma ja veren eosinofiilipitoisuuksien suureneminen potilailla, joilla oli suuret lääkevasta-ainetitterit verrattuna vasta-ainenegatiivisiin potilaisiin. Harvoissa tapauksissa veren eosinofiilipitoisuudet palautuivat hoitoa edeltäville tasoille. Tällä hetkellä saatavilla olevien seurantatietojen perusteella ei ole havaittu näyttöä lääkevasta-aineiden yhteydestä tehoon tai turvallisuuteen.
Näiden vaiheen 3 lumekontrolloituihin tutkimuksiin osallistuneiden, astmaa sairastavien potilaiden toisen hoitovuoden jälkeen vielä 18 potilaalle 510:stä (4 %:lle) oli muodostunut hoidon tuottamia vasta-aineita. Kaiken kaikkiaan potilailla, joilla oli edeltävissä tutkimuksissa todettu lääkevasta-aineita, titterit pysyivät vakaina tai pienenivät toisena hoitovuonna. Lääkevasta-aineiden yhteydestä tehoon tai turvallisuuteen ei havaittu näyttöä.
Eosinofiilista granulomatoottista polyangiittia sairastavien potilaiden joukossa todettiin hoidon aikana kehittynyt lääkevasta-ainevaste 6 potilaalla 67 potilaasta (9 %), jotka saivat benralitsumabia vaiheen 3 aktiivikontrolloidun, 52 viikon pituisen hoitojakson aikana. Neutraloivien vasta-aineiden aktiivisuutta todettiin yhdellä lääkevasta-ainepositiivisista potilaista.
Hypereosinofiilista oireyhtymää sairastavien potilaiden joukossa todettiin hoidon aikana kehittynyt lääkevasta-ainevaste 7 potilaalla 66 potilaasta (11 %), jotka saivat benralitsumabia vaiheen 3 lumekontrolloidun, 24 viikon pituisen hoitojakson aikana. Yhteensä 2 potilaalle (3 %) benralitsumabiryhmässä kehittyi neutraloivia vasta-aineita.
Pediatriset potilaat
Astma
Vaiheen 3 tutkimuksiin osallistui 108 iältään 12–17-vuotiasta, astmaa sairastavaa nuorta (tutkimus 1: n = 53, tutkimus 2: n = 55). Näistä potilaista 46 sai lumelääkettä ja 40 sai benralitsumabia 3 ensimmäistä annosta 4 viikon välein ja sen jälkeen 8 viikon välein ja 22 potilasta sai benralitsumabia 4 viikon välein. Näissä tutkimuksissa astman pahenemisvaiheiden esiintymistiheys oli suositelluilla annoksilla benralitsumabia saaneilla nuorilla 0,70 (n = 40, 95 %:n luottamusväli 0,42, 1,18) ja lumelääkettä saaneilla nuorilla 0,41 (n = 46, 95 %:n luottamusväli 0,23, 0,73) [esiintymistiheyksien suhde 1,70, 95 %:n luottamusväli 0,78, 3,69].
Tutkimuksiin 1 ja 2 osallistuneet nuoret 12–17-vuotiaat potilaat (n = 86) jatkoivat benralitsumabihoitoa tutkimuksessa 4 enintään 108 viikon ajan. Teho ja turvallisuus vastasivat edeltävissä tutkimuksissa todettua tehoa ja turvallisuutta.
Farmakokinetiikkaa ja farmakodynamiikkaa arvioitiin avoimessa, kontrolloimattomassa, 48 viikon mittaisessa tutkimuksessa pienellä joukolla 6–11‑vuotiaita potilaita (n = 28). Potilailla oli vaikea astma, joka ei ollut hoitotasapainossa. Tämän tutkimuksen potilailla eosinofiilien määrä veressä pieneni saman verran kuin aikuisilla ja nuorilla.
Valmisteen tehosta pediatristen potilaiden astman hoidossa ei voida tehdä johtopäätöksiä (ks. kohta Annostus ja antotapa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset benralitsumabin käytöstä astman hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Eosinofiilinen granulomatoottinen polyangiitti
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset benralitsumabin käytöstä eosinofiilisen granulomatoottisen polyangiitin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Hypereosinofiilinen oireyhtymä (HES)
Vaiheen 3 tutkimukseen otettiin mukaan 4 iältään 12–17-vuotiasta nuorta, joilla oli hypereosinofiilinen oireyhtymä. 3 nuorta sai benralitsumabia ja 1 sai lumelääkettä 4 viikon välein 24 viikon ajan. Kummassakin hoitoryhmässä yhdellä nuorella potilaalla ilmeni hypereosinofiilisen oireyhtymän pahenemisvaihe 24 viikon pituisen hoitojakson aikana.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset benralitsumabin käytöstä hypereosinofiilisen oireyhtymän hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Jäljempänä kuvattavat tiedot benralitsumabin farmakokinetiikasta perustuvat astmatutkimusten pohjalta tehtyihin populaatiofarmakokineettisiin analyyseihin. Astmapotilaille annetun benralitsumabin farmakokinetiikka oli annoksesta riippuvaista ihon alle annetuilla 2−200 mg:n annoksilla.
