
SKYRIZI injektionsvätska, lösning i förfylld spruta 55 mg
Kvalitativ och kvantitativ sammansättning
Varje förfylld spruta innehåller 55 mg risankizumab i 0,37 ml lösning.
Risankizumab är en humaniserad monoklonal antikropp av typen immunglobulin G1 (IgG1) som produceras i ovarieceller från kinesisk hamster (CHO) med rekombinant DNA-teknik.
Hjälpämnen med känd effekt
Detta läkemedel innehåller 0,07 mg polysorbat 20 per 55 mg dos.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning (injektionsvätska).
Kliniska uppgifter
Terapeutiska indikationer
Plackpsoriasis hos barn
Skyrizi är avsett för behandling av måttlig till svår plackpsoriasis hos barn och ungdomar från 6 års ålder som behöver systemisk behandling.
Villkor
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Dosering och administreringssätt
Detta läkemedel är avsett att användas under vägledning och överinseende av läkare med erfarenhet av diagnos och behandling av de sjukdomar som Skyrizi är avsett för.
Dosering
Plackpsoriasis hos barn (i åldern 6 till under 18 år)
Den rekommenderade dosen för pediatriska patienter baseras på kroppsvikt (tabell 1) och administreras via subkutan injektion vid vecka 0, vecka 4 och därefter var 12:e vecka.
Tabell 1. Rekommenderad dos för plackpsoriasishos barn
Kroppsvikt vid behandlingstidfället | Rekommenderad dos |
< 40 kg | 55 mg (ges som en injektion med förfylld spruta à 55 mg) |
≥ 40 kg | 150 mg (ges som två injektioner med förfyllda sprutor à 75 mg, eller som en injektion med förfylld injektionspenna eller förfylld spruta à 150 mg)* |
*För barn och ungdomar som väger minst 40 kg, se produktresumén för Skyrizi 150 mg förfylld penna, 150 mg förfylld spruta och 75 mg förfylld spruta.
För ovanstående indikation bör behandlingsavbrott övervägas för patienter som inte har visat något svar efter 16 veckors behandling. Vissa patienter med plackpsoriasis, som till en början uppvisar ett partiellt svar på behandlingen, kan senare uppnå ytterligare förbättring med fortsatt behandling i mer än 16 veckor.
Missad dos
Om en dos missas ska dosen tas så snart som möjligt. Därefter ska doseringen återupptas enligt ordinarie schema.
Särskilda populationer
Nedsatt njur- eller leverfunktion
Inga särskilda studier har utförts för att utvärdera vilken effekt nedsatt lever- eller njurfunktion har på farmakokinetiken hos risankizumab. Dessa tillstånd förväntas generellt sett inte ha någon signifikant inverkan på farmakokinetiken hos monoklonala antikroppar och inga dosjusteringar anses vara nödvändiga (se avsnitt Farmakokinetiska egenskaper).
Pediatrisk population
Det finns ingen relevant användning av risankizumab hos barn yngre än 6 år för indikationen måttlig till svår plackpsoriasis.
Överviktiga patienter
Ingen dosjustering krävs (se avsnitt Farmakokinetiska egenskaper).
Administreringssätt
Skyrizi administreras via subkutan injektion.
Injektionen ska ges i låret eller buken. Patienter ska inte injicera i områden där huden är öm, har blåmärken, är erytematös, har förhårdnader eller påverkad av psoriasis.
Patienter kan själva injicera Skyrizi efter att ha tränats i subkutan injektionsteknik. För barn och ungdomar i åldern 10 till under 18 år rekommenderas att Skyrizi administreras av, eller under överinseende av, en vuxen. För barn i åldern 6 till under 10 år ska Skyrizi administreras av en vuxen. Patienter ska instrueras att läsa de ”Instruktioner för användning” som följer med bipacksedeln före administrering.
Administrering av Skyrizi på utsidan av överarmen får bara utföras av sjukvårdspersonal eller vårdgivare.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Kliniskt betydelsefulla aktiva infektioner (t.ex. aktiv tuberkulos, se avsnitt Varningar och försiktighet).
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Infektioner
Risankizumab kan öka risken för infektion.
För patienter med kronisk infektion, återkommande infektioner eller kända riskfaktorer för infektion, bör risankizumab användas med försiktighet. Behandling med risankizumab bör inte påbörjas hos patienter med klinisk betydelsefull aktiv infektion förrän infektionen gått över eller behandlats på lämpligt sätt.
Patienter som behandlas med risankizumab ska instrueras att söka medicinsk rådgivning om tecken eller symtom på kliniskt betydelsefull kronisk eller akut infektion uppstår. Om en patient utvecklar en sådan infektion eller inte svarar på insatt standardbehandling för infektionen ska patienten övervakas noga och risankizumab ska inte administreras förrän infektionen har gått över.
Tuberkulos
Innan behandling med risankizumab påbörjas bör patienterna undersökas med avseende på
tuberkulosinfektion (TB). Patienter som får risankizumab bör övervakas avseende tecken och
symtom på aktiv TB. Behandling mot TB bör övervägas innan behandling med risankizumab påbörjas hos patienter med anamnes på latent eller aktiv TB för vilka en adekvat behandlingskur inte kan bekräftas.
Immuniseringar
Överväg att slutföra alla lämpliga vaccinationer enligt aktuella riktlinjer för vaccinering innan behandling med risankizumab påbörjas. Om en patient har blivit vaccinerad med levande viralt eller bakteriellt vaccin är rekommendationen att vänta minst 4 veckor innan behandling med risankizumab påbörjas. Patienter som behandlas med risankizumab bör inte få levande vaccin under pågående behandling och i minst 21 veckor efter avslutad behandling (se avsnitt Farmakokinetiska egenskaper).
Överkänslighet
Allvarliga överkänslighetsreaktioner, inklusive anafylaxi, har rapporterats vid användning av risankizumab (se avsnitt Biverkningar). Om en allvarlig överkänslighetsreaktion inträffar ska administreringen av risankizumab omedelbart avbrytas och lämplig behandling påbörjas.
Hjälpämnen med känd effekt
Polysorbat
Detta läkemedel innehåller 0,07 mg polysorbat 20 per 55 mg dos. Polysorbater kan orsaka allergiska reaktioner.
Natrium
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per förfylld spruta, d.v.s. är näst intill ”natriumfritt”.
Interaktioner
Risankizumab förväntas inte genomgå metabolism med hjälp av leverenzymer eller renal eliminering. Inga interaktioner förväntas mellan risankizumab och inhibitorer, inducerare eller substrat till läkemedelsmetaboliserande enzymer och ingen dosjustering behövs (se avsnitt Farmakokinetiska egenskaper).
Samtidig immunsuppressiv behandling eller ljusbehandling
Säkerhet och effekt för risankizumab i kombination med immunsuppressiva läkemedel, inklusive biologiska läkemedel eller ljusbehandling, har inte utvärderats.
Fertilitet, graviditet och amning
Fertila kvinnor
Fertila kvinnor ska använda en effektiv preventivmetod under pågående behandling och i minst 21 veckor efter avslutad behandling.
Graviditet
Det finns ingen eller begränsad mängd data (färre än 300 graviditeter) från användning av risankizumab hos gravida kvinnor. Djurstudier tyder inte på direkta eller indirekta reproduktionstoxikologiska effekter. Som en försiktighetsåtgärd bör man undvika användning av risankizumab under graviditet.
Amning
Det är okänt om risankizumab utsöndras i bröstmjölk. Det är dock känt att humant IgG utsöndras i bröstmjölk under de första dagarna efter födseln, vilket sjunker till låga koncentrationer kort därefter. En risk för det ammande spädbarnet kan därför inte uteslutas under denna korta period. Ett beslut måste tas huruvida man ska avbryta/avstå från behandling med risankizumab efter att man tagit hänsyn till fördelen med amning för barnet och fördelen med behandlingen med risankizumab för kvinnan.
Fertilitet
Effekten av risankizumab på fertiliteten hos människa har inte utvärderats. Djurstudier visar inga direkta eller indirekta skadliga effekter avseende fertilitet.
Effekter på förmågan att framföra fordon och använda maskiner
Risankizumab har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligast rapporterade biverkningarna var övre luftvägsinfektioner (13,0 % vid psoriasis).
Biverkningstabell
Biverkningar för risankizumab från kliniska studier (tabell 2) är listade enligt MedDRA-organsystemklass och är baserade på följande konvention: mycket vanliga (≥1/10), vanliga (≥1/100, <1/10), mindre vanliga (≥1/1 000, <1/100), sällsynta (≥1/10 000, <1/1 000), mycket sällsynta (<1/10 000) och ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp anges biverkningarna i fallande allvarlighetsgrad.
Tabell 2: Biverkningstabell
Organsystemklass | Frekvens | Biverkningar |
Infektioner och infestationer | Mycket vanliga | Övre luftvägsinfektionera |
Vanliga | Tinea-infektionerb | |
Mindre vanliga | Follikulit | |
Immunsystemet | Sällsynta | Anafylaktiska reaktioner |
Centrala och perifera nervsystemet | Vanliga | Huvudvärkc |
Hud och subkutan vävnad | Vanliga | Klåda Hudutslag Eksem |
Mindre vanliga | Urtikaria | |
Allmänna symtom och/eller symtom vid administreringsstället | Vanliga | Trötthetd Reaktioner vid injektionsställete |
a Inklusive: luftvägsinfektion (viral, bakteriell eller ospecificerad), sinuit (inklusive akut), rinit, nasofaryngit, faryngit (inklusive viral), tonsillit, laryngit, trakeit. b Inklusive: tinea pedis, tinea cruris, tinea corporis, tinea versicolor, tinea manuum, onykomykos, tinea infektion c Inklusive: huvudvärk, spänningshuvudvärk, bihålerelaterad huvudvärk d Inklusive: trötthet, asteni e Inklusive: blåmärken vid injektionsstället, erytem, hematom, blödning, irritation, smärta, klåda, reaktion, svullnad, induration, utslag | ||
Beskrivning av utvalda biverkningar
Infektioner
Infektionsfrekvensen var 75,5 händelser per 100 patientår i det kliniska studieprogrammet för psoriasis och 43,0 händelser per 100 patientår i det kliniska studieprogrammet för psoriasisartrit, inklusive långtidsexponering för risankizumab. Majoriteten av fallen var icke-allvarliga och milda till måttliga i svårighetsgrad och ledde inte till utsättning av risankizumab. Frekvensen av allvarliga infektioner var 1,7 händelser per 100 patientår i psoriasisstudierna och 2,6 händelser per 100 patientår i psoriasisartritstudierna (se avsnitt Varningar och försiktighet).
Immunogenicitet
Hos vuxna patienter behandlade med rekommenderad klinisk dos av risankizumab i upp till 52 veckor i de kliniska psoriasisstudierna upptäcktes behandlingsutlösta antikroppar mot läkemedlet och neutraliserande antikroppar hos 24 % (263/1 079) respektive 14 % (150/1 079) av de utvärderade patienterna. För patienter som exponerades för långtidsbehandling med risankizumab i förlängningsstudien var den observerade immunogenicitetsprofilen upp till 204 veckors behandling, jämförbar med de första 52 veckorna av behandlingen.
För de flesta vuxna patienterna med psoriasis förknippades förekomsten av antikroppar mot risankizumab inklusive neutraliserande antikroppar inte med förändringar i klinisk respons eller säkerhet. Bland de få patienter (ungefär 1 %; 7/1 000 vid vecka 16 och 6/598 vid vecka 52) med hög antikroppstiter (> 128), verkade det kliniska svaret vara reducerat. Förekomsten av reaktioner vid injektionsstället är numeriskt högre i grupperna som är positiva för antikroppar mot läkemedlet jämfört med grupperna som är negativa för antikroppar mot läkemedlet på kort sikt (16 veckor: 2,7 % mot 1,3 %) och vid behandling på lång sikt (52 veckor: 5,0 % mot 3,3 %). Reaktionerna vid injektionsstället var alla milda till måttliga i svårighetsgrad, ingen var allvarlig och ingen ledde till utsättning av risankizumab.
Hos pediatriska patienter i åldern 6 till under 18 år behandlade med rekommenderad klinisk dos av risankizumab i upp till 52 veckor i de kliniska studierna av psoriasis, upptäcktes behandlingsutlösta antikroppar mot läkemedlet och neutraliserande antikroppar hos 14,8 % (13/88) respektive 2,3 % (2/88) av de utvärderade patienterna. Antikroppar mot risankizumab påverkade inte klinisk respons eller säkerhet. Antalet patienter som var positiva för antikroppar mot risankizumab är dock för litet för att dra definitiva slutsatser om påverkan på risankizumabs effekt och säkerhet.
Hos vuxna patienter behandlade med rekommenderad klinisk dos av risankizumab i upp till 28 veckor i de kliniska studierna av psoriasisartrit, upptäcktes behandlingsutlösta antikroppar mot läkemedlet och neutraliserande antikroppar hos 12,1 % (79/652) respektive 0 % (0/652) av de utvärderade patienterna. Antikroppar mot risankizumab påverkade inte klinisk respons eller säkerhet vid psoriasisartrit.
Psoriasisartrit
Den övergripande säkerhetsprofilen hos patienter med psoriasisartrit som behandlades med risankizumab överensstämde med säkerhetsprofilen hos patienter med plackpsoriasis.
Pediatrisk population
I en fyrdelad prövning med pediatriska patienter med måttlig till svår plackpsoriasis utvärderades säkerheten i upp till 52 veckor hos 137 pediatriska patienter i åldern 6 till under 18 år. Den övergripande säkerhetsprofilen hos patienter med plackpsoriasis som behandlades med risankizumab överensstämde med säkerhetsprofilen hos vuxna patienter med plackpsoriasis.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
I händelse av överdosering är rekommendationen att patienten övervakas med avseende på tecken eller symtom på biverkningar och att lämplig symtomatisk behandling sätts in omedelbart.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: immunsuppressiva medel, interleukinhämmare, ATC-kod: L04AC18
Verkningsmekanism
Risankizumab är en humaniserad monoklonal antikropp av typen immunglobulin G1 (IgG1) som med hög affinitet binder selektivt till p19-subenheten på humant interleukin-23 (IL-23) cytokin, utan att binda till IL-12 och hämmar dess interaktion med IL-23 receptorkomplexet. IL-23 är ett cytokin som är inblandat i inflammatoriska reaktioner och immunsvar. Genom att blockera IL-23 från att binda till dess receptor hämmar risankizumab IL-23-beroende cellsignalering och frisättning av pro-inflammatoriska cytokiner.
Farmakodynamisk effekt
I en studie av psoriasispatienter nedreglerades genuttrycket i huden för de gener som är förknippade med IL-23/IL-17-axeln efter enkeldoser med risankizumab. Minskad epidermal tjocklek, infiltration av inflammatoriska celler och uttryck av sjukdomsmarkörer för psoriasis observerades också i psoriasislesioner.
Klinisk effekt och säkerhet
Plackpsoriasis hos vuxna
Effekt och säkerhet för risankizumab utvärderades hos 2 109 patienter med måttlig till svår plackpsoriasis i fyra randomiserade, dubbelblinda multicenterstudier (ULTIMMA-1, ULTIMMA-2, IMMHANCE och IMMVENT). De patienter som deltog var 18 år och äldre med plackpsoriasis som täckte ≥ 10 % av kroppsytan (Body Surface Area, BSA), ett sPGA-värde (static Physician Global Assessment) på ≥ 3 i den övergripande bedömningen (tjocklek/induration av plack, erytem och fjällning) av psoriasis på en skala med svårighetsgrad 0 till 4, ett PASI-värde (Psoriasis Area and Severity Index) på ≥ 12 och var kandidater för systemisk behandling eller ljusbehandling.
Totalt sett hade patienterna ett medianvärde för PASI vid baslinjen på 17,8, ett medianvärde för BSA på 20,0 % och ett medianvärde för DLQI vid baslinjen på 13,0. 19,3 % av patienterna hade ett sPGA-värde av ”svår psoriasis” vid baslinjen och 80,7 % av patienterna hade ”måttlig psoriasis” vid baslinjen. Totalt hade 9,8 % av studiepatienterna anamnes på en diagnosticerad psoriasisartrit.
I alla studier var 30,9 % av patienterna naiva för någon form av systemisk behandling (inklusive icke-biologisk och biologisk), 38,1 % hade tidigare fått fototerapi eller fotokemoterapi, 48,3 % hade tidigare fått icke-biologisk systemisk behandling, 42,1 % hade tidigare fått biologisk behandling och 23,7 % hade fått minst ett anti-TNF-alfa-läkemedel för behandling av psoriasis. Patienter som slutförde dessa studier och andra fas 2/3 studier hade möjlighet att ingå i en öppen förlängningsstudie, LIMMITLESS.
ULTIMMA-1 och ULTIMMA-2
I ULTIMMA-1 och ULTIMMA-2 deltog 997 patienter (598 randomiserade till risankizumab 150 mg, 199 till ustekinumab 45 mg eller 90 mg [utifrån vikt vid baslinjen] och 200 till placebo). Patienterna fick behandling vid vecka 0, vecka 4 och därefter var 12:e vecka. De två co-primära effektmåtten i ULTIMMA-1 och ULTIMMA-2 var andelen patienter som uppnådde 1) PASI 90-respons och 2) sPGA-värde utläkt eller nästan utläkt (sPGA 0 eller 1) vid vecka 16 jämfört med placebo. Resultaten för de co-primära effektmåtten presenteras i tabell 3 och figur 1.
Tabell 3: Effekt- och livskvalitetsresultat hos vuxna med plackpsoriasis i ULTIMMA‑1 och ULTIMMA‑2
ULTIMMA‑1 | ULTIMMA‑2 | |||||
Risankizumab(N = 304)n (%) | Ustekinumab (N = 100) n (%) | Placebo(N = 102)n (%) | Risankizumab (N = 294)n (%) | Ustekinumab (N = 99) n (%) | Placebo(N = 98)n (%) | |
sPGA utläkt eller nästan utläkt (0 eller 1) | ||||||
Vecka 16a | 267 (87,8) | 63 (63,0) | 8 (7,8) | 246 (83,7) | 61 (61,6) | 5 (5,1) |
Vecka 52 | 262 (86,2) | 54 (54,0) | -- | 245 (83,3) | 54 (54,5) | -- |
sPGA utläkt (0) | ||||||
Vecka 16 | 112 (36,8) | 14 (14,0) | 2 (2,0) | 150 (51,0) | 25 (25,3) | 3 (3,1) |
Vecka 52 | 175 (57,6) | 21 (21,0) | -- | 175 (59,5) | 30 (30,3) | -- |
PASI 75 | ||||||
Vecka 12 | 264 (86,8) | 70 (70,0) | 10 (9,8) | 261 (88,8) | 69 (69,7) | 8 (8,2) |
Vecka 52 | 279 (91,8) | 70 (70,0) | -- | 269 (91,5) | 76 (76,8) | -- |
PASI 90 | ||||||
Vecka 16a | 229 (75,3) | 42 (42,0) | 5 (4,9) | 220 (74,8) | 47 (47,5) | 2 (2,0) |
Vecka 52 | 249 (81,9) | 44 (44,0) | -- | 237 (80,6) | 50 (50,5) | -- |
PASI 100 | ||||||
Vecka 16 | 109 (35,9) | 12 (12,0) | 0 (0,0) | 149 (50,7) | 24 (24,2) | 2 (2,0) |
Vecka 52 | 171 (56,3) | 21 (21,0) | -- | 175 (59,5) | 30 (30,3) | -- |
DLQI 0 eller 1b | ||||||
Vecka 16 | 200 (65,8) | 43 (43,0) | 8 (7,8) | 196 (66,7) | 46 (46,5) | 4 (4,1) |
Vecka 52 | 229 (75,3) | 47 (47,0) | -- | 208 (70,7) | 44 (44,4) | -- |
PSS 0 (symtomfri)c | ||||||
Vecka 16 | 89 (29,3) | 15 (15,0) | 2 (2,0) | 92 (31,3) | 15 (15,2) | 0 (0,0) |
Vecka 52 | 173 (56,9) | 30 (30,0) | -- | 160 (54,4) | 30 (30,3) | -- |
Alla jämförelser av risankizumab jämfört med ustekinumab och placebo uppnådde p<0,001 förutom PASI 75 vid vecka 52 i ULTIMMA-2 där p = 0,001 a Co-primära effektmått jämfört med placebo b Ingen inverkan på hälsorelaterad livskvalitet c Psoriasis Symptom Scale (PSS) värde på 0 betyder inga symtom av smärta, klåda, rodnad och sveda under de senaste 24 timmarna | ||||||
Figur 1: Genomsnittlig procentuell förändring från baslinjen för PASI över tid i ULTIMMA-1 och ULTIMMA-2
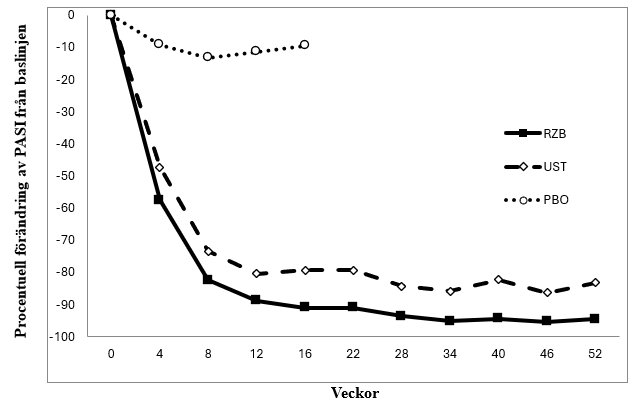
RZB = risankizumab
UST = ustekinumab
PBO = placebo
p<0,001 vid varje tidpunkt
Undersökning av olika subgrupper som ålder, kön, etnicitet, kroppsvikt ≤ 130 kg, PASI-värde vid baslinjen, samtidig psoriasisartrit, tidigare icke-biologisk systemisk behandling, tidigare biologisk behandling och tidigare misslyckad biologisk behandling visade inga skillnader i respons på risankizumab.
Förbättringar observerades av psoriasis lokaliserade till hårbotten, naglar, handflator och fotsulor vid vecka 16 och vecka 52 hos patienter behandlade med risankizumab.
Tabell 4: Genomsnittlig förändring av NAPSI, PPASI och PSSI från baslinjen
ULTIMMA-1 | ULTIMMA-2 | IMMHANCE | ||||
Risankizumab | Placebo | Risankizumab | Placebo | Risankizumab | Placebo | |
NAPSI: Förändring vid vecka 16 (SE) | N = 178; -9,0 (1,17) | N = 56; 2,1 (1,86) *** | N = 177; -7,5 (1,03) | N = 49; 3,0 (1,76) *** | N = 235; -7,5 (0,89) | N = 58; 2,5 (1,70) *** |
PPASI: Förändring vid vecka 16 (SE) | N = 95; -5,93 (0,324) | N = 34; -3,17 (0,445) *** | N = 86; -7,24 (0,558) | N = 23; -3,74 (1,025) ** | N = 113; -7,39 (0,654) | N = 26; -0,27 (1,339) *** |
PSSI: Förändring vid vecka 16 (SE) | N = 267; -17,6 (0.47) | N = 92; -2,9 (0,69) *** | N = 252; -18,4 (0,52) | N = 83; -4,6 (0,82) *** | N = 357; -20,1 (0,40) | N = 88; -5,5 (0,77) *** |
NAPSI: Förändring vid vecka 52 (SE) | N = 178; -15,7 (0,94) | - | N = 183; -16,7 (0,85) | - | - | - |
PPASI: Förändring vid vecka 52 (SE) | N = 95; -6,16 (0,296) | - | N = 89; -8,35 (0,274) | - | - | - |
PSSI: Förändring vid vecka 52 (SE) | N = 269; -179 (0,34) | - | N = 259; -18,8 (0,24) | - | - | - |
Nail Psoriasis Severity Index (NAPSI), Palmoplantar Psoriasis Severity Index (PPASI), Psoriasis Scalp Severity Index (PSSI) och Standardfel (SE) ** P < 0,01 jämfört med risankizumab *** P < 0,001 jämfört med risankizumab | ||||||
Ångest och depression, mätt med självskattningsskalan HADS (Hospital Anxiety and Depression Scale), förbättrades i risankizumabgruppen vid vecka 16 jämfört med placebogruppen.
Upprätthållande av respons
I en integrerad analys av patienterna som hade PASI 100 vid vecka 16 med risankizumab i ULTIMMA-1 och ULTIMMA-2, bibehöll 79,8 % (206/258) samma respons vid vecka 52. För PASI 90-responders vid vecka 16 bibehöll 88,4 % (398/450) av patienterna samma respons vid vecka 52.
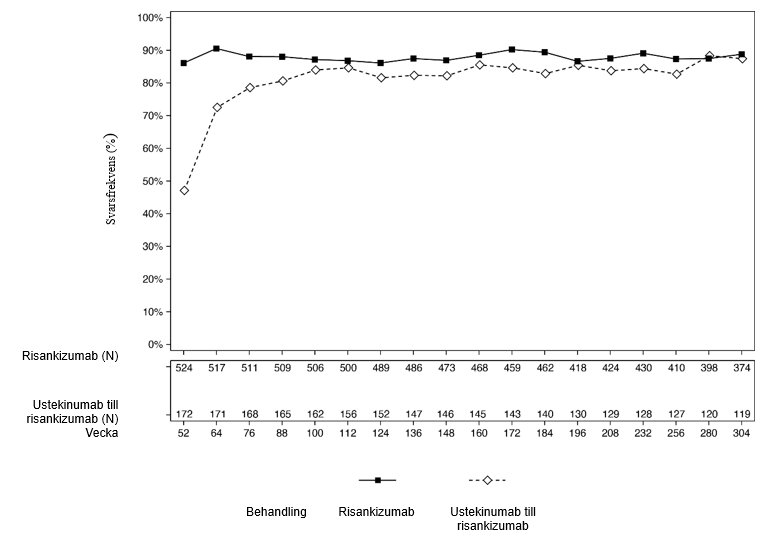
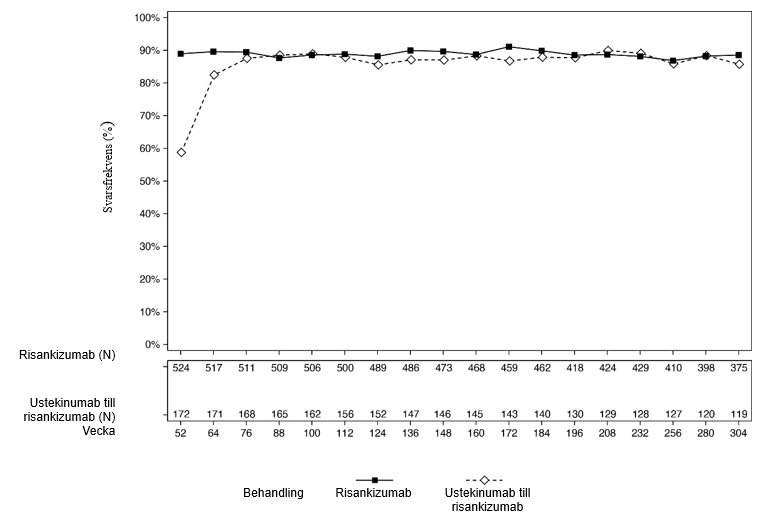
Av de patienter som fick risankizumab i ULTIMMA-1 och ULTIMMA-2 fortsatte 525 att få risankizumab var 12:e vecka i LIMMITLESS. Av dessa slutförde 376 patienter (71,6 %) ytterligare 252 veckors öppen behandling. De förbättringar som uppnåtts med risankizumab vid vecka 52, andelen PASI 90 responders och sPGA-värde utläkt eller nästan utläkt, bibehölls till och med vecka 304 för de patienter som kvarstod i studien.
Av de patienter som fick ustekinumab i ULTIMMA-1 och ULTIMMA-2 fick 172 risankizumab var 12:e vecka i LIMMITLESS. Av dessa slutförde 116 (67,4 %) studien, inklusive 252 veckors öppen behandling med risankizumab och uppföljning i slutet av studien. Av de patienter som kvarstod i studien ökade andelen som uppnådde PASI 90 och sPGA-värde utläkt eller nästan utläkt från vecka 52 till och med vecka 76 och bibehölls därefter till och med vecka 304.
Figur 2 och 3 visar andelen som uppnådde PASI 90 respektive sPGA-värde utläkt eller nästan utläkt hos patienter som slutförde 252 veckors öppen behandling i LIMMITLESS.
Figur 2: Procentandel patienter som uppnådde PASI 90-respons (OC) i LIMMITLESS
Figur 3: Procentandel patienter som uppnådde sPGA-värde utläkt eller nästan utläkt per besök (OC) i LIMMITLESS
Förbättringarna på Dermatology Life Quality Index (DLQI 0 eller 1) bibehölls hos patienter som fick kontinuerlig behandling med risankizumab till och med vecka 304 i den öppna förlängningsstudien LIMMITLESS.
Säkerhetsprofilen för risankizumab med mer än 5 års exponering överensstämde med profilen som observerades i upp till 16 veckor.
IMMHANCE
I IMMHANCE deltog 507 patienter (407 randomiserade till risankizumab 150 mg och 100 till placebo). Patienterna fick behandling vid vecka 0, vecka 4 och därefter var 12:e vecka. Patienter som ursprungligen fick risankizumab och som hade sPGA utläkt eller nästan utläkt vid vecka 28 randomiserades på nytt till att fortsätta med risankizumab var 12:e vecka till och med vecka 88 (med uppföljning 16 veckor efter den sista dosen risankizumab) eller få behandlingen utsatt.
Vid vecka 16 var risankizumab överlägsen placebo gällande de co-primära effektmåtten sPGA utläkt eller nästan utläkt (83,5 % risankizumab jämfört med 7,0 % placebo) och PASI 90 (73,2 % risankizumab jämfört med 2,0 % placebo).
Av de 31 patienter i IMMHANCE-studien med latent tuberkulos (TB) som inte fick profylax under studien var det ingen som utvecklade aktiv TB under den genomsnittliga uppföljningen på 55 veckor av risankizumab.
Bland patienter med sPGA utläkt eller nästan utläkt vid vecka 28 i IMMHANCE hade 81,1 % (90/111) av de som återrandomiserats till fortsatt behandling med risankizumab kvar samma resultat vid vecka 104 jämfört med 7,1 % (16/225) av de som återrandomiserats till att risankizumab skulle sättas ut. Av dessa patienter, uppnådde 63,1 % (70/111) av de som återrandomiserats till fortsatt behandling med risankizumab ett sPGA av utläkt respons vid vecka 104 jämfört med 2,2 % (5/225) av de som återrandomiserats till att risankizumab skulle sättas ut.
Bland patienter som nådde sPGA utläkt eller nästan utläkt vid vecka 28 och fick återfall till ett sPGA-värde av måttlig eller svår psoriasis till följd av utsättande av risankizumab, återfick 83,7 % (128/153) ett sPGA-värde av utläkt eller nästan utläkt efter 16 veckors återinsättning. Uteblivna sPGA-värden av utläkt och nästan utläkt observerades så tidigt som 12 veckor efter en missad dos. Av de patienter som återrandomiserats till att behandlingen skulle sättas ut fick 80,9 % (182/225) återfall och mediantiden till återfall var 295 dagar. Inga karakteristika identifierades för att förutsäga tiden till utebliven respons eller sannolikheten för att återfå respons på den individuella patientnivån.
IMMVENT
I IMMVENT deltog 605 patienter (301 randomiserade till risankizumab och 304 till adalimumab). Patienter randomiserade till risankizumab fick 150 mg behandling vid vecka 0, vecka 4 och därefter var 12:e vecka. Patienter randomiserade till adalimumab fick 80 mg vecka 0, 40 mg vecka 1 och 40 mg varannan vecka till och med vecka 15. Från vecka 16 fick adalimumab-patienter fortsätta eller byta behandling beroende på uppnådd effekt:
-
<PASI 50 fick byta till risankizumab
-
PASI 50 till <PASI 90 blev återrandomiserade till att antingen fortsätta med adalimumab eller byta till risankizumab
-
PASI 90 fortsatte med adalimumab
Resultat presenteras i tabell 5.
Tabell 5: Effekt- och livskvalitetsresultat vid vecka 16 hos vuxna med plackpsoriasis i IMMVENT
Risankizumab(N = 301)n (%) | Adalimumab (N = 304) n (%) | |
sPGA utläkt eller nästan utläkta | 252 (83,7) | 183 (60,2) |
PASI 75 | 273 (90,7) | 218 (71,7) |
PASI 90a | 218 (72,4) | 144 (47,4) |
PASI 100 | 120 (39,9) | 70 (23,0) |
DLQI 0 eller 1b | 198 (65,8) | 148 (48,7) |
Alla jämförelser uppnådde p<0,001 a Co-primära effektmått b Ingen inverkan på hälsorelaterad livskvalitet | ||
För patienter med adalimumab som hade PASI 50 till
Resultat 28 veckor efter återrandomisering presenteras i tabell 6 och figur 4.
Tabell 6: Effektresultat 28 veckor efter återrandomiseringen i IMMVENT
Bytte till risankizumab(N = 53)n (%) | Fortsatte med adalimumab (N = 56) n (%) | |
PASI 90 | 35 (66,0) | 12 (21,4) |
PASI 100 | 21 (39,6) | 4 (7,1) |
Alla jämförelser uppnådde p<0,001 | ||
Figur 4: PASI 90 över tid efter återrandomisering i IMMVENT
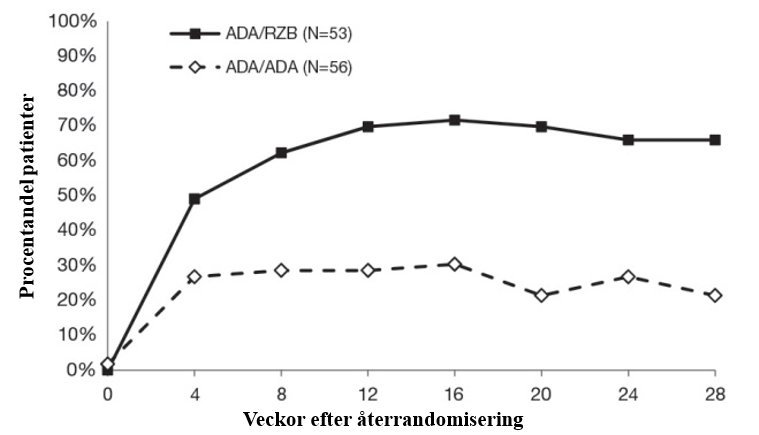
ADA/ADA: Patienter randomiserade till adalimumab och återrandomiserade till att fortsätta med adalimumab
ADA/RZB: Patienter randomiserade till adalimumab och återrandomiserade till att byta till risankizumab
p < 0,05 vid vecka 4 och p < 0,001 vid varje tidpunkt med start vid vecka 8
Hos de 270 patienter som bytte från adalimumab till risankizumab utan en washout-period var säkerhetsprofilen för risankizumab liknande den hos patienter som började med risankizumab efter en washout-period från eventuella tidigare systemiska behandlingar.
Plackpsoriasis lokaliserad i hårbotten eller genitala delar
Effekt och säkerhet för risankizumab bedömdes i en randomiserad, dubbelblind, placebokontrollerad multicenterstudie (UNLIMMITED). Patienterna som deltog var 18 år och äldre med måttlig till svår psoriasis i hårbotten (UNLIMMITED-S), definierad som ett PSSI-värde (Psoriasis Scalp Severity Index) på ≥ 12, ett IGA-värde (Investigator Global Assessment) på ≥ 3 och som täckte ≥ 30 % av hårbotten, eller måttlig till svår genital psoriasis (UNLIMMITED-G), definierad som sPGA-G (static Physician’s Global Assessment of Genitalia) ≥ 3 vid baslinjen. Alla patienter hade BSA ≥ 1 % och PGA ≥ 3 vid baslinjen.
I UNLIMMITED randomiserades patienterna till att få antingen risankizumab 150 mg eller placebo subkutant vecka 0 och 4. Från och med vecka 16 fick alla patienter risankizumab 150 mg var 12:e vecka fram till den sista dosen vecka 40.
Psoriasis i hårbotten (UNLIMMITED-S)
I UNLIMMITED-S deltog 105 patienter. BSA vid baslinjen var ≥ 10 % för 61,9 % av patienterna och < 10 % för 38,1% av patienterna. Genomsnittlig BSA vid baslinjen var 16,8 %. Vid baslinjen hade 76,2 % av patienterna sPGA = 3 och 23,8 % hade sPGA = 4.
Vid baslinjen var 54,3 % av patienterna naiva för någon form av systemisk behandling (inklusive icke-biologisk och biologisk), 0 % hade tidigare fått ljusbehandling, 15,2 % hade tidigare fått icke-biologisk systemisk behandling och 37,1 % hade tidigare fått biologisk behandling.
Resultaten för de primära och de viktigaste sekundära effektmåtten presenteras i tabell 7.
Tabell 7. Effektresultat hos vuxna med psoriasis i hårbotten i UNLIMMITED-S vid vecka 16
Effektmått | Risankizumab(N = 51)n (%) | Placebo(N = 54)n (%) | Behandlingsskillnad (95 % KI) |
IGA för hårbotten utläkt eller nästan utläkt (0 or 1)a | 31 (60,8) | 7 (13,0) | 47,0 [31,2; 62,8] |
PSSI 75b | 38 (74,5) | 12 (22,2) | 52,9 [37,5; 68,3] |
PSSI 90c | 27 (52,9) | 7 (13,0) | 39,8 [24,4; 55,2] |
PSSI 100d | 23 (45,1) | 7 (13,0) | 31,2 [15,4; 46,9] |
Genomsnittlig förändring av PSS från baslinjen | N = 44 -6,0 | N = 49 -1,0 | -5,0 [-6,6; -3,3] |
Alla jämförelser uppnådde p<0,001, justerad behandlingsskillnad (95 % KI) a Primärt effektmått b Uppnådde ≥ 75 % förbättring i PSSI från baslinjen c Uppnådde ≥ 90 % förbättring i PSSI från baslinjen d Uppnådde ≥ 100 % förbättring i PSSI från baslinjen | |||
En större andel patienter som behandlades med risankizumab uppnådde en IGA-poäng för hårbotten på 0 vecka 16 jämfört med placebo (41,2 % respektive 11,1 %).
Respons på NRS-skalan (Scalp Itch Numeric rating scale), definierad som ≥ 4 poängs förbättring (minskning) från baslinjen på NRS-skalan för klåda hos patienter med poäng ≥ 4 vid baslinjen uppnåddes hos ett större antal patienter som behandlats med risankizumab vecka 16 jämfört med de som fått placebo (50,0 % respektive 11,1%).
En större andel patienter som behandlades med risankizumab uppnådde en DLQI-poäng på 0 eller 1 (ingen effekt på hälsorelaterad livskvalitet) vecka 16 jämfört med placebo (47,1 % respektive 11,1 %).
Genital psoriasis (UNLIMMITED-G)
I UNLIMMITED-G deltog 109 patienter. BSA vid baslinjen var ≥ 10 % för 63,3 % av patienterna och < 10 % för 36,7 % av patienterna. Genomsnittlig BSA vid baslinjen var 17,2 %. Vid baslinjen hade 80,7 % av patienterna sPGA = 3 och 19,3 % hade sPGA = 4.
Vid baslinjen var 61,5 % av patienterna naiva för någon form av systemisk behandling (inklusive icke-biologisk och biologisk), 2,8 % hade tidigare fått ljusbehandling, 16,5 % hade tidigare fått icke-biologisk systemisk behandling och 25,7 % hade tidigare fått biologisk behandling.
Resultaten för de viktigaste sekundära effektmåtten presenteras i tabell 8.
Tabell 8. Effektresultat hos vuxna med genital psoriasis i UNLIMMITED-G vid vecka 16
Effektmått | Risankizumab(N = 55)n (%) | Placebo(N = 54)n (%) | Behandlingsskillnad (95 % KI) |
sPGA-G utläkt eller nästan utläkt (0 eller 1)a | 38 (69,1) | 7 (13,0) | 57,0 [42,3; 71,7] |
sPGA-G utläkt (0) | 28 (50,9) | 3 (5,6) | 46,7 [32,6; 60,8] |
DLQI 0 eller 1b | 33 (60,0) | 2 (3,7) | 56,5 [43,0; 70,0] |
GPI-NRS-minskning på ≥ 4 poäng från baslinjenc | N = 41 20 (48,8) | N = 45 3 (6,7) | 43,0 [26,6; 59,3] |
Poäng för punkt 2 i GenPs-SFQ på 0 (aldrig) eller 1 (sällsynt)d,e | N = 31 22 (71,0) | N = 32 7 (21,9) | 46,1 [26,7; 65,6] |
Alla jämförelser uppnådde p<0,001, justerad behandlingsskillnad (95 % KI) a Primärt effektmått b Total DLQI-poäng på 0 eller 1 anger att hudtillståndet inte påverkar patientens hälsorelaterade livskvalitet c Förbättring i svårighetsgraden av genital klåda mätt som en minskning på minst 4 poäng på den 11-gradiga numeriska skalan Genital Psoriasis Itch (GPI) Numeric Rating Scale (NRS) från Genital Psoriasis Symptom Scale (GPSS) hos patienter med en poäng på ≥ 4 vid baslinjen. d I punkt 2 i enkäten Genital Psoriasis Sexual Frequency Questionnaire (GenPs-SFQ) mäts patientupplevd effekt på sexuell hälsa på grund av genital psoriasis avseende frekvens av sexuell aktivitet (samlag eller andra aktiviteter) den senaste veckan (på en skala från 0 till 4, där en högre poäng anger större begränsningar) e Hos patienter med en poäng på ≥ 2 vid baslinjen | |||
Patienter behandlande med risankizumab uppnådde större minskning av psoriasissymtomens allvarlighetsgrad i det genitala området (klåda, smärta, obehag, sveda, brännande känsla, rodnad, fjällning och sprickor) från baslinjen mätt med GPSS vecka 16 jämfört med placebo. Förändringen från baslinjen i GPSS-totalpoäng vecka 16 var -26,5 med risankizumab och -1,0 med placebo.
En större andel patienter som behandlades med risankizumab jämfört med placebo uppnådde minst 2-poängs minskning av PatGA-Genital (Patient’s Global Assessment of Genital Psoriasis), av patienter med en poäng på ≥ 2 vid baslinjen ≥ (71,7 % respektive 22,9 %).
Säkerhetsprofilen för risankizumab i studierna UNLIMMITED-S och UNLIMMITED-G överensstämde med säkerhetsprofilen som observerats i de tidigare studierna hos patienter med plackpsoriasis.
Pediatrisk population
Plackpsoriasis hos barn
Effekt, säkerhet och farmakokinetik för risankizumab utvärderades hos totalt 137 pediatriska patienter i åldern 6 till under 18 år i en fyrdelad studie (OptIMMize-1) som inkluderade 12, 82, 13 respektive 30 patienter i del 1, 2, 3 och 4. Patienter som slutförde studien hade möjlighet att ingå i den öppna förlängningsstudien, OptIMMize-2.
OptIMMize-1
Del 2 var en randomiserad kohort som var prövarblindad för effekt, aktivt behandlingskontrollerad och inkluderade pediatriska patienter i åldern 12 till under 18 år. Del 4 var en öppen kohort utan kontrollgrupp som inkluderade pediatriska patienter i åldern 6 till under 12 år. Deltagande patienter hade måttlig till svår plackpsoriasis, definierad som ≥ 10 % BSA med ett sPGA-värde på ≥ 3 eller ett PASI-värde på ≥ 12.
I del 2 och del 4 fick patienter som vägde ≥ 40 kg risankizumab 150 mg och patienter som vägde < 40 kg fick risankizumab 55 mg vecka 0, vecka 4 och var 12:e vecka därefter. De co-primära effektmåtten var ett sPGA-värde motsvarande utläkt eller nästan utläkt (0 eller 1) och PASI 75-respons vecka 16.)
I del 2 hade patienter vid baslinjen ett medianvärde för PASI på 15,7 och ett medianvärde för BSA på 18 %. Totalt 3,7 % hade fått tidigare biologisk behandling. Patienter randomiserades i förhållandet 2:1 till att få risankizumab (N = 54) eller ustekinumab (N = 28). Vecka 0 och vecka 4 fick patienter som randomiserats till att få ustekinumab 0,75 mg/kg om de vägde < 60 kg, 45 mg om de vägde 60 till < 100 kg respektive 90 mg om de vägde ≥ 100 kg. Vid vecka 16 bytte patienterna som fått ustekinumab till att få risankizumab var 12:e vecka därefter. Behandlingstiden var upp till 68 veckor.
I del 4 hade patienter vid baslinjen ett medianvärde för PASI på 14,7 och ett medianvärde för BSA på 14,5 %. Totalt 3,3 % hade fått tidigare biologisk behandling. Behandlingstiden var 52 veckor.
Effektresultaten baserade på deskriptiv statistik i OptIMMize-1 efter 16 veckors initial behandling presenteras nedan (se tabell 9).
Tabell 9. Effektresultat i OptIMMize-1 vid vecka 16
Del 2 | Del 4 | ||
Risankizumab (N = 54) n (%) | Ustekinumab (N = 28) n (%) | Risankizumab(N = 30) n (%) | |
sPGA-värde utläkt eller nästan utläkt (0 eller 1)a | 43 (79,6) | 21 (75,0) | 27 (90,0) |
PASI 75 a | 46 (85,2) | 24 (85,7) | 26 (86,7) |
PASI 90 | 35 (64,8) | 17 (60,7) | 23 (76,7) |
PASI 100 | 22 (40,7) | 5 (17,9) | 13 (43,3) |
sPGA-värde utläkt (0) | 22 (40,7) | 5 (17,9) | 13 (43,3) |
a Co-primära effektmått | |||
Effekt bibehölls till och med vecka 52 enligt de effektmått som presenteras i tabell 9.
I del 2 randomiserades patienter som ursprungligen fått risankizumab och som uppnått ett sPGA-värde motsvarande utläkt eller nästan utläkt vecka 16 på nytt till att antingen fortsätta med risankizumab var 12:e vecka till vecka 52 (N = 22) eller avbryta behandlingen (N = 21). Vecka 52 hade 95,5 % (21/22) av patienterna som fortsatte med risankizumab bibehållit ett sPGA-värde motsvarande utläkt eller nästan utläkt jämfört med 42,9 % (9/21) av de som avbröt behandlingen.
I del 2 rapporterades förbättringar från baslinjen i hälsorelaterad livskvalitet hos patienter behandlade med risankizumab respektive ustekinumab, mätt med Children’s Dermatology Life Quality Index (CDLQI: –7,4 respektive –6,8) efter 16 veckor med inledande behandling.
I del 2, bland patienter med ett klådvärde vid baslinjen på minst 4 poäng, upplevde 64,9 % (24/37) av de som behandlades med risankizumab och 57,1 % (8/14) av dem som behandlades med ustekinumab en förbättring av klådan efter 16 veckor med inledande behandling, definierad som en minskning med minst 4 poäng från baslinjen på en 11‑gradig numerisk skala för klåda.
OptIMMize-2
I OptIMMize‑2 utvärderades långtidseffekt, säkerhet och tolerabilitet för risankizumab 150 mg eller 55 mg (viktbaserat) givet var 12:e vecka hos 129 pediatriska patienter i åldern 6 till under 18 år med måttlig till svår plackpsoriasis som hade fullföljt OptIMMize-1. I OptIMMize-2 bibehölls svarsfrekvenserna för PASI 75/90/100 och en sPGA-poäng motsvarande utläkt eller nästan utläkt hos de 36 patienter som fortsatte med behandlingen med risankizumab fram till vecka 108.
Farmakokinetiska egenskaper
Absorption
Risankizumab uppvisade linjär farmakokinetik med dosproportionell exponeringsökning för dosintervallen 18 till 300 mg och 0,25 till 1 mg/kg administrerat subkutant, och 200 till 1 200 mg och 0,01 till 5 mg/kg administrerat intravenöst.
Vid subkutan dosering av risankizumab uppnåddes maximala plasmakoncentrationer efter 3–14 dagar med en uppskattad absolut biotillgänglighet på 89 %. Vid dosering med 150 mg vid vecka 0, vecka 4 och därefter var 12:e vecka uppskattas den maximala steady-state-koncentrationen och dalvärdeskoncentrationen i plasma till 12 respektive 2 µg/ml.
Bioekvivalens påvisades mellan en enstaka 150 mg injektion av risankizumab och två 75 mg injektioner av risankizumab i förfylld spruta. Bioekvivalens påvisades även mellan risankizumab 150 mg förfylld spruta och förfylld injektionspenna.
Distribution
Den genomsnittliga (±standardavvikelse) steady-state-distributionsvolymen (Vss) för risankizumab var 11,4 (±2,7) liter i fas 3-studier hos patienter med psoriasis, vilket tyder på att distributionen av risankizumab huvudsakligen är begränsad till de vaskulära och interstitiella utrymmena.
Metabolism
Terapeutiska monoklonala antikroppar av typen IgG bryts vanligen ned till små peptider och aminosyror via katabola vägar på samma sätt som endogent IgG. Risankizumab förväntas inte metaboliseras av cytokrom P450-enzymer.
Eliminering
Genomsnittlig (±standardavvikelse) systemisk clearance (CL) för risankizumab var 0,3 (±0,1) l/dag i fas 3-studier hos patienter med psoriasis. Den genomsnittliga terminala elimineringshalveringstiden för risankizumab varierade mellan 28 och 29 dagar i fas 3-studier hos patienter med psoriasis.
Som en monoklonal antikropp av typen IgG1 förväntas inte risankizumab filtreras via glomerulär filtrering i njurarna eller utsöndras som en intakt molekyl i urinen.
Linjäritet/icke-linjäritet
Risankizumab uppvisade linjär farmakokinetik med ungefärliga dosproportionella ökningar i systemisk exponering (Cmax och AUC) för det utvärderade dosintervallet på 18 till 300 mg och 0,25 till 1 mg/kg efter subkutan administrering till friska individer eller psoriasispatienter.
Interaktioner
En interaktionsstudie genomfördes på patienter med plackpsoriasis för att bedöma effekten av upprepad administrering av risankizumab på farmakokinetiken hos cytokrom P450-känsliga probsubstrat. Exponeringen av koffein (CYP1A2-substrat), warfarin (CYP2C9-substrat), omeprazol (CYP2C19-substrat), metoprolol (CYP2D6-substrat) och midazolam (CYP3A4-substrat) efter behandling med risankizumab var jämförbar med exponering före behandling med risankizumab, vilket indikerar att det inte finns några kliniskt betydelsefulla interaktioner av dessa enzymer.
Populationsfarmakokinetiska analyser visade att risankizumabexponering inte påverkades av annan samtidig behandling som vissa patienter med plackpsoriasis använde under de kliniska studierna.
Särskilda populationer
Pediatrisk population
Exponeringen för risankizumab hos patienter i åldern 6 till under 18 år med plackpsoriasis var liknande den som observerats hos vuxna. Med de rekommenderade doseringsregimer som utvärderades hos dessa patienter var de uppskattade medianvärdena för topp- respektive dalplasmakoncentration vid steady state 15,7 µg/ml och 2,3 µg/ml hos patienter som vägde ≥ 40 kg, och 11,1 µg/ml respektive 1,6 µg/ml hos patienter som vägde < 40 kg.
Patienter med nedsatt njur- eller leverfunktion
Inga specifika studier har genomförts för att fastställa effekten av nedsatt njur- eller leverfunktion på farmakokinetiken för risankizumab. Baserat på populationsfarmakokinetiska analyser hade kreatininnivåer i serum, kreatininclearance eller leverfunktionsmarkörer (ASAT/ALAT/bilirubin) ingen betydelsefull påverkan på risankizumabs clearance hos patienter med plackpsoriasis.
Som en monoklonal antikropp av typen IgG1 elimineras risankizumab huvudsakligen via intracellulär katabolism och förväntas inte genomgå metabolism via cytokrom P450-leverenzymer eller renal eliminering.
Kroppsvikt
Clearance och distributionsvolym för risankizumab ökar när kroppsvikten ökar vilket kan leda till minskad effekt hos patienter med hög kroppsvikt (> 130 kg). Denna observation är dock baserad på ett begränsat antal patienter. Ingen dosjustering med hänsyn till kroppsvikt rekommenderas för närvarande för vuxna patienter.
Kön eller etnicitet
Clearance för risankizumab påverkades inte signifikant av kön eller etnicitet hos vuxna patienter med plackpsoriasis. Inga kliniskt betydelsefulla skillnader i risankizumabexponering observerades hos kinesiska eller japanska patienter jämfört med kaukasiska patienter i en klinisk farmakokinetisk studie hos friska frivilliga.
Prekliniska säkerhetsuppgifter
Gängse studier avseende allmäntoxicitet inklusive utvärderingar av säkerhetsfarmakologi, och en förstärkt pre- och postnatal utvecklingstoxicitetsstudie hos cynomolgusapor vid doser på upp till 50 mg/kg/vecka (vilket ger exponeringar på ca 70 gånger den kliniska exponeringen vid maximal rekommenderad human dos [MRHD]) visade inte på några särskilda risker tänkbara för människa.
Mutagenicitets- och karcinogenicitetsstudier har inte utförts med risankizumab. I en 26 veckor lång kronisk toxikologistudie hos cynomolgusapor vid doser på upp till 50 mg/kg/vecka (ungefär 70 gånger den kliniska exponeringen vid MRHD) observerades inga preneoplastiska eller neoplastiska lesioner och inga ogynnsamma immunotoxiska eller kardiovaskulära effekter noterades.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Natriumacetattrihydrat
Ättiksyra
Trehalosdihydrat
Polysorbat 20
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas ska detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
2 år
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C–8 °C). Får ej frysas.
Förvara den förfyllda sprutan i ytterförpackningen. Ljuskänsligt.
Den förfyllda sprutan kan förvaras utanför kylskåp (högst 25 °C) i upp till 24 timmar i ytterförpackningen för att skydda mot ljus.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
Förfylld glasspruta med en fast nål och nålskydd, monterad med ett automatiskt stickskydd.
Skyrizi 55 mg tillhandahålls i förpackningar som innehåller 1 förfylld spruta.
Läkemedlets utseende:
Lösningen är färglös till gul och klar till svagt opalescent.
Särskilda anvisningar för destruktion och övrig hantering
Före injicering bör patienten ta ut förpackningen från kylskåpet och låta den nå rumstemperatur (15 till 30 minuter), ej i direkt solljus, utan att ta ut den förfyllda sprutan ur förpackningen.
Lösningen ska vara färglös till gul och klar till svagt opalescent.
Allmänna särskilda försiktighetsåtgärder
Före användning rekommenderas en visuell inspektion av varje förfylld spruta. Lösningen kan innehålla ett fåtal genomskinliga till vita produktrelaterade partiklar. Skyrizi ska inte användas om lösningen är grumlig, missfärgad, eller innehåller stora partiklar. Skaka inte den förfyllda sprutan.
Fullständiga instruktioner för administrering finns i bipacksedeln.
Varje förfylld spruta är endast för engångsbruk.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
SKYRIZI injektioneste, liuos, esitäytetty ruisku
55 mg 1 kpl
- Ei korvausta.
Atc-kod
L04AC18
Datum för översyn av produktresumén
19.06.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi