
VORANIGO filmdragerad tablett 10 mg, 40 mg
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Voranigo 10 mg filmdragerade tabletter
En filmdragerad tablett innehåller 10 mg vorasidenib (som hemicitronsyra, hemihydrat).
Hjälpämne med känd effekt
En filmdragerad tablett innehåller laktosmonohydrat motsvarande 0,60 mg laktos (se avsnitt Varningar och försiktighet).
Voranigo 40 mg filmdragerade tabletter
En filmdragerad tablett innehåller 40 mg vorasidenib (som hemicitronsyra, hemihydrat).
Hjälpämne med känd effekt
En filmdragerad tablett innehåller laktosmonohydrat motsvarande 2,39 mg laktos (se avsnitt Varningar och försiktighet).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Filmdragerad tablett
Kliniska uppgifter
Terapeutiska indikationer
Voranigo är indicerat som monoterapi vid behandling av företrädesvis icke kontrastuppladdande astrocytom eller oligodendrogliom av grad 2 med en IDH1 R132- eller IDH2 R172-mutation, hos vuxna och ungdomar från 12 års ålder, med en kroppsvikt på minst 40 kg, som endast genomgått kirurgiskt ingrepp och inte är i omedelbart behov av strålbehandling eller kemoterapi (se avsnitt Farmakodynamiska egenskaper).
Villkor
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Dosering och administreringssätt
Behandlingen ska inledas och övervakas av läkare med erfarenhet av läkemedelsbehandling mot cancer.
Innan patienter tar Voranigo måste isocitratdehydrogenas-1 (IDH1) R132-mutationen eller isocitratdehydrogenas-2 (IDH2) R172-mutationen ha bekräftats med ett lämpligt diagnostiskt test. Förekomst av IDH1 R132- eller IDH2 R172-mutation ska bedömas med en CE-märkt medicinteknisk produkt för in vitro-diagnostik (IVD) avsedd för detta ändamål. Om det inte finns någon CE-märkt IVD‑produkt tillgänglig ska IDH1 R132- eller IDH2 R172-mutation bedömas med ett annat validerat test.
Dosering
Rekommenderad dos av Voranigo till vuxna och ungdomar från 12 års ålder är 40 mg en gång om dagen till patienter som väger minst 40 kg. Ingen dosrekommendation kan ges för patienter som väger mindre än 40 kg eftersom det saknas kliniska data för denna population.
Behandlingen ska fortsätta så länge som klinisk nytta kan observeras eller tills behandlingen inte längre tolereras av patienten.
Missade eller försenade doser
Om en dos missas eller inte tas på vanlig tid ska den tas så snart som möjligt inom sex timmar efter den missade dosen. Påföljande dos ska tas på vanlig schemalagd tid.
Om en dos missas med mer än sex timmar ska den hoppas över och påföljande dos tas på vanlig schemalagd tid.
Om en dos kräks upp ska inga ersättande tabletter tas. Tabletterna ska tas som vanligt under påföljande dag.
Försiktighetsåtgärder som ska vidtas före administrering samt övervakning
Bedömning av fullständig blodstatus och blodkemi (elektrolyter, glukos, leverenzymer och njurfunktion) ska utföras innan behandlingen påbörjas, varannan vecka under de två första månaderna och därefter en gång per månad under de två första åren av behandlingen och därefter enligt klinisk indikation. Vissa patienter kan behöva mer frekvent och fortlöpande övervakning (se avsnitt Varningar och försiktighet).
Dosjustering vid biverkningar
Utsättning, tillfällig eller permanent, eller dosminskning kan behövas beroende på individuell säkerhet och tolerans. Rekommenderade dosminskningar framgår av tabell 1.
Tabell 1: Rekommenderade dosminskningar | ||
Dosnivå | Dos och doseringsschema | Antal tabletter och styrka |
Startdos | 40 mg en gång om dagen | En 40 mg-tablett en gång om dagen |
Första dosminskningen | 20 mg en gång om dagen | Två 10 mg-tabletter en gång om dagen |
Andra dosminskningen | 10 mg en gång om dagen | En 10 mg-tablett en gång om dagen |
Rekommendationer för dosjustering av Voranigo samt åtgärder vid biverkningar återfinns i tabell 2.
Tabell 2: Rekommendationer för dosjustering av Voranigo samt åtgärder vid biverkningar
Biverkning | Svårighetsgrada | Åtgärder och dosjustering |
Levertoxicitet (förhöjt ALAT eller ASAT) (se avsnitt Varningar och försiktighet) | Grad 1 förhöjt ALAT eller ASAT > ULN till 3 × ULN utan samtidig totalt bilirubin > 2 × ULN | Fortsätt behandlingen med nuvarande dos av Voranigo. Leverenzymer kontrolleras veckovis fram till återhämtning till < grad 1. |
Grad 2 ALAT eller ASAT > 3 till 5 × ULN utan samtidig totalt bilirubin > 2 × ULN | Första händelsen: sätt ut Voranigo och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje.
Vid återkommande biverkningar: sätt ut Voranigo och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje och fortsätt behandlingen med minskad dos av Voranigo (se tabell 1). | |
Grad 3 ALAT eller ASAT > 5 till 20 × ULN utan samtidig totalt bilirubin > 2× ULN | Första händelsen: sätt ut Voranigo och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje.
Om ingen återhämtning skett på ≤ 28 dagar, sätt ut Voranigo permanent. Vid återkommande biverkningar: sätt ut Voranigo permanent och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje. | |
Grad 2 eller 3 endera ALAT eller ASAT > 3 till 20 × ULN med samtidig totalt bilirubin > 2 × ULN i frånvaro av tydlig, alternativ förklaring. b | Sätt ut Voranigo permanent och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje. | |
Grad 4 endera ALAT eller ASAT > 20 × ULN | Sätt ut Voranigo permanent och kontrollera leverenzymer två gånger per vecka fram till återhämtning till ≤ grad 1 eller baslinje. | |
Övriga biverkningar | Grad 3 | Första händelsen: sätt ut Voranigo fram till återhämtning till ≤ grad 1 eller baslinje.
Vid återkommande biverkningar: sätt ut Voranigo permanent. |
Grad 4 | Sätt ut Voranigo permanent. |
Förkortningar: ALAT = alaninaminotransferas; ASAT = aspartataminotransferas; ULN (Upper Limit of Normal) = övre normalgränsen
a Biverkningarna är graderade enligt National Cancer Institute Common Terminology Criteria for Adverse Event (NCI-CTCAE) version 5.0.
b Överväg att återuppta behandlingen med lägre dos av Voranigo (se tabell 1) efter återhämtning till grad 1 eller baslinje, om en alternativ etiologi kan identifieras.
Särskilda populationer
Äldre
Ingen dosjustering rekommenderas för patienter ≥ 65 års ålder (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Ingen justering av startdosen rekommenderas för patienter med nedsatt njurfunktion (beräknad glomerulär filtrationshastighet [eGFR] > 40 ml/min/1,73 m²). Farmakokinetiken för vorasidenib och metaboliten AGI-69460 har inte studerats hos patienter med eGFR ≤ 40 ml/min/1,73 m² eller med nedsatt njurfunktion som kräver dialys. Vorasidenib ska inte användas till patienter med eGFR ≤ 40 ml/min/1,73 m² eller till patienter som är i behov av dialys (se avsnitt Varningar och försiktighet och Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen justering av startdosen rekommenderas för patienter med lindrigt eller måttligt (Child-Pugh-klass A eller B) nedsatt leverfunktion. Farmakokinetiken för vorasidenib och AGI-69460 har inte studerats hos patienter med svårt nedsatt leverfunktion (Child-Pugh-klass C). Vorasidenib ska användas med försiktighet hos patienter med svårt nedsatt leverfunktion, och denna patientgrupp ska övervakas noggrant (se avsnitt Varningar och försiktighet och Farmakokinetiska egenskaper).
Pediatrisk population
Det finns inga kliniska data för barn och ungdomar i åldrarna 12 till < 18 år (se avsnitt Farmakodynamiska egenskaper).
Administreringssätt
Voranigo är avsett för oral användning.
Tabletterna ska tas en gång om dagen vid ungefär samma tidpunkt varje dag. Patienterna ska avstå från mat i minst två timmar före och en timme efter att ha tagit Voranigo (se avsnitt Farmakokinetiska egenskaper). Tabletterna ska sväljas hela med ett glas vatten och får inte delas, krossas eller tuggas, eftersom det saknas data som bekräftar likvärdig biotillgänglighet av läkemedlet när tabletterna har påverkats.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Levertoxicitet
Läkemedelsinducerad levertoxicitet, inklusive allvarliga fall av leversvikt, levernekros och akut hepatit har rapporterats hos patienter som behandlats med vorasidenib i den pivotala kliniska studien eller efter godkännande för försäljning (se avsnitt Biverkningar).
Leverenzymer (däribland ALAT, ASAT och gammaglutamyltransferas [GGT]) samt totalt bilirubin måste kontrolleras innan behandlingen påbörjas, varannan vecka under de två första månaderna av behandlingen, en gång per månad under de två första åren av behandlingen och därefter enligt klinisk indikation. Överväg veckovisa kontroller vid förhöjningar av ALAT eller ASAT på ≤ 3 gånger ULN. Beroende på svårighetsgrad av avvikande leverenzymer ska doseringen avbrytas, minskas eller sättas ut permanent (se avsnitt Dosering och administreringssätt).
Karcinogenicitetsrisk
Djurfynd kan tyda på en möjlig risk för karcinogenicitet (särskilt i levern, se avsnitt Prekliniska säkerhetsuppgifter). Karcinogenicitetsstudier har ännu inte utförts och långsiktiga kliniska säkerhetsdata är inte heller tillräckliga. En risk för karcinogenicitet hos människor kan därför inte uteslutas.
Fertila kvinnor och preventivmedel
Vorasidenib kan orsaka fosterskada om det administreras till en gravid kvinna. Graviditetstestning rekommenderas för fertila kvinnor innan behandling inleds. Fertila kvinnor ska använda effektivt preventivmedel under behandlingens gång och i minst två månader efter den sista dosen. Kvinnor som planerar att skaffa barn ska rekommenderas att söka reproduktionsrådgivning.
Vorasidenib kan minska koncentrationerna av hormonella preventivmedel. Samtidig användning av ett barriärpreventivmedel rekommenderas därför under behandlingen och i minst två månader efter den sista dosen (se avsnitt Interaktioner och Fertilitet, graviditet och amning).
Manliga patienter
Män med fertil kvinnlig partner ska använda effektivt preventivmedel under behandlingen och i minst två månader efter den sista dosen. Män ska söka råd kring att frysförvara sperma innan behandlingen inleds (se avsnitt Fertilitet, graviditet och amning).
Nedsatt leverfunktion
Säkerhet och effekt för vorasidenib har inte fastställts hos patienter med svårt nedsatt leverfunktion (Child-Pugh-klass C). Vorasidenib ska användas med försiktighet hos patienter med befintlig svårt nedsatt leverfunktion (Child-Pugh-klass C), och denna patientgrupp ska övervakas noggrant (se avsnitt Dosering och administreringssätt och Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Farmakokinetik och säkerhet för vorasidenib har inte studerats hos patienter med nedsatt njurfunktion (eGFR ≤ 40 ml/min/1,73 m²) eller med nedsatt njurfunktion som kräver dialys. Vorasidenib ska inte användas till dessa patienter (se avsnitt Dosering och administreringssätt och Farmakokinetiska egenskaper).
Laktos
Voranigo innehåller laktos. Patienter med något av följande sällsynta ärftliga tillstånd bör inte använda detta läkemedel: galaktosintolerans, total laktasbrist eller glukos-galaktosmalabsorption.
Natrium
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per tablett, d.v.s. är näst intill ”natriumfritt”.
Interaktioner
Baserat på experiment in vitro utgör vorasidenib en stark inducerare, genom aktivering av receptorn för pregnan X (PXR), och kan därmed påverka exponeringen i plasma för samtidigt administrerade läkemedel som metaboliseras eller transporteras av enzymer eller transportörer vars uttryck medieras av PXR.
Andra läkemedels inverkan på vorasidenib
Starka hämmare av CYP1A2
Administrering av vorasidenib tillsammans med starka hämmare av CYP1A2 (fluvoxamin och ciprofloxacin) kan öka plasmakoncentrationen av vorasidenib. Samtidig användning av starka CYP1A2-hämmare ska undvikas. Överväg alternativa behandlingar utan starka hämmare av CYP1A2 under behandlingen med vorasidenib.
I en läkemedelsinteraktionsstudie in vivo medförde samtidig administrering av 20 mg vorasidenib tillsammans med en stark hämmare av CYP1A2 (500 mg ciprofloxacin två gånger om dagen i 14 dagar) en förhöjning av den maximala plasmakoncentrationen av vorasidenib (Cmax) med 29 % och av arean under tid/koncentrationskurvan (AUC) för plasma med 153 %.
Måttliga inducerare av CYP1A2
Administrering av vorasidenib tillsammans med måttliga inducerare av CYP1A2 (fenytoin och rifampicin) kan minska plasmakoncentrationen av vorasidenib. Överväg alternativ behandling utan måttliga inducerare av CYP1A2 vid behandling med vorasidenib.
Magsyrahämmande medel
Inga kliniskt signifikanta farmakokinetiska skillnader observerades för vorasidenib efter samtidig administrering av vorasidenib och det magsyrahämmande medlet omeprazol.
Inverkan av vorasidenib på andra läkemedel
Substrat för cytokrom P450 (CYP) med smalt terapeutiskt index
Samtidig administrering av vorasidenib och CYP2B6, CYP2C8, CYP2C9, CYP2C19 eller CYP3A4-substrat med smalt terapeutiskt index (inkluderande, men inte begränsat till, amitriptylin, alfentanil, karbamazepin, ciklosporin, dosulepin, everolimus, fentanyl, fosfenytoin, ifosfamid, imipramin, fenobarbital, fenytoin, pimozid, kinidin, sirolimus , takrolimus, tamoxifen, trimipramin, valproinsyra och warfarin) kan minska dessa läkemedels koncentrationer i plasma. Samtidig användning av substrat för dessa enzymer med smalt terapeutiskt index ska undvikas till patienter som tar vorasidenib.
Känsliga CYP-enzymsubstrat som inte har smalt terapeutiskt index
Samtidig administrering av vorasidenib och känsliga CYP2B6, CYP2C8, CYP2C9, CYP2C19 eller CYP3A4-substrat som inte har smalt terapeutiskt index (inkluderande, men inte begränsat till, bupropion, buspiron, celecoxib, darunavir, ibrutinib, midazolam, repaglinid, sakvinavir, tipranavir och triazolam) kan minska plasmakoncentrationerna av dessa läkemedel. Överväg alternativa behandlingar som inte är känsliga substrat för dessa enzymer vid behandling med vorasidenib.
Interaktioner med transportörer
In vitro är vorasidenib en hämmare av BCRP (breast cancer resistance protein) (se avsnitt Farmakokinetiska egenskaper). Försiktighet ska iakttas vid administrering av vorasidenib tillsammans med BCRP-substrat (inkluderande, men inte begränsat till, rosuvastatin).
Hormonella preventivmedel
Vorasidenib kan minska koncentrationerna av hormonella preventivmedel. Samtidig användning av ett barriärpreventivmedel rekommenderas därför under behandlingen och i minst två månader efter den sista dosen (se avsnitt Varningar och försiktighet och Fertilitet, graviditet och amning).
Fertilitet, graviditet och amning
Fertila kvinnor och preventivmedel
Graviditetstestning rekommenderas för fertila kvinnor före behandling med vorasidenib inleds (se avsnitt Varningar och försiktighet).
Fertila kvinnor och män med fertil kvinnlig partner ska använda effektivt preventivmedel under behandlingen och i minst två månader efter den sista dosen. Eftersom det inte har undersökts hur vorasidenib inverkar på hormonella preventivmedels metabolism och effekt ska barriärmetoder användas som en ytterligare preventivmetod för att undvika graviditet (se avsnitt Varningar och försiktighet och Interaktioner).
Graviditet
Det finns inga eller begränsad mängd data från användningen av vorasidenib hos gravida kvinnor. Data från djurstudier har visat toxikologiska effekter på utveckling hos embryo/foster (se avsnitt Prekliniska säkerhetsuppgifter).
Vorasidenib ska inte användas under graviditet eller till fertila kvinnor som inte använder preventivmedel. Fertila kvinnor eller män med fertil kvinnlig partner ska upplysas om den möjliga risken för fosterskador.
Amning
Det är okänt om vorasidenib och dess metaboliter utsöndras i bröstmjölk. Amning ska avbrytas under behandlingen och i minst två månader efter den sista dosen.
Fertilitet
Det finns inga data om vorasidenibs inverkan på fertilitet hos människa. I toxicitetsstudier med upprepad dosering till hondjur och handjur observerades fynd på reproduktionsorganen (se avsnitt Prekliniska säkerhetsuppgifter). Den kliniska relevansen av dessa effekter är inte känd. Manliga och kvinnliga patienter som planerar att skaffa barn ska rekommenderas att söka reproduktionsrådgivning, och män ska söka råd kring att frysförvara sperma innan behandlingen inleds (se avsnitt Varningar och försiktighet).
Effekter på förmågan att framföra fordon och använda maskiner
Vorasidenib har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste biverkningarna, inklusive laboratorieavvikelser, var förhöjt ALAT (59,3 %), förhöjt ASAT (45,5 %), förhöjt GGT (37,7 %), utmattning (36,5 %) samt diarré (24,6 %).
De vanligaste biverkningarna av grad ≥ 3 var förhöjt ALAT (9,6 %), förhöjt ASAT (4,2 %) och förhöjt GGT (3,0 %).
Leversvikt, autoimmun leverinflammation och förhöjt ALAT förekom var och en som allvarlig biverkning hos 0,6 % av de patienter som fick Voranigo.
Permanent utsättning av vorasidenib, till följd av förhöjt ALAT av grad ≥ 3 eller autoimmun leverinflammation, rapporterades hos 3,0 % respektive 0,6 % av patienterna.
Utsättning till följd av biverkning förekom hos 18,6 % av de patienter som behandlades med vorasidenib. De vanligaste biverkningar som krävde utsättning var förhöjt ALAT (14,4 %) och förhöjt ASAT (6,0 %).
Minskad dos av vorasidenib till följd av biverkning förekom hos 9,6 % av patienterna. Den vanligaste biverkning som krävde dosminskning var förhöjt ALAT (7,8 %).
Tabell över biverkningar
Biverkningarna som beskrivs i detta avsnitt grundar sig på studiedata (INDIGO-studie [N = 167]) och erfarenhet efter godkännande för försäljning av Voranigo.
Biverkningarna som rapporterats hos patienter som behandlats med vorasidenib framgår av tabell 3, nedan, indelade enligt MedDRA-organsystem och frekvens.
Frekvenserna definieras som mycket vanliga (≥ 1/10); vanliga (≥ 1/100, < 1/10); mindre vanliga (≥ 1/1 000, < 1/100); sällsynta (≥ 1/10 000, < 1/1 000); mycket sällsynta (< 1/10 000); ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom respektive frekvensgruppering presenteras biverkningarna i fallande allvarlighetsgrad.
Tabell 3: Läkemedelsbiverkningar rapporterade hos patienter behandlade med vorasidenib
| Organsystem | Frekvens | Biverkningar |
| Blodet och lymfsystemet | mycket vanliga | minskat trombocytantala |
| Metabolism och nutrition | vanliga | hyperglykemi |
| minskad aptit | ||
| hypofosfatemi | ||
| Centrala och perifera nervsystemet | mycket vanliga | yrsel |
| Andningsvägar, bröstkorg och mediastinum | vanliga | andnöd |
| Magtarmkanalen | mycket vanliga | diarré |
| buksmärta | ||
| vanliga | gastroesofagal refluxsjukdom | |
| Lever och gallvägar | mycket vanliga | förhöjt alaninaminotransferasa |
| förhöjt aspartataminotransferasa | ||
| förhöjt gammaglutamyltransferasa | ||
| vanliga | förhöjt alkaliskt fosfatasa | |
| förhöjt bilirubin i bloda | ||
| mindre vanliga | leversvikt | |
| autoimmun leverinflammation | ||
| levernekros | ||
| ingen känd frekvens | läkemedelsinducerad leverskada* | |
| akut hepatit* | ||
| Allmänna symtom och/eller symtom vid administreringsstället | mycket vanliga | utmattning |
* Identifierade vid användning efter godkännande.
a Laboratorieavvikelser definieras som nya eller förvärrade symtom med minst en grad från baslinjen eller när baslinjen är okänd.
Beskrivning av ett urval av biverkningar
Levertoxicitet
Av de 167 patienter som behandlats med vorasidenib i INDIGO-studien fick 18,6 % förhöjt ALAT > 3 gånger ULN och 8,4 % fick förhöjt ASAT > 3 gånger ULN. Bland dessa patienter hade 1,2 % samtidigt förhöjt ALAT eller ASAT > 3 gånger ULN och totalt bilirubin > 2 gånger ULN. Förhöjda leverenzymer och bilirubin var övergående och gick tillbaka delvis eller helt efter dosjustering eller permanent utsättning av behandlingen (se avsnitt Dosering och administreringssätt och Varningar och försiktighet). Mediantiden till första debut för förhöjt ASAT och ALAT (alla grader) var 85,0 dagar (intervall: 14‑451 dagar) respektive 57,0 dagar (intervall: 1‑506 dagar). Var och en av följande biverkningar, leversvikt, levernekros och autoimmun leverinflammation, rapporterades en gång med vorasidenib i INDIGO-studien (se avsnitt Varningar och försiktighet).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
I händelse av överdosering är det sannolikt att toxiciteten visar sig som en försämring av de biverkningar som är förknippade med vorasidenib (se avsnitt Biverkningar). Patienterna ska övervakas noga och ges lämplig stödjande vård (se avsnitt Dosering och administreringssätt och Varningar och försiktighet). Det finns ingen specifik antidot mot överdosering av vorasidenib.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel; övriga antineoplastiska medel
ATC-kod: L01XM04
Verkningsmekanism
Vorasidenib är en hämmare som verkar på muterade IDH1- och IDH2-enzymer. Hos patienter med astrocytom eller oligodendrogliom kan IDH1- och IDH2-mutationer medföra överproduktion av den onkogena metaboliten 2-hydroxiglutarat (2-HG), vilket medför en nedsatt celldifferentiering som bidrar till onkogenes. Genom hämning av de IDH1- och IDH2-muterade proteinerna med hjälp av vorasidenib hämmas den onormala produktionen av 2-HG, vilket leder till differentiering och minskad förökning av maligna celler. Inga prekliniska studier av har utförts avseende vorasidenibs förmåga att minska tumörers storlek.
Farmakodynamisk effekt
En terapeutisk daglig dos av vorasidenib sågs minska 2-HG-koncentrationen i tumörer hos försökspersoner med IDH1- eller IDH2-muterat gliom.
Klinisk effekt och säkerhet
Effekt och säkerhet för vorasidenib utvärderades i prövningen INDIGO, en randomiserad (1:1) dubbelblind placebokontrollerad multicenterstudie i fas 3 som omfattade 331 vuxna och ungdomar ≥ 12 år med en kroppsvikt på ≥ 40 kg. För att vara lämplig som patient i studien krävdes astrocytom eller oligodendrogliom av grad 2 enligt WHO:s kriterier från 2016 med IDH1 R132- eller IDH2 R172-mutation, tidigare kirurgi, inklusive biopsi och delvis eller fullständig resektion, som enda behandling och att inte, enligt prövarens bedömning, vara i behov av omedelbar kemoterapi eller strålbehandling. De patienter som skrevs in hade sjukdom som var MR-bedömbar, mätbar och icke kontrastförstärkt, vilket hade bekräftats av en blindad oberoende granskningskommitté (BIRC). Patienter med centralt bekräftad kontrastförstärkt sjukdom fick skrivas in förutsatt att progressionen var minimal, icke-nodulär, icke-mätbar och oförändrad mellan de två senaste avbildningarna. INDIGO-prövningen exkluderade patienter som fått någon tidigare cancerbehandling, inklusive kemoterapi- eller strålbehandling. IDH1- eller IDH2-mutationsstatus bestämdes prospektivt med hjälp av Oncomine Dx Target Test.
Patienterna randomiserades till att få antingen 40 mg vorasidenib peroralt en gång om dagen, eller motsvarande placebo, fram till radiografiskt konstaterad sjukdomsprogression eller oacceptabel toxicitet. Randomiseringen stratifierades efter lokal 1p19q-status (kodeletion eller utan kodeletion) samt tumörens storlek vid baslinjen (diameter ≥ 2 cm eller < 2 cm). Patienter som randomiserats till placebo gavs möjlighet att gå över till att få vorasidenib, efter radiografiskt konstaterad sjukdomsprogression, förutsatt att de enligt prövarens bedömning inte var i behov av omedelbar kemoterapi- eller strålbehandling.
Det primära effektmåttet var radiografiskt konstaterad progressionsfri överlevnad (PFS) bedömd av en BIRC i enlighet med en modifierad version (endast radiografiskt konstaterad sjukdomsprogression) av RANO–LGG-kriterierna (Response Assessment in Neuro-Oncology for Low Grade Glioma).
Patientdemografi och sjukdomsrelaterade karakteristika var balanserade mellan behandlingsarmarna. Hos de 168 patienter som randomiserats till vorasidenib var medianåldern 41 år (intervall: 21 till 71 år), varav 98,8 % var i åldrarna 18–64 år. En enda pediatrisk patient, 16 år, randomiserades till placebo och ingen patient under 18 år randomiserades till vorasidenib. En majoritet av patienterna var män (60,1 %), 74,4 % var vita, 3,0 % asiater, 1,2 % svarta, 1,2 % övriga, 19,6 % ej angivna och 53,6 % hade en KPS-poäng (Karnofsky Performance Status) på 100, 45,8 % hade en KPS-poäng på 90–80 och 0,6 % hade en KPS-poäng på 70–60. De flesta patienterna hade genomgått minst 1 kirurgiskt ingrepp för gliom (75 %) och 25 % hade genomgått ≥ 2 kirurgiska ingrepp. Båda behandlingsarmarna inräknat hade 95 % av patienterna IDH1 R132-mutation och 5 % IDH2 R172-mutation.
Effektutfallen för PFS sammanfattas i tabell 4 och figur 1.
Tabell 4: Effektutfall för prövningen INDIGO (studie AG881-C-004)
Effektparameter | Voranigo 40 mg per dag (n = 168) | Placebo (n = 163) |
Progressionsfri överlevnad (PFS) | ||
Antal händelser, n (%) progredierande sjukdom dödsfall | 47 (28,0) 0 | 88 (54,0) 0 |
Median-PFS, månader (95 % KI)a | 27,7 (17,0; NE) | 11,1 (11,0; 13,7) |
Riskkvot (95 % KI)b | 0,39 (0,27; 0,56) | |
p-värdec | 0,000000067 | |
Förkortningar: KI = konfidensintervall; NE = obestämbart
Det fullständiga analysurvalet omfattade samtliga patienter som genomgått randomisering.
- Konfidensintervallet 95 % för medianen beräknades enligt Brookmeyers och Crowleys metod.
- Beräknad med Cox proportionella riskmodell justerad för följande stratifieringsfaktorer: 1p19q-status och tumörstorlek vid baslinjen.
- Beräknad utifrån ett ensidigt stratifierat log–ranktest. PFS testades med en ensidig effekt-α-nivå på 0,000359, baserat på en uppdaterad effektgräns motsvarande 82 % informationsfraktion.
Figur 1:Kaplan–Meier-kurva för progressionsfri överlevnad per BIRC-granskning i prövningen INDIGO
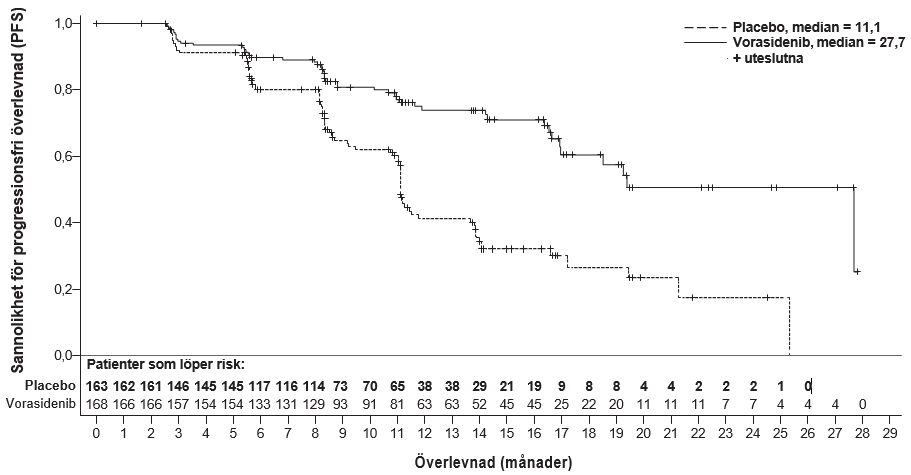
En uppdaterad analys av PFS per BIRC-granskning, utförd på 96 % (N = 158) av händelserna, bekräftade nyttan av vorasidenib jämfört med placebo (riskkvot: 0,35 [95 % KI: 0,25; 0,49]). Vid 24 månader var andelen progressionsfri överlevnad 59 % (95 % KI: 48,4; 67,8) i vorasidenibgruppen och 26 % (95 % KI: 17,9; 35,3) i placebogruppen. Median-PFS gick inte att beräkna (95 % KI: 22,1, NE) för vorasidenibgruppen och var 11,4 månader (95 % KI: 11,1; 13,9) för placebogruppen.
Pediatrisk population
Ungdomar från 12 års ålder till yngre än 18 år
Det finns stöd för användning av vorasidenib till patienter från 12 år till yngre än 18 år, med IDH1- eller IDH2-muterat astrocytom eller oligodendrogliom, eftersom farmakokinetiska data visade att ålder inte hade någon kliniskt betydelsefull inverkan på farmakokinetiken hos vorasidenib (se avsnitt Farmakokinetiska egenskaper).
Farmakokinetiska egenskaper
Farmakokinetiken för vorasidenib har karakteriserats för patienter med låggradigt gliom med en IDH1- eller IDH2-mutation samt för friska försökspersoner. Den farmakokinetiska profilen för vorasidenib är likvärdig för patienter med låggradigt gliom och friska försökspersoner.
Absorption
Efter en enstaka peroral dos på 40 mg var mediantiden till Cmax (Tmax) för vorasidenib 2,0 timmar, geometriskt medelvärde för Cmax var 75,4 ng/ml (CV%: 44) och geometriskt medelvärde för AUC var 2 860 h∙ng/ml (CV%: 56). Vid steady-state var det geometriska medelvärdet Cmax för vorasidenib 133 ng/ml (CV%: 73) och geometriskt medelvärde för AUC var 1 988 h∙ng/ml (CV%: 95). Hos flertalet patienter nådde plasmakoncentrationen en ny höjdpunkt inom 24 timmar efter administrering av läkemedlet, men denna var lägre än det Cmax som observerats två timmar efter dosering. Även om den absoluta biotillgängligheten inte har fastställts direkt bedöms vorasidenib ha måttlig till hög absorptionsgrad vid intag av 40 mg filmdragerade tabletter.
Ackumulationskvoterna var cirka 3,8 för Cmax och 4,4 för AUC. Steady state-nivåer i plasma uppnåddes efter två till tre veckor med dosering en gång om dagen.
Genomsnittlig Cmax och AUC för vorasidenib ökade med 3,1 respektive 1,4 gånger när vorasidenib administrerades tillsammans med en fettrik måltid. Administrering av vorasidenib tillsammans med en fettsnål måltid medförde en ökning av Cmax och AUC med 2,3 respektive 1,4 gånger (se avsnitt Dosering och administreringssätt).
Distribution
Vorasidenib har en genomsnittlig skenbar distributionsvolym på 3 930 l (CV%: 40). Distributionsvolymen för vorasidenib, efter en enstaka intravenös mikrodos på 0,1 mg, är 1 110 l. Plasmaproteinbindningskvoten var 97 procent för vorasidenib och 87 procent för AGI-69460. Både vorasidenib och AGI-69460 uppvisar en preferens för bindning till serumalbumin före surt alfa-1-glykoprotein. För vorasidenib är blod/plasmakvoten 0,87, för AGI-69460 är blod‑plasmakvoten 1,38 och för vorasidenib är hjärntumör‑plasmakvoten 1,6.
Metabolism
Vorasidenib metaboliseras företrädesvis av CYP1A2 med obetydliga till ringa bidrag från CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 och CYP3A4/5. Andra metaboliseringsvägar än CYP kan bidra med upp till 30 % av clearance av vorasidenib via levern.
AGI-69460 är en aktiv nedströmsmetabolit av vorasidenib. Efter en enstaka peroral dos på 40 mg vorasidenib var observerad Tmax för metaboliten AGI-69460 336 timmar, observerat geometriskt medelvärde för Cmax 3,32 ng/ml (CV%: 55,6) och geometriskt medelvärde för AUC0-t var 1 172 h∙ng/ml (CV%: 61). Vid steady-state var det geometriska medelvärdet för AGI-69460 Cmin,ss 111 ng/ml (CV%: 58) och geometriskt medelvärde för AUC0–4 på dag 1 i cykel 2 var 190 h∙ng/ml (CV%: 90).
Interaktioner
In vitro har vorasidenib en starkt inducerande verkan på känsliga CYP3A4-substrat och en måttligt inducerande verkan på känsliga substrat för CYP2B6 och CYP2C19 (se avsnitt Interaktioner).
In vitro-data tyder på att vorasidenib är en hämmare av BCRP. Vorasidenib hämmar inte P-glykoprotein (P-gp) och levertransportören organisk anjonisk transportpolypeptid (OATP)1B1. AGI-69460 inhiberar BCRP och OATP1B3 in vitro.
Vorasidenib är inte ett substrat för P-gp, BCRP eller levertransportörerna OATP1B1 och OATP1B3.
Eliminering
Cirka 89 % av den administrerade dosen av radioaktivt vorasidenib, formulerat som pulverfylld kapsel med en absolut biotillgänglighet på < 34 %, återfanns på 44 dagar, varav 85 % i feces och 4,5 % i urin. Större delen av den administrerade radioaktivitet som återfanns i feces utgjordes av oförändrat vorasidenib (55 %) medan inget oförändrat vorasidenib detekterades i urin.
Den genomsnittliga terminala halveringstiden för vorasidenib är 238 timmar (CV%: 57), effektiv halveringstid är 63,2 timmar (CV%: 75) och skenbar clearance är 14,0 l/h (CV%: 56).
Linjäritet/icke-linjäritet
Efter administrering av Voranigo ökade Cmax och AUC för vorasidenib proportionellt mellan 10 och 40 mg.
Särskilda populationer
Äldre
Inga kliniskt betydelsefulla effekter på vorasidenibs farmakokinetik har observerats hos äldre patienter upp till 75 år (se avsnitt Dosering och administreringssätt).
Nedsatt njurfunktion
Nedsatt njurfunktion (eGFR > 40 ml/min/1,73 m²) hade ingen kliniskt signifikant inverkan på farmakokinetiken för vorasidenib. Farmakokinetiken för vorasidenib hos patienter med eGFR ≤ 40 ml/min/1,73 m² eller nedsatt njurfunktion som kräver dialys är inte känd.
Nedsatt leverfunktion
Måttligt nedsatt leverfunktion (Child-Pugh-klass B) hade ingen kliniskt signifikant inverkan på farmakokinetiken för vorasidenib och AGI-69460. Det förekom inga kliniskt relevanta förändringar av totala eller fria (obundna) vorasidenibkoncentrationer (liknande Cmax-värden för vorasidenib och en ökning med 26,0 % av AUC0-t för vorasidenib observerades samtidigt med minskad exponering för AGI-69460) hos patienter med måttligt nedsatt leverfunktion efter en enstaka peroral dos på 20 mg vorasidenib. Farmakokinetiken för vorasidenib och AGI-69460 hos patienter med svårt nedsatt leverfunktion (Child-Pugh-klass C) är inte känd (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Övrigt
Inga kliniskt signifikanta effekter på farmakokinetiken för vorasidenib kunde observeras baserat på ålder (16 till 75 år), ras, etnicitet och kroppsvikt (43,5 till 168 kg). Kvinnliga patienter observerades ha 1,6 gånger högre exponering för vorasidenib jämfört med manliga patienter.
Pediatrisk population
Farmakokinetiska data visade att ålder inte hade någon kliniskt betydelsefull inverkan på farmakokinetiken för vorasidenib. Exponeringen för vorasidenib väntas vara densamma hos vuxna som hos unga patienter från 12 års ålder.
Prekliniska säkerhetsuppgifter
De huvudsakliga måltoxiciteterna identifierades under toxicitetsstudier med upprepad dosering, avseende lever, magtarmkanal, hud, njurar, skelettmuskler, reproduktionsorgan och bröstkörtel.
Vorasidenib befanns inte vara gentoxisk i det omvända bakteriella mutationstestet (Ames test) in vitro, mikrokärntest in vitro på lymfocyter från människa samt mikrokärntest in vivo på benmärg från råtta. AGI-69460, dess främsta cirkulerande metabolit, befanns inte vara gentoxisk i Ames test, mikrokärntest in vitro på lymfocyter från människa samt mikrokärntest in vivo på benmärg från råtta och Comet‑analys.
Under en 13 veckor lång studie på apor vid åttadubbel klinisk exponering, observerades hyperplasi av Kupfferceller vid primär obduktion, vilken förvärrades efter en återhämtningsperiod. Toxicitetsstudier på råtta tydde dessutom på hormonrubbningar. Dylika fynd kan tyda på en möjlig karcinogen risk. Inga karcinogenicitetsstudier har ännu utförts med vorasidenib.
Inga fertilitetsstudier på djur har utförts med vorasidenib. Inverkan på reproduktionsorganen konstaterades efter administrering av vorasidenib till råtta under toxicitetsstudier med upprepad dosering. Biverkningar på reproduktionsorganen hos honor omfattade atrofi i äggstockar, livmoder, livmoderhals och vagina samt variationer i östruscykeln. Hos hanråttor konstaterades effekter på epididymis (celldebris), sädesblåsa/prostata (atrofi) samt testiklar (vikt, tubulär degeneration). Dessa fynd observerades vid den lägsta testade dosen på 5 mg/kg/dag i en 13 veckor lång studie på råtta, vilket medförde en exponeringsnivå 26 gånger högre än exponeringen hos människa vid en daglig dos på 50 mg.
Vorasidenib medförde embryofetal toxicitet hos dräktiga råttor och kaniner (högre incidens av resorptioner, försenad benbildning, viscerala missbildningar av njurar och testiklar hos råtta). Dessa effekter uppkom vid doser som var högre än vad patienter får som en terapeutisk daglig dos. Exponeringskvoterna vid NOAEL för embryofetal utveckling hos råtta och kanin var 8,0 till 28,5 respektive 1,1 till 4,9 på dräktighetsdag 6 och 17 för råtta respektive 6 och 19 för kanin.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Tablettkärna
mikrokristallin cellulosa (E460)
kroskarmellosnatrium
silicifierad mikrokristallin cellulosa (innehåller mikrokristallin cellulosa och kolloidal vattenfri kiseldioxid)
magnesiumstearat (E470b)
natriumlaurilsulfat (E487)
Tablettens filmdragering
hypromellos
titandioxid (E171)
laktosmonohydrat
makrogol (E1521)
Tryckfärg
svart järnoxid (E172)
propylenglykol (E1520)
hypromellos (E464)
Inkompatibiliteter
Ej relevant.
Hållbarhet
36 månader.
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
VORANIGO tabletti, kalvopäällysteinen
10 mg (L:kyllä) 30 kpl (10129,22 €)
40 mg (L:kyllä) 30 kpl (20077,83 €)
PF-selosteen tieto
Vit burk av HDPE med barnskyddande förslutning av polypropen (PP) och induktionsvärmeförseglad film av polyeten (PE) innehållande tre HDPE-behållare med torkmedel av kiselgel. Förpackning med 30 filmdragerade tabletter.
Läkemedlets utseende:
Voranigo 10 mg filmdragerade tabletter
Vita till benvita, runda tabletter, 6 mm i diameter, präglade med ”10” på ena sidan.
Voranigo 40 mg filmdragerade tabletter
Vita till benvita, avlånga tabletter, 14,8 mm långa och 6,3 mm breda, präglade med ”40” på ena sidan.
Särskilda anvisningar för destruktion och övrig hantering
Patienterna ska avrådas från att svälja det torkmedel av kiselgel som finns i tablettburken.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
VORANIGO tabletti, kalvopäällysteinen
10 mg 30 kpl
40 mg 30 kpl
- Ei korvausta.
Atc-kod
L01XM04
Datum för översyn av produktresumén
10.04.2026
Yhteystiedot
Äyritie 12 A
01510 Vantaa
040 901 2978
www.servierfinland.fi
info.finland@servier.com