
VORANIGO tabletti, kalvopäällysteinen 10 mg, 40 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Voranigo 10 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 10 mg vorasidenibia (vorasidenib) (hemisitruunahappona, hemihydraattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää laktoosimonohydraattia määrän, joka vastaa 0,60 mg:aa laktoosia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Voranigo 40 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 40 mg vorasidenibia (vorasidenib) (hemisitruunahappona, hemihydraattina).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää laktoosimonohydraattia määrän, joka vastaa 2,39 mg:aa laktoosia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti
Kliiniset tiedot
Käyttöaiheet
Voranigo on tarkoitettu monoterapiana pääosin tehostumattoman graduksen 2 astrosytooman tai oligodendrogliooman hoitoon, kun kasvaimessa on IDH1 R132 ‑mutaatio tai IDH2 R172 ‑mutaatio, aikuisille ja vähintään 12-vuotiaille, vähintään 40 kg painaville nuorille potilaille, joille on aiemmin tehty vain kirurginen toimenpide ja jotka eivät tarvitse välitöntä sädehoitoa tai solunsalpaajahoitoa (ks. kohta Farmakodynamiikka).
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtynyt lääkäri, ja hänen pitää myös valvoa hoitoa.
Potilaan isositraattidehydrogenaasi-1 (IDH1) R132- tai isositraattidehydrogenaasi-2 (IDH2) R172 ‑mutaatio on varmistettava asianmukaisella diagnostisella testillä ennen Voranigo-valmisteen ottamista. IDH1 R132- tai IDH2 R172 ‑mutaation esiintyminen on selvitettävä tähän tarkoitukseen soveltuvalla CE-merkityllä in vitro -diagnostisella lääkinnällisellä laitteella. Jos CE-merkittyä in vitro ‑diagnostista lääkinnällistä laitetta ei ole saatavilla, IDH1 R132- tai IDH2 R172 ‑mutaatio on selvitettävä tähän käyttötarkoitukseen validoidulla vaihtoehtoisella testillä.
Annostus
Voranigo-valmisteen suositeltu annos aikuisille ja vähintään 12-vuotiaille, vähintään 40 kg painaville nuorille on 40 mg kerran vuorokaudessa. Alle 40 kg painavien potilaiden annostuksesta ei voida antaa suosituksia, koska tästä potilaspopulaatiosta ei ole kliinisiä tietoja.
Hoitoa jatketaan niin pitkään kuin siitä havaitaan potilaalle kliinistä hyötyä tai kunnes potilas ei enää siedä hoitoa.
Väliin jääneet tai myöhästyneet annokset
Jos annos unohtuu tai sitä ei oteta tavanomaiseen aikaan, se on otettava mahdollisimman pian 6 tunnin kuluessa väliin jääneestä annoksesta. Seuraava annos otetaan sen tavanomaisena ottoajankohtana.
Jos annos unohtuu ja tavanomaisesta ottoajankohdasta on kulunut yli 6 tuntia, annos jätetään väliin ja seuraava annos otetaan sen tavanomaisena ottoajankohtana.
Jos potilas oksentaa annoksen, korvaavia tabletteja ei pidä ottaa. Tabletit on otettava tavalliseen tapaan seuraavana päivänä.
Varotoimet ennen antoa ja seuranta
Täydellinen verenkuva ja veren kemiallinen koostumus (mukaan lukien maksaentsyymit) on tutkittava ennen hoidon aloittamista, joka toinen viikko kahden ensimmäisen hoitokuukauden aikana, sitten vähintään kerran kuukaudessa kahden ensimmäisen hoitovuoden aikana ja sen jälkeen kliinisen tarpeen mukaan. Joitakin potilaita saattaa olla tarpeen seurata jatkuvasti ja edellä mainittua tiheämmin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annosmuutokset haittavaikutusten vuoksi
Hoito voi olla tarpeen keskeyttää tai annosta voi olla tarpeen pienentää yksilöllisen turvallisuuden ja sietokyvyn mukaan. Annostason suositellut pienennykset on esitetty alla taulukossa 1.
Taulukko 1: Annostason suositellut pienennykset | ||
Annostaso | Annos ja ajoitus | Tablettien lukumäärä ja vahvuus |
Aloitusannos | 40 mg kerran päivässä | Yksi 40 mg:n tabletti / kerran päivässä |
Ensimmäinen annoksen pienennyskerta | 20 mg kerran päivässä | Kaksi 10 mg:n tablettia / kerran päivässä |
Toinen annoksen pienennyskerta | 10 mg kerran päivässä | Yksi 10 mg:n tabletti / kerran päivässä |
Suositellut Voranigo-valmisteen annosmuutokset ja haittavaikutusten hallinta on esitetty taulukossa 2.
Taulukko 2: Suositellut Voranigo-valmisteen annosmuutokset ja haittavaikutusten hallinta
Haittavaikutus | Vaikeusastea | Hoito ja annosmuutokset |
Maksatoksisuus (ALAT- tai ASAT-arvojen nousu) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Aste 1 ALAT- tai ASAT-arvon nousu tasolle > ULN – 3 x ULN ilman samanaikaista kokonaisbilirubiinipitoisuutta > 2 x ULN | Jatka nykyisen Voranigo-annoksen käyttöä. Seuraa maksaentsyymiarvoja viikoittain, kunnes ne palautuvat asteeseen < 1. |
Aste 2 ALAT tai ASAT > 3–5 x ULN ilman samanaikaista kokonaisbilirubiinipitoisuutta > 2 x ULN | Ensimmäinen kerta: Keskeytä Voranigo-valmisteen käyttö ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle.
Uusiutuminen: Keskeytä Voranigo-valmisteen käyttö ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle, ja jatka Voranigo-valmisteen käyttöä pienemmällä annoksella (ks. taulukko 1). | |
Aste 3 ALAT tai ASAT > 5–20 x ULN ilman samanaikaista kokonaisbilirubiinipitoisuutta > 2 x ULN | Ensimmäinen kerta: Keskeytä Voranigo-valmisteen käyttö ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle.
Uusiutuminen: Lopeta Voranigo-valmisteen käyttö pysyvästi ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle. | |
Aste 2 tai 3 Mikä tahansa ALAT- tai ASAT-arvo > 3–20 x ULN ja samanaikaisesti kokonaisbilirubiinipitoisuus > 2 x ULN, jos selkeää vaihtoehtoista selitystä ei ole. b | Lopeta Voranigo-valmisteen käyttö pysyvästi ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle. | |
Aste 4 Mikä tahansa ALAT- tai ASAT-arvo > 20 x ULN | Lopeta Voranigo-valmisteen käyttö pysyvästi ja seuraa maksaentsyymiarvoja kahdesti viikossa, kunnes ne palautuvat asteeseen ≤ 1 tai lähtötasolle. | |
Muut haittavaikutukset | Aste 3 | Ensimmäinen kerta: Keskeytä Voranigo-valmisteen käyttö, kunnes haittavaikutus lievenee asteeseen ≤ 1 tai lähtötasolle.
Uusiutuminen: Lopeta Voranigo-valmisteen käyttö pysyvästi. |
Aste 4 | Lopeta Voranigo-valmisteen käyttö pysyvästi. |
Lyhenteet: ALAT = alaniiniaminotransferaasi; ASAT = aspartaattiaminotransferaasi; ULN = viitearvon yläraja
a Haittavaikutukset on luokiteltu National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‑CTCAE) -kriteerien version 5.0 mukaan.
b Jos vaihtoehtoinen syy tunnistetaan, harkitse Voranigo-valmisteen käytön jatkamista pienemmällä annoksella (ks. taulukko 1), kun kohonnut arvo on palautunut asteeseen 1 tai lähtötasolle.
Erityisryhmät
Iäkkäät
Annoksen muuttamista ≥ 65-vuotiaille potilaille ei suositella (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Aloitusannoksen muuttamista ei suositella, jos potilaalla on munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] > 40 ml/min/1,73 m2). Vorasidenibin ja AGI-69460-metaboliitin farmakokinetiikkaa ei ole tutkittu potilailla, joiden eGFR on ≤ 40 ml/min/1,73 m2 tai joilla on dialyysihoitoa vaativa munuaisten vajaatoiminta. Vorasidenibia ei pidä käyttää, jos potilaan eGFR on ≤ 40 ml/min/1,73 m2 tai jos potilas tarvitsee dialyysihoitoa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet. ja 5.2.).
Maksan vajaatoiminta
Aloitusannoksen muuttamista ei suositella, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta (Child–Pugh-luokka A tai B). Vorasidenibin ja AGI-69460:n farmakokinetiikkaa ei ole tutkittu potilailla, joilla on vaikea-asteinen maksan vajaatoiminta (Child–Pugh-luokka C). Vorasidenibia on käytettävä varoen potilaille, joilla on vaikea-asteinen maksan vajaatoiminta, ja näitä potilaita on seurattava tarkoin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet. ja 5.2).
Pediatriset potilaat
Kliinisiä tietoja 12- – < 18-vuotiaiden lasten hoidosta ei ole saatavilla (ks. kohta Farmakodynamiikka).
Antotapa
Voranigo otetaan suun kautta.
Tabletit pitää ottaa kerran päivässä suunnilleen samaan aikaan joka päivä. Potilaiden pitää olla syömättä vähintään kahden tunnin ajan ennen Voranigo-valmisteen ottamista sekä tunnin ajan valmisteen ottamisen jälkeen (ks. kohta Farmakokinetiikka). Tabletit niellään kokonaisina vesilasillisen kanssa. Tabletteja ei saa paloitella, murskata eikä pureskella, koska saatavilla ei ole tietoja, jotka vahvistaisivat lääkevalmisteen biologisen hyötyosuuden olevan vastaava, jos tabletteja manipuloidaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Maksatoksisuus
Vorasidenibihoitoa kliinisessä pivotaalitutkimuksessa tai valmisteen markkinoille tulon jälkeen saaneilla potilailla on raportoitu lääkkeen aiheuttamaa maksatoksisuutta, mukaan lukien vakavaa maksan vajaatoimintaa, maksanekroosia ja akuuttia hepatiittia (ks. kohta Haittavaikutukset).
Maksaentsyymiarvoja (mukaan lukien ALAT, ASAT ja glutamyylitransferaasi [GT]) ja kokonaisbilirubiiniarvoja on seurattava ennen hoidon aloittamista, joka toinen viikko kahden ensimmäisen hoitokuukauden aikana, sitten kerran kuussa kahden ensimmäisen hoitovuoden aikana ja sen jälkeen kliinisen tarpeen mukaan. Jos ALAT- tai ASAT-arvot suurenevat tasolle ≤ 3 x ULN, harkitse viikoittaista seurantaa. Keskeytä hoito, pienennä annosta tai lopeta hoito pysyvästi maksaentsyymipitoisuuden poikkeavuuksien vaikeusasteen mukaan (ks. kohta Annostus ja antotapa).
Karsinogeenisuusriski
Eläimistä todetut havainnot saattavat viitata mahdolliseen karsinogeenisuusriskiin (erityisesti maksassa, ks. kohta Prekliiniset tiedot turvallisuudesta). Karsinogeenisuustutkimuksia ei ole vielä tehty, ja kliinistä turvallisuutta koskevat pitkäaikaistiedot ovat riittämättömiä. Sen vuoksi karsinogeenisuusriskiä ihmisillä ei voitu sulkea pois.
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Vorasidenibin käyttö raskauden aikana saattaa vahingoittaa sikiötä. Naisten, jotka voivat tulla raskaaksi, on suositeltavaa tehdä raskaustesti ennen hoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään kahden kuukauden ajan viimeisen annoksen jälkeen. Lapsen hankkimista suunnittelevia naisia on kehotettava hakeutumaan lisääntymislääketieteelliseen neuvontaan.
Vorasidenibi saattaa pienentää hormonaalisten ehkäisyvalmisteiden pitoisuuksia, minkä vuoksi hoidon aikana ja vähintään kahden kuukauden ajan viimeisen annoksen jälkeen on suositeltavaa käyttää lisäksi jotakin estemenetelmää (ks. kohdat Yhteisvaikutukset ja Raskaus ja imetys).
Miespotilaat
Miespotilaan, jonka naiskumppani voi tulla raskaaksi, pitää käyttää tehokasta ehkäisyä hoidon aikana ja vähintään kahden kuukauden ajan viimeisen annoksen jälkeen. Miespotilaiden pitää kysyä neuvoa siemennesteen pakastamisesta ennen hoidon aloittamista (ks. kohta Raskaus ja imetys).
Maksan vajaatoiminta
Vorasidenibin turvallisuutta ja tehoa vaikea-asteista maksan vajaatoimintaa (Child‑Pugh-luokka C) sairastavien potilaiden hoidossa ei ole varmistettu. Vorasidenibia on käytettävä varoen potilaille, joilla on ennestään vaikea-asteinen maksan vajaatoiminta (Child‑Pugh-luokka C), ja näitä potilaita on seurattava tarkoin (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Vorasidenibin farmakokinetiikkaa ja turvallisuutta ei ole tutkittu potilailla, joilla on munuaisten vajaatoiminta (eGFR ≤ 40 ml/min/1,73 m2) tai dialyysihoitoa vaativa munuaisten vajaatoiminta. Vorasidenibia ei pidä käyttää näillä potilailla (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Laktoosi
Voranigo sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
In vitro ‑kokeiden perusteella vorasidenibi on voimakas indusoija pregnaani-X-reseptorin (PXR) välityksellä ja saattaa vaikuttaa sellaisten samanaikaisesti annettujen lääkkeiden pitoisuuteen plasmassa, jotka metaboloituvat tai jotka kulkeutuvat sellaisten entsyymien tai kuljettajaproteiinien välityksellä, joiden ilmentyminen on PXR-välitteistä.
Muiden lääkevalmisteiden vaikutus vorasidenibiin
Voimakkaat CYP1A2:n estäjät
Vorasidenibin ja voimakkaiden CYP1A2:n estäjien (fluvoksamiini ja siprofloksasiini) samanaikainen käyttö saattaa suurentaa vorasidenibin pitoisuutta plasmassa. Voimakkaiden CYP1A2:n estäjien samanaikaista käyttöä vorasidenibihoidon aikana pitää välttää, ja hoitoon on harkittava muita vaihtoehtoja, jotka eivät ole voimakkaita CYP1A2:n estäjiä.
Lääkkeiden yhteisvaikutuksia selvittäneessä in vivo -tutkimuksessa vorasidenibin (20 mg) ja voimakkaan CYP1A2:n estäjän (500 mg siprofloksasiinia kahdesti päivässä 14 päivän ajan) samanaikainen käyttö suurensi vorasidenibin maksimipitoisuutta plasmassa (Cmax) 29 % sekä plasman pitoisuus-aikakuvaajan alainen pinta-alaa (AUC) 153 %.
Keskivoimakkaat CYP1A2:n indusoijat
Vorasidenibin ja keskivoimakkaiden CYP1A2:n indusoijien (fenytoiini ja rifampisiini) samanaikainen käyttö saattaa pienentää vorasidenibin pitoisuutta plasmassa. Harkitse vorasidenibihoidon aikana muita vaihtoehtoja, jotka eivät ole keskivoimakkaita CYP1A2:n indusoijia.
Mahahapon eritystä vähentävät aineet
Vorasidenibin farmakokinetiikassa ei todettu kliinisesti merkittäviä eroja, kun vorasidenibia käytettiin samanaikaisesti mahahapon eritystä vähentävän omepratsolin kanssa.
Vorasidenibin vaikutukset muihin lääkevalmisteisiin
Sytokromi P450 -entsyymien (CYP) substraatit, joiden terapeuttinen indeksi on kapea
Vorasidenibin samanaikainen käyttö sellaisten CYP2B6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n tai CYP3A4:n substraattien kanssa, joilla on kapea terapeuttinen indeksi (mm. amitriptyliini, alfentaniili, karbamatsepiini, siklosporiini, dosulepiini, everolimuusi, fentanyyli, fosfenytoiini, ifosfamidi, imipramiini, fenobarbitaali, fenytoiini, pimotsidi, kinidiini, sirolimuusi, takrolimuusi, tamoksifeeni, trimipramiini, valproiinihappo ja varfariini), saattaa pienentää näiden lääkevalmisteiden pitoisuuksia plasmassa. Näiden entsyymien sellaisten substraattien, joiden terapeuttinen indeksi on kapea, samanaikaista käyttöä pitää välttää, jos potilas saa vorasidenibihoitoa.
Herkät CYP-entsyymien substraatit, joiden terapeuttinen indeksi ei ole kapea
Vorasidenibin samanaikainen käyttö sellaisten herkkien CYP2B6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n tai CYP3A4:n substraattien kanssa, joiden terapeuttinen indeksi ei ole kapea (mm. bupropioni, buspironi, selekoksibi, darunaviiri, ibrutinibi, midatsolaami, repaglinidi, sakinaviiri, tipranaviiri ja triatsolaami), saattaa pienentää näiden lääkevalmisteiden pitoisuuksia plasmassa. Harkitse vorasidenibihoidon aikana muita vaihtoehtoja, jotka eivät ole näiden entsyymien herkkiä substraatteja.
Yhteisvaikutukset kuljettajaproteiinien kanssa
Vorasidenibi estää in vitro rintasyövän resistenssiproteiinia (BCRP) (ks. kohta Farmakokinetiikka).
Vorasidenibin antamisessa yhdessä BCRP:n substraattien (mm. rosuvastatiini) kanssa on oltava varovainen.
Hormonaaliset ehkäisyvalmisteet
Vorasidenibihoito saattaa pienentää hormonaalisten ehkäisyvalmisteiden pitoisuuksia, joten hoidon aikana ja vähintään kahden kuukauden ajan viimeisen annoksen jälkeen on suositeltavaa käyttää lisäksi jotakin estemenetelmää (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Raskaus ja imetys).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Naisten, jotka voivat tulla raskaaksi, on suositeltavaa tehdä raskaustesti ennen vorasidenibihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Naisten, jotka voivat tulla raskaaksi, sekä miesten, joiden naispuolinen kumppani voi tulla raskaaksi, pitää käyttää tehokasta ehkäisyä hoidon aikana ja vähintään kahden kuukauden ajan viimeisen annoksen jälkeen. Koska vorasidenibin vaikutusta systeemisesti vaikuttavien hormonaalisten ehkäisyvalmisteiden metaboliaan ja tehoon ei ole tutkittu, on käytettävä lisäksi jotakin estemenetelmää raskauden ehkäisemiseksi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Raskaus
Vorasidenibin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu alkion ja sikiön kehitykseen liittyvää toksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vorasidenibia ei pidä käyttää raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi, mutta eivät käytä ehkäisyä. Naisille, jotka voivat tulla raskaaksi, tai miehille, joiden naiskumppani voi tulla raskaaksi, on kerrottava vorasidenibista sikiölle mahdollisesti aiheutuvista riskeistä.
Imetys
Ei tiedetä, erittyvätkö vorasidenibi ja sen metaboliitit ihmisillä äidinmaitoon. Imetys on lopetettava hoidon ajaksi ja vähintään kahdeksi kuukaudeksi viimeisen annoksen jälkeen.
Hedelmällisyys
Vorasidenibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja. Toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten aikana havaittiin muutoksia sekä uros- että naaraspuolisten eläinten lisääntymiselimissä (ks. kohta Prekliiniset tiedot turvallisuudesta). Näiden vaikutusten kliinistä merkitystä ei tiedetä. Lapsen hankkimista suunnittelevia mies- ja naispotilaita on kehotettava hakeutumaan lisääntymislääketieteelliseen neuvontaan, ja miesten tulisi pyytää neuvoja siemennesteen pakastamisesta ennen hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vorasidenibilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset, mukaan lukien laboratoriotutkimusten tulosten poikkeavuudet, olivat suurentunut ALAT-arvo (59,3 %), suurentunut ASAT-arvo (45,5 %), suurentunut GT-arvo (37,7 %), väsymys (36,5 %) ja ripuli (24,6 %).
Yleisimmät ≥ 3. asteen haittavaikutukset olivat suurentunut ALAT-arvo (9,6 %), suurentunut ASAT-arvo (4,2 %) ja suurentunut GT-arvo (3,0 %).
Maksan vajaatoimintaa, autoimmuunihepatiittia ja ALAT-arvon suurenemista ilmeni kutakin vakavana haittavaikutuksena 0,6 %:lla Voranigo-valmistetta saaneista potilaista.
Vorasidenibihoidon pysyvä lopettaminen asteeseen ≥ 3 suurentuneen ALAT-arvon vuoksi raportoitiin 3,0 %:lla potilaista tai autoimmuunihepatiitin vuoksi raportoitiin 0,6 %:lla potilaista.
Vorasidenibihoitoa saaneista potilaista 18,6 % keskeytti hoidon haittavaikutusten vuoksi. Yleisimmät haittavaikutukset, joiden vuoksi hoito jouduttiin keskeyttämään, olivat suurentunut ALAT-arvo (14,4 %) ja suurentunut ASAT-arvo (6,0 %).
Vorasidenibin annosta pienennettiin haittavaikutuksen vuoksi 9,6 %:lla potilaista. Yleisin haittavaikutus, jonka vuoksi annosta jouduttiin pienentämään, oli ALAT-arvon suureneminen (7,8 %).
Haittavaikutustaulukko
Tässä kohdassa kuvatut haittavaikutukset perustuvat Voranigo-valmistetta koskeviin tutkimustietoihin (INDIGO-tutkimus [N = 167]) ja valmisteen markkinoille tulon jälkeiseen kokemukseen.
Vorasidenibihoitoa saaneilla potilailla raportoidut haittavaikutukset on esitetty alla taulukossa 3 MedDRA-elinjärjestelmäluokituksen ja esiintyvyyden mukaan.
Esiintyvyydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä yleisyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Vorasidenibihoitoa saaneilla potilailla raportoidut haittavaikutukset
| Elinjärjestelmäluokka | Esiintymistiheys | Haittavaikutukset |
| Veri ja imukudos | Hyvin yleinen | Pienentynyt verihiutalemääräa |
| Aineenvaihdunta ja ravitsemus | Yleinen | Hyperglykemia |
| Heikentynyt ruokahalu | ||
| Hypofosfatemia | ||
| Hermosto | Hyvin yleinen | Heitehuimaus |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Hengenahdistus |
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli |
| Vatsakipu | ||
| Yleinen | Ruokatorven refluksitauti | |
| Maksa ja sappi | Hyvin yleinen | Suurentunut alaniiniaminotransferaasiarvoa |
| Suurentunut aspartaattiaminotransferaasiarvoa | ||
| Suurentunut glutamyylitransferaasiarvoa | ||
| Yleinen | Suurentunut alkalisen fosfataasin arvoa | |
| Suurentunut veren bilirubiinipitoisuusa | ||
| Melko harvinainen | Maksan vajaatoiminta | |
| Autoimmuunihepatiitti | ||
| Maksanekroosi | ||
| Tuntematon | Lääkkeen aiheuttama maksavaurio* | |
| Akuutti maksatulehdus* | ||
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Väsymys |
*Tunnistettu myyntiluvan saamisen jälkeisessä käytössä.
aLaboratoriotutkimuksen tuloksen poikkeavuudeksi on määritelty uusi poikkeava tulos tai tulos, joka on huonontunut vähintään yhden asteen verran lähtötasosta, tai tilanne, jossa lähtötaso on tuntematon.
Valikoitujen haittavaikutusten kuvaus
Maksatoksisuus
INDIGO-tutkimuksessa vorasidenibihoitoa saaneista 167 potilaasta 18,6 %:lla ALAT-arvo suureni tasolle > 3 x viitevälin yläraja ja 8,4 %:lla potilaista ASAT-arvo suureni tasolle > 3 x viitevälin yläraja. Näistä potilaista 1,2 %:lla ALAT- tai ASAT-arvo suureni tasolle > 3 x viitevälin yläraja ja kokonaisbilirubiiniarvo suureni samanaikaisesti tasolle > 2 x viitevälin yläraja. Maksaentsyymi- ja bilirubiiniarvojen suureneminen oli väliaikaista ja korjautui osittain tai kokonaan annosmuutoksen tai hoidon lopettamisen myötä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.). Suurentuneita ASAT-arvoja koskevaan ensimmäiseen tapahtumaan (mikä tahansa vaikeusaste) kuluneen ajan mediaani oli 85,0 päivää (vaihteluväli: 14–451 päivää), ja suurentuneita ALAT-arvoja koskevaan ensimmäiseen tapahtumaan (mikä tahansa vaikeusaste) kuluneen ajan mediaani oli 57,0 päivää (vaihteluväli: 1–506 päivää). Kutakin seuraavista haittavaikutuksista raportoitiin INDIGO-tutkimuksessa vorasidenibihoitoa yhden kerran: maksan vajaatoiminta, maksanekroosi ja autoimmuunihepatiitti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa toksisuus ilmenee todennäköisesti vorasidenibiin liittyvien haittavaikutusten pahenemisena (ks. kohta Haittavaikutukset). Potilaita on seurattava tarkoin ja heille on annettava asianmukaista tukihoitoa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Vorasidenibin yliannostukseen ei ole spesifistä antidoottia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet
ATC-koodi: L01XM04
Vaikutusmekanismi
Vorasidenibi on mutatoituneisiin IDH1- ja IDH2-entsyymeihin kohdentuva estäjä. Potilailla, joilla on astrosytooma tai oligodendrogliooma, IDH1- ja IDH2 ‑mutaatiot johtavat onkogeenisen 2‑hydroksiglutaraattimetaboliitin (2‑HG) ylituotantoon, mikä haittaa solujen erilaistumista, joka puolestaan edistää kasvainten muodostumista. Vorasidenibi estää IDH1‑ ja IDH2‑mutatoituneita proteiineja, mikä estää 2‑HG:n poikkeavaa tuotantoa ja johtaa siten pahanlaatuisten solujen erilaistumiseen ja niiden jakautumisen vähenemiseen. Vorasidenibin kyvystä pienentää kasvainta ei tehty prekliinisiä tutkimuksia.
Farmakodynaamiset vaikutukset
Potilailla, joilla oli IDH1- tai IDH2-mutatoitunut gliooma, päivittäisen vorasidenibihoidon todettiin pienentävän kasvaimen 2‑HG:n pitoisuutta.
Kliininen teho ja turvallisuus
Vorasidenibin tehoa ja turvallisuutta arvioitiin vaiheen 3 satunnaistetussa (1:1), kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (INDIGO), johon osallistui 331 aikuista ja vähintään 12-vuotiasta, vähintään 40 kg:n painoista nuorta. Edellytykset tutkimukseen soveltuneille potilaille olivat: potilaalla WHO:n vuoden 2016 kriteerien mukainen graduksen 2 astrosytooma tai oligodendrogliooma, jossa oli IDH1 R132 ‑mutaatio tai IDH2 R172 ‑mutaatio, potilasta oli hoidettu ainoastaan leikkauksella, mukaan lukien biopsia, subtotaalinen resektio tai täydellinen resektio, ja potilas ei tutkijan arvion mukaan tarvinnut välitöntä solunsalpaajahoitoa tai sädehoitoa. Tutkimukseen otettiin mukaan potilaita, joilla oli sokkoutetun riippumattoman keskitetyn arvioijatahon (BIRC) vahvistama magneettikuvantamisen perusteella arvioitavissa oleva, tehostumaton tauti. Potilaat, joilla oli keskitetysti varmistettu tehostuva tauti, otettiin mukaan tutkimukseen sillä edellytyksellä, että tehostuminen oli minimaalista, se ei ollut nodulaarista eikä mitattavaa eikä se ollut muuttunut kahden viimeisimmän kuvantamistutkimuksen välillä. INDIGO-tutkimuksesta suljettiin pois potilaat, jotka olivat saaneet aiempaa syöpähoitoa, mukaan lukien solunsalpaajahoitoa tai sädehoitoa. IDH1- tai IDH2-mutaatiostatus määritettiin prospektiivisesti Oncomine Dx Target Test ‑testillä.
Potilaat satunnaistettiin saamaan joko 40 mg vorasidenibia suun kautta kerran päivässä tai vastaavaa lumevalmistetta sairauden radiologiseen etenemiseen saakka tai kunnes ilmeni toksisuutta, joka ei ollut hyväksyttävissä. Satunnaistaminen ositettiin paikallisen 1p19q-statuksen (ko-deletoitunut tai ei ko-deletoitunut) ja kasvaimen lähtötilanteen koon (läpimitta ≥ 2 cm tai < 2 cm) mukaan. Lumehoitoon satunnaistettujen potilaiden oli sallittua siirtyä saamaan vorasidenibihoitoa, kun sairauden radiologinen eteneminen oli dokumentoitu edellyttäen, että he eivät tutkijan arvion mukaan tarvinneet välitöntä solunsalpaajahoitoa tai sädehoitoa.
Ensisijainen tehoa koskevan päätetapahtuman mittari oli radiologinen etenemättömyysaika (PFS), joka perustui sokkoutetun riippumattoman keskitetyn arvioijatahon modifioitujen RANO‑LGG-kriteerien (Response Assessment in Neuro‑Oncology for Low Grade Glioma) mukaiseen (vain radiologinen eteneminen) arvioon.
Potilaiden demografiset tiedot ja sairauden ominaisuudet olivat tasapainossa tutkimusryhmien kesken. Vorasidenibihoitoon satunnaistettujen 168 potilaan iän mediaani oli 41 vuotta (vaihteluväli 21–71 vuotta). Näistä potilaista 98,8 % oli 18–64-vuotiaita. Yksi 16-vuotias pediatrinen potilas satunnaistettiin saamaan lumelääkettä, eikä vorasidenibihoitoa saaneeseen ryhmään satunnaistettu lainkaan alle 18-vuotiaita potilaita. Valtaosa potilaista oli miehiä (60,1 %), 74,4 % oli valkoihoisia, 3,0 % aasialaisia, 1,2 % mustaihoisia, 1,2 % kuului muuhun etniseen ryhmään ja 19,6 %:n osalta etnistä taustaa ei ollut ilmoitettu. 53,6 %:lla potilaista Karnofsky-toimintakykymittarin pistemäärä (KPS-pistemäärä) oli 100, 45,8 %:lla KPS-pistemäärä oli 90–80 ja 0,6 %:lla se oli 70–60. Suurimmalle osalle potilaista (75 %) oli tehty aiemmin vähintään kerran glioomaa koskenut leikkaus, ja 25 %:lle oli aiemmin tehty vähintään kaksi leikkausta. Kummassakin hoitoryhmässä 95 %:lla potilaista oli IDH1 R132 ‑mutaatio ja 5 %:lla oli IDH2 R172 ‑mutaatio.
Etenemättömyysaikaa koskevat tehon tulokset esitetään taulukossa 4 ja kuvassa 1.
Taulukko 4: INDIGO-tutkimuksen (tutkimus AG881‑C‑004) tehoa koskevat tulokset
Tehoa koskeva parametri | Voranigo 40 mg vuorokaudessa (n = 168) | Lumelääke (n = 163) |
Etenemättömyysaika (PFS) | ||
Tapahtumien lukumäärä, n (%) Etenevä sairaus Kuolema | 47 (28,0) 0 | 88 (54,0) 0 |
Etenemättömyysajan mediaani, kk(95 %:n luottamusväli)a | 27,7 (17,0–NE) | 11,1 (11,0–13,7) |
Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,39 (0,27–0,56) | |
p‑arvoc | 0,000000067 | |
Lyhenne: NE = ei arvioitavissa (Not estimable)
Koko analyysijoukko käsitti kaikki satunnaistetut potilaat.
- Mediaanin 95 %:n luottamusväli laskettiin Brookmeyerin ja Crowleyn menetelmällä.
- Arvioitu Coxin suhteellisen vaaran mallilla, jossa on huomioitu seuraavat stratifiointitekijät: 1p19q-status ja kasvaimen koko lähtötilanteessa.
- Arvioitu yksisuuntaisella ositetulla log-rank-testillä. Etenemättömyysaika testattiin yksisuuntaisella tehon α-tasolla 0,000359, joka perustui 82 % informaatiosta vastaavaan päivitettyyn tehon raja-arvoon.
Kuva 1: Sokkoutetun riippumattoman keskitetyn arvioijatahon INDIGO-tutkimuksessa arvioiman etenemättömyysajan Kaplan‑Meier-kuvaaja
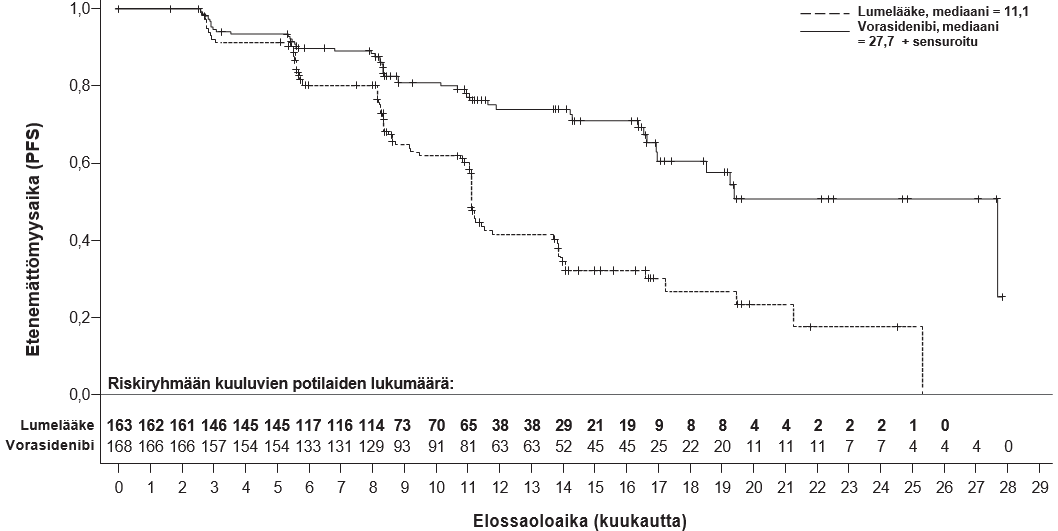
Päivitetty etenemättömyysaika, jonka sokkoutettu riippumaton keskitetty arvioijataho arvioi, kun oli todettu 96 % (N = 158) tapahtumista, vahvisti vorasidenibin hyödyn lumelääkkeeseen verrattuna (riskitiheyksien suhde: 0,35 [95 %:n luottamusväli: 0,25–0,49]). Kahdenkymmenenneljän (24) kuukauden kuluttua etenemättömyysosuus oli vorasidenibiryhmässä 59 % (95 %:n luottamusväli: 48,4–67,8) ja lumelääkeryhmässä 26 % (95 %:n luottamusväli: 17,9–35,3). Etenemättömyysajan mediaani ei ollut vorasidenibiryhmässä arvioitavissa (95 %:n luottamusväli: 22,1–NE), ja lumelääkeryhmässä se oli 11,4 (95 %:n luottamusväli: 11,1–13,9) kuukautta.
Pediatriset potilaat
12- – alle 18-vuotiaat nuoret
Farmakokineettiset tiedot tukevat vorasidenibin käyttöä potilaille, joiden ikä on 12 vuodesta alle 18 vuoteen ja joilla on IDH1- tai IDH2-mutatoitunut astrosytooma tai oligodendrogliooma. Tiedot osoittavat, ettei iällä ollut kliinisesti merkittävää vaikutusta vorasidenibin farmakokinetiikkaan (ks. kohta Farmakokinetiikka).
Farmakokinetiikka
Vorasidenibin farmakokinetiikkaa on tutkittu potilailla, joilla on matalan maligniteettiasteen gliooma, jossa on IDH1- tai IDH2 ‑mutaatio, sekä terveillä tutkittavilla. Vorasidenibin farmakokineettinen profiili on samankaltainen sekä potilailla, joilla on matalan maligniteettiasteen gliooma, että terveillä tutkittavilla.
Imeytyminen
Suun kautta otetun 40 mg:n kerta-annoksen jälkeen vorasidenibin huippupitoisuuden (Cmax) saavuttamiseen kulunut aika (Tmax) oli 2,0 tuntia (mediaani). Cmax-arvon geometrinen keskiarvo oli 75,4 ng/ml (variaatiokerroin, CV%: 44), ja AUC-arvon geometrinen keskiarvo oli 2 860 h∙ng/ml (CV%: 56). Vakaassa tilassa vorasidenibin Cmax-arvon geometrinen keskiarvo oli 133 ng/ml (CV%: 73) ja AUC-arvon geometrinen keskiarvo oli 1 988 h∙ng/ml (CV%: 95). Useimmilla potilailla todettiin plasmassa toinen pitoisuushuippu 24 tunnin kuluttua lääkkeen antamisesta, mutta se oli pienempi kuin 2 tunnin kuluttua annoksen antamisesta havaittu Cmax. Vaikka absoluuttista biologista hyötyosuutta ei ole suoraan määritetty, vorasidenibin imeytymisen 40 mg:n kalvopäällysteisistä tableteista arvioidaan olevan kohtalaista tai suurta.
Cmax:n kumuloitumiskerroin oli noin 3,8 ja AUC-arvon kumuloitumiskerroin oli noin 4,4. Vakaan tilan pitoisuudet plasmassa saavutettiin 2–3 viikon jälkeen, kun annos otettiin kerran päivässä.
Vorasidenibin keskimääräinen Cmax suureni 3,1-kertaiseksi ja keskimääräinen AUC-arvo 1,4-kertaiseksi, kun vorasidenibia otettiin runsasrasvaisen aterian yhteydessä. Kun vorasidenibiannos otettiin vähärasvaisen aterian yhteydessä, vorasidenibin Cmax suureni 2,3-kertaiseksi ja AUC-arvo 1,4‑kertaiseksi (ks. kohta Annostus ja antotapa).
Jakautuminen
Vorasidenibin keskimääräinen näennäinen jakautumistilavuus on 3 930 l (CV%: 40).Vorasidenibin jakautumistilavuus yhden laskimoon annetun mikroannoksen (0,1 mg) antamisen jälkeen on 1 110 l. Plasman proteiineihin sitoutunut fraktio oli vorasidenibin osalta 97 % ja AGI-69460:n osalta 87 %. Sekä vorasidenibi että AGI‑69460 sitoutuvat mieluummin seerumin albumiiniin kuin happamaan alfa-1-glykoproteiiniin. Vorasidenibin vereen ja plasmaan jakautumisen suhde on 0,87, AGI-69460:n vereen ja plasmaan jakautumisen suhde on 1,38, ja aivokasvaimen ja plasman välisen vorasidenibipitoisuuden suhde on 1,6.
Biotransformaatio
Vorasidenibi metaboloituu pääasiassa CYP1A2:n välityksellä, ja CYP2B6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n, CYP2D6:n ja CYP3A4/5:n osuudet ovat merkityksettömiä tai vähäisiä. Vorasidenibin metabolisesta puhdistumasta maksassa enimmillään 30 % tapahtuu muiden kuin CYP-reittien kautta.
AGI‑69460 on vorasidenibin loppupään aktiivinen metaboliitti. Suun kautta otetun 40 mg:n vorasidenibikerta-annoksen jälkeen todettu AGI‑69460-metaboliitin Tmax oli 336 tuntia. Cmax-arvon geometrinen keskiarvo oli 3,32 ng/ml (CV%: 55,6) ja AUC0-t-arvon geometrinen keskiarvo oli 1 172 h∙ng/ml (CV%: 61). Vakaassa tilassa AGI‑69460:n Cmin,ss-arvon geometrinen keskiarvo oli 111 ng/ml (CV%: 58) ja AUC0-4-arvon geometrinen keskiarvo 2. syklin 1. päivänä oli 190 h∙ng/ml (CV%: 90).
Yhteisvaikutukset
Vorasidenibilla on in vitro voimakas induktiovaikutus herkkiin CYP3A4:n substraatteihin sekä kohtalainen induktiovaikutus herkkiin CYP2B6:n ja CYP2C19:n substraatteihin (ks. kohta Yhteisvaikutukset).
In vitro ‑tiedot osoittavat, että vorasidenibi on BCRP:n estäjä. Vorasidenibi ei estä P‑glykoproteiinia (P‑gp) eikä maksan OATP1B1-kuljettajaproteiinia. AGI-69460 on BCRP:n ja OATP1B3:n estäjä in vitro.
Vorasidenibi ei ole P-gp:n, BCRP:n eikä maksan kuljettajaproteiinien OATP1B1 ja OATP1B3 substraatti.
Eliminaatio
Käytettäessä kapseliin pakattua jauhevalmistemuotoa, jonka absoluuttinen biologinen hyötyosuus on < 34 %, noin 89 % annetusta radioaktiivisesta vorasidenibiannoksesta havaittiin 44 vuorokauden kuluessa, 85 % ulosteessa ja 4,5 % virtsassa. Valtaosa ulosteessa havaitusta radioaktiivisuudesta oli muuttumatonta vorasidenibia (55 %), kun taas virtsassa ei havaittu muuttumatonta vorasidenibia.
Vorasidenibin keskimääräinen terminaalinen puoliintumisaika on 238 tuntia (CV%: 57), efektiivinen puoliintumisaika on 63,2 tuntia (CV%: 75) ja keskimääräinen näennäinen puhdistuma on 14,0 l/h (CV%: 56).
Lineaarisuus/ei-lineaarisuus
Voranigo-valmisteen antamisen jälkeen vorasidenibin Cmax- ja AUC-arvo suurenevat suhteessa annokseen, kun annos on 10–40 mg.
Erityispotilasryhmät
Iäkkäät
Iäkkäillä (enintään 75-vuotiailla) potilailla ei ole havaittu kliinisesti merkittäviä vaikutuksia vorasidenibin farmakokinetiikkaan (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnalla (eGFR > 40 ml/min/1,73 m2) ei ollut kliinisesti merkittävää vaikutusta vorasidenibin farmakokinetiikkaan. Vorasidenibin farmakokinetiikkaa potilailla, joiden eGFR on ≤ 40 ml/min/1,73 m2 tai joilla on dialyysihoitoa vaativa munuaisten vajaatoiminta, ei tunneta.
Maksan vajaatoiminta
Keskivaikealla maksan vajaatoiminnalla (Child–Pugh-luokka B) ei ollut kliinisesti merkittävää vaikutusta vorasidenibin ja AGI‑69460:n farmakokinetiikkaan. Potilailla, joilla oli keskivaikea maksan vajaatoiminta, ei havaittu kliinisesti oleellisia muutoksia vorasidenibin kokonaispitoisuuksissa tai vapaan (sitoutumattoman) vorasidenibin pitoisuuksissa (samankaltaisia vorasidenibin Cmax-arvoja ja vorasidenibin AUC0-t-arvon suureneminen 26,0 % havaittiin, kun taas altistus AGI‑69460:lle väheni) suun kautta otetun 20 mg:n vorasidenibikerta-annoksen jälkeen. Vorasidenibin ja AGI‑69460:n farmakokinetiikkaa potilailla, joilla on vaikea-asteinen maksan vajaatoiminta (Child–Pugh-luokka C) ei tunneta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Muut erityisryhmät
Iän (16–75 vuotta), etnisen taustan tai painon (43,5–168 kg) ei todettu vaikuttavan kliinisesti merkittävästi vorasidenibin farmakokinetiikkaan. Vorasidenibialtistuksen todettiin olevan naispotilailla 1,6 kertaa suurempi kuin miespotilailla.
Pediatriset potilaat
Farmakokineettiset tiedot osoittavat, ettei iällä ollut kliinisesti merkittävää vaikutusta vorasidenibin farmakokinetiikkaan. Vorasidenibialtistus on oletettavasti samankaltainen aikuisilla ja vähintään 12-vuotiailla nuorilla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa tunnistetut pääasialliset toksiset vaikutukset liittyvät maksaan, maha-suolikanavaan, ihoon, munuaisiin, luustolihaksiin, sukupuolielimiin ja rintarauhaseen.
Vorasidenibi ei ollut genotoksinen Amesin testissä in vitro, ihmisen lymfosyyttien mikrotumatestissä in vitro eikä rotan luuytimen mikrotumatestissä in vivo. Sen tärkein verenkierrossa esiintyvä metaboliitti AGI-69460 ei ollut genotoksinen Amesin testissä, ihmisen lymfosyyttien mikrotumatestissä in vitro eikä rotan luuytimen mikrotumatestissä in vivo ja Comet-testissä.
Apinoilla tehdyssä 13 viikkoa kestäneessä tutkimuksessa havaittiin primaarisessa ruumiinavauksessa Kupfferin solujen hyperplasiaa, joka paheni toipumisjakson jälkeen, kun altistus oli 8-kertainen kliiniseen altistukseen verrattuna. Lisäksi rotilla tehtyjen toksisuustutkimusten havainnot viittasivat hormonaaliseen häiriöön. Tällaiset havainnot voivat viitata karsinogeenisuuden riskiin. Vorasidenibilla ei ole vielä tehty karsinogeenisuustutkimuksia.
Vorasidenibilla ei ole tehty hedelmällisyyttä koskevia eläintutkimuksia. Kun vorasidenibia annettiin rotille toistuvan altistuksen toksisuutta koskeneissa tutkimuksissa, todettiin sukupuolielimiin kohdistuvia vaikutuksia. Naaraiden sukupuolielimiin kohdistuvia haittavaikutuksia olivat muun muassa munasarjojen, kohdun, kohdunkaulan ja emättimen atrofia sekä kiimakierron vaihtelut. Urosrotilla todettiin vaikutuksia lisäkiveksiin (solujäte), rakkularauhaseen/eturauhaseen (atrofia) sekä kiveksiin (paino, tiehyiden rappeutuminen). Näitä löydöksiä todettiin pienimmällä tutkitulla annoksella 5 mg/kg/vrk (13 viikon tutkimus rotilla), joka saatu altistus oli 26-kertainen verrattuna ihmisen altistukseen 50 mg:n vuorokausiannoksen yhteydessä.
Vorasidenibi aiheutti tiineille rotille ja kaniineille alkio- ja sikiötoksisuutta (suurempi resorptioiden ilmaantuvuus, luutumisen viivästyminen, viskeraaliset munuaisten ja kivesten epämuodostumat rotilla). Näitä vaikutuksia ilmeni annoksilla, jotka olivat suurempia kuin potilaiden saamat päivittäiset hoitoannokset. Altistussuhde alkion tai sikiön kehitykselle haitattomalla annoksella (NOAEL) oli gestaatiopäivinä 6 ja 17 rotilla 8,0–28,5 ja gestaatiopäivinä 6 ja 19 kaniineilla 1,1-4,9.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa (E 460)
Kroskarmelloosinatrium
Silisifioitu mikrokiteinen selluloosa (sisältää mikrokiteistä selluloosaa ja vedetöntä kolloidista piidioksidia)
Magnesiumstearaatti (E 470b)
Natriumlauryylisulfaatti (E 487)
Tabletin kalvopäällyste
Hypromelloosi
Titaanidioksidi (E 171)
Laktoosimonohydraatti
Makrogoli (E 1521)
Painoväri
Musta rautaoksidi (E 172)
Propyleeniglykoli (E 1520)
Hypromelloosi (E 464)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
36 kuukautta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VORANIGO tabletti, kalvopäällysteinen
10 mg (L:kyllä) 30 kpl (10129,22 €)
40 mg (L:kyllä) 30 kpl (20077,83 €)
PF-selosteen tieto
Valkoinen HDPE-purkki, jossa on polypropeenista (PP) valmistettu turvasuljin ja polyeteenillä (PE) pinnoitettu, induktiokuumennuksella saumattu tiiviste sekä kolme HDPE-säiliötä silikageelikuivatusainetta. Pakkauskoko: 30 kalvopäällysteistä tablettia.
Valmisteen kuvaus:
Voranigo 10 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, pyöreä tabletti, jonka läpimitta on 6 mm ja jonka toisella puolella on merkintä 10.
Voranigo 40 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, pitkänomainen tabletti, jonka pituus on 14,8 mm, leveys 6,3 mm ja jonka toisella puolella on merkintä 40.
Käyttö- ja käsittelyohjeet
Potilaille on kerrottava, ettei tablettipurkissa olevaa silikageelisäiliötä saa niellä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VORANIGO tabletti, kalvopäällysteinen
10 mg 30 kpl
40 mg 30 kpl
- Ei korvausta.
ATC-koodi
L01XM04
Valmisteyhteenvedon muuttamispäivämäärä
10.04.2026
Yhteystiedot
Äyritie 12 A
01510 Vantaa
040 901 2978
www.servierfinland.fi
info.finland@servier.com