
CARVYKTI infuusioneste, dispersio 3,2 x 10exp6 - 1 x 10exp8 solua
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Riskienhallintamateriaalit apteekeille, solulaboratorioille ja päteville hoitokeskuksille
Riskienhallintamateriaalit CAR-T-hoitokeskuksen hoitohenkilöstölle
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
CARVYKTI (siltakabtageeniautoleuseeli) on muuntogeeninen autologinen solupohjainen valmiste. Se sisältää T-soluja, jotka on transdusoitu ex vivo käyttäen replikaatiokyvytöntä lentivirusvektoria, joka koodaa B-solujen kypsymisantigeenia (BCMA) tunnistavaa kimeeristä antigeenireseptoria (chimeric antigen receptor, CAR). Tämä reseptori koostuu kahdesta yhden domeenin vasta-aineesta, johon on liitetty kostimulatorinen 4‑1BB-domeeni ja signaloiva CD3-zeeta-domeeni.
Jokainen potilaskohtainen CARVYKTI-infuusiopussi sisältää siltakabtageeniautoleuseelia, jossa on tietty eräkohtainen pitoisuus autologisia, geneettisen muuntelun vuoksi kimeeristä anti-BCMA-antigeenireseptoria ilmentäviä T-soluja (CAR-positiivisia, elinkykyisiä T-soluja) (ks. kohta Annostus ja antotapa). Lääkevalmiste on pakattu yhteen infuusiopussiin, jonka sisältämässä infuusionesteessä, soludispersiossa, on 3,2 × 106 – 1 × 108 CAR-positiivista elinkykyistä T‑solua suspendoituna pakastusliuokseen.
Infuusiopussi sisältää 30 ml tai 70 ml infuusionestettä, dispersiota.
Solukoostumus ja solujen lopullinen määrä ovat riippuvaisia potilaan painosta, ja määrä vaihtelee yksilöllisen potilaskohtaisen erän mukaan. Valmisteessa voi olla T-solujen lisäksi luonnollisia tappajasoluja (NK-soluja).
Lääkevalmisteen määrälliset tiedot, mukaan lukien elinkykyisten solujen kokonaispitoisuus, dispersiotilavuus ja CAR+-solujen kokonaismäärä infuusiopussia kohden sekä toimitettu annos mainitaan eräselosteessa (Lot Information Sheet, LIS), joka on CARVYKTI-valmisteen kuljetuksessa käytetyssä pakastussäiliössä.
Apuaine(et), joiden vaikutus tunnetaan
Yksi CARVYKTI-annos sisältää 0,05 ml dimetyylisulfoksidia (DMSO) per ml ja kanamysiinijäämiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, dispersio
Kliiniset tiedot
Käyttöaiheet
CARVYKTI on tarkoitettu uusiutuneen ja hoitoon reagoimattoman multippelin myelooman hoitoon aikuisille, jotka ovat saaneet vähintään yhtä aiempaa hoitoa, mukaan lukien immuunivasteen muuntajaa ja proteasomin estäjää, joilla sairauden on todettu edenneen viimeisimmän hoidon aikana ja joiden sairaus ei ole reagoinut lenalidomidiin.
Ehto
CAR-T-soluhoito annetaan kvalifioidussa hoitokeskuksessa hematologisten syöpien hoitoon perehtyneen ja CAR-T-soluhoitojen antamiseen koulutetun lääkärin ohjauksessa ja valvonnassa.
Annostus ja antotapa
CARVYKTI on annettava pätevässä hoitokeskuksessa.
Hoito pitää aloittaa sellaisen hematologisten syöpien hoidosta kokemusta omaavan terveydenhoidon ammattilaisen ohjauksessa ja valvonnassa, joka on saanut koulutuksen CARVYKTI-valmisteen antamiseen ja sillä hoidettujen potilaiden hoitamiseen.
Pätevässä hoitokeskuksessa on ennen infusointia oltava käytettävissä ainakin yksi annos tosilitsumabia sytokiinioireyhtymän varalta, ja lisäannos on oltava saatavilla aina 8 tunnin kuluessa edellisestä annoksesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Poikkeuksellisessa tilanteessa, jossa tosilitsumabia ei ole Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön vuoksi saatavissa, tosilitsumabin sijaan on ennen infuusiota oltava käytettävissä sopivia vaihtoehtoisia menetelmiä sytokiinioireyhtymän hoitoon.
Ensihoitovälineistön on oltava saatavilla ennen infuusiota ja toipumisjakson aikana.
Annostus
CARVYKTI on tarkoitettu autologiseen käyttöön (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoito koostuu yhdestä infuusioannoksesta. Yksi annos on CAR‑positiivisia, elinkykyisiä T-soluja sisältävä dispersio, joka on yhdessä infuusiopussissa.
Tavoiteannos on 0,75 × 106 CAR-positiivista elinkykyistä T-solua painokiloa (kg) kohden (enintään 1 × 108 CAR-positiivista elinkykyistä T-solua).
Enintään 100 kg:n painoiset potilaat: 0,5–1 × 106 CAR-positiivista elinkykyistä T-solua painokiloa (kg) kohden.
Yli 100 kg:n painoiset potilaat: 0,5–1 × 108 CAR-positiivista elinkykyistä T-solua (annostus ei perustu painoon).
Ks. annokseen liittyvät lisätiedot valmisteen mukana tulevasta eräselosteesta.
Siltahoito
Ennen CARVYKTI-infuusiota annetaan siltahoito lääkkeen määräävän lääkärin harkinnan ja valinnan mukaan, jotta vähennetään kasvaintaakkaa tai vakautetaan sairaus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esihoito (lymfosyyttejä vähentävä kemoterapia)
Lymfosyyttejä vähentävää esihoitoa on siirrettävä myöhemmäksi, jos potilaalla on vakavia haittavaikutuksia edeltävistä siltahoidoista (mukaan lukien kliinisesti merkittävä aktiivinen infektio, sydäntoksisuus ja keuhkotoksisuus) (ks. kohta Farmakodynamiikka).
CARVYKTI-hoidon saatavuus pitää varmistaa ennen lymfosyyttejä vähentävän esihoidon aloittamista.
Lymfosyyttejä vähentävää esihoitoa, joka käsittää 300 mg/m2 syklofosfamidia laskimoon ja 30 mg/m2 fludarabiinia laskimoon, pitää antaa päivittäin 3 päivän ajan. CARVYKTI-infuusio annetaan 5–7 päivää lymfosyyttejä vähentävän esihoidon aloittamisen jälkeen. Jos lymfosyyttejä vähentävästä esihoidosta aiheutuvan toksisuuden lieveneminen 1. asteeseen tai lievemmäksi kestää yli 14 päivää ja siten viivästyttää CARVYKTI-hoitoa, lymfosyyttejä vähentävä esihoito pitää antaa uudelleen vähintään 21 päivää ensimmäisen lymfosyyttejä vähentävän esihoidon ensimmäisen annoksen jälkeen.
Ks. syklofosfamidin ja fludarabiinin annosmuutokset niiden valmisteyhteenvedoista.
Esilääkitys
Kaikille potilaille pitää antaa 30–60 minuuttia ennen CARVYKTI-infuusiota seuraava infuusion esilääkitys:
- kuumetta alentavaa lääkettä (650–1 000 mg parasetamolia suun kautta tai laskimoon)
- antihistamiinia (25–50 mg difenhydramiinia suun kautta tai laskimoon tai vastaava lääkitys).
Systeemisten kortikosteroidien profylaktista käyttöä pitää välttää, sillä se voi muuttaa CARVYKTI-valmisteen vaikutuksia.
Erityisryhmät
Iäkkäät
≥ 65-vuotiaiden potilaiden annosta ei tarvitse muuttaa.
Hepatiitti B ‑virus- (HBV), hepatiitti C ‑virus- (HCV) tai ihmisen immuunikatovirus (HIV) ‑seropositiiviset potilaat
CARVYKTI-valmisteen valmistamisesta potilaille, jotka ovat HIV-positiivisia tai joilla on aktiivinen HBV- tai HCV-infektio, ei ole toistaiseksi kokemusta. HBV-, HCV- ja HIV-infektion ja muiden tartunnanaiheuttajien seulontatutkimus on tehtävä ennen solujen keräämistä valmisteen valmistusta varten.
Pediatriset potilaat
CARVYKTI-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
CARVYKTI on tarkoitettu annettavaksi vain laskimoon.
Leukosyyttejä poistavaa suodatinta EI SAA käyttää.
CARVYKTI-valmisteen valmistelu infuusiota varten
Ennen infuusiota ja toipumisjakson aikana on varmistettava, että tosilitsumabia on saatavilla tai sellaisessa poikkeuksellisessa tilanteessa, jossa tosilitsumabia ei ole Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön vuoksi saatavissa, on käytettävissä sopivia vaihtoehtoisia lääkkeitä. Lisäksi on varmistettava, että ensiapuvälineet ovat saatavilla.
Ennen infuusiota on varmistettava, että potilaan henkilöllisyys vastaa CARVYKTI-pakastussäiliössä, infuusiopussissa ja eräselosteessa ilmoitettuja yksilöllisiä potilastietoja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääkevalmistetta ei saa sulattaa ennen kuin se on tarkoitus käyttää. CARVYKTI-valmisteen sulatuksen ja infuusion ajankohdat on koordinoitava; infuusion ajankohta on vahvistettava etukäteen, ja sulatuksen aloitus on ajoitettava siten, että CARVYKTI on käytettävissä infuusioon, kun potilas on valmis. Lääkevalmiste pitää antaa heti sulatuksen jälkeen, ja infuusion pitää olla annettuna 2,5 tunnin kuluessa sulatuksesta.
Yksityiskohtaiset ohjeet, jotka koskevat valmistelua, antamista sekä vahinkoaltistumisen tapahduttua ja CARVYKTI-valmistetta hävitettäessä tehtäviä toimenpiteitä, on esitetty kohdassa Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle (vaikuttaville aineille) tai kohdassa Apuaineet mainituille apuaineille.
Lymfosyyttejä vähentävän kemoterapian ja tukihoidon vasta-aiheet pitää huomioida.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Jäljitettävyysvaatimuksia, jotka koskevat solupohjaisia pitkälle kehitetyissä hoidoissa käytettäviä lääkkeitä, on noudatettava. Jäljitettävyyden varmistamiseksi valmisteen nimeä, eränumeroa ja hoidetun potilaan nimeä on säilytettävä 30 vuoden ajan valmisteen viimeisen käyttöpäivän jälkeen.
Yleistä
Autologinen käyttö
CARVYKTI on tarkoitettu ainoastaan autologiseen käyttöön, eikä sitä saa missään tapauksessa antaa muille potilaille. CARVYKTI-infuusiota ei saa antaa, jos valmisteen merkinnöissä ja eräselosteessa olevat tiedot eivät vastaa potilaan henkilötietoja.
Potilaan kliininen tutkiminen ennen CARVYKTI-infuusiota
CARVYKTI-infuusion antoa pitää siirtää myöhemmäksi, jos potilaalla on jokin seuraavista sairauksista:
- kliinisesti merkittävä aktiivinen infektio tai tulehdustiloja
- lymfosyyttejä vähentävästä syklofosfamidi- ja fludarabiiniesihoidosta aiheutunut ≥ 3. asteen ei-hematologinen toksisuus, lukuun ottamatta 3. asteen pahoinvointia, oksentelua, ripulia tai ummetusta. CARVYKTI-infuusiota pitää siirtää, kunnes nämä tapahtumat lievenevät ≤ 1. asteeseen
- aktiivinen käänteishyljintä.
Potilaat, joilla on aktiivinen tai on anamneesissa merkittävä keskushermostosairaus tai munuaisten, maksan, keuhkojen tai sydämen riittämätön toiminta, ovat todennäköisesti alttiimpia jäljempänä kuvattujen haittavaikutusten seurauksille ja tarvitsevat siten erityistä huomiota. CARVYKTI-valmisteen käytöstä potilaille, joilla myelooma on levinnyt keskushermostoon tai joilla on ennestään muu kliinisesti oleellinen keskushermostosairaus, ei ole kokemusta.
CARVYKTI-valmisteen tehoa/turvallisuutta muille anti-BCMA-hoidoille aiemmin altistuneille potilaille ei tunneta.
CARVYKTI-valmisteen tehosta/turvallisuudesta uudelleen hoitoa saaneille potilaille on vähän näyttöä saatavissa.
Nopeasti etenevä sairaus
Lääkärin on CARVYKTI-hoitoa potilaalle harkitessaan arvioitava nopeasti etenevän sairauden vaikutusta siihen, voidaanko potilaalle antaa CAR-T-infuusio. Jotkut potilaat eivät välttämättä hyödy CARVYKTI-hoidosta, sillä varhaisen kuoleman riski voi olla lisääntynyt, jos sairaus etenee nopeasti siltahoidon aikana.
Seuranta infuusion jälkeen
Potilaiden pitää olla pätevän hoitokeskuksen päivittäisessä seurannassa 14 päivän ajan CARVYKTI-infuusion jälkeen ja sen jälkeen säännöllisin väliajoin vielä 2 viikkoa CARVYKTI-infuusion jälkeen, jotta sytokiinioireyhtymän oireet ja löydökset, neurologiset tapahtumat ja muu toksisuus voidaan havaita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaita on ohjeistettava olemaan pätevän hoitokeskuksen lähiseudulla vähintään 4 viikon ajan infuusion jälkeen.
Sytokiinioireyhtymä
CARVYKTI-infuusion jälkeen voi ilmetä sytokiinioireyhtymä, mukaan lukien kuolemaan johtavia tai henkeä uhkaavia reaktioita.
Sytokiinioireyhtymä ilmaantui lähes kaikille CARVYKTI-infuusion saaneille potilaille, ja valtaosassa niistä vaikeusaste oli 1. tai 2. aste (ks. kohta Haittavaikutukset). CARVYKTI-infuusiosta (päivä 1) sytokiinioireyhtymän ilmaantumiseen kuluneen ajan mediaani oli 7 päivää (vaihteluväli: 1–23 päivää). Noin 83 %:lla potilaista sytokiinioireyhtymä ilmaantui kolmannen CARVYKTI-infuusion jälkeisen päivän jälkeen.
Sytokiinioireyhtymän kesto oli lähes kaikissa tapauksissa 1–18 päivää (keston mediaani 4 päivää). Kahdeksallakymmenelläyhdeksällä prosentilla potilaista sytokiinioireyhtymä kesti ≤ 7 päivää.
Sytokiinioireyhtymän kliinisiä oireita ja löydöksiä voivat olla mm. kuume (johon voi liittyä vilunpuistatuksia), vilunväristykset, hypotensio, hypoksia ja maksaentsyymiarvojen suureneminen. Sytokiinioireyhtymän mahdollisesti henkeä uhkaavia komplikaatioita voivat olla mm. sydämen toimintahäiriöt, neurologinen toksisuus ja hemofagosyyttinen lymfohistiosytoosi (HLH). Potilailla, joille hemofagosyyttinen lymfohistiosytoosi kehittyy, voi olla lisääntynyt vaikea-asteisten verenvuotojen riski. Potilaita pitää seurata tarkoin näiden tapahtumien oireiden ja löydösten, mukaan lukien kuumeen, havaitsemiseksi. Vaikea-asteisen sytokiinioireyhtymän riskitekijöitä ovat suuri kasvaintaakka ennen infuusiota, aktiivinen infektio ja varhaisessa vaiheessa ilmenevä kuume tai kuumeen jatkuminen, kun oireenmukaista hoitoa on annettu 24 tunnin ajan.
CARVYKTI-infuusiota pitää siirtää, jos potilaan edeltävästä lymfosyyttejä vähentävästä hoidosta tai siltahoidosta aiheutuvat vakavat haittavaikutukset (mukaan lukien sydäntoksisuus tai keuhkotoksisuus) eivät ole hävinneet, jos potilaan tauti etenee nopeasti tai potilaalla on kliinisesti merkittävä aktiivinen infektio (ks. kohta Annostus ja antotapa). Infektioihin pitää antaa asianmukainen hoito tai estohoito, ja aktiivisten infektioiden paraneminen täysin pitää varmistaa ennen CARVYKTI-infuusiota. Infektioita voi ilmetä myös samanaikaisesti sytokiinioireyhtymän kanssa, ja ne voivat lisätä kuolemaan johtavan tapahtuman riskiä.
Vähintään yhden tosilitsumabiannoksen saatavuus sytokiinioireyhtymän varalta pitää varmistaa ennen infuusiota. Pätevässä hoitokeskuksessa on oltava lisäannos tosilitsumabia saatavissa aina 8 tunnin kuluessa kustakin aiemmasta annoksesta. Poikkeuksellisessa tilanteessa, jossa tosilitsumabia ei ole Euroopan lääkeviraston saatavuushäiriöluettelossa mainitun saatavuushäiriön vuoksi saatavissa, hoitokeskuksessa on oltava tosilitsumabin sijaan käytettävissä sopivia vaihtoehtoisia menetelmiä sytokiinioireyhtymän hoitoon. Potilaiden pitää olla pätevän hoitokeskuksen päivittäisessä seurannassa 14 päivän ajan sytokiinioireyhtymän oireiden ja löydösten varalta CARVYKTI-infuusion jälkeen ja sen jälkeen säännöllisin väliajoin vielä 2 viikkoa CARVYKTI-infuusion jälkeen.
Potilaita pitää neuvoa hakeutumaan välittömästi lääkärinhoitoon, jos heille ilmaantuu milloin tahansa sytokiinioireyhtymän oireita ja löydöksiä. Potilas pitää tutkia heti sytokiinioireyhtymän ensimmäisten oireiden ilmetessä sairaalahoidon ja tukihoidon sekä tosilitsumabihoidon tarpeen suhteen tai tosilitsumabi- ja kortikosteroidihoito pitää aloittaa jäljempänä olevan taulukon 1 mukaisesti.
Jos potilaalla on vaikea-asteinen tai hoitoon reagoimaton sytokiinioireyhtymä, hemofagosyyttisen lymfohistiosytoosin tutkimista pitää harkita. Jos potilaalla on ennen infuusiota suuri kasvaintaakka, jos kuumetta ilmaantuu varhaisessa vaiheessa tai jos kuume jatkuu 24 tunnin jälkeen, tosilitsumabin varhaista antoa pitää harkita. Myelooisten kasvutekijöiden, etenkin granulosyytti-makrofagikasvutekijöiden (GM-CSF) käyttöä pitää välttää sytokiinioireyhtymän aikana. Jos potilaalla on suuri kasvaintaakka, harkitse lähtötilanteen kasvaintaakan vähentämistä siltahoidolla ennen CARVYKTI-infuusiota (ks. kohta Annostus ja antotapa).
CARVYKTI-hoitoon liittyvän sytokiinioireyhtymän hoito
Jos sytokiinioireyhtymää epäillään, hoito on taulukon 1 suositusten mukainen. Sytokiinioireyhtymän tukihoitoa (mm. antipyreettejä, laskimonsisäistä nestehoitoa, vasopressoreita, lisähappea) on annettava tarpeen mukaan. Laboratoriotutkimuksia pitää harkita disseminoituneen intravaskulaarisen koagulaation (DIC) varalta sekä hematologisten parametrien ja keuhkojen, sydämen, munuaisten ja maksan toiminnan seuraamiseksi. Muita sytokiineihin kohdentuvia monoklonaalisia vasta-aineita (kuten anti-IL‑1 ja/tai anti-TNFα) tai CAR-T-solujen määrää vähentäviä ja eliminoivia hoitoja voidaan harkita, jos potilaalle ilmaantuu korkean asteen sytokiinioireyhtymä ja hemofagosyyttinen lymfohistiosytoosi, joka jatkuu vaikea-asteisena tai henkeä uhkaavana tosilitsumabin ja kortikosteroidien annon jälkeen.
Jos sytokiinioireyhtymän aikana epäillään samanaikaista neurologista toksisuutta, anna
- kortikosteroideja taulukoissa 1 ja 2 mainittujen sytokiinioireyhtymän ja neurologisen toksisuuden vaikeusasteiden perusteella aggressiivisemman toimenpiteen mukaisesti
- tosilitsumabia taulukossa 1 mainitun sytokiinioireyhtymän vaikeusasteen mukaisesti
- kohtauksia estävää lääkitystä taulukossa 2 mainitun neurologisen toksisuuden mukaisesti.
| Taulukko 1. Sytokiinioireyhtymän vaikeusaste ja hoito-ohjeet | ||
| Sytokiinioireyhtymän vaikeusastea | Tosilitsumabib | Kortikosteroiditf |
| 1. aste Ruumiinlämpö ≥ 38 °Cc | 8 mg/kg tosilitsumabia laskimoon (i.v.) 1 tunnin aikana (enintään 800 mg) voidaan harkita. | Ei oleellinen |
2. aste Oireet edellyttävät kohtalaisia toimenpiteitä ja vastaavat niihin. Ruumiinlämpö ≥ 38 °Cc ja hypotensio, joka ei vaadi vasopressoreita, ja/tai hypoksia, joka vaatii happihoitoa happiviiksilläe tai blow-by-menetelmällä tai 2. asteen elintoksisuus. | Anna 8 mg/kg tosilitsumabia laskimoon 1 tunnin aikana (enintään 800 mg). Toista tosilitsumabin anto tarvittaessa 8 tunnin välein, jos potilas ei vastaa enintään 1 litran laskimonsisäiseen nestehoitoon tai lisähapen lisäämiseen. | Harkitse 1 mg/kg metyyliprednisolonia laskimoon (i.v.) kahdesti päivässä tai deksametasonia (esim. 10 mg laskimoon 6 tunnin välein). |
Ellei paranemista havaita 24 tunnin kuluessa tai jos eteneminen on nopeaa, toista tosilitsumabihoito ja suurenna deksametasoniannosta (20 mg laskimoon 6–12 tunnin välein). Harkitse kahden tosilitsumabiannoksen jälkeen vaihtoehtoisia antisytokiinilääkeaineita.d Älä ylitä kolmea tosilitsumabiannosta 24 tunnissa tai yhteensä neljää annosta. | ||
3. aste Oireet edellyttävät aggressiivisia toimenpiteitä ja vastaavat niihin. Ruumiinlämpö ≥ 38 °Cc ja hypotensio, joka edellyttää yhtä vasopressoria ja mahdollisesti vasopressiinia, ja/tai hypoksia, joka edellyttää happihoitoa suurivirtauksisilla happiviiksilläe, happimaskilla, varaajapussillisella maskilla tai venturimaskilla tai 3. asteen elintoksisuus tai 4. asteen hypertransaminasemia. | 2. asteen mukaisesti | Anna 1 mg/kg metyyliprednisolonia laskimoon kahdesti päivässä tai deksametasonia (esim. 10 mg laskimoon 6 tunnin välein). |
Ellei paranemista havaita 24 tunnin kuluessa tai jos eteneminen on nopeaa, toista tosilitsumabihoito ja suurenna deksametasoniannosta (20 mg laskimoon 6–12 tunnin välein). Ellei paranemista havaita 24 tunnin kuluessa tai jos nopea eteneminen jatkuu, vaihda hoidoksi 2 mg/kg metyyliprednisolonia laskimoon 12 tunnin välein. Harkitse kahden tosilitsumabiannoksen jälkeen vaihtoehtoisia antisytokiinilääkeaineita.d Älä ylitä kolmea tosilitsumabiannosta 24 tunnissa tai yhteensä neljää annosta. | ||
4. aste Henkeä uhkaavia oireita. Respiraattorituen tarve, jatkuva venovenoosinen hemodialyysi (CVVHD). Ruumiinlämpö ≥ 38 °Cc ja hypotensio, joka edellyttää useita vasopressoreita (vasopressiini pois lukien), ja/tai hypoksia, joka edellyttää ylipainehengityshoitoa (esim. CPAP, BiPAP, intubaatio ja hengityskonehoito) tai 4. asteen elintoksisuus (hypertransaminasemia pois lukien). | 2. asteen mukaisesti | Anna 20 mg deksametasonia laskimoon 6 tunnin välein. |
Harkitse kahden tosilitsumabiannoksen jälkeen vaihtoehtoisia antisytokiinilääkeaineitad. Älä ylitä kolmea tosilitsumabiannosta 24 tunnissa tai yhteensä neljää annosta. Ellei paranemista havaita 24 tunnin kuluessa, harkitse metyyliprednisolonia (1–2 g laskimoon, toista tarvittaessa 24 tunnin välein; lopeta hoito vähentämällä lääkitystä asteittain kliinisen tarpeen mukaan) tai muita immunosuppressantteja (esim. muita T‑soluhoitoja). | ||
| a Perustuu ASTCT 2019 ‑luokitusjärjestelmään (Lee et al. 2019), jota on muokattu lisäämällä elintoksisuus. b Ks. tarkemmat tiedot tosilitsumabin valmistetiedoista. Harkitse vaihtoehtoisia menetelmiä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). c Liittyy sytokiinioireyhtymään. Hypotension tai hypoksian yhteydessä ei aina esiinny kuumetta, sillä toimenpiteet, kuten kuumetta alentavat lääkkeet tai antisytokiinihoito (esim. tosilitsumabi tai steroidit), voivat peittää sen. Kuumeettomuus ei vaikuta sytokiinioireyhtymän hoitopäätökseen. Siinä tapauksessa sytokiinioireyhtymän hoitoa ohjaavat hypotensio ja/tai hypoksia sekä vaikeampiasteiset oireet, jotka eivät liity mihinkään muuhun syyhyn. d Hoitoon vastaamattomaan sytokiinioireyhtymään voidaan harkita hoitokeskuksen käytännön mukaan sytokiineihin kohdentuvia monoklonaalisia vasta-aineita (esimerkiksi IL-1:n antagonisteja, kuten anakinraa). e Pienivirtauksiset happiviikset ≤ 6 l/min; suurivirtauksiset happiviikset > 6 l/min. f Jatka kortikosteroidien käyttöä, kunnes tapahtuma on vaikeusasteeltaan 1. aste tai lievempi; lopeta steroidihoito annosta vähitellen pienentämällä, jos kortikosteroidin kokonaisaltistus ylittää 3 päivää. | ||
Neurologinen toksisuus
Neurologista toksisuutta ilmenee yleisesti CARVYKTI-hoidon jälkeen, ja se voi johtaa potilaan kuolemaan tai olla henkeä uhkaavaa (ks. kohta Haittavaikutukset). Neurologista toksisuutta olivat mm. ICANS-oireyhtymä sekä liikkumiseen vaikuttava ja neurokognitiivinen toksisuus, joihin liittyi parkinsonismin oireita ja löydöksiä, Guillain-Barrén oireyhtymä, perifeeriset neuropatiat ja aivohermohalvaukset. Potilaille pitää kertoa tällaisten neurologisten toksisuuksien oireista ja löydöksistä sekä joidenkin tällaisten toksisuuksien luonteeseen liittyvästä viivästyneestä ilmaantumisesta. Potilaita pitää neuvoa hakeutumaan välittömästi lääkärinhoitoon jatkotutkimuksia ja ‑hoitoa varten, jos mitään tällaisen neurologisen toksisuuden oireita ja löydöksiä ilmaantuu milloin tahansa.
ICANS-oireyhtymä (Immune effector cell-associated neurotoxicity syndrome)
CARVYKTI-hoitoa saaville potilaille voi CARVYKTI-hoidon jälkeen ilmaantua kuolemaan johtava tai henkeä uhkaava ICANS-oireyhtymä, joka voi ilmetä ennen sytokiinioireyhtymän ilmaantumista, samaan aikaan sytokiinioireyhtymän kanssa, sytokiinioireyhtymän häviämisen jälkeen tai ilman sytokiinioireyhtymää. Oireita olivat mm. afasia, puheen hitaus, dysgrafia, enkefalopatia, alentunut tajunnantaso ja sekavuustila.
Potilaille, joilla on suuri kasvaintaakka, pitää harkita lähtötilanteen tautikuorman vähentämistä siltahoidolla ennen CARVYKTI-infuusiota, jotta voidaan vähentää neurologisen toksisuuden kehittymisen riskiä (ks. kohta Haittavaikutukset). Potilaiden pitää olla seurannassa neljä viikkoa infuusion jälkeen ICANS-oireyhtymän oireiden ja löydösten havaitsemiseksi. Potilas pitää heti ICANS-oireyhtymän ensimmäisten merkkien ilmetessä tutkia sairaalahoidon ja tukihoidon tarpeen suhteen jäljempänä olevan taulukon 2 mukaisesti. Sytokiinioireyhtymän tai ICANS-oireyhtymän varhainen havaitseminen ja aggressiivinen hoito voivat olla tärkeitä neurologisen toksisuuden ilmaantumisen tai sen pahenemisen estämiseksi. Jatka potilaiden neurologisen toksisuuden oireiden ja löydösten seurantaa sytokiinioireyhtymästä ja/tai ICANS-oireyhtymästä toipumisen jälkeen.
CARVYKTI-hoitoon liittyvän neurologisen toksisuuden hoito
Neurologisen toksisuuden, mukaan lukien ICANS-oireyhtymän, ensimmäisten oireiden ilmetessä pitää harkita neurologisia tutkimuksia. Sulje pois neurologisten oireiden muut syyt. Anna vaikea-asteiseen tai henkeä uhkaavaan neurologiseen toksisuuteen tehohoitoa ja tukihoitoa.
Jos neurologisen toksisuustapahtuman aikana epäillään samanaikaista sytokiinioireyhtymää, anna
- kortikosteroideja taulukoissa 1 ja 2 mainittujen sytokiinioireyhtymän ja neurologisen toksisuuden vaikeusasteiden perusteella aggressiivisemman toimenpiteen mukaisesti
- tosilitsumabia taulukossa 1 mainitun sytokiinioireyhtymän vaikeusasteen mukaisesti
- kohtauksia estävää lääkitystä taulukossa 2 mainitun neurologisen toksisuuden mukaisesti.
| Taulukko 2. ICANS-oireyhtymän hoito-ohjeet | |
| ICANS-oireyhtymän vaikeusastea | Kortikosteroidit |
1. aste ICE-pisteet 7–9b tai alentunut tajunnantaso: herää spontaanisti. | Harkitse 10 mg:aa deksametasoniac laskimoon 6–12 tunnin välein 2–3 päivän ajan. Harkitse kohtausten estoon ei-sedatiivista kohtauslääkitystä (esim. levetirasetaamia). |
2. aste ICE-pisteet 3–6b tai alentunut tajunnantaso: herää puhuteltaessa | Anna 10 mg deksametasoniac laskimoon 6 tunnin välein 2–3 päivän ajan tai pidempään, jos oireet pitkittyvät. Harkitse steroidihoidon lopettamista annosta vähitellen pienentämällä, jos kortikosteroidien kokonaisaltistus on yli 3 päivää. Harkitse kohtausten estoon ei-sedatiivista kohtauslääkitystä (esim. levetirasetaamia). |
3. aste ICE-pisteet 0–2b (Jos ICE-pisteet ovat 0, mutta potilas on herätettävissä [esim. hereillä, mutta globaali afasia] ja on arvioitavissa) tai alentunut tajunnantaso: herää vain kosketuksesta tai kohtauksia joko
tai kohonnut kallonsisäinen paine: neurokuvantamisessa näkyvä fokaalinen/paikallinen turvotusd. | Anna 10–20 mg deksametasoniac laskimoon 6 tunnin välein. Ellei paranemista havaita 48 tunnin kuluttua tai jos neurologinen toksisuus pahenee, suurenna deksametasoniannosc vähintään 20 mg:aan laskimoon 6 tunnin välein; lopeta hoito pienentämällä annosta asteittain 7 päivän kuluessa TAI siirry suuren metyyliprednisoloniannoksen käyttöön (1 g/vrk, toista tarvittaessa 24 tunnin välein; lopeta hoito kliinisen tarpeen mukaan annosta vähitellen pienentämällä). Harkitse kohtausten estoon ei-sedatiivista kohtauslääkitystä (esim. levetirasetaamia). |
4. aste ICE-pisteet 0b (potilas ei ole heräteltävissä eikä ICE-arviointia voida tehdä) tai alentunut tajunnantaso joko
tai kohtauksia joko
tai motorisia löydöksiäe:
tai kohonnut kallonsisäinen paine / aivoturvotus, johon liittyy esim. seuraavia oireita/löydöksiä:
| Anna 10–20 mg deksametasoniac laskimoon 6 tunnin välein. Ellei paranemista havaita 24 tunnin kuluttua tai jos neurologinen toksisuus pahenee, siirry suuren metyyliprednisoloniannoksen käyttöön (1–2 g/vrk, toista tarvittaessa 24 tunnin välein; lopeta hoito kliinisen tarpeen mukaan annosta vähitellen pienentämällä). Harkitse kohtausten estoon ei-sedatiivista kohtauslääkitystä (esim. levetirasetaamia). Jos epäillään kohonnutta kallonsisäistä painetta / aivoturvotusta, harkitse hyperventilaatiohoitoa ja hyperosmolaarista hoitoa. Anna suuria metyyliprednisoloniannoksia (1–2 g/vrk, toista tarvittaessa 24 tunnin välein; lopeta hoito kliinisen tarpeen mukaan annosta vähitellen pienentämällä), ja harkitse neurologin ja/tai neurokirurgin konsultointia. |
| EEG = aivosähkökäyrä; ICE = immuunijärjestelmän efektorisoluihin liittyvä enkefalopatia (Immune Effector Cell-Associated Encephalopathy) Huom.: ICANS-oireyhtymän vaikeusaste ja hoito määräytyvät vaikea-asteisimman tapahtuman perusteella (ICE-pisteet, tajunnantaso, kohtaus, motoriset löydökset, kohonnut kallonsisäinen paine / aivoturvotus), johon ei liity mitään muuta syytä. a Neurologisen toksisuuden vaikeusastetta koskevat ASTCT 2019 ‑kriteerit (Lee et al. 2019). b Jos potilas on herätettävissä ja ICE-arviointi on mahdollista tehdä, arvioi jäljempänä olevan taulukon 3 mukaisesti. c Kaikki deksametasonin antoa koskevat viittaukset tarkoittavat deksametasonia tai vastaavaa. d Kallonsisäistä verenvuotoa, johon liittyy tai ei liity turvotusta, ei katsota neurologisen toksisuuden piirteeksi ja jätetään pois ICANS-oireyhtymän vaikeusasteesta. Sen vaikeusaste voidaan arvioida CTCAE v5.0 ‑kriteerien mukaisesti. e Immuunijärjestelmän efektorisoluhoitoihin liittyvän vapinan ja myoklonuksen vaikeusaste voidaan arvioida CTCAE v5.0 ‑kriteerien mukaisesti, mutta ne eivät vaikuta ICANS-oireyhtymän vaikeusasteen luokitteluun. | |
| Taulukko 3. ICE (Immune Effector Cell-Associated Encephalopathy) ‑arviointi | |
| ICE (Immune Effector Cell-Associated Encephalopathy) ‑työkalua | |
| Pisteet | |
| Orientoituminen: orientoituu vuoteen, kuukauteen, kaupunkiin, sairaalaan | 4 |
| Nimeäminen: nimeää/osoittaa 3 esinettä (esim. osoita kelloa, kynää, nappia) | 3 |
| Käskyjen noudattaminen: (esim. ”Näytä minulle 2 sormea” tai ”Sulje silmäsi ja työnnä kielesi ulos suusta”) | 1 |
| Kirjoittaminen: kykenee kirjoittamaan tavanomaisen virkkeen | 1 |
| Tarkkaavuus: laskee sadasta taaksepäin kymmenen numeron välein | 1 |
a ICE-pisteytystyökalu:
| |
Liikkumiseen vaikuttava ja neurokognitiivinen toksisuus, johon liittyy parkinsonismin oireita ja löydöksiä
CARVYKTI-tutkimuksissa on raportoitu liikkumiseen vaikuttavaa neurologista toksisuutta ja neurokognitiivista toksisuutta, joihin liittyy parkinsonismin oireita ja löydöksiä. Ne olivat oireryväs, joka ilmaantui vaihtelevasti ulottuen useammalle kuin yhdelle oirealueelle, joita olivat mm. liikkuminen (esim. mikrografia, vapina, bradykinesia, jäykkyys, kumara asento, laahaava kävely), kognitio (esim. muistamattomuus, tarkkaavuuden häiriöt, sekavuus) ja persoonallisuuden muutos (esim. vähentyneet kasvojen ilmeet, tunteiden latistuminen, kasvojen ilmeettömyys, apatia), joka alkoi usein hiipien (esim. mikrografia, tunteiden latistuminen) ja joka eteni joillakin potilailla työkyvyttömyydeksi tai kyvyttömyydeksi huolehtia itsestään. Valtaosalla näistä potilaista oli kahden tai useamman tekijän yhdistelmä, kuten suuri kasvaintaakka lähtötilanteessa (luuytimen plasmasolut ≥ 80 % tai seerumin M‑piikki ≥ 5 g/dl tai seerumin vapaa kevytketju ≥ 5 000 mg/l), aiempi vähintään 2. asteen sytokiinioireyhtymä, aiempi ICANS-oireyhtymä ja suuri CAR-T-solujen ekspansio ja pysyvyys. Levodopa-/karbidopahoito (n = 4) ei vähentänyt näiden potilaiden oireita.
Potilaita pitää seurata parkinsonismin oireiden ja löydösten havaitsemiseksi. Ne voivat ilmetä viivästyneesti, ja ne hoidetaan tukihoitotoimenpitein.
Guillain-Barrén oireyhtymä
CARVYKTI-hoidon jälkeen on raportoitu Guillain-Barrén oireyhtymää. Raportoituja oireita ovat olleet mm. Guillain-Barrén oireyhtymän Miller-Fisher-varianttiin sopivat oireet, motorinen heikkous, puhehäiriöt ja polyradikuloneuriitti (ks. kohta Haittavaikutukset).
Potilaita pitää seurata Guillain-Barrén oireyhtymän havaitsemiseksi. Potilailta, joilla on perifeeristä neuropatiaa, on tutkittava Guillain-Barrén oireyhtymä. Hoitoa laskimoon annettavalla immunoglobuliinilla (IVIG) ja hoidon laajentamista plasmafereesiin pitää harkita toksisuuden vaikeusasteesta riippuen.
Perifeerinen neuropatia
CARVYKTI-tutkimuksissa on raportoitu perifeerisen neuropatian, mukaan lukien sensorisen, motorisen tai sensomotorisen neuropatian, ilmaantumista.
Potilaita pitää seurata perifeerisen neuropatian oireiden ja löydösten havaitsemiseksi. Hoitona pitää harkita lyhyttä systeemistä kortikosteroidikuuria oireiden ja löydösten vaikeusasteesta ja etenemisestä riippuen.
Aivohermohalvaukset
CARVYKTI-tutkimuksissa on raportoitu 7., 3., 5. ja 6. aivohermon halvauksia, joista osa on ollut molemminpuolisia, sekä aivohermohalvauksen pahenemista ensin tapahtuneen lievenemisen jälkeen ja aivohermohalvauspotilailla ilmennyttä perifeeristä neuropatiaa.
Potilaita on seurattava aivohermohalvauksen oireiden ja löydösten havaitsemiseksi. Hoitona pitää harkita lyhyttä systeemistä kortikosteroidikuuria oireiden ja löydösten vaikeusasteesta ja etenemisestä riippuen.
Pitkittynyt ja uusiutunut sytopenia
Potilaille voi ilmetä sytopeniaa useiden viikkojen ajan lymfosyyttejä vähentävän kemoterapian ja CARVYKTI-infuusion jälkeen; sytopenia on hoidettava paikallisten suositusten mukaisesti. CARVYKTI-tutkimuksissa lähes kaikille potilaille ilmaantui haittavaikutuksena 3. tai 4. asteen sytopeniaa. Useimmilla potilailla aika (mediaani) infuusiosta 3. tai 4. asteen sytopenian ilmaantumiseen oli alle kaksi viikkoa, ja useimpien potilaiden tila lieveni ≤ 2. asteeseen päivään 30 mennessä (ks. kohta Haittavaikutukset).
Verenkuvaa pitää seurata ennen CARVYKTI-infuusiota ja sen jälkeen. Trombosytopenian ilmetessä on harkittava tukihoitona verensiirtoa. Pitkittyneeseen neutropeniaan liittyy suurentunut infektioiden riski. Myelooiset kasvutekijät, etenkin granulosyytti-makrofagikasvutekijä (GM-CSF), saattavat pahentaa sytokiinioireyhtymän oireita, eikä niiden käyttö ole suositeltavaa kolmen ensimmäisen viikon aikana CARVYKTI-infuusion jälkeen tai ennen kuin sytokiinioireyhtymä on hävinnyt.
Vakavat infektiot ja kuumeinen neutropenia
Potilailla on ilmennyt CARVYKTI-infuusion jälkeen vakavia infektioita, mukaan lukien henkeä uhkaavia tai kuolemaan johtaneita infektioita (ks. kohta Haittavaikutukset).
Potilaita pitää seurata infektioiden oireiden ja löydösten varalta ennen CARVYKTI-hoitoa ja sen aikana, ja infektiot pitää hoitaa asianmukaisesti. Mikrobilääkkeitä on annettava estolääkityksenä paikallisten suositusten mukaisesti. Infektioiden tiedetään komplisoivan samanaikaisen sytokiinioireyhtymän kulkua ja hoitoa. CARVYKTI-hoitoa ei pidä aloittaa potilaille, joilla on kliinisesti merkittävä aktiivinen infektio, ennen kuin infektio on saatu hallintaan.
Jos potilaalla on kuumeinen neutropenia, infektio on arvioitava ja hoidettava asianmukaisesti laajakirjoisilla antibiooteilla, nestehoidolla ja muilla tukitoimilla, jotka lääkäri katsoo aiheellisiksi.
CARVYKTI-hoitoa saaneilla potilailla voi olla tavanomaista suurempi vaikea-asteisen tai kuolemaan johtavan covid-19-infektion riski. Potilaille pitää kertoa, että sen ehkäisytoimenpiteet ovat tärkeitä.
Virusten uudelleenaktivoituminen
B-solutoimintaa estäviä lääkevalmisteita saaneilla potilailla voi esiintyä hepatiitti B ‑viruksen (HBV) uudelleenaktivoitumista, mikä voi joissakin tapauksissa johtaa fulminanttiin hepatiittiin, maksan vajaatoimintaan ja kuolemaan.
CARVYKTI-valmisteen valmistamisesta potilaille, jotka ovat HIV-positiivisia tai joilla on aktiivinen hepatiitti B (HBV) -virus tai aktiivinen hepatiitti C (HCV) -virus, ei ole toistaiseksi kokemusta. HBV-, HCV- ja HIV-infektion ja muiden tartunnanaiheuttajien seulontatutkimus on tehtävä ennen solujen keräämistä valmisteen valmistusta varten (ks. kohta Annostus ja antotapa).
CARVYKTI-hoitoa saaneilla potilailla, jotka ovat saaneet aiemmin myös muita immunosuppressiivisia lääkkeitä, on ilmoitettu John Cunningham -viruksen (JC) uudelleenaktivoitumisesta, joka johtaa etenevään multifokaaliseen leukoenkefalopatiaan (PML:ään). Ilmoituksia on saatu kuolemaan johtaneista tapauksista.
Hypogammaglobulinemia
CARVYKTI-hoitoa saaneille potilaille voi ilmaantua hypogammaglobulinemiaa.
Immunoglobuliinien pitoisuuksia pitää seurata CARVYKTI-hoidon jälkeen; IVIG-hoitoa pitää antaa, jos IgG < 400 mg/dl. Hoida tavanomaisten hoito-ohjeiden mukaisesti, mukaan lukien antibiootti- tai viruslääkeprofylaksi ja potilaan seuranta infektioiden varalta.
Immuunivälitteinen enterokoliitti
Potilaille voi kehittyä immuunivälitteinen enterokoliitti, joka voi ilmaantua useita kuukausia CARVYKTI-infuusion jälkeen. Joissakin tapauksissa kortikosteroideilla annettu hoito ei tehoa, ja muita hoitovaihtoehtoja voi olla tarpeen harkita. Maha-suolikanavan perforaatiotapauksia, myös kuolemaan johtaneita, on havaittu.
Sekundaariset maligniteetit mukaan luettuina myeloidi- ja T-soluperäiset sekundaariset syövät
CARVYKTI-hoitoa saaneille potilaille voi ilmaantua sekundaarisia maligniteetteja. T-soluperäisiä syöpiä on raportoitu sen jälkeen, kun hematologisia syöpiä on hoidettu BCMA- tai CD19-kohdennetulla CAR-T-soluhoidolla, kuten CARVYKTI-valmisteella. T-soluperäisiä syöpiä, myös CAR-positiivisia syöpiä, on raportoitu viikkoja ja jopa useita vuosia BCMA- tai CD19-kohdennetun CAR-T-soluhoidon jälkeen. Kuolemaan johtaneita tapauksia on esiintynyt.
Potilaita pitää seurata loppuelämän ajan sekundaaristen maligniteettien varalta. Sekundaarisen maligniteetin ilmaantuessa on otettava yhteyttä lääkeyhtiöön asiasta ilmoittamiseksi ja potilaalta otettavia näytteitä koskevien ohjeiden saamiseksi T-soluperäisen sekundaarisen maligniteetin tutkimiseksi. Jos potilaalla on HIV-infektio, on otettava yhteyttä lääkeyhtiöön sekundaaristen maligniteettien, mukaan lukien muiden kuin T-soluperäisten sekundaaristen maligniteettien, tutkimiseksi.
Potilaille on CARVYKTI-infuusion jälkeen ilmaantunut myelodysplastista oireyhtymää (MDS) ja akuuttia myelooista leukemiaa (AML), mukaan lukien kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset).
Interferenssi virustestauksessa
Koska CARVYKTI-valmisteen valmistuksessa käytettävässä lentivirusvektorissa on pieni määrä lyhyitä, HI-viruksen kanssa identtisiä geneettisen tiedon jaksoja, jotkin HIV-nukleiinihappotestit (NAT-testit) saattavat antaa väärän positiivisen tuloksen.
Veren, elinten, kudosten ja solujen luovutus
CARVYKTI-hoitoa saaneet potilaat eivät saa luovuttaa verta eivätkä elimiä, kudoksia tai soluja transplantaatiota varten. Tämä tieto kerrotaan myös potilaskortissa, joka on annettava potilaalle.
Yliherkkyys
CARVYKTI-infuusion yhteydessä voi ilmetä allergisia reaktioita. CARVYKTI-valmisteen sisältämä dimetyylisulfoksidi (DMSO) tai kanamysiinijäämät voivat aiheuttaa vakavia yliherkkyysreaktioita, anafylaksia mukaan lukien. Potilaita pitää seurata tarkoin 2 tunnin ajan infuusion jälkeen vaikea-asteisen reaktion oireiden ja löydösten havaitsemiseksi. Hoida potilasta viipymättä asianmukaisesti yliherkkyysreaktion vaikeusasteen mukaan.
Pitkäaikainen seuranta
Potilaiden odotetaan kirjautuvan rekisteriin heidän vointinsa seuraamiseksi, jotta voidaan selvittää paremmin CARVYKTI-valmisteen turvallisuutta ja tehoa pitkällä aikavälillä.
Yhteisvaikutukset
CARVYKTI-valmisteella ei ole tehty farmakokineettisiä eikä farmakodynaamisia yhteisvaikutustutkimuksia.
T-solujen toimintaa tunnetusti estävien lääkeaineiden samanaikaista antoa ei ole varsinaisesti tutkittu. T-solujen toimintaa tunnetusti stimuloivien lääkeaineiden samanaikaista antoa ei ole tutkittu eikä vaikutuksia tunneta.
Jotkut potilaat tarvitsivat kliinisissä CARVYKTI-tutkimuksissa tosilitsumabia, kortikosteroideja ja anakinraa sytokiinioireyhtymän hoitoon. CARVYKTI-valmisteen ekspansio jatkuu ja se pysyy elimistössä edelleen tosilitsumabin annon jälkeen. MMY2001-tutkimuksessa tosilitsumabia saaneilla potilailla (n = 68) CARVYKTI-valmisteen Cmax oli 81 % ja AUC0-28d oli 72 % suurempi verrattuna potilaisiin (n = 29), jotka eivät saaneet tosilitsumabia. Kortikosteroideja saaneilla potilailla (n = 28) Cmax oli 75 % ja AUC0-28d oli 112 % suurempi verrattuna potilaisiin (n = 69), jotka eivät saaneet kortikosteroideja. Anakinraa saaneilla potilailla (n = 20) Cmax oli 41 % ja AUC0-28d oli 72 % suurempi verrattuna potilaisiin (n = 77), jotka eivät saaneet anakinraa. Tosilitsumabiin ja kortikosteroideihin liittyvät MMY3002-tutkimuksen tulokset olivat yhdenmukaisia MMY2001-tutkimuksen kanssa.
Eläviä taudinaiheuttajia sisältävät rokotteet
Eläviä viruksia sisältävien rokotteiden käytön turvallisuutta CARVYKTI-hoidon aikana tai sen jälkeen ei ole tutkittu. Varotoimenpiteenä suositellaan, että eläviä taudinaiheuttajia sisältäviä rokotteita ei anneta vähintään 6 viikkoon ennen lymfosyyttejä vähentävän kemoterapian aloittamista, CARVYKTI-hoidon aikana eikä ennen kuin immuniteetti on elpynyt hoidon jälkeen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy miehillä ja naisilla
Naisilta, jotka voivat tulla raskaaksi, pitää varmistaa raskausstatus ennen CARVYKTI-hoidon aloittamista.
Altistusta koskevia tietoja ei ole riittävästi suositusten antamiseksi siitä, miten kauan ehkäisyä on käytettävä CARVYKTI-hoidon jälkeen.
Kliinisissä tutkimuksissa naispotilaita, jotka voivat tulla raskaaksi, kehotettiin käyttämään erittäin tehokasta ehkäisymenetelmää. Miespotilaita, joiden kumppani voi tulla raskaaksi tai joiden kumppani oli raskaana, ohjeistettiin käyttämään ehkäisynä estemenetelmää, kunnes CARVYKTI-hoidon saamisesta oli kulunut vuosi.
Katso lymfosyyttejä vähentävän kemoterapian valmistetiedoista, tarvitsevatko lymfosyyttejä vähentävää kemoterapiaa saavat potilaat ehkäisyä.
Raskaus
Ei ole olemassa tietoja CARVYKTI-valmisteen käytöstä raskaana oleville naisille. CARVYKTI-valmisteen lisääntymis- ja kehitystoksisuuden tutkimiseksi ei ole tehty eläinkokeita. Ei tiedetä, voiko CARVYKTI-valmiste siirtyä sikiöön ja aiheuttaa sikiötoksisuutta.
CARVYKTI-valmisteen käyttöä ei siksi suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä. Raskaana oleville naisille on kerrottava, että sikiölle voi aiheutua riskejä. CARVYKTI-hoidon jälkeisestä raskaudesta on keskusteltava hoitavan lääkärin kanssa.
Raskaana olevilla naisilla, jotka ovat saaneet CARVYKTI-hoitoa, voi olla hypogammaglobulinemiaa. CARVYKTI-hoitoa saaneille naisille syntyneiden vastasyntyneiden immunoglobuliinipitoisuudet on aiheellista arvioida.
Imetys
Ei tiedetä, erittyykö CARVYKTI-valmiste ihmisen rintamaitoon. Imettäville naisille pitää kertoa imeväiseen mahdollisesti kohdistuvasta riskistä.
Harkittaessa imettämistä CARVYKTI-valmisteen annon jälkeen päätöksestä on keskusteltava hoitavan lääkärin kanssa.
Hedelmällisyys
CARVYKTI-valmisteen vaikutuksista hedelmällisyyteen ei ole tietoja. CARVYKTI-valmisteen vaikutusta miesten ja naisten hedelmällisyyteen ei ole tutkittu eläinkokeiden avulla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
CARVYKTI-valmisteella on huomattava vaikutus ajokykyyn ja koneidenkäyttökykyyn.
CARVYKTI-valmiste saattaa aiheuttaa neurologisia tapahtumia, joten sitä saavilla potilailla on tajunnantason tai koordinaatiokyvyn muutosten tai heikkenemisen riski CARVYKTI-infuusion jälkeisten 8 viikon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaita on kehotettava välttämään moottoriajoneuvon kuljettamista ja vaaraa aiheuttavia toimintoja, kuten raskaan tai mahdollisesti vaaraa aiheuttavan kaluston käyttöä, tämän hoidon alkuvaiheen aikana ja neurologisten oireiden ilmaantuessa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
CARVYKTI-valmisteen turvallisuutta arvioitiin 396:lla multippelia myeloomaa sairastavalla aikuispotilaalla, jotka saivat CARVYKTI-valmistetta kolmessa avoimessa kliinisessä tutkimuksessa: MMY2001-tutkimuksessa (n = 106), johon otettiin potilaita pääasiallisesta vaiheen 1b/2 kohortista (Yhdysvallat; n = 97) sekä lisäkohortista (Japani; n = 9), vaiheen 2 MMY2003-tutkimuksessa (n = 94) ja vaiheen 3 MMY3002-tutkimuksessa (n = 196). Potilaat, jotka ovat MMY2001-, MMY2003- tai MMY3002-tutkimuksessa mukana tutkimuksen päättymiseen saakka, soveltuvat otettavaksi mukaan erilliseen pitkäkestoiseen seurantatutkimukseen (MMY4002).
CARVYKTI-valmisteen yleisimpiä haittavaikutuksia (≥ 20 %) olivat neutropenia (90 %), kuume (85 %), sytokiinioireyhtymä (83 %), trombosytopenia (60 %), anemia (60 %), luuston ja lihasten kipu (40 %), lymfopenia (38 %), uupumus (35 %), leukopenia (34 %), hypotensio (34 %), hypogammaglobulinemia (33 %), ripuli (32 %), ylähengitysteiden infektio (32 %), transaminaasipitoisuuden kohoaminen (26 %), päänsärky (25 %), pahoinvointi (23 %) ja yskä (22 %).
Vakavia haittavaikutuksia ilmaantui 44 %:lle potilaista; vakavia ≥ 2 %:lla potilaista raportoituja haittavaikutuksia olivat sytokiinioireyhtymä (11 %), keuhkokuume (9 %), sepsis (5 %), virusinfektio (5 %), neutropenia (4 %), aivohermohalvaus (4 %), ICANS-oireyhtymä (4 %), enkefalopatia (3 %), ylähengitysteiden infektio (3 %), bakteeri-infektiot (2 %), maha-suolitulehdus (2 %), kuumeinen neutropenia (2 %), trombosytopenia (2 %), hemofagosyyttinen lymfohistiosytoosi (2 %), motorinen toimintahäiriö (2 %), hengenahdistus (2 %), ripuli (2 %) ja munuaisten vajaatoiminta (2 %).
Yleisimpiä (≥ 5 %) ≥ 3. asteen ei-hematologisia haittavaikutuksia olivat transaminaasipitoisuuden kohoaminen (11 %), keuhkokuume (11 %), kuumeinen neutropenia (8 %), sepsis (7 %), kuume (7 %), suurentunut gammaglutamyylitransferaasipitoisuus (6 %), hypotensio (6 %), bakteeri-infektio (5 %) ja hypogammaglobulinemia (5 %).
Yleisimpiä (≥ 20 %) ≥ 3. asteen hematologisia poikkeavuuksia olivat neutropenia (89 %), trombosytopenia (45 %), anemia (44 %), lymfopenia (36 %) ja leukopenia (33 %).
Haittavaikutustaulukko
Taulukossa 4 esitetään yhteenveto CARVYKTI-hoitoa saaneilla potilailla esiintyneistä haittavaikutuksista.
Haittavaikutukset esitetään kussakin elinjärjestelmässä yleisyyden mukaan. Kussakin yleisyysryhmässä haittavaikutukset esitetään oleellisilta osin vakavuuden mukaan alenevassa järjestyksessä seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
| Taulukko 4. Haittavaikutukset CARVYKTI-valmistetta multippelin myelooman hoitoon saaneilla potilailla | ||||
| Elinjärjestelmä | Esiintyvyys | Haittavaikutus | Ilmaantuvuus (%) | |
| Kaikki vaikeus-asteet | ≥ 3. aste | |||
| Infektiot | Hyvin yleinen | Bakteeri-infektio*# | 14 | 5 |
| Ylähengitysteiden infektio* | 32 | 2 | ||
| Virusinfektio* | 19 | 4 | ||
| Keuhkokuume*# | 14 | 11 | ||
| Yleinen | Sepsis1# | 9 | 7 | |
| Maha-suolitulehdus2 | 6 | 1 | ||
| Virtsatieinfektio3 | 5 | 2 | ||
| Sieni-infektio* | 3 | < 1 | ||
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen | Myeloidiperäinen sekundaarinen syöpä# | 4 | 4 |
| Melko harvinainen | T-soluperäinen sekundaarinen syöpä | 1 | 1 | |
| Veri ja imukudos | Hyvin yleinen | Neutropenia* | 90 | 89 |
| Trombosytopenia | 60 | 45 | ||
| Anemia4 | 60 | 44 | ||
| Leukopenia | 34 | 33 | ||
| Lymfopenia | 38 | 36 | ||
| Hyytymishäiriö5 | 12 | 3 | ||
| Yleinen | Kuumeinen neutropenia | 8 | 8 | |
| Lymfosytoosi | 3 | 1 | ||
| Immuunijärjestelmä | Hyvin yleinen | Hypogammaglobulinemia* | 33 | 5 |
| Sytokiinioireyhtymä# | 83 | 4 | ||
| Yleinen | Hemofagosyyttinen lymfohistiosytoosi# | 3 | 2 | |
| Infuusioon liittyvät reaktiot | 5 | 0 | ||
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hypokalsemia | 16 | 3 |
| Hypofosfatemia | 17 | 4 | ||
| Heikentynyt ruokahalu | 16 | 1 | ||
| Hypokalemia | 17 | 2 | ||
| Hypoalbuminemia | 11 | < 1 | ||
| Hyponatremia | 10 | 2 | ||
| Hypomagnesemia | 12 | < 1 | ||
| Hyperferritinemia6 | 10 | 2 | ||
| Psyykkiset häiriöt | Yleinen | Delirium7 | 3 | < 1 |
| Persoonallisuuden muutokset8 | 3 | 1 | ||
| Hermosto | Hyvin yleinen | Enkefalopatia9# | 14 | 3 |
| ICANS-oireyhtymä# | 11 | 2 | ||
| Motorinen toimintahäiriö10 | 13 | 2 | ||
| Huimaus* | 13 | 1 | ||
| Päänsärky | 25 | 0 | ||
| Unihäiriöt11 | 10 | 1 | ||
| Yleinen | Afasia12 | 5 | < 1 | |
| Aivohermohalvaus13 | 7 | 1 | ||
| Pareesi14 | 1 | < 1 | ||
| Ataksia15 | 4 | < 1 | ||
| Vapina* | 5 | < 1 | ||
| Neurotoksisuus# | 1 | 1 | ||
| Perifeerinen neuropatia16 | 7 | 1 | ||
| Melko harvinainen | Guillain-Barrén oireyhtymä | < 1 | < 1 | |
| Sydän | Hyvin yleinen | Takykardia* | 14 | 1 |
| Yleinen | Sydämen rytmihäiriöt17 | 4 | 2 | |
| Verisuonisto | Hyvin yleinen | Hypotensio* | 34 | 6 |
| Hypertensio | 11 | 4 | ||
| Verenvuoto18# | 11 | 2 | ||
| Yleinen | Tromboosi* | 4 | 1 | |
| Hiussuonivuoto-oireyhtymä | 1 | 0 | ||
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Hypoksia* | 13 | 4 |
| Hengenahdistus19# | 14 | 3 | ||
| Yskä* | 22 | 0 | ||
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli20 | 32 | 3 |
| Pahoinvointi | 23 | < 1 | ||
| Oksentelu | 12 | 0 | ||
| Ummetus | 15 | 0 | ||
| Yleinen | Vatsakipu* | 9 | 0 | |
| Immuunivälitteinen enterokoliitti | ||||
| Maksa ja sappi | Yleinen | Hyperbilirubinemia | 3 | 1 |
| Iho ja ihonalainen kudos | Yleinen | Ihottuma* | 9 | 0 |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Luuston ja lihasten kipu* | 40 | 3 |
| Munuaiset ja virtsatiet | Yleinen | Munuaisten vajaatoiminta21 | 7 | 4 |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Kuume | 85 | 7 |
| Uupumus* | 35 | 4 | ||
| Vilunväreet | 15 | 0 | ||
| Turvotus22 | 16 | 1 | ||
| Kipu* | 11 | 1 | ||
| Tutkimukset | Hyvin yleinen | Kohonnut transaminaasipitoisuus* | 26 | 11 |
| Suurentunut gammaglutamyylitransferaasi-pitoisuus | 10 | 6 | ||
| Yleinen | Suurentunut C-reaktiivisen proteiinin pitoisuus | 7 | 1 | |
| Suurentunut veren alkalisen fosfataasin pitoisuus | 8 | 3 | ||