Imeytyminen
Ihon alle antamisen jälkeen imeytymisen puoliintumisaika oli astmapotilailla 3,5 vuorokautta. Populaatiofarmakokineettisen analyysin perusteella arvioitu absoluuttinen hyötyosuus oli noin 59 %, eikä suhteellisessa hyötyosuudessa todettu kliinisesti merkittäviä eroja, kun benralitsumabi-injektio annettiin vatsaan, reiteen tai olkavarteen.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella benralitsumabin sentraalinen jakautumistilavuus on 3,1 l ja perifeerinen jakautumistilavuus 2,5 l henkilöllä, joka painaa 70 kg.
Biotransformaatio
Benralitsumabi on humanisoitu monoklonaalinen IgG1-vasta-aine, jota hajottavat proteolyyttiset entsyymit. Näitä entsyymejä on laajalti elimistössä, ei pelkästään maksakudoksessa.
Eliminaatio
Populaatiofarmakokineettisessa analyysissä benralitsumabin farmakokinetiikka oli lineaarista eikä kohdereseptorivälitteisestä puhdistumareitistä havaittu näyttöä. Benralitsumabin arvioitu systeeminen puhdistuma oli 0,29 l/vrk. Eosinofiilista granulomatoottista polyangiittia tai hypereosinofiilista oireyhtymää sairastavilla mallin perusteella arvioitu systeeminen puhdistuma oli noin 0,22 l/vrk. Ihon alle annetun benralitsumabin eliminaation puoliintumisaika oli noin 15,5 vuorokautta.
Erityisryhmät
Iäkkäät (vähintään 65-vuotiaat)
Populaatiofarmakokineettisen analyysin perusteella iällä ei ole vaikutusta benralitsumabin puhdistumaan. Yli 75-vuotiaista ei kuitenkaan ole saatavilla tietoja.
Pediatriset potilaat
Populaatiofarmakokineettisen analyysin ja kliinisten tutkimustietojen perusteella benralitsumabin farmakokinetiikka astmaa sairastavilla, 6−17‑vuotiailla lapsilla ja nuorilla vastasi farmakokinetiikkaa aikuisilla, kun potilaiden paino otettiin asianmukaisesti huomioon (ks. kohta Annostus ja antotapa).
Benralitsumabin farmakokinetiikka hypereosinofiilista oireyhtymää sairastavilla 12–17-vuotiailla nuorilla (n = 3) vastasi yleisesti ottaen aikuisilla todettua farmakokinetiikkaa (ks. kohta Annostus ja antotapa). Nuorilla farmakokinetiikkaa tutkittiin myös mallinnuksella ja simulaatioilla.
Sukupuoli, rotu
Populaatiofarmakokineettinen analyysi osoitti, ettei sukupuolella tai rodulla ole merkittävää vaikutusta benralitsumabin puhdistumaan.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutusta benralitsumabiin ei ole arvioitu kliinisissä tutkimuksissa. Populaatiofarmakokineettisen analyysin perusteella benralitsumabin puhdistuma tutkittavilla, joiden kreatiniinipuhdistuma-arvot olivat 30–80 ml/min, oli verrannollinen puhdistumaan potilailla, joiden munuaiset toimivat normaalisti. Tutkittavista, joiden kreatiniinipuhdistuma-arvot ovat alle 30 ml/min, on saatavilla vain vähän tietoa. Benralitsumabi ei kuitenkaan poistu munuaisten kautta.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta benralitsumabiin ei ole arvioitu kliinisissä tutkimuksissa. Monoklonaaliset IgG-vasta-aineet eivät ensisijaisesti poistu maksan kautta, joten maksan toiminnan muutosten ei odoteta vaikuttavan benralitsumabin puhdistumaan. Populaatiofarmakokineettisen analyysin perusteella maksan toimintaa kuvaavien biomerkkiaineiden (ASAT, ALAT ja bilirubiini) pitoisuuksilla lähtötilanteessa ei ollut kliinisesti merkittävää vaikutusta benralitsumabin puhdistumaan.
Yhteisvaikutukset
Populaatiofarmakokineettisen analyysin perusteella tavallisesti samanaikaisesti annetuilla lääkevalmisteilla (montelukastilla, parasetamolilla, protonipumpun estäjillä, makrolideilla ja teofylliinillä/aminofylliinillä) ei ollut vaikutusta benralitsumabin puhdistumaan astmapotilailla.
Prekliiniset tiedot turvallisuudesta
Koska benralitsumabi on monoklonaalinen vasta-aine, sillä ei ole tehty geenitoksisuutta tai karsinogeenisuutta koskevia tutkimuksia.
Toksikologia ja farmakologia koe-eläimillä
Farmakologista turvallisuutta tai toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten, apinoilla tehtyjen tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Cynomolgus-apinoille laskimoon tai ihon alle antamiseen liittyi eosinofiilipitoisuuksien pienenemistä ääreisverenkierrossa ja luuytimessä, toksikologisia löydöksiä ei ollut.
Raskaus
Tiineillä Cynomolgus-apinoilla tehdyissä pre- ja postnataalista kehitystä koskeneissa tutkimuksissa ei havaittu benralitsumabiin liittyviä emoihin tai alkioihin/sikiöihin kohdistuvia tai postnataalisia vaikutuksia.
Hedelmällisyys
Eläinkokeita ei ole tehty. Benralitsumabin käytön yhteydessä ei havaittu lisääntymistä koskevien parametrien huononemista uros- tai naaraspuolisilla Cynomolgus-apinoilla. Hedelmällisyyteen liittyvien korvikeparametrien (kuten elinten massan ja lisääntymiselinten kudosten histopatologian) tarkastelussa benralitsumabia saaneilla eläimillä ei havaittu viitteitä hedelmällisyyden heikentymisestä. Eosinofiilien vähenemistä havaittiin kuitenkin apinoiden poikasilla, joiden emot olivat saaneet benralitsumabia tiineyden aikana.
Farmaseuttiset tiedot
Apuaineet
Histidiini (pH:n säätämiseen)
Histidiinihydrokloridimonohydraatti (pH:n säätämiseen)
Trehaloosidihydraatti
Polysorbaatti 20 (E 432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Fasenra-valmistetta voidaan säilyttää huoneenlämmössä enintään 14 vuorokautta korkeintaan 25 °C:n lämpötilassa. Jääkaapista ottamisen jälkeen Fasenra on käytettävä 14 vuorokauden kuluessa tai hävitettävä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Ei saa jäätyä. Älä ravista. Älä altista kuumuudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FASENRA injektioneste, liuos, esitäytetty kynä
30 mg (L:ei) 1 kpl (1 ml) (2269,30 €)
FASENRA injektioneste, liuos, esitäytetty ruisku
30 mg (L:ei) 1 kpl (1 ml) (2269,30 €)
PF-selosteen tieto
Esitäytetty ruisku
Yksi ml liuosta kertakäyttöisessä, tyypin I lasista valmistetussa ruiskussa, jossa on 29 gaugen n. 12 mm:n pituinen (12,7 mm), ruostumattomasta teräksestä valmistettu kiinteä neula, kova neulansuojus ja FluroTec-päällystetty männän pysäytin. Esitäytetyssä ruiskussa on neulan suojamekanismi, sormituki ja mäntä.
Pakkaus sisältää yhden esitäytetyn ruiskun.
Esitäytetty kynä
Yksi ml liuosta kertakäyttöisessä, tyypin I lasista valmistetussa ruiskussa, jossa on 29 gaugen n. 12 mm:n pituinen (12,7 mm), ruostumattomasta teräksestä valmistettu kiinteä neula, kova neulansuojus ja FluroTec-päällystetty männän pysäytin. Esitäytetty kynä koostuu ruiskusta ja käsikäyttöisestä mekaanisesta (jousella toimivasta) injektiolaitteesta.
Pakkaus sisältää yhden esitäytetyn kynän.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai keltainen liuos, joka saattaa sisältää läpikuultavia, valkoisia tai luonnonvalkoisia hiukkasia.
Käyttö- ja käsittelyohjeet
Ennen antamista ota kotelo pois jääkaapista noin 30 minuutiksi, jotta esitäytetty ruisku tai esitäytetty kynä ehtii lämmetä huoneenlämpöön (20 °C – 25 °C).
Tarkasta Fasenra silmämääräisesti hiukkasten ja värimuutosten varalta ennen antamista. Fasenra on kirkas tai opaalinhohtoinen, väritön tai keltainen liuos, joka saattaa sisältää läpikuultavia, valkoisia tai luonnonvalkoisia hiukkasia. Älä käytä Fasenra-valmistetta, jos neste on sameaa, värjäytynyttä tai jos se sisältää suuria hiukkasia tai vierasaineita.
Lue pakkausselosteesta ja käyttöohjeesta lisätietoja ja ohjeet Fasenra esitäytetyn ruiskun tai esitäytetyn kynän valmistelemisesta käyttöön ja antamisesta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
FASENRA injektioneste, liuos, esitäytetty kynä
30 mg 1 kpl
FASENRA injektioneste, liuos, esitäytetty ruisku
30 mg 1 kpl
- Alempi erityiskorvaus (65 %). Benralitsumabi: Aikuisten vaikean eosinofiilisen astman hoito erityisin edellytyksin (251).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Benralitsumabi: Vaikean eosinofiilisen astman hoito erityisin edellytyksin (3027).
ATC-koodi
R03DX10
Valmisteyhteenvedon muuttamispäivämäärä
03.07.2026
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi