
ORSERDU tabletti, kalvopäällysteinen 86 mg, 345 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
ORSERDU 86 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää elasestranttidihydrokloridia vastaten 86,3 mg elasestranttia.
ORSERDU 345 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää elasestranttidihydrokloridia vastaten 345 mg elasestranttia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti
Kliiniset tiedot
Käyttöaiheet
ORSERDU-monoterapia on tarkoitettu postmenopausaalisten naisten ja sellaisten miesten hoitoon, joilla on estrogeenireseptori (ER)‑positiivinen, HER2-negatiivinen, paikallisesti edennyt tai metastasoitunut rintasyöpä, jossa on aktivoiva ESR1-mutaatio, ja joiden tauti on edennyt vähintään yhden endokriinihoitolinjan jälkeen, mukaan lukien CDK 4/6:n estäjä.
Ehto
Ainoastaan syöpälääkkeiden antoon perehtyneen lääkärin tulee aloittaa hoito.
Annostus ja antotapa
ORSERDU-hoidon saa aloittaa vain lääkäri, jolla on kokemusta syöpälääkkeiden käytöstä.
Potilaat, joilla on ER-positiivinen, HER2-negatiivinen pitkälle edennyt rintasyöpä, on valittava ORSERDU-hoitoon perustuen aktivoivan ESR1-mutaation läsnäoloon plasmanäytteissä, käyttäen CE-merkinnän saanutta in vitro -diagnostiikkaa (IVD), jolla on vastaava käyttötarkoitus. Jos CE-merkinnän saanut IVD ei ole saatavilla, aktivoivan ESR1-mutaation läsnäolo plasmanäytteissä on arvioitava vaihtoehtoisellavalidoidulla testillä.
Annostus
Suositeltu annos on 345 mg (yksi 345 mg kalvopäällysteinen tabletti) kerran vuorokaudessa.
ORSERDU-valmisteen suositeltu enimmäisannos on 345 mg vuorokaudessa.
Hoitoa on jatkettava niin kauan kuin kliinistä hyötyä havaitaan tai kunnes ei-hyväksyttävää toksisuutta ilmenee.
Väliin jäänyt annos
Jos annos jää väliin, se voidaan ottaa heti 6 tunnin kuluessa tavanomaisesta annoksen ottoajasta. Yli 6 tunnin jälkeen annos tulee jättää väliin siltä päivältä. Seuraavana päivänä ORSERDU tulee ottaa tavanomaiseen aikaan.
Oksentelu
Jos potilas oksentaa ORSERDU-annoksen ottamisen jälkeen, potilaan ei pidä ottaa ylimääräistä annosta kyseisenä päivänä ja hänen pitää jatkaa tavanomaisen annosteluaikataulun mukaisesti seuraavana päivänä tavanomaiseen aikaan.
Annoksen muuttaminen
Suositellut elasestrantin annoksen muutokset potilaille, joilla esiintyy haittavaikutuksia (ks. kohta Haittavaikutukset), on annettu taulukoissa 1 ja 2:
Taulukko 1: ORSERDU-valmisteen annoksen pienentäminen haittavaikutusten ilmetessä
ORSERDU-annostaso | Annos ja aikataulu | Tablettien lukumäärä ja vahvuus |
Annoksen pienentäminen | 258 mg kerran vuorokaudessa | Kolme 86 mg:n tablettia |
Jos annosta on vielä tarpeen pienentää alle 258 mg kerran vuorokaudessa, keskeytä ORSERDU-valmisteen käyttö.
Taulukko 2: ORSERDU-valmisteen muuttamista koskevat ohjeet haittavaikutusten ilmetessä
Vaikeusaste | Annoksen muuttaminen |
Aste 2 | Harkitse ORSERDU-valmisteen käytön keskeytystä, kunnes potilaan haittavaikutuksen vaikeusaste palautuu arvoon ≤ 1 tai lähtötilanteeseen. Jatka sen jälkeen ORSERDU-valmisteen käyttöä samalla annostasolla. |
Aste 3 | Keskeytä ORSERDU-valmisteen käyttö, kunnes potilaan haittavaikutuksen vaikeusaste palautuu arvoon ≤ 1 tai lähtötilanteeseen. Annosta pitää pienentää 258 mg:aan hoitoa jatkettaessa. Mikäli asteen 3 toksisuus palaa, keskeytä ORSERDU-valmisteen käyttö, kunnes potilaan haittavaikutuksen vaikeusaste palautuu arvoon ≤ 1 tai lähtötilanteeseen. Pienennettyä 258 mg:n annosta voidaan jatkaa hoitavan lääkärin harkinnan mukaisesti, jos potilas hyötyy hoidosta. Jos asteen 3 tai sietämätön haittavaikutus toistuu, lopeta ORSERDU-valmisteen käyttö pysyvästi. |
Aste 4 | Keskeytä ORSERDU-valmisteen käyttö, kunnes potilaan haittavaikutuksen vaikeusaste palautuu arvoon ≤ 1 tai lähtötilanteeseen. Annosta pitää pienentää 258 mg:aan verran hoitoa jatkettaessa. Jos asteen 4 tai sietämätön haittavaikutus ilmenee jälleen, lopeta ORSERDU-valmisteen käyttö pysyvästi. |
ORSERDU-valmisteen käyttö CYP3A4:n estäjien kanssa
Voimakkaiden tai kohtalaisten CYP3A4:n estäjien samanaikaista käyttöä on vältettävä ja on harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vain vähän potentiaalia estää CYP3A4:ää.
Jos voimakasta CYP3A4:n estäjää täytyy käyttää, elasestrantin annos pitää pienentää 86 mg:aan kerran vuorokaudessa, ja siedettävyyttä on tarkkailtava huolellisesti. Jos kohtalaista CYP3A4:n estäjää täytyy käyttää, elasestrantin annos pitää pienentää 172 mg:aan kerran vuorokaudessa, ja siedettävyyttä on tarkkailtava huolellisesti. Myöhemmin voidaan harkita annoksen pienentämistä 86 mg:aan kerran vuorokaudessa kohtalaisten CYP3A4:n estäjien siedettävyyden perusteella.
Jos CYP3A4:n estäjän käyttö keskeytetään, elasestrantin annosta on suurennettava annokseen, jota käytettiin ennen CYP3A4:n estäjän käytön aloittamista (CYP3A4:n estäjän 5 puoliintumisajan jälkeen) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka).
Annosta ei tarvitse muuttaa, jos ORSERDU-valmistetta annetaan samanaikaisesti heikkojen CYP3A4:n estäjien kanssa (ks. kohta Yhteisvaikutukset).
ORSERDU-valmisteen käyttö CYP3A4:n induktorien kanssa
Voimakkaiden tai kohtalaisten CYP3A4:n induktorien samanaikaista käyttöä on vältettävä ja harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vähän potentiaalia indusoida CYP3A4:ää.
Jos voimakasta tai kohtalaista CYP3A4:n induktoria on käytettävä lyhyen ajan (ts. ≤ 3 vuorokautta) tai ajoittaisesti (ts. hoitojaksojen pituus on ≤ 3 vuorokautta ja näiden välillä on vähintään 2 viikkoa, tai 1 viikko + 5 CYP3A4:n induktorin puoliintumisaikaa, kumpi tahansa on pitempi), jatka elasestrantin käyttöä suurentamatta annosta.
Annosta ei tarvitse muuttaa, jos ORSERDU-valmistetta annetaan samanaikaisesti heikkojen CYP3A4:n induktorien kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka).
Erityiset potilasryhmät
Iäkkäät
Annoksen mukauttaminen potilaan iän perusteella ei ole tarpeen. 75-vuotiaita tai sitä vanhempia potilaita koskevia tietoja on vain vähän (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole suositeltavaa lievää maksan vajaatoimintaa sairastaville potilaille (Child-Pugh A). ORSERDU-annos on pienennettävä 258 mg:aan, jos potilaalla on kohtalainen maksan vajaatoiminta (Child-Pugh B). Elasestranttia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh C), joten annostusohjeita ei voida antaa vaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa munuaisten vajaatoimintaa sairastaville potilaille. Elasestranttia ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla, joten annossuosituksia ei voida antaa vaikeaa munuaisten vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
ORSERDU-valmisteen turvallisuutta ja tehoa alle 18-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
ORSERDU annetaan suun kautta.
Tabletit tulee niellä kokonaisina. Niitä ei saa pureskella, murskata tai jakaa ennen nielemistä. Potilaiden on otettava ORSERDU-annoksensa suunnilleen samaan aikaan joka päivä. ORSERDU pitää ottaa kevyen aterian yhteydessä. Annostelu ruoan kanssa voi myös vähentää pahoinvointia ja oksentelua (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Maksan vajaatoiminta
ORSERDU-valmiste metaboloituu maksan toiminnan tuloksena ja maksan vajaatoiminta voi suurentaa haittavaikutusten riskiä. Siksi ORSERDU-valmistetta pitää käyttää varovasti potilaille, joilla on maksan vajaatoiminta, ja potilaita on seurattava säännöllisesti ja huolellisesti haittavaikutusten varalta. Elasestrantin annostelu on aloitettava varovasti, 258 mg:n annoksella kerran vuorokaudessa potilaille, joilla on kohtalainen maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Koska kliinisiä tietoja ei ole, elasestranttia ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (Child-Pugh C) (ks. kohta Annostus ja antotapa).
Samanaikainen käyttö CYP3A4:n estäjien kanssa
ORSERDU-valmisteen samanaikaista annostelua voimakkaiden CYP3A4:n estäjien kanssa, kuten klaritromysiini, indinaviiri, itrakonatsoli, ketokonatsoli, lopinaviiri/ritonaviiri, nefatsodoni, nelfinaviiri, posakonatsoli, sakinaviiri, telapreviiri, telitromysiini, vorikonatsoli, sekä greippien tai greippimehun kanssa on vältettävä. On harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vähän potentiaalia estää CYP3A4:ää. Jos voimakasta CYP3A4:n estäjää ei voida välttää, ORSERDU-annosta on muutettava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
ORSERDU-valmisteen samanaikaista annostelua kohtalaisten CYP3A4:n estäjien kanssa, kuten aprepitantti, siprofloksasiini, konivaptaani, kritsotinibi, siklosporiini, diltiatseemi, dronedaroni, erytromysiini, flukonatsoli, fluvoksamiini, greippimehu, imatinibi, isavukonatsoli, tofisopaami ja verapamiili, on vältettävä. On harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vähän potentiaalia estää CYP3A4:ää. Jos kohtalaista CYP3A4:n estäjää ei voida välttää, ORSERDU-annosta on muutettava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Samanaikainen käyttö CYP3A4:n induktorien kanssa
ORSERDU-valmisteen samanaikaista annostelua voimakkaiden CYP3A4:n induktorien kanssa, kuten fenytoiini, rifampisiini, karbamatsepiini ja mäkikuisma (Hypericum perforatum), on vältettävä. On harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vähän potentiaalia indusoida CYP3A4:ää. Jos voimakasta CYP3A4:n induktoria ei voida välttää, ORSERDU-annosta on muutettava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
ORSERDU-valmisteen samanaikaista antoa kohtalaisten CYP3A4:n induktorien kanssa, kuten bosentaani, senobamaatti, dabrafenibi, efavirentsi, etraviriini, lorlatinibi, fenobarbitaali, primidoni ja sotorasibi, on vältettävä. On harkittava vaihtoehtoista samanaikaista lääkevalmistetta, jolla ei ole tai on vähän potentiaalia indusoida CYP3A4:ää. Jos kohtalaista CYP3A4:n induktoria ei voida välttää, ORSERDU-annosta on muutettava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Tromboemboliset tapahtumat
Tromboembolisia tapahtumia havaitaan usein edennyttä rintasyöpää sairastavilla potilailla ja niitä on esiintynyt ORSERDU-valmisteella tehdyissä kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Tämä tulee ottaa huomioon määrättäessä ORSERDU-valmistetta riskipotilaille.
Yhteisvaikutukset
ORSERDU metaboloituu ensisijaisesti CYP3A4-entsyymin vaikutuksesta ja se on orgaanisia anioneja kuljettavan polypeptidin 2B1 (OATP2B1) substraatti. ORSERDU on P-glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) ulospumppausta tekevien kuljetusproteiinien estäjä.
Muiden lääkevalmisteiden vaikutus ORSERDU-valmisteeseen
CYP3A4:n estäjät
Voimakkaan CYP3A4:n estäjän, itrakonatsolin, samanaikainen anto (200 mg kerran vuorokaudessa 7 vuorokauden ajan) ORSERDU-valmisteen kanssa (172 mg kerran vuorokaudessa 7 vuorokauden ajan) suurensi terveillä tutkittavilla elasestrantin plasma-altistusta (AUCinf) 5,3-kertaisesti ja huippupitoisuutta (Cmax) 4,4-kertaisesti.
Fysiologiaan perustuvat farmakokinetiikan (PBPK) simulaatiot syöpäpotilailla viittasivat siihen, että useiden päivittäisten elasestrantin 345 mg:n ja itrakonatsolin 200 mg:n annosten anto samanaikaisesti saattaa suurentaa elasestrantin vakaan tilan AUC-arvoa 5,5-kertaisesti ja Cmax-arvoa 3,9-kertaisesti. Tämä saattaa suurentaa haittavaikutusten riskiä.
PBPK-simulaatiot syöpäpotilailla viittasivat siihen, että useiden päivittäisten elasestrantin 345 mg:n annosten samanaikainen anto kohtalaisten CYP3A4:n estäjien kanssa saattaa suurentaa elasestrantin vakaan tilan AUC-arvoa ja Cmax-arvoa (flukonatsoli 200 mg kerran vuorokaudessa suurensi AUC-arvoa 2,3-kertaisesti ja Cmax-arvoa 1,9-kertaisesti), ja erytromysiini (500 mg neljä kertaa vuorokaudessa) suurensi AUC-arvoa 3,9 ja Cmax-arvoa 3,0-kertaisesti. Tämä saattaa suurentaa haittavaikutusten riskiä.
CYP3A4:n induktorit
Voimakkaan CYP3A4:n induktorin, rifampisiinin, samanaikainen anto (600 mg kerran vuorokaudessa 7 vuorokauden ajan) 345 mg:n ORSERDU-kerta-annoksen kanssa alensi elasestrantin plasma-altistusta (AUCinf) 86 % ja huippupitoisuutta (Cmax) 73 % terveillä tutkittavilla. Tämä saattaa vähentää elasestrantin aktiivisuutta.
PBPK-simulaatiot syöpäpotilailla viittasivat siihen, että useiden päivittäisten elasestrantin 345 mg:n annosten samanaikainen anto rifampisiinin 600 mg:n annoksen kanssa saattaa alentaa elasestrantin vakaan tilan AUC-arvoa 84 % ja Cmax-arvoa 77 %. Tämä saattaa heikentää elasestrantin aktiivisuutta.
PBPK-simulaatiot syöpäpotilailla viittasivat siihen, että useiden päivittäisten elasestrantin 345 mg:n annosten samanaikainen anto kohtalaisen CYP3A4- induktorin, efavirentsin (600 mg) kanssa saattaa alentaa elasestrantin vakaan tilan AUC-arvoa 57 % ja Cmax-arvoa 52 %. Tämä saattaa heikentää elasestrantin aktiivisuutta.
OATP2B1:n estäjät
Elasestrantti on OATP2B1:n substraatti in vitro. Koska ei voi sulkea pois sitä mahdollisuutta, että samaan aikaan annettavat OATP2B1:n estäjät saattavat suurentaa altistumista elasestrantille, mikä saattaa suurentaa haittavaikutusten riskiä, suositellaan varovaisuutta, mikäli ORSERDU-valmistetta käytetään OATP2B1:n estäjien kanssa.
ORSERDU-valmisteen vaikutus muihin lääkevalmisteisiin
P-gp:n substraatit
ORSERDU-valmisteen (345 mg:n kerta-annos) samanaikainen anto digoksiinin (0,5 mg:n kerta-annos) kanssa suurensi digoksiinille altistumista 27 % (Cmax) ja AUC:tä 13 %. Digoksiinin antoa on seurattava ja annosta pienennettävä tarvittaessa.
ORSERDU-valmisteen samanaikainen anto muiden P‑gp:n substraattien kanssa saattaa suurentaa niiden pitoisuuksia, mikä saattaa lisätä P‑gp-substraatteihin liittyviä haittavaikutuksia. Samaan aikaan annettavien P‑gp:n substraattien annosta on pienennettävä niiden valmisteyhteenvedon mukaisesti.
BCRP:n substraatit
ORSERDU-valmisteen (345 mg:n kerta-annos) samanaikainen anto rosuvastatiinin (20 mg:n kerta-annos) kanssa suurensi rosuvastatiinille altistumista 45 % (Cmax) ja AUC:tä 23 %. Rosuvastatiinin antoa on seurattava ja sen annosta pienennettävä tarvittaessa.
ORSERDU-valmisteen samanaikainen anto muiden BCRP-substraattien kanssa saattaa suurentaa niiden pitoisuuksia, mikä voi lisätä BCRP-substraatteihin liittyviä haittavaikutuksia. Samaan aikaan annettavien BCRP-substraattien annosta on pienennettävä niiden valmisteyhteenvedon mukaisesti.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehillä ja naisilla
ORSERDU-valmistetta ei pidä käyttää raskauden aikana eikä naisille, jotka voivat tulla raskaaksi, mutta eivät käytä ehkäisyä. Elasestrantin vaikutusmekanismin ja eläimillä tehdyissä lisääntymistoksisuutta koskevissa tutkimuksissa havaittujen löydösten perusteella ORSERDU voi vahingoittaa sikiötä, kun sitä annetaan raskaana oleville naisille. Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä ORSERDU-hoidon aikana ja viikon ajan viimeisen annoksen jälkeen.
Raskaus
Elasestrantin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). ORSERDU-valmistetta ei pidä käyttää raskauden aikana eikä naisille, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä. Naisten, jotka voivat tulla raskaaksi, raskaustila on varmistettava ennen ORSERDU-hoidon aloittamista. Jos potilas tulee raskaaksi ORSERDU-hoidon aikana, on potilaalle kerrottava mahdollisesta sikiölle aiheutuvasta vaarasta ja raskauden keskeytymisriskistä.
Imetys
Ei tiedetä, erittyvätkö elasestrantti tai sen metaboliitit äidinmaitoon. Koska imetettävään lapseen kohdistuvat vakavat haittavaikutukset ovat mahdollisia, suositellaan että imettävät naiset eivät imetä ORSERDU-hoidon aikana eivätkä yhden viikon ajan viimeisen ORSERDU-annoksen jälkeen.
Hedelmällisyys
Eläimillä tehdyissä tutkimuksissa havaittujen löydösten (ks. kohta Prekliiniset tiedot turvallisuudesta) ja ORSERDU-valmisteen vaikutusmekanismin perusteella se saattaa heikentää hedelmällisessä iässä olevien naisten ja miesten hedelmällisyyttä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ORSERDU-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Koska joillakin elasestranttia käyttävillä potilailla on kuitenkin raportoitu uupumusta, voimattomuutta ja unettomuutta (ks. kohta Haittavaikutukset), näitä haittavaikutuksia havaitsevien potilaiden on noudatettava varovaisuutta ajaessaan tai käyttäessään koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät (≥ 10 %) haittavaikutukset ORSERDU-valmistetta käytettäessä olivat pahoinvointi, kohonneet triglyseridiarvot, kohonneet kolesteroliarvot, oksentelu, uupumus, dyspepsia, ripuli, alentuneet kalsiumarvot, selkäkipu, kohonneet kreatiniiniarvot, nivelkipu, alentuneet natriumarvot, ummetus, päänsärky, kuumat aallot, vatsakipu, anemia, alentuneet kaliumarvot, ja kohonneet alaniiniaminotransferaasiarvot. Elasestrantin yleisimmät asteen ≥ 3 (≥ 2 %) haittavaikutukset olivat pahoinvointi (2,7 %), kohonneet ASAT-arvot (2,7 %), kohonneet ALAT-arvot (2,3 %), anemia (2 %), selkäkipu (2 %) ja luukipu (2 %).
Vakavia haittavaikutuksia, joita raportoitiin ≥ 1 %:lla potilaista, olivat pahoinvointi, dyspnea ja tromboembolia (laskimon).
Haittavaikutuksia, jotka johtivat käytön keskeyttämiseen ≥ 1 %:lla potilaista, olivat mm. Pahoinvointi ja vähentynyt ruokahalu.
Annoksen pienentämiseen johtaneita haittavaikutuksia, pahoinvointi mukaan lukien, esiintyi ≥ 1 %:lla potilaista.
Haittavaikutuksia, jotka johtivat annostelun keskeyttämiseen ≥ 1 %:lla potilaista, olivat mm. Pahoinvointi, vatsakipu, kohonneet alaniiniaminotransferaasiarvot, oksentelu, ihottuma, luukipu, ruokahalun heikkeneminen, kohonneet aspartaattiaminotransferaasiarvot, ja ripuli.
Haittavaikutustaulukko
Alla olevassa luettelossa esitetyt haittavaikutukset kuvaavat altistumista elasestrantille 301 rintasyöpäpotilaalla kolmessa avoimessa tutkimuksessa (RAD1901-005, RAD1901-106 ja RAD1901-308), joissa potilaat saivat pelkkää elasestranttia 400 mg kerran vuorokaudessa. Haittavaikutusten esiintyvyydet perustuvat kaikista syistä johtuvien haittatapahtumien esiintyvyyteen, jotka todettiin potilailla, jotka altistuivat elasestrantille suositellulla annoksella kohdekäyttöaiheessa, kun taas muutokset laboratorioparametrien esiintyvyydessä perustuvat pahenemiseen lähtötilanteesta vähintään 1 asteella ja siirtymiin ≥ asteeseen 3. Hoidon keston mediaani oli 85 vuorokautta (vaihteluväli: 5 – 1 288)
Kliinisissä tutkimuksissa saadut haittavaikutusten esiintymistiheydet perustuvat kaikista syistä aiheutuviin haittatapahtumien esiintymistiheyteen, jossa osalla haittavaikutuksiin liittyvistä tapahtumista voi olla muita syitä kuin lääkkeellä, kuten sairaus, muu lääkitys tai siihen liittymättömät syyt.
Seuraavaa tapaa käytetään haittavaikutuksen (ADR) esiintyvyyden luokitteluun, ja se perustuu Council for International Organizations of Medical Sciences (CIOMS) -neuvoston suosituksiin: hyvin yleinen (≥1/10); yleinen (≥1/100, <1/10); melko harvinainen (≥1/1 000, < 1/100); harvinainen (≥1/10 000, <1/1 000); hyvin harvinainen (<1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3. Haittavaikutukset potilailla, joita hoidettiin elasestranttimonoterapialla 345 mg ja joilla oli metastasoitunut rintasyöpä
Elasestrantti N= 301 | ||
| Infektiot | Yleinen | Virtsatietulehdus |
| Veri ja imukudos | Hyvin yleinen | Anemia |
| Yleinen | Alentunut lymfosyyttimäärä | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Ruokahalun heikkeneminen |
| Psyykkiset häiriöt | Yleinen | Unettomuus |
| Hermosto | Hyvin yleinen | Päänsärky |
| Yleinen | Heitehuimaus, pyörtyminen | |
| Verisuonisto | Hyvin yleinen | Kuumat aallot* |
| Melko harvinainen | Tromboembolia (laskimon)* | |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Hengenahdistus, yskä* |
| Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi, oksentelu, ripuli, ummetus, vatsakipu*, dyspepsia* |
| Yleinen | Suutulehdus | |
| Maksa ja sappitiet | Melko harvinainen | Akuutti maksan vajaatoiminta |
| Iho ja ihonalainen kudos | Yleinen | Ihottuma* |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu, selkäkipu |
| Yleinen | Raajan kipu, muskuloskeletaalinen rintakipu*, luukipu | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus |
| Yleinen | Voimattomuus | |
| Tutkimukset | Hyvin yleinen | Kohonneet aspartaattiaminotransferaasiarvot, kohonneet triglyseridiarvot, kohonneet kolesteroliarvot, kohonneet alaniiniaminotransferaasiarvot, alentuneet kalsiumarvot, kohonneet kreatiniiniarvot, alentuneet natriumarvot, alentuneet kaliumarvot |
| Yleinen | Kohonneet veren alkalisen fosfataasin arvot | |
*Ilmaantuvuus edustaa samankaltaisten termien ryhmittelyä
Haittavaikutukset luetellaan elinjärjestelmäluokituksen ja alenevan yleisyyden perusteella.
Valikoitujen haittavaikutusten kuvaus
Pahoinvointi
Pahoinvointia raportoitiin 35 %:lla potilaista. Vaikeusasteen 3–4 pahoinvointia raportoitiin 2,5 %:lla potilaista. Pahoinvointi raportoitiin yleensä varhain, mediaaniaika ensimmäiseen ilmenemiseen oli 14 vuorokautta (vaihteluväli: 1–490 vuorokautta). Pahoinvointi ilmeni useammin ensimmäisen syklin aikana ja syklistä 2 eteenpäin, ja pahoinvoinnin ilmaantuvuus oli yleensä pienempi myöhemmissä sykleissä (eli ajan mittaan). Profylaktista hoitoa pahoinvointiin määrättiin 12 (5 %) tutkittavalle elasestranttihaarassa, ja 28 (11,8 %) sai antiemeettiä pahoinvoinnin hoitoon hoitojakson aikana.
Iäkkäät
RAD1901-308-tutkimuksessa elasestranttia saaneet 104 potilasta olivat ≥ 65-vuotiaita, ja 40 potilasta oli ≥ 75-vuotiaita. Ruoansulatuselimistön häiriöitä raportoitiin yleisemmin potilailla, jotka olivat ≥ 75-vuotiaita. Hoidon aikana ilmeneviä haittavaikutuksia seuratessaan hoitavan lääkärin on otettava huomioon potilaan ikä ja samanaikaiset sairaudet potilaalle mukautettuja hoitoja valittaessa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‐haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurin kliinisissä tutkimuksissa annettu ORSERDU-annos oli 1 000 mg vuorokaudessa. Haittavaikutukset, joita raportoitiin suositeltua annosta suuremmilla annoksilla, olivat yhdenmukaisia vakiintuneen turvallisuusprofiilin kanssa (ks. kohta Haittavaikutukset). Ruoansulatuselimistön häiriöiden (vatsakipu, pahoinvointi, dyspepsia ja oksentelu) esiintymistiheys ja vaikeusaste vaikuttivat olevan annosriippuvaisia. ORSERDU-valmisteen yliannostukselle ei ole tunnettua vastalääkettä. Potilaita pitää seurata huolellisesti ja yliannostuksen hoidon tulee koostua tukihoitotoimenpiteistä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Endokrinologiset lääkeaineet, antiestrogeenit, ATC-koodi: L02BA04
Vaikutusmekanismi
Elasestrantti, tetrahydronaftaleeniyhdiste, on voimakas, selektiivinen ja suun kautta vaikuttava estrogeenireseptori-α:n (ERα) antagonisti ja hajottaja.
Farmakodynaamiset vaikutukset
Elasestrantti estää estradiolista riippuvaista ja riippumatonta ERα-positiivisten rintasyöpäsolujen kasvua, mukaan lukien mallit, jotka sisältävät estrogeenireseptori 1 (ESR1) -geenimutaatioita. Elasestrantilla ilmeni voimakasta kasvainten kasvua estävää vaikutusta potilailta peräisin olevissa ksenograftimalleissa, jotka oli aikaisemmin altistettu useille endokriinihoidoille, ja jotka sisälsivät villityypin ESR1:n tai ESR1-geenimutaatioita ligandia sitovassa domeenissa.
Potilailla, joilla oli pitkälle edennyt ER+ rintasyöpä, ja joilla oli 2,5 (mediaani) aikaisempaa endokriinihoitolinjaa, ja joille annosteltiin elasestranttidihydrokloridia 400 mg (345 mg elasestranttia) vuorokaudessa, kasvaimen 16α-18F-fluori-17β-estradiolin (FES) sisäänotto väheni lähtötilanteesta päivään 14 88,7 % (mediaani). Tämä osoitti, että ER:n saatavuus oli heikentynyt ja että kasvaimen kasvu oli estynyt, mitattuna FES-PET/TT:llä potilailta, joita oli aiemmin hoidettu endokriinihoidolla.
Kliininen teho ja turvallisuus
ORSERDU-valmisteen teho ja turvallisuus potilailla, joilla oli pitkälle edennyt ER+/HER2- rintasyöpä, aikaisemman endokriinihoidon jälkeen yhdistettynä CDK4/6:n estäjään, arvioitiin RAD1901-308-tutkimuksessa. Tämä oli satunnaistettu, avoin, vaikuttavalla lääkkeellä kontrolloitu monikeskustutkimus, jossa vertailtiin ORSERDU-valmistetta käypään hoitoon (fulvestrantti potilaille, jotka saivat aiemmin aromataasin estäjiä metastasoituneeseen tautiin, tai aromataasin estäjiä potilaille, jotka saivat fulvestranttia metastasoituneeseen tautiin). Soveltuvia potilaita olivat postmenopausaaliset naiset ja sellaiset miehet, joiden tauti oli uusiutunut tai edennyt käytettäessä vähintään yhtä ja enintään kahta aikaisempaa endokriinihoitolinjaa. Kaikilla potilailla edellytettiin olevan Eastern Cooperative Oncology Group (ECOG) -toimintakykyluokka 0 tai 1, ja heillä oli arvioitavissa olevia leesioita käytettäessä Response Evaluation Criteria in Solid Tumors (RECIST) -versiota 1.1, ts. mitattavissa oleva tauti tai pelkästään luussa oleva tauti, jossa on arvioitavissa olevia leesioita. Aikaisemman endokriinihoidon oli täytynyt sisältää yhdistelmän CDK4/6:n estäjää käyttävän hoidon kanssa eikä enempää kuin yhden aikaisemman hoitolinjan, jossa käytettiin sytotoksista solunsalpaajahoitoa metastasoituneeseen rintasyöpään. Potilaiden edellytettiin olevan sopivia ehdokkaita endokriiniseen monoterapiaan. Potilaat, joilla ilmeni oireileva metastasoitunut sisäelinsairaus, potilaat, joilla oli samanaikainen sydänsairaus, ja potilaat, joilla oli vaikea maksan vajaatoiminta, suljettiin pois tutkimuksesta.
Yhteensä 478 potilasta satunnaistettiin 1:1 saamaan päivittäin suun kautta 400 mg elasestranttidihydrokloridia (345 mg elasestranttia) tai käypää hoitoa (239 sai elasestranttia ja 239 sai käypää hoitoa), tähän sisältyi 228 potilasta (47,7 %), joilla oli lähtötilanteessa ESR1-mutaatioita (115 potilasta sai elasestranttia ja 113 potilasta sai käypää hoitoa). Niiden 239 potilaan joukossa, jotka satunnaistettiin käypä hoito -haaraan, 166 sai fulvestranttia ja 73 sai aromataasin estäjää, johon sisältyi anastrotsoli, letrotsoli tai eksemestaani. Satunnaistaminen ositettiin ESR1-mutaatiostatuksen perusteella (ESR1-mut vs. ESR1-mut-nd [ESR1-mutaatioita ei havaittu]), aikaisempi hoito fulvestrantilla (kyllä tai ei), ja sisäelinmetastaasi (kyllä tai ei). ESR1-mutaatiostatus määritettiin verenkierrossa olevan kasvaimen deoksiribonukleiinihapon (ctDNA) perusteella käyttämällä Guardant360 CDx -määritystä, ja se rajoittui ESR1-missense-mutaatioihin ligandia sitovassa domeenissa (kodonien 310–547 välissä).
Potilaiden mediaani-ikä (ORSERDU vs. käypä hoito) lähtötilanteessa oli 63,0 vuotta (vaihteluväli 24–89) vs. 63,5 (vaihteluväli 32–83), ja 45,0 % oli yli 65-vuotiaita (43,5 vs. 46,4). Useimmat potilaat olivat naisia (97,5 % vs. 99,6 %) ja useimmat potilaat olivat valkoihoisia (88,4 % vs. 87,2 %), seuraavaksi yleisimpiä potilaita olivat aasialaiset (8,4 % vs. 8,2 %), mustaihoiset tai afroamerikkalaiset (2,6 % vs. 4,1 %) ja muu/tuntematon (0,5 % vs. 0,5 %). Lähtötilanteen ECOG-toimintakykyluokka oli 0 (59,8 % vs. 56,5 %), 1 (40,2 % vs. 43,1 %) tai > 1 (0 % vs. 0,4 %). Niiden potilaiden demografiset tiedot, joilla oli ESR1-mutatoituneita kasvaimia, edustivat yleisesti laajempaa tutkimuspopulaatiota. ORSERDU-altistumisen keston mediaani oli 2,8 kuukautta (vaihteluväli: 0,4–24,8).
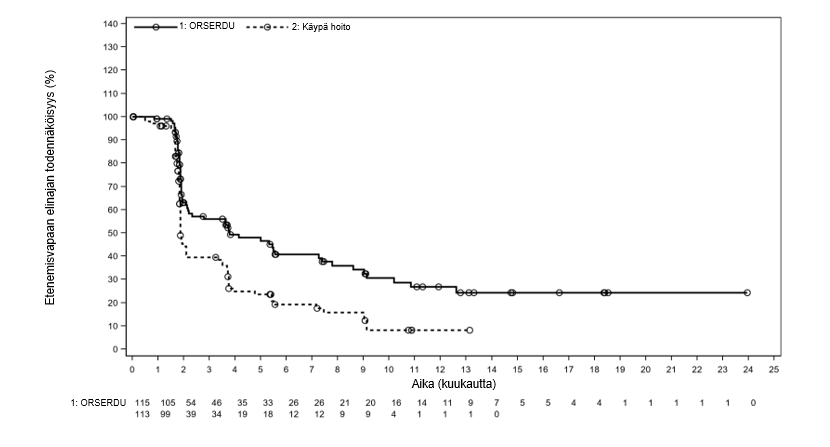
Ensisijainen tehon päätetapahtuma oli etenemisvapaa elinaika (PFS), jonka arvioi IRC (riippumaton arviointikomitea) kaikissa potilaissa ts. mukaan lukien potilaat, joilla oli ESR1-mutaatio, ja potilaissa, joilla oli ESR1-mutaatioita. Tilastollisesti merkitsevä PFS-hyöty havaittiin kaikilla potilailla, joiden PFS-mediaani oli 2,79 kuukautta Orserdu-haarassa verrattuna 1,91 kuukauteen käypä hoito -haarassa (HR=0,70, 95 % CI: 0,55, 0,88). Tehoa koskevat tulokset on esitetty taulukossa 4 ja kuvassa 1 potilaille, joilla oli ESR1-mutaatioita.
Taulukko 4: Tehoa koskevat tulokset potilailla, joilla oli ESR1-mutaatioita (sokkoutetun kuvantamisen arviointikomitean arvioima)
| ORSERDU | Käypä hoito | |
| Etenemisvapaa elinaika (PFS) | N = 115 | N = 113 |
| PFS-tapahtumien määrä, n (%) | 62 (53,9) | 78 (69,0) |
| PFS-kuukausien mediaani* (95 % CI) | 3,78 (2,17, 7,26) | 1,87 (1,87, 2,14) |
| Riskitiheyksien suhde** (95 % CI) | 0,546 (0,387, 0,768) | |
| p-arvo (ositettu log-rank) | 0,0005 | |
| Kokonaiselinaika (OS) | N = 115 | N = 113 |
| OS-tapahtumien määrä, n (%) | 61 (53) | 60 (53,1) |
| OS-kuukausien mediaani* (95 % CI) | 24,18 (20,53, 28,71) | 23,49 (15,64, 29,90) |
| Riskitiheyksien suhde** (95 % CI) | 0,903 (0,629, 1,298) | |
CI=luottamusväli; ESR1=estrogeenireseptori 1; PFS=etenemisvapaa elinaika.
*Kaplan-Meir-estimaatti; 95 % CI perustuu Brookmeyer-Crowleyn menetelmään käyttämällä lineaarista transformaatiota.
**Saatu Coxin suhteellisten riskitiheyksien mallista, joka on ositettu aikaisemman fulvestranttihoidon (kyllä tai ei) ja sisäelinmetastaasin perusteella (kyllä tai ei).
Tiedon keruun katkaisupäivät ovat 6. syyskuuta 2021 (PFS) ja 2. syyskuuta 2022 (OS).
Kuva 1: PFS potilailla, joilla oli ESR1-mutaatio (sokkoutetun kuvantamiskomitean arvioima)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset ORSERDU-valmisteen käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohta Annostus ja antotapa).
Farmakokinetiikka
Suun kautta annetun elasestrantin biologinen hyötyosuus on noin 10 %. Vakaa tila saavutetaan päivään 6 mennessä kerran vuorokaudessa tapahtuvan annostelun jälkeen. Cmax ja AUC suurenevat hieman enemmän kuin suhteessa annokseen, kun annokset ovat ≥ 50 mg (suolamuoto).
Imeytyminen
Suun kautta annettuna elasestrantti imeytyi nopeasti, Cmax saavutettiin 1–4 tunnin sisällä. Cmax:n geometrinen keskiarvo oli 52,86 ng/ml (35,2 % variaatiokerroin [CV %]) ja AUCinf oli 1 566 ng*h/ml (38,4 % CV) elasestrantin 345 mg:n kerta-annoksen ja ruokailun jälkeen. Vakaassa tilassa mediaanin [minimi, maksimi] plasmapitoisuuden 4 tunnin kohdalla annoksen jälkeen (C4h) ja AUC:n ennustetaan olevan 108 ng/ml [27,5 – 351] ja vastaavasti 2 190 ng*h/ml [461 – 8 470].
Ruokailun vaikutus
Elasestrantin 345 mg:n tabletin annostelu runsasrasvaisen, runsaskalorisen aterian kanssa suurensi Cmax- ja AUC-arvoja 40 %:lla ja vastaavasti 20 %:lla, paastonneille tutkittaville annettuun lääkeaineeseen verrattuna. Kun tabletti annettiin samaan aikaan kevyen aterian kanssa, Cmax ja AUC suurenivat samalla tavalla, eli Cmax 30 % ja AUC 20 %. Nieleminen ruoan kanssa saattaa vähentää ruoansulatuselimistöön kohdistuvia haittavaikutuksia.
Kuljettajaproteiini P-gp:n vaikutus elasestranttiin
Elasestrantti on P-gp:n substraatti. Kuljettaminen saturoituu 258 mg:n ja 345 mg:n annoksilla. Koska elasestrantin pienempien, 86 mg:n ja 172 mg:n annosten samanaikaisesta annosta P-gp:n estäjän kanssa ei ole saatavilla kliinisiä tietoja, ei voida sulkea pois, että samanaikainen anto P-gp:n estäjän kanssa saattaa lisätä imeytymistä pienempiä elasestranttiannoksia käytettäessä.
Jakautuminen
Elasestrantista > 99 % sitoutuu plasman proteiineihin eikä se riipu pitoisuudesta eikä maksan vajaatoimintastatuksesta. Elasestrantti läpäisee veri-aivoesteen annosriippuvaisella tavalla. Kun elasestranttia oli annettu kerran vuorokaudessa 7 peräkkäisen päivän ajan, elasestrantin mediaanipitoisuudet aivo-selkäydinnesteessä olivat 0,0966 ng/ml ja 0,155 ng/ml annoksilla 200 ja vastaavasti 500 mg.
Populaatiofarmakokineettisen analyysin perusteella elasestrantti jakautuu laajasti kudoksiin ja näennäinen perifeerinen jakautumistilavuus on 5 411 l. Elasestrantin näennäinen keskeinen jakautumistilavuus vakaassa tilassa on 422 l.
Biotransformaatio
Elasestrantti oli vähäinen (< 10 % plasman radioaktiivisuudesta) komponentti ihmisen plasmassa. 4-[2-(etyyliamino)etyyli]bentsoehappo (EAEBA) -glukuronidi oli merkittävä metaboliitti ihmisen plasmassa (noin 41 % plasman radioaktiivisuudesta). Elasestrantti metaboloituu pääasiassa CYP3A4:n vaikutuksesta ja mahdollisesti vähäisessä määrin myös CYP2A6:n ja CYP2C9:n myötävaikutuksesta.
Eliminaatio
Elasestrantin puoliintumisajan ennustetaan olevan noin 30 tuntia. Kerta-annoksen jälkeen elasestrantin keskimääräinen (% CV) puhdistuma oli 220,3 l/h (38,4 %). Vakaassa tilassa elasestrantin keskimääräisen (% CV) puhdistuman ennustetaan olevan 186 l/h (43,5 %).
Radioleimatun elasestrantin 345 mg:n oraalisen kerta-annoksen jälkeen 81,5 % (valtaosa muuttumatonta) löydettiin ulosteesta ja 7,53 % (vähäinen määrä muuttumatonta) virtsasta. Elasestrantin munuaispuhdistuma on hyvin vähäistä (≤ 2,3 ml/min), ja se eliminoitui oksidatiivisen metabolian ja ulosteeseen erittymisen kautta.
Erityiset potilasryhmät
Iän, painon ja sukupuolen vaikutus
Syöpäpotilaita koskevien populaatiofarmakokineettisten tietojen analyysien perusteella annosta ei tarvitse muuttaa painon, iän tai sukupuolen mukaan.
Maksan vajaatoiminta
Tutkittavien väliset Cmax- ja AUC-arvot olivat yhdenmukaiset maksan lievän vajaatoiminnan (Child-Pugh A) ja maksan normaalin toiminnan ryhmässä, kun elasestranttia oli annettu 176 mg:n kerta-annos. Merkitseviä nousuja todettiin AUC0–t (76 %) ja AUC0–∞ -arvoissa (83 %) maksan kohtalaisen vajaatoiminnan ryhmässä (Child-Pugh B) verrattuna maksan normaalin toiminnan ryhmään. Normaaliryhmän ja kohtalaisen vajaatoiminnan ryhmän Cmax-arvot olivat yhdenmukaisia.
Eliminaation puoliintumisajan (t1/2) geometrisella keskiarvolla oli taipumusta suurentua maksan vajaatoiminnan vaikeutumisen myötä. Elasestranttia ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (Child-Pugh C).
Elasestrantin PBPK-mallinnussimulaatiossa 345 mg:n annoksella vakaan tilan AUC- ja Cmax-arvojen ennustettiin nousevan 2,14- ja vastaavasti 1,92-kertaisiksi tutkittavilla, joilla on kohtalainen maksan vajaatoiminta, verrattuna potilaisiin, joilla maksan toiminta on normaali.
Prekliiniset tiedot turvallisuudesta
Elasestrantin akuutti toksisuus on vähäinen. Toksisuustutkimuksissa, joissa valmistetta annettiin toistuvina annoksina rotille ja apinoille, elasestrantin antiestrogeenivaikutus oli syynä havaittuihin vaikutuksiin, erityisesti naaraiden sukupuolielimissä, mutta myös muissa hormoneille herkissä elimissä, kuten maitorauhanen, aivolisäke ja kivekset. Apinoilla havaittiin ajoittaista oksentelua ja ripulia. Lisäksi pitkäaikaisissa tutkimuksissa (26 viikkoa rotilla ja 39 viikkoa jaavanmakakeilla) rotilla havaittiin rauhasia sisältämättömän mahan osan limakalvoepiteelin lisääntynyttä vakuolisoitumista, ja vakuolisoituneita makrofagi-infiltraatteja havaittiin ohutsuolessa sekä rotilla että apinoilla. Apinoilla tämä vaikutus ilmeni tasolla, jossa systeeminen altistuminen oli noin 70 % ihmisen altistumisesta.
Elasestrantilla ei todettu genotoksisia ominaisuuksia Amesin testissä eikä kromosomipoikkeavuuksia testeissä ihmisen lymfosyyteillä ja mikrotumamäärityksessä rotilla.
Hedelmällisyyttä koskevia eläinkokeita ei ole tehty. Toistuvien annosten aiheuttamaa toksisuutta koskevissa tutkimuksissa hedelmällisyyteen liittyviä vaikutuksia havaittiin naarasrottien ja -apinoiden sukuelimissä; nämä vaikutukset ilmenivät alle ihmisen altistumisen suurimmalla suositellulla annoksella (MRHD). Leydigin solujen vähentynyttä solukkuutta rottien kiveksissä havaittiin myös altistumistasoilla, jotka olivat 2,7-kertaisia ihmisten altistumistasoihin verrattuna.
Rotilla suoritetuissa alkion ja sikiön kehittymistä koskevissa tutkimuksissa elasestrantin anto suun kautta aiheutti toksisuutta emolle (painon lasku, vähäinen ruoan kulutus, punaista eritettä vulvasta), ja enemmän resorptioita, implantaation jälkeisen sikiökuolleisuuden lisääntymistä, ja elävien sikiöiden pienempiä määriä ja sikiöiden muutoksia ja epämuodostumia annoksilla, jotka olivat pienempiä kuin ihmisen altistumiset suurimmalla suositellulla annoksella.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa [E460]
Silikonoitu mikrokiteinen selluloosa
Krospovidoni [E1202]
Magnesiumstearaatti [E470b]
Kolloidinen piidioksidi [E551]
Kalvopäällyste
Opadry II 85F105080 Blue sisältäen polyvinyylialkoholia [E1203], titaanidioksidia [E171], makrogolia [E1521], talkkia [E553b] ja briljanttisinistä FCF alumiinilakkaa [E133]
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ORSERDU tabletti, kalvopäällysteinen
86 mg (L:ei) 28 fol (1821,81 €)
345 mg (L:ei) 28 fol (6763,60 €)
PF-selosteen tieto
ORSERDU on pakattu alumiini-alumiini-läpipainopakkauksiin, jotka on pakattu pahvirasiaan.
ORSERDU 86 mg kalvopäällysteiset tabletit
Pakkaukset, joissa on 28 kalvopäällysteistä tablettia: 4 läpipainopakkausta, joissa kussakin on 7 tablettia
ORSERDU 345 mg kalvopäällysteiset tabletit
Pakkaukset, joissa on 28 kalvopäällysteistä tablettia: 4 läpipainopakkausta, joissa kussakin on 7 tablettia
Valmisteen kuvaus:
ORSERDU 86 mg kalvopäällysteiset tabletit
Sininen tai vaaleansininen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka yhdellä puolella on painanteena ME ja vastakkainen puoli on ilman merkintää. Likimääräinen halkaisija: 8,8 mm.
ORSERDU 345 mg kalvopäällysteiset tabletit
Sininen tai vaaleansininen, kaksoiskupera, soikea kalvopäällysteinen tabletti, jonka yhdellä puolella on painanteena MH ja vastakkainen puoli on ilman merkintää. Likimääräinen koko: 19,2 mm (pituus), 10,8 mm (leveys).
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ORSERDU tabletti, kalvopäällysteinen
86 mg 28 fol
345 mg 28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Elasestrantti: Postmenopausaalisten naisten sekä miesten hormonireseptoripositiivisen ja HER2-negatiivisen paikallisesti edenneen tai etäpesäkkeisen rintasyövän hoito monoterapiana erityisin edellytyksin (3105).
ATC-koodi
L02BA04
Valmisteyhteenvedon muuttamispäivämäärä
Yhteystiedot
Basisweg 10
1043 AP Amsterdam
Noord-Holland, Nederland
+45 2815 7816
www.menarini.com
srana@menarinistemline.com