| Haittavaikutukset raportoitu MedDRA-version 26.1 mukaisesti # Sisältää kuolemaan johtaneita lopputuloksia. * Perustuu ryhmiteltyyn termiin 1 Sepsis käsittää seuraavat: bakteremia, bakteerisepsis, kandidasepsis, laitteeseen liittyvä bakteremia, enterokokin aiheuttama bakteremia, enterokokkisepsis, Haemophilus-bakteerin aiheuttama sepsis, neutropeeninen sepsis, pseudomonaksen aiheuttama bakteremia, pseudomonaksen aiheuttama sepsis, sepsis, septinen sokki, stafylokokin aiheuttama bakteremia, streptokokkisepsis, systeeminen kandidiaasi ja urosepsis. 2 Maha-suolitulehdus käsittää seuraavat: bakteerin aiheuttama enterokoliitti, infektioosi enterokoliitti, viruksen aiheuttama enterokoliitti, enterovirusinfektio, maha-suolitulehdus, Cryptosporidium-alkueläimen aiheuttama maha-suolitulehdus, rotaviruksen aiheuttama maha-suolitulehdus, Salmonella-bakteerin aiheuttama maha-suolitulehdus, viruksen aiheuttama maha-suolitulehdus, Escherichia coli ‑bakteerin aiheuttama maha-suolitulehdus, maha-suolikanavan infektio ja paksusuolen infektio. 3 Virtsatieinfektio käsittää seuraavat: virtsarakkotulehdus, Escherichia-bakteerin aiheuttama virtsatieinfektio, virtsatieinfektio, bakteerin aiheuttama virtsatieinfektio ja viruksen aiheuttama virtsatieinfektio. 4 Anemia käsittää seuraavat: anemia, hypokrominen anemia, raudanpuutosanemia ja kalpeus. 5 Hyytymishäiriö käsittää seuraavat: pidentynyt aktivoitu partiaalinen tromboplastiiniaika, vähentynyt veren fibrinogeenipitoisuus, poikkeavuudet veren hyytymiskokeissa, pidentynyt hyytymisaika, hyytymishäiriö, disseminoitunut intravaskulaarinen koagulaatio, hypofibrinogenemia, suurentunut INR-arvo (international normalised ratio), suurentunut protrombiinipitoisuus ja pidentynyt protrombiiniaika. 6 Hyperferritinemia käsittää seuraavat: hyperferritinemia ja suurentunut seerumin ferritiinipitoisuus. 7 Delirium käsittää seuraavat: agitaatio, delirium, desorientaatio, euforinen mielentila, hallusinaatiot, ärtyisyys ja levottomuus. 8 Persoonallisuuden muutokset käsittävät seuraavat: affektilabiilius, apatia, tunteiden latistuminen, välinpitämättömyys, persoonallisuuden muutos ja vähentyneet kasvojen ilmeet. 9 Enkefalopatia käsittää seuraavat: muistinmenetys, bradyfrenia, kognitiivinen häiriö, sekavuustila, alentunut tajunnantaso, tarkkaavuuden häiriöt, enkefalopatia, letargia, muistin heikkeneminen, mielenterveyden heikkeneminen, mielentilan muutos, psykomotorinen taantuma ja hidas reagointi ärsykkeisiin. 10 Motoriset toimintahäiriöt käsittävät seuraavat: agrafia, bradykinesia, hammasratasilmiö, poikkeava koordinaatio, dysgrafia, ekstrapyramidaalihäiriö, silmäluomen ptoosi, mikrografia, motorinen toimintahäiriö, lihasjäykkyys, lihasspasmit, lihasten kireys, lihasheikkous, myoklonus, parkinsonismi, poikkeava ryhti ja kaavamaisuus. 11 Unihäiriöt käsittävät seuraavat: liikaunisuus, unettomuus, unihäiriöt ja uneliaisuus. 12 Afasia käsittää seuraavat: afasia, dysartria, puheen hitaus ja puheen häiriöt. 13 Aivohermohalvaus käsittää seuraavat: Bellin pareesi, aivohermohalvaus, kasvohermon toimintahäiriö, kasvohalvaus, kasvojen pareesi, kolmannen hermon halvaus, kolmoishermon halvaus ja kuudennen hermon halvaus. 14 Pareesi käsittää seuraavat: pareesi, hemipareesi ja pohjehermon halvaus. 15 Ataksia käsittää seuraavat: ataksia, tasapainohäiriö, dysmetria ja kävelyn häiriöt. 16 Perifeerinen neuropatia käsittää seuraavat: perifeerinen neuropatia, perifeerinen motorinen neuropatia, perifeerinen sensomotorinen neuropatia, perifeerinen sensorinen neuropatia ja polyneuropatia. 17 Sydämen rytmihäiriöt käsittävät seuraavat: eteisvärinä, eteislepatus, täydellinen eteis-kammiokatkos, toisen asteen eteis-kammiokatkos, supraventrikulaarinen takykardia, kammiolisälyönnit ja kammiotakykardia. 18 Verenvuoto käsittää seuraavat: katetrin kiinnityskohdan verenvuoto, aivoverenvuoto, sidekalvon verenvuoto, kontuusio, nenäverenvuoto, silmäruhje, maha-suolikanavan verenvuoto, verioksennus, veriuloste, hematooma, verivirtsaisuus, veriyskä, infuusiokohdan hematooma, maha-suolikanavan loppupään verenvuoto, suun ruhje, toimenpiteen jälkeinen verenvuoto, keuhkoverenvuoto, verkkokalvon verenvuoto, retroperitoneaalinen verenvuoto, lukinkalvonalainen verenvuoto ja kovakalvonalainen verenpurkauma. 19 Hengenahdistus käsittää seuraavat: akuutti hengityksen vajaatoiminta, hengenahdistus, rasitushengenahdistus, hengityksen vajaatoiminta, takypnea ja hengityksen vinkuminen. 20 Ripuli käsittää seuraavat: koliitti ja ripuli. 21 Munuaisten vajaatoiminta käsittää seuraavat: akuutti munuaisvaurio, suurentunut veren kreatiniinipitoisuus, krooninen munuaissairaus, munuaisten vajaatoiminta ja munuaisten toiminnan heikkeneminen. 22 Turvotus käsittää seuraavat: kasvojen turvotus, nesteen kertyminen elimistöön, yleistynyt turvotus, hypervolemia, paikallinen turvotus, turvotus, perifeerinen turvotus, suulaen turvotus, silmäkuoppaa ympäröivä turvotus, raajojen turpoaminen, keuhkolaskimoverentungos, keuhkoedeema, kivespussin edeema ja kielen turpoaminen. | ||||
MMY3002-tutkimuksen 196 potilaasta 20 potilaalla, joilla oli suuremman riskin sairaus, sairaus eteni nopeasti CARVYKTI-infuusiota edeltävässä siltahoidon varhaisvaiheessa, joten he saivat seuraavana hoitona CARVYKTI-valmistetta (ks. kohta Farmakodynamiikka). Näistä potilaista yhdellä (5 %) raportoitiin liikkumiseen vaikuttavaa ja neurokognitiivista toksisuutta, joka oli vaikeusasteeltaan lievää (1. tai 2. aste). Sytokiinioireyhtymää raportoitiin yleisemmin vaikeusasteina 3. aste ja 4. aste (25 %), mukaan lukien sytokiinioireyhtymätapahtumat, joihin liittyi komplisoivina tekijöinä hemofagosyyttinen lymfohistiosytoosi (10 %) tai disseminoitunut intravaskulaarinen koagulaatio (10 %). ICANS-oireyhtymää raportoitiin 3. asteen osalta yleisemmin (35 %) ja vaikeampiasteisena (10 %). Viisi potilasta kuoli CARVYKTI-valmisteeseen liittyvien kuolemaan johtaneiden tapahtumien seurauksena (kaksi heistä verenvuotoon hemofagosyyttisen lymfohistiosytoosin tai disseminoituneen intravaskulaarisen koagulaation yhteydessä ja kolme heistä kuolemaan johtaneen infektion seurauksena).
Valikoitujen haittavaikutusten kuvaus
Sytokiinioireyhtymä
Sytokiinioireyhtymää raportoitiin 83 %:lla potilaista (n = 330); 79 %:lla potilaista (n = 314) oli 1. asteen tai 2. asteen sytokiinioireyhtymätapahtumia, 4 %:lla (n = 15) potilaista oli 3. asteen tai 4. asteen sytokiinioireyhtymätapahtumia ja < 1 %:lla (n = 1) potilaista oli 5. asteen sytokiinioireyhtymätapahtuma. Yhdeksänkymmentäkahdeksan prosenttia potilaista (n = 324) toipui sytokiinioireyhtymästä. Sytokiinioireyhtymän kesto oli ≤ 18 päivää, paitsi yhdellä potilaalla. Tällä yhdellä potilaalla sytokiinioireyhtymä kesti 97 päivää ja siihen liittyi komplisoivana tekijänä sekundaarinen hemofagosyyttinen lymfohistiosytoosi, joka johti myöhemmin potilaan kuolemaan. Yleisimpiä (≥ 10 %) sytokiinioireyhtymään liittyviä oireita ja löydöksiä olivat kuume (82 %), hypotensio (28 %), suurentunut aspartaattiaminotransferaasipitoisuus (ASAT-arvo) (12 %) ja hypoksia (10 %). Ks. ohjeet seurantaan ja hoitoon kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Neurologinen toksisuus
Neurologista toksisuutta esiintyi 23 %:lla potilaista (n = 90); 6 %:lla (n = 22) potilaista oli 3. asteen tai 4. asteen neurologista toksisuutta ja 1 %:lla (n = 3) potilaista oli 5. asteen neurologista toksisuutta (yhdellä ICANS-oireyhtymän seurauksena, yhdellä neurologisen toksisuuden ja sen aikaan potilaalla olleen parkinsonismin seurauksena ja yhdellä enkefalopatian seurauksena). Lisäksi yksitoista potilasta kuoli ja heillä oli kuoleman aikaan neurologista toksisuutta; kahdeksan kuolemaa johtui infektiosta (näistä kahdella potilaalla oli kuoleman aikaan jäljempänä kuvattuja parkinsonismin oireita ja löydöksiä) ja yksi kuolema aiheutui kustakin seuraavista: hengityksen vajaatoiminta, sydämen toiminnan ja hengityksen pysähtyminen ja parenkyyminsisäinen verenvuoto. Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet ohjeet seurantaan ja hoitoon.
ICANS-oireyhtymä (Immune effector cell-associated neurotoxicity syndrome)
ICANS-oireyhtymää esiintyi yhdistetyissä tutkimuksissa (n = 396) 11 %:lla potilaista (n = 45); 2 %:lla (n = 8) potilaista oli 3. tai 4. asteen ICANS-oireyhtymä ja < 1 %:lla (n = 1) oli 5. asteen ICANS-oireyhtymä. Oireita olivat afasia, puheen hitaus, dysgrafia, enkefalopatia, alentunut tajunnantaso ja sekavuustila. CARVYKTI-infuusiosta ICANS-oireyhtymän ensimmäiseen ilmaantumiskertaan kuluneen ajan mediaani oli 8 päivää (vaihteluväli: 2–15 päivää, paitsi yhdellä potilaalla, jolle se ilmaantui 26. päivänä), ja sen keston mediaani oli 3 päivää (vaihteluväli: 1–29 päivää, paitsi yhdellä potilaalla, joka kuoli 40. päivänä).
Liikkumiseen vaikuttava ja neurokognitiivinen toksisuus, johon liittyy parkinsonismin oireita ja löydöksiä
Niistä yhdistettyjen tutkimusten 90:stä (n = 396) potilaasta, joille ilmaantui neurotoksisuutta, yhdeksällä miespotilaalla oli neurologista toksisuutta, johon liittyi useita ICANS-oireyhtymästä erottuvia parkinsonismin oireita ja löydöksiä. Parkinsonismin korkeimmat vaikeusasteet olivat 1. aste (n = 1), 2. aste (n = 2) ja 3. aste (n = 6). Parkinsonismin ilmaantumiseen kuluneen ajan mediaani oli 38,0 päivää (vaihteluväli: 14–914 päivää) CARVYKTI-infuusiosta. Yksi potilas (3. aste) kuoli 247 päivää CARVYKTI-valmisteen annon jälkeen neurologisen toksisuuden seurauksena ja hänellä oli kuoleman aikaan parkinsonismia. Lisäksi kaksi potilasta (2. aste ja 3. aste) kuoli 162 päivää ja 119 päivää CARVYKTI-valmisteen annon jälkeen infektion seurauksena ja heillä oli kuoleman aikaan parkinsonismia. Yksi potilas (3. aste) toipui. Lopuilla 5 potilaalla oli parhaillaan parkinsonismia, kun CARVYKTI-valmisteen annosta oli kulunut enimmillään 996 päivää. Kaikilla 9 potilaalla oli anamneesissa aiempi sytokiinioireyhtymä (1. asteen n = 1; 2. asteen n = 6; 3. asteen n = 1; 4. asteen n = 1), kun taas 6 potilaalla 9:stä oli aiemmin ollut ICANS-oireyhtymä (1. aste n = 5; 3. aste n = 1).
Guillain-Barrén oireyhtymä
Guillain-Barrén oireyhtymää raportoitiin yhdistetyissä tutkimuksissa (n = 396) yhdellä potilaalla CARVYKTI-hoidon jälkeen. Vaikka Guillain-Barrén oireyhtymän oireet lievenivät steroidi- ja IVIG-hoidon jälkeen, potilas kuoli 139 päivää CARVYKTI-valmisteen annon jälkeen gastroenteriitin jälkeisen enkefalopatian vuoksi, ja hänellä oli kuoleman aikaan Guillain-Barrén oireyhtymän oireita.
Perifeerinen neuropatia
Perifeeristä neuropatiaa kehittyi yhdistetyissä tutkimuksissa (n = 396) 28 potilaalle, ja se ilmeni sensorisena, motorisena tai sensomotorisena neuropatiana. Oireiden ilmaantumiseen kuluneen ajan mediaani oli 58 päivää (vaihteluväli: 1–914 päivää), ja perifeerisen neuropatian keston mediaani oli 142 päivää (vaihteluväli: 1–1 062 päivää), mukaan lukien niillä, joilla oli parhaillaan neuropatiaa. Näistä 28 potilaasta 5:llä oli 3. asteen tai 4. asteen perifeeristä neuropatiaa (joka hävisi 1 potilaalla ilman raportoitua hoitoa; muilla 4 potilaalla perifeerinen neuropatia jatkui, mukaan lukien yhdellä potilaalla, jonka tila koheni deksametasonihoidon jälkeen). Muista 23 potilaasta, joilla oli ≤ 2. asteen perifeeristä neuropatiaa, perifeerinen neuropatia hävisi 7 potilaalla ilman raportoitua hoitoa ja 3 potilaalla duloksetiinihoidon jälkeen, ja lopuilla 9 potilaalla se parhaillaan jatkui.
Aivohermohalvaus
Aivohermohalvaus ilmaantui yhdistetyissä tutkimuksissa (n = 396) 27 potilaalle. Sen ilmaantumiseen kuluneen ajan mediaani oli 22 päivää (vaihteluväli: 17–101 päivää) CARVYKTI-infuusion jälkeen, ja sen häviämiseen kuluneen ajan mediaani oli 61 päivää (vaihteluväli: 1–443 päivää) oireiden ilmaantumisen jälkeen.
Pitkittynyt ja uusiutunut sytopenia
Valmisteen annon jälkeisenä päivänä (päivä 1) esiintyneitä 3. tai 4. asteen sytopenioita, jotka eivät lieventyneet päivään 30 mennessä 2. asteeseen tai lievemmiksi, olivat trombosytopenia (33 %), neutropenia (28 %), lymfopenia (25 %) ja anemia (3 %). Päivänä 60 CARVYKTI-hoidon jälkeen 23 %:lla potilaista oli 3. tai 4. asteen lymfopeniaa, 21 %:lla oli 3. tai 4. asteen neutropeniaa, 7 %:lla oli 3. tai 4. asteen anemiaa ja 4 %:lla oli 3. tai 4. asteen trombosytopeniaa sen jälkeen, kun he olivat aluksi toipuneet 3. tai 4. asteen sytopeniasta.
Taulukossa 5 luetellaan valmisteen annon jälkeen ilmaantuneiden sellaisten 3. asteen ja 4. asteen sytopenioiden ilmaantuvuus, jotka eivät olleet lieventyneet 2. asteeseen tai lievemmäksi päivään 30 ja päivään 60 mennessä.
| Taulukko 5. Pitkittyneiden ja uusiutuneiden sytopenioiden ilmaantuvuus CARVYKTI-hoidon jälkeen (n = 396) | ||||
| 3./4. aste (%) päivän 1 hoidon jälkeen | Alkuvaiheen 3./4. aste (%) ei lieventynyta ≤ 2. asteeseen päivään 30 mennessä | Alkuvaiheen 3./4. aste (%) ei lieventynyta ≤ 2. asteeseen päivään 60 mennessä | 3./4. asteen ilmaantuvuus (%) päivän 60 jälkeen (3./4. asteen alkuvaiheen lievenemisena jälkeen) | |
| Trombosytopenia | 191 (48 %) | 132 (33 %) | 76 (19 %) | 14 (4 %) |
| Neutropenia | 381 (96 %) | 111 (28 %) | 44 (11 %) | 81 (21 %) |
| Lymfopenia | 394 (99 %) | 97 (25 %) | 45 (11 %) | 91 (23 %) |
| Anemia | 184 (47 %) | 10 (3 %) | 10 (3 %) | 26 (7 %) |
| a Kalenteripäivälle käytetään laboratoriotulosta, jonka toksisuusaste on huonoin. Lievenemisen määritelmä: jos lievenemisjakso ≤ 10 päivää, eri päivinä on oltava kaksi peräkkäistä ≤ 2. asteen tulosta. Huom.: Analyysiin on otettu mukaan MMY2001- ja MMY2003-tutkimuksissa päivästä 1 päivään 100 tai MMY3002-tutkimuksessa päivään 112 tai seuraavan hoidon alkaessa (sen mukaan, mikä näistä toteutuu ensimmäisenä) arvioidut laboratoriotulokset. Trombosytopenia: 3./4. aste – trombosyyttimäärä < 50 000 solua/mikrol. Neutropenia: 3./4. aste – neutrofiilimäärä < 1 000 solua/mikrol. Lymfopenia: 3./4. aste – lymfosyyttimäärä < 0,5 × 109 solua/l. Anemia: 3. aste – hemoglobiinipitoisuus < 80 g/l. 4. astetta ei NCI-CTCAE v5 ‑kriteerien mukaisesti määritelty laboratoriossa määritetyn määrän perusteella. Prosenttiosuudet perustuvat hoitoa saaneiden potilaiden lukumäärään. | ||||
Vakavat infektiot
Infektioita ilmaantui 54 %:lle potilaista (n = 213); 18 %:lla potilaista (n = 73) oli 3. asteen tai 4. asteen infektioita, ja 4 %:lla potilaista (n = 17) oli kuolemaan johtanut infektio (COVID-19-infektioon liittyvä keuhkokuume, keuhkokuume, sepsis, Clostridium difficile ‑koliitti, septinen sokki, bronkopulmonaalinen aspergilloosi, pseudomonaksen aiheuttama sepsis, neutropeeninen sepsis ja keuhkopaise). Yleisimmin raportoituja (≥ 2 %) 3. asteen tai vaikeampiasteisia infektioita olivat keuhkokuume, COVID-19-infektioon liittyvä keuhkokuume ja sepsis. Kuumeista neutropeniaa havaittiin 6 %:lla potilaista, ja 2 %:lla oli vakava kuumeinen neutropenia.
Ks. ohjeet seurantaan ja hoitoon kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Hypogammaglobulinemia
Hypogammaglobulinemiaa ilmaantui yhdistetyissä tutkimuksissa (n = 396) 34 %:lle potilaista, ja 5 %:lla potilaista oli 3. asteen hypogammaglobulinemia. IgG:n laboratorioarvot pienenivät infuusion jälkeen alle tason 500 mg/dl 91 %:lla (360/396) CARVYKTI-hoitoa saaneista potilaista. Hypogammaglobulinemiaa ilmaantui infuusion jälkeen joko haittavaikutuksena tai IgG:n laboratorioarvon pienenemisenä alle tason 500 mg/dl 92 %:lla (364/396) potilaista. Viisikymmentäkahdeksan prosenttia potilaista sai CARVYKTI-hoidon jälkeen IVIG-hoitoa joko haittavaikutuksen hoitoon tai estohoitona. Ks. ohjeet seurantaan ja hoitoon kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Immunogeenisuus
CARVYKTI-valmisteen immunogeenisuutta on arvioitu CARVYKTI-valmisteeseen sitoutuvien vasta-aineiden havaitsemiseen tarkoitetulla validoidulla määrityksellä ennen annosta sekä useissa aikapisteissä infuusion jälkeen. Yhdistetyissä tutkimuksissa (n = 363) 23 %:lla (83/363) potilaista, joilta oli tarkoituksenmukaisia näytteitä, todettiin hoidonaikaisten CAR-vasta-aineiden positiivinen testitulos. Selkeää näyttöä ei ollut siitä, että CAR:n vasta-aineet vaikuttaisivat CARVYKTI-valmisteen kinetiikkaan alkuvaiheen ekspansion ja elimistössä pysymisen osalta tai tehoon tai turvallisuuteen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
CARVYKTI-valmisteen yliannoksen oireista ja jälkiseurauksista ei ole tietoja.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC-koodi: L01XL05.
Vaikutusmekanismi
CARVYKTI on B-solujen maturaatioantigeeniin (BCMA) kohdennettu muuntogeenisiä autologisia T-soluja sisältävä immuunihoito, jossa potilaan omat T-solut on ohjelmoitu uudelleen kimeeristä antigeenireseptoria (CAR) koodaavalla transgeenillä, joka tunnistaa ja eliminoi BCMA:ta ilmentävät solut. Pääasiassa pahanlaatuiset B-linjan myeloomasolut sekä myöhäisvaiheen B-solut ja plasmasolut ilmentävät BCMA:ta pinnallaan. CARVYKTI-valmisteen CAR-proteiinissa on kaksi BCMA:han kohdentuvaa yhden domeenin vasta-ainetta, joilla on korkea aviditeetti ihmisen BCMA:han; kostimulatorinen 4-1BB-domeeni ja sytoplasminen CD3‑zeeta (CD3ζ)-signaalidomeeni. BCMA:ta ilmentäviin soluihin sitoutunut CAR-proteiini edistää T-solujen aktivaatiota ja ekspansiota sekä kohdesolujen eliminaatiota.
Farmakodynaamiset vaikutukset
Yhteisviljelykokeet in vitro osoittivat, että siltakabtageeniautoleuseelivälitteinen sytotoksisuus ja sytokiinin vapautuminen (interferonigamma [IFN-γ], tuumorinekroositekijäalfa [TNF-α], interleukiini [IL]-2) olivat BCMA:sta riippuvaisia.
Kliininen teho ja turvallisuus
CARTITUDE-1 (MMY2001-tutkimus)
MMY2001 oli avoin, yhden hoitoryhmän, faasin 1b/2 monikeskustutkimus, jossa arvioitiin CARVYKTI-hoidon tehoa ja turvallisuutta aikuisille potilaille, joilla oli uusiutunut tai hoitoon reagoimaton multippeli myelooma ja jotka olivat saaneet vähintään kolmea myeloomaan tarkoitettua hoitolinjaa, mukaan lukien proteasomin estäjää, immuunivasteen muuntajaa ja CD38:n vasta-ainetta, ja joiden sairaus oli edennyt viimeisimmän hoidon aikana tai 12 kuukauden kuluessa sen jälkeen. Tutkimukseen ei otettu mukaan potilaita, joilla tiedettiin olevan aktiivinen keskushermoston sairaus tai joilla oli aiemmin ollut merkittävä keskushermoston sairaus, mukaan lukien keskushermoston multippeli myelooma, jotka olivat aiemmin altistuneet muille anti-BCMA-hoidoille, joille oli tehty allogeeninen kantasolusiirto afereesia edeltäneiden 6 kuukauden aikana tai joilla oli parhaillaan käynnissä oleva immunosuppressiivinen hoito, kreatiniinipuhdistuma oli < 40 ml/min, absoluuttinen lymfosyyttipitoisuus oli < 300/mikrol, maksan transaminaasit olivat > 3-kertaiset normaalien viitearvojen ylärajaan nähden, sydämen ejektiofraktio oli < 45 % tai joilla oli aktiivinen vakava infektio.
Leukafereesi tehtiin yhteensä 113 potilaalle, ja CARVYKTI-hoito valmistettiin kaikille potilaille. Kuusitoista potilasta ei saanut CARVYKTI-hoitoa (n = 12 leukafereesin jälkeen ja n = 4 lymfosyyttejä vähentävän hoidon jälkeen), koska potilas joko vetäytyi tutkimuksesta (n = 5), sairaus eteni (n = 2) tai potilas kuoli (n = 9).
Hoitoa saaneilla 97 potilaalla leukafereesimateriaalin vastaanottopäivämäärästä valmistuspaikassa infuusiolääkevalmisteen vapauttamiseen kulunut aika (mediaani) oli 29 päivää (vaihteluväli: 23–64 päivää), ja ensimmäisestä leukafereesistä CARVYKTI-infuusioon kulunut aika (mediaani) oli 47 päivää (vaihteluväli: 41–167 päivää).
Leukafereesin jälkeen ennen CARVYKTI-infuusiota 73 potilasta yhteensä 97 potilaasta (75 %) sai siltahoitoa. Yleisimpiä siltahoitoon (≥ 20 %:lla potilaista) käytettyjä lääkeaineita olivat deksametasoni 62 potilaalla (63,9 %), bortetsomibi 26 potilaalla (26,8 %), syklofosfamidi 22 potilaalla (22,7 %) ja pomalidomidi 21 potilaalla (21,6 %).
CARVYKTI annettiin kertainfuusiona laskimoon 5–7 päivää lymfosyyttejä vähentävän kemoterapian (300 mg/m2 syklofosfamidia laskimoon päivittäin ja 30 mg/m2 fludarabiinia laskimoon päivittäin kolmen päivän ajan) aloittamisen jälkeen. Yhdeksänkymmentäseitsemän potilasta sai CARVYKTI-hoitoa annoksina (mediaani) 0,71 × 106 CAR-positiivista elinkykyistä T-solua/kg (vaihteluväli: 0,51–0,95 × 106 solua/kg). Kaikki potilaat otettiin sairaalahoitoon CARVYKTI-infuusiota varten ja vähintään 10 päiväksi sen jälkeen.
| Taulukko 6. Tiivistelmä potilaiden demografisista ja lähtötilanteen ominaisuuksista | ||
| Analyysitietue | Kaikki hoitoa saaneet (n = 97) | Potilaat, joille tehtiin leukafereesi (n = 113) |
| Ikä (vuotta) | ||
| Ikäryhmä n (%) | ||
| < 65 | 62 (64) | 70 (62) |
| 65–75 | 27 (28) | 34 (30) |
| > 75 | 8 (8) | 9 (8) |
| Mediaani (vaihteluväli) | 61,0 (43–78) | 62 (29–78) |
Sukupuoli Miehiä n (%) Naisia n (%) |
57 (59) 40 (41) |
65 (57,5) 48 (42,5) |
| Etninen tausta | ||
| Amerikan intiaani tai Alaskan alkuperäiskansa | 1 (1) | 1 (1) |
| Aasialainen | 1 (1) | 1 (1) |
| Mustaihoinen tai afroamerikkalainen | 17 (17,5) | 17 (15) |
| Havaijin tai muiden Tyynenmeren saarten alkuperäiskansa | 1 (1) | 1 (1) |
| Valkoihoinen | 69 (71) | 83 (73,5) |
| Useita | 0 | 0 |
| Ei raportoitu | 8 (8) | 10 (9) |
| ECOG-pisteet ennen infuusiota n (%) | ||
| 0 | 39 (40) | 55 (49) |
| 1 | 54 (56) | 58 (51) |
| 2 | 4 (4) | - |
| ISS-luokitus tutkimuksen lähtötilanteessa n (%) | ||
| N | 97 | 58 |
| I | 61 (63) | 32 (55) |
| II | 22 (23) | 21 (36) |
| III | 14 (14) | 5 (9) |
Kreatiniinipuhdistuma/eGFR (MDRD) (ml/min/1,73 m2) Mediaani (vaihteluväli) |
88,44 (41,8–242,9) |
73,61 (36,2–177,8) |
| Multippelin myelooman diagnoosin saamisesta tutkimukseen mukaan tuloon kulunut aika (vuotta) | ||
| Mediaani (vaihteluväli) | 5,94 (1,6–18,2) | 5,73 (1,0–18,2) |
| Ekstramedullaarisia plasmasytoomia n (%) | ||
| Kyllä | 13 (13) | NAa |
| Ei | 84 (87) | NAa |
| Sytogeneettinen riski tutkimuksen lähtötilanteessa n (%) | ||
| Tavanomainen riski | 68 (70) | 70 (62) |
| Suuri riski | 23 (24) | 28 (25) |
| Del17p | 19 (20) | 22 (19,5) |
| t (4;14) | 3 (3) | 5 (4) |
| t (14;16) | 2 (2) | 3 (3) |
| Ei tiedossa | 6 (6) | 15 (13) |
| BCMA:ta ilmentävä kasvain (%) | ||
| Mediaani (vaihteluväli) | 80 (20–98) | 80 (20–98) |
Multippeliin myeloomaan aiemmin saatujen hoitolinjojen lukumäärä Mediaani (vaihteluväli) |
6 (3–18) |
5 (3–18) |
Aiempi hoito proteaasin estäjän, immuunivasteen muuntajan ja CD38:n vasta-aineen yhdistelmällä n (%) | 97 (100) | 113 (100) |
| Aiempi autologinen kantasolusiirto n (%) | 87 (90) | 99 (88) |
| Aiempi allogeeninen kantasolusiirto n (%) | 8 (8) | 8 (7) |
| Hoitoon reagoimaton jossakin hoitovaiheessa n (%) | 97 (100) | 113 (100) |
| Reagoimaton proteaasin estäjän, immuunivasteen muuntajan ja CD38:n vasta-aineen yhdistelmään n (%) | 85 (88) | 100 (88,5) |
| Reagoimaton edelliseen hoitolinjaan n (%) | 96 (99) | 112 (99) |
ECOG = Eastern Cooperative Oncology Group; ISS = International Staging System; NA = ei sovellu (not applicable). a Plasmasytoomia ei arvioitu ennen lymfosyyttien vähentämistä. | ||
Tehoa koskevat tulokset perustuivat riippumattoman arviointikomitean (Independent Review Committee) IMWG-kriteerien perusteella arvioimaan kokonaisvastelukuun (ks. taulukko 7).
| Taulukko 7. MMY2001-tutkimuksen tehoa koskevat tulokset | ||
| Analyysitietue | Kaikki hoitoa saaneet (n = 97) | Potilaat, joille tehtiin leukafereesi (n = 113) |
| Kokonaisvasteluku (sCRa + VGPR + PR) n (%) | 95 (97,9) | 95 (84,1) |
| 95 %:n luottamusväli (%) | (92,7–99,7) | (76,0–90,3) |
| Täydellinen vaste lisäehdoin (stringent complete response, sCR)a n (%) | 80 (82,5) | 80 (70,8) |
| Erittäin hyvä osittainen vaste (very good partial response, VGPR) n (%) | 12 (12,4) | 12 (10,6) |
| Osittainen vaste (partial response, PR) n (%) | 3 (3,1) | 3 (2,7) |
Vasteen kesto (kuukautta)b Mediaani (95 %:n luottamusväli) |
NE (28,3–NE) |
- |
Vasteen kesto, jos paras vaste on täydellinen vaste lisäehdoina (kuukautta) Mediaani (95 %:n luottamusväli) |
NE (28,3–NE) |
- |
Vasteeseen kulunut aika (kuukautta) Mediaani (vaihteluväli) |
0,95 (0,9–10,7) |
- |
| MRD-negatiivisia n (%)c | 56 (57,7) | 56 (49,6) |
| 95 %:n luottamusväli (%) | (47,3–67,7) | (40,0–59,1) |
| MRD-negatiivisia potilaita, joilla täydellinen vaste lisäehdoin n (%)c | 42 (43,3) | 42 (37,2) |
| 95 %:n luottamusväli (%) | (33,3–53,7) | (28,3–46,8) |
MRD = minimaalinen jäännöstauti (Minimal Residual Disease), NE = ei arvioitavissa (not estimable) a Kaikki täydelliset vasteet olivat täydellisiä vasteita lisäehdoin. | ||
CARTITUDE-4 (MMY3002-tutkimus)
MMY3002 on avoin, satunnaistettu faasin 3 monikeskustutkimus, jossa arvioidaan CARVYKTI-valmisteen tehoa potilaille, joilla on uusiutunut ja lenalidomidihoitoon reagoimaton multippeli myelooma ja jotka ovat aiemmin saaneet vähintään yhtä aiempaa hoitolinjaa, mukaan lukien proteasomin estäjää ja immuunivasteen muuntajaa. Yhteensä 419 potilasta satunnaistettiin saamaan joko afereesin, siltahoidon, lymfosyyttejä vähentävän hoidon ja CARVYKTI-hoidon sekvenssiä (n = 208) tai tavanomaista hoitoa, joka käsitti lääkärin valinnan mukaan daratumumabia, pomalidomidia ja deksametasonia tai bortetsomibia, pomalidomidia ja deksametasonia (n = 211).
Tutkimukseen ei otettu mukaan potilaita, joilla tiedettiin olevan aktiivinen keskushermoston affisio tai oli aiemmin ollut keskushermoston affisio, joilla oli kliinisiä merkkejä multippelista myeloomasta aiheutuvasta aivokalvojen affisiosta, joilla anamneesissa oli Parkinsonin tauti tai muu hermostoa rappeuttava sairaus, jotka olivat aiemmin altistuneet muille anti‑BCMA-hoidoille tai joille oli tehty mihin tahansa kohteeseen kohdentunut CAR‑T-soluhoito, joille oli tehty allogeeninen kantasolusiirto afereesia edeltäneiden 6 kuukauden aikana tai joilla oli parhaillaan käynnissä immunosuppresiivinen hoito tai joille oli tehty autologinen kantasolusiirto afereesia edeltäneiden 12 viikon aikana.
Näistä satunnaistetuista 419 potilaasta (208 satunnaistettiin saamaan CARVYKTI-hoitoa ja 211 saamaan tavanomaista hoitoa) 57 % oli miehiä, 75 % oli valkoihoisia, 3 % oli mustaihoisia tai afroamerikkalaisia ja 7 % oli latinalaisamerikkalaisia. Potilaiden iän mediaani oli 61 vuotta (vaihteluväli: 28–80 vuotta). Potilaat olivat saaneet kahta aiempaa hoitolinjaa (mediaani 2; vaihteluväli: 1–3) ja 85 % potilaista oli saanut aiemmin autologisen kantasolusiirron. Yhdeksälläkymmenelläyhdeksällä prosentilla potilaista sairaus ei ollut reagoinut viimeisimmän hoitolinjan hoitoon. Neljälläkymmenelläkahdeksalla prosentilla sairaus ei ollut reagoinut johonkin proteasomin estäjään ja 100 %:lla sairaus ei ollut reagoinut johonkin immuunivasteen muuntajaan.
Kaikille CARVYKTI-ryhmään satunnaistetuille 208 potilaalle tehtiin afereesi. Kaikki 208 satunnaistettua potilasta sai afereesin jälkeen, ennen CARVYKTI-valmisteen saamista, tutkimussuunnitelman mukaista siltahoitoa (tavanomainen hoito). Näistä 208 potilaasta 12 potilasta ei saanut CARVYKTI-hoitoa sairauden etenemisen (n = 10) tai kuoleman (n = 2) vuoksi ja 20 potilaalla sairaus eteni ennen CARVYKTI-infuusiota, mutta heille voitiin antaa CARVYKTI-valmistetta seuraavana hoitona.
Niillä 176 potilaalla, jotka saivat CARVYKTI-valmistetta tutkimushoitona, afereesimateriaalin vastaanottopäivämäärästä valmistuspaikassa infuusiolääkevalmisteen vapauttamiseen kulunut aika (mediaani) oli 44 päivää (vaihteluväli: 25–127 päivää), ja ensimmäisestä afereesista CARVYKTI-infuusioon kulunut aika (mediaani) oli 79 päivää (vaihteluväli: 45–246 päivää).
CARVYKTI annettiin kertainfuusiona laskimoon 5–7 päivää lymfosyyttejä vähentävän kemoterapian (300 mg/m2 syklofosfamidia laskimoon päivittäin ja 30 mg/m2 fludarabiinia laskimoon päivittäin kolmen päivän ajan) aloittamisen jälkeen annoksena (mediaani) 0,71 × 106 CAR-positiivista elinkykyistä T-solua/kg (vaihteluväli: 0,39–1,07 × 106 solua/kg).
Ensisijainen tehon mittari oli etenemättömyysaika (progression-free survival, PFS), joka analysoitiin hoitoaikeen mukaisen (Intent-To-Treat, ITT) analyysitietueen perusteella. Seurannan kestettyä 15,9 kuukautta (mediaani) etenemättömyysajan mediaani oli tavanomaista hoitoa saaneessa ryhmässä 11,8 kuukautta (95 %:n luottamusväli: 9,7–13,8) ja CARVYKTI-hoitoa saaneessa ryhmässä se ei ollut arvioitavissa (95 %:n luottamusväli: 22,8 – ei arvioitavissa) (riskitiheyksien suhde [HR]: 0,26 [95 %:n luottamusväli: 0,18–0,38]; p‑arvo < 0,0001). Arvioitu etenemättömyysaikaosuus 12 kuukauden aikapisteessä oli CARVYKTI-ryhmässä 75,9 % (95 %:n luottamusväli: 69,4–81,1 %) ja tavanomaista hoitoa saaneessa ryhmässä 48,6 % (95 %:n luottamusväli: 41,5–55,3 %). Arvioitua vasteen keston mediaania ei ole saavutettu CARVYKTI-ryhmässä. Tavanomaisen hoidon ryhmässä arvioitu vasteen keston mediaani oli 16,6 kuukautta (95 %:n luottamusväli: 12,9 – ei arvioitavissa). 15,9 kuukauden (mediaani) seurannan jälkeen kokonaiselossaolon mediaani ei ollut CARVYKTI-ryhmässä arvioitavissa (95 %:n luottamusväli: ei arvioitavissa – ei arvioitavissa) ja oli tavanomaista hoitoa saaneessa ryhmässä 26,7 kuukautta (95 %:n luottamusväli: 22,5 – ei arvioitavissa) (riskitiheyksien suhde: 0,78 [95 %:n luottamusväli: 0,50–1,20]; p-arvo = 0,2551).
Niillä 176 potilaalla, jotka saivat CARVYKTI-valmistetta tutkimushoitona, etenemättömyysajan (PFS) mediaani ei ollut arvioitavissa (95 %:n luottamusväli: ei arvioitavissa – ei arvioitavissa) ja 12 kuukauden etenemättömyysaikaosuus oli 89,7 %. Näiden potilaiden kokonaisvasteluku (ORR) oli 99,4 % (95 %:n luottamusväli: 96,9–100,0 %). Täydellisen vasteen (CR) / täydellisen vasteen lisäehdoin (sCR) osuus oli 86,4 % (95 %:n luottamusväli: 80,4–91,1 %).
MMY3002-tutkimuksen päivitetyt tehoa koskevat tulokset
MMY3002-tutkimuksessa 33,6 kuukauden (mediaani) seurannan aikapisteessä tehdyssä tutkimussuunnitelmassa määritellyssä toisessa välianalyysissa CARVYKTI-ryhmässä ei ollut saavutettu etenemättömyysajan mediaania. Kummassakaan ryhmässä ei ollut saavutettu kokonaiselossaolon mediaania. Yhden kerran annettavasta CARVYKTI-infuusiosta todetaan CARVYKTI-hoitoa saaneilla tutkittavilla tilastollisesti merkitsevä kokonaiselossaolon piteneminen tavanomaiseen hoitoon verrattuna. Etenemättömyysajan tulokset esitetään taulukossa 8 ja kuvassa 1. Kokonaiselossaolon tulokset esitetään taulukossa 8 ja kuvassa 2.
| Taulukko 8. Yhteenveto MMY3002-tutkimuksen tehoa koskevista tuloksista (hoitoaikeen mukainen [ITT] analyysitietue) | ||
| CARVYKTI (n = 208) | Tavanomainen hoito (n = 211) | |
| Etenemättömyysaikaa,b | ||
| Tapahtumien lukumäärä, n (%) | 89 (42,8) | 153 (72,5) |
| Mediaani, kuukautta (95 %:n luottamusväli)c | NE (34,5–NE) | 11,8 (9,7–14,0) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli)d | 0,29 (0,22–0,39) | |
| Täydellisen tai sitä paremman vasteen osuus (Complete Response or Better Rate)b,e, % (95 %:n luottamusväli) | 73,1 (66,5–79,0) | 21,8 (16,4–28,0) |
| p-arvof | < 0,0001 | |
| Kokonaisvasteluku (ORR)b,e, % (95 %:n luottamusväli) | 84,6 (79,0–89,2) | 67,3 (60,5–73,6) |
| p-arvof | < 0,0001 | |
| MRD-negatiivisten kokonaisosuuse, % (95 %:n luottamusväli) | 60,6 (53,6–67,3) | 15,6 (11,0–21,3) |
| p-arvog | < 0,0001 | |
| Kokonaiselossaolo (OS)a | ||
| Tapahtumien lukumäärä (%) | 50 (24,0 %) | 83 (39,3 %) |
| Sensoroitujen lukumäärä (%) | 158 (76,0 %) | 128 (60,7 %) |
| Mediaani, kuukautta (95 %:n luottamusväli)c | NE (NE–NE) | NE (37,75–NE) |
| Riskitiheyksien suhde (HR) (95 %:n luottamusväli)h | 0,55 (0,39–0,79) | |
| p-arvoi | 0,0009 | |
Selite: NE = ei arvioitavissa (not estimable). a Toinen välianalyysi (tiedonkeruu katkaistu: 1.5.2024), jossa seurannan keston mediaani oli 33,6 kuukautta. | ||
Kuva 1. Päivitetyn etenemättömyysajan tulosten Kaplan–Meierin käyrä; hoitoaikeen mukainen analyysitietue (MMY3002-tutkimus)
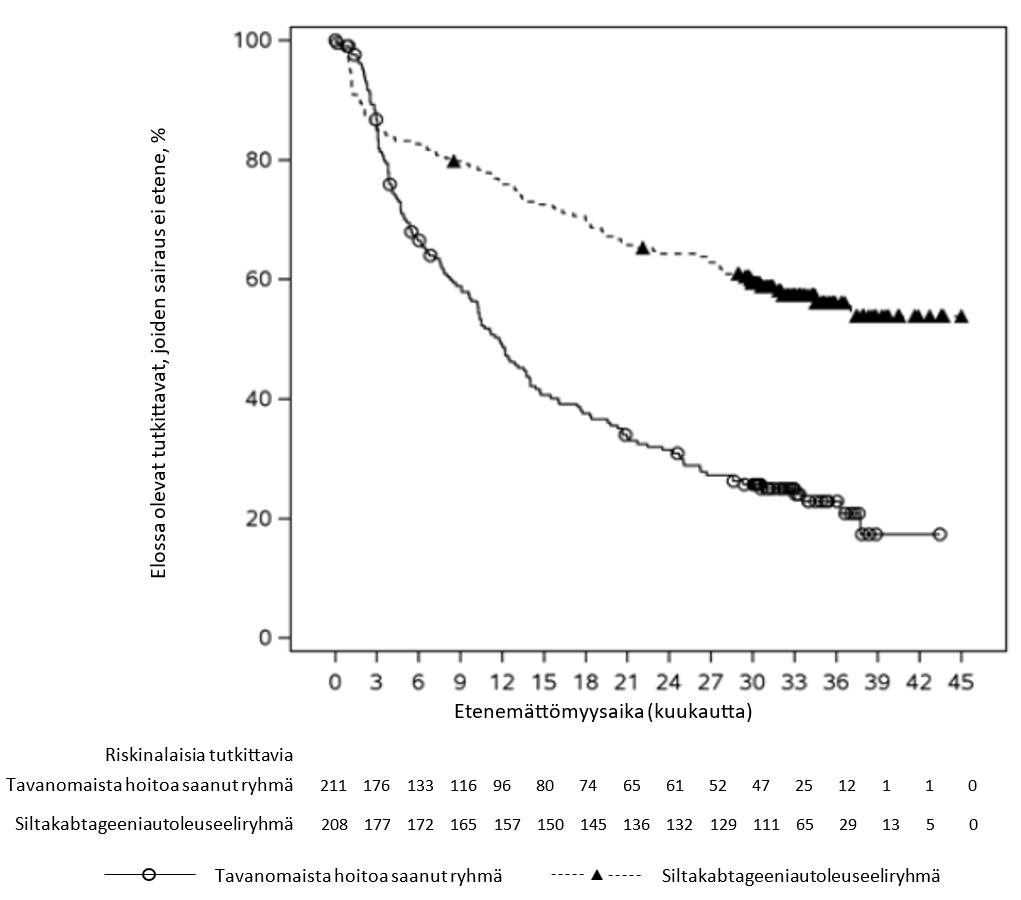
Huom.: Etenemättömyysaika perustuu toiseen välianalyysiin, jossa seurannan keston mediaani oli 33,6 kuukautta. Hoitoaikeen mukainen analyysitietue koostuu tutkimuksessa satunnaistetuista tutkittavista.
Selite: tavanomaista hoitoa saanut ryhmä = PVd tai DPd; siltakabtageeniautoleuseeliryhmä = afereesisekvenssi, siltahoito (PVd tai DPd), esihoito (syklofosfamidi ja fludarabiini) ja siltakabtageeniautoleuseeli-infuusio.
Selite: PVd = pomalidomidi-bortetsomibi-deksametasoni; DPd = daratumumabi-pomalidomidi-deksametasoni.
Kuva 2. Päivitettyjen kokonaiselossaolon tulosten Kaplan–Meierin käyrä; hoitoaikeen mukainen analyysitietue (MMY3002-tutkimus)
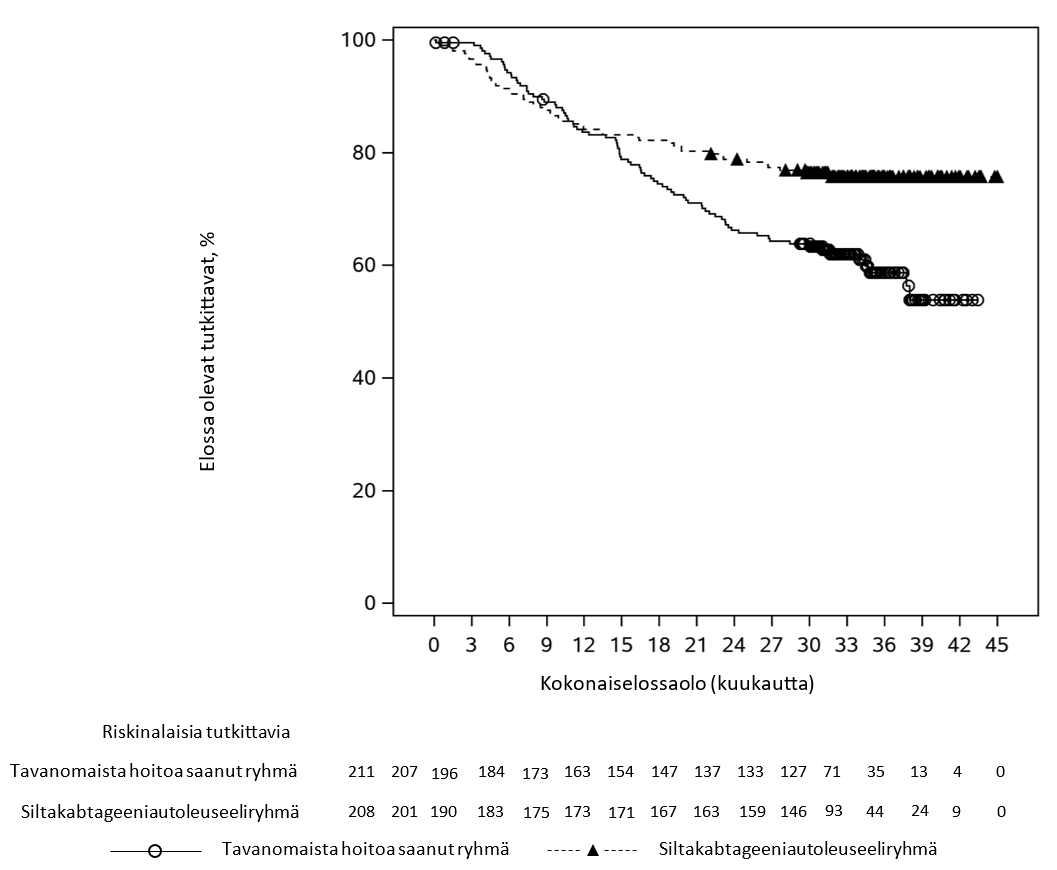
Huom.: Kokonaiselossaolo perustuu toiseen välianalyysiin, jossa seurannan keston mediaani oli 33,6 kuukautta. Hoitoaikeen mukainen analyysitietue koostuu tutkimuksessa satunnaistetuista tutkittavista.
Selite: tavanomaista hoitoa saanut ryhmä = PVd tai DPd; siltakabtageeniautoleuseeliryhmä = afereesisekvenssi, siltahoito (PVd tai DPd), esihoito (syklofosfamidi ja fludarabiini) ja siltakabtageeniautoleuseeli-infuusio.
Selite: PVd = pomalidomidi-bortetsomibi-deksametasoni; DPd = daratumumabi-pomalidomidi-deksametasoni.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset CARVYKTI-valmisteen käytöstä multippelin myelooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
CARVYKTI-valmisteen farmakokinetiikkaa arvioitiin MMY2001-tutkimuksessa 97:llä uusiutunutta tai hoitoon reagoimatonta multippelia myeloomaa sairastavalla aikuisella potilaalla, jotka saivat CARVYKTI-kertainfuusion annoksena (mediaani) 0,71 × 106 CAR-positiivista elinkykyistä T-solua/kg (vaihteluväli: 0,51–0,95 × 106 solua/kg).
CARVYKTI-kertainfuusion antamista seurasi aluksi ekspansiovaihe ja sen jälkeen nopea väheneminen ja sitten hitaampi väheneminen. Yksilöiden välillä havaittiin kuitenkin suurta vaihtelua.
| Taulukko 9. CARVYKTI-valmisteen farmakokineettiset parametrit multippelia myeloomaa sairastavilla potilailla | ||
| Parametri | Tilastollinen tiivistelmä | n = 97 |
| Cmax (kopiota/mikrog genomista DNA:ta) | Keskiarvo (keskihajonta), n | 48 692 (27 174), 97 |
| tmax (vrk) | Mediaani (vaihteluväli), n | 12,71 (8,73–329,77), 97 |
| AUC0-28d (kopiota*vrk/mikrog genomista DNA:ta) | Keskiarvo (keskihajonta), n | 504 496 (385 380), 97 |
| AUC0-last (kopiota*vrk/mikrog genomista DNA:ta) | Keskiarvo (keskihajonta), n | 1 098 030 (1 387 010), 97 |
| AUC0- 6m (kopiota*vrk/mikrog genomista DNA:ta) | Keskiarvo (keskihajonta), n | 1 033 373 (1 355 394), 96 |
| t1/2 (vrk) | Keskiarvo (keskihajonta), n | 23,5 (24,2), 42 |
| tlast (vrk) | Mediaani (vaihteluväli), n | 125,90 (20,04–702,12), 97 |
Kaikilla potilailla havaittiin solujen ekspansion jälkeen CARVYKTI-valmisteen elimistössä pysymisjakso. Analyysiajankohtana (n = 65) ääreisveren CAR-transgeenipitoisuuden palautumiseen annosta edeltävälle lähtötilanteen tasolle kulunut aika (mediaani) oli noin 100 päivää (vaihteluväli: 28–365 päivää) infuusion jälkeen. CARVYKTI-valmisteen farmakokinetiikkaa arvioitiin MMY3002-tutkimuksessa 176:lla lenalidomidiin reagoimatonta multippelia myeloomaa sairastavalla aikuisella potilaalla, ja arviot olivat yleisesti yhdenmukaiset MMY2001-tutkimuksen arvioiden kanssa.
Luuytimessä havaittavissa oleva CARVYKTI-altistus osoittaa, että CARVYKTI jakautuu systeemisestä verenkierrosta luuytimeen. Veren transgeenipitoisuuden tavoin myös luuytimen transgeenipitoisuus pieneni ajan mittaan ja siinä oli suurta vaihtelua yksilöiden välillä.
Erityisryhmät
Ikä (vaihteluväli: 27–78 vuotta) ei vaikuttanut CARVYKTI-valmisteen farmakokinetiikkaan (Cmax ja AUC0-28d) < 65-vuotiaat (n = 215; 64,8 %), 65–75-vuotiaat (n = 105; 31,6 %) ja > 75-vuotiaat (n = 12; 3,6 %) potilaat mukaan lukien.
Myöskään sukupuoli, paino ja etninen tausta eivät vaikuttaneet CARVYKTI-valmisteen farmakokinetiikkaan (Cmax ja AUC0-28d).
Munuaisten vajaatoiminta
CARVYKTI-valmisteella ei ole tehty munuaisten vajaatoimintaa koskevia tutkimuksia. Potilailla, joilla oli lievä munuaisten toimintahäiriö (60 ml/min ≤ kreatiniinipuhdistuma [CrCl] < 90 ml/min) tai keskivaikea munuaisten toimintahäiriö (30 ml/min ≤ kreatiniinipuhdistuma [CrCl] < 60 ml/min), CARVYKTI-valmisteen Cmax ja AUC0-28d olivat samankaltaiset kuin potilailla, joiden munuaisten toiminta oli normaali (CrCl ≥ 90 ml/min).
Maksan vajaatoiminta
CARVYKTI-valmisteella ei ole tehty maksan vajaatoimintaa koskevia tutkimuksia. CARVYKTI-valmisteen Cmax ja AUC0-28d olivat samankaltaiset potilailla, joilla oli lievä maksan toimintahäiriö ([kokonaisbilirubiini ≤ normaaliarvojen yläraja eli ULN ja aspartaattiaminotransferaasi > ULN] tai [ULN < kokonaisbilirubiini ≤ 1,5 kertaa ULN]) ja joiden maksan toiminta oli normaali.
Prekliiniset tiedot turvallisuudesta
CARVYKTI muodostuu muokatuista ihmisen T-soluista, minkä vuoksi ei ole edustavia in vitro ‑määrityksiä, ex vivo ‑malleja eikä in vivo ‑malleja, joilla ihmisperäisen valmisteen toksikologisia ominaisuuksia voitaisiin selvittää tarkasti. Näin ollen lääkevalmisteen kehityksen aikana ei tehty perinteisiä toksikologisia tutkimuksia.
Karsinogeenisuus ja mutageenisuus
Genotoksisuus- tai karsinogeenisuustutkimuksia ei ole tehty.
CARVYKTI-valmisteen valmistuksenaikaista insertiomutageneesin riskiä autologisten ihmisen T‑solujen integroituvaan lentivirusvektoriin transduktion jälkeen selvitettiin arvioimalla vektorin integroitumista CARVYKTI-valmisteessa ennen infuusiota. Tämä genomin insertiokohdan analyysi tehtiin 6 multippelia myeloomaa sairastavan potilaan 7 näytteestä ja 3 terveen luovuttajan 3 näytteestä valmistetuista CARVYKTI-valmisteista. Huolenaiheena olleiden geenien läheisyydessä ei havaittu preferentiaalista integraatiota.
Lisääntymistoksikologia
CARVYKTI-valmisteella ei ole tehty lisääntymis- ja kehitystoksisuutta koskevia eläinkokeita.
CARVYKTI-valmisteen vaikutuksista hedelmällisyyteen ei ole tehty tutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Cryostor CS5 (sisältää dimetyylisulfoksidia)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
9 kuukautta
Sulatettu valmiste: enintään 2,5 tuntia huoneenlämmössä (20–25 °C). CARVYKTI-infuusio on annettava heti sulatuksen jälkeen, ja infuusio pitää saattaa loppuun 2,5 tunnin kuluessa sulattamisesta.
Säilytys
CARVYKTI-valmistetta on säilytettävä ja kuljetettava nestetypen höyryfaasissa (≤ −120 °C), ja valmisteen on pysyttävä jäätyneenä, kunnes potilas on valmis hoitoon. Näin varmistetaan, että potilaalle on käytettävissä elinkykyisiä soluja.
Sulatettua lääkevalmistetta ei saa ravistaa, pakastaa uudelleen eikä säilyttää jääkaapissa.
Pidä infuusiopussi alumiinisessa pakastussäiliössä.
Sulatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
Eteenivinyyliasetaatti (EVA) ‑infuusiopussi, jossa sinetöity lisäysletku ja käytettävissä kaksi neulaporttia; infuusiopussit sisältävät joko 30 ml (50 ml:n pussi) tai 70 ml (250 ml:n pussi) soludispersiota.
Jokainen infuusiopussi on pakattu alumiiniseen pakastussäiliöön.
Valmisteen kuvaus:
Väritön tai valkoinen (käsittää myös valkoisen, keltaisen ja pinkin vivahteet) dispersio.
Käyttö- ja käsittelyohjeet
CARVYKTI-valmistetta ei saa sädettää, sillä sädetys voi inaktivoida lääkevalmisteen.
Ennen lääkevalmisteen käsittelyä tai antamista tehtävät varotoimet
CARVYKTI on kuljetettava hoitopaikan sisällä suljetuissa, hajoamattomissa ja vuotamattomissa pakkauksissa.
Tämä lääkevalmiste sisältää ihmisen verisoluja. CARVYKTI-valmistetta käsittelevien terveydenhuollon ammattilaisten on noudatettava asianmukaisia varotoimia (käytettävä käsineitä, suojavaatetusta ja silmiensuojaimia), jotta vältetään infektiotautien mahdollinen tarttuminen.
CARVYKTI on pidettävä ≤ −120 °C:ssa koko ajan siihen saakka, kunnes pussin sisältö sulatetaan infuusiota varten.
Valmistelu ennen antamista
CARVYKTI-valmisteen sulatus ja infuusio pitää koordinoida. Infuusion antoajankohta pitää varmistaa etukäteen, ja sulatuksen aloittamisaika on ajoitettava siten, että CARVYKTI on saatavissa infuusiota varten, kun potilas on valmiina. Lääkevalmiste pitää antaa välittömästi sulatuksen jälkeen, ja infuusio pitää saattaa loppuun 2,5 tunnin kuluessa sulatuksesta.
- Ennen CARVYKTI-valmisteen valmistelua on varmistettava, että potilaan henkilöllisyys vastaa CARVYKTI-pakastussäiliöön ja eräselosteeseen merkittyjä potilaan tunnistetietoja. CARVYKTI-infuusiopussia ei pidä ottaa pakastussäiliöstä, jos sen potilaskohtaiset tiedot eivät vastaa hoidettavan potilaan tietoja.
- Kun potilaan henkilöllisyys on varmistettu, CARVYKTI-infuusiopussi otetaan pakastussäiliöstä.
- Infuusiopussi on ennen sulatusta ja sulatuksen jälkeen tarkistettava murtumien ja halkeamien varalta. Jos pussi ei ole täysin ehjä, älä käytä valmistetta, vaan ota yhteyttä Janssen Cilag International NV:hen.
Sulatus
- Ennen sulatusta infuusiopussi asetetaan suljettavaan muovipussiin.
- Sulata CARVYKTI 37 °C ± 2 °C:ssa joko vesihauteessa tai kuivasulatuslaitteessa, kunnes infuusiopussissa ei näy enää jäätä. Sulatuksen kokonaiskesto (sulatuksen aloittamisesta sulatuksen loppuun) saa olla enintään 15 minuuttia.
- Infuusiopussi poistetaan suljettavasta muovipussista ja pyyhitään kuivaksi. Infuusiopussin sisältöä sekoitetaan varovasti paakkuuntuneen soluaineksen hajottamiseksi. Jos valmisteessa näkyy solupaakkuuntumia, infuusiopussin sisällön varovaista sekoittamista jatketaan. Pienten soluainespaakkujen pitäisi hajota varovaisella manuaalisella sekoituksella. CARVYKTI-valmistetta ei saa esisuodattaa toiseen astiaan, pestä, sentrifugoida eikä suspendoida muuhun elatusaineeseen ennen infuusiota.
- Sulatettu lääkevalmiste ei saa jäätyä uudelleen eikä sitä saa säilyttää kylmässä.
Antaminen
- CARVYKTI on tarkoitettu vain autologiseen kertakäyttöön.
- Ennen infuusiota ja toipumisjakson aikana on varmistettava, että saatavilla on tosilitsumabia sekä ensiapuvälineet.
- Varmista, että potilaan henkilöllisyys vastaa CARVYKTI-infuusiopussiin ja eräselosteeseen merkittyjä potilaan tunnistetietoja. Älä infusoi CARVYKTI-valmistetta, jos valmisteen potilaskohtaiset tiedot eivät vastaa hoidettavan potilaan tietoja.
- Sulatetun CARVYKTI-infuusiopussin koko sisältö pitää antaa huoneenlämmössä (20–25 °C) infuusiona laskimoon 2,5 tunnin kuluessa sulatuksesta infuusiolaitteistolla, jossa on kiinteä (in-line) suodatin. Infuusion kesto on tavallisesti alle 60 minuuttia.
- Leukosyyttejä poistavaa suodatinta EI SAA käyttää.
- Sekoita pussin sisältöä varovasti CARVYKTI-infuusion aikana solupaakkujen hajottamiseksi.
- Kun pussin koko sisältö on infusoitu, huuhtele infuusioletku ja kiinteä (in-line) suodatin 9 mg/ml (0,9 %) natriumkloridi-injektionesteellä. Näin varmistetaan, että kaikki lääkevalmiste on annettu.
Lääkevalmisteen hävittämiseen liittyvät varotoimet
Käyttämätön lääkevalmiste ja kaikki materiaalit, jotka ovat olleet kosketuksissa CARVYKTI-valmisteen kanssa (kiinteä ja nestemäinen jäte), on käsiteltävä, ja se on hävitettävä mahdollisesti tartuntavaarallisena jätteenä ihmisperäisen materiaalin käsittelyä koskevien paikallisten ohjeiden mukaisesti.
Toimenpiteet vahinkoaltistumisen tapahtuessa
Vahinkoaltistumisen tapahduttua on noudatettava ihmisperäisen materiaalin käsittelyä koskevia paikallisia ohjeita. Työtasot ja materiaalit, jotka ovat mahdollisesti joutuneet kosketuksiin CARVYKTI-valmisteen kanssa, on dekontaminoitava asianmukaisella desinfiointiaineella.
ATC-koodi
L01XL05
Valmisteyhteenvedon muuttamispäivämäärä
28.05.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com