
ELAHERE koncentrat till infusionsvätska, lösning 5 mg/ml
Observera
▼ Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
1 ml koncentrat till infusionsvätska, lösning, innehåller 5 mg mirvetuximabsoravtansin.
En injektionsflaska innehåller 100 mg mirvetuximabsoravtansin i 20 ml.
Mirvetuximabsoravtansin är ett antikroppsläkemedelskonjugat (ADC) riktat mot folatreceptor-alfa (FRα). Antikroppsläkemedelskonjugatet består av en anti-FRα monoklonal antikropp av IgG1-subtyp som framställts med rekombinant DNA-teknik i ovarieceller från kinesisk hamster och som fästs via klyvbar bindning (butansyra, 4-(2-pyridinylditio)-2-sulfo-1-(2,5-dioxo-1-pyrrolidinyl)-ester) till en maytansinoid-DM4, ett anti-tubulinmedel. Mirvetuximabsoravtansin innehåller i genomsnitt 3,4 DM4-payload-molekyler som är bundna till anti-FRα antikroppen.
Hjälpämnen med känd effekt
Detta läkemedel innehåller 2,11 mg polysorbat 20 per injektionsflaska.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Koncentrat till infusionsvätska, lösning (sterilt koncentrat).
Kliniska uppgifter
Terapeutiska indikationer
ELAHERE som monoterapi är avsedd för behandling av vuxna patienter med folatreceptor-alfa (FRα)-positiv, platinumresistent höggradig serös epitelial äggstockscancer, äggledarcancer eller primär peritonealcancer som har fått en till tre tidigare systemiska behandlingsregimer (se avsnitt Dosering och administreringssätt).
Villkor
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Dosering och administreringssätt
ELAHERE måste sättas in och övervakas av en läkare med erfarenhet av användning av cancerläkemedel.
Val av patienter
Patienter som behandlas med mirvetuximabsoravtansin ska ha FRα-positiv tumörstatus definierad som måttlig (2+) och/eller stark (3+) membranfärgning med immunhistokemi (IHC) i ≥75 % viabla tumörceller bedömd med CE-märkt in vitro-diagnostisk (IVD) avsedd att användas i detta syfte. Om CE-märkt in vitro-diagnostik inte finns tillgänglig ska ett validerat alternativt test användas.
Dosering
Rekommenderad dos av ELAHERE är 6 mg/kg justerad ideal kroppsvikt (adjusted ideal body weight; AIBW), som administreras en gång var tredje vecka (21‑dagarscykel) som intravenös infusion fram till sjukdomsprogression eller oacceptabel toxicitet. Dosering baserad på AIBW minskar exponeringsvariabiliteten för patienter som antingen är underviktiga eller överviktiga.
Den totala dosen av ELAHERE beräknas baserat på den individuella patientens AIBW med följande formel:
Kvinna IBW (ideal kroppsvikt [kg]) = 0,9 × längden [cm] – 92
AIBW = IBW [kg] + 0,4 × (faktisk vikt [kg] – IBW)
Till exempel, för en kvinnlig patient som är 165 cm lång och väger 80 kg
| Beräkna först IBW: | IBW = 0,9 × 165 – 92 = 56,5 kg |
| Beräkna sedan AIBW: | AIBW = 56,5 + 0,4 × (80 – 56,5) = 65,9 kg |
Premedicinering
Premedicinering för infusionsrelaterade reaktioner (IRR), illamående och kräkningar
Administrera premedicineringen i tabell 1 före varje infusion av ELAHERE för att minska incidensen och svårighetsgraden av infusionsrelaterade reaktioner, illamående och kräkningar.
Tabell 1: Premedicinering före varje ELAHERE-infusion
| Premedicinering | Administreringsväg | Exempel (eller motsvarighet) | Administreringstid före ELAHERE-infusion |
| Kortikosteroid | intravenöst | dexametason 10 mg | minst 30 minuter före |
| Antihistamin | oralt eller intravenöst | difenhydramin 25 mg till 50 mg | |
| Febernedsättande medel | oralt eller intravenöst | paracetamol 325 mg till 650 mg | |
| Antiemetika | oralt eller intravenöst | 5-HT3-serotoninreceptorblockerare eller lämpliga alternativ | före varje dos och efter administrering av annan premedicinering |
För patienter med illamående och/eller kräkningar kan därefter ytterligare antiemetika övervägas efter behov.
När det gäller patienter med en IRR-grad ≥ 2 bör man överväga att ge ytterligare premedicinering med dexametason 8 mg två gånger dagligen (eller motsvarande) dagen före ELAHERE-administrering.
Ögonundersökning och premedicinering
Ögonundersökning: En ögonundersökning inklusive test av synskärpa och undersökning med spaltlampa ska genomföras innan behandlingen med ELAHERE påbörjas och inför nästkommande dos om en patient utvecklar nya eller förvärrade okulära symtom. Hos patienter med okulära biverkningar av ≥ grad 2 ska ytterligare ögonundersökningar göras minst varannan cykel och när det är kliniskt indicerat tills biverkningarna har upphört eller återgått till baslinjen.
Oftalmologiska topikala steroider: För patienter som konstaterats ha tecken på hornhinnebiverkningar (keratopati) av ≥ grad 2 vid undersökning med spaltlampa rekommenderas sekundär profylax med oftalmologiska topikala steroider för efterföljande ELAHERE-cykler, såvida inte patientens ögonläkare anser att riskerna med en sådan behandling överväger nyttan.
- Patienterna ska instrueras att använda ögondroppar med steroider på infusionsdagen och i ytterligare 7 dagar efter varje påföljande ELAHERE-cykel (se tabell 3).
- Patienterna ska uppmanas att vänta minst 15 minuter efter administrering av oftalmologiska topikala steroider innan de använder smörjande ögondroppar.
Under behandling med oftalmologiska topikala steroider ska mätning av det intraokulära trycket samt undersökning med spaltlampa utföras regelbundet.
Smörjande ögondroppar: Det rekommenderas att instruera patienter att använda smörjande ögondroppar under hela behandlingen med ELAHERE.
Dosändringar
Före start av varje behandlingscykel ska patienten uppmanas att rapportera eventuella nya eller förvärrade symtom till den behandlande läkaren eller en kvalificerad person.
Hos patienter som utvecklar nya eller förvärrade okulära symtom ska en ögonundersökning utföras före dosering. Den behandlande läkaren ska gå igenom rapporten från patientens ögonundersökning före dosering och fastställa dosen av ELAHERE baserat på allvarlighetsgraden på fynden i det svårast drabbade ögat.
I tabell 2 och tabell 3 finns information om dosreduktioner och dosjusteringar vid biverkningar. Administreringsschemat med 3 veckors intervall mellan doserna bör bibehållas.
Tabell 2: Schema för dosreduktion
| Dosnivåer av ELAHERE | |
| Startdos | 6 mg/kg AIBW |
| Första dosreduktion | 5 mg/kg AIBW |
| Andra dosreduktion | 4 mg/kg AIBW* |
* Avsluta behandlingen permanent hos patienter som inte tolererar 4 mg/kg AIBW.
Tabell 3: Dosjusteringar vid biverkningar
| Biverkning | Biverkningens allvarlighetsgrad* | Dosjustering |
Keratit/keratopati (se avsnitt Varningar och försiktighet och Biverkningar) | Icke-konfluerande ytlig keratit/keratopati | Övervaka |
| Konfluerande ytlig keratit/keratopati, defekt i hornhinnans epitel eller förlust av bästa korrigerade synskärpa på 3 rader eller mer. | Avvakta med dosen tills tillståndet har förbättrats till icke-konfluerande ytlig keratit/keratopati eller bättre, eller tills det har upphört. Fortsätt sedan med samma dos. Överväg dosreduktion för patienter som har återkommande konfluerande keratit/keratopati trots att de fått bästa understöjdande vård, eller för patienter med okulär toxicitet som varar längre än 14 dagar. | |
| Hornhinnesår eller stromal opacitet eller bästa korrigerade synskärpa på avstånd 6/60 eller sämre | Avvakta med dosen tills tillståndet har förbättrats till icke-konfluerande ytlig keratit/keratopati eller bättre, eller tills det har upphört. Minska sedan med en dosnivå. | |
| Hornhinneperforation | Sätt ut permanent | |
Pneumonit (se avsnitt Varningar och försiktighet och Biverkningar) | Grad 1 | Övervaka |
| Grad 2 | Avvakta med dosen tills tillståndet har förbättrats till grad 1 eller lägre. Fortsätt sedan med samma dos eller överväg dosreduktion om tillståndet är recidiverande, varar längre än 28 dagar eller enligt läkares bedömning. | |
| Grad 3 eller 4 | Sätt ut permanent | |
Perifer neuropati (se avsnitt Varningar och försiktighet och Biverkningar) | Grad 2 | Avvakta med dosen till grad 1 eller lägre, minska sedan med en dosnivå. |
| Grad 3 eller 4 | Sätt ut permanent | |
Infusionsrelaterade reaktioner/överkänslighet (se avsnitt Varningar och försiktighet och Biverkningar) | Grad 1 | Bibehåll infusionshastigheten |
| Grad 2 |
| |
| Grad 3 eller 4 |
| |
Hematologiska (se avsnitt Biverkningar) | Grad 3 eller 4 | Avvakta med dosen till grad 1 eller lägre och återuppta sedan vid en dosnivå lägre. |
Andra biverkningar (se avsnitt Biverkningar) | Grad 3 | Avvakta med dosen till grad 1 eller lägre och återuppta sedan vid en dosnivå lägre. |
| Grad 4 | Sätt ut permanent |
*: Om inte annat anges, National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0.
Särskilda patientgrupper
Pediatrisk population
Det finns ingen relevant användning av ELAHERE för behandling av epitelial äggstockscancer, äggledarcancer eller primär peritonealcancer i den pediatriska populationen (se avsnitt Farmakodynamiska egenskaper).
Äldre
Ingen dosjustering av ELAHERE rekommenderas till patienter ≥65 år (se avsnitt Farmakokinetiska egenskaper).
Nedsatt njurfunktion
Ingen dosjustering av ELAHERE rekommenderas för patienter med lindrigt till måttligt nedsatt njurfunktion (kreatininclearance [CLcr] 30 till <90 ml/minut). ELAHERE har inte utvärderats hos patienter med svårt nedsatt njurfunktion (CLcr 15 till < 30 ml/min) eller njursjukdom i slutstadiet och det potentiella behovet av dosjustering hos dessa patienter kan inte fastställas (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Ingen dosjustering av ELAHERE rekommenderas för patienter med lindrigt nedsatt leverfunktion (totalt bilirubin ≤ övre normalvärdet (ULN) och aspartataminotransferas [ASAT] > ULN eller totalt bilirubin > 1 till 1,5 gånger ULN oavsett ASAT) (se avsnitt Farmakokinetiska egenskaper).
ELAHERE ska undvikas hos patienter med måttligt till svårt nedsatt leverfunktion (totalt bilirubin > 1,5 ULN oavsett ASAT).
Administreringssätt
ELAHERE är avsett för intravenös infusion vid en hastighet på 1 mg/min. Om detta tolereras väl efter 30 minuter kan infusionshastigheten ökas till 3 mg/min. Om detta tolereras väl efter 30 minuter vid 3 mg/min kan infusionshastigheten ökas till 5 mg/min.
För inkompatibiliteter, se avsnitt Inkompatibiliteter.
ELAHERE måste spädas med 50 mg/ml (5 %) glukos för intravenös infusion. Anvisningar om spädning av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
ELAHERE får endast administreras som intravenös infusion med ett inline-filter av polyetersulfon (PES) på 0,2 eller 0,22 µm (se Särskilda anvisningar för destruktion och övrig hantering i avsnitt Särskilda anvisningar för destruktion och övrig hantering).
Försiktighetsåtgärder före hantering eller administrering av läkemedlet
Detta läkemedel innehåller en cytotoxisk komponent som är kovalent bunden till den monoklonala antikroppen (se särskilda anvisningar för destruktion och övrig hantering i avsnitt Särskilda anvisningar för destruktion och övrig hantering).
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Ögonsjukdomar
Mirvetuximabsoravtansin kan orsaka allvarliga okulära biverkningar, inklusive nedsatt syn (främst dimsyn), keratopati (hornhinnesjukdomar), torrt öga, fotofobi och ögonsmärta (se avsnitt Effekter på förmågan att framföra fordon och använda maskiner och Biverkningar).
Patienterna ska remitteras till en ögonläkare för ögonundersökning innan behandling med mirvetuximabsoravtansin påbörjas.
Före starten av varje behandlingscykel ska patienten uppmanas att rapportera eventuella nya eller förvärrade okulära symtom till den behandlande läkaren eller en kvalificerad person.
Om patienten utvecklar okulära symtom ska en ögonundersökning göras. Rapporten från ögonundersökningen ska gås igenom och dosen av mirvetuximabsoravtansin kan justeras beroende på fyndens svårighetsgrad (se avnitt Dosering och administreringssätt).
Användning av smörjande ögondroppar under behandling med mirvetuximabsoravtansin rekommenderas. Hos patienter som utvecklar hornhinnebiverkningar av ≥ grad 2 rekommenderas oftalmologiska topikala steroider för efterföljande cykler av mirvetuximabsoravtansin (se avsnitt Dosering och administreringssätt).
Läkaren ska övervaka patienterna avseende okulär toxicitet och avvakta med, minska eller permanent sätta ut mirvetuximabsoravtansin baserat på de okulära biverkningarnas allvarlighetsgrad och varaktighet (se avsnitt Dosering och administreringssätt)
Patienterna bör rådas att undvika användning av kontaktlinser under behandlingen med mirvetuximabsoravtansin, såvida inte sjukvårdspersonalen säger annat.
Pneumonit
Allvarlig, livshotande eller dödlig interstitiell lungsjukdom, inklusive pneumonit, kan förekomma hos patienter som behandlas med mirvetuximabsoravtansin (se avsnitt Biverkningar).
Patienter ska övervakas avseende pulmonella tecken och symtom på pneumonit, vilket kan inkludera hypoxi, hosta, dyspné eller interstitiella infiltrat på röntgenundersökningar. Infektiösa, neoplastiska och andra orsaker till sådana symtom ska uteslutas genom lämplig utredning.
Uppehåll bör göras i behandlingen med mirvetuximabsoravtansin om patienten utvecklar ihållande eller recidiverande pneumonit av grad 2 till dess att symtomen går tillbaka till ≤ grad 1, och dosreduktion bör övervägas. Mirvetuximabsoravtansin ska sättas ut permanent hos alla patienter med pneumonit av grad 3 eller 4 (se avsnitt Dosering och administreringssätt). Patienter som är asymtomatiska kan fortsätta med doseringen av mirvetuximabsoravtansin med noggrann övervakning.
Perifer neuropati
Perifer neuropati har förekommit med mirveutximabsoravtansin, inklusive reaktioner av grad ≥ 3 (se avsnitt Biverkningar).
Patienter bör övervakas avseende tecken och symtom på neuropati, såsom parestesi, pirrningar eller en brännande känsla, neuropatisk smärta, muskelsvaghet eller dysestesi. Om patienter får ny eller förvärrad perifer neuropati bör läkaren avvakta med, minska eller permanent sätta ut mirvetuximabsoravtansin baserat på den perifera neuropatins svårighetsggrad (se avsnitt Dosering och administreringssätt).
Embryofetal toxicitet
Baserat på dess verkningsmekanism kan mirvetuximabsoravtansin orsaka embryofetal skada när det administreras till en gravid patient eftersom det innehåller en gentoxisk förening (DM4) och påverkar celler som delar sig aktivt.
Fertila patienter ska använda effektivt preventivmedel under behandling med mirvetuximabsoravtansin och i sju månader efter den sista dosen (se avsnitt Fertilitet, graviditet och amning).
Hjälpämnen med känd effekt
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per dos, d.v.s. är näst intill ”natriumfritt”.
Detta läkemedel innehåller 2,11 mg polysorbat 20 per injektionsflaska.
Interaktioner
Inga kliniska interaktionsstudier har utförts med ELAHERE.
DM4 är ett CYP3A4-substrat. Samtidig användning av ELAHERE och starka CYP3A4-hämmare kan öka exponeringen för okonjugerat DM4 (se avsnitt Farmakokinetiska egenskaper). Detta kan öka risken för biverkningar av ELAHERE (se avsnitt Biverkningar). Om samtidig användning av starka CYP3A4-hämmare (t.ex. ceritinib, klaritromycin, kobicistat, idelalisib, itrakonazol, ketokonazol, nefazodon, posakonazol, ritonavir, telitromycin, vorikonazol) inte kan undvikas bör patienterna övervakas noga avseende biverkningar. Starka CYP3A4-inducerare (t ex fenytoin, rifampicin, karbamazepin) kan minska exponeringen för okonjugerat DM4.
Fertilitet, graviditet och amning
Fertila kvinnor/preventivmedel
Graviditetsstatus hos fertila patienter ska kontrolleras innan behandling med mirvetuximabsoravtansin påbörjas.
Fertila patienter ska använda effektivt preventivmedel under behandling med mirvetuximabsoravtansin och i minst sju månader efter den sista dosen.
Graviditet
Baserat på dess verkningsmekanism kan mirvetuximabsoravtansin orsaka embryofetal skada när det administreras till en gravid patient eftersom det innehåller en gentoxisk förening (DM4) och påverkar celler som delar sig aktivt (se avsnitt Farmakodynamiska egenskaper och Prekliniska säkerhetsuppgifter). Det är känt att humant immunglobulin G (IgG) passerar placentabarriären. Därför har mirvetuximabsoravtansin potential att överföras från den gravida patienten till det växande fostret. Det finns inga tillgängliga data om användning av mirvetuximabsoravtansin hos gravida patienter som underbygger en läkemedelsassocierad risk. Inga studier av reproduktions- eller utvecklingstoxicitet hos djur har genomförts med mirvetuximabsoravtansin.
Administrering av ELAHERE till gravida patienter rekommenderas inte och patienter ska informeras om de potentiella riskerna för fostret om de blir eller vill bli gravida. Patienter som blir gravida måste omedelbart kontakta sin läkare. Om en patient blir gravid under behandling med ELAHERE eller inom 7 månader efter den sista dosen rekommenderas noggrann övervakning.
Amning
Det är okänt om mirvetuximabsoravtansin/metaboliter från mirvetuximabsoravtansin utsöndras i bröstmjölk. En risk för det nyfödda barnet/spädbarnet kan inte uteslutas eftersom det är känt att humant immunglobulin G (IgG) överförs till bröstmjölk. ELAHERE ska inte användas under amning och i 1 månad efter den sista dosen.
Fertilitet
Inga fertilitetsstudier har genomförts med mirvetuximabsoravtansin eller DM4. Det finns inga data om effekten av ELAHERE på fertiliteten hos människa. Med tanke på att verkningsmekanismen för ELAHERE leder till störning av mikrotubuli och avdödning av celler som delar sig snabbt finns det dock risk för läkemedelsrelaterade effekter på fertiliteten.
Effekter på förmågan att framföra fordon och använda maskiner
ELAHERE har måttlig effekt på förmågan att framföra fordon och använda maskiner. Om patienterna får synstörningar, perifer neuropati, trötthet eller yrsel under behandling med mirvetuximabsoravtansin ska de instrueras att inte köra bil eller använda maskiner förrän det har bekräftats att symtomen har upphört.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste biverkningarna av mirvetuximabsoravtansin var dimsyn (43 %), illamående (41 %), lös avföring (39 %), trötthet (35 %), buksmärta (30 %), keratopati (29 %), torrt öga (27 %), förstoppning (26 %), kräkningar (23 %), minskad aptit (22 %), perifer neuropati (20 %), huvudvärk (19%), asteni (18 %), förhöjt ASAT (16 %) och artralgi (16 %).
De vanligast rapporterade allvarliga biverkningarna var pneumonit (4 %), tunntarmsobstruktion (3 %), tarmobstruktion (3 %), vätskeutgjutning i lungsäcken (2 %), buksmärta (2 %), uttorkning (1 %), förstoppning (1 %), illamående (1 %), ascites (1 %) och trombocytopeni (˂1 %).
De biverkningar som oftast ledde till dosreduktion eller dosfördröjning var dimsyn (17 %), keratopati (10 %), torrt öga (5 %), neutropeni (5 %), keratit (4 %), grå starr (3 %), nedsatt synskärpa (3 %), trombocytopeni (3 %), perifer neuropati (3 %) och pneumonit (3 %).
Hos 12 % av patienterna som fick mirvetuximabsoravtansin avslutades behandlingen permanent på grund av en biverkning. De vanligaste inkluderade magtarmkanalen (4 %), andningsvägar, bröstkorg och mediastinum (3 %), blodet och lymfsystemet (1 %), centrala och perifera nervsystemet (1 %) och ögon (1 %).
Lista över biverkningar i tabellform
Biverkningsfrekvenserna bygger på sammanslagna data från 4 kliniska studier som inkluderade 682 patienter med epitelial äggstockscancer, äggledarcancer eller primär peritonealcancer (gemensamt kallat epitelial äggstockscancer (EOC), som behandlades med mirvetuximabsoravtansin 6 mg/kg AIBW var tredje vecka. Mediantiden för behandling med mirvetuximabsoravtansin var 19,1 veckor (intervall: 3, 132 veckor).
Biverkningsfrekvenserna från kliniska studier är baserade på frekvenser av biverkningar oavsett orsak, där det efter noggrann bedömning har fastställts att det åtminstone finns en rimlig möjlighet för ett orsakssamband mellan läkemedlet och biverkningen.
Frekvenserna definieras som: mycket vanliga (≥1/10), vanliga (≥1/100, <1/10), mindre vanliga (≥1/1 000, <1/100), sällsynta (≥1/10 000, <1/1 000), mycket sällsynta (<1/10 000). Inom varje frekvensgrupp där det är relevant presenteras de observerade biverkningarna i fallande allvarlighetsgrad.
Tabell 4: Lista i tabellform över biverkningar av alla grader hos patienter som behandlats med mirvetuximabsoravtansin i kliniska studier
| Organsystem | Frekvenskategori | Biverkningar |
| Infektioner och infestationer | Mycket vanliga | Urinvägsinfektion |
| Blodet och lymfsystemet | Mycket vanliga | Anemi, trombocytopeni |
| Vanliga | Neutropeni | |
| Metabolism och nutrition | Mycket vanliga | Minskad aptit, hypomagnesemi |
| Vanliga | Onormalt låga halter av kalium i blodet, uttorkning | |
| Psykiatriska tillstånd | Vanliga | Insomni |
| Centrala och perifera nervsystemet | Mycket vanliga | Perifer neuropati1, huvudvärk |
| Vanliga | Dysgeusi, yrsel | |
| Ögon | Mycket vanliga | Keratopati2, grå starr3, episod av dimsyn4, fotofobi, ögonsmärta, torrt öga5 |
| Vanliga | Obehag i ögat6 | |
| Blodkärl | Vanliga | Hypertoni |
| Andningsvägar, bröstkorg och mediastinum | Mycket vanliga | Pneumonit7, dyspné, hosta |
| Magtarmkanalen | Mycket vanliga | Lös avföring, buksmärta8, förstoppning, bukdistension, kräkningar, illamående |
| Vanliga | Ascites, gastroesofageal refluxsjukdom, stomatit, dyspepsi | |
| Lever och gallvägar | Vanliga | Hyperbilirubinemi |
| Hud och subkutan vävnad | Vanliga | Pruritus |
| Muskuloskeletala systemet och bindväv | Mycket vanliga | Artralgi |
| Vanliga | Myalgi, ryggsmärta, smärta i extremitet, muskelspasmer | |
| Allmänna symtom och/eller symtom vid administreringsstället | Mycket vanliga | Trötthet |
| Vanliga | Pyrexi | |
| Undersökningar och provtagningar | Mycket vanliga | Förhöjt asparatataminotransferas, förhöjt alaninaminotransferas |
| Vanliga | Förhöjt alkaliskt fosfatas i blod, förhöjt gamma-glutamyltransferas, minskad vikt | |
| Skador, förgiftningar och behandlingskomplikationer | Vanliga | Infusionsrelaterad reaktion/överkänslighet9 |
1 Den sammanslagna termen perifer neuropati inkluderar hypoestesi, perifer neuropati, neurotoxicitet, parestesi, perifer motorisk neuropati, perifer sensorimotorisk neuropati, perifer senorisk neuropati och polyneuropati (se avsnittet Beskrivning av utvalda biverkningar).
2 Den sammanslagna termen keratopati inkluderar hornhinnecysta, hornhinneutfällningar, hornhinnesjukdom, mikrocystor i hornhinneepitel, hornhinnepiteldefekt, hornhinneerosion, hornhinnegrumling, hornhinnepigmentering, keratit, interstitiell keratit, keratopati, limbal stamcellsbrist och punktatakeratit (se avsnittet Beskrivning av utvalda biverkningar).
3 Den sammanslagna termen grå starr inkluderar grå starr; katarakt, kortikal och kärnkatarakt (se avsnittet Beskrivning av utvalda biverkningar).
4 Den sammanslagna termen dimsyn inkluderar ackommodationssjukdom, diplopi, hypermetropi, presbyopi, refraktionsrubbning, dimsyn, nedsatt syn, nedsatt synskärpa och grumlingar i glaskropp (se avsnittet Beskrivning av utvalda biverkningar).
5 Den sammanslagna termen torrt öga inkluderar torrt öga och minskat tårflöde (se avsnittet Beskrivning av utvalda biverkningar).
6 Den sammanslagna termen okulärt obehag inkluderar ögonirritation, ögonpruritus, känsla av främmande kropp i öga och okulärt obehag (se avsnittet Beskrivning av utvalda biverkningar).
7 Den sammanslagna termen pneumonit inkluderar interstitiell lungsjukdom, organiserande pneumoni, pneumonit, pulmonell fibros och andningssvikt (se avsnittet Beskrivning av utvalda biverkningar).
8 Den sammanslagna termen buksmärta inkluderar obehag i buken, buksmärta, lågt sittande buksmärta och högt sittande buksmärta.
9 Den sammanslagna termen infusionsrelaterad reaktion/överkänslighet inkluderar överkänslighet (SMQ, smal) och hudrodnad, erytem, ögonlocksrodnad.
Beskrivning av utvalda biverkningar
Ögonsjukdomar
Okulära biverkningar (sammanslagen term) förekom hos 59 % av patienterna med epitelial äggstockscancer som behandlades med mirvetuximabsoravtansin. Elva procent (11 %) av patienterna fick okulära biverkningar av grad 3 och < 1 % fick biverkningar av grad 4. De vanligaste okulära biverkningarna av ≥ grad 3 var dimsyn och keratopati (båda 5 %, sammanslagna termer) samt grå starr (4 %).
Mediantiden till debut för den första okulära biverkningen var 5,1 veckor (intervall: 0,1 till 68,6). Av de patienter som fick okulära biverkningar hade 53 % fullständig förbättring (grad 0) och 38 % partiell förbättring (definierat som en minskning av allvarlighetsgraden med en eller flera grader från den allvarligaste graden). Vid den sista uppföljningen hade 0,3 % (2/682) av patienterna okulära biverkningar av ≥ grad 3 (1 patient med nedsatt synskärpa av grad 3 och 1 patient med grå starr av grad 4).
Okulära biverkningar ledde till dosfördröjningar hos 24 % av patienterna och dosreduktioner hos 15 % av patienterna. Okulära biverkningar ledde till permanent utsättning av mirvetuximabsoravtansin hos 1 % av patienterna.
Pneumonit
Pneumonit (sammanslagen term) förekom hos 10 % av patienterna med epitelial äggstockscancer som behandlades med mirvetuximabsoravtansin, inklusive 0,9 % (6/682) patienter med biverkningar av grad 3 och 0,2 % (1/682) patienter med biverkningar av grad 4. Två patienter (0,3 %) dog till följd av andningssvikt. En patient (0,2 %) dog till följd av andningssvikt i samband med pneumonit av grad 1 och lungmetastaser som bekräftades vid obduktion. En patient (0,2 %) dog till följd andningssvikt av okänd etiologi utan samtidig pneumonit.
Mediantiden till debut av pneumonit var 18,1 veckor (intervall: 1,6 till 97,0). Pneumonit ledde till dosfördröjningar av mirvetuximabsoravtansin hos 3 %, dosreduktioner hos 1 % och permanent utsättning hos 3% av patienterna.
Perifer neuropati
Perifer neuropati (sammanslagen term) förkom hos 36 % av patienterna med epitelial äggstockscancer som behandlades med mirvetuximabsoravtansin i kliniska studier. 3 % av patienterna fick perifer neuropati av grad 3.
Mediantiden till debut av perifer neuropati var 5,9 veckor (intervall 0,1 till 126,7). Perifer neuropati ledde till dosfördröjningar av mirvetuximabsoravtansin hos 2 %, dosreduktioner hos 4 % och permanent utsättning hos 0,7 % av patienterna.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via det nationella rapporteringssystemet
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns ingen känd behandling/antidot för överdosering av mirvetuximabsoravtansin. Vid överdos måste patienterna övervakas noga avseende tecken eller symtom på biverkningar och lämplig symtomatisk behandling sättas in.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska och immunmodulerande medel, monoklonala antikroppar och antikroppsläkemedelskonjugat, övriga monoklonala antikroppar och antikroppsläkemedelskonjugat. ATC-kod: L01FX26
Verkningsmekanism
Mirvetuximabsoravtansin är ett antikroppsläkemedelskonjugat. Antikroppen är ett framställt IgG1 som är riktat mot folatreceptor alfa (FRα). Antikroppsdelens funktion är att binda till FRα som uttrycks på ytan av äggstockscancercellerna. DM4 är en mikrotubulihämmare som är fäst till antikroppen via klyvbar bindning Vid bindning till FRα internaliseras mirvetuximabsoravtansin följt av intracellulär frisättning av DM4 via proteolytisk klyvning. DM4 förstör det mikrotubulära nätverket i cellen, vilket leder till cellcykelarrest och apoptotisk celldöd.
Farmakodynamisk effekt
Kardiell elektrofysiologi
Vid den godkända rekommenderade dosen orsakade inte mirvetuximabsoravtansin medelökningar av QTc-intervallet på >10 msek, baserat på resultaten av koncentrations-QTc-analys.
Klinisk effekt och säkerhet
Studie IMGN853-0416 (MIRASOL)
Effekt och säkerhet för mirvetuximabsoravtansin undersöktes i studie IMGN853-0416, en multicenter, öppen, aktivt kontrollerad, randomiserad fas 3-studie med två behandlingsgrupper. Till studien rekryterades platinumresistenta patienter med framskriden höggradig serös epitelial äggstockscancer, primär peritonealcancer eller äggledarcancer vars tumörer (inklusive arkiverad vävnad) var FRα-positiva enligt bestämning genom FOLR1 (FOLR1-2.1) RxDx-analys (≥ 75 % av de viabla tumörcellerna med måttlig (2+) och/eller stark (3+) membranfärgningsintensitet med immunhistokemi (IHC)).
Platinumresistent sjukdom definierades som epitelial äggstockscancer som recidiverade inom 6 månader efter den sista platinumdosen.
I studien exkluderades patienter med primär platinumrefraktär sjukdom, patienter med ECOG ≥ 2 och patienter med aktiva eller kroniska hornhinnesjukdomar, okulära tillstånd som kräver pågående behandling, perifer neuropati av grad ≥ 2 samt icke-infektiös ILD/pneumonit.
Patienterna randomiserades 1:1 till antingen ELAHERE 6 mg/kg AIBW iv (N=227) dag 1 i varje 3‑veckorscykel eller någon av följande kemoterapier (N=226) enligt prövarens beslut före randomisering:
- Paclitaxel (Pac) 80 mg/m2 administrerat en gång i veckan inom en 4‑veckorscykel;
- Pegylerad liposomal doxorubicin (PLD) 40 mg/m2 administrerat en gång var 4:e vecka;
- Topotecan (Topo) 4 mg/m2 administrerat dag 1, 8 och 15 var 4:e vecka eller i 5 dagar i rad vid 1,25 mg/m2 från dag 1–5 i varje 21‑dagarscykel
Randomiseringen stratifierades efter antalet tidigare behandlingslinjer (1 jämfört med 2 jämfört med 3) och genom prövarens val av kemoterapi (Pac jämfört med PLD jämfört med Topo). Behandling administrerades fram till sjukdomsprogression, död, tillbakadraget samtycke eller oacceptabel toxicitet.
Det primära effektmåttet var progressionsfri överlevnad (PFS) baserat på prövarens bedömning enligt RECIST 1.1-kriterierna. Viktiga sekundära effektmått var objektiv responsfrekvens (objective response rate; ORR) och total överlevnad (OS).
Sammanlagt randomiserades 453 patienter. Medianåldern var 63 år (intervall: 29 till 88 år) och patienterna var främst vita (66 %; 12 % asiater). De flesta patienter (80 %) hade äggstockscancer av epitelialt ursprung, 11 % av patienterna hade äggledarcancer och 8 % av patienterna hade primär peritonealcancer. Samtliga (100 %) hade höggradig serös histologi. Ungefär hälften av patienterna (47 %) hade fått 3 tidigare systemiska behandlingar, 39 % hade fått 2 tidigare behandlingslinjer och 14 % av patienterna hade fått 1 tidigare behandlingslinje. Majoriteten av patienterna hade tidigare fått en Poly(ADPribos) polymeras (PARP)-hämmare (55 %) och bevacizumab (62 %). Det platinumfria intervallet efter den senaste behandlingslinjen var ≤ 3 månader hos 41 % av patienterna och 3 till 6 månader hos 58 % av patienterna. 55 % av patienterna hade ECOG-funktionsstatus 0 och 44 % hade ECOG 1.
Den primära analysen visade en statistiskt signifikant förbättring av PFS och OS hos patienter som randomiserats till ELAHERE jämfört med prövarens val av kemoterapi.
I tabell 5 sammanfattas effektresultaten för studie IMGN853-0416 (MIRASOL).
Tabell 5: Effektresultat i studie IMGN853-0416
| Effektmått | ELAHERE N=227 | Prövarens val av kemoterapier N=226 |
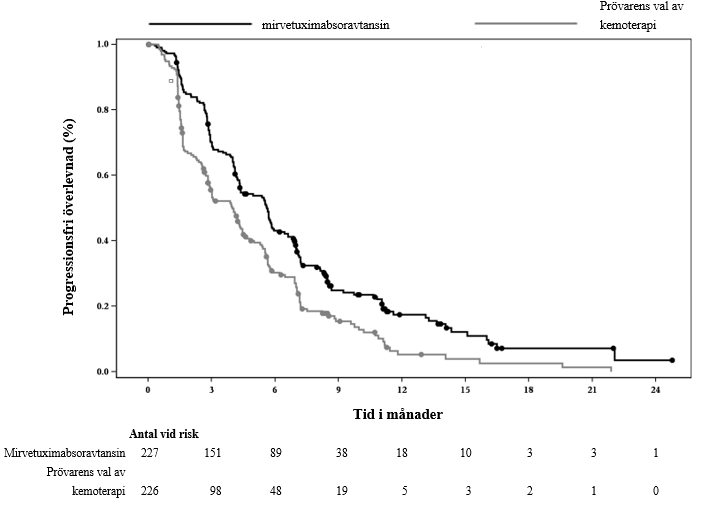
| Progressionsfri överlevnad (PFS) enligt prövarens bedömning | ||
| Antal händelser (%) | 176 (77,5) | 166 (73,5) |
| Median, månader (95 % KI) | 5,62 (4,34; 5,95) | 3,98 (2,86; 4,47) |
| Riskkvot (95 % KI) | 0,65 (0,521; 0,808) | |
| p-värde | < 0,0001 | |
| Total överlevnad (OS) | ||
| Antal händelser (%) | 90 (39,6) | 114 (50,4) |
| Median, månader (95 % KI) | 16,46 (14,46; 24,57) | 12,75 (10,91; 14,36) |
| Riskkvot (95 % KI) | 0,67 (0,504; 0,885) | |
| p-värde | 0,0046* | |
Sista datum för datainsamling (data cut-off) 6 mars 2023.
*: fördefinierad effektgräns = 0,01313; 2-sidig (justerad efter observerat antal dödsfall 204).
Kaplan Meier-kurvorna för prövarbedömd PFS (medianuppföljning på 11,2 månader) och OS (medianuppföljning på 13,1 månader) presenteras i figur 1 och figur 2.
Figur 1: Kaplan-Meier-kurva för progressionsfri överlevnad per behandlingsgrupp i MIRASOL (intent-to-treat-population)
Figur 2: Kaplan-Meier-kurva för total överlevnad per behandlingsgrupp i MIRASOL (intent-to-treat-population)
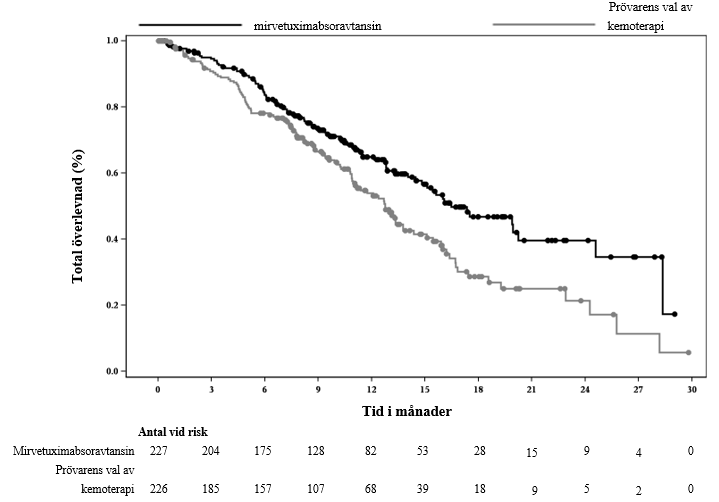
Vid en ytterligare deskriptiv analys med medianuppföljning på 20,3 månader överensstämde resultat för OS med primäranalysen.
Immunogenicitet
Antikroppar mot läkemedlet (anti-drug antibodies, ADA) detekterades med frekvensen vanlig. Det finns ingen evidens för ADA:s påverkan på farmakokinetik, effekt eller säkerhet, men det finns fortfarande endast begränsade data.
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för ELAHERE för alla grupper av den pediatriska populationen för behandling av äggstockscancer, behandling av äggledarcancer och behandling av peritonealcancer (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Farmakokinetiken karakteriserades efter att patienterna hade fått mirvetuximabsoravtansin i doser om 0,161 mg/kg till 8,71 mg/kg AIBW (dvs. 0,0268 gånger till 1,45 gånger den godkända rekommenderade dosen 6 mg/kg AIBW), om inte annat noterats.
I tabell 6 sammanfattas exponeringsparametrarna för mirvetuximabsoravtansin, okonjugerat DM4 och dess metabolit S-metyl-DM4 efter administrering efter den första cykeln (3-veckorscykel) av mirvetuximabsoravtansin 6 mg/kg till patienter. Toppkoncentrationerna av mirvetuximabsoravtansin observerades nära slutet av den intravenösa infusionen, medan toppkoncentrationerna av okonjugerat DM4 observerades den andra dagen efter administrering av mirvetuximabsoravtansin. Toppkoncentrationerna av S-metyl-DM4 observerades cirka 3 dagar efter administrering av mirvetuximabsoravtansin. Steady-state-koncentrationer av mirvetuximabsoravtansin, DM4 och S‑metyl-DM4 nåddes efter en behandlingscykel. Ackumuleringen av mirvetuximabsoravtansin, DM4, och S-metyl-DM4 var minimal efter upprepad administrering av mirvetuximabsoravtansin.
Tabell 6: Exponeringsparametrar av mirvetuximabsoravtansin, okonjugerat DM4 och S-metyl-DM4 efter första behandlingscykeln på 6 mg/kg mirvetuximabsoravtansin
| Mirvetuximabsoravtansin Medelvärde (±SD) | Okonjugerat DM4 Medelvärde (±SD) | S-metyl-DM4 Medelvärde (±SD) | |
| Cmax | 137,3 (±62,3) µg/ml | 4,11 (±2,29) ng/ml | 6,98 (±6,79) ng/ml |
| AUCtau | 20,65 (±6,84) h × mg/ml | 530 (±245) h × ng/ml | 1 848 (±1585) h × ng/ml |
Cmax = maximal koncentration, AUCtau = area under kurvan för koncentration i förhållande till tid under doseringsintervallet (21 dagar).
Absorption
Mirvetuximabsoravtansin administreras som intravenös infusion. Inga studier har genomförts med andra administreringsvägar.
Distribution
Medelvärdet (±SD) för distributionsvolymen vid steady-state för mirvetuximabsoravtansin var 2,63 (±2,98) liter. Bindningen av DM4 och S-metyl-DM4 till humant plasmaprotein var > 99 %, in vitro.
Metabolism
Den monoklonala antikroppsdelen av mirvetuximabsoravtansin förväntas metaboliseras till små peptider genom katabola vägar. Okonjugerat DM4 och S-metyl-DM4 metaboliseras av CYP3A4. I human plasma identifierades DM4 och S-metyl-DM4 som de huvudsakliga cirkulerande metaboliterna och stod för cirka 0,4 % respektive 1,4 % av AUC för mirvetuximabsoravtansin.
Eliminering
Medelvärdet (± SD) för total plasmaclearance för mirvetuximabsoravtansin var 18,9 (± 9,8) ml/timme. Medelvärdet för den terminala halveringstiden för mirvetuximabsoravtansin efter den första dosen var 4,9 dagar. För okonjugerat DM4 var medelvärdet (± SD) för total plasmaclearance 14,5 (± 4,5) liter/timme och medelvärdet för den terminala halveringstiden var 2,8 dagar. För S-metyl-DM4 var medelvärdet (± SD) för total plasmaclearance 5,3 (± 3,4) liter/timme och medelvärdet för den terminala halveringstiden var 5,1 dagar. In vitro- och prekliniska in vivo-studier tyder på att DM4 och S-metyl-DM4 främst metaboliseras av CYP3A4 och elimineras via biliär utsöndring i avföring.
Särskilda patientgrupper
Inga kliniskt signifikanta skillnader i farmakokinetiken för mirvetuximabsoravtansin observerades baserat på ålder (32 till 89 år), etnicitet (vit, svart eller asiatisk), kroppsvikt (36 till 136 kg), lindrigt nedsatt leverfunktion (totalt bilirubin ≤ ULN oavsett ASAT > ULN eller totalt bilirubin > 1 till 1,5 gånger ULN oavsett ASAT), eller lindrigt till måttligt nedsatt njurfunktion (CLcr ≥ 30 och <90 ml/min).
Farmakokinetiken för mirvetuximabsoravtansin är okänd hos patienter med måttligt till svårt nedsatt leverfunktion (totalt bilirubin > 1,5 ULN oavsett ASAT) eller svårt nedsatt njurfunktion (CLcr 15 till 30 ml/min).
Studier av läkemedelsinteraktioner
In vitro-studier
Cytokrom P450(CYP)-enzymer: Okonjugerat DM4 är en tidsberoende hämmare av CYP3A4. Okonjugerat DM4 och S-metyl-DM4 är inte direkta hämmare av CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 eller CYP3A. DM4 och S-metyl-DM4 är inte inducerare av CYP1A2, CYP2B6 eller CYP3A4.
Transportörsystem: Okonjugerat DM4 och S-metyl-DM4 är substrat för P-gp, men är inte hämmare av P-gp.
Prekliniska säkerhetsuppgifter
Målorgan som identifierades vid administrering av en engångsdos av mirvetuximabsoravtansin hos krabbmakaker var begränsade till hud och cellförlust i benmärg och lymfvävnad. Upprepad dosering hos krabbmakaker och holländska kaniner visade också oftalmologiska fynd, inklusive mikrocystor i hornhinnan, pigmentering, försvagning och degeneration/nekros i hornhinneepitelet. Dessa fynd var beroende av dosintensitet (dos och schema), med färre övergripande fynd och återhämtning av de fynd som observerades i 3-veckorsschemat (det kliniska doseringsschemat).
Studier av karcinogenicitet har inte genomförts med mirvetuximabsoravtansin eller DM4.
DM4 och S-metyl-DM4 var inte mutagena i en analys av bakteriell omvänd mutation (Ames). DM4 och S-metyl-DM4 resulterade i mikronuklei i polykromatiska erytrocyter.
Inga studier av reproduktions- eller utvecklingstoxicitet hos djur har genomförts med mirvetuximabsoravtansin.
Fertilitetsstudier har inte genomförts med mirvetuximabsoravtansin eller DM4. Det finns inga data om effekten av ELAHERE på fertiliteten hos människa. Med tanke på att verkningsmekanismen för ELAHERE leder till störning av mikrotubuli och avdödning av celler som delar sig snabbt finns det dock risk för läkemedelsrelaterade effekter på fertiliteten.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Koncentrerad ättiksyra (E260)
Natriumacetat (E262)
Sackaros
Polysorbat 20 (E432)
Vatten för injektionsvätskor
Inkompatibiliteter
ELAHERE är inte kompatibel med natriumklorid 9 mg/ml (0,9 %) infusionsvätska, lösning. Detta läkemedel får inte blandas med andra läkemedel förutom de som nämns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Hållbarhet
Oöppnad injektionsflaska
5 år
Spädd lösning
Efter spädning har kemisk och fysikalisk stabilitet påvisats mellan 1 mg/ml och 2 mg/ml i 8 timmar vid 15 °C–25 °C eller i 24 timmar vid 2 °C–8 °C och därefter i 8 timmar vid 15 °C–25 °C.
Ur ett mikrobiologiskt perspektiv ska läkemedlet användas omedelbart, såvida inte spädningsmetoden utesluter risken för mikrobiell kontaminering. Om det inte används omedelbart är förvaringstider och förvaringsförhållanden vid användning användarens ansvar.
Särskilda förvaringsanvisningar
Förvaras upprätt i kylskåp (2 °C–8 °C).
Får ej frysas.
Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter spädning finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
ELAHERE infuusiokonsentraatti, liuosta varten
5 mg/ml (L:ei) 20 ml (3629,33 €)
PF-selosteen tieto
Injektionsflaska av typ I-glas med butylgummipropp och aluminiumförsegling med kungsblått fliplock av polypropylen, innehållande 20 ml koncentrat till lösning.
Förpackningsstorlek: 1 injektionsflaska.
Läkemedlets utseende:
Klar till lätt opalskimrande, färglös lösning.
Särskilda anvisningar för destruktion och övrig hantering
ELAHERE är ett cytotoxiskt läkemedel. Följ tillämpliga särskilda förfaranden för hantering och destruktion.
Beredning
- Beräkna dosen (mg) (baserat på patientens AIBW), total volym (ml) lösning som krävs och antalet ELAHERE-injektionsflaskor som behövs (se avsnitt Dosering och administreringssätt). Mer än en injektionsflaska behövs för en full dos.
- Ta ut injektionsflaskorna med ELAHERE ur kylskåpet och låt lösningen anta rumstemperatur.
- Parenterala läkemedel ska inspekteras visuellt avseende partiklar och missfärgning före administrering, när lösningen och behållaren gör det möjligt. ELAHERE är en klar till lätt opalskimrande, färglös lösning.
- Läkemedlet ska inte användas om lösningen är missfärgad eller grumlig, eller om den innehåller främmande partiklar.
- Snurra försiktigt varje injektionsflaska och inspektera lösningen innan du drar upp den beräknade dosvolymen av ELAHERE för ytterligare spädning. Injektionsflaskan får inte skakas.
- Dra med aseptisk teknik upp den beräknade dosvolymen av ELAHERE för ytterligare spädning. Varje injektionsflaska innehåller en överfyllning som gör det möjligt att dra upp den angivna mängden.
- ELAHERE innehåller inte konserveringsmedel och är endast avsedd för engångsdosering. Kassera eventuell oanvänd lösning som finns kvar i injektionsflaskan.
Spädning
- ELAHERE måste spädas med 50 mg/ml (5 %) glukos före administrering till en slutkoncentration på 1 mg/ml till 2 mg/ml.
- ELAHERE är inte kompatibel med natriumklorid 9 mg/ml (0,9 %) infusionsvätska, lösning. ELAHERE får inte blandas med andra läkemedel eller intravenösa vätskor.
- Fastställ hur stor volym 50 mg/ml (5 %) glukos som krävs för att uppnå den slutliga koncentrationen av spädd aktiv substans. Ta antingen bort överskottet av 50 mg/ml (5 %) glukos från en förfylld intravenös påse eller tillsätt den beräknade volymen av 50 mg/ml (5 %) glukos till en steril tom intravenös påse. Tillsätt sedan den beräknade dosvolymen av ELAHERE till den intravenösa påsen.
- Blanda den spädda lösningen försiktigt genom att långsamt vända på påsen flera gånger för att säkerställa att lösningen blir jämnt blandad. Skaka inte och rör inte om .
- Om den spädda infusionslösningen inte används omedelbart ska lösningen förvaras enligt avsnitt Hållbarhet. Om infusionspåsen är kyld, låt den uppnå rumstemperatur före administrering. Efter kylning ska spädda infusionslösningar administreras inom 8 timmar (inklusive infusionstiden).
- Den beredda infusionslösningen får inte frysas.
Administrering
- Inspektera ELAHERE-påsen för intravenös infusion visuellt avseende partiklar och missfärgning före administrering.
- Administrera premedicinering innan administrering av ELAHERE (se avsnitt Dosering och administreringssätt).
- Administrera endast ELAHERE som intravenös infusion med ett inline-filter av polyetersulfon (PES) på 0,2 eller 0,22 µm. Byt inte ut andra membranmaterial.
- Användning av administreringstillbehör som innehåller di-2-etylhexyl-ftalat (DEHP) ska undvikas.
- Administrera den initiala dosen som intravenös infusion vid en hastighet på 1 mg/min. Om den tolereras väl efter 30 minuter vid 1 mg/min kan infusionshastigheten ökas till 3 mg/min. Om den tolereras väl efter 30 minuter vid 3 mg/min kan infusionshastigheten ökas till 5 mg/min.
- Om inga infusionsrelaterade reaktioner uppkom vid den föregående dosen bör efterföljande infusioner startas vid den maximalt tolererade hastigheten och kan ökas till en maximal infusionshastighet på 5 mg/min, beroende på tolerans.
- Efter infusionen ska den intravenösa slangen spolas med 50 mg/ml (5 %) glukos för att säkerställa att hela dosen ges. Använd inte andra intravenösa vätskor för spolning.
Kassering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
ELAHERE infuusiokonsentraatti, liuosta varten
5 mg/ml 20 ml
- Ei korvausta.
Atc-kod
L01FX26
Datum för översyn av produktresumén
09.01.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi