
SKYRIZI injektioneste, liuos, esitäytetty ruisku 55 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 55 mg risankitsumabia 0,37 millilitrassa liuosta.
Risankitsumabi on humanisoitu immunoglobuliini G1 ‑luokan (IgG1) monoklonaalinen vasta-aine, joka valmistetaan kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,07 mg polysorbaattia 20 per 55 mg:n annos.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (inj.)
Kliiniset tiedot
Käyttöaiheet
Pediatrinen läiskäpsoriaasi
Skyrizi on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon vähintään 6 vuoden ikäisille lapsille ja nuorille, joille harkitaan systeemistä hoitoa.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi kohdassa Käyttöaiheet mainittujen käyttöaiheiden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja valvonnassa.
Annostus
Pediatrinen läiskäpsoriaasi (6 – < 18‑vuotiaat potilaat)
Suositeltu annos pediatrisille potilaille perustuu potilaan painoon (taulukko 1) ja se annetaan injektiona ihon alle viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein.
Taulukko 1. Suositeltu annos pediatrisessa läiskäpsoriaasissa
Paino antoajankohtana | Suositeltu annos |
< 40 kg | 55 mg (yksi 55 mg:n injektio esitäytetyllä ruiskulla) |
≥ 40 kg | 150 mg (kaksi 75 mg:n injektiota esitäytetyllä ruiskulla tai yksi 150 mg:n injektio esitäytetyllä kynällä tai esitäytetyllä ruiskulla)* |
*Lapset ja nuoret, jotka painavat vähintään 40 kg, katso Skyrizi 150 mg esitäytetyn kynän, 150 mg esitäytetyn ruiskun ja 75 mg esitäytetyn ruiskun valmisteyhteenveto.
Jos edellä mainitussa käyttöaiheessa ei todeta vastetta 16 hoitoviikon jälkeen, on harkittava hoidon lopettamista. Joillakin läiskäpsoriaasipotilailla aluksi saavutettu osittainen vaste saattaa parantua, kun hoitoa jatketaan yli 16 viikon ajan.
Annoksen unohtuminen
Jos annos jää ottamatta, se on otettava mahdollisimman pian. Tämän jälkeen lääkkeen annostelua jatketaan tavanomaisella aikataululla.
Erityisryhmät
Munuaisten tai maksan vajaatoiminta
Maksan tai munuaisten vajaatoiminnan vaikutusta risankitsumabin farmakokinetiikkaan ei ole tutkittu spesifisissä tutkimuksissa. Näiden tilojen ei yleisesti odoteta vaikuttavan merkitsevästi monoklonaalisten vasta-aineiden farmakokinetiikkaan, eikä annosmuutoksia pidetä tarpeellisina (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää risankitsumabia alle 6 vuoden ikäisille lapsille keskivaikean tai vaikean läiskäpsoriaasin hoitoon.
Ylipainoiset potilaat
Annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Antotapa
Skyrizi annetaan injektiona ihon alle.
Injektio annetaan reiteen tai vatsan alueelle. Potilas ei saa antaa injektiota alueelle, jonka iho on arka, mustelmilla, punoittava, kovettunut tai psoriaasioireinen.
Potilaat voivat pistää Skyrizi-injektiot itse saatuaan ihon alle annettavia injektioita koskevaa pistosohjausta. Iältään 10 – < 18‑vuotiaiden lasten ja nuorten hoidossa on suositeltavaa, että Skyrizi-injektiot antaa tai antamista valvoo aikuinen. Iältään 6 – < 10‑vuotiaiden lasten hoidossa aikuisen on annettava Skyrizi-injektiot. Potilaita on ohjattava lukemaan pakkausselosteessa olevat käyttöohjeet ennen valmisteen antoa.
Skyrizi voidaan antaa olkavarren ulkosyrjään vain, jos injektion antaa terveydenhuollon ammattilainen tai potilasta hoitava henkilö.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio (esim. aktiivinen tuberkuloosi, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Risankitsumabi voi suurentaa infektion riskiä.
Jos potilaalla on krooninen infektio, toistuvia infektioita tai tiedossa olevia infektion riskitekijöitä, risankitsumabia on käytettävä varoen. Jos potilaalla on mikä tahansa kliinisesti merkittävä aktiivinen infektio, risankitsumabihoitoa ei saa aloittaa ennen kuin infektio on parantunut tai hoidettu asianmukaisesti.
Risankitsumabihoitoa saaneita potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on kliinisesti merkittävän kroonisen tai akuutin infektion oireita tai merkkejä. Jos potilaalle kehittyy tällainen infektio tai infektion standardihoito ei tuota vastetta, potilaan tilannetta on seurattava tarkoin eikä risankitsumabia saa antaa ennen kuin infektio on parantunut.
Tuberkuloosi
Ennen risankitsumabihoidon aloittamista potilaat on arvioitava tuberkuloosi-infektion varalta. Risankitsumabia saavien potilaiden vointia on seurattava aktiivisen tuberkuloosin oireiden ja löydösten varalta. Tuberkuloosilääkityksen käyttöä on harkittava ennen risankitsumabihoidon aloittamista, jos potilaalla on anamneesissa latentti tai aktiivinen tuberkuloosi eikä asianmukaisen hoitojakson toteutumista pystytä vahvistamaan.
Immunisaatiot
Ennen risankitsumabihoidon aloittamista on harkittava kaikkien asianmukaisten rokotusten antamista ajankohtaisten rokotussuositusten mukaisesti. Jos potilas on saanut eläviä rokotteita (virus- tai bakteerirokotteita), on suositeltavaa odottaa vähintään 4 viikkoa ennen risankitsumabihoidon aloittamista. Risankitsumabihoitoa saaville potilaille ei pidä antaa eläviä rokotteita hoidon aikana eikä ainakaan 21 viikkoon hoidon jälkeen (ks. kohta Farmakokinetiikka).
Yliherkkyys
Vakavia yliherkkyysreaktioita, mukaan lukien anafylaksiaa, on raportoitu risankitsumabin käytön yhteydessä (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy vakava yliherkkyysreaktio, risankitsumabin anto on lopetettava heti ja asianmukainen hoito on aloitettava.
Apuaineet, joiden vaikutus tunnetaan
Polysorbaatti
Tämä lääkevalmiste sisältää 0,07 mg polysorbaattia 20 per 55 mg:n annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per esitäytetty ruisku eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Risankitsumabi ei oletettavasti metaboloidu maksaentsyymien välityksellä eikä eliminoidu munuaisteitse. Risankitsumabilla ei odoteta olevan interaktioita lääkevalmisteiden metaboliaan osallistuvien entsyymien estäjien, indusorien tai substraattien kanssa, eikä annosta tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Samanaikainen immunosuppressiivinen hoito tai valohoito
Risankitsumabin ja immunosuppressanttien (myös biologisten lääkkeiden) tai valohoidon yhdistelmän turvallisuutta ja tehoa ei ole arvioitu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja vähintään 21 viikon ajan sen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta) risankitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia. Varmuuden vuoksi risankitsumabin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö risankitsumabi ihmisen rintamaitoon. Ihmisen IgG:tä tiedetään erittyvän rintamaitoon muutaman päivän ajan synnytyksen jälkeen, minkä jälkeen pitoisuudet pienevät nopeasti matalalle tasolle. Rintaruokittavaan imeväiseen kohdistuvia riskejä ei siis voida poissulkea tämän lyhyen jakson aikana. On päätettävä, lopetetaanko risankitsumabihoito tai pidättäydytäänkö risankitsumabihoidosta, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja risankitsumabihoidosta koituvat hyödyt äidille.
Hedelmällisyys
Risankitsumabin vaikutusta ihmisen hedelmällisyyteen ei ole arvioitu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria hedelmällisyyteen kohdistuvia haittavaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Risankitsumabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia olivat ylähengitystieinfektiot (13,0 % psoriaasipotilailla).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa risankitsumabin käytön yhteydessä todetut haittavaikutukset (taulukko 2) luetellaan MedDRA-elinjärjestelmäluokittain seuraavan käytännön mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on kussakin yleisyysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Haittavaikutukset
Elinjärjestelmä | Esiintymistiheys | Haittavaikutukset |
Infektiot | Hyvin yleinen | Ylähengitystieinfektioa |
Yleinen | Silsainfektiob | |
Melko harvinainen | Karvatupen tulehdus | |
Immuunijärjestelmä | Harvinainen | Anafylaktiset reaktiot |
Hermosto | Yleinen | Päänsärkyc |
Iho ja ihonalainen kudos | Yleinen | Kutina Ihottuma Ekseema |
Melko harvinainen | Nokkosihottuma | |
Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Uupumusd Pistoskohdan reaktiote |
a Mukana: hengitystieinfektio (virusperäinen, bakteeriperäinen tai määrittelemätön), sinuiitti (myös akuutti), riniitti, nenänielutulehdus, nielutulehdus (myös virusperäinen), tonsilliitti, laryngiitti, trakeiitti b Mukana: jalkasilsa, nivussilsa, vartalosilsa, tinea versicolor, käsisilsa, kynsisilsa, ihon sieni‑infektio c Mukana: päänsärky, jännityspäänsärky, sivuontelopäänsärky d Mukana: uupumus, voimattomuus e Mukana: pistoskohdan mustelma, punoitus, hematooma, verenvuoto, ärsytys, kipu, kutina, reaktio, turvotus, kovettuma, ihottuma | ||
Valikoitujen haittavaikutusten kuvaus
Infektiot
Infektioiden esiintyvyys kliinisissä psoriaasitutkimuksissa oli 75,5 tapahtumaa / 100 potilasvuotta ja kliinisissä nivelpsoriaasitutkimuksissa 43,0 tapahtumaa / 100 potilasvuotta. Tutkittavat altistuivat risankitsumabille myös pitkäaikaisesti. Valtaosa tapauksista oli ei-vakavia ja vaikeusasteeltaan lieviä tai keskivaikeita, eikä valtaosa tapauksista johtanut risankitsumabihoidon lopettamiseen. Vakavien infektioiden esiintyvyys oli 1,7 tapahtumaa / 100 potilasvuotta psoriaasitutkimuksissa ja 2,6 tapahtumaa / 100 potilasvuotta nivelpsoriaasitutkimuksissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Kun risankitsumabia käytettiin kliinisissä psoriaasitutkimuksissa aikuisille kliinisinä suositusannoksina enintään 52 viikon ajan, hoidon aikana kehittyneitä vasta-aineita lääkettä kohtaan todettiin 24 %:lla arvioiduista tutkittavista (263/1 079) ja neutraloivia vasta-aineita 14 %:lla arvioiduista tutkittavista (150/1 079). Jatkotutkimuksessa pitkäaikaiselle risankitsumabihoidolle altistuneilla henkilöillä enintään 204 viikon hoidon aikana todettu immunogeenisuusprofiili oli yhdenmukainen verrattuna ensimmäisiin 52 hoitoviikkoon.
Risankitsumabia kohtaan muodostuneisiin vasta-aineisiin, myöskään neutraloiviin vasta-aineisiin, ei useimpien psoriaasia sairastavien aikuisten tutkittavien kohdalla liittynyt kliinisen vasteen eikä turvallisuuden muutoksia. Kliininen vaste vaikutti olevan tavallista alhaisempi niillä harvoilla tutkittavilla (noin 1 %; 7/1 000 viikolla 16 ja 6/598 viikolla 52), joilla vasta-ainetitterit olivat suuret (> 128). Injektiokohdan reaktioiden ilmaantuvuus oli numeerisesti suurempi lääkevasta-ainepositiivisilla tutkittavilla verrattuna lääkevasta-ainenegatiivisiin tutkittaviin sekä lyhyellä aikavälillä (16 viikkoa: lääkevasta-ainepositiivisilla 2,7 % ja lääkevasta-ainenegatiivisilla 1,3 %) että pitkällä aikavälillä (52 viikkoa: lääkevasta-ainepositiivisilla 5,0 % ja lääkevasta-ainenegatiivisilla 3,3 %). Kaikki injektiokohdan reaktiot olivat vaikeusasteeltaan lieviä tai keskivaikeita, yksikään ei ollut vakava eikä yksikään johtanut risankitsumabihoidon lopettamiseen.
Kun risankitsumabia käytettiin kliinisessä psoriaasitutkimuksessa 6 – < 18‑vuotiaille pediatrisille tutkittaville kliinisinä suositusannoksina enintään 52 viikon ajan, hoidon aikana kehittyneitä vasta-aineita lääkettä kohtaan todettiin 14,8 %:lla arvioiduista tutkittavista (13/88) ja neutraloivia vasta-aineita 2,3 %:lla arvioiduista tutkittavista (2/88). Risankitsumabia kohtaan muodostuneisiin vasta‑aineisiin ei liittynyt kliinisen vasteen eikä turvallisuuden muutoksia; risankitsumabin vasta-aineille positiivisten potilaiden määrä on kuitenkin liian pieni, jotta voitaisi tehdä lopullisia päätelmiä vaikutuksesta risankitsumabin tehoon ja turvallisuuteen.
Kun risankitsumabia käytettiin kliinisissä nivelpsoriaasitutkimuksissa aikuisille kliinisinä suositusannoksina enintään 28 viikon ajan, hoidon aikana kehittyneitä vasta‑aineita lääkettä kohtaan todettiin 12,1 %:lla arvioiduista tutkittavista (79/652) ja neutraloivia vasta‑aineita 0 %:lla arvioiduista tutkittavista (0/652). Risankitsumabia kohtaan muodostuneisiin vasta‑aineisiin ei nivelpsoriaasia sairastavilla tutkittavilla liittynyt kliinisen vasteen eikä turvallisuuden muutoksia.
Nivelpsoriaasi
Risankitsumabia käyttäneillä nivelpsoriaasipotilailla todettu turvallisuusprofiili vastasi yleisesti ottaen läiskäpsoriaasipotilailla todettua turvallisuusprofiilia.
Pediatriset potilaat
Risankitsumabin turvallisuutta keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavilla pediatrisilla potilailla arvioitiin neliosaisessa tutkimuksessa, jossa turvallisuutta arvioitiin enintään 52 viikon ajan 137:llä iältään 6 – < 18‑vuotiaalla pediatrisella tutkittavalla. Turvallisuusprofiili risankitsumabihoitoa saaneilla läiskäpsoriaasia sairastavilla pediatrisilla tutkittavilla vastasi läiskäpsoriaasia sairastavilla aikuisilla tutkittavilla todettua turvallisuusprofiilia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden ja löydösten varalta ja aloittaa viipymättä asianmukainen oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC18
Vaikutusmekanismi
Risankitsumabi on humanisoitu immunoglobuliini G1 ‑luokan (IgG1) monoklonaalinen vasta-aine, joka sitoutuu selektiivisesti ja suurella affiniteetilla ihmisen interleukiini-23-sytokiinin (IL-23) p19-alayksikköön sitoutumatta IL-12-sytokiiniin ja estää IL-23:n vuorovaikutusta IL-23-reseptorikompleksin kanssa. IL-23 on inflammaatioon ja immuunivasteisiin osallistuva sytokiini. Estämällä IL-23:n sitoutumisen reseptoriinsa risankitsumabi estää IL-23-riippuvaista solujen signalointia ja inflammaatiota edistävien sytokiinien vapautumista.
Farmakodynaamiset vaikutukset
Psoriaasia sairastaneilla tutkittavilla tehdyissä tutkimuksissa todettiin, että IL-23/IL-17-akseliin liittyvien geenien ilmentyminen ihossa väheni risankitsumabikerta-annoksen jälkeen. Psoriaasimuutoksissa todettiin myös epidermiksen ohenemista, tulehdussoluinfiltraation vähenemistä ja psoriaasin tautimarkkerien ilmentymisen vähenemistä.
Kliininen teho ja turvallisuus
Aikuisten läiskäpsoriaasi
Risankitsumabin tehoa ja turvallisuutta arvioitiin 2 109 tutkittavalla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi, neljässä satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (ULTIMMA-1, ULTIMMA-2, IMMHANCE ja IMMVENT). Tutkittavat olivat 18 vuotta täyttäneitä läiskäpsoriaasipotilaita, joilla psoriaasi-ihottuma peitti vähintään 10 % kehon pinta-alasta (BSA, Body Surface Area ≥10 %), lääkärin staattinen kokonaisarvio psoriaasin vaikeusasteesta (static Physician Global Assessment, sPGA) oli ≥ 3 asteikolla 0–4 (läiskien paksuus/kovettumat, punoitus ja hilseily), Psoriasis Area and Severity Index ‑pisteet (PASI) olivat ≥ 12 ja joille harkittiin systeemistä hoitoa tai valohoitoa.
Tutkittavien PASI-pisteiden mediaani oli lähtötilanteessa 17,8 pistettä, BSA-arvon mediaani 20,0 % ja DLQI-pisteiden mediaani oli 13,0. Lähtötilanteen sPGA-arvo oli ”vaikea” 19,3 %:lla tutkittavista ja ”kohtalainen” 80,7 %:lla tutkittavista. Yhteensä 9,8 %:lla tutkittavista oli diagnosoitu nivelpsoriaasi.
Kaikkien tutkimusten tutkittavista yhteensä 30,9 % ei ollut saanut aiemmin mitään systeemisiä hoitoja (kuten ei-biologisia tai biologisia lääkkeitä). 38,1 % oli saanut aiemmin valohoitoa tai fotokemoterapiaa, 48,3 % oli saanut aiemmin ei-biologista systeemistä hoitoa, 42,1 % oli saanut aiemmin biologista hoitoa ja 23,7 % oli saanut vähintään yhtä TNF‑alfan estäjähoitoa psoriaasin hoitoon. Näissä ja muissa vaiheen 2/3 tutkimuksissa loppuun saakka mukana olleet potilaat saivat mahdollisuuden osallistua avoimeen LIMMITLESS-jatkotutkimukseen.
ULTIMMA-1 ja ULTIMMA-2
ULTIMMA-1- ja ULTIMMA-2-tutkimuksiin otettiin 997 tutkittavaa (598 tutkittavaa satunnaistettiin saamaan 150 mg risankitsumabia, 199 tutkittavaa saamaan 45 mg tai 90 mg ustekinumabia [lähtöpainon mukaan] ja 200 tutkittavaa saamaan lumelääkettä). Tutkittavat saivat hoitoa viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein. ULTIMMA-1- ja ULTIMMA-2-tutkimusten kaksi rinnakkaista ensisijaista päätetapahtumaa olivat 1) PASI 90 ‑vasteen saavuttaneiden tutkittavien osuus ja 2) sPGA-arvon 0 tai 1 (”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”) saavuttaneiden tutkittavien osuus viikolla 16 verrattuna lumelääkkeeseen. Rinnakkaisten ensisijaisten päätetapahtumien ja muiden päätetapahtumien tulokset esitetään taulukossa 3 ja kuvassa 1.
Taulukko 3: Teho- ja elämänlaatutulokset ULTIMMA‑1-ja ULTIMMA‑2-tutkimusten aikuisilla läiskäpsoriaasipotilailla
ULTIMMA‑1 | ULTIMMA‑2 | |||||
Risankitsumabi(N = 304)n (%) | Ustekinumabi (N = 100) n (%) | Lume(N = 102)n (%) | Risankitsumabi(N = 294)n (%) | Ustekinumabi (N = 99) n (%) | Lume(N = 98)n (%) | |
sPGA 0 tai 1 (”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”) | ||||||
Vk 16a | 267 (87,8) | 63 (63,0) | 8 (7,8) | 246 (83,7) | 61 (61,6) | 5 (5,1) |
Vk 52 | 262 (86,2) | 54 (54,0) | -- | 245 (83,3) | 54 (54,5) | -- |
sPGA 0 (”ei ihomuutoksia”) | ||||||
Vk 16 | 112 (36,8) | 14 (14,0) | 2 (2,0) | 150 (51,0) | 25 (25,3) | 3 (3,1) |
Vk 52 | 175 (57,6) | 21 (21,0) | -- | 175 (59,5) | 30 (30,3) | -- |
PASI 75 | ||||||
Vk 12 | 264 (86,8) | 70 (70,0) | 10 (9,8) | 261 (88,8) | 69 (69,7) | 8 (8,2) |
Vk 52 | 279 (91,8) | 70 (70,0) | -- | 269 (91,5) | 76 (76,8) | -- |
PASI 90 | ||||||
Vk 16a | 229 (75,3) | 42 (42,0) | 5 (4,9) | 220 (74,8) | 47 (47,5) | 2 (2,0) |
Vk 52 | 249 (81,9) | 44 (44,0) | -- | 237 (80,6) | 50 (50,5) | -- |
PASI 100 | ||||||
Vk 16 | 109 (35,9) | 12 (12,0) | 0 (0,0) | 149 (50,7) | 24 (24,2) | 2 (2,0) |
Vk 52 | 171 (56,3) | 21 (21,0) | -- | 175 (59,5) | 30 (30,3) | -- |
DLQI 0 tai 1b | ||||||
Vk 16 | 200 (65,8) | 43 (43,0) | 8 (7,8) | 196 (66,7) | 46 (46,5) | 4 (4,1) |
Vk 52 | 229 (75,3) | 47 (47,0) | -- | 208 (70,7) | 44 (44,4) | -- |
PSS 0 (ei oireita)c | ||||||
Vk 16 | 89 (29,3) | 15 (15,0) | 2 (2,0) | 92 (31,3) | 15 (15,2) | 0 (0,0) |
Vk 52 | 173 (56,9) | 30 (30,0) | -- | 160 (54,4) | 30 (30,3) | -- |
Kaikissa risankitsumabin vertailuissa ustekinumabiin ja lumelääkkeeseen saavutettiin merkitsevyystaso p < 0,001; poikkeuksena oli PASI 75 ‑vasteen saavuttaneiden osuus ULTIMMA-2-tutkimuksen viikolla 52, jossa p = 0,001. a Rinnakkaiset ensisijaiset päätetapahtumat verrattuna lumelääkkeeseen b Ei vaikutusta terveyteen liittyvään elämänlaatuun c Psoriasis Symptoms Scale (PSS) arvo 0 tarkoittaa ei oireita; kipua, kutinaa, punoitusta eikä poltetta viimeisen 24 tunnin aikana | ||||||
Kuva 1: PASI-pisteiden prosentuaalinen keskimuutos lähtötilanteesta ULTIMMA-1- ja ULTIMMA-2-tutkimuksissa eri ajankohtina
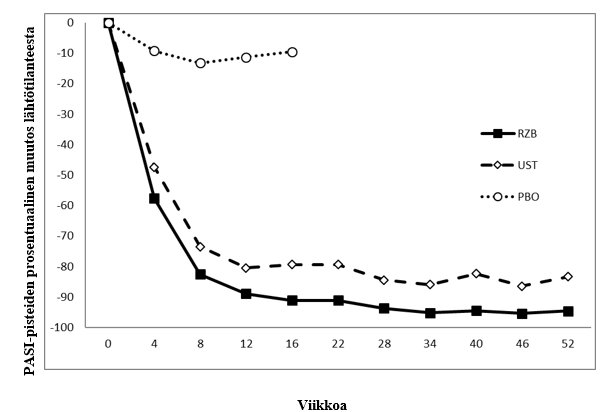
RZB = risankitsumabi
UST = ustekinumabi
PBO = lumelääke
p < 0,001 kunakin ajankohtana
Kun tietojen analyysissä huomioitiin ikä, sukupuoli, etninen tausta, paino ≤ 130 kg, lähtötilanteen PASI-pisteet, samanaikainen nivelpsoriaasi, aiempi ei-biologinen systeemilääkitys, aiempi biologinen hoito ja biologisen hoidon aiempi epäonnistuminen, risankitsumabin tuottamassa vasteessa ei todettu eroja näiden alaryhmien välillä.
Päänahan, kynsien, kämmenten ja jalkapohjien psoriaasin todettiin lieventyneen risankitsumabihoitoa saaneilla tutkittavilla viikolla 16 ja viikolla 52.
Taulukko 4: NAPSI-, PPASI- ja PSSI-indeksien keskimuutokset lähtötilanteesta
ULTIMMA-1 | ULTIMMA-2 | IMMHANCE | ||||
Risankitsumabi | Lume | Risankitsumabi | Lume | Risankitsumabi | Lume | |
NAPSI: Muutos viikon 16 kohdalla (SE) | N = 178; -9,0 (1,17) | N = 56; 2,1 (1,86) *** | N = 177; -7,5 (1,03) | N = 49; 3,0 (1,76) *** | N = 235; -7,5 (0,89) | N = 58; 2,5 (1,70) *** |
PPASI: Muutos viikon 16 kohdalla (SE) | N = 95; -5,93 (0,324) | N = 34; -3,17 (0,445) *** | N = 86; -7,24 (0,558) | N = 23; -3,74 (1,025) ** | N = 113; -7,39 (0,654) | N = 26; -0,27 (1,339) *** |
PSSI: Muutos viikon 16 kohdalla (SE) | N = 267; -17,6 (0,47) | N = 92; -2,9 (0,69) *** | N = 252; -18,4 (0,52) | N = 83; -4,6 (0,82) *** | N = 357; -20,1 (0,40) | N = 88; -5,5 (0,77) *** |
NAPSI: Muutos viikon 52 kohdalla (SE) | N = 178; -15,7 (0,94) | - | N = 183; -16,7 (0,85) | - | - | - |
PPASI: Muutos viikon 52 kohdalla (SE) | N = 95; -6,16 (0,296) | - | N = 89; -8,35 (0,274) | - | - | - |
PSSI: Muutos viikon 52 kohdalla (SE) | N = 269; -17,9 (0,34) | - | N = 259; -18,8 (0,24) | - | - | - |
NAPSI: Nail Psoriasis Severity Index, PPASI: Palmoplantar Psoriasis Severity Index, PSSI: Psoriasis Scalp Severity Index ja SE: keskivirhe ** P < 0,01 risankitsumabiin verrattuna *** P < 0,001 risankitsumabiin verrattuna | ||||||
Hospital Anxiety and Depression Scale ‑asteikolla (HADS) mitatun ahdistuneisuuden ja masennuksen todettiin lievittyneen risankitsumabiryhmässä viikolla 16 verrattuna lumeryhmään.
Vasteen säilyminen
Risankitsumabia ULTIMMA-1- ja ULTIMMA-2-tutkimuksissa saaneiden tutkittavien integroidussa analyysissä todettiin, että PASI 100 ‑vasteen viikolla 16 saavuttaneiden tutkittavien joukossa sama vaste säilyi 79,8 %:lla (206/258) risankitsumabihoitoa jatkaneista tutkittavista viikolla 52. Tutkittavista, jotka saavuttivat PASI 90 ‑vasteen viikolla 16, tämä vaste säilyi 88,4 %:lla (398/450) viikolla 52.
ULTIMMA-1- ja ULTIMMA-2-tutkimuksissa risankitsumabia saaneista potilaista 525 jatkoi LIMMITLESS-tutkimuksessa saaden risankitsumabihoitoa 12 viikon välein. Heistä 376 (71,6 %) jatkoi avointa hoitoa vielä 252 viikon ajan. Tutkittavilla, jotka pysyivät mukana tutkimuksessa, risankitsumabihoidolla viikolla 52 saavutetut paranemat PASI 90 -vasteessa ja sPGA-vasteessa ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” säilyivät viikon 304 loppuun asti.
ULTIMMA-1- ja ULTIMMA-2-tutkimuksissa ustekinumabia saaneista potilaista 172 sai LIMMITLESS-tutkimuksessa risankitsumabia 12 viikon välein. Heistä 116 (67,4 %) pysyi mukana tutkimuksen loppuun, mihin sisältyi 252 viikkoa kestänyt avoin risankitsumabihoito ja seuranta tutkimuksen päättyessä. Tutkittavilla, jotka pysyivät mukana tutkimuksessa, PASI 90 -vasteet ja sPGA-vasteet ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” lisääntyivät viikolta 52 viikolle 76 ja säilyivät sitten viikon 304 loppuun asti.
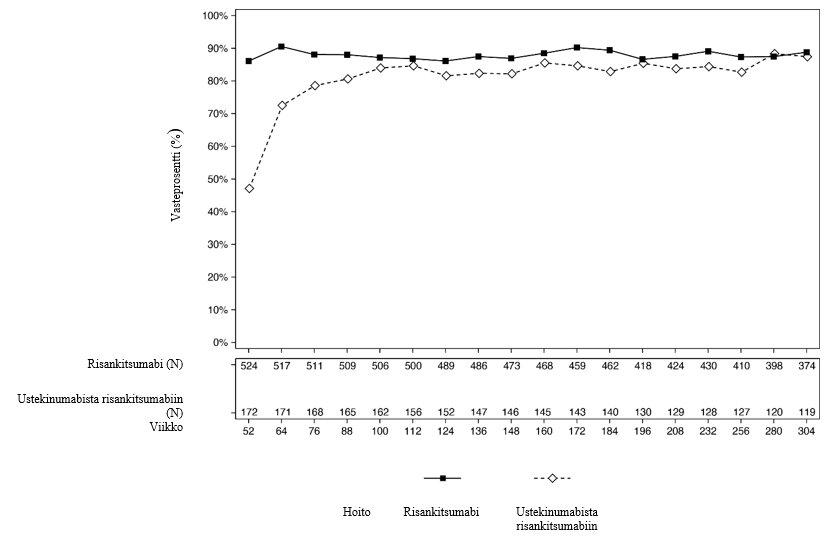
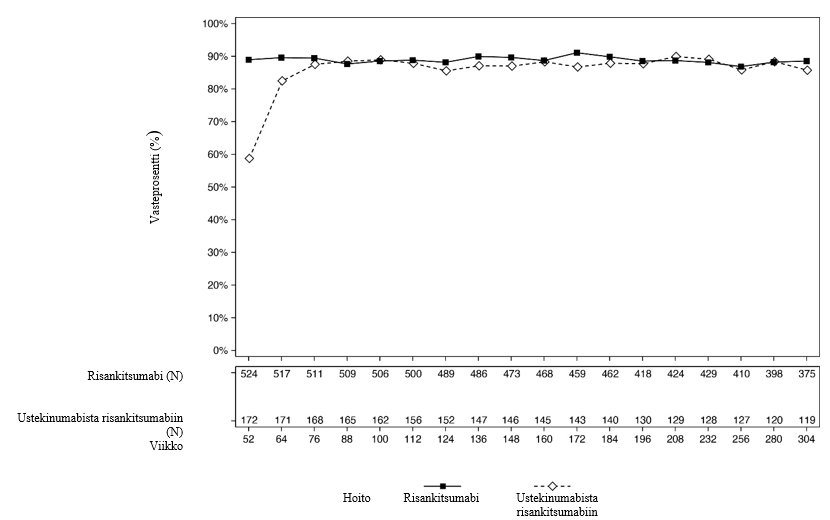
Kuvissa 2 ja 3 esitetään PASI 90 -vasteprosentit ja sPGA-vasteprosentit ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” tutkittavilla, jotka olivat mukana LIMMITLESS-tutkimuksen 252 viikkoa kestäneen avoimen hoitojakson loppuun.
Kuva 2: Niiden potilaiden prosenttiosuudet, jotka saavuttivat PASI 90 -vasteen (havaitut tapaukset) LIMMITLESS-tutkimuksessa
Kuva 3: Niiden tutkittavien prosenttiosuudet, jotka saavuttivat sPGA-vasteen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” käyntikohtaisesti (havaitut tapaukset) LIMMITLESS-tutkimuksessa
Dermatology Life Quality Index (DLQI 0 tai 1) -pisteiden paranemat säilyivät jatkuvaa risankitsumabihoitoa saaneilla potilailla viikon 304 loppuun asti avoimessa LIMMITLESS-jatkotutkimuksessa.
Risankitsumabin turvallisuusprofiili yli viiden vuoden altistuksen aikana vastasi sen turvallisuusprofiilia viikkoon 16 mennessä.
IMMHANCE
IMMHANCE-tutkimukseen otettiin 507 tutkittavaa (407 tutkittavaa satunnaistettiin saamaan 150 mg risankitsumabia ja 100 tutkittavaa lumeryhmään). Tutkittavat saivat hoitoa viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein. Tutkittavat, jotka saivat aluksi risankitsumabia ja saavuttivat sPGA:n”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” viikolla 28, satunnaistettiin uudelleen joko jatkamaan risankitsumabihoitoa 12 viikon välein viikkoon 88 asti (seuranta 16 viikkoa viimeisen risankitsumabiannoksen jälkeen) tai lopettamaan hoito.
Viikolla 16 risankitsumabi oli lumelääkettä parempi rinnakkaisten ensisijaisten päätetapahtumien suhteen eli sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” saavuttaneiden tutkittavien osuuden suhteen (osuus 83,5 % risankitsumabiryhmässä ja 7,0 % lumeryhmässä) ja PASI 90 ‑vasteen saavuttaneiden tutkittavien osuuden suhteen (73,2 % risankitsumabiryhmässä ja 2,0 % lumeryhmässä).
IMMHANCE-tutkimukseen osallistui 31 tutkittavaa, joilla oli latentti tuberkuloosi ja jotka eivät saaneet tutkimuksen aikana estolääkitystä. Kenellekään heistä ei kehittynyt aktiivista tuberkuloosia risankitsumabihoidon aikana, kun seurannan keskipituus oli 55 viikkoa.
Tutkittavat, jotka saavuttivat sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” IMMHANCE-tutkimuksen viikolla 28, satunnaistettiin uudelleen joko jatkamaan risankitsumabihoitoa tai lopettamaan se. 81,1 % (90/111) risankitsumabihoitoa jatkamaan satunnaistetuista säilytti tämän vasteen viikolla 104, kun taas risankitsumabihoidon lopettamiseen satunnaistetuilla vastaava osuus oli 7,1 % (16/225). Näiden tutkittavien joukossa 63,1 % (70/111) risankitsumabihoitoa jatkamaan satunnaistetuista saavutti sPGA-tuloksen ”ei ihomuutoksia” viikolla 104, kun taas risankitsumabihoidon lopettamiseen satunnaistetuilla vastaava osuus oli 2,2 % (5/225).
Tutkittavat, jotka saavuttivat sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” viikolla 28 ja joiden sPGA-tulos palautui ”kohtalaiseksi” tai ”vaikeaksi” risankitsumabihoidon lopettamisen jälkeen, 83,7 % (128/153) saavutti uudelleen sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” 16 viikon kuluttua hoidon uudelleenaloituksesta. sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” menettämistä todettiin jo viikolla 12 unohtuneen annoksen jälkeen. Tutkittavista, jotka satunnaistettiin uudelleen lopettamaan hoito, 80,9 %:lla (182/225) oireet palasivat ja keskimääräinen aika oireiden paluuseen oli 295 päivää. Sellaisia ominaisuuksia ei tunnistettu, jotta voitaisiin ennakoida aika vasteen menetykseen tai todennäköisyyttä vasteen palautumiseen yksittäisen potilaan kohdalla.
IMMVENT
IMMVENT-tutkimukseen otettiin 605 tutkittavaa (301 tutkittavaa satunnaistettiin saamaan risankitsumabia ja 304 tutkittavaa saamaan adalimumabia). Risankitsumabiryhmään satunnaistetut tutkittavat saivat 150 mg risankitsumabia viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein. Adalimumabiryhmään satunnaistetut tutkittavat saivat 80 mg adalimumabia viikolla 0, 40 mg adalimumabia viikolla 1 ja 40 mg adalimumabia joka toinen viikko viikkoon 15 asti. Viikosta 16 alkaen adalimumabia saavat tutkittavat jatkoivat adalimumabihoitoaan tai vaihtoivat hoitoa vasteesta riippuen seuraavasti:
-
Jos vaste oli huonompi kuin PASI 50, tutkittavat siirtyivät risankitsumabihoitoon;
-
Jos vaste oli vähintään PASI 50 mutta alle PASI 90, tutkittavat satunnaistettiin uudelleen joko jatkamaan adalimumabihoitoa tai siirtymään risankitsumabihoitoon;
-
Jos PASI 90 ‑vaste saavutettiin, adalimumabihoitoa jatkettiin.
Tulokset esitetään taulukossa 5.
Taulukko 5: Teho- ja elämänlaatutulokset läiskäpsoriaasia sairastavilla aikuisilla IMMVENT-tutkimuksen viikolla 16
Risankitsumabi (N = 301) n (%) | Adalimumabi (N = 304) n (%) | |
sPGA ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”a | 252 (83,7) | 183 (60,2) |
PASI 75 | 273 (90,7) | 218 (71,7) |
PASI 90a | 218 (72,4) | 144 (47,4) |
PASI 100 | 120 (39,9) | 70 (23,0) |
DLQI 0 tai 1b | 198 (65,8) | 148 (48,7) |
Kaikissa vertailuissa p < 0,001. a Rinnakkaiset ensisijaiset päätetapahtumat b Ei vaikutusta terveyteen liittyvään elämänlaatuun | ||
Kun adalimumabilla PASI 50 ‑tasoa paremman mutta PASI 90 ‑tasoa huonomman vasteen viikolla 16 saavuttaneet tutkittavat satunnaistettiin uudelleen, PASI 90 ‑vasteprosenteissa todettiin eroja risankitsumabihoitoon siirtyneen ja adalimumabihoitoa jatkaneen ryhmän välillä 4 viikon kuluttua uudelleen satunnaistamisesta (PASI 90 ‑vasteprosentti risankitsumabihoitoon siirtyneillä 49,1 % ja adalimumabia jatkaneilla 26,8 %).
28 viikon kuluttua uudelleen satunnaistamisesta saavutetut tulokset esitetään taulukossa 6 ja kuvassa 4.
Taulukko 6: Tehotulokset IMMVENT-tutkimuksessa 28 viikon kuluttua uudelleen satunnaistamisesta
Risankitsumabiin siirtyneet (N = 53)n (%) | Adalimumabia jatkaneet (N = 56) n (%) | |
PASI 90 | 35 (66,0) | 12 (21,4) |
PASI 100 | 21 (39,6) | 4 (7,1) |
Kaikissa vertailuissa p < 0,001 | ||
Kuva 4: PASI 90 ‑vasteet IMMVENT-tutkimuksen eri ajankohtina uudelleen satunnaistamisen jälkeen
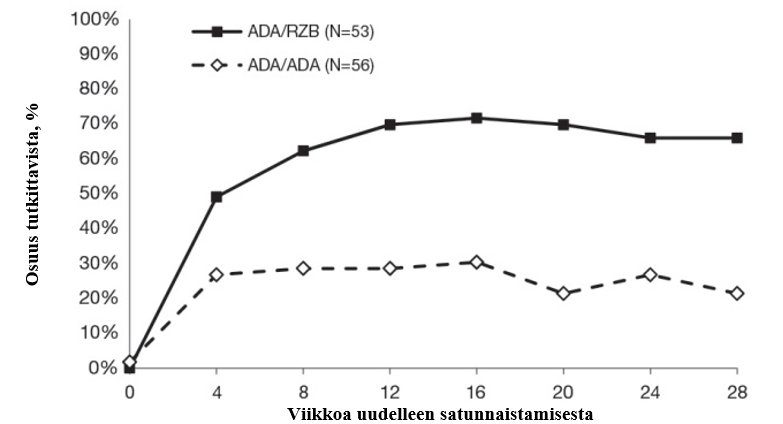
ADA/ADA: Adalimumabiryhmään satunnaistetut tutkittavat, jotka jatkoivat adalimumabihoitoa
ADA/RZB: Adalimumabiryhmään satunnaistetut tutkittavat, jotka siirtyivät risankitsumabihoitoon
p < 0,05 viikolla 4 ja p < 0,001 kunakin ajankohtana viikolta 8 alkaen
270 tutkittavaa siirtyi adalimumabista risankitsumabihoitoon ilman lääkkeetöntä hoitotaukoa. Risankitsumabin turvallisuusprofiili oli tässä ryhmässä samankaltainen kuin tutkittavilla, jotka aloittivat risankitsumabihoidon mahdollisten aiempien systeemisten hoitojen jälkeen pidetyn hoitotauon jälkeen.
Päänahan ja genitaalialueen läiskäpsoriaasi
Risankitsumabin tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (UNLIMMITED), johon otettiin vähintään 18 vuoden ikäisiä tutkittavia, joilla oli keskivaikea tai vaikea päänahan psoriaasi (UNLIMMITED‑S), joka määriteltiin PSSI (Psoriasis Scalp Severity Index) -pistemääränä ≥ 12, päänahan osalta tutkijan yleisarvion (scalp Investigator Global Assessment, scalp IGA) pistemääränä ≥ 3 sekä psoriaasin ≥ 30 %:n laajuutena päänahan pinta-alasta, tai keskivaikea tai vaikea sukupuolielinten psoriaasi (UNLIMMITED‑G), joka määriteltiin sukupuolielinten osalta tutkijan staattisen yleisarvion (static Physician’s Global Assessment of Genitalia, sPGA) pistemääränä ≥ 3 lähtötilanteessa. Kaikkien tutkittavien BSA-arvo oli ≥ 1 % ja sPGA-pistemäärä ≥ 3 lähtötilanteessa.
UNLIMMITED-tutkimuksen tutkittavat satunnaistettiin saamaan ihon alle joko 150 mg risankitsumabia tai lumelääkettä viikolla 0 ja 4. Viikolta 16 alkaen kaikki tutkittavat saivat 150 mg risankitsumabia 12 viikon välein viikolla 40 annettavaan viimeiseen annokseen asti.
Päänahan alue (UNLIMMITED‑S)
UNLIMMITED‑S-alatutkimukseen otettiin 105 tutkittavaa. Lähtötilanteen BSA-arvo oli 61,9 %:lla tutkittavista ≥ 10 % ja 38,1 %:lla tutkittavista < 10 %. Lähtötilanteen keskimääräinen BSA-arvo oli 16,8 %. Lähtötilanteessa 76,2 %:lla tutkittavista sPGA-pistemäärä = 3 ja 23,8 %:lla sPGA pistemäärä = 4.
Lähtötilanteessa 54,3 % tutkittavista ei ollut saanut aiempaa systeemistä hoitoa (ei‑biologiset ja biologiset hoidot mukaan lukien), eikä kukaan tutkittavista (0 %) ollut saanut aiempaa valohoitoa. 15,2 % oli saanut aiempaa ei‑biologista systeemistä hoitoa ja 37,1 % oli saanut aiempaa biologista hoitoa.
Ensisijaisen ja tärkeimpien toissijaisten päätetapahtumien tulokset on esitetty taulukossa 7.
Taulukko 7. Tehotulokset päänahan psoriaasia sairastavilla aikuisilla UNLIMMITED‑S-alatutkimuksessa viikolla 16
Päätetapahtuma | Risankitsumabi (N = 51) n (%) | Lumelääke (N = 54) n (%) | Hoitoero (95 %:n luottamusväli) |
scalp IGA -vaste ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” (0 tai 1)a | 31 (60,8) | 7 (13,0) | 47,0 (31,2, 62,8) |
PSSI 75b | 38 (74,5) | 12 (22,2) | 52,9 (37,5, 68,3) |
PSSI 90c | 27 (52,9) | 7 (13,0) | 39,8 (24,4, 55,2) |
PSSI 100d | 23 (45,1) | 7 (13,0) | 31,2 (15,4, 46,9) |
PSS-arvon keskimuutos lähtötilanteesta | N = 44 -6,0 | N = 49 -1,0 | -5,0 (-6,6, -3,3) |
Kaikissa vertailuissa saavutettiin merkitsevyystaso p < 0,001, korjattu hoitoero (95 %:n luottamusväli) a Ensisijainen päätetapahtuma b PSSI-pistemäärän ≥ 75 %:n paranema lähtötilanteesta c PSSI-pistemäärän ≥ 90 %:n paranema lähtötilanteesta d PSSI-pistemäärän 100 %:n paranema lähtötilanteesta | |||
Scalp IGA -pistemäärän 0 viikolla 16 saavuttaneiden tutkittavien osuus oli suurempi risankitsumabihoitoa saaneilla tutkittavilla verrattuna lumelääkettä saaneisiin tutkittaviin (41,2 % vs. 11,1 %).
Päänahan kutinaa koskevaan numeeriseen arviointiasteikkoon (Scalp Itch Numeric Rating Scale) perustuvan vasteen – jonka määritelmä oli ≥ 4 pisteen paranema (vähenemä) Scalp Itch NRS -pistemäärässä lähtötilanteesta – saavuttaneiden osuus tutkittavilla, joiden lähtötilanteen pistemäärä oli ≥ 4, oli suurempi risankitsumabihoitoa saaneilla tutkittavilla verrattuna lumelääkettä saaneisiin tutkittaviin viikolla 16 (50,0 % vs. 11,1 %).
DLQI-pistemäärän 0 tai 1 (ei vaikutusta terveyteen liittyvään elämänlaatuun) viikolla 16 saavuttaneiden tutkittavien osuus oli suurempi risankitsumabihoitoa saaneilla tutkittavilla verrattuna lumelääkettä saaneisiin tutkittaviin (47,1 % vs. 11,1 %).
Genitaalialue (UNLIMMITED‑G)
UNLIMMITED‑G-alatutkimukseen otettiin 109 tutkittavaa. Lähtötilanteen BSA-arvo oli 63,3 %:lla tutkittavista ≥ 10 % ja 36,7 %:lla tutkittavista < 10 %. Lähtötilanteen keskimääräinen BSA-arvo oli 17,2 %. Lähtötilanteessa 80,7 %:lla tutkittavista sPGA pistemäärä = 3 ja 19,3 %:lla sPGA pistemäärä = 4.
Lähtötilanteessa 61,5 % tutkittavista ei ollut saanut aiempaa systeemistä hoitoa (ei‑biologiset ja biologiset hoidot mukaan lukien), ja 2,8 % tutkittavista oli saanut aiempaa valohoitoa. 16,5 % oli saanut aiempaa ei‑biologista systeemistä hoitoa ja 25,7 % oli saanut aiempaa biologista hoitoa.
Ensisijaisen ja kaikkien toissijaisten päätetapahtumien tulokset on esitetty taulukossa 8.
Taulukko 8. Tehotulokset sukupuolielinten psoriaasia sairastavilla aikuisilla UNLIMMITED‑G-alatutkimuksessa viikolla 16
Päätetapahtuma | Risankitsumabi (N = 55) n (%) | Lumelääke (N = 54) n (%) | Hoitoero (95 %:n luottamusväli) |
sPGA‑G-vaste ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” (0 tai 1)a | 38 (69,1) | 7 (13,0) | 57,0 (42,3, 71,7) |
sPGA‑G-vaste ”ei ihomuutoksia” (0) | 28 (50,9) | 3 (5,6) | 46,7 (32,6, 60,8) |
DLQI-pistemäärä 0 tai 1b | 33 (60,0) | 2 (3,7) | 56,5 (43,0, 70,0) |
≥ 4 pisteen vähenemä GPI-NRS-pistemäärässä lähtötilanteestac | N = 41 20 (48,8) | N = 45 3 (6,7) | 43,0 (26,6, 59,3) |
GenPs-SFQ-kyselyn osion 2 pistemäärä 0 (ei koskaan) tai 1 (harvoin)d, e | N = 31 22 (71,0) | N = 32 7 (21,9) | 46,1 (26,7, 65,6) |
Kaikissa vertailuissa saavutettiin merkitsevyystaso p < 0,001, korjattu hoitoero (95 %:n luottamusväli) a Ensisijainen päätetapahtuma b DLQI-kokonaispistemäärä 0 tai 1 tarkoittaa, ettei ihosairaus vaikuta lainkaan potilaan terveyteen liittyvään elämänlaatuun c Sukupuolielinten kutinan vaikeusasteen lievittyminen, joka määriteltiin vähintään 4 pisteen vähenemänä GPSS (Genital Psoriasis Symptom Scale) -kyselyyn kuuluvan 11‑asteisen numeerisen GPI-NRS (Genital Psoriasis Itch Numeric Rating Scale) ‑arviointiasteikon pistemäärässä tutkittavilla, joiden lähtötilanteen pistemäärä oli ≥ 4 d GenPs-SFQ (Genital Psoriasis Sexual Frequency Questionnaire) -kyselyn osion 2 avulla mitataan missä määrin potilas kokee genitaalialueen psoriaasin vaikuttaneen seksuaaliseen terveyteensä seksuaalitoiminnan (yhdyntöjen tai muun toiminnan) tiheyden osalta viimeisen viikon aikana (kyselyssä käytetään asteikkoa 0–4, ja suurempi pistemäärä tarkoittaa suurempia toiminnan rajoituksia). e Tutkittavilla, joiden lähtötilanteen pistemäärä oli ≥ 2 | |||
Risankitsumabia saaneiden tutkittavien genitaalialueen psoriaasioireiden (kutina, kipu, epämukavuus, pistely, polttelu, punoitus, hilseily ja halkeilu) vaikeusaste oli lievittynyt enemmän lähtötilanteesta GPSS-kyselyllä mitattuna viikolla 16 verrattuna lumelääkettä saaneisiin tutkittaviin. GPSS-kokonaispisteiden muutos lähtötilanteesta viikolla 16 oli risankitsumabiryhmässä -26,5 ja lumelääkeryhmässä -1,0.
Suurempi osuus risankitsumabia saaneista tutkittavista verrattuna lumelääkettä saaneisiin tutkittaviin (71,7 % vs. 22,9 %) saavutti vähintään 2 pisteen vähenemän sukupuolielinten psoriaasia koskevassa potilaan yleisarviossa (Patient’s Global Assessment of Genital Psoriasis, PatGA-Genital) niiden potilaiden osalta, joiden lähtötilanteen pistemäärä oli ≥ 2.
Risankitsumabin turvallisuusprofiili UNLIMMITED‑S- ja UNLIMMITED‑G-tutkimuksissa oli yhdenmukainen aiemmissa läiskäpsoriaasipotilailla tehdyissä tutkimuksissa havaitun turvallisuusprofiilin kanssa.
Pediatriset potilaat
Pediatrinen läiskäpsoriaasi
Risankitsumabin tehoa, turvallisuutta ja farmakokinetiikkaa arvioitiin yhteensä 137:llä iältään 6 – < 18‑vuotiaalla pediatrisella tutkittavalla neliosaisessa tutkimuksessa (OptIMMize‑1), jonka osaan 1 osallistui 12 tutkittavaa, osaan 2 82 tutkittavaa, osaan 3 13 tutkittavaa ja osaan 4 30 tutkittavaa. Tutkittavat, jotka olivat mukana tutkimuksen loppuun asti, saivat mahdollisuuden osallistua avoimeen OptIMMize‑2-jatkotutkimukseen.
OptIMMize‑1
Osa 2 oli satunnaistettu, tehon suhteen arvioijasokkoutettu, aktiivikontrolloitu kohortti, johon otettiin iältään 12 – < 18‑vuotiaita pediatrisia tutkittavia. Osa 4 oli yksihaarainen, avoin kohortti, johon otettiin iältään 6 – < 12‑vuotiaita pediatrisia tutkittavia. Mukaan otetuilla tutkittavilla oli keskivaikea tai vaikea läiskäpsoriaasi, jonka määritelmänä oli BSA-arvo ≥ 10 % sekä sPGA-pistemäärä ≥ 3 tai PASI-pistemäärä ≥ 12.
Osissa 2 ja 4 vähintään 40 kg painavat tutkittavat saivat 150 mg risankitsumabia ja alle 40 kg painavat tutkittavat 55 mg risankitsumabia viikolla 0, viikolla 4 ja sen jälkeen 12 viikon välein. Rinnakkaiset ensisijaiset päätetapahtumat olivat sPGA-tulos ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” (0 tai 1) ja PASI-pistemäärä 75 viikolla 16.
Osassa 2 tutkittavien lähtötilanteen PASI-pistemäärän mediaani oli 15,7 ja lähtötilanteen BSA-arvon mediaani 18,0 %. Yhteensä 3,7 % tutkittavista oli saanut aiemmin biologista hoitoa. Tutkittavat satunnaistettiin suhteessa 2:1 saamaan joko risankitsumabia (N = 54) tai ustekinumabia (N = 28). Ustekinumabihoitoon satunnaistetut < 60 kg painavat tutkittavat saivat 0,75 mg/kg ustekinumabia, 60 – < 100 kg painavat tutkittavat 45 mg ustekinumabia ja ≥ 100 kg painavat tutkittavat 90 mg ustekinumabia viikolla 0 ja viikolla 4. Viikolla 16 ustekinumabiryhmän tutkittavat siirrettiin saamaan risankitsumabia 12 viikon välein siitä eteenpäin. Hoidon kesto oli enimmillään 68 viikkoa.
Osassa 4 tutkittavien lähtötilanteen PASI-pistemäärän mediaani oli 14,7 ja lähtötilanteen BSA-arvon mediaani 14,5 %. Yhteensä 3,3 % tutkittavista oli saanut aiemmin biologista hoitoa. Hoidon kesto oli 52 viikkoa.
Seuraavassa (taulukko 9) on esitetty OptIMMize‑1-tutkimuksen tehotulokset alkuperäisen hoitojakson viikolla 16 kuvaavia tilastoja käyttäen.
Taulukko 9. OptIMMize-1-tutkimuksen tehotulokset viikolla 16
Osa 2 | Osa 4 | ||
Risankitsumabi (N = 54) n (%) | Ustekinumabi (N = 28) n (%) | Risankitsumabi(N = 30) n (%) | |
sPGA-tulos ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”(0 tai 1)a | 43 (79,6) | 21 (75,0) | 27 (90,0) |
PASI-pistemäärä 75 a | 46 (85,2) | 24 (85,7) | 26 (86,7) |
PASI-pistemäärä 90 | 35 (64,8) | 17 (60,7) | 23 (76,7) |
PASI-pistemäärä 100 | 22 (40,7) | 5 (17,9) | 13 (43,3) |
sPGA-tulos ”ei ihomuutoksia” (0) | 22 (40,7) | 5 (17,9) | 13 (43,3) |
a Rinnakkaiset ensisijaiset päätetapahtumat | |||
Teho säilyi viikolle 52 asti taulukossa 9 esitettyjen päätetapahtumien perusteella arvioituna.
Osassa 2 risankitsumabiryhmään satunnaistetut tutkittavat, jotka saavuttivat sPGA-tuloksen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” viikolla 16, satunnaistettiin uudelleen joko jatkamaan risankitsumabin saamista 12 viikon välein viikolle 52 asti (N = 22) tai lopettamaan hoito (N = 21). Viikolla 52 risankitsumabihoitoa jatkaneista tutkittavista 95,5 %:lla (21/22) sPGA-tulos oli edelleen ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset”, verrattuna 42,9 %:iin (9/21) tutkittavista, joiden risankitsumabihoito oli lopetettu.
Osan 2 alkuperäisen hoitojakson viikolla 16 risankitsumabia ja ustekinumabia saaneilla potilailla raportoitiin terveyteen liittyvän elämänlaadun parantumista lähtötilanteeseen nähden CDLQI (Children’s Dermatology Life Quality Index) -indeksillä mitattuna (CDLQI-pisteiden parannus oli risankitsumabia saaneilla -7,4 ja ustekinumabia saaneilla -6,8).
Niillä osan 2 tutkittavista, joiden kutinaa koskeva pistemäärä 11-asteisella numeerisella arviointiasteikolla oli lähtötilanteessa vähintään 4, kutinan todettiin alkuperäisen hoitojakson viikolla 16 lievittyneen 64,9 %:lla (24/37) risankitsumabia saaneista ja 57,1 %:lla (8/14) ustekinumabia saaneista tutkittavista, kun lievittyminen määriteltiin vähintään 4 pisteen vähenemänä lähtötilanteesta.
OptIMMize‑2
OptIMMize‑2-tutkimuksessa arvioitiin risankitsumabihoidon pitkän aikavälin tehoa, turvallisuutta ja siedettävyyttä tutkittavan painoon perustuvalla annoksella 150 mg tai 55 mg joka 12. viikko 129:llä iältään 6 – < 18‑vuotiaalla pediatrisella tutkittavalla, joilla oli keskivaikea tai vaikea läiskäpsoriaasi ja jotka olivat olleet mukana OptIMMize‑1-tutkimuksessa loppuun asti. OptIMMize‑2-tutkimuksessa PASI-pistemääriä 75/90/100 ja sPGA-tuloksia ”ei ihomuutoksia” tai ”minimaaliset ihomuutokset” koskevat vasteosuudet säilyivät niillä 36 tutkittavalla, jotka jatkoivat risankitsumabihoitoa viikolle 108 asti.
Farmakokinetiikka
Imeytyminen
Risankitsumabin farmakokinetiikka oli lineaarinen ja altistus suureni suhteessa annokseen, kun ihon alle annettu annos oli 18–300 mg ja 0,25–1 mg/kg ja laskimoon annettu annos oli 200–1 200 mg ja 0,01–5 mg/kg.
Kun risankitsumabia annettiin ihon alle, huippupitoisuudet plasmassa saavutettiin 3–14 päivän kuluttua annostelusta ja arvioitu absoluuttinen biologinen hyötyosuus oli 89 %. Kun valmistetta annettiin 150 mg:n annoksina viikolla 0, viikolla 4 ja tämän jälkeen 12 viikon välein, arvioidut vakaan tilan huippu- ja jäännöspitoisuudet olivat 12 µg/ml ja 2 µg/ml.
Bioekvivalenssi osoitettiin esitäytetyistä ruiskuista annettujen yhden 150 mg:n risankitsumabi-injektion ja kahden 75 mg:n risankitsumabi-injektion välillä. Bioekvivalenssi osoitettiin myös esitäytetystä ruiskusta ja esitäytetystä kynästä annettujen 150 mg:n risankitsumabi-injektioiden välillä.
Jakautuminen
Risankitsumabin vakaan tilan jakautumistilavuuden (Vss) keskiarvo (± keskihajonta) oli 11,4 (± 2,7) l vaiheen 3 tutkimuksissa psoriaasipotilailla. Tämä viittaa siihen, että risankitsumabin jakautuminen rajoittuu lähinnä vaskulaari- ja interstitiaalitiloihin.
Biotransformaatio
Monoklonaaliset IgG-vasta-ainelääkkeet pilkkoutuvat tyypillisesti pieniksi peptideiksi ja aminohapoiksi kataboliareittien välityksellä samaan tapaan kuin endogeeniset IgG-molekyylit. Risankitsumabi ei oletettavasti metaboloidu sytokromi P450 ‑entsyymivälitteisesti.
Eliminaatio
Risankitsumabin systeemisen puhdistuman (CL) keskiarvo (± keskihajonta) oli 0,3 (± 0,1) l/vrk vaiheen 3 tutkimuksissa psoriaasipotilailla. Risankitsumabin terminaalisen eliminaation puoliintumisajan keskiarvo vaihteli 28 vuorokaudesta 29 vuorokauteen vaiheen 3 tutkimuksissa psoriaasipotilailla.
Risankitsumabi on IgG1-luokan monoklonaalinen vasta-aine, joten se ei oletettavasti suodatu munuaisissa glomerulussuodatuksen kautta eikä erity pilkkoutumattomana virtsaan.
Lineaarisuus/ei-lineaarisuus
Risankitsumabin farmakokinetiikka oli lineaarinen ja systeeminen altistus (Cmax ja AUC) suureni suunnilleen suhteessa annokseen arvioiduilla annosalueilla eli 18–300 mg:n annoksilla ja 0,25–1 mg/kg:n annoksilla ihon alle terveillä henkilöillä ja psoriaasipotilailla.
Interaktiot
Läiskäpsoriaasia sairastaneilla tutkittavilla toteutetussa interaktiotutkimuksessa arvioitiin toistuvien risankitsumabiannosten vaikutusta sytokromi P450 (CYP) ‑toiminnan herkkien testisubstraattien farmakokinetiikkaan. Altistus kofeiinille (CYP1A2-substraatti), varfariinille (CYP2C9-substraatti), omepratsolille (CYP2C19-substraatti), metoprololille (CYP2D6-substraatti) ja midatsolaamille (CYP3A-substraatti) oli risankitsumabihoidon jälkeen verrattavissa tutkittavien altistukseen näille aineille ennen risankitsumabihoitoa. Tämä viittaa siihen, että näiden entsyymien kautta välittyviä kliinisesti merkittäviä interaktioita ei ole.
Populaatiofarmakokineettiset analyysit viittasivat siihen, että joidenkin kliinisiin tutkimuksiin osallistuneiden läiskäpsoriaasipotilaiden käyttämä samanaikainen hoito ei vaikuttanut risankitsumabialtistukseen.
Erityisryhmät
Pediatriset potilaat
Risankitsumabialtistus 6 – < 18‑vuotiailla läiskäpsoriaasia sairastavilla tutkittavilla oli samankaltainen kuin aikuisilla. Näillä tutkittavilla arvioiduilla suositelluilla annostuksilla vakaan tilan huippu- ja jäännöspitoisuuksien mediaanien plasmassa arvioitiin olevan ≥ 40 kg painavilla tutkittavilla 15,7 µg/ml ja 2,3 µg/ml ja < 40 kg painavilla tutkittavilla 11,1 µg/ml ja 1,6 µg/ml.
Potilaat, joilla on munuaisten tai maksan vajaatoiminta
Munuaisten tai maksan vajaatoiminnan vaikutusta risankitsumabin farmakokinetiikkaan ei ole arvioitu spesifisissä tutkimuksissa. Populaatiofarmakokineettisten analyysien perusteella seerumin kreatiniinipitoisuus, kreatiniinipuhdistuma ja maksan toiminnan merkkiaineet (ALAT/ASAT/bilirubiini) eivät vaikuttaneet merkittävästi risankitsumabin puhdistumaan läiskäpsoriaasipotilailla.
Risankitsumabi on IgG1-luokan monoklonaalinen vasta-aine, joten se eliminoituu lähinnä solunsisäisen katabolian kautta eikä todennäköisesti metaboloidu maksan sytokromi P450 ‑entsyymien kautta eikä eliminoidu munuaisteitse.
Paino
Risankitsumabin puhdistuma ja jakautumistilavuus suurenevat painon myötä, mikä voi johtaa tehon heikkenemiseen potilailla, joilla on merkittävä ylipaino (> 130 kg). Tämä havainto perustuu kuitenkin rajalliseen määrään potilaita. Tämänhetkisen suosituksen mukaan annosta ei tarvitse muuttaa painon perusteella aikuispotilaille.
Sukupuoli tai etninen tausta
Sukupuoli ja etninen tausta eivät vaikuttaneet merkitsevästi risankitsumabin puhdistumaan aikuisilla läiskäpsoriaasipotilailla. Risankitsumabialtistuksessa ei todettu kliinisesti merkittäviä eroja, kun kiinalaisia ja japanilaisia tutkittavia verrattiin valkoihoisiin tutkittaviin terveillä vapaaehtoisilla tehdyssä kliinisessä farmakokinetiikan tutkimuksessa.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Mukana on farmakologisen turvallisuuden arviointeja sekä tehostettu pre- ja postnataalista kehitystoksisuutta selvittänyt tutkimus, jossa jaavanmakakiapinoille annetut annokset olivat enintään 50 mg/kg/viikko (jolloin altistus on noin 70-kertainen verrattuna ihmisellä käytettyihin suurimpiin kliinisiin suositusannoksiin).
Risankitsumabilla ei ole tehty mutageenisuus- eikä karsinogeenisuustutkimuksia. Jaavanmakakiapinoilla toteutetussa 26 viikon pituisessa pitkäaikaistoksisuuden tutkimuksessa, jossa annokset olivat enintään 50 mg/kg/viikko (noin 70-kertainen verrattuna ihmisellä käytettyihin suurimpiin kliinisiin suositusannoksiin), ei todettu preneoplastisia tai neoplastisia muutoksia eikä immunotoksisia tai kardiovaskulaarisia haittavaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaattitrihydraatti
Etikkahappo
Trehaloosidihydraatti
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta
Säilytys
Säilytä jääkaapissa (2 °C–8 °C). Ei saa jäätyä.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Esitäytettyä ruiskua voidaan säilyttää huoneenlämmössä (enintään 25 °C:ssa) enintään 24 tunnin ajan alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
Esitäytetty lasiruisku, jossa on kiinteä neula ja neulansuojus sekä automaattinen turvamekanismi.
Skyrizi 55 mg on pakattu pakkauksiin, joissa on 1 esitäytetty ruisku.
Valmisteen kuvaus:
Liuos on väritön tai kellertävä ja kirkas tai hieman opalisoiva.
Käyttö- ja käsittelyohjeet
Ennen lääkkeen pistämistä potilas voi ottaa pakkauksen jääkaapista ja antaa sen lämmetä huoneenlämpöiseksi poissa suorasta auringonvalosta (15–30 minuutin ajan). Esitäytettyä ruiskua ei oteta tällöin ulos pakkauksesta.
Liuoksen pitää olla väritöntä tai kellertävää ja kirkasta tai hieman opalisoivaa.
Yleiset varotoimet
On suositeltavaa tarkastaa jokainen esitäytetty ruisku silmämääräisesti ennen käyttöä. Liuoksessa voi olla muutama läpikuultava tai valkoinen valmistehiukkanen. Skyriziä ei saa käyttää, jos liuos on sameaa tai siinä on värimuutoksia tai suuria hiukkasia. Esitäytettyä ruiskua ei saa ravistaa.
Pakkausselosteessa on valmisteen käyttöä koskevat laajat ohjeet.
Kukin esitäytetty ruisku on tarkoitettu vain yhtä käyttökertaa varten.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SKYRIZI injektioneste, liuos, esitäytetty ruisku
55 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AC18
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi