
EBGLYSS injektioneste, liuos, esitäytetty kynä 250 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Ebglyss 250 mg injektioneste, liuos, esitäytetty kynä
Yksi kertakäyttöinen esitäytetty kynä sisältää 250 mg lebrikitsumabia 2 ml:ssa liuosta (125 mg/ml).
Lebrikitsumabi tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa (CHO).
Apuaine, jonka vaikutus tunnetaan
Yksi Ebglyss 250 mg injektioneste, liuos, esitäytetty kynä sisältää 0,6 mg polysorbaatti 20:tä (E 432).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Ebglyss on tarkoitettu keskivaikean tai vaikean atooppisen ihottuman hoitoon aikuisille ja vähintään 12-vuotiaille ja vähintään 40 kg painaville nuorille, joille voidaan antaa systeemistä hoitoa.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus ja antotapa
Hoidon saa aloittaa terveydenhuollon ammattilainen, jolla on kokemusta atooppisen ihottuman diagnosoinnista ja hoidosta.
Annostus
Lebrikitsumabin suositeltu annos on 500 mg (kaksi 250 mg:n injektiota) sekä hoitoviikolla 0 että hoitoviikolla 2, jonka jälkeen annetaan 250 mg ihon alle joka toinen viikko hoitoviikkoon 16 asti.
Jos potilaalle ei ole ilmennyt kliinistä vastetta 16 hoitoviikon jälkeen, on harkittava hoidon lopettamista. Joillakin potilailla, joilla on alkuvaiheessa osittainen vaste, tilanne saattaa kohentua edelleen, kun hoitoa jatketaan joka toinen viikko hoitoviikkoon 24 asti.
Kun kliininen vaste on saatu, lebrikitsumabin suositeltu ylläpitoannos on 250 mg neljän viikon välein.
Lebrikitsumabia voidaan käyttää paikallisesti käytettävien kortikosteroidien kanssa tai ilman niitä. Paikallisesti käytettäviä kalsineuriinin estäjiä voidaan käyttää, mutta niiden käyttö tulisi rajata ainoastaan ongelma-alueille, kuten kasvoille, kaulalle, taive- ja genitaalialueille.
Annoksen jääminen väliin
Jos annos jää väliin, se on annettava mahdollisimman pian. Tämän jälkeen annostusta on jatkettava normaalin hoitoaikataulun mukaisesti.
Erityispotilaat
Iäkkäät (≥ 65-vuotiaat)
Iäkkäiden potilaiden annoksen muuttamista ei suositella (ks. kohta Farmakokinetiikka).
Munuaisten ja maksan vajaatoiminta
Munuaisten tai maksan vajaatoimintaa sairastavien potilaiden annoksen muuttamista ei suositella (ks. kohta Farmakokinetiikka).
Paino
Annoksen muuttamista painon perusteella ei suositella (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Lebrikitsumabin turvallisuutta ja tehoa 6 kuukauden – alle 12 vuoden ikäisten lasten tai alle 40 kg:n painoisten 12–17-vuotiaiden nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Ihon alle.
Lebrikitsumabi annetaan injektiona ihon alle reiteen tai vatsaan, lukuun ottamatta navan ympärillä olevaa 5 cm:n aluetta. Jos joku muu antaa injektion, myös olkavartta voidaan käyttää.
Ensimmäistä 500 mg:n annosta varten annetaan kaksi 250 mg:n injektiota peräkkäin eri pistoskohtiin.
On suositeltavaa vaihdella pistoskohtaa jokaisen pistoksen yhteydessä. Lebrikitsumabia ei saa pistää ihoon, joka on arka tai vaurioitunut tai jossa on mustelmia tai arpia.
Potilas voi pistää lebrikitsumabin itse tai potilasta hoidossa avustava henkilö voi antaa lebrikitsumabi-injektion, jos hoitava terveydenhuollon ammattilainen katsoo sen asianmukaiseksi. Potilaille ja/tai hoidossa avustaville henkilöille on annettava asianmukainen opastus lebrikitsumabin antoon ennen käyttöä. Yksityiskohtaiset käyttöohjeet ovat pakkausselosteen lopussa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
Jos systeeminen yliherkkyysreaktio (välitön tai viivästynyt) ilmenee, lebrikitsumabin anto on lopetettava ja asianmukainen hoito on aloitettava.
Sidekalvotulehdus
Jos lebrikitsumabihoitoa saaneelle potilaalle kehittyy sidekalvotulehdus, joka ei häviä tavanomaisen hoidon jälkeen, potilaalle on tehtävä silmätutkimus (ks. kohta Haittavaikutukset).
Matoinfektio
Potilaat, joilla oli todettu matoinfektio, suljettiin pois kliinisiin tutkimuksiin osallistumisesta. Ei tiedetä, vaikuttaako lebrikitsumabi matoinfektioita vastaan syntyvään immuunivasteeseen estämällä IL-13:n signalointia.
Potilaan, jolla on todettu matoinfektio, on saatava siihen hoitoa ennen lebrikitsumabihoidon aloittamista. Jos potilas saa infektion saadessaan lebrikitsumabihoitoa eikä reagoi matolääkehoitoon, lebrikitsumabihoito on keskeytettävä siihen saakka, kunnes infektio on hävinnyt.
Rokotukset
Ennen lebrikitsumabihoidon aloittamista on suositeltavaa, että potilaiden kaikki iänmukaiset rokotukset saatetaan ajan tasalle voimassa olevien ohjeistojen mukaisesti. Lebrikitsumabin kanssa samanaikaisesti ei pidä antaa eläviä ja eläviä heikennettyjä taudinaiheuttajia sisältäviä rokotteita, koska kliinistä turvallisuutta ja tehoa ei ole varmistettu. Immuunivastetta rokotteille, jotka eivät sisällä eläviä taudinaiheuttajia, arvioitiin dtap-rokotteella (kurkkumätä, jäykkäkouristus ja soluton hinkuyskä) sekä meningokokkipolysakkaridirokotteella (ks. kohta Yhteisvaikutukset).
Apuaineet
Tämä lääkevalmiste sisältää 0,6 mg polysorbaatti 20:tä (E 432) per 250 mg:n esitäytetty kynä, joka vastaa 0,3 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Eläviä taudinaiheuttajia sisältävät rokotteet
Lebrikitsumabin ja eläviä tai eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden samanaikaisen käytön turvallisuutta ja tehoa ei ole tutkittu. Eläviä ja eläviä heikennettyjä taudinaiheuttajia sisältäviä rokotteita ei pidä antaa samanaikaisesti lebrikitsumabin kanssa.
Rokotteet, jotka eivät sisällä eläviä taudinaiheuttajia
Immuunivasteita rokotteille, jotka eivät sisällä eläviä taudinaiheuttajia, arvioitiin tutkimuksessa (ADopt-VA), jossa atooppista ihottumaa sairastavia aikuisia potilaita hoidettiin 500 mg:lla lebrikitsumabia hoitoviikoilla 0 ja 2, minkä jälkeen annettiin 250 mg lebrikitsumabia joka toinen viikko. 12 viikon lebrikitsumabihoidon jälkeen potilaat rokotettiin dtap-rokotteella (kurkkumätä, jäykkäkouristus ja soluton hinkuyskä; T-soluriippuvainen) sekä meningokokkipolysakkaridirokotteella (T-soluista riippumaton) ja immuunivasteet arvioitiin 4 viikkoa myöhemmin. Samanaikainen lebrikitsumabihoito ei vaikuttanut negatiivisesti kummankaan sellaisen rokotteen vasta-ainevasteisiin, joka ei sisällä eläviä taudinaiheuttajia. Tutkimuksessa rokotteiden, jotka eivät sisällä eläviä taudinaiheuttajia, ja lebrikitsumabin välillä ei havaittu haitallisia yhteisvaikutuksia. Näin ollen lebrikitsumabia saaville potilaille voidaan antaa samanaikaisesti inaktivoituja rokotteita tai rokotteita, jotka eivät sisällä eläviä taudinaiheuttajia. Lisätietoja eläviä taudinaiheuttajia sisältävistä rokotteista on kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Samanaikaiset hoidot
Koska lebrikitsumabi on monoklonaalinen vasta-aine, farmakokineettisiä yhteisvaikutuksia ei ole odotettavissa.
Raskaus ja imetys
Raskaus
On vain vähän tietoja lebrikitsumabin käytöstä raskaana oleville naisille. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi lebrikitsumabin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö lebrikitsumabi ihmisen rintamaitoon tai imeytyykö se systeemisesti nielemisen jälkeen. Äidin IgG:tä tiedetään esiintyvän rintamaidossa. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko lebrikitsumabihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläinkokeissa ei havaittu hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lebrikitsumabilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset ovat sidekalvotulehdus (6,9 %), injektiokohdan reaktiot (2,6 %), allerginen sidekalvotulehdus (1,8 %) ja kuivasilmäisyys (1,4 %).
Haittavaikutustaulukko
Lebrikitsumabia annettiin kaikissa atooppista ihottumaa koskevissa kliinisissä tutkimuksissa yhteensä 1 720 potilaalle. Näistä 891 potilasta altistui lebrikitsumabille vähintään vuoden ajan. Ellei toisin mainita, esiintyvyydet perustuvat yhdistettyihin tietoihin neljästä satunnaistetusta, kaksoissokkoutetusta tutkimuksesta, joissa oli mukana keskivaikeaa tai vaikeaa atooppista ihottumaa sairastavia potilaita ja jossa 783 potilasta hoidettiin ihon alle annetulla lebrikitsumabilla lumelääkekontrolloidun jakson aikana (ensimmäiset 16 hoitoviikkoa).
Taulukko 1 on lueteltu kliinisissä tutkimuksissa havaitut haittavaikutukset elinjärjestelmäluokittain ja esiintyvyyksittäin seuraavien kategorioiden mukaan: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Luettelo haittavaikutuksista
| MedDRA-elinjärjestelmäluokka | Esiintyvyys | Haittavaikutus |
| Infektiot | Yleinen | Sidekalvotulehdus |
| Melko harvinainen | Vyöruusu | |
| Veri ja imukudos | Melko harvinainen | Eosinofilia |
| Silmät | Yleinen | Allerginen sidekalvotulehdus Kuivasilmäisyys |
| Melko harvinainen | Keratiitti Silmäluomitulehdus | |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Injektiokohdan reaktio |
Valikoitujen haittavaikutusten kuvaus
Sidekalvotulehdus ja siihen liittyvät tapahtumat
Ensimmäisten 16 hoitoviikon aikana sidekalvotulehdusta, allergista sidekalvotulehdusta, silmäluomitulehdusta ja keratiittia raportoitiin yleisemmin lebrikitsumabilla hoidetuilla potilailla (sidekalvotulehdus 6,9 %, allerginen sidekalvotulehdus 1,8 %, silmäluomitulehdus 0,8 % ja keratiitti 0,6 %) verrattuna lumelääkkeeseen (sidekalvotulehdus 1,8 %, allerginen sidekalvotulehdus 0,7 %, silmäluomitulehdus 0,2 % ja keratiitti 0,3 %).
Ylläpitohoitojakson aikana (hoitoviikot 16–52) lebrikitsumabihoidon yhteydessä sidekalvotulehduksen ilmaantuvuus oli 5,0 % ja allergisen sidekalvotulehduksen ilmaantuvuus oli 5,9 %.
Kaikissa kliinisissä tutkimuksissa lebrikitsumabihoitoa saaneilla potilailla hoito keskeytyi sidekalvotulehduksen takia 0,7 %:lla ja allergisen sidekalvotulehduksen takia 0,3 %:lla tapauksista. Vaikea-asteisia sidekalvotulehduksen tapauksia ilmeni 0,1 %:lla potilaista ja vaikea-asteisia allergisen sidekalvotulehduksen tapauksia 0,2 %:lla potilaista. 72 % potilaista toipui, joista 57 % toipui 90 vuorokauden sisällä.
Eosinofilia
Lebrikitsumabilla hoidettujen potilaiden eosinofiilimäärä lisääntyi lähtötilanteesta keskimäärin enemmän kuin lumelääkettä saaneilla potilailla. Eosinofiilimäärä lisääntyi 20,3 %:lla lebrikitsumabilla hoidetuista potilaista verrattuna 11,7 %:lla lumelääkkeellä hoidetuista potilaista. Lebrikitsumabilla hoidettujen potilaiden eosinofiilimäärän lisääntyminen oli yleensä lievää tai kohtalaista ja ohimenevää. Eosinofiliaa (> 5 000 solua/mikrol) havaittiin 0,4 %:lla lebrikitsumabihoitoa saaneista potilaista eikä yhdelläkään lumelääkehoitoa saaneista potilaista. Eosinofiliaa raportoitiin haittavaikutuksena 0,6 %:lla lebrikitsumabihoitoa saaneista potilaista ja samanlaisella esiintyvyydellä lumelääkkeellä hoidetuista potilaista alkuvaiheen hoitojakson aikana. Eosinofilia ei johtanut hoidon lopettamiseen eikä eosinofiileihin liittyviä häiriöitä raportoitu.
Injektiokohdan reaktiot
Injektiokohdan reaktioita (mukaan lukien kipu ja punoitus) raportoitiin lebrikitsumabia saaneilla potilailla yleisemmin (2,6 %) kuin lumelääkettä saaneilla potilailla (1,5 %). Suurin osa (95 %) injektiokohdan reaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita, ja harvat potilaat (< 0,5 %) lopettivat lebrikitsumabihoidon.
Vyöruusu
Vyöruusua raportoitiin 0,6 %:lla lebrikitsumabihoitoa saaneista potilaista eikä yhdelläkään lumelääkeryhmän potilaista. Kaikki raportoidut vyöruusutapahtumat olivat vaikeusasteeltaan lieviä tai keskivaikeita, eikä mikään niistä johtanut hoidon pysyvään lopettamiseen.
Pitkäaikainen turvallisuus
Lebrikitsumabin turvallisuutta arvioitiin aikuisilla ja nuorilla, jotka olivat saaneet jatkuvaa hoitoa enintään kolmen vuoden ajan. ADjoin-jatkotutkimukseen otettiin mukaan yhteensä 1 153 potilasta joko suoraan tai ADvocate-1-, ADvocate-2- ja ADore-tutkimuksista, joissa altistus kesti enintään 1 vuoden, tai ADhere- ja Adopt-VA-tutkimuksista, joissa altistus kesti enintään 16 viikkoa. ADjoin-tutkimuksessa 771 potilasta sai lebrikitsumabijatkohoitoa 2 vuotta. Turvallisuusprofiili pysyi pidemmän lebrikitsumabialtistuksen yhteydessä yhdenmukaisena eikä uusia turvallisuushuolia havaittu.
Pediatriset potilaat
12–17-vuotiaat nuoret
Lebrikitsumabin turvallisuutta arvioitiin 372:lla 12–17-vuotiaalla potilaalla, joilla oli keskivaikea tai vaikea atooppinen ihottuma, mukaan lukien 270 potilasta, joiden altistus kesti vähintään vuoden ajan. Lebrikitsumabin turvallisuusprofiili näillä potilailla oli samankaltainen kuin turvallisuusprofiili atooppista ihottumaa sairastavilla aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ihmisille on annettu enintään 10 mg painokiloa kohti kerta-annoksena laskimoon ja useita enintään 500 mg:n annoksia ihon alle ilman annosta rajoittavaa toksisuutta. Lebrikitsumabiyliannostukseen ei ole spesifistä hoitoa. Yliannostuksen sattuessa potilasta on tarkkailtava haittavaikutusten oireiden tai löydösten varalta, ja asianmukainen oireenmukainen hoito on aloitettava välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut ihotautien lääkkeet, ihottumalääkkeet, lukuun ottamatta kortikosteroideja, ATC-koodi: D11AH10
Vaikutusmekanismi
Lebrikitsumabi on monoklonaalinen immunoglobuliini (IgG4) vasta-aine, joka sitoutuu suurella affiniteetilla interleukiini (IL) 13:een ja joka estää selektiivisesti IL-13:n signaloinnin IL4-reseptori alfa (IL4Rα)/ IL-13-reseptori alfa 1 (IL13Rα1) -heterodimeerin kautta ja estää siten IL-13:n välittämiä vaikutuksia. IL-13:n signaloinnin estymisestä oletetaan olevan hyötyä sairauksissa, joissa IL-13 on keskeinen taudin patogeneesiin vaikuttava tekijä. Lebrikitsumabi ei estä IL-13:n sitoutumista IL-13-reseptori alfa 2:een (IL-13Rα2 eli decoy-reseptori), mikä mahdollistaa IL-13:n viemisen soluun.
Farmakodynaamiset vaikutukset
Kliinisissä lebrikitsumabitutkimuksissa lebrikitsumabi vähensi seerumin periostiinipitoisuutta, immunoglobuliini E:n (IgE) kokonaispitoisuutta, CC-kemokiiniligandin (CCL)17 (kateenkorvan ja aktivaation säätelemä kemokiini [TARC]) pitoisuutta, CCL18:n (keuhkojen ja aktivaation säätelemä kemokiini [PARC]) pitoisuutta ja CCL13:n (monosyyttien kemotaktinen proteiini 4 [MCP-4]) pitoisuutta. Tyypin 2 tulehdusvälittäjäaineiden vähentyminen osoittaa epäsuorasti, että lebrikitsumabi estää IL-13-reittiä.
Immunogeenisuus
Lääkevasta-aineita havaittiin yleisesti. Lääkevasta-aineiden ei havaittu vaikuttavan farmakokinetiikkaan, tehoon eikä turvallisuuteen.
Kliininen teho ja turvallisuus
Atooppista ihottumaa sairastavat aikuiset ja nuoret
Lebrikitsumabin tehoa ja turvallisuutta monoterapiana (ADvocate-1, ADvocate-2) ja samanaikaisen paikallisesti käytettävän kortikosteroidihoidon kanssa (ADhere) arvioitiin kolmessa satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa pivotaalitutkimuksessa 1 062 aikuisella ja nuorella (12–17-vuotiaita, joiden paino ≥ 40 kg), joilla oli keskivaikea tai vaikea atooppinen ihottuma, jonka määritelmä oli EASI-mittarin (Eczema Area and Severity Index) pistemäärä ≥ 16, IGA-pisteet (Investigator’s Global Assessment) ≥ 3 ja atooppisen ihottuman laajuus ≥ 10 % kehon pinta-alasta. Näihin kolmeen tutkimukseen mukaan otetuilla potilailla oli aiemmin ollut riittämätön vaste paikallisesti käytettävään lääkitykseen tai paikallisesti käytettävät hoidot oli muutoin katsottu lääketieteelliseltä kannalta sopimattomiksi potilaalle.
Kaikissa kolmessa tutkimuksessa potilaat saivat aloitusannoksena 500 mg lebrikitsumabia (kaksi 250 mg:n injektiota) hoitoviikoilla 0 ja 2, minkä jälkeen annettiin 250 mg joka toinen viikko hoitoviikkoon 16 asti tai kaltaistettua lumelääkettä suhteessa 2:1. ADhere-tutkimuksessa tutkimuspotilaat saivat aktiivisiin leesioihin myös samanaikaista mietoa tai keskivahvaa paikallisesti käytettävää kortikosteroidia tai paikallisesti käytettävää kalsineuriinin estäjää. Potilaiden oli sallittua saada oirelääkitystä tutkijan harkinnan mukaan atooppisen ihottuman sietämättömien oireiden saamiseksi hoitotasapainoon. Systeemistä oirelääkitystä tarvinneiden potilaiden tutkimushoito keskeytettiin.
Potilaat, jotka saavuttivat IGA-pistemäärän 0 tai 1 tai vähintään 75 %:n vähenemän EASI-mittarilla (EASI-75) mitattuna ilman oirelääkitystä, satunnaistettiin uudelleen sokkoutetusti saamaan (i) 250 mg lebrikitsumabia joka toinen viikko, (ii) 250 mg lebrikitsumabia joka neljäs viikko tai (iii) kaltaistettua lumelääkettä enintään 52 viikon ajan.
ADvocate-1- ja -2-tutkimuksissa potilaat, jotka eivät olleet saavuttaneet IGA-pistemäärää 0 tai 1 tai EASI-75-vastetta hoitoviikon 16 aikapisteessä tai jotka saivat oirelääkitystä ennen hoitoviikkoa 16, otettiin mukaan Escape-haaraan, jossa he saivat avoimena hoitona 250 mg lebrikitsumabia joka toinen viikko hoitoviikkoon 52 asti.
ADvocate-1- ja ADvocate-2-tutkimuksissa 52 viikon pituisen tutkimuksen loppuun saakka mukana olleille ja ADhere-tutkimuksessa 16 viikon pituisen tutkimuksen loppuun saakka mukana olleille potilaille tarjottiin mahdollisuus jatkaa hoitoa erillisessä pitkäkestoisessa jatkotutkimuksessa (ADjoin).
Päätetapahtumat
Kaikissa kolmessa tutkimuksessa ensisijaisia päätetapahtumia olivat niiden potilaiden prosenttiosuus, joiden IGA-pistemäärä oli 0 tai 1 (”täysin parantunut” tai ”lähes parantunut”) ja joilla oli ≥ 2 pisteen vähenemä lähtötilanteesta, sekä niiden potilaiden prosenttiosuus, jotka saavuttivat EASI-75-vasteen lähtötilanteeseen verrattuna hoitoviikon 16 aikapisteessä. Keskeisiä toissijaisia päätetapahtumia (joille tehtiin multiplisiteettikorjaus) olivat niiden potilaiden prosenttiosuus, jotka saavuttivat vähintään 90 %:n vähenemän EASI-mittarilla (EASI-90), niiden potilaiden prosenttiosuus, joilla oli kutinan arviointiin käytettävällä numeerisella NRS-mittarilla (Pruritus NRS) mitattu vähintään 4 pisteen paraneminen lähtötilanteesta, niiden potilaiden prosenttiosuus, joilla oli vähintään 4 pisteen paraneminen lähtötilanteesta DLQI-kyselyllä (Dermatology Life Quality Index) mitattuna, sekä kutinan unta häiritsevä vaikutus (Sleep–Loss Scale -asteikko), joka on potilaiden raportoima yhden osion sisältävä päivittäin täytettävä asteikko, jolla mitataan kutinan häiritsevää vaikutusta uneen edellisenä yönä viisikohtaisella Likert-asteikolla. Lisäksi toissijaisena päätetapahtumana (ei multiplisiteettikorjausta) käytettiin myös POEM-kysymyssarjalla (Patient Oriented Eczema Measure) mitattua muutosta lähtötilanteesta.
Potilaat
Lähtötilanteen ominaisuudet
Monoterapiatutkimukseen ADvocate-1 otettiin mukaan 424 potilasta ja monoterapiatutkimukseen ADvocate2 otettiin mukaan 427 potilasta. Tutkimuksissa keskimääräinen ikä oli 35,8 vuotta ja keskimääräinen paino 77,1 kg. 49,9 % potilaista oli naisia, 63,7 % oli valkoisia, 22,6 % oli aasialaisia ja 9,9 % oli mustia. 12,0 % potilaista oli nuoria (12–17-vuotiaita). Lähtötilanteen IGA-pistemäärä oli 3 (keskivaikea atooppinen ihottuma) 61,5 %:lla potilaista ja 4 (vaikea-asteinen atooppinen ihottuma) 38,5 %:lla potilaista, ja 54,8 % potilaista oli saanut aiemmin systeemistä hoitoa. EASI-mittarin lähtötilanteen keskiarvo oli 29,6, kutinaa kuvaavan NRS-mittarin lähtötilanteen keskiarvo oli 7,2 ja DLQI-mittarin lähtötilanteen keskiarvo oli 15,5.
Samanaikaista paikallisesti käytettävää kortikosteroidihoitoa koskevaan tutkimukseen ADhere otettiin mukaan 211 potilasta. Keskimääräinen ikä oli 37,2 vuotta ja keskimääräinen paino 76,2 kg. 48,8 % potilaista oli naisia, 61,6 % oli valkoisia, 14,7 % oli aasialaisia, 13,3 % oli mustia ja 21,8 % oli nuoria. Tässä tutkimuksessa lähtötilanteen IGA-pistemäärä oli 3 (keskivaikea atooppinen ihottuma) 69,2 %:lla potilaista ja 4 (vaikea-asteinen atooppinen ihottuma) 30,8 %:lla potilaista, ja 47,4 % potilaista oli saanut aiemmin systeemistä hoitoa. EASI-mittarin lähtötilanteen keskiarvo oli 27,3, kutinaa kuvaavan NRS-mittarin lähtötilanteen keskiarvo oli 7,1 ja DLQI-mittarin lähtötilanteen keskiarvo oli 14,4.
Kliininen vaste
Monoterapiatutkimukset (ADvocate-1 ja ADvocate-2) – induktiojakso, hoitoviikot 0–16
ADvocate-1- ja ADvocate-2-tutkimuksissa merkittävästi suurempi osa potilaista, jotka satunnaistettiin saamaan 250 mg lebrikitsumabia joka toinen viikko, oli saavuttanut hoitoviikon 16 aikapisteessä IGA-pistemäärän 0 tai 1 ja ≥ 2 pisteen paranemisen lähtötilanteesta, EASI-75-vasteen, EASI-90-vasteen ja ≥ 4 pisteen paranemisen kutinaa kuvaavalla NRS-mittarilla ja DLQI-mittarilla lumelääkkeeseen verrattuna (ks. taulukko 2).
Molemmissa monoterapiatutkimuksissa lebrikitsumabi vähensi päivittäisen vaikeimman kutinan vaikeusastetta lumelääkkeeseen verrattuna kutinaa kuvaavalla NRS-mittarin prosentuaalisena muutoksena lähtötilanteesta jo hoitoviikon 1 aikapisteessä. Kutinaa kuvaavalla NRS-mittarilla mitattu paraneminen tapahtui samanaikaisesti atooppiseen ihottumaan liittyvän ihotulehduksen lievittymisen ja elämänlaadun paranemisen kanssa.
Taulukko 2. Lebrikitsumabimonoterapian tehoa koskevat tulokset ADvocate-1- ja ADvocate-2-tutkimusten hoitoviikon 16 aikapisteessä
| ADvocate-1 | ADvocate-2 | |||
| Viikko 16 | ||||
Lumelääke N = 141 | 250 mg lebrikitsumabia joka 2. viikko N = 283 | Lumelääke N = 146 | 250 mglebrikitsumabia joka 2. viikko N = 281 | |
| IGA 0 tai 1, %a | 12,7 | 43,1*** | 10,8 | 33,2*** |
| EASI-75, %b | 16,2 | 58,8*** | 18,1 | 52,1*** |
| EASI-90, %b | 9,0 | 38,3*** | 9,5 | 30,7*** |
| Kutinaa kuvaava NRS-mittari (≥ 4 pisteen paraneminen), %c | 13,0 | 45,9*** | 11,5 | 39,8*** |
| DLQI (aikuiset) (≥ 4 pisteen paraneminen), %d | 33,8 | 75,6*** | 33,6 | 66,3*** |
N = potilaiden lukumäärä
a Potilaat, joilla oli IGA-pistemäärä 0 tai 1 (”täysin parantunut” tai ”lähes parantunut”) ja ≥ 2 pisteen vähenemä lähtötilanteesta IGA-asteikolla 0–4.
b Potilaat, joiden EASI-mittarin pistemäärän vähenemä hoitoviikon 16 aikapisteestä oli 75 % tai 90 % lähtötilanteesta.
c Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden kutinaa kuvaavan NRS-mittarin lähtötilanteen pistemäärä ≥ 4.
d Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden lähtötilanteen DLQI-mittarin pistemäärä ≥ 4.
*** p < 0,001 lumelääkkeeseen verrattuna.
Näissä kahdessa tutkimuksessa oirelääkitystä (paikallisesti käytettävät kortikosteroidit, systeemiset kortikosteroidit, immuunisalpaajat) tarvitsivat harvemmat lebrikitsumabia saamaan satunnaistetut potilaat (14,7 % molempien tutkimusten yhdistetyissä tiedoissa) lumelääkettä saamaan satunnaistettuihin potilaisiin (36,6 % molempien tutkimusten yhdistetyissä tiedoissa) verrattuna.
Monoterapiatutkimukset (ADvocate-1 ja ADvocate-2) – ylläpitojakso, hoitoviikot 16–52
Vasteen säilymistä arvioitiin siten, että 157 potilasta ADvocate-1-tutkimuksesta ja 134 potilasta ADvocate-2-tutkimuksesta, jotka olivat saaneet 250 mg lebrikitsumabia joka toinen viikko ja jotka saavuttivat IGA-pistemäärän 0 tai 1 tai EASI-75-vasteen hoitoviikon 16 aikapisteessä ilman paikallista tai systeemistä oirelääkitystä, satunnaistettiin uudelleen suhteessa 2:2:1 sokkoutetusti 36 viikon pituiseen jatkohoitoon, joka oli (i) 250 mg lebrikitsumabia joka toinen viikko, (ii) 250 mg lebrikitsumabia joka neljäs viikko tai (iii) kaltaistettua lumelääkettä kumulatiivisen 52 viikon tutkimushoidon ajan (ks. taulukko 3).
Taulukko 3. Lebrikitsumabimonoterapian tehoa koskevat tulokset hoitoviikon 52 aikapisteessä potilailla, jotka olivat saaneet hoitovasteen ADvocate-1- ja ADvocate-2-tutkimusten hoitoviikon 16 aikapisteessä (yhdistetty analyysi)
| ADvocate-1 ja ADvocate-2 (yhdistetty) | ||
| Viikko 52 | ||
Lumelääked (lebrikitsumabin lopettaminen) N = 60 | 250 mg lebrikitsumabia joka 4. viikko N = 118 | |
| IGA 0 tai 1, %a | 47,9 | 76,9** |
| EASI-75, %b | 66,4 | 81,7* |
| EASI-90, %b | 41,9 | 66,4** |
| Kutinaa kuvaava NRS-mittari (≥ 4 pisteen paraneminen), %c | 66,3 | 84,7 |
a Potilaat, joilla oli IGA-pistemäärä 0/1 ja ≥ 2 pisteen paraneminen viikon 16 aikapisteessä lähtötilanteesta ja joiden IGA-pistemäärä oli edelleen 0/1 ja ≥ 2 pistettä lähtötilannetta parempi hoitoviikon 52 aikapisteessä.
b Potilaat, jotka olivat saavuttaneet EASI-75-vasteen hoitoviikon 16 aikapisteessä ja joilla oli edelleen EASI-75-vaste hoitoviikon 52 aikapisteessä, tai potilaat, jotka olivat saavuttaneet EASI-75-vasteen hoitoviikon 16 aikapisteessä ja EASI-90-vasteen hoitoviikon 52 aikapisteessä.
c Prosenttiosuus on laskettu suhteessa niiden potilaiden määrään, joiden kutinaa kuvaavan NRS-mittarin lähtötilanteen pistemäärä ≥ 4.
d Potilaat, jotka olivat saaneet hoitovasteen hoitoviikon 16 aikapisteessä annettaessa 250 mg lebrikitsumabia joka toinen viikko (IGA-pistemäärä 0 tai 1 tai EASI-75-vaste) ja jotka satunnaistettiin uudelleen lumelääkehoitoon.
*p < 0,05; ** p < 0,01 lumelääkkeeseen verrattuna.
Niistä potilaista, jotka saivat lebrikitsumabia induktiojakson aikana ja saivat edelleen avoimena hoitona 250 mg lebrikitsumabia joka toinen viikko viikkoon 52 asti Escape-haarassa, 58 % oli saavuttanut EASI-75-vasteen ja 28 % oli saavuttanut IGA-pistemäärän 0 tai 1 ja ≥ 2 pisteen paranemisen lähtötilanteesta ADvocate-1- ja ADvocate-2-tutkimusten (yhdistetty) viikon 52 aikapisteessä.
Monoterapiatutkimukset (ADvocate-1 ja ADvocate-2) – pitkäkestoinen jatkotutkimus (ADjoin), viikot 52–152
Kliinisen vasteen säilymistä arvioitiin potilailla, jotka saavuttivat IGA-pistemäärän 0 tai 1 tai EASI-75-vasteen viikolla 16 ilman paikallista tai systeemistä varalääkitystä ja jotka saivat ADvocate-1- tai ADvocate-2-tutkimuksessa lebrikitsumabimonoterapiaa 52 viikon ajan ja jatkoivat hoitoa 100 viikkoa kestäneessä ADjoin-jatkotutkimuksessa. (Kuva 1).
Kuva 1. IGA-pistemäärä 0 tai 1, EASI-75-vaste ja kutinaa kuvaavan NRS-mittarin pistemäärä vasteen viikolla 16 saaneilla potilailla, jotka olivat ADvocate-1- ja ADvocate-2-tutkimuksessa mukana 52 viikon ajan ja jotka saivat ADjoin-tutkimuksessa hoitona 250 mg lebrikitsumabia neljän viikon välein*
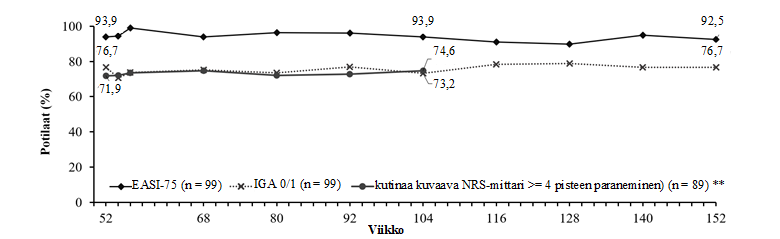
* Puuttuvat tiedot on imputoitu Markov Chain-Monte Carlo -moni-imputoinnin avulla.
** Vain potilaat, joiden kutinaa kuvaavan NRS-mittarin pistemäärä on lähtötilanteessa ≥ 4. Kutinaa arvioitiin vain ADjoin-tutkimuksen ensimmäisenä vuotena.
Samanaikaisen paikallisesti käytettävän kortikosteroidihoidon tutkimus (ADhere), viikot 0–16
ADhere-tutkimuksen lähtötilanteesta hoitoviikkoon 16 merkittävästi suurempi osa potilaista, jotka satunnaistettiin saamaan ja joille annettiin 250 mg lebrikitsumabia joka toinen viikko yhdistelmänä paikallisesti käytettävän kortikosteroidihoidon kanssa, saavutti IGA-pistemäärän 0 tai 1, EASI-75-vasteen ja ≥ 4 pisteen paranemisen kutinaa kuvaavalla NRS-mittarilla ja DLQI-mittarilla lumelääkkeen ja paikallisesti käytettävän kortikosteroidihoidon yhdistelmään verrattuna (ks. Taulukko 4).
Taulukko 4. Lebrikitsumabista ja paikallisesti käytettävästä kortikosteroidista koostuvan yhdistelmähoidon tehoa koskevat tulokset ADhere-tutkimuksen hoitoviikon 16 aikapisteessä
| ADhere | ||
| Viikko 16 | ||
Lumelääke + paikallisesti käytettävä kortikosteroidi N = 66 | 250 mg lebrikitsumabia joka 2. viikko + paikallisesti käytettävä kortikosteroidi N = 145 | |
| IGA 0 tai 1, %a | 22,1 | 41,2* |
| EASI-75, %b | 42,2 | 69,5*** |
| EASI-90, %b | 21,7 | 41,2** |
| Kutinaa kuvaava NRS-mittari (≥ 4 pisteen paraneminen), %c | 31,9 | 50,6* |
| DLQI (aikuiset) (≥ 4 pisteen paraneminen), %d | 58,7 | 77,4* |
a Potilaat, joilla oli IGA-pistemäärä 0 tai 1 (”täysin parantunut” tai ” lähes parantunut”) ja ≥ 2 pisteen vähenemä lähtötilanteesta IGA-asteikolla 0–4.
b Potilaat, joiden EASI-mittarin pistemäärän vähenemä lähtötilanteesta oli hoitoviikon 16 aikapisteessä 75 % tai 90 %.
c Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden kutinaa kuvaavan NRS-mittarin lähtötilanteen pistemäärä ≥ 4.
d Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden lähtötilanteen DLQI-mittarin pistemäärä ≥ 4.
* p < 0,05; ** p < 0,01; *** p < 0,001 lumelääkkeeseen verrattuna.
ADhere-tutkimuksessa potilaat, jotka saivat 250 mg lebrikitsumabia joka toinen viikko yhdistelmänä paikallisesti käytettävän kortikosteroidihoidon kanssa hoitoviikosta 0 hoitoviikkoon 16, käyttivät voimakasta paikallisesti käytettävää kortikosteroidihoitoa oirelääkityksenä harvemmin (1,4 %) kuin potilaat, jotka saivat lumelääkkeen ja paikallisesti käytettävän kortikosteroidihoidon yhdistelmää (4,5 %).
Samanaikaisen paikallisesti käytettävän kortikosteroidihoidon tutkimus (ADhere) – pitkäaikainen jatkotutkimus (ADjoin), viikot 16–116
Kliininen vaste säilyi niillä potilailla, jotka saavuttivat ADhere-tutkimuksessa IGA-pistemäärän 0 tai 1 tai EASI-75-vasteen hoitoviikolla 16 ja jotka jatkoivat 100 viikkoa kestäneessä ADjoin-jatkotutkimuksessa hoitoa 250 mg:n lebrikitsumabiannoksilla 4 viikon välein.
Kliininen vaste potilailla, joilla ei ole riittävää hoitotasapainoa siklosporiinihoidon avulla, jotka eivät siedä siklosporiinia tai joille siklosporiinin ei katsottu lääketieteelliseltä kannalta sopivan (ADvantage)
ADvantage-tutkimuksessa arvioitiin lebrikitsumabin tehoa lumelääkkeeseen verrattuna aikuisilla ja nuorilla (≥ 12-vuotiaat – < 18-vuotiaat, paino ≥ 40 kg), joilla oli keskivaikea tai vaikea atooppinen ihottuma, jota hoidettiin samanaikaisesti paikallisesti käytettävällä kortikosteroidilla, ja joilla ei ollut riittävää hoitotasapainoa siklosporiinihoidon avulla tai joille siklosporiinin ei katsottu lääketieteelliseltä kannalta sopivan.
Tutkimukseen otettiin yhteensä 331 potilasta, joiden iän keskiarvo oli 33,8 vuotta. 52,9 % potilaista oli miehiä, 93,7 % potilaista identifioitui valkoihoisiksi, ja nuorten osuus potilasjoukosta oli 11,8 %. Lähtötilanteessa keskimääräinen EASI-pistemäärä oli 28,1, kutinaa kuvaavan NRS-mittarin pistemäärän keskiarvo oli 6,9 ja 38,7 %:lla potilaista IGA-pistemäärä oli 4. Kaikkiaan 53,2 % oli aiemmin altistunut siklosporiinille (ks. taulukko 5).
Taulukko 5. Lebrikitsumabista ja paikallisesti käytettävästä kortikosteroidista koostuvan yhdistelmähoidon tehoa koskevat tulokset ADvantage-tutkimuksen hoitoviikon 16 aikapisteessä
| ADvantage | ||
| Viikko 16 | ||
Lumelääke + paikallisesti käytettävä kortikosteroidi N = 111 | 250 mg lebrikitsumabia joka 2. viikko + paikallisesti käytettävä kortikosteroidi N = 220 | |
| EASI-75, %a | 40,8 | 68,4*** |
| IGA 0 tai 1, %b | 24,5 | 42,0** |
| EASI-90, %c | 20,8 | 42,9*** |
| Kutinaa kuvaava NRS-mittari (≥ 4 pisteen paraneminen), %d | 29,7 | 49,9* |
| DLQI (aikuiset) (≥ 4 pisteen paraneminen), %e | 69,6 | 78,0 |
a Potilaat, joiden EASI-mittarin pistemäärän vähenemä lähtötilanteesta oli hoitoviikon 16 aikapisteessä 75 %.
b Potilaat, joilla oli IGA-pistemäärä 0 tai 1 (”täysin parantunut” tai ”lähes parantunut”) ja ≥ 2 pisteen vähenemä lähtötilanteesta IGA-asteikolla 0–4.
c Potilaat, joiden EASI-mittarin pistemäärän vähenemä lähtötilanteesta oli hoitoviikon 16 aikapisteessä 90 %.
d Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden kutinaa kuvaavan NRS-mittarin lähtötilanteen pistemäärä oli ≥ 4.
e Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden lähtötilanteen DLQI-mittarin pistemäärä ≥ 4.
* p < 0,05; **p < 0,01; *** p < 0,001 lumelääkkeeseen verrattuna. IGA-pistemäärän 0 tai 1, EASI-90:n, kutinaa kuvaavan NRS-mittarin ja DLQI:n p-arvot ovat nimellisiä.
Muut potilaiden raportoimat hoitotulokset
Molemmissa monoterapiatutkimuksissa (ADvocate-1 ja ADvocate-2) ja samanaikaisen paikallisesti käytettävän steroidihoidon tutkimuksissa (ADhere ja ADvantage) 250 mg lebrikitsumabia joka toinen viikko paransi merkittävästi potilaiden raportoimia sairauden vaikeusastetta koskevia hoitotuloksia (POEM) ja vähensi unen häiriintymistä kutinan takia (Sleep-Loss Scale) hoitoviikon 16 aikapisteessä lumelääkkeeseen verrattuna.
Nuoret (12–17-vuotiaat)
Monoterapiatutkimuksissa ADvocate 1 ja ADvocate 2 nuorten potilaiden keski-ikä oli 14,6 vuotta, keskimääräinen paino oli 68,2 kg ja 56,9 % oli tyttöjä. Näissä tutkimuksissa 63,7 %:lla lähtötilanteen IGA-pistemäärä oli 3 (keskivaikea atooppinen ihottuma), 36,3 %:lla lähtötilanteen IGA-pistemäärä oli 4 (vaikea atooppinen ihottuma) ja 47,1 % oli saanut aiempaa systeemistä hoitoa. Samanaikaista paikallisesti käytettävää kortikosteroidihoitoa koskeneessa ADhere-tutkimuksessa nuorten potilaiden keski-ikä oli 14,6 vuotta, keskimääräinen paino oli 62,2 kg ja 50,0 % oli tyttöjä. Tässä tutkimuksessa 76,1 %:lla lähtötilanteen IGA-pistemäärä oli 3 (keskivaikea atooppinen ihottuma), 23,9 %:lla lähtötilanteen IGA-pistemäärä oli 4 (vaikea atooppinen ihottuma) ja 23,9 % oli saanut aiempaa systeemistä hoitoa.
Tehoa koskevat tulokset nuorilla potilailla hoitoviikon 16 aikapisteessä on esitetty taulukossa 6.
Taulukko 6. Lebrikitsumabimonoterapian tehoa koskevat tulokset nuorilla potilailla ADvocate-1- ja ADvocate-2-tutkimuksissa sekä lebrikitsumabin ja paikallisesti käytettävän kortikosteroidihoidon yhdistelmää koskeneessa ADhere-tutkimuksessa hoitoviikon 16 aikapisteessä
| ADvocate-1 | ADvocate-2 | ADhere | ||||||
| Viikko 16 | ||||||||
Lume-lääke N = 18 | 250 mg lebrikitsumabia joka 2. viikko N = 37 | Lume-lääke N = 17 | 250 mg lebrikitsumabia joka 2. viikko N = 30 | Lumelääke + paikallisesti käytettävä kortikosteroidi N = 14 | 250 mg lebrikitsumabia joka 2. viikko + paikallisesti käytettävä kortikosteroidi N = 32 | |||
| IGA 0 tai 1, %a | 22,2 | 48,6 | 5,9 | 44,1** | 28,6 | 57,3 | ||
| EASI-75, %a | 22,2 | 62,2** | 12,0 | 61,7** | 57,1 | 88,0* | ||
| EASI-90, %a | 16,7 | 45,9* | 6,1 | 34,3* | 28,6 | 55,1 | ||
| Kutinaa kuvaava NRS-mittari (≥ 4 pisteen paraneminen), %b | 22,8 | 54,3* | 0,3 | 42,1 | 13,8 | 45,8 | ||
a Potilaat, joilla oli hoitoviikon 16 aikapisteessä IGA-pistemäärä 0 tai 1 (”täysin parantunut” tai ”lähes parantunut”) ja ≥ 2 pisteen vähenemä lähtötilanteesta IGA-asteikolla 0–4 tai hoitoviikon 16 aikapisteessä EASI-mittarin pistemäärän vähenemä 75 % tai 90 % lähtötilanteesta. b Prosenttiosuus on laskettu suhteessa niiden potilaiden lukumäärään, joiden kutinaa kuvaavan NRS-mittarin lähtötilanteen pistemäärä ≥ 4. *p < 0,05; **p < 0,01 lumelääkkeeseen verrattuna. | ||||||||
Lebrikitsumabia ja lebrikitsumabin yhdistelmää paikallisesti käytettävän kortikosteroidihoidon kanssa saaneilla nuorilla potilailla sairauden vaikeusaste lieveni kliinisesti merkittävästi ja vaste säilyi hoitoviikkoon 52 asti. Lisätiedot yhden hoitohaaran ADore-tutkimuksesta, jossa 206 nuorta sai lebrikitsumabia, tukevat lebrikitsumabin tehoa nuorten potilaiden hoidossa hoitoviikkoon 52 asti.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset lebrikitsumabin käytöstä atooppisen ihottuman hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ihon alle annetun 250 mg:n lebrikitsumabiannoksen jälkeen huippupitoisuudet seerumissa saavutettiin noin 7–8 vuorokautta annoksen antamisen jälkeen.
Hoitoviikolla 0 ja hoitoviikolla 2 annettujen 500 mg:n latausannosten jälkeen vakaan tilan pitoisuudet seerumissa oli saavutettu ensimmäisen 250 mg:n kahden viikon välein annetun annoksen yhteydessä hoitoviikon 4 aikapisteessä.
Populaatiofarmakokineettisen analyysin perusteella ennustetut vakaan tilan minimipitoisuudet (Ctrough,ss) atooppista ihottumaa sairastavilla potilailla, jotka saivat 250 mg lebrikitsumabia kahden viikon tai neljän viikon välein (mediaani ja 5.–95. persentiili), olivat 87 (46–159) mikrog/ml (kahden viikon välein) ja 36 (18–68) mikrog/ml (neljän viikon välein).
Populaatiofarmakokineettisen analyysin perusteella absoluuttiseksi biologiseksi hyötyosuudeksi arvioitiin 86 %. Injektiokohdan sijainti ei vaikuttanut merkittävästi lebrikitsumabin imeytymiseen.
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella vakaan tilan kokonaisjakautumistilavuus oli 5,14 l.
Biotransformaatio
Erityisiä metaboliatutkimuksia ei ole tehty, koska lebrikitsumabi on proteiini. Lebrikitsumabi hajoaa oletettavasti katabolisten reittien kautta pieniksi peptideiksi ja yksittäisiksi aminohapoiksi samalla tavalla kuin endogeeninen IgG.
Eliminaatio
Populaatiofarmakokineettisessä analyysissa puhdistuma oli 0,154 l/vrk ja se oli annoksesta riippumaton. Eliminaation keskimääräinen puoliintumisaika oli noin 24,5 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Lebrikitsumabin farmakokinetiikka oli lineaarista, ja altistus suureni 37,5–500 mg:n annosalueella suhteessa annokseen, kun annos annettiin injektiona atooppista ihottumaa sairastavien potilaiden tai terveiden vapaaehtoisten ihon alle.
Erityisryhmät
Sukupuoli, ikä ja etninen tausta
Sukupuolella, iällä (12–93 vuotta) ja etnisellä taustalla ei ollut merkittävää vaikutusta lebrikitsumabin farmakokinetiikkaan.
Munuaisten ja maksan vajaatoiminta
Erityisiä kliinistä farmakologiaa koskevia tutkimuksia munuaisten tai maksan vajaatoiminnasta lebrikitsumabin farmakokinetiikkaan aiheutuvien vaikutusten arvioimiseksi ei ole tehty. Lebrikitsumabi ei oletettavasti eliminoidu merkittävästi munuaisten tai maksan kautta, koska se on monoklonaalinen vasta-aine. Populaatiofarmakokineettiset analyysit osoittavat, että munuaisten tai maksan toimintaa kuvastavat markkerit eivät vaikuttaneet lebrikitsumabin farmakokinetiikkaan.
Paino
Lebrikitsumabialtistus oli vähäisempää potilailla, joiden paino oli suurempi, mutta tällä ei ollut merkittävää vaikutusta kliiniseen tehoon.
Pediatriset potilaat
Populaatiofarmakokineettisen analyysin perusteella 12–17-vuotiailla nuorilla, joilla oli atooppista ihottumaa, lebrikitsumabin minimipitoisuudet seerumissa olivat hieman suuremmat kuin aikuisilla, mikä liittyi heidän pienempään painojakaumaansa.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta (mukaan lukien turvallisuutta koskevia farmakologisia päätetapahtumia) sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Lebrikitsumabin mutageenisuutta ei ole arvioitu. Monoklonaaliset vasta-aineet eivät kuitenkaan oletettavasti aiheuta muutoksia DNA:han tai kromosomeihin.
Lebrikitsumabilla ei ole tehty karsinogeenisuustutkimuksia. Saatavilla olevan IL-13:n estoon liittyvän näytön arviointi ja eläintoksikologiaa koskevat tiedot lebrikitsumabista eivät viittaa siihen, että lebrikitsumabilla olisi karsinogeenisia vaikutuksia.
Sukukypsillä apinoilla ei havaittu pitkäaikaisesti laskimoon (naaraat) tai ihon alle (urokset) annetun lebrikitsumabihoidon jälkeen hedelmällisyysparametreihin kohdistuvia vaikutuksia. Lebrikitsumabilla ei ollut vaikutuksia alkion ja sikiön kehitykseen eikä syntymänjälkeiseen kehitykseen.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Väkevä etikkahappo (E 260)
Sakkaroosi
Polysorbaatti 20 (E 432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Jääkaapista ottamisen jälkeen Ebglyss on käytettävä 7 vuorokauden kuluessa (enintään 30 °C) tai hävitettävä. Kun sitä on säilytetty poissa jääkaapista, sitä ei saa palauttaa takaisin jääkaappiin.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EBGLYSS injektioneste, liuos, esitäytetty kynä
250 mg (L:ei) 2 x 1 kpl (2 ml (125 mg/ml)) (2209,99 €)
PF-selosteen tieto
Ebglyss 250 mg injektioneste, liuos, esitäytetty kynä
2 ml liuosta tyypin 1 kirkkaasta lasista valmistetussa 2,25 ml:n ruiskussa, joka on esitäytetyssä kynässä ja jossa on erityisen pieni pyöreä ulkoneva reunus, 27 G:n erityinen ohutseinämäinen x 8 mm:n kiinteästi ruiskuun kuuluva neula, joka on valmistettu ruostumattomasta teräksestä ja on suljettu laminoidulla bromibutyylielastomeerisella männällä ja jäykällä neulansuojalla.
Pakkauskoot:
2 esitäytettyä kynää
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai hieman kellertävä tai hieman ruskehtava liuos, jossa ei ole näkyviä hiukkasia.
Käyttö- ja käsittelyohjeet
Tarkemmat ohjeet Ebglyss-esitäytetyn kynän antoon ovat pakkausselosteen lopussa.
Liuoksen on oltava kirkasta tai opaalinhohtoista, väritöntä tai hieman kellertävää tai hieman ruskehtavaa eikä siinä saa olla näkyviä hiukkasia. Jos liuos on sameaa, värjääntynyttä tai siinä on näkyviä hiukkasia, liuosta ei saa käyttää.
Esitäytettyä kynää ei saa altistaa korkeille lämpötiloille eikä suoralle auringonvalolle, eikä sitä saa ravistaa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
EBGLYSS injektioneste, liuos, esitäytetty kynä
250 mg 2 x 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Lebrikitsumabi: Aikuisten ja vähintään 12-vuotiaiden vaikean atooppisen ihottuman hoito erityisin edellytyksin (3102).
ATC-koodi
D11AH10
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
 ORION OYJ ORION PHARMA
ORION OYJ ORION PHARMA Orionintie 1, PL 65
02101 Espoo
010 4261
www.orion.fi
etunimi.sukunimi@orionpharma.com