
OCREVUS infuusiokonsentraatti, liuosta varten 300 mg
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 300 mg okrelitsumabia 10 ml:ssa pitoisuutena 30 mg/ml. Laimennetun lääkevalmisteen lopullinen pitoisuus on noin 1,2 mg/ml.
Okrelitsumabi on kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla tuotettu humanisoitu monoklonaalinen vasta-aine.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Ocrevus on tarkoitettu aaltomaisen MS-taudin (RMS) hoitoon aikuispotilailla, joilla on kliinisten piirteiden tai kuvantamislöydösten perusteella määriteltyä MS-taudin aktiivisuutta (ks. kohta Farmakodynamiikka).
Ocrevus on tarkoitettu varhaisvaiheen primaaristi etenevän MS-taudin (PPMS) hoitoon aikuispotilailla määriteltynä taudin keston ja toimintakykytason sekä tulehdusaktiivisuudelle tyypillisten kuvantamislöydösten perusteella (ks. kohta Farmakodynamiikka).
Ehto
Hoito aloitetaan ja toteutetaan neurologisten sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin valvonnassa yksikössä, jossa on tarvittavat valmiudet vaikeiden haittavaikutusten, kuten vakavien infuusioreaktioiden, hoitamiseen.
Annostus ja antotapa
Hoito aloitetaan ja toteutetaan neurologisten sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin valvonnassa yksikössä, jossa on tarvittavat valmiudet vaikeiden haittavaikutusten, kuten vakavien infuusioreaktioiden, hoitamiseen.
Esilääkitys infuusioon liittyvien reaktioiden varalta
Potilaalle on annettava seuraavat kaksi esilääkitystä ennen jokaista okrelitsumabi-infuusiota vähentämään infuusioreaktioiden esiintyvyyttä ja vaikeusastetta (ks. lisätoimenpiteet infuusioreaktioiden vähentämiseksi kohdassa Varoitukset ja käyttöön liittyvät varotoimet):
- 100 mg metyyliprednisolonia (tai vastaavaa) laskimoon noin 30 minuuttia ennen jokaista infuusiota
- antihistamiinia noin 30–60 minuuttia ennen jokaista infuusiota.
Esilääkitykseksi voidaan lisäksi harkita jotakin kuumetta alentavaa lääkettä (esim. parasetamolia) noin 30–60 minuuttia ennen jokaista infuusiota.
Annostus
Aloitusannos
Aloitusannos 600 mg laskimoon annetaan kahteen erilliseen infuusioon jaettuna: ensin 300 mg:n infuusio ja tästä kaksi viikkoa myöhemmin toinen 300 mg:n infuusio (ks. taulukko 1).
Seuraavat annokset
Seuraavat okrelitsumabiannokset annetaan 600 mg:n kerta-annoksina infuusiona laskimoon kuuden kuukauden välein (ks. taulukko 1). Ensimmäinen 600 mg:n kerta-annos pitäisi antaa kuusi kuukautta aloitusannoksen ensimmäisen infuusion jälkeen.
Okrelitsumabiannosten välisen ajan pitää olla aina vähintään viisi kuukautta.
Infuusioihin tehtävät muutokset infuusioon liittyvien reaktioiden vuoksi
Hengenvaaralliset infuusioon liittyvät reaktiot
Jos infuusion aikana ilmenee merkkejä hengenvaarallisesta tai invalidisoivasta infuusioreaktiosta, kuten akuutti yliherkkyys tai akuutti hengitysvaikeusoireyhtymä, on infuusion anto lopetettava heti ja potilaalle on annettava tarkoituksenmukaista hoitoa. Infuusio on tällöin lopetettava pysyvästi (ks. kohta Vasta-aiheet).
Vaikea-asteiset infuusioreaktiot
Jos potilaalle ilmaantuu vaikea-asteinen infuusioreaktio (esim. hengenahdistus) tai kaulan ja kasvojen punoituksesta, kuumeesta ja kurkkukivusta koostuva oireisto, infuusion antaminen on keskeytettävä heti ja potilaalle pitää antaa oireenmukaista hoitoa. Infuusiota saa jatkaa vasta, kun kaikki oireet ovat hävinneet. Hoitoa jatkettaessa infuusio aloitetaan puolella siitä antonopeudesta, jota käytettiin reaktion ilmaantuessa. Seuraavien uusien infuusioiden yhteydessä ei tarvitse tehdä muutoksia infuusion antoon, mikäli potilaalle ei ilmaannu infuusioreaktiota.
Lievät tai keskivaikeat infuusioreaktiot
Jos potilaalla on lievä tai keskivaikea infuusioreaktio (esim. päänsärky), infuusionopeus pitää hidastaa puoleen siitä, joka oli käytössä tapahtuman ilmaantuessa. Hidastettua antonopeutta pitää jatkaa vähintään 30 minuutin ajan. Infuusionopeus voidaan tämän jälkeen palauttaa alkuperäiseksi, jos potilas sietää sen. Seuraavien uusien infuusioiden yhteydessä ei tarvitse tehdä muutoksia, jos potilaalle ei ilmaannu infuusioreaktioita.
Annosmuutokset hoidon aikana
Edellä esitetyissä esimerkeissä annoksen keskeyttämisestä ja hidastamisesta (lievät tai keskivaikeat ja vaikea-asteiset infuusioreaktiot) infuusionopeus muuttuu ja infuusion kokonaiskestoaika pitenee, mutta kokonaisannos ei muutu. Annoksen pienentämistä ei suositella.
Annosten viivästyminen tai antamatta jääminen
Jos infuusio jää antamatta, se on annettava mahdollisimman pian. Älä odota seuraavaa suunniteltua antoajankohtaa. Annosten välisen ajan tulisi olla kuusi kuukautta, kuitenkin vähintään viisi kuukautta (ks. taulukko 1).
Erityispotilasryhmät
Yli 55-vuotiaat aikuiset
Saatavissa olevien rajallisten tietojen perusteella (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka) yli 55-vuotiaiden potilaiden annostusta ei tarvitse muuttaa. Käynnissä oleviin kliinisiin tutkimuksiin mukaan otetut potilaat jatkavat 55 vuotta täytettyään okrelitsumabihoitoa 600 mg:n annoksin kuuden kuukauden välein.
Munuaisten vajaatoiminta
Okrelitsumabin turvallisuutta ja tehoa ei ole virallisesti tutkittu potilailla, joilla on munuaisten vajaatoiminta. Kliinisissä tutkimuksissa oli mukana lievää munuaisten vajaatoimintaa sairastavia potilaita. Keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole kokemusta. Okrelitsumabi on monoklonaalinen vasta-aine, ja se poistuu elimistöstä kataboloitumalla (eli hajoamalla peptideiksi ja aminohapoiksi), joten annoksen säätäminen ei oletettavasti ole munuaisten vajaatoimintaa sairastaville potilaille tarpeen (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Okrelitsumabin turvallisuutta ja tehoa ei ole virallisesti tutkittu potilailla, joilla on maksan vajaatoiminta. Kliinisissä tutkimuksissa oli mukana lievää maksan vajaatoimintaa sairastavia potilaita. Keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole kokemusta. Okrelitsumabi on monoklonaalinen vasta-aine, ja se poistuu elimistöstä kataboloitumalla (ei niinkään maksan kautta metaboloitumalla), joten annoksen säätäminen ei oletettavasti ole maksan vajaatoimintaa sairastaville potilaille tarpeen (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Okrelitsumabin turvallisuutta ja tehoa 0–18 vuoden ikäisten lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Ocrevus 300 mg infuusiokonsentraatti (laskimoon annettava lääkemuoto) ei ole tarkoitettu annettavaksi ihon alle. Sitä saa antaa vain infuusiona laskimoon.
On tärkeää varmistaa valmisteen etiketistä, että potilaalle annetaan lääkemääräyksen mukaista oikeaa lääkemuotoa (laskimoon tai ihon alle).
Hoito voidaan aloittaa laskimoon tai ihon alle annettavalla okrelitsumabilla.
Valmiste annetaan laimentamisen jälkeen infuusiona laskimoon erillisen infuusiolinjan kautta. Infuusiota ei saa antaa laskimoon paineella eikä boluksena.
Jos potilaalla ei ole yhdenkään aiemman okrelitsumabi-infuusion aikana esiintynyt vakavaa infuusioreaktiota, seuraavat annokset voidaan antaa lyhytkestoisempana (2 tuntia) infuusiona (ks. taulukko 1, vaihtoehto 2).
Taulukko 1. Annos ja hoitoaikataulu
| Annettavaokrelitsumabimäärä | Infuusio-ohjeet | ||
Aloitusannos (600 mg) 2 infuusioon jaettuna | 1. infuusio | 300 mg 250 ml:ssa | • Aloita infuusio nopeudella 30 ml/tunti 30 minuutin ajan. • Nopeutta voidaan lisätä 30 ml/tunti lisäyksinä 30 minuutin välein enimmäisnopeuteen 180 ml/tunti saakka. • Jokainen infuusio tulisi antaa noin 2,5 tunnin kestoisena. |
2. infuusio (2 viikkoa myöhemmin) | 300 mg 250 ml:ssa | ||
Seuraavat annokset (600 mg) kerta-annos 6 kuukauden välein | Vaihtoehto 1 Infuusion kesto noin 3,5 tuntia | 600 mg 500 ml:ssa | • Aloita infuusio nopeudella 40 ml/tunti 30 minuutin ajan. • Nopeutta voidaan lisätä 40 ml/tunti lisäyksinä 30 minuutin välein enimmäisnopeuteen 200 ml/tunti saakka. • Jokainen infuusio tulisi antaa noin 3,5 tunnin kestoisena. |
| TAI | |||
Vaihtoehto 2 Infuusion kesto noin 2 tuntia | 600 mg 500 ml:ssa | • Aloita infuusio nopeudella 100 ml/tunti ensimmäisten 15 minuutin ajan. • Lisää infuusionopeus 200 ml:aan/tunti seuraavien 15 minuutin ajaksi. • Lisää infuusionopeus 250 ml:aan/tunti seuraavien 30 minuutin ajaksi. • Lisää infuusionopeus 300 ml:aan/tunti jäljellä olevien 60 minuutin ajaksi. • Jokainen infuusio tulisi antaa noin 2 tunnin kestoisena. | |
Laskimoon infuusiona annettava liuos valmistetaan laimentamalla konsentraatti infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta, lopulliseen okrelitsumabipitoisuuteen noin 1,2 mg/ml.
Ks. ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa kohdasta Käyttö- ja käsittelyohjeet.
Potilasta pitää tarkkailla infuusion aikana sekä vähintään tunnin ajan infuusion päättymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Parhaillaan sairastettava aktiivinen infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Potilaalla oleva vaikea-asteinen immuunipuutostila (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Tiedossa oleva aktiivinen syöpä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioreaktiot
Okrelitsumabihoitoon liittyy infuusioreaktioita, jotka saattavat olla yhteydessä sytokiinien vapautumiseen ja/tai muihin kemiallisiin välittäjäaineisiin.
Infuusioreaktioiden oireita saattaa esiintyä minkä tahansa okrelitsumabi-infuusion aikana, mutta niitä on raportoitu yleisemmin ensimmäisen infuusion aikana. Infuusioreaktioita voi esiintyä 24 tunnin ajan infuusion jälkeen (ks. kohta Haittavaikutukset). Reaktiot saattavat ilmetä kutinana, ihottumana, urtikariana, eryteemana, kurkun ärsytyksenä, suunielun kipuna, hengenahdistuksena, nielun tai kurkunpään turvotuksena, kaulan ja kasvojen punoituksena, hypotensiona, kuumeena, uupumuksena, päänsärkynä, heitehuimauksena, pahoinvointina, takykardiana ja anafylaksiana.
Ennen infuusiota
Vaikea-asteisten reaktioiden hoito
Tarkoituksenmukaiset resurssit vaikea-asteisten reaktioiden, kuten vakavien infuusioreaktioiden, yliherkkyysreaktioiden ja/tai anafylaktisten reaktioiden, hoitoon pitää olla saatavilla.
Hypotensio
Infuusioiden aikana saattaa esiintyä hypotensiota infuusioreaktiona. Siksi on harkittava verenpainelääkityksen keskeyttämistä 12 tunniksi ennen kutakin infuusiota sekä infuusion annon ajaksi. Potilaita, joilla on aiemmin ollut kongestiivista sydämen vajaatoimintaa (New York Heart Association III & IV; NYHA III-IV), ei tutkittu.
Esilääkitys
Potilaille on annettava esilääkitys infuusioon liittyvien reaktioiden ja niiden vaikeusasteen vähentämiseksi (ks. kohta Annostus ja antotapa).
Infuusion aikana
Jos potilaalla on vaikea-asteisia keuhko-oireita, kuten bronkospasmeja tai astman pahenemista, on ryhdyttävä seuraaviin toimenpiteisiin:
- infuusion anto on keskeytettävä heti pysyvästi
- oireenmukaista hoitoa on annettava
- potilasta on seurattava, kunnes keuhko-oireet ovat hävinneet, koska kliiniset oireet voivat pahentua alkuvaiheen paranemisen jälkeen.
Yliherkkyysreaktioita saattaa olla mahdotonta kliinisesti erottaa infuusioreaktioista niiden oireiden perusteella. Jos infuusion aikana epäillään yliherkkyysreaktiota, infuusion anto on lopetettava heti pysyvästi (ks. jäljempänä Yliherkkyysreaktiot).
Infuusion jälkeen
Potilasta pitää tarkkailla infuusioreaktioiden havaitsemiseksi vähintään tunnin ajan infuusion päättymisen jälkeen.
Lääkärin pitää kertoa potilaalle, että 24 tunnin sisällä infuusiosta voi ilmaantua jokin infuusioon liittyvä reaktio.
Ohjeet infuusioreaktion yhteydessä infuusioon tehtävistä muutoksista, ks. kohta Annostus ja antotapa.
Yliherkkyysreaktiot
Yliherkkyysreaktioita (akuutti allerginen reaktio lääkevalmisteelle) voi myös ilmetä. Tyypin 1 (IgE‑välitteiset) akuutit yliherkkyysreaktiot eivät välttämättä ole kliinisesti erotettavissa infuusioreaktioista.
Yliherkkyysreaktio voi ilmaantua minkä tahansa antokerran yhteydessä, mutta tyypillisesti niitä ei esiinny ensimmäisellä antokerralla. Seuraavilla antokerroilla aiempaa vaikeampiasteisten tai uudenlaisten vaikeiden oireiden ilmaantuessa on syytä huomioida yliherkkyysreaktion mahdollisuus. Potilasta, jolla tiedetään olevan IgE‑välitteinen yliherkkyys okrelitsumabille tai jollekin apuaineelle, ei saa hoitaa (ks. kohta Vasta-aiheet).
Infektiot
Jos potilaalla on aktiivinen infektio, okrelitsumabin antamista pitää siirtää myöhemmäksi, kunnes infektio on parantunut.
Potilaan immuniteetin tila suositellaan varmistamaan ennen infuusion antoa potilaalle, koska valmistetta ei tule antaa potilaille, joilla on vaikea-asteinen immuunipuutostila (esim. lymfopenia, neutropenia, hypogammaglobulinemia) (ks. kohdat Vasta-aiheet ja Haittavaikutukset).
Niiden potilaiden kokonaisosuus, joilla oli jokin vakava infektio, oli samankaltainen kuin verrokeilla (ks. kohta Haittavaikutukset). Hengenvaarallisten (4. asteen) ja kuolemaan johtavien (5. asteen) infektioiden esiintyvyys oli kaikissa hoitoryhmissä pieni, mutta primaaristi etenevää MS-tautia sairastavilla potilailla hengenvaaralliset ja kuolemaan johtaneet infektiot olivat yleisempiä okrelitsumabihoitoa saaneilla kuin lumehoitoa saaneilla potilailla (hengenvaaralliset: 1,6 % [Ocrevus-hoito] vs 0,4 % [lumehoito], ja kuolemaan johtaneet: 0,6 % [Ocrevus-hoito] vs 0 % [lumehoito]). Kaikki hengenvaaralliset infektiot paranivat ilman okrelitsumabihoidon lopettamista.
Primaaristi etenevää MS-tautia sairastavilla potilailla, joilla on nielemisvaikeuksia, on tavanomaista suurempi aspiraatiopneumonian riski. Näillä potilailla okrelitsumabihoito saattaa entisestään lisätä vaikea-asteisen pneumonian riskiä. Lääkärin pitää ryhtyä nopeasti toimenpiteisiin, jos potilaalla on pneumonia.
Progressiivinen multifokaalinen leukoenkefalopatia (PML)
Progressiivisen multifokaalisen leukoenkefalopatian aiheuttavaa JC‑virusinfektiota on havaittu hyvin harvoin anti-CD20-vasta-aineilla, mukaan lukien okrelitsumabilla, hoidetuilla potilailla. Infektioon on tällöin liittynyt useimmiten riskitekijöitä (potilasryhmä, esim. lymfopenia, korkea ikä, usean immunosuppressiivisen lääkkeen käyttö).
Lääkärin pitää tarkkailla PML:n varhaisvaiheen oireita ja löydöksiä, joita voivat olla uudenlaisten neurologisten oireiden ilmaantuminen tai aiempien löydösten ilmaantuminen tai paheneminen, sillä ne voivat olla samankaltaisia kuin MS-taudissa.
Jos PML:a epäillään, okrelitsumabihoito pitää keskeyttää. Tutkimuksia, kuten magneettikuvaus (MK) mieluiten varjoainetehosteisena (vertailu ennen hoitoa tehtyyn MK:n), JCV-DNA-määritys aivo-selkäydinnesteestä sekä toistuvat neurologiset tutkimukset, on harkittava. Jos PML varmistuu, hoito on lopetettava pysyvästi.
B-hepatiitin uudelleen aktivoituminen
Anti-CD20-vasta-aineilla hoitoa saaneilla potilailla on raportoitu hepatiitti B ‑viruksen (HBV) uudelleen aktivoitumista, mikä on johtanut joissain tapauksissa fulminantin hepatiitin kehittymiseen, maksan vajaatoimintaan ja kuolemaan.
Kaikille potilaille pitää ennen hoidon aloittamista tehdä HBV-seulonta paikallisten ohjeistojen mukaisesti. Aktiivista HBV-infektiota (eli aktiivinen infektio varmistunut positiivisilla HBsAg- ja anti-HB-testituloksilla) sairastaville potilaille ei pitäisi antaa okrelitsumabihoitoa (ks. kohta Vasta-aiheet). Jos potilaan serologinen testitulos on positiivinen ([eli negatiivinen HBsAg ja positiivinen HB-c-antigeeni (HBcAB +]; HBV:n kantaja [pinta-antigeenipositiivinen, HBsAg+]), maksatautien erikoislääkäriä pitää konsultoida ennen hoidon aloittamista, ja potilasta pitää seurata ja hoitaa paikallisten hoitokäytäntöjen mukaisesti B-hepatiitin uudelleenaktivoitumisen estämiseksi.
Viivästynyt neutropenia
Viivästyneesti ilmenevää neutropeniaa on raportoitu aikaisintaan 4 viikkoa viimeisimmän okrelitsumabi-infuusion jälkeen (ks. kohta Haittavaikutukset). Vaikka jotkut tapaukset olivat vaikeusasteen 3. tai 4. tapauksia, oli valtaosa tapauksista vaikeusasteen 1. tai 2. asteen tapauksia. Jos potilaalla on infektion oireita ja löydöksiä, veren neutrofiilimäärä suositellaan määrittämään.
Syövät
Kliinisten pivotaalitutkimusten kontrolloidulla aikajaksolla on havaittu syöpien (mukaan lukien rintasyöpien) määrän lisääntymistä okrelitsumabihoitoa saaneilla potilailla vertailuryhmään verrattuna. Esiintyvyys oli MS-potilaiden oletetun taustaesiintyvyyden mukainen. Kliinisten pivotaalitutkimusten kontrolloidulla jaksolla ja avoimessa jatkovaiheessa annetun okrelitsumabihoidon jatkuttua yhtäjaksoisesti noin 10 vuotta syöpien ilmaantuvuus oli edelleen MS-potilaiden oletetun taustaesiintyvyyden mukainen. Jos potilaalla tiedetään olevan aktiivinen syöpä, okrelitsumabihoitoa ei saa antaa (ks. kohta Vasta-aiheet). Jos potilaalla tiedetään olevan syöpien riskitekijöitä tai potilaan sairastaman syövän uusiutumista seurataan aktiivisesti, potilaan hoidon yksilölliset hyödyt ja riskit pitää arvioida. Potilaan pitää noudattaa tavanomaisia paikallisia suosituksia rintasyöpäseulonnoista.
Vaikea-asteisesti immuunipuutteisten potilaiden hoito
Vaikea-asteisesti immuunipuutteisia potilaita ei saa hoitaa ennen kuin immuunipuutostila korjautuu (ks. kohta Vasta-aiheet).
Okrelitsumabin käyttö muiden autoimmuunisairauksien yhteydessä samanaikaisesti käytettävien immunosuppressiivisten lääkkeiden (esim. pitkäaikaisesti käytettävien kortikosteroidien, ei-biologisten ja biologisten taudinkulkua muuntavien reumalääkkeiden [DMARD], mykofenolaattimofetiilin, syklofosfamidin, atsatiopriinin) kanssa lisäsi vakavia infektioita, mukaan lukien opportunisti-infektioita. Infektioita olivat epätyypillinen keuhkokuume ja pneumocystis jirovecin aiheuttama keuhkokuume, vesirokkoviruksen aiheuttama keuhkokuume, tuberkuloosi ja histoplasmoosi näihin kuitenkaan rajoittumatta. Jotkut näistä infektioista johtivat harvinaisissa tapauksissa potilaan kuolemaan. Lisäanalyysissa todettiin seuraavien tekijöiden liittyvän vakavien infektioiden riskiin: MS-taudin hoitoon suositeltua suuremmat okrelitsumabiannokset, muut samanaikaiset sairaudet ja immunosuppressiivisten lääkkeiden/kortikosteroidien pitkäaikaiskäyttö.
Muiden immunosuppressiivisten lääkkeiden samanaikaista käyttöä okrelitsumabin kanssa ei suositella, lukuun ottamatta pahenemisvaiheiden oireenmukaiseen hoitoon käytettäviä kortikosteroideja. Tiedot siitä, liittyykö kliinisessä hoidossa pahenemisvaiheiden samanaikaiseen oireenmukaiseen steroidihoitoon lisääntynyt infektioriski, ovat suppeita. Okrelitsumabilla tehdyissä MS-tautia koskevissa pivotaalitutkimuksissa pahenemisvaiheiden kortikosteroidihoitoon ei liittynyt vakavien infektioiden riskin lisääntymistä.
Kun okrelitsumabihoito aloitetaan immunosuppressiivisen hoidon jälkeen tai kun immunosuppressiivista hoitoa aloitetaan okrelitsumabihoidon jälkeen, on otettava huomioon päällekkäisten farmakodynaamisten vaikutusten mahdollisuus (ks. kohta Farmakodynamiikka). Okrelitsumabin määräämisessä on oltava varovainen ja huomioitava muiden taudinkulkua muuntavien MS-lääkkeiden farmakodynamiikka.
Rokotukset
Eläviä tai heikennettyjä eläviä viruksia sisältävillä rokotteilla annettujen rokotusten turvallisuutta okrelitsumabihoidon jälkeen ei ole tutkittu, joten rokotuksia eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla ei suositella hoidon aikana eikä ennen kuin B-solut ovat palautuneet. Kliinisissä tutkimuksissa ajan mediaani B-solujen palautumiseen oli 72 viikkoa (ks. kohta Farmakodynamiikka).
Satunnaistetussa avoimessa tutkimuksessa aaltomaista MS-tautia sairastavia potilaita rokotettiin seuraavilla rokotteilla: tetanustoksoidi, 23-valenttinen pneumokokkipolysakkaridi (PPV23) ilman tehosteannosta tai tehosteannoksen kanssa, KLH (keyhole limpet haemocyanin) ‑neoantigeeni ja kausi-influenssarokote. Potilaat saivat humoraalisen, tosin alentuneen vasteen rokotteille (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka).
Okrelitsumabihoitoa saavien potilaiden kausi-influenssarokotukseen suositellaan käyttämään inaktivoitua virusta sisältävää rokotetta.
Lääkärin pitää tarkistaa potilaan rokotustilanne, kun potilaalle harkitaan okrelitsumabihoitoa. Jos potilas tarvitsee rokotuksia, ne pitää antaa viimeistään 6 viikkoa ennen hoidon aloittamista.
Sikiöaikainen altistuminen okrelitsumabille sekä vastasyntyneiden ja imeväisikäisten rokottaminen eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla
Okrelitsumabille raskauden aikana altistuneen äidin imeväisikäisellä lapsella saattaa olla B‑solupuutos. Tästä syystä rokottamista eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla suositellaan siirtämään siihen asti, kunnes B‑solumäärä on korjautunut normaaliksi. Vastasyntyneiltä ja imeväisikäisiltä suositellaan tämän vuoksi mittaamaan CD19-positiivisten B‑solujen määrä ennen rokotuksen antamista.
Kaikki muut kuin eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävät rokotteet suositellaan antamaan paikallisen rokotusohjelman mukaisesti. Rokotteista saadun vasta-ainetiitterin mittaamista pitää harkita, jotta voidaan tarkistaa, onko henkilö saanut suojaavan immuunivasteen, sillä rokotuksen teho saattaa olla tavanomaista heikompi.
Rokotuksen turvallisuudesta ja ajoituksesta pitää keskustella lasta hoitavan lääkärin kanssa (ks. kohta Raskaus ja imetys).
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli se on olennaisesti natriumiton.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty, koska yhteisvaikutuksia sytokromi P450 ‑entsyymien, muiden metaboloivien entsyymien tai kuljettajaproteiinien kanssa ei oletettavasti esiinny.
Rokotukset
Eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla annettujen rokotusten turvallisuutta okrelitsumabihoidon jälkeen ei ole tutkittu.
Seuraavien rokotteiden vaikutuksista okrelitsumabihoitoa saavilla potilailla on olemassa tietoa: tetanustoksoidi-, 23-valenttinen pneumokokkipolysakkaridi- (PPV23), KLH (keyhole limpet haemocyanin) -neoantigeenia sisältävä rokote ja kausi-influenssarokote (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Kahden vuoden hoidon jälkeen niiden potilaiden osuus, joiden vasta-ainetiitteri S. pneumoniae ‑bakteeria, sikotautia, vihurikokkoa ja vesirokkoa vastaan oli positiivinen, oli yleisesti samankaltainen kuin lähtötilanteessa.
Immunosuppressiiviset lääkkeet
Muiden immunosuppressiivisten lääkkeiden käyttöä samanaikaisesti okrelitsumabihoidon kanssa ei suositella, lukuun ottamatta pahenemisvaiheiden oireenmukaiseen hoitoon käytettäviä kortikosteroideja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Hedelmällisessä iässä olevien naisten on käytettävä ehkäisyä okrelitsumabihoidon aikana ja 4 kuukauden ajan viimeisen annetun okrelitsumabiannoksen jälkeen.
Raskaus
Okrelitsumabin käytöstä raskaana oleville naisille on vain vähän tietoja. Okrelitsumabi on immunoglobuliini G (IgG). IgG:n tiedetään läpäisevän istukkaesteen. Okrelitsumabille sikiöaikana altistuneiden vastasyntyneiden tai imeväisikäisten rokotus eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla pitää harkita siirrettäväksi myöhemmäksi. Okrelitsumabille kohdussa altistuneista vastasyntyneistä ja imeväisikäisistä ei ole kerätty B‑solumäärää koskevia tietoja, joten vastasyntyneen ja imeväisen B‑solupuutoksen mahdollista kestoaikaa ei tiedetä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muille anti-CD20-vasta-aineille raskauden aikana altistuneille äideille syntyneillä lapsilla on raportoitu ohimenevää perifeeristä B-solupuutosta ja lymfopeniaa. Myös eläimillä tehdyissä tutkimuksissa havaittiin in utero B‑solupuutosta.
Eläinkokeet (alkio- ja sikiötoksisuus) eivät osoittaneet teratogeenisia vaikutuksia. Pre- ja postnataalista kehitystä koskevissa tutkimuksissa havaittiin lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Okrelitsumabin käyttöä tulee välttää raskauden aikana elleivät mahdolliset hyödyt äidille ole sikiölle mahdollisesti aiheutuvia riskejä suuremmat.
Imetys
Ihmisen immunoglobuliinien G (IgG) tiedetään erittyvän äidinmaitoon muutaman päivän ajan synnytyksen jälkeen (ensimaitojakso). Pian sen jälkeen pitoisuudet laskevat mataliksi.
Prospektiivisessa, avoimessa MN42989-monikeskustutkimuksessa (SOPRANINO) 13 imettävää naista sai okrelitsumabia 2,0 kuukautta (mediaani) synnytyksen jälkeen (vaihteluväli 0,5–5,0 kuukautta). Äidinmaidossa havaittiin pieniä okrelitsumabipitoisuuksia 60 päivän ajan ensimmäisen infuusion jälkeen, jonka äiti sai synnytyksen jälkeen (vauvan suhteellisen annoksen mediaani 0,27 % [vaihteluväli 0,0–1,8 %]), mikä osoittaa okrelitsumabia siirtyvän minimaalisesti äidinmaitoon. 30 päivää ensimmäisen infuusion jälkeen, jonka äiti sai synnytyksen jälkeen, okrelitsumabia ei ollut enää havaittavissa imetettyjen vauvojen saatavissa olleissa seeruminäytteissä (n = 9) ja vauvojen B‑solumäärä oli kaikissa saatavissa olleissa verinäytteissä (n = 10) normaaliarvoissa. Okrelitsumabin ei havaittu vaikuttavan imetettyjen vauvojen terveyteen, kasvuun ja kehitykseen 44,6 viikon seurantajakson aikana (vaihteluväli 8,6–62,7 viikkoa).
Vaikka okrelitsumabille äidinmaidon kautta mahdollisesti altistuneista ja eläviä tai heikennettyjä eläviä rokotteita saaneista vauvoista ei ole saatavissa kliinisiä tietoja, riskejä ei oletettavasti ole, sillä näiden vauvojen B‑solupitoisuuksien havaittiin olevan normaalit eikä seerumissa ollut havaittavia okrelitsumabipitoisuuksia.
Erillisessä prospektiivisessa kliinisessä tutkimuksessa 29 imettävällä naisella, jotka saivat okrelitsumabia 4,3 kuukautta (mediaani) (vaihteluväli 0,1–36 kuukautta) synnytyksen jälkeen, havaittiin äidinmaidossa pieniä okrelitsumabipitoisuuksia (vauvan suhteellisen annoksen mediaani 0,1 % [vaihteluväli 0,07–0,7 %]) 90 päivän ajan ensimmäisen infuusion jälkeen, jonka äiti sai synnytyksen jälkeen. 21:n imetetyn vauvan vähintään kaksi viikkoa kestäneessä seurannassa todettiin normaali kasvu ja kehitys yhteen ikävuoteen saakka.
Okrelitsumabia voidaan käyttää imetyksen aikana aloittamalla sen käyttö muutama päivä synnytyksen jälkeen.
Hedelmällisyys
Cynomolgus-apinauroksilla ja -naarailla tehtyjen hedelmällisyyttä koskevien prekliinisten tutkimusten tulokset eivät viittaa erityiseen haittaan ihmisille.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ocrevus-valmisteella ei ole haitallista vaikutusta ajokykyyn tai koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tärkeimmät ja yleisimmin raportoidut haittavaikutukset kliinisten pivotaalitutkimusten kontrolloidulla jaksolla olivat infuusioreaktiot (34,3 %:lla aaltomaista MS-tautia sairastavista ja 40,1 %:lla primaaristi etenevää MS‑tautia sairastavista) ja infektiot (58,5 %:lla aaltomaista MS-tautia sairastavista ja 72,2 %:lla primaaristi etenevää MS-tautia sairastavista) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisten pivotaalitutkimusten kontrolloidulla jaksolla oli mukana yhteensä 2 376 potilasta, joista 1 852 potilasta osallistui avoimeen jatkovaiheeseen. Kaikki potilaat siirtyivät okrelitsumabihoitoon avoimen jatkovaiheen aikana. 1 155 potilasta oli mukana avoimen jatkovaiheen loppuun asti, jolloin kontrolloidun jakson ja avoimen jatkovaiheen aikana annettu okrelitsumabihoito kesti yhtäjaksoisesti noin 10 vuotta (15 515 potilasvuoden altistus). Kontrolloidulla jaksolla ja avoimessa jatkovaiheessa todettu turvallisuusprofiili oli yhdenmukainen kontrolloidulla jaksolla todetun yleisen turvallisuusprofiilin kanssa.
Haittavaikutustaulukko
Kliinisten pivotaalitutkimusten kontrolloidulla jaksolla raportoidut ja spontaaniraportointiin perustuvat haittavaikutukset luetellaan jäljempänä taulukossa 2. Haittavaikutukset luetellaan MedDRA-elinjärjestelmäluokituksen ja esiintyvyysluokkien mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin elinjärjestelmäluokassa haittavaikutuksen yleisyyden mukaan alenevassa järjestyksessä.
Taulukko 2. Haittavaikutukset
MedDRA Elinjärjestelmäluokka | Hyvin yleiset | Yleiset | Tuntematon |
| Infektiot | Ylähengitysteiden infektio, nasofaryngiitti, influenssa | Sinuiitti, bronkiitti, huuliherpes, gastroenteriitti, hengitystie-infektio, virusinfektio, herpes zoster ‑infektio, konjunktiviitti, selluliitti | |
| Veri ja imukudos | Neutropenia | Viivästynyt neutropenia2 | |
| Hengityselimet, rintakehä ja välikarsina | Yskä, katarri | ||
| Tutkimukset | Veren pienentynyt immunoglobuliini M ‑pitoisuus | Veren pienentynyt immunoglobuliini G ‑pitoisuus | |
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvät reaktiot1 |
1 Ks. Valikoitujen haittavaikutusten kuvaus.
2 Havaittu valmisteen markkinoille tulon jälkeen.
Valikoitujen haittavaikutusten kuvaus
Infuusioreaktiot
Aaltomaista MS-tautia ja primaaristi etenevää MS-tautia koskeneissa tutkimuksissa infuusioreaktioiden oireita olivat, näihin kuitenkaan rajoittumatta: kutina, ihottuma, urtikaria, eryteema, kasvojen ja kaulan punoitus, hypotensio, kuume, uupumus, päänsärky, heitehuimaus, kurkun ärsytys, suunielun kipu, hengenahdistus, nielun tai kurkunpään turvotus, pahoinvointi, takykardia. Kontrolloiduissa tutkimuksissa ei esiintynyt kuolemaan johtaneita infuusioon liittyneitä reaktioita. Valmisteen markkinoille tulon jälkeen infuusioreaktioiden oireena on esiintynyt lisäksi anafylaksiaa.
Aktiivisella vertailuvalmisteella kontrolloiduissa kliinisissä tutkimuksissa (aaltomainen MS-tauti) infuusioreaktio oli okrelitsumabihoitoryhmässä yleisin haittavaikutus. Sen kokonaisilmaantuvuus oli 34,3 %, kun ilmaantuvuus interferonibeeta-1a:ta saaneessa hoitoryhmässä (lumeinfuusio) oli 9,9 %. Infuusioreaktioiden ilmaantuvuus oli suurin 1. annoksen 1. infuusion aikana (27,5 %) ja väheni ajan mittaan alle 10 %:iin 4. annoksen yhteydessä. Valtaosa infuusioreaktioista oli kummassakin hoitoryhmässä lieviä tai keskivaikeita. Okrelitsumabihoitoa saaneiden potilaiden infuusioreaktioista 21,7 % oli lieviä ja 10,1 % keskivaikeita. 2,4 % infuusioreaktioista oli vaikea-asteisia ja 0,1 % hengenvaarallisia.
Lumekontrolloidussa kliinisessä tutkimuksessa (primaaristi etenevä MS-tauti) infuusioreaktio oli okrelitsumabihoitoryhmässä yleisin haittavaikutus. Sen kokonaisilmaantuvuus oli 40,1 %, kun ilmaantuvuus lumeryhmässä oli 25,5 %. Infuusioon liittyvien reaktioiden ilmaantuvuus oli suurin 1. annoksen 1. infuusion aikana (27,4 %) ja väheni seuraavien annosten yhteydessä alle 10 %:iin 4. annoksen yhteydessä. Kummassakin ryhmässä suuremmalla osalla potilaista esiintyi infuusioreaktio kummankin annoksen ensimmäisen infuusion yhteydessä verrattuna saman annoksen toiseen infuusioon. Valtaosa infuusioreaktioista oli lieviä tai keskivaikeita. Okrelitsumabihoitoa saaneiden potilaiden infuusioreaktioista 26,7 % oli lieviä ja 11,9 % keskivaikeita; 1,4 % infuusioon liittyvistä reaktioista oli vaikea-asteisia. Hengenvaarallisia infuusioreaktioita ei esiintynyt. Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Aaltomaista MS-tautia ja primaaristi etenevää MS-tautia koskeneiden kliinisten tutkimusten kontrolloidun jakson ja avoimen jatkovaiheen aikana potilaat saivat noin 20 annosta okrelitsumabia. Infuusioreaktioiden ilmaantuvuus väheni aaltomaista MS-tautia sairastavilla potilailla alle 4 %:iin avoimen jatkovaiheen 4. annokseen mennessä ja primaaristi etenevää MS-tautia sairastavilla potilailla alle 5 %:iin avoimen jatkovaiheen 5. annokseen mennessä. Infuusioon liittyvien reaktioiden ilmaantuvuus pysyi pienenä seuraavien avoimessa jatkovaiheessa annettujen annosten yhteydessä. Valtaosa avoimessa jatkovaiheessa ilmenneistä infuusioreaktioista oli lieviä.
Seuraavien annosten vaihtoehtoinen lyhytkestoisempi infuusio
Tutkimuksessa (MA30143 lyhytkestoisempaa infuusiota koskeva alatutkimus) selvitettiin lyhytkestoisemman (2 tuntia) okrelitsumabi-infuusion turvallisuusprofiilia aaltomaista MS-tautia sairastavilla potilailla. Siinä todettiin, että infuusioreaktioiden oireiden ilmaantuvuus, voimakkuus ja tyyppi olivat samankaltaiset kuin annettaessa infuusio 3,5 tunnin kestoisena (ks. kohta Farmakodynamiikka) . Tarvittavien interventoiden kokonaismäärä oli matala molemmassa infuusioryhmässä, lyhytkestoisemmassa infuusioryhmässä (2 tuntia) tarvittiin kuitenkin enemmän interventoita (infuusion hidastaminen tai väliaikainen keskeyttäminen) infuusioreaktioiden hoitamiseen kuin 3,5 tunnin kestoisena infuusiona saaneiden ryhmässä (8,7% verrattuna 4,8%).
Infektio
Aktiivisella vertailuvalmisteella kontrolloiduissa aaltomaista MS-tautia koskeneissa tutkimuksissa infektioita esiintyi 58,5 %:lla okrelitsumabihoitoa saaneista potilaista ja 52,5 %:lla interferonibeeta-1a:ta saaneista potilaista. Vakavia infektioita esiintyi 1,3 %:lla okrelitsumabihoitoa saaneista potilaista ja 2,9 %:lla interferonibeeta-1a:ta saaneista potilaista. Primaaristi etenevää MS-tautia koskeneessa lumekontrolloidussa tutkimuksessa infektioita esiintyi 72,2 %:lla okrelitsumabihoitoa saaneista potilaista ja 69,9 %:lla lumehoitoa saaneista potilaista. Vakavia infektioita esiintyi 6,2 %:lla okrelitsumabihoitoa saaneista potilaista ja 6,7 %:lla lumehoitoa saaneista potilaista.
Sekä aaltomaista MS-tautia että primaaristi etenevää MS-tautia koskeneissa tutkimuksissa kaikki potilaat siirtyivät okrelitsumabihoitoon tutkimuksen avoimen jakson aikana. Aaltomaista MS-tautia ja primaaristi etenevää MS-tautia sairastavilla potilailla vakavien infektioiden kokonaisriski ei ollut avoimen jatkovaiheen aikana suurempi kuin kontrolloidulla jaksolla havaittu riski. Kuten kontrolloidulla jaksolla havaittiin, vakavien infektioiden esiintyvyys primaaristi etenevää MS-tautia sairastavilla potilailla oli edelleen yleisempää kuin aaltomaista MS-tautia sairastavilla potilailla.
Muihin autoimmuunisairauksiin kuin MS-tautiin liittyvien vakavien infektioiden riskitekijöistä aiemmin tehdyn analyysin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) mukaisesti vakavien infektioiden riskitekijöistä tehtiin monen muuttujan analyysi noin 10 vuoden kumulatiivisista altistustiedoista, jotka oli saatu kliinisten pivotaalitutkimusten kontrolloidusta jaksosta ja avoimesta jatkovaiheesta. Vakavien infektioiden riskitekijöitä aaltomaista MS-tautia sairastavilla potilailla ovat vähintään yksi muu samanaikainen sairaus, äskettäinen kliininen pahenemisvaihe ja liikunta- ja toimintakyvyn heikentymistä osoittavat pisteet (Expanded Disability Status Scale (EDSS) -pisteet) ≥ 6,0. Vakavien infektioiden riskitekijöitä primaaristi etenevää MS-tautia sairastavilla potilailla ovat painoindeksi yli 25 kg/m2, vähintään kaksi muuta samanaikaista sairautta, EDSS-pisteet ≥ 6,0 ja IgM viitevälin alarajan alapuolella (< LLN). Muita samanaikaisia sairauksia olivat muun muassa sydän- ja verisuonitaudit, munuais- ja virtsatiesairaudet, aiemmat infektiot ja masennus.
Hengitystieinfektiot
Hengitystieinfektioiden osuus oli suurempi okrelitsumabihoitoa saaneilla potilailla verrattuna interferonibeeta-1a- ja lumehoitoa saaneisiin potilaisiin.
Aaltomaista MS-tautia koskeneissa tutkimuksissa ylähengitystieinfektioita esiintyi 39,9 %:lla okrelitsumabihoitoa saaneista potilaista ja 33,2 %:lla interferonibeeta-1a:ta saaneista potilaista, ja alahengitystieinfektioita esiintyi 7,5 %:lla okrelitsumabihoitoa saaneista potilaista ja 5,2 %:lla interferonibeeta-1a:ta saaneista potilaista.
Primaaristi etenevää MS-tautia koskeneessa tutkimuksessa ylähengitystieinfektioita esiintyi 48,8 %:lla okrelitsumabihoitoa saaneista potilaista ja 42,7 %:lla lumehoitoa saaneista potilaista, ja alahengitystieinfektioita esiintyi 9,9 %:lla okrelitsumabihoitoa saaneista potilaista ja 9,2 %:lla lumehoitoa saaneista potilaista.
Okrelitsumabihoitoa saaneilla potilailla raportoidut hengitystieinfektiot olivat pääasiassa lieviä tai keskivaikeita (80–90 %).
Herpes
Aktiivisella vertailuvalmisteella kontrolloiduissa (aaltomainen MS-tauti) kliinisissä tutkimuksissa herpesinfektioita raportoitiin yleisemmin okrelitsumabihoitoa saaneilla potilailla kuin interferonibeeta-1a:ta saaneilla potilailla, mukaan lukien herpes zoster (2,1 % vs 1,0 %), herpes simplex (0,7 % vs 0,1 %), huuliherpes (3,0 % vs 2,2 %), sukupuolielinherpes (0,1 % vs 0 %) ja herpesvirusinfektio (0,1 % vs 0 %). Kaikki infektiot olivat lieviä tai keskivaikeita, yhtä kolmannen asteen tapahtumaa lukuun ottamatta, ja potilaat paranivat normaalin hoitokäytännön mukaisella hoidolla.
Lumekontrolloidussa (primaaristi etenevä MS-tauti) kliinisessä tutkimuksessa suurempi osa huuliherpesinfektioista (2,7 % vs 0,8 %) todettiin okrelitsumabihoitoryhmässä.
Laboratorioarvojen poikkeavuudet
Immunoglobuliinit
Okrelitsumabihoito pienensi kliinisten pivotaalitutkimusten kontrolloitujen jaksojen aikana immunoglobuliinien kokonaispitoisuutta siten, että IgM-pitoisuus pieneni voimakkaimmin.
Tiedot kliinisten pivotaalitutkimusten kontrolloidusta jaksosta ja avoimesta jatkovaiheesta ovat osoittaneet yhteyden IgG:n (ja harvemmin IgM:n tai IgA:n) pitoisuuksien pienenemisen ja vakavien infektioiden lisääntymisen välillä. Aaltomaista MS-tautia sairastavista potilaista 2,1 %:lla ja primaaristi etenevää MS-tautia sairastavista potilaista 2,3 %:lla ilmeni vakava infektio, kun IgG oli viitevälin alarajan alapuolella (< LLN). Ero vakavien infektioiden esiintyvyydessä potilailla, joilla IgG oli viitevälin alarajan alapuolella (< LLN), verrattuna potilaisiin, joilla IgG oli viitevälin alarajalla tai sen yläpuolella (≥ LLN), ei kasvanut ajan mittaan. Sellaisten jaksojen aikana, jolloin immunoglobuliinipitoisuus oli viitevälin alarajan alapuolella, todettujen vakavien infektioiden tyyppi, vaikeusaste, piilevyys, kesto ja lopputulos olivat yhdenmukaisia okrelitsumabihoitoa kontrolloidulla jaksolla ja avoimessa jatkovaiheessa saaneilla potilailla todettujen kaikkien vakavien infektioiden kanssa. Aaltomaista MS-tautia ja primaaristi etenevää MS-tautia sairastavien potilaiden keskimääräiset IgG-pitoisuudet pysyivät viitevälin alarajan yläpuolella koko 10 vuotta kestäneen yhtäjaksoisen okrelitsumabihoidon aikana.
Lymfosyytit
Aaltomaisen MS-taudin yhteydessä lymfosyyttien vähenemistä viitevälin alarajan alapuolelle (< LLN) havaittiin 20,7 %:lla okrelitsumabihoitoa saaneista potilaista ja 32,6 %:lla interferonibeeta-1a:ta saaneista potilaista. Primaaristi etenevän MS-taudin yhteydessä lymfosyyttien vähenemistä viitevälin alarajan alapuolelle (< LLN) havaittiin 26,3 %:lla okrelitsumabihoitoa saaneista potilaista ja 11,7 %:lla lumehoitoa saaneista potilaista.
Valtaosa raportoidusta lymfosyyttien vähenemisestä okrelitsumabihoitoa saaneilla potilailla oli vaikeusasteeltaan 1 (< LLN – 800 solua/mm3) ja 2 (500–800 solua/mm3). Okrelitsumabiryhmässä noin 1 %:lla potilaista oli 3. asteen lymfopenia (200–500 solua/mm3). Yhdelläkään potilaalla ei raportoitu 4. asteen lymfopeniaa (< 200 solua/mm3).
Okrelitsumabihoitoa saaneilla potilailla havaittiin vakavien infektioiden lisääntymistä sellaisten jaksojen aikana, jolloin lymfosyyttien kokonaismäärän väheneminen varmistui. Vakavia infektioita oli liian vähän, jotta siitä voitaisiin tehdä varmoja päätelmiä.
Neutrofiilit
Aktiivisella vertailuvalmisteella kontrolloidulla (aaltomainen MS-tauti) hoitojaksolla neutrofiilien vähenemistä normaaliarvojen alarajan alapuolelle (< LLN) havaittiin 14,7 %:lla okrelitsumabihoitoa saaneista potilaista ja 40,9 %:lla interferonibeeta-1a:ta saaneista potilaista. Lumekontrolloidussa (primaaristi etenevä MS-tauti) kliinisessä tutkimuksessa niiden okrelitsumabihoitoa saaneiden potilaiden osuus, joiden neutrofiilimäärä väheni, oli suurempi (12,9 %) kuin lumehoitoa saaneilla potilailla (10,0 %). Toisen tai vaikeampiasteisen neutropenian osuus oli suurempi okrelitsumabiryhmässä (4,3 %) kuin lumeryhmässä (1,3 %). Neljännen asteen neutropeniaa oli okrelitsumabiryhmässä noin 1 %:lla potilaista ja 0 %:lla lumeryhmässä.
Valtaosa neutrofiilimäärän vähenemisistä oli ohimenevää (havaittiin tietyllä okrelitsumabihoitoa saavalla potilaalla vain kerran) ja vaikeusasteeltaan 1 (< LLN – 1500 solua/mm3) tai 2 (1000−1500 solua/mm3). Kolmannen tai neljännen asteen neutropeniaa oli kaikkiaan noin 1 %:lla okrelitsumabiryhmän potilaista. Yksi potilas, jolla oli 3. asteen neutropenia (500−1000 solua/mm3) ja yksi potilas, jolla oli 4. asteen neutropenia (< 500 solua/mm3), tarvitsivat spesifistä hoitoa granulosyyttejä stimuloivilla kasvutekijöillä, minkä jälkeen potilaat jatkoivat okrelitsumabihoitoa. Neutropenia voi ilmetä useita kuukausia okrelitsumabin annon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muuta
Yksi 2000 mg:n okrelitsumabiannoksen saanut potilas kuoli tuntemattomasta syystä kehittyneen tulehdusreaktio-oireyhtymän (SIRS) seurauksena 12 viikkoa viimeisestä infuusiosta tehdyn magneettikuvauksen jälkeen. Tulehdusreaktio-oireyhtymän kehittymiseen saattoi vaikuttaa anafylaktinen reaktio magneettikuvauksessa käytetylle gadoliniumia sisältäneelle varjoaineelle.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Hyväksyttyä okrelitsumabiannosta suuremmista annoksista on vain vähän kliinistä kokemusta. Tähän mennessä suurin tutkittu annos MS-potilailla on 2000 mg annosteltuna kahtena 1000 mg:n infuusiona laskimoon kahden viikon välein (aaltomaista MS-tautia sairastavilla potilailla tehty vaiheen II annoshakututkimus) sekä 1200 mg annosteltuna injektiona ihon alle (vaiheen Ib annoshakututkimus). Haittavaikutukset sopivat kliinisissä pivotaalitutkimuksissa todettuun turvallisuusprofiiliin.
Yliannokseen ei ole spesifistä vasta-ainetta. Infuusio on keskeytettävä heti, ja potilasta on tarkkailtava infuusioon liittyvien reaktioiden havaitsemiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, monoklonaaliset vasta-aineet, ATC-koodi: L04AG08.
Vaikutusmekanismi
Okrelitsumabi on yhdistelmä-DNA-tekniikalla tuotettu humanisoitu monoklonaalinen vasta-aine, jonka vaikutus kohdistuu selektiivisesti CD20:ta ilmentäviin B-soluihin.
CD20 pinta-antigeenia esiintyy pre-B-solujen, kypsien ja B-muistisolujen pinnalla, mutta ei lymfoidisissa kantasoluissa tai plasmasoluissa.
Tarkkaa mekanismia, johon okrelitsumabin kliininen hoitovaikutus MS-taudissa perustuu, ei täysin tunneta, mutta sen oletetaan liittyvän CD20:ta ilmentävien B-solujen määrän ja toiminnan vähenemisestä aiheutuvaan immunomodulaatioon. Solun pintaan sitoutunut okrelitsumabi hävittää CD20:ta ilmentäviä B-soluja selektiivisesti vasta-aineriippuvaisen solujen fagosytoosin (antibody-dependent cellular phagocytosis, ADCP), vasta-aineriippuvaisen soluvälitteisen solutuhon (antibody-dependent cellular cytotoxicity, ADCC), komplementtivälitteisen solutuhon (complement-dependent cytotoxicity, CDC) ja ohjelmoidun solukuoleman välityksellä. B-solujen elpymiskyky ja aiempi humoraalinen immuunivaste säilyvät. Myöskään luonnollinen immuniteetti ja T-solujen kokonaismäärä eivät muutu.
Farmakodynaamiset vaikutukset
Okrelitsumabihoito aiheuttaa oletettuna farmakologisena vaikutuksena veressä nopean CD19-positiivisten B-solujen vähenemisen 14. hoidon jälkeiseen päivään mennessä (ensimmäinen arviointiajankohta). Tämä säilyi koko hoitojakson ajan. B-solumäärän laskentaan käytetään CD19-positiivisia B-soluja, sillä okrelitsumabi häiritsee määrityksessä CD20-positiivisten B-solujen tunnistamista.
Vaiheen III tutkimuksissa enintään 5 %:lla potilaista todettiin okrelitsumabiannosten välillä vähintään yhden kerran B-solujen palautumista yli viitevälin alarajan [LLN] tai lähtötilanteen. B-solujen vähenemisen laajuus ja kesto oli ensisijaisesti etenevää MS-tautia ja MS-tautia koskeneissa tutkimuksissa yhdenmukainen.
Pisin seuranta-aika viimeisen infuusion jälkeen (vaiheen II tutkimus WA21493, N = 51) osoittaa, että mediaaniaika B-solujen palautumiseen (takaisin lähtötasolle tai viitevälin alarajalle sen mukaan, kumpi näistä tapahtui ensin) oli 72 viikkoa (vaihteluväli 27–175 viikkoa). B-solut olivat palautuneet viitevälin alarajalle tai lähtötilanteeseen 90 %:lla kaikista potilaista noin kahden ja puolen vuoden kuluessa viimeisen infuusion jälkeen.
Kliininen teho ja turvallisuus
Aaltomainen MS-tauti (RMS)
Okrelitsumabin tehoa ja turvallisuutta selvitettiin kahdessa satunnaistetussa, kaksoissokkoutetussa (double-blind, double-dummy), aktiivisella vertailuvalmisteella kontrolloidussa kliinisessä tutkimuksessa (WA21092 ja WA21093), joiden tutkimusasetelma oli identtinen ja joissa mukana olleet potilaat sairastivat aaltomaista MS-tautityyppiä (McDonaldin vuoden 2010 kriteerien mukaisesti) ja heillä oli näyttöä taudin aktiivisuudesta kliinisten piirteiden tai kuvantamislöydösten perusteella viimeisen kahden vuoden aikana. Tiivistelmä tutkimusasetelmasta ja tutkimuksen potilasjoukon lähtötilanteen ominaisuuksista on esitetty taulukossa 3.
Demografiset tiedot ja lähtötilanteen ominaisuudet olivat tasapainossa kahden hoitoryhmän välillä. Okrelitsumabihoitoa saaville potilaille (ryhmä A) annettiin 600 mg kuuden kuukauden välein (ensimmäinen annos kahtena 300 mg:n infuusiona laskimoon kahden viikon välein, jonka jälkeen seuraavat annokset 600 mg:n kertainfuusiona laskimoon). Ryhmän B potilaat saivat 44 mikrog interferonibeeta-1a:ta injektiona ihon alle kolme kertaa viikossa.
Taulukko 3. Tutkimusasetelma sekä demografiset ja lähtötilanteen tiedot
| Tutkimus 1 | Tutkimus 2 | |||
| Tutkimuksen nimi | WA21092 (OPERA I) (n = 821) | WA21093 (OPERA II) (n = 835) | ||
| Tutkimusasetelma | ||||
| Tutkimuksen potilasjoukko | Aaltomaista (RMS) MS-tautia sairastavat potilaat | |||
| Sairaushistoria seulonnassa | Vähintään kaksi pahenemisvaihetta edellisten kahden vuoden aikana tai yksi pahenemisvaihe edellisen vuoden aikana; EDSS* 0–5,5, raja-arvot mukaan lukien | |||
| Tutkimuksen kesto | 2 vuotta | |||
| Hoitoryhmät | Ryhmä A: 600 mg okrelitsumabia Ryhmä B: 44 mikrog interferonibeeta-1a s.c. (IFN) | |||
| Lähtötilanteen ominaisuudet | Okrelitsumabi 600 mg (n = 410) | IFN 44 mikrog (n = 411) | Okrelitsumabi 600 mg (n = 417) | IFN 44 mikrog (n = 418) |
| Keskimääräinen ikä (vuotta) | 37,1 | 36,9 | 37,2 | 37,4 |
| Iän vaihteluväli (vuotta) tutkimuksen sisäänottovaiheessa | 18–56 | 18–55 | 18–55 | 18–55 |
| Sukupuolijakauma (% miehiä/% naisia) | 34,1/65,9 | 33,8/66,2 | 35,0/65,0 | 33,0/67,0 |
| Sairauden kestoajan keskiarvo/mediaani diagnoosin jälkeen (vuotta) | 3,82/1,53 | 3,71/1,57 | 4,15/2,10 | 4,13/1,84 |
| Taudin kulkua muuntavilla lääkkeillä aiemmin hoitamattomia potilaita (%)** | 73,4 | 71,0 | 72,7 | 74,9 |
| Keskimääräinen pahenemisvaiheiden määrä kuluneena vuonna | 1,31 | 1,33 | 1,32 | 1,34 |
| Niiden potilaiden osuus, joilla gadoliniumilla tehostuvia T1-muutoksia | 42,5 | 38,1 | 39,0 | 41,4 |
| Keskimääräiset EDSS-pisteet* | 2,82 | 2,71 | 2,73 | 2,79 |
* Liikunta- ja toimintakyvyn heikentymistä osoittava pisteytys, Expanded Disability Status Scale
** Potilaat, jotka eivät olleet käyttäneet mitään taudinkulkua muuntavaa hoitoa 2 vuoden aikana ennen satunnaistamista.
Keskeiset kliiniset ja magneettikuvauksella todetut tehotulokset esitetään taulukossa 4 ja kuvassa 1.
Näiden tutkimusten tulokset osoittavat, että okrelitsumabi vähentää merkitsevästi pahenemisvaiheita, magneettikuvauksella mitattavaa subkliinistä tautiaktiivisuutta ja taudin etenemistä verrattuna ihon alle annettuun 44 mikrog:n interferonibeeta-1a-hoitoon.
Taulukko 4. Tutkimusten WA21092 ja WA21093 keskeiset kliiniset ja magneettikuvaukseen liittyvät päätetapahtumat (aaltomainen MS-tauti)
| Päätetapahtumat | Tutkimus 1: WA21092 (OPERA I) | Tutkimus 2: WA21093 (OPERA II) | |||||
Okrelitsumabi 600 mg (n = 410) | IFN 44 mikrog (n = 411) | Okrelitsumabi 600 mg (n = 417) | IFN 44 mikrog (n = 418) | ||||
| Kliiniset päätetapahtumat | |||||||
| Vuosittainen pahenemisvaiheiden määrä (ARR, ensisijainen päätetapahtuma) | 0,156 | 0,292 | 0,155 | 0,290 | |||
| Suhteellinen vähenemä | 46 % (p < 0,0001) | 47 % (p < 0,0001) | |||||
Niiden potilaiden osuus, joilla oli 12 viikon vahvistettu toimintakyvyn heikkeneminen3 Riskin alenema (yhdistetty analyysi1) Riskin alenema (yksittäiset tutkimukset2) | 9,8 % okrelitsumabi vs 15,2 % IFN 40 % (p = 0,0006)7 | ||||||
| 43 % (p = 0,0139)7 | 37 % (p = 0,0169)7 | ||||||
Niiden potilaiden osuus, joilla oli 24 viikon vahvistettu toimintakyvyn heikkeneminen3 Riskin alenema (yhdistetty analyysi1) Riskin alenema (yksittäiset tutkimukset2) | 7,6 % okrelitsumabi vs 12,0 % IFN 40 % (p = 0,0025)7 | ||||||
| 43 % (p = 0,0278)7 | 37 % (p = 0,0370)7 | ||||||
| Niiden potilaiden osuus, joilla oli vähintään 12 viikon vahvistettu toimintakyvyn parannus4 | 20,7 % okrelitsumabi vs 15,6 % IFN | ||||||
Suhteellinen lisäys (yhdistetty analyysi1) Suhteellinen lisäys (yksittäiset tutkimukset2) | 33 % (p = 0,0194) | ||||||
| 61 % (p = 0,0106) | 14 % (p = 0,4019) | ||||||
| Niiden potilaiden osuus, joilla ei ollut pahenemisvaihetta hoitoviikkoon 96 mennessä2 | 80,4 % | 66,7 % | 78,9 % | 64,3 % | |||
| (p < 0,0001) | (p < 0,0001) | ||||||
| Niiden potilaiden osuus, joilla ei ollut näyttöä taudin aktiivisuudesta (NEDA)5 | 48 % | 29 % | 48 % | 25 % | |||
| Suhteellinen lisäys2 | 64 % (p < 0,0001) | 89 % (p < 0,0001) | |||||
| Magneettikuvauksen (MKn) päätetapahtumat | |||||||
| Gadoliniumilla tehostuvien T1-muutosten määrä (keskiarvo) magneettikuvauskertaa kohden | 0,016 | 0,286 | 0,021 | 0,416 | |||
| Suhteellinen vähenemä | 94 % (p < 0,0001) | 95 % (p < 0,0001) | |||||
| Uusien ja/tai suurentuneiden T2-hyperintensiivisten muutosten määrä (keskiarvo) magneettikuvauskertaa kohden | 0,323 | 1,413 | 0,325 | 1,904 | |||
| Suhteellinen vähenemä | 77 % (p < 0,0001) | 83 % (p < 0,0001) | |||||
| Aivojen tilavuuden prosentuaalinen muutos viikosta 24 viikkoon 96 | -0,572 | -0,741 | -0,638 | -0,750 | |||
| Aivojen tilavuuden menetyksen suhteellinen vähenemä | 22,8 % (p = 0,0042)6 | 14,9 % (p = 0,0900) | |||||
1 Tutkimusten 1 ja 2 prospektiivisesti yhdistetyt tiedot
2 Ei-konfirmatorinen p-arvoanalyysi; ei osa ennalta määriteltyä testaushierarkiaa
3 Vahvistettu toimintakyvyn heikkeneminen (Confirmed Disability Progression, CDP) määritelty EDSS-pisteiden (Expanded Disability Status Scale) ≥ 1,0 pisteen suurenemiseksi lähtötilanteen pisteistä, jos potilaan lähtötilanteen pisteet ovat 5,5 tai vähemmän, tai ≥ 0,5 pisteen suurenemiseksi, kun lähtötilanteen pisteet ovat > 5,5; Kaplan–Meierin estimaatit viikolla 96
4 Määritelty EDSS-pisteiden ≥ 1,0 pisteen vähenemiseksi lähtötilanteesta, jos potilaan lähtötilanteen EDSS-pisteet ≥ 2 ja ≤ 5,5, tai ≥ 0,5 pisteen vähenemiseksi, kun lähtötilanteen pisteet ovat > 5,5. Potilaita, joiden lähtötilanteen pisteet olivat < 2, ei otettu mukaan analyysiin.
5 Tilanteeksi, jossa ei näyttöä taudin aktiivisuudesta (NEDA), on määritelty tutkimussuunnitelmassa määriteltyjen pahenemisvaiheiden puuttuminen, ei merkkiä 12 viikon vahvistetusta toimintakyvyn heikkenemisestä eikä mitään MK:ssa todettua aktiivisuutta (joko gadoliniumilla tehostuvia T1-muutoksia tai uusia tai laajenevia T2-muutoksia) koko 96 viikon hoidon aikana. Koko hoitoaikeen mukaiseen (ITT) potilasjoukkoon perustuvat eksploratiiviset tulokset.
6 Ei-konfirmatorinen p-arvo; hierarkkinen testaus lopetettu ennen päätetapahtuman saavuttamista.
7 Log-rank-testi
8 Varmistetut relapsit (joihin liittyy kliinisesti oleellinen EDSS-pisteiden muutos)
Kuva 1. Kaplan–Meier-kuvaaja ajankohtaan, jolloin todetaan toimintakyvyn heikkeneminen vähintään 12 viikon ajaksi ja jossa ensimmäinen havainto neurologisesta heikkenemisestä tehtiin kaksoissokkoutetun hoitojakson aikana (tutkimusten WA21092 ja WA21093 yhdistetty hoitoaikeen mukainen potilasjoukko)*
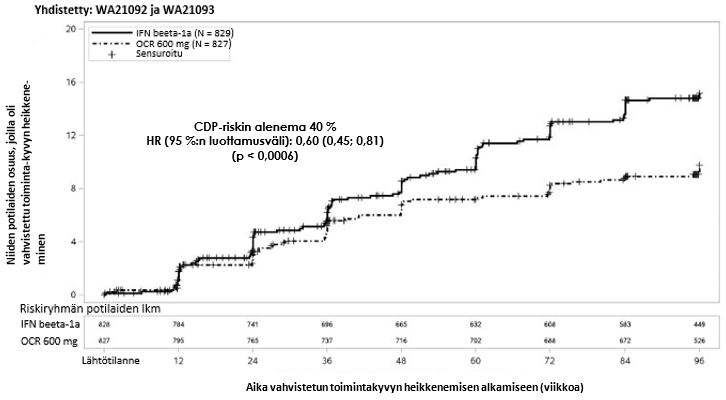
*Tutkimusten WA21092 ja WA21093 ennalta määritelty yhdistetty analyysi
Ennalta määritettyjen yhdistettyjen analyysien tulokset ajankohtaan, jolloin todetaan vähintään 12 viikkoa kestänyt vahvistettu toimintakyvyn heikkeneminen (riskin alenema okrelitsumabihoidossa 40 % verrattuna interferonibeeta-1a-hoitoon [p = 0,0006]) olivat erittäin yhdenmukaiset vähintään 24 viikon ajan kestäneen toimintakyvyn heikkenemisen tulosten kanssa (riskin alenema okrelitsumabihoidossa 40 % verrattuna interferonibeeta-1a-hoitoon, p = 0,0025).
Tutkimuksiin otettiin mukaan potilaita, joilla oli aktiivinen tauti. Potilaat eivät olleet aiemmin saaneet aktiivista hoitoa tai hoidon vaste oli ollut riittämätön kliinisten piirteiden tai kuvantamislöydösten perusteella. Analyysi potilasjoukoista, joissa taudin aktiivisuudessa oli lähtötilanteessa eroja, mukaan lukien aktiivinen tai erittäin aktiivinen tauti, osoitti okrelitsumabihoidon tehon vuosittaiseen pahenemisvaiheiden määrään ja 12 viikon vahvistettuun toimintakyvyn heikkenemiseen olevan yhdenmukainen koko potilasjoukossa.
Primaaristi etenevä MS-tauti
Okrelitsumabin tehoa ja turvallisuutta selvitettiin myös satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa potilailla, jotka sairastavat primaaristi etenevää MS-tautia (tutkimus WA25046), joka oli tärkeimpien sisäänottokriteerien perusteella varhaisvaiheessa, eli potilaan ikä oli 18–55 vuotta, EDSS-pisteet olivat seulonnassa 3,0–6,5 pistettä, taudin kesto MS-oireiden alusta oli alle 10 vuotta, jos potilaan EDSS-pisteet olivat seulonnassa ≤ 5,0, tai alle 15 vuotta, jos potilaan EDSS-pisteet olivat seulonnassa > 5,0. Myös etenevässä MS-taudissa taudin aktiivisuudelle tyypillinen tulehdusaktiivisuus voi olla olla todettavissa kuvantamisella (eli Gd-tehosteiset T1‑muutokset ja/tai aktiiviset [uudet tai laajenevat] T2‑muutokset]). Tulehdusaktiivisuus pitäisi varmistaa kaikilla potilailla magneettikuvauksella. Yli 55-vuotiaita potilaita ei tutkittu. Tutkimusasetelma ja tutkimuksen potilasjoukon lähtötilanteen ominaisuudet esitetään taulukossa 5.
Demografiset tiedot ja lähtötilanteen ominaisuudet olivat hyvin tasapainossa näiden kahden hoitoryhmän välillä. Pään magneettikuvauksessa todettiin joko Gd-tehosteisten T1‑muutosten tai T2‑muutosten perusteella tulehdusaktiivisuudelle tyypillisiä piirteitä.
Primaaristi etenevää MS-tautia koskeneessa vaiheen III tutkimuksessa potilaat saivat 600 mg:n okrelitsumabiannoksen kuuden kuukauden välein kahtena kahden viikon välein annettuna 300 mg:n infuusiona koko hoitojakson ajan. Aaltomaista MS-tautia sairastaville potilaille annettujen 600 mg:n infuusioiden ja ensisijaisesti etenevää MS-tautia sairastaville potilaille kahtena 300 mg:n infuusiona annetun hoidon farmakokineettiset/farmakodynaamiset profiilit olivat yhdenmukaiset. Infuusioreaktiot olivat infuusiota kohden myös samankaltaiset riippumatta siitä, annettiinko 600 mg:n annos yhtenä 600 mg:n infuusiona vai kahtena 300 mg:n infuusiona kahden viikon välein (ks. kohdat Haittavaikutukset ja Farmakokinetiikka). Koska kahden 300 mg:n infuusion hoito-ohjelmassa annettiin kokonaisuudessaan enemmän infuusioita, infuusioreaktioiden kokonaismäärä oli kuitenkin suurempi. Näin ollen suositellaan, että 1. annoksen jälkeen okrelitsumabi annetaan 600 mg:n kertainfuusiona (ks. kohta Annostus ja antotapa) infuusioiden kokonaismäärän (sekä samanaikaisen altistuksen estohoitona annetulle metyyliprednisolonille ja antihistamiinille) sekä niihin liittyvien infuusioreaktioiden vähentämiseksi.
Taulukko 5. Tutkimuksen WA25046 tutkimusasetelma sekä demografiset ja lähtötilanteen tiedot
| Tutkimuksen nimi | Tutkimus WA25046 ORATORIO (n = 732) | |
| Tutkimusasetelma | ||
| Tutkimuksen potilasjoukko | Primaaristi etenevää MS-tautia sairastavat potilaat | |
| Tutkimuksen kesto | Tapahtumaperusteinen (vähintään 120 viikkoa ja 253 vahvistettua toimintakyvyn heikkenemiseen liittyvää tapahtumaa) (Seuranta-ajan mediaani: okrelitsumabi 3,0 vuotta, lumehoito 2,8 vuotta | |
| Sairaushistoria seulonnassa | Ikä 18–55 vuotta, EDSS-pisteet 3,0–6,5 | |
| Hoitoryhmät | Ryhmä A: okrelitsumabi 600 mg Ryhmä B: lumehoito, satunnaistettu suhteessa 2:1 | |
| Lähtötilanteen tiedot | Okrelitsumabi 600 mg (n = 488) | Lumehoito (n = 244) |
| Keskimääräinen ikä (vuotta) | 44,7 | 44,4 |
| Iän vaihteluväli (vuotta) tutkimuksen sisäänottovaiheessa | 20–56 | 18–56 |
| Sukupuolijakauma (% miehiä/% naisia) | 51,4/48,6 | 49,2/50,8 |
| Sairauden kestoajan keskiarvo/mediaani primaaristi etenevän MS-taudin diagnoosin jälkeen (vuotta) | 2,9/1,6 | 2,8/1,3 |
| Keskimääräiset EDSS-pisteet | 4,7 | 4,7 |
Keskeiset kliiniset ja magneettikuvauksella todetut tehon tulokset esitetään taulukossa 6 ja kuvassa 2.
Tämän tutkimuksen tulokset osoittavat, että okrelitsumabi hidastaa taudin etenemistä ja vähentää kävelynopeuden hidastumista merkitsevästi lumehoitoon verrattuna.
Taulukko 6. Tutkimuksen WA25046 (ensisijaisesti etenevä MS-tauti) keskeiset kliiniset ja magneettikuvauksen päätetapahtumat
| Tutkimus 3 | ||
| Päätetapahtumat | WA25046 (Oratorio) | |
Okrelitsumabi 600 mg (n = 488) | Lumehoito (n = 244) | |
| Kliiniset päätetapahtumat | ||
Ensisijainen tehon päätetapahtuma Niiden potilaiden osuus, joilla oli 12 viikon vahvistettu toimintakyvyn heikkeneminen (ensisijainen päätetapahtuma) Riskin alenema | 30,2 % | 34,0 % |
24 % (p = 0,0321) | ||
| Niiden potilaiden osuus, joilla oli 24 viikon vahvistettu toimintakyvyn heikkeneminen1 | 28,3 % | 32,7 % |
| Riskin alenema | 25 % (p = 0,0365) | |
| Kävelynopeustestin (Timed 25-Foot Walk) prosentuaalinen muutos lähtötilanteesta viikkoon 120 | 38,9 | 55,1 |
| Kävelynopeuden hidastumisen suhteellinen vähenemä | 29,4 % (p = 0,0404) | |
| Magneettikuvauksen päätetapahtumat | ||
| T2-hyperintensiivisten muutosten tilavuuden prosentuaalinen muutos lähtötilanteesta viikkoon 120 | -3,4 | 7,4 |
| (p < 0,0001) | ||
| Aivojen tilavuuden prosentuaalinen muutos viikosta 24 viikkoon 120 | -0,902 | -1,093 |
| Aivojen tilavuuden menetyksen suhteellinen vähenemä | 17,5 % (p = 0,0206) | |
1 Määritelty EDSS-pisteiden (Expanded Disability Status Scale) ≥ 1,0 pisteen suurenemiseksi potilailla, joilla lähtötilanteen pisteet ≤ 5,5 tai ≥ 0,5 pisteen suurenemiseksi, kun lähtötilanteen pisteet ovat > 5,5; Kaplan–Meierin estimaatit viikolla 120
Kuva 2. Kaplan–Meier-kuvaaja* ajankohtaan, jolloin todetaan toimintakyvyn heikkeneminen vähintään 12 viikon ajaksi ja jossa ensimmäinen havainto neurologisesta heikkenemisestä tehtiin kaksoissokkoutetun hoitojakson aikana (tutkimuksen WA25046 hoitoaikeen mukainen potilasjoukko)*
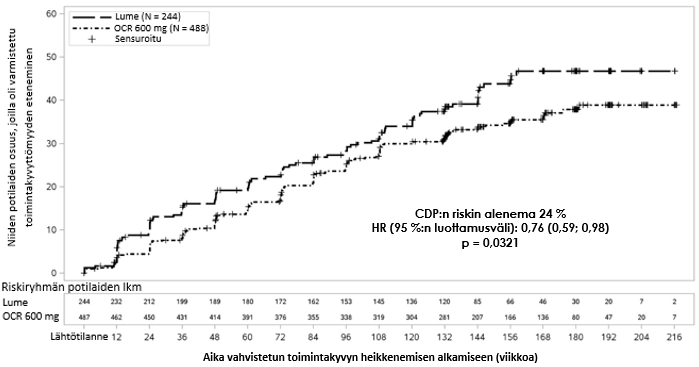
* Kaikkien tässä analyysissa mukana olevien potilaiden seuranta-aika oli vähintään 120 viikkoa. Ensisijainen analyysi perustuu kaikkiin kertyneisiin tapahtumiin.
Ennalta määritellyn, mutta tilastolliselta voimaltaan riittämättömän ensisijaisen päätetapahtuman alaryhmäanalyysi viittaa siihen, että nuoremmat potilaat tai potilaat, joilla oli lähtötilanteessa Gd‑tehosteisia T1‑muutoksia, saavat hoidosta suuremman hyödyn kuin iäkkäämmät potilaat tai potilaat, joilla ei ole Gd‑tehosteisia T1‑muutoksia (≤ 45 vuoden iässä: HR 0,64 [0,45, 0,92], > 45 vuoden iässä: HR 0,88 [0,62, 1,26]), joilla on Gd‑tehosteisia T1-muutoksia lähtötilanteessa (HR 0,65 [0,40–1,06]) tai joilla ei ole Gd‑tehosteisia T1‑muutoksia lähtötilanteessa (HR 0,84 [0,62–1,13]).
Post-hoc-analyysit viittasivat lisäksi siihen, että hoidon teho on parempi nuoremmilla potilailla, joilla on lähtötilanteessa Gd‑tehosteisia T1‑muutoksia (≤ 45 vuoden iässä: HR 0,52 [0,27–1,00], ≤ 46 vuoden iässä [iän mediaani tutkimuksessa WA25046]; HR 0,48 [0,25–0,92], < 51 vuoden iässä: HR 0,53 [0,31–0,89]).
Jatketun kontrolloidun jakson (Extended Controlled Period, ECP) osalta, joka käsitti kaksoissokkoutetun hoitojakson ja noin 9 lisäkuukauden pituisen kontrolloidun seurantajakson ennen siirtymistä avoimeen jatkovaiheeseen (Open-Label Extension, OLE) tai tutkimushoidosta vetäytymiseen saakka, tehtiin post-hoc-analyysit. Niiden potilaiden osuus, joilla 24 viikon vahvistettua toimintakyvyn heikkenemistä (24-week Confirmed Disease Progression, 24W-CDP) osoittavat EDSS-pisteet olivat ≥ 7,0 (EDSS 24W-CDP ≥ 7,0, pyörätuolin tarpeeseen kuluva aika), oli lumeryhmässä 9,1 % verrattuna 4,8 %:iin okrelitsumabiryhmässä viikolla 144, mikä tarkoittaa 47 % riskin alenemista (HR 0,53, [0,31–0,92]) pyörätuolin tarpeeseen kuluvan ajan osalta jatketun kontrolloidun jakson aikana. Nämä tulokset olivat luonteeltaan eksploratiivisia ja sisälsivät sokkouttamisen avaamisen jälkeiset tiedot, joten tuloksia pitää tulkita harkiten.
Lyhytkestoisempaa infuusiota koskeva alatutkimus
Lyhytkestoisemman, 2 tuntia kestävän okrelitsumabi-infuusion turvallisuutta arvioitiin prospektiivisessa, satunnaistetussa, kaksoissokkoutetussa, kontrolloidussa, rinnakkaisryhmillä tehdyssä monikeskusalatutkimuksessa MA30143 (Ensemble) potilailla, joilla oli aaltomainen MS-tauti ja jotka eivät olleet aiemmin saaneet muita taudinkulkua muuntavia lääkkeitä. Ensimmäinen annos annettiin kahteen erilliseen infuusioon jaettuna: ensin 300 mg:n infuusio ja tästä kaksi viikkoa myöhemmin toinen 300 mg:n infuusio (yhteensä 600 mg). Potilaat satunnaistettiin toisesta annoksesta eteenpäin (annokset 2–6) suhteessa 1:1 joko tavanomaisen infuusion ryhmään tai lyhytkestoisemman infuusion ryhmään. Tavanomaisen infuusion ryhmässä okrelitsumabi-infuusiot annettiin noin 3,5 tunnin kestoisina 24 viikon välein ja lyhytkestoisemman infuusion ryhmässä noin 2 tunnin kestoisina 24 viikon välein. Satunnaistaminen stratifioitiin asuinalueen ja annoksen perusteella. Annoksena käytettiin sitä, johon potilaan ensimmäiseksi satunnaistettiin.
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joille ilmaantui infuusioreaktio ensimmäisen satunnaistetun infuusion aikana tai 24 tunnin kuluessa sen jälkeen. Ensisijainen analyysi tehtiin, kun 580 potilasta oli satunnaistettu. Niiden potilaiden osuus, joille ilmaantui infuusioreaktio ensimmäisen satunnaistetun infuusion aikana tai 24 tunnin kuluessa sen jälkeen, oli lyhytkestoisempien infuusioiden ryhmässä 24,6 % verrattuna 23,1 %:iin tavanomaisten infuusioiden ryhmässä. Stratifioitujen ryhmien ero oli samankaltainen. Valtaosa kaikkien satunnaistettujen annosten yhteydessä havatuista infuusioreaktioista oli lieviä tai keskivaikeita. Vain kaksi infuusioreaktiota oli vaikea-asteisia, ja kummassakin ryhmässä ilmeni yksi vaikea-asteinen infuusioreaktio. Hengenvaarallisia, kuolemaan johtaneita tai vakavia infuusioreaktioita ei esiintynyt.
Immunogeenisuus
MS-tutkimuksissa (WA21092, WA21093 ja WA25046) mukana olleilta potilailta testattiin useana ajankohtana (lähtötilanteessa ja kuuden kuukauden välein annetun hoidon jälkeen koko tutkimuksen keston ajan) vasta-aineet lääkevalmisteelle. Okrelitsumabihoitoa saaneista 1 311 potilaasta 12 potilasta (~1 %) kehitti vasta-aineita lääkettä kohtaan, ja näistäkahdella potilaalla oli neutraloivia vasta-aineita. Hoidosta aiheuttamien lääkevasta-aineiden vaikutusta turvallisuuteen ja tehoon ei voida arvioida, koska okrelitsumabihoitoon liittyneiden vasta-aineiden ilmaantuvuus on pieni.
Immunisaatio
Aaltomaista MS-tautia sairastavilla potilailla (N = 102) tehdyssä satunnaistetussa avoimessa tutkimuksessa niiden potilaiden prosenttiosuus, jotka saivat positiivisen vasteen tetanusrokotteeseen kahdeksan viikkoa rokotuksen jälkeen, oli okrelitsumabiryhmässä 23,9 % ja vertailuryhmässä 54,5 % (ei taudinkulkua muuntavaa hoitoa paitsi interferonibeeta). Antitetanustoksoidispesifisten vasta-ainetiitterien geometrinen keskiarvo viikolla kahdeksan oli okrelitsumabiryhmässä 3,74 IU/ml ja vertailuryhmässä 9,81 IU/ml. Positiivinen vaste vähintään viidelle PPV23-rokotteen sisältämälle serotyypille todettiin 71,6 %:lla potilaista okrelitsumabiryhmässä ja 100 %:lla potilaista vertailuryhmässä neljä viikkoa rokotuksen jälkeen. Okrelitsumabihoitoa saaneille potilaille neljä viikkoa PPV23-rokotuksen jälkeen annettu tehosterokotus (PCV13) ei lisännyt merkittävästi vastetta niille 12 serotyypille, jotka olivat samoja kuin PPV23-rokotteessa. Potilaiden prosenttiosuus, joilla oli suojaava serologinen tiitteri viittä influenssakantaa vastaan, oli ennen rokotusta okrelitsumabiryhmässä 20,0−60,0 % ja vertailuryhmässä 16,7−43,8 %. Neljä viikkoa rokotuksen jälkeen prosenttiosuus oli okrelitsumabiryhmässä 55,6−80,0 % ja vertailuryhmässä 75,0−97,0 %. Ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Ocrevus-valmisteen käytöstä yhden tai useamman pediatrisen potilasryhmän MS-taudin hoidossa. Ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Okrelitsumabin farmakokinetiikkaa MS-tutkimuksissa kuvasi kaksitilamalli, jossa puhdistuma on aikariippuvainen, ja jossa oli IgG1-monoklonaaliselle vasta-aineelle tyypilliset farmakokineettiset parametrit. Primaaristi etenevää MS-tautia koskeneen tutkimuksen kahden 300 mg:n annoksen ja aaltomaista MS-tautia koskeneen tutkimuksen yhden 600 mg:n annoksen kokonaisaltistus (AUC 24 viikon antovälin aikana) oli identtinen, mikä oli käytetyllä identtisellä annoksella oletettavissa. Käyrän alle jäävä pinta-ala (AUCτ) neljännen 600 mg:n okrelitsumabiannoksen jälkeen oli 3510 µg/ml•vrk, ja suurimman pitoisuuden (Cmax) keskiarvo oli 212 µg/ml aaltomaisen MS-taudin yhteydessä (600 mg:n infuusio) ja 141 µg/ml primaaristi etenevän MS-taudin yhteydessä (300 mg:n infuusiot).
Imeytyminen
Okrelitsumabi annetaan infuusiona laskimoon.
Jakautuminen
Keskusjakautumistilavuuden populaatiofarmakokineettinen arvio oli 2,78 l. Perifeeriseksi tilavuudeksi arvioitiin 2,68 l, ja tilojen väliseksi puhdistumaksi arvioitiin 0,294 l/vrk.
Biotransformaatio
Okrelitsumabin metaboliaa ei ole tutkittu suoraan, sillä vasta-aineet puhdistuvat pääasiassa katabolian kautta (eli hajoavat peptideiksi ja aminohapoiksi).
Eliminaatio
Vakiopuhdistumaksi arvioitiin 0,17 l/vrk, ja alkuvaiheen aikariippuvaiseksi puhdistumaksi arvioitiin 0,0489 l/vrk, mikä väheni 33 viikon puoliintumisajan mukaan. Okrelitsumabin terminaalisen eliminaation puoliintumisaika oli 26 vuorokautta.
Erityisryhmät
Pediatriset potilaat
Okrelitsumabin farmakokinetiikkaa ei ole tutkittu alle 18-vuotiailla lapsilla ja nuorilla.
Iäkkäät
Okrelitsumabilla ei ole tehty erityisesti vähintään 55-vuotiaita potilaita koskevia farmakokineettisiä tutkimuksia, koska kliinistä kokemusta on vain vähän (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Varsinaisia farmakokineettisiä tutkimuksia ei ole tehty. Kliinisissä tutkimuksissa oli mukana lievää munuaisten vajaatoimintaa sairastavia potilaita eikä okrelitsumabin farmakokinetiikassa havaittu tässä potilasryhmässä muutoksia. Keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavista potilaista ei ole farmakokineettisiä tietoja saatavissa.
Maksan vajaatoiminta
Varsinaisia farmakokineettisiä tutkimuksia ei ole tehty. Kliinisissä tutkimuksissa oli mukana lievää maksan vajaatoimintaa sairastavia potilaita eikä farmakokinetiikassa havaittu tässä potilasryhmässä muutoksia. Keskivaikeaa ja vaikeaa maksan vajaatoimintaa sairastavista potilaista ei ole farmakokineettisiä tietoja saatavissa.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta sekä alkion ja sikiön kehitystä koskevien perinteisten ei-kliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Okrelitsumabilla ei ole tehty karsinogeenisuus- eikä mutageenisuustutkimuksia.
Kahdessa Cynomolgus-apinoilla tehdyssä pre- ja postnataalista kehitystä koskevassa tutkimuksessa okrelitsumabin antoon tiineyspäivästä 20 vähintään synnytykseen saakka liittyi jälkeläisillä glomerulopatiaa, lymfoidisten follikkelien muodostumista luuytimeen, lymfoplasmasyyttistä munuaistulehdusta ja kivesten painon vähenemistä. Näissä tutkimuksissa emolle annetuista annoksista aiheutuneet suurimmat keskiarvopitoisuudet (Cmax) seerumissa olivat 4,5–21-kertaisia kliinisessä käytössä oletettaviin pitoisuuksiin nähden.
Viiteen tapaukseen liittyi neonataalikuolleisuutta. Yhdessä tapauksessa kyse oli keskosuuteen liittyvästä heikkoudesta, mihin liittyi opportunistinen bakteeri-infektio, yhdessä tapauksessa emon aktiivisesta bakteeri-infektiosta (mastiitti) aiheutui vastasyntyneelle jälkeläiselle pikkuaivojen infektiivinen meningoenkefaliitti ja kolmessa tapauksessa havaittiin ikterusta ja maksavaurio, joiden epäiltiin olleen virusperäisiä, mahdollisesti polyoomaviruksen aiheuttamia. B-solujen puutos on saattanut vaikuttaa näihin viiteen varmistettuun tai epäiltyyn infektioon. Okrelitsumabille altistuneiden emojen vastasyntyneillä jälkeläisillä havaittiin vähentyneitä B-solupopulaatioita heti syntymän jälkeen.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaattitrihydraatti (E 262)
Väkevä etikkahappo
Trehaloosidihydraatti
Polysorbaatti 20 (E 432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Lääkevalmistetta ei tule sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
2 vuotta
Laimennettu laskimoon annettava infuusioliuos
Kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 24 tuntia 2–8 °C:n lämpötilassa ja sen jälkeen 8 tuntia huoneenlämmössä.
Mikrobiologisesta näkökulmasta käyttövalmis infuusioliuos tulisi käyttää heti. Jos valmistetta ei käytetä heti, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24 tuntia 2–8 °C:n lämpötilassa ja sen jälkeen 8 tuntia huoneenlämmössä, ellei valmistetta ole laimennettu valvotuissa ja validoiduissa aseptisissa olosuhteissa.
Jos laskimoon annettavaa infuusiota ei voida antaa loppuun samana päivänä, käyttämättä jäävä liuos on hävitettävä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 ºC).
Ei saa jäätyä.
Pidä injektiopullot ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OCREVUS infuusiokonsentraatti, liuosta varten
300 mg (L:ei) 1 kpl (10 ml (30 mg/ml)) (6360,68 €)
PF-selosteen tieto
10 ml konsentraattia injektiopullossa (väritöntä tyypin I lasia).
Pakkauskoot 1 tai 2 injektiopulloa. Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai hieman opaalinhohtoinen, ja väritön tai vaaleanruskehtava liuos.
Käyttö- ja käsittelyohjeet
Laimennusohjeet
Terveydenhuollon ammattilaisen pitää valmistaa valmiste käyttökuntoon aseptista tekniikkaa noudattaen. Älä ravista injektiopulloa. Laimennetun infuusioliuoksen valmistuksessa tulee käyttää steriiliä neulaa ja ruiskua.
Valmiste on tarkoitettu yhteen käyttökertaan.
Älä käytä konsentraattia, jos sen väri on muuttunut tai jos konsentraatissa on vierashiukkasia (ks. kohta 3).
Lääkevalmiste on laimennettava ennen antoa. Laskimoon annettavat liuokset valmistetaan laimentamalla konsentraatti infuusiopussiin, joka sisältää isotonista 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta (300 mg/250 ml tai 600 mg/500 ml) lopulliseen okrelitsumabipitoisuuteen noin 1,2 mg/ml.
Tämän lääkevalmisteen ja polyvinyylikloridista (PVC) tai polyolefiinista (PO) valmistettujen infuusiopussien ja laskimonsisäiseen antoon tarkoitettujen antolaitteiden välillä ei ole havaittu yhteensopimattomuuksia.
Laimennettu infuusioliuos on annettava infuusiovälineistöllä, jossa on 0,2 mikronin tai 0,22 mikronin letkunsisäinen suodatin.
Infuusiopussin sisällön pitää olla huoneenlämpöistä ennen laskimoon annettavan infuusion aloittamista.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OCREVUS infuusiokonsentraatti, liuosta varten
300 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AG08
Valmisteyhteenvedon muuttamispäivämäärä
13.02.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com