
DUVYZAT oral suspension 8,86 mg/ml
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar i produktresumén om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
En ml innehåller 8,86 mg givinostat (som hydrokloridmonohydrat).
Hjälpämne(n) med känd effekt
En ml innehåller 4,4 mg natriumbensoat (E211).
En ml innehåller 400 mg sorbitol (E420).
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Oral suspension.
Kliniska uppgifter
Terapeutiska indikationer
Duvyzat är avsett för behandling av Duchennes muskeldystrofi (DMD) hos patienter med bevarad gångförmåga från 6 års ålder som får samtidig behandling med kortikosteroider.
Villkor
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Dosering och administreringssätt
Behandling med givinostat ska påbörjas av läkare med erfarenhet av att behandla patienter med Duchennes muskeldystrofi.
Dosering
Trombocytantal och triglyceridnivåer ska kontrolleras innan behandling med givinostat inleds. Behandling med givinostat ska inte påbörjas hos patienter med ett trombocytantal under 150 × 109/l. Under behandling ska trombocytantal och triglyceridnivåer övervakas enligt rekommendation nedan för att avgöra eventuellt behov av dosändringar (se avsnitt Varningar och försiktighet samt nedanstående anvisningar för dosjustering).
Hos patienter med underliggande hjärtsjukdom eller de som har samtidig behandling med läkemedel som orsakar QT-förlängning ska ett EKG tas i samband med att behandling med givinostat inleds, under samtidig behandling med QT-förlängare samt vid kliniskt behov (se avsnitt Varningar och försiktighet).
Rekommenderad dos givinostat är baserad på kroppsvikt och ska ges peroralt två gånger dagligen (se tabell 1).
Tabell 1 – Doseringsrekommendationer
| Vikt(a) | Dos | Mängd oral suspension |
| 15 kg till mindre än 20 kg | 22,2 mg två gånger dagligen | 2,5 ml två gånger dagligen |
| 20 kg till mindre än 40 kg | 31 mg två gånger dagligen | 3,5 ml två gånger dagligen |
| 40 kg till mindre än 60 kg | 44,3 mg två gånger dagligen | 5 ml två gånger dagligen |
| 60 kg eller mer | 53,2 mg två gånger dagligen | 6 ml två gånger dagligen |
(a) Baserat på faktisk kroppsvikt
Beslut om fortsatt behandling av patienter som förlorat gångförmågan ska fattas av behandlande läkare baserat på en övergripande nytta-riskbedömning.
Dosjustering för trombocytopeni, diarré eller hypertriglyceridemi
Dosen ska sänkas (se tabell 2) för patienter med:
- mindre än 150 × 109 trombocyter/l, verifierat med två mätningar med en veckas mellanrum
- måttlig till svår diarré (fler än 4 tarmtömningar per dag)
- mer än 300 mg triglycerider/dl vid fastande, verifierat med två mätningar med en veckas mellanrum.
Baserat på biverkningarnas allvarlighetsgrad ska behandlingsuppehåll övervägas före dosändring.
Tabell 2 – Dosändringar vid biverkningar
| Första dosändring(b) | Andra dosändring(c) | |||
| Vikt(a) | Dos | Mängd oral suspension | Dos | Mängd oral suspension |
| 15 kg till mindre än 20 kg | 17,7 mg två gånger dagligen | 2 ml två gånger dagligen | 13,3 mg två gånger dagligen | 1,5 ml två gånger dagligen |
| 20 kg till mindre än 40 kg | 22,2 mg två gånger dagligen | 2,5 ml två gånger dagligen | 17,7 mg två gånger dagligen | 2 ml två gånger dagligen |
| 40 kg till mindre än 60 kg | 31 mg två gånger dagligen | 3,5 ml två gånger dagligen | 26,6 mg två gånger dagligen | 3 ml två gånger dagligen |
| 60 kg eller mer | 39,9 mg två gånger dagligen | 4,5 ml två gånger dagligen | 35,4 mg två gånger dagligen | 4 ml två gånger dagligen |
(a) Baserat på faktisk kroppsvikt
(b) Om biverkningen(-arna) kvarstår efter den första dosändringen, fortsätt med den andra dosändringen.
(c) Om biverkningen(-arna) kvarstår efter den andra dosändringen ska Duvyzat sättas ut.
Patienter ska inte ta dubbel dos eller en extrados om en dos har missats.
Pediatrisk population
Effekt och säkerhet för Duvyzat hos barn under 6 års ålder har inte fastställts. Inga data finns tillgängliga.
Administreringssätt
För oral användning.
Före användning måste suspensionen skakas i minst 30 sekunder genom att vända på flaskan 180 grader upp och ned cirka 40 gånger, och man ska därefter kontrollera att suspensionen ser homogen ut.
Felaktig skakning kan orsaka över- eller underdosering.
Duvyzat måste tas som det är (dvs. inte spädas med vatten eller andra vätskor).
Suspensionen ska administreras med hjälp av den medföljande graderade orala doseringssprutan för att mäta upp den mängd suspension som motsvarar patientens förskrivna dos.
Duvyzat ska tas tillsammans med mat för att motverka den beska smaken hos givinostat.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Hematologiska effekter
Givinostat är förknippat med dosrelaterad trombocytopeni och andra tecken på myelosuppression, inklusive minskat hemoglobinvärde och neutropeni.
Effekten är mest uttalad på trombocytantalet (se avsnitt Biverkningar).
Fullständig blodstatus ska tas innan behandling med givinostat inleds. Trombocytantalet ska övervakas noggrant under behandling med givinostat, dvs. varannan vecka de första 2 månaderna, i månad 3 och var 3:e månad därefter.
Vid ihållande trombocytopeni ska dosen givinostat justeras. Behandlingen ska avbrytas om besvären kvarstår (se avsnitt Dosering och administreringssätt).
Hos patienter vars dos höjts på grund av viktökning ska trombocytantalet övervakas noggrant varannan vecka under de första 2 månaderna efter doshöjningen.
Förhöjda triglyceridnivåer
Givinostat är förknippat med förhöjda triglyceridnivåer.
Triglyceridnivåer ska mätas innan behandling med givinostat inleds.
Triglyceridnivåer ska övervakas som minst efter tre månader, efter sex månader och därefter varje halvår.
För DMD-patienter med ihållande förhöjda triglyceridnivåer i fastande tillstånd (> 300 mg/dl) ska dosen av givinostat justeras enligt anvisningarna i avsnitt Dosering och administreringssätt.
Behandling med givinostat ska avbrytas om triglyceridnivåer förblir förhöjda trots adekvat kostintervention och dosjustering (se avsnitt Dosering och administreringssätt).
Mag-tarmrubbningar
Diarré och kräkningar var mycket vanliga biverkningar i kliniska prövningar med givinostat inom DMD (se avsnitt Biverkningar).
Diarré och kräkningar är ofta förekommande de första veckorna efter att behandling med givinostat har inletts.
Antiemetika eller läkemedel mot diarré kan övervägas under behandling med givinostat.
Vätska och elektrolyter ska ersättas efter behov för att förebygga uttorkning.
Dosen givinostat ska justeras vid måttlig till svår diarré (fler än 4 tarmtömningar per dag) (se avsnitt Dosering och administreringssätt).
Behandlingen ska avbrytas om besvären kvarstår (se avsnitt Dosering och administreringssätt).
QTc-förlängning
Givinostat kan orsaka förlängt QTc-intervall vid doser som är 5 gånger högre än rekommenderad dos.
Givinostat ska användas med försiktighet hos patienter med ökad risk för ventrikulära arytmier (inklusive torsades de pointes), patienter med medfött långt QT-syndrom, kransartärsjukdom, elektrolytrubbningar eller vid samtidig användning av andra läkemedel som man vet orsakar QT-förlängning. Hos dessa patienter ska EKG tas i samband med att behandling med Duvyzat inleds, under samtidig behandling med QT-förlängare samt vid kliniskt behov.
Hos patienter med hypokalemi ska detta korrigeras innan behandling med givinostat inleds och övervakas i händelse av uttorkning på grund av diarré.
Duvyzat ska tillfälligt sättas ut om QT-intervallet är > 500 ms eller om förändringen från baseline är > 60 ms.
Hjälpämnen med känd effekt
Patienter med hereditär fruktosintolerans bör inte använda detta läkemedel.
Detta läkemedel innehåller 400 mg sorbitol per ml vilket motsvarar 40 mg/kg.
Additiv effekt av samtidigt administrerade läkemedel som innehåller sorbitol (eller fruktos) och födointag av sorbitol (eller fruktos) ska beaktas.
Innehåll av sorbitol i läkemedel för oralt bruk kan påverka biotillgängligheten av andra läkemedel för oralt bruk som administreras samtidigt.
Detta läkemedel innehåller 4,4 mg natriumbensoat per ml vilket motsvarar 0,44 mg/kg.
Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per dos, d.v.s. är näst intill ”natriumfritt”.
Interaktioner
Försiktighet rekommenderas när Duvyzat förskrivs med läkemedel med känd QT-förlängande effekt med känd eller möjlig risk för torsades de pointes, t.ex. anestetika (t.ex. sevofluran, propofol), antiarytmika klass III (t.ex. amiodaron, sotalol), antiemetika (ondasetron), antibiotika (azitromycin, klaritromycin, ciprofloxacin), antimykotika (flukonazol), antipsykotika (aripiprazol, risperidon) och antihistaminer (t.ex. famotidin). Listan är vägledande och inte fullständig.
Effekten av samtidig användning av Dyvyzat och antitrombotiska medel på trombocytantalet är okänd.
Duvyzat ska användas med försiktighet hos patienter som får samtidig behandling med läkemedel kända för att öka triglyceridvärden eftersom detta kan öka risken för hypertriglyceridemi.
Givinostats effekter på andra läkemedels farmakokinetik
En svag CYP3A4-hämning, framför allt i tarmen, har visats i en läkemedelsinteraktionsstudie (DDI) på människa. Försiktighet bör iakttas vid samtidig administrering av givinostat och läkemedel som är substrat för CYP3A4 och har ett smalt terapeutiskt intervall.
En potentiell risk för hämning av transportproteinet P-gp i tarmen kan inte uteslutas. Läkemedel som är kända substrat för P-gp-transportören och har ett smalt terapeutiskt intervall bör användas med försiktighet tillsammans med givinostat.
En svag hämning av den renala upptagstransportören OCT2 har observerats in vitro och i kliniska prövningar med givinostat genom kreatininmätningar. Läkemedel som är kända substrat för OCT2-transportören och som har ett smalt terapeutiskt intervall bör användas med försiktighet tillsammans med givinostat.
Fertilitet, graviditet och amning
Graviditet
Det finns inga data från användning av givinostat hos gravida kvinnor. Djurstudier har visat reproduktionstoxicitet (se avsnitt Prekliniska säkerhetsuppgifter).
Som en försiktighetsåtgärd bör användning av givinostat undvikas under graviditet.
Amning
Det är okänt om givinostat eller dess metaboliter utsöndras i bröstmjölk. En risk för det ammande barnet kan inte uteslutas. Givinostat ska inte användas under amning.
Fertilitet
Det finns inga data om effekten av givinostat på fertilitet hos människa. Hos hanråttor visades biverkningar av givinostat på accessoriska könskörtlar, men fertiliteten hos djur påverkades inte (se avsnitt Prekliniska säkerhetsuppgifter). Relevansen för människa är okänd.
Effekter på förmågan att framföra fordon och använda maskiner
Givinostat kan ha mindre påverkan på förmågan att framföra fordon och använda maskiner.
Yrsel och utmattning kan förekomma efter administrering av givinostat (se avsnitt Biverkningar).
Biverkningar
Sammanfattning av säkerhetsprofilen
Säkerhetsprofilen för Duvyzat är baserad på en dubbelblind, placebokontrollerad, 18 månader lång fas 3-studie med totalt 179 DMD-patienter med bevarad gångförmåga i åldern 6 år eller äldre med samtidig kortikosteroidbehandling. Av dessa fick 118 patienter upp till 62 mg givinostat två gånger dagligen och 61 fick placebo (EPIDYS-studien).
De vanligaste rapporterade händelserna i den placebokontrollerade studien (baserat på aggregerade termer där så är tillämpligt) var diarré (38,1 %), buksmärtor (33,9 %), trombocytopeni (32,2 %), kräkningar (28,8 %) och hypertriglyceridemi (22,9 %).
Tabell över biverkningar
Biverkningarna presenteras enligt MedDRA:s klassificering av organsystem och frekvens (se tabell 3). Tabellen innehåller biverkningar rapporterade hos givinostatbehandlade patienter med en frekvens över 2 % jämfört med placebobehandlade patienter i EPIDYS-studien.
Frekvenserna anges enligt följande: Mycket vanliga (≥1/10), vanliga (≥1/100, <1/10), mindre vanliga (≥1/1000, <1/100), sällsynta (≥1/10 000, <1/1000), mycket sällsynta (<1/10 000), ingen känd frekvens (kan inte beräknas från tillgängliga data).
Tabell 3 – Biverkningar rapporterade med en frekvens över 2 % hos givinostatbehandlade patienter jämfört med placebo i den placebokontrollerade EPIDYS-studien
Organsystem | Mycket vanliga | Vanliga |
Infektioner och infestationer |
| Gastroenterit |
Blodet och lymfsystemet | Trombocytopeni(a) |
|
Metabolism och nutrition | Hypertriglyceridemi(b) | Minskad aptit |
Psykiatriska störningar |
| Ångest |
Centrala och perifera nervsystemet |
| Yrsel |
Blodkärl |
| Hematom |
Magtarmkanalen | Diarré(c), kräkningar, buksmärta(d) | Förstoppning |
Hud och subkutan vävnad |
| Erytem, hudutslag |
Muskuloskeletala systemet och bindväv |
| Myalgi, artralgi, muskelsvaghet |
Allmänna symtom och/eller symtom vid administreringsstället | Pyrexi | Utmattning |
Undersökningar och provtagningar |
| Förhöjt tyreoideastimulerande hormon i blod(e) |
(a) Trombocytopeni omfattar minskat trombocytantal och trombocytopeni
(b) Hypertriglyceridemi omfattar hypertriglyceridemi och förhöjda triglyceridnivåer i blodet
(c) Diarré omfattar diarré och mjuk avföring
(d) Buksmärta omfattar buksmärta och smärta i övre del av buk
(e) Förhöjt tyreoideastimulerande hormon i blod omfattar onormalt tyreoideafunktionstest och förhöjt tyreoideastimulerande hormon i blod.
Beskrivning av utvalda biverkningar
Hematologiska förändringar
Givinostat har visats minska trombocytantalet, med den största minskningen observerad efter cirka 88 dagar. Trombocytantalet förblev lågt under hela behandlingstiden. Inga allvarliga blödningshändelser relaterade till trombocytopeni rapporterades. Efter dosreduktion återgick trombocytantalet till normala nivåer inom cirka 3‑4 veckor.
Trombocytopeni förekom hos 32,2 % av patienterna som behandlades med Duvyzat, jämfört med inga fall i placebogruppen. Av dessa händelser rapporterades 86,8 % som lindriga och 13,2 % som måttliga.
Lågt trombocytantal ledde till dosreduktion hos 28 % av patienterna. Patienter med trombocytantal under den nedre normalgränsen vid baseline exkluderades från studien.
Minskade nivåer av hemoglobin och neutrofila granulocyter observerades också hos patienter som behandlades med givinostat jämfört med placebo.
Förändrade triglyceridnivåer
Givinostat har visats öka triglyceridnivåer, med den största ökningen observerad efter cirka 221 dagar. Efter utsättning av givinostat återgick triglyceridnivåerna till baselinevärden inom cirka 90 dagar.
Höga triglyceridnivåer (dvs. > 300 mg/dl) ledde till utsättning hos 2 % och dosjustering hos 8 % av patienterna som behandlades med Duvyzat.
Hypertriglyceridemi förekom hos 22,9 % av patienterna som behandlades med Duvyzat. Av dessa händelser rapporterades 70,4 % som lindriga, 25,9 % som måttliga och i ett fall (3,7 %) som allvarlig.
Gastrointestinala störningar
Gastrointestinala störningar inklusive diarré, kräkningar och buksmärta förekom hos patienter behandlade med givinostat.
Diarré rapporterades hos 38 % av patienter behandlade med Duvyzat (med 1 allvarligt fall rapporterat) jämfört med 18 % av patienter på placebo. Diarré förekom vanligtvis under de första veckorna efter behandlingsstart med givinostat.
Kräkning förekom hos 29 % av patienter behandlade med Duvyzat (med 2 allvarliga fall rapporterade) jämfört med 13 % av patienter på placebo. Kräkning förekom vanligtvis under de första 2 behandlingsmånaderna.
Buksmärta förekom hos 34 % av patienter behandlade med Duvyzat jämfört med 23 % av patienter på placebo. Ett fall av buksmärta var allvarligt.
Beskrivning av andra avvikande laboratorievärden
Biverkningar i form av hypotyreos och/eller förhöjda nivåer av tyreoideastimulerande hormon (TSH) förekom hos 5 % av patienter som behandlades med Duvyzat jämfört med 2 % av patienter som fick placebo.
Vid långtidsbehandling observerades dessutom hypotyreos (vanlig biverkning).
Nivåer av tyreoideastimulerande hormoner i blod var generellt inom 2 gånger den övre normalgränsen, med inga eller endast marginella förändringar av tyreoideahormoner.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
I händelse av en misstänkt överdos ska understödjande behandling, inklusive hjärtövervakning, sättas in.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Övriga medel för sjukdomar i rörelseapparaten, ATC-kod: M09AX14.
Verkningsmekanism
Givinostat är en hämmare av histondeacetylaser (HDAC) klass I och II, som modulerar den okontrollerade HDAC-aktiviteten i dystrofisk muskulatur, vilket bidrar till patologin vid Duchennes muskeldystrofi (DMD).
Givinostats HDAC-hämning har visats reducera muskelfiberskador, kronisk muskelinflammation, fibros, fettinlagring samt främja mitokondriell biogenes.
Givinostats verkningsmekanism är oberoende av den bakomliggande mutationen i dystrofingenen som orsakar sjukdomen.
Muskelfettfraktion bedömd med MR-spektroskopi
Andelen fettfraktion i musculus vastus lateralis (VLM) i låret mättes i EPIDYS-studien med hjälp av magnetresonansspektroskopi (MR-spektroskopi). Efter 18 månader, hos patienter med en VLM-fettandel i intervallet > 5 % till ≤ 30 % vid baseline, var ökningen i LS-medelvärde av VLM-fettandelen 7,63 % hos patienter behandlade med Duvyzat jämfört med 10,56 % hos patienter som fick placebo.
Klinisk effekt och säkerhet
Säkerhet och effekt av Duvyzat hos DMD-patienter utvärderades i EPIDYS-studien. EPIDYS var en 18 månader lång, randomiserad (2:1), dubbelblind, placebokontrollerad fas 3-studie som inkluderade 179 DMD-patienter med bevarad gångförmåga i åldern 6 år eller äldre. Givinostat eller placebo gavs som tillägg till en stabil dos kortikosteroider under hela studien. Patienterna rekryterades till 2 grupper:
- Grupp A (120 patienter): patienter med baslinjevärde för VL MFF i intervallet > 5 % till ≤ 30 %, fastställd med MRS.
- Grupp B (59 patienter): patienter med baslinjevärde för VL MFF utanför ovanstående intervall (övriga kriterier var samma).
En viktbaserad doseringsregim tillämpades. Startdosen var initialt 17,7‑62 mg peroralt givinostat två gånger dagligen, med en reducerad dos på 11,8‑41,4 mg två gånger dagligen. Protokollet ändrades därefter för att sänka startdosen för nya studiedeltagare till 11,8‑41,4 mg två gånger dagligen, med möjlighet till ytterligare dosreduktion på 9,4‑33,1 mg två gånger dagligen.
Det primära effektmåttet i grupp A (förutbestämd primär analysgrupp) var förändringen i tid för att gå upp fyra trappsteg (4SC) efter 18 månader.
Det primära effektmåttet uppfylldes, givinostat reducerade minskningen i 4SC signifikant (p = 0,035) jämfört med placebo baserat på den i förutbestämda analysen med logaritmisk skala (tabell 4). När resultaten analyserades med en icke-logaritmisk skala ökade genomsnittlig tid för 4SC med 1,25 sekunder i givinostatgruppen jämfört med 3,03 sekunder i placebogruppen (se tabell 4). Behandlingseffekten (förändring från baseline, givinostat minus placebo) var -1,78 sekunder (p = 0,037).
Tabell 4 – EPIDYS-studien: Tid (sekunder) för 4SC, förändring från baseline till 18 månader (grupp A)
| Tid för 4SC | Givinostat§(N = 81) | Placebo§(N = 39) |
| Analys med logaritmisk skala* | ||
| GLS-medelvärde (log-skala SE) (95 % KI) | 1,27 (0,040) (1,172; 1,372) | 1,48 (0,058) (1,317; 1,657) |
GLS-medelvärde (givinostat/placebo) (log-skala SE) (95 % KI) p-värde | 0,86 (0,071) (0,745; 0,989) 0,0345 | |
| Analys utan logaritmisk skala | ||
| LS-medelvärde (95 % KI) | 1,25 (0,311; 2,181) | 3,03 (1,666; 4,394) |
Skillnad i LS-medelvärden (givinostat–placebo) (95 % KI) p-värde | -1,78 (-3,462; -0,106) 0,0374 | |
*Analys utfördes med logaritmisk skala eftersom data inte var normalfördelade.
§ Givinostat eller placebo administrerades i tillägg till en fast dos kortikosteroider under hela studien.
Obs! LS-medelvärden, KI och p-värden erhölls från en kovariansanalys (ANCOVA)-modell av förändring i 4SC från baseline efter 18 månader.
Förändring i GLS-medelvärdet från baseline ska tolkas som en frekvensförändring (EOS/baseline).
I figur 1 beskrivs observerad genomsnittlig tid för 4SC under de 72 behandlingsveckorna i de två grupperna.
Figur 1 – studie 48: Observerad genomsnittlig förändring i sekunder för 4SC enligt behandling över tid (grupp A)
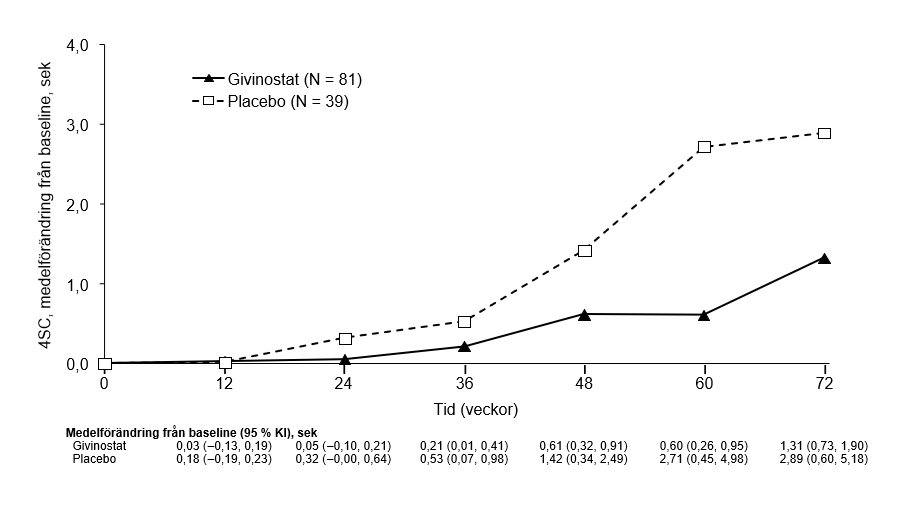
*Givinostat eller placebo gavs som tillägg till en stabil dos kortikosteroider under hela studien.
De viktiga sekundära effektmåtten i grupp A var förändring från baseline efter 18 månader i: fysisk förmåga bedömd med skalan North Star Ambulatory Assessment (NSAA), tid att resa sig från golvet (TTR), gångsträcka under sex minuter (6MWT), muskelstyrka utvärderad genom knäextension och armbågsflexion mätt med handhållen myometri (HHM) samt fettandel i vastus lateralis-muskeln utvärderad med magnetresonansspektroskopi (MRS). Sammantaget uppnådde resultaten för de viktiga sekundära effektmåtten avseende funktion, styrka och muskelmorfologi inte formell statistisk signifikans baserat på den förutbestämda Hochberg-analysen, men alla utfall var till fördel för givinostat (figur 2).
Figur 2 – EPIDYS-studien: Primära och viktiga sekundära effektmåttför givinostat jämfört med placebo (grupp A)§
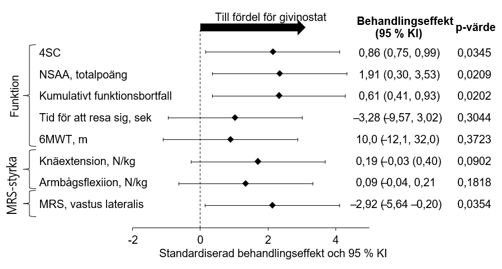
§ Givinostat eller placebo gavs som tillägg till en stabil dos kortikosteroider under hela studien.
Långtidssäkerhet, tolerabilitet och effekt för givinostat utvärderas i en pågående prospektiv, öppen långtidsförlängningsstudie (OLE) kallad STUDY 51. Patienter som slutfört givinostat fas 2-studien (STUDY 43) och patienter som slutfört givinostat fas 3-studier (EPIDYS) inkluderades i STUDY 51. Dessutom inkluderades 30 patienter utan tidigare exponering för givinostat i OLE-kohorten. Totalt inkluderades 207 manliga patienter som fick givinostat enligt en viktbaserad dosregim från 9,4 mg två gånger dagligen upp till 62 mg två gånger dagligen. Alla patienter stod på en stabil dos kortikosteroider före inskrivning och fortsatte med kortikosteroidbehandling under hela studien.
Nytta-riskförhållandet för givinostat utan samtidig kortikosteroidbehandling hos DMD-patienter har inte fastställts.
Nytta-riskförhållandet för givinostat hos patienter utan bevarad gångförmåga har inte fastställts.
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för Duvyzat för en eller flera undergrupper av den pediatriska populationen för DMD.
Detta läkemedel har godkänts enligt reglerna om”villkorat godkännande för försäljning”. Detta innebär att det ska inkomma ytterligare evidens för detta läkemedel.
Europeiska läkemedelsmyndigheten går igenom ny information om detta läkemedel minst varje år och uppdaterar denna produktresumé när så behövs.
Farmakokinetiska egenskaper
Absorption
Givinostat absorberas väl efter peroral administrering. Medelkoncentrationerna i plasma ökar på ett dosproportionellt sätt och maximal plasmakoncentration uppnås cirka 2‑3 timmar efter administrering. En standardiserad fettrik måltid resulterade i en viss ökning i exponering (cirka 30 % ökning i arean under kurvan [AUC] för plasmakoncentration över tid, cirka 20 % ökning i högsta plasmakoncentration [Cmax]) och en förlängning av tid till högsta koncentration (Tmax) från 2 till 3 timmar. Steady state-koncentrationer uppnås inom 5 till 7 dagar både efter dosering en gång och två gånger dagligen. En måttlig ackumulering på under det dubbla observerades efter administrering två gånger dagligen.
En fysiologiskt baserad farmakokinetisk analys, inklusive data från friska försökspersoner, förutspådde en oral biotillgänglighet hos människa på ≥ 50 % efter engångsdos oralt i intervallet 44,3 till 177,2 mg.
Distribution
Givinostat är cirka 96 % bundet till humana plasmaproteiner och fördelas i viss mån till röda blodkroppar (blod/plasma-kvot = 1,3).
Metabolism
In vitro-studier med humana enzympreparat samt studier av djurmetabolism både in vitro och in vivo har visat att givinostat genomgår omfattande metabolism och bildar flerta metaboliter. CYP450 och UGT-enzymer är inte involverade i de huvudsakliga metabola reaktionerna. Enzymerna som bildar de primära metaboliterna har endast delvis identifierats. Fyra huvudsakliga metaboliter, som är inaktiva har karaktäriserats hos både människa och djurarter, även om mängdförhållandena skiljer sig åt.
Eliminering
Givinostat uppvisar i plasma en tvåfasisk eliminationsprofil med en genomsnittlig uppenbar terminal eliminationsfas (halveringstid) på cirka 6 timmar. Elimineringen av givinostat är sannolikt beroende av metabolism följt av renal och biliär utsöndring. Utsöndringen av givinostat och dess huvudsakliga metaboliter i urin hos människa har utvärderats hos friska försökspersoner efter både engångs- och upprepad dosering av givinostat. Andelen oförändrat givinostat som återfanns i urinen var mycket låg efter administrering som både engångsdos och upprepad dos två gånger dagligen (< 3 % av given dos).
Linjäritet/icke-linjäritet
Farmakokinetiken för givinostat är linjär, eftersom AUC∞ efter engångsadministrering är jämförbar med den efter upprepad daglig dosering, med en möjlig minimal synlig ackumulering av den aktiva substansen över tid (ackumulationskvoter i intervallet 1,0‑1,7).
Linjäritet testades efter engångsadministrering av doser från 44,3 till 354,4 mg och upprepad administrering av doser från 44,3 till 177,2 mg.
Vikt
Baserat på populationens farmakokinetiska analyser visade sig vikten ha en signifikant effekt på givinostats clearance.
Effekten är inte linjär, dvs. effekten är större vid lägre vikter och mindre vid vikter på 30 kg och däröver. Därför rekommenderas dosering baserad på vikt.
Egenskaper i specifika grupper
Populationens farmakokinetiska analyser visar att ålder eller samtidig administrering av kortikosteroider inte har någon effekt på givinostats farmakokinetik.
Farmakokinetiken för givinostat har utvärderats hos manliga pediatriska DMD-patienter från 6 års ålder.
Nedsatt leverfunktion
Givinostat har inte studerats hos patienter med nedsatt leverfunktion. Försiktighet bör iakttas vid administrering och övervakning av läkemedlet hos dessa patienter.
Nedsatt njurfunktion
Givinostat har inte studerats hos patienter med nedsatt njurfunktion. Nedsatt njurfunktion förväntas dock inte påverka exponeringen av givinostat eftersom renal utsöndring inte är en signifikant elimineringsväg för givinostat.
Prekliniska säkerhetsuppgifter
I upprepade perorala toxicitetsstudier med givinostat i råttor och apor observerades en dosberoende minskning av antalet vita blodkroppar med tillhörande atrofi av lymfoida organ (thymus, lymfkörtlar och mjälte), minskning av antalet röda blodkroppar och blodplättar samt minskad cellhalt i benmärgen. En ökning av leverenzymer observerades också. Hos apor inducerades dessutom gallgångshyperplasi. Dessa toxiska effekter var i allmänhet reversibla efter avslutad behandling, men uppträdde vid lägre exponeringar för givinostat hos djur än de som uppnås vid högsta rekommenderade dos för människa (MRHD).
Gentoxicitet och karcinogenicitet
Givinostat var vid höga doser positivt för läsramsmutationer in vitro i bakterier (Ames test), negativt i däggdjursceller (TK+/- i muslymfomceller) och negativt in vivo i transgena Big Blue-råttor samt i pig-a-lokuset.
Sammantaget bedöms givinostat inte ha någon relevant genotoxisk potential in vivo.
För närvarande finns inga tillgängliga data från karcinogenicitetsstudier med givinostat.
Reproduktions- och utvecklingstoxicitet
Givinostat orsakade dosberoende minskningar i storlek och vikt på manliga accessoriska könsorgan redan vid den lägsta dosen. Djur som fick medelhög och hög dos uppvisade ett förlängt prekoitalt intervall och färre kopulationspluggar, troligen till följd av störd ejakulatbildning. Spermieparametrar och antal dräktiga honor påverkades dock inte.
Moderns biverkningar observerades vid höga dosnivåer i studier av embryofetal samt pre-och postnatal utvecklingstoxicitet. Effekter på dräktighet, embryofetal utveckling och kullparametrar bedömdes vara sekundära till moderns toxicitet. Effekter på embryofetal utveckling och kullparametrar observerades dock redan vid medelhöga doser i den embryofetala studien på råtta och kanin samt vid låg dos i pre-/postnatal utvecklingsstudie. Ingen negativ påverkan sågs på avkommornas beteende, neurologiska utveckling, könsmognad och reproduktionsförmåga.
Sammantaget observerades reproduktionstoxikologiska effekter vid lägre exponeringar för givinostat hos djur än de som uppnås vid MRHD, med undantag för embryofetalstudien i kanin där säkerhetsmarginalen var cirka 10 i förhållande till human exponering vid MRHD.
Juvenil toxicitet
Hos råttor observerades vissa effekter på hematologiska parametrar och lymfoida organ vid höga dosnivåer, vilka var helt eller delvis reversibla. Dessa effekter observerades vid lägre givinostatexponeringar hos djur än de som uppnås vid MRHD. Inga behandlingsrelaterade effekter på tillväxt, könsmognad, reproduktionsförmåga eller neurobeteendemässig utveckling observerades hos djur.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Polysorbat 20 (E432)
Glycerol (E422)
Dragantgummi (E413)
Natriumbensoat (E211)
Persikosmak: naturliga aromämnen, aromämnen, propylenglykol (E1520)
Gräddsmak: naturliga aromämnen, aromämnen, propylenglykol (E1520)
Natriumsackarin (E954)
Flytande sorbitol (E420)
Vinsyra (E334)
Natriumhydroxid (E524)
Renat vatten
Inkompatibiliteter
Ej relevant.
Hållbarhet
3 år.
Efter första öppnande: 60 dagar.
Särskilda förvaringsanvisningar
Inga särskilda temperaturanvisningar.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
DUVYZAT oraalisuspensio
8,86 mg/ml (L:ei) 140 ml (asteikollinen 10 ml mittaruisku) (19026,14 €)
PF-selosteen tieto
Bärnstensfärgad flaska av polyetentereftalat innehållande 140 ml oral suspension med en barnskyddande förslutning av högdensitetspolyeten och en sprutadapter av lågdensitetspolyeten.
Varje förpackning innehåller en flaska och en graderad 10 ml oral doseringsspruta.
10 ml-sprutan är graderad från 1 till 10 ml i steg om 0,5 ml.
Läkemedlets utseende:
Vit till benvit eller svagt rosa, homogen suspension efter blandning.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
DUVYZAT oraalisuspensio
8,86 mg/ml 140 ml
- Ei korvausta.
Atc-kod
M09AX14
Datum för översyn av produktresumén
01.06.2026
Yhteystiedot
Karl Gustavsgatan 1A
SE-411 25 Göteborg
Sweden
+46 3120 5020
www.campuspharma.se
info@campuspharma.se