
VYVGART injektioneste, liuos 1000 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo sisältää 1 000 mg efgartigimodi alfaa 5,6 millilitrassa (180 mg/ml).
Efgartigimodi alfa on rekombinantti ihmisen immunoglobuliini G1:stä (IgG1) johdettu Fc-fragmentti, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos
Kliiniset tiedot
Käyttöaiheet
Vyvgart on tarkoitettu
- lisälääkkeeksi tavanomaiseen hoitoon aikuispotilaille, joilla on yleistynyt myasthenia gravis (gMG) ja jotka ovat asetyylikoliinireseptori (AChR) -vasta-ainepositiivisia.
- monoterapiana aikuispotilaiden hoitoon, joilla on etenevä tai relapsoiva aktiivinen krooninen inflammatorinen demyelinoiva polyneuropatia (CIDP), aiemman kortikosteroidi- tai immunoglobuliinihoidon jälkeen.
Ehto
Valmistetta saa antaa vain terveydenhoitoalan ammattilainen hyväksytyn käyttöaiheen hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Neuromuskulaarisista sairauksista kärsivien potilaiden hoitoon perehtyneen lääkärin tulee aloittaa hoito ja valvoa hoitoa.
Annostus
Yleistynyt myasthenia gravis
Ensimmäinen hoitojakso ja toisen hoitojakson ensimmäinen antokerta on toteutettava joko terveydenhuollon ammattilaisen toimesta tai valvonnassa. Seuraavat hoitokerrat antaa terveydenhuollon ammattilainen tai vaihtoehtoisesti potilas tai potilasta hoitava henkilö kotona saatuaan riittävän opastuksen ihonalaisen injektion antotekniikkaan.
Suositeltu annos on 1 000 mg ihon alle kerran viikossa annettavina injektioina toteutettavana hoitojaksona 4 viikon ajan. Seuraavat hoitojaksot tulee antaa kliinisen arvion mukaan. Hoitojaksojen tiheys voi vaihdella potilaittain (ks. kohta Farmakodynamiikka).
Kliinisessä kehitysohjelmassa varhaisin aika seuraavan hoitojakson aloittamiselle oli 7 viikkoa edellisen hoitojakson ensimmäisestä infuusiosta.
Ihon alle annettavaa injektionestettä voidaan käyttää vaihtoehtona potilaille, jotka saavat tällä hetkellä efgartigimodi alfaa laskimoon. Lääkemuotojen välillä vaihtaminen on suositeltavaa toteuttaa uuden hoitojakson alussa. Saman hoitojakson sisällä lääkemuotoa vaihtavista potilaista ei ole olemassa turvallisuutta ja tehoa koskevia tietoja.
Krooninen inflammatorinen demyelinoiva polyneuropatia
Ensimmäiset neljä injektiota annetaan joko terveydenhuollon ammattilaisen toimesta tai valvonnassa. Seuraavat injektiot antaa terveydenhuollon ammattilainen tai vaihtoehtoisesti potilas tai potilasta hoitava henkilö kotona saatuaan riittävän opastuksen ihonalaisen injektion antotekniikkaan.
Suositeltu annos on 1 000 mg ihon alle kerran viikossa annettavina injektioina.
Hoito aloitetaan viikoittaisella annostuksella, ja sitä voidaan muuttaa joka toinen viikko annettavaksi kliinisen arvioinnin perusteella. Oireiden pahentuessa on palattava kerran viikossa annettaviin injektioihin.
Niiden potilaiden kohdalla, jotka siirtyvät nykyisistä CIDP-hoidoistaan, Vyvgart-hoito on mieluiten aloitettava ennen näiden aiempien hoitojen kliinisen vaikutuksen vähentymistä.
Kliininen vaste saavutetaan yleensä 3 kuukauden kuluessa ihon alle annettavan efgartigimodi alfa -hoidon aloittamisesta. Kliinistä arviointia on harkittava 3–6 kuukauden kuluttua hoidon aloittamisesta hoitovaikutuksen arvioimiseksi ja sen jälkeen säännöllisin väliajoin.
Väliin jäänyt annos
Kahden peräkkäisen lääkkeenannon välillä on oltava vähintään 3 vuorokautta. Jos lääkkeenantoa ei voida toteuttaa aikataulunmukaisena ajankohtana, se on tehtävä mahdollisimman pian ja vähintään 3 vuorokautta ennen seuraavaa antokertaa. Jos seuraavaan antokertaan on alle 3 vuorokautta, väliin jäänyt annos on jätettävä väliin ja seuraava annos on annettava aikataulunmukaisena ajankohtana.
Erityisryhmät
Iäkkäät potilaat
65-vuotiaiden ja sitä vanhempien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Tiedot turvallisuudesta ja tehosta potilailla, joilla on lievä munuaisten vajaatoiminta, ovat vähäisiä. Annosta ei tarvitse muuttaa potilaille, joilla on lievä munuaisten vajaatoiminta. Turvallisuutta ja tehoa koskevia tietoja on hyvin vähän potilaista, joilla on kohtalainen tai vaikea munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Tietoja maksan vajaatoimintaa sairastavista potilaista ei ole saatavilla. Annosta ei tarvitse muuttaa potilaille, joilla on maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Efgartigimodi alfan turvallisuutta ja tehoa pediatristen potilaiden hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste annetaan vain injektiona ihon alle. Ei saa antaa laskimoon.
Sen jälkeen kun valmiste on otettu pois jääkaapista, odota vähintään 15 minuutin ajan ennen injisoimista, jotta valmiste lämpiää huoneenlämpöiseksi. Käytä aseptista tekniikkaa lääkevalmisteen valmistelussa ja annossa. Injektiopulloa ei saa ravistaa.
Injektioneste voidaan antaa polypropeeniruiskulla, ruostumattomasta teräksestä valmistetuilla siirtoneuloilla ja polyvinyylikloridista valmistetulla siivekkeellisellä infuusiosetillä, jonka suurin esitäyttötilavuus on 0,4 ml.
- Vedä koko efgartigimodi alfa -liuoksen sisältö injektiopullosta siirtoneulan avulla.
- Vaihda ruiskun neula siivekkeelliseen infuusiosettiin.
- Ennen antoa ruiskun tilavuus on säädettävä 5,6 ml:aan.
Ensimmäisten efgartigimodi alfa -valmisteen antokertojen aikana (ks. kohta Annostus ja antotapa) injektioon ja yliherkkyyteen liittyvien reaktioiden asianmukaisen hoidon on oltava helposti saatavilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Suositeltuja injektiokohtia (vatsan alueella) on kierrätettävä, ja injektioita ei pidä koskaan antaa luomiin tai arpiin eikä alueille, joissa iho on arka, mustelmilla, punoittava tai kovettunut. 5,6 ml:n tilavuus on injisoitava 30 - 90 sekunnin aikana. Injektiota voidaan hidastaa, jos potilaalle tulee epämukavuutta.
Ensimmäinen itseannostelu on aina toteutettava terveydenhuollon ammattilaisen valvonnassa. Kun potilas tai potilasta hoitava henkilö on saanut riittävän opastuksen ihonalaisen injektion antotekniikkaan, hän voi injisoida lääkevalmisteen kotona, mikäli terveydenhuollon ammattilainen katsoo tämän olevan asianmukaista. Potilasta tai potilasta hoitavaa henkilöä on ohjeistettava injisoimaan Vyvgart-valmistetta sen pakkausselosteessa annettujen ohjeiden mukaisesti.
Kattavammat lääkevalmisteen anto-ohjeet löytyvät pakkausselosteessa olevista käyttöohjeista.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Myasthenia Gravis Foundation of American (MGFA) -luokan V potilaat
Efgartigimodi alfan käyttöä ei ole tutkittu MGFA:n luokka V -potilailla (eli myasteeninen kriisi), jonka määritelmänä on intubaatio mekaanisen ventilaation kanssa tai ilman sitä, paitsi tavanomaisen postoperatiivisen hoidon yhteydessä. Ajallista järjestystä myasthenia graviksen kriisin vakiintuneiden hoitojen ja efgartigimodi alfan välillä on harkittava, ja hoitojen mahdolliset yhteisvaikutukset on huomioitava (ks. kohta Yhteisvaikutukset).
Infektiot
Koska efgartigimodi alfa aiheuttaa ohimenevää IgG-pitoisuuksien pienenemistä, infektioriski voi suurentua (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Yleisimmät kliinisissä tutkimuksissa havaitut infektiot olivat ylähengitystieinfektiot ja virtsatieinfektiot (ks. kohta Haittavaikutukset). Potilasta on seurattava infektioiden kliinisten merkkien ja oireiden varalta Vyvgart-hoidon aikana. Jos potilaalla on aktiivinen infektio, efgartigimodi alfa -hoidon jatkamisen tai keskeyttämisen hyöty-haittasuhdetta on arvioitava, kunnes infektio on hävinnyt. Jos vakava infektio ilmenee, efgartigimodi alfa -hoidon lykkäämistä on harkittava, kunnes infektio on parantunut.
Injektioreaktiot ja yliherkkyysreaktiot
Injektioreaktioita, kuten ihottumaa tai kutinaa, on raportoitu kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Nämä reaktiot olivat lieviä tai kohtalaisia. Anafylaktisen reaktion tapauksia on raportoitu laskimoon annetun efgartigimodi alfa -hoidon yhteydessä markkinoille tulon jälkeen. Ensimmäiset Vyvgart-valmisteen antokerrat on toteutettava terveydenhuollon ammattilaisen valvonnassa (ks. kohta Annostus ja antotapa). Potilasta on seurattava 30 minuutin ajan lääkkeenannon jälkeen injektioreaktioiden kliinisten merkkien ja oireiden havaitsemiseksi. Jos reaktio ilmenee, on ryhdyttävä asianmukaisiin tukitoimiin reaktion vaikeusasteen mukaisesti. Seuraavat injektiot voidaan antaa varoen kliinisen arvioinnin perusteella.
Jos anafylaktista reaktiota epäillään, Vyvgart-lääkkeen antaminen on lopetettava välittömästi ja asianmukainen hoito aloitettava. Potilaille on kerrottava yliherkkyys- ja anafylaktisten reaktioiden merkeistä ja oireista, ja heitä on neuvottava ottamaan yhteyttä terveydenhuollon ammattilaiseen välittömästi niiden ilmetessä.
Immunisaatiot
Kaikki rokotteet on annettava rokotusohjeiden mukaisesti.
Eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävien rokotteiden käytön turvallisuutta ja näillä rokotteilla efgartigimodi alfa -hoidon aikana toteutettavan immunisaation vastetta ei tunneta. Efgartigimodi alfa -hoitoa saaville potilaille ei yleisesti ottaen suositella rokottamista eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla. Jos rokottaminen eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla on tarpeen, nämä rokotteet on annettava vähintään 4 viikkoa ennen hoitoa ja vähintään 2 viikkoa viimeisen efgartigimodi alfa -annoksen jälkeen.
Muut rokotteet voidaan antaa tarpeen mukaan milloin tahansa efgartigimodi alfa -hoidon aikana.
Immunogeenisuus
Aktiivikontrolloidussa ARGX‑113‑2001-tutkimuksessa 12/110 gMG-potilaalla (11 %:lla) todettiin olemassa olevia efgartigimodi alfaan sitoutuvia vasta-aineita. Anti-efgartigimodi alfan vasta-aineita havaittiin 19/55 efgartigimodi alfa -hoitoa ihon alle saaneella potilaalla (35 %:lla) ja 11/55 laskimoon annettavaa lääkemuotoa saaneella potilaalla (20 %:lla). Neutraloivia vasta-aineita havaittiin kahdella efgartigimodi alfa -hoitoa ihon alle saaneella potilaalla (4 %:lla) ja kahdella efgartigimodi alfaa laskimoon saaneella potilaalla (4 %:lla).
ARGX‑113‑1802-tutkimuksessa 13/317 CIDP-potilaalla (4,1 %:lla) todettiin olemassa olevia efgartigimodi alfaan sitoutuvia vasta-aineita. Anti-efgartigimodi alfan vasta-aineita havaittiin 20/317 potilaalla (6,3 %:lla), jotka saivat hoitoa tutkimuksen avoimessa osassa (vaihe A) ja 2/111 potilaalla (1,8 %:lla), jotka saivat hoitoa tutkimuksen lumekontrolloidussa osassa (vaihe B). Neutraloivia vasta-aineita havaittiin vain yhdellä potilaalla (0,3 %:lla) tutkimuksen avoimessa osassa (ks. kohta Farmakodynamiikka).
Efgartigimodi alfan vasta-aineiden vaikutusta kliiniseen tehoon, turvallisuuteen, farmakokinetiikkaan tai farmakodynamiikkaan ei voida arvioida neutraloivien vasta-aineiden vähäisestä esiintyvyydestä johtuen.
Immunosuppressantti- ja antikoliiniesteraasihoito
Kun ei-steroidaalista immunosuppressantti-, kortikosteroidi- ja antikoliiniesteraasihoitoa vähennetään tai hoito lopetetaan, potilasta on seurattava tarkoin sairauden pahenemisen merkkien varalta.
Apuaineet, joiden vaikutus tunnetaan
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo, eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatit
Tämä lääkevalmiste sisältää 2,7 mg polysorbaatti 20:tä per injektiopullo, joka vastaa 0,4 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Efgartigimodi alfa saattaa pienentää humaaniin neonataali-Fc-reseptoriin (FcRn) sitoutuvien yhdisteiden, eli immunoglobuliinivalmisteiden, monoklonaalisten vasta-aineiden tai IgG-alaluokan ihmisen Fc-domeenia sisältävien vasta-ainejohdannaisten, pitoisuuksia. Mahdollisuuksien mukaan on suositeltavaa lykätä näillä valmisteilla toteutettavan hoidon aloittamista, kunnes viimeisestä Vyvgart-annoksesta on kulunut 2 viikkoa. Varotoimena potilaita, jotka saavat Vyvgart-valmistetta näiden valmisteiden käytön aikana, on seurattava tarkoin näiden valmisteiden tavoitellun tehovasteen suhteen.
Plasmanvaihto, immunoadsorptio ja plasmafereesi voivat pienentää efgartigimodi alfan pitoisuutta verenkierrossa.
Kaikki rokotteet on annettava rokotusohjeiden mukaisesti.
Mahdollisia yhteisvaikutuksia rokotteiden kanssa tutkittiin ei-kliinisessä mallissa, jossa antigeenina käytettiin Keyhole limpet -kotilon hemosyaniinia (KLH). Viikoittainen 100 mg/kg:n annos apinoille ei vaikuttanut KLH-immunisaation synnyttämään immuunivasteeseen.
Efgartigimodi alfa -hoitoa saaville potilaille ei yleisesti ottaen suositella rokottamista eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla. Jos rokottaminen eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävillä rokotteilla on tarpeen, nämä rokotteet on annettava vähintään 4 viikkoa ennen hoitoa ja vähintään 2 viikkoa viimeisen efgartigimodi alfa -annoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Raskaus
Efgartigimodi alfan käytöstä raskauden aikana ei ole tietoja. Vasta-aineiden, mukaan lukien terapeuttiset monoklonaaliset vasta-aineet, tiedetään kulkeutuvan aktiivisesti istukan läpi (30 raskausviikon jälkeen) FcRn-reseptoriin sitoutumalla.
Efgartigimodi alfa voi siirtyä äidistä kehittyvään sikiöön. Koska efgartigimodi alfan odotetaan alentavan äidin vasta-ainepitoisuuksia ja myös estävän äidin vasta-aineiden siirtymistä sikiöön, vastasyntyneen passiivisen suojan heikkeneminen on odotettavissa. Näin ollen on otettava huomioon riskit ja hyödyt, jotka liittyvät elävien/elävien heikennettyjen rokotteiden antamiseen imeväisille, jotka ovat altistuneet efgartigimodi alfalle kohdussa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaana olevien naisten Vyvgart-hoitoa on harkittava vain, jos hoidosta saatava kliininen hyöty on riskejä suurempi.
Imetys
Ei ole olemassa tietoja efgartigimodi alfan erittymisestä ihmisen rintamaitoon, vaikutuksista imetettävään lapseen eikä vaikutuksista maidontuotantoon. Efgartigimodi alfan siirtymisestä rintamaitoon ei ole tehty eläinkokeita, joten erittymistä rintamaitoon ei voida sulkea pois. Äidin IgG:n tiedetään erittyvän ihmisen rintamaitoon. Imettävien naisten efgartigimodi alfa -hoitoa tulee harkita vain, jos hoidosta saatava kliininen hyöty on riskejä suurempi.
Hedelmällisyys
Ei ole olemassa tietoja efgartigimodi alfan vaikutuksesta ihmisen hedelmällisyyteen. Eläinkokeissa efgartigimodi alfalla ei havaittu olevan vaikutusta urosten ja naaraiden hedelmällisyysparametreihin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vyvgart-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisimmin havaittuja haittavaikutuksia olivat injektiokohdan reaktiot (33 %), ylähengitystieinfektiot (10,7 %) ja virtsatieinfektiot (9,5 %).
Ihon alle annettavan Vyvgart-valmisteen yleinen turvallisuusprofiili, sekä hoitojaksoina annettavan että jatkuvan hoito-ohjelman osalta, oli yhdenmukainen laskimoon annettavan lääkemuodon tunnetun turvallisuusprofiilin kanssa.
Luettelo haittavaikutuksista taulukkomuodossa
Tässä kohdassa kuvattavat haittavaikutukset ovat peräisin kliinisistä tutkimuksista ja markkinoille tulon jälkeisistä raporteista. Nämä reaktiot on esitetty elinjärjestelmän ja suositellun termin mukaan jaoteltuina. Esiintyvyysluokkien määritelmät: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin esiintyvyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutukset
| Elinjärjestelmä | Haittavaikutus | Esiintyvyysluokka |
| Infektiot* | Ylähengitystieinfektiot | Hyvin yleinen |
| Virtsatieinfektiot | Yleinen | |
| Bronkiitti | Yleinen | |
| Immuunijärjestelmä | Anafylaktinen reaktio a | Tuntematon |
| Ruoansulatuselimistö | Pahoinvointi b | Yleinen |
| Luusto, lihakset ja sidekudos | Myalgia | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat* | Injektiokohdan reaktiot c, d | Hyvin yleinen |
| Vammat, myrkytykset ja hoitokomplikaatiot* | Toimenpidepäänsärky e | Yleinen |
* Ks. kohta ”Valikoitujen haittavaikutusten kuvaukset”
a Markkinoille tulon jälkeisistä spontaaneista raporteista laskimoon annon yhteydessä
b Markkinoille tulon jälkeisistä spontaaneista raporteista.
c Vain ihon alle annon yhteydessä
d (esim. injektiokohdan ihottuma, injektiokohdan punoitus, injektiokohdan kutina, injektiokohdan kipu)
e Vain laskimoon annon yhteydessä
Valikoitujen haittavaikutusten kuvaukset
Injektiokohdan reaktiot
Yhdistetyssä tietoaineistossa, joka oli peräisin kahdesta ihon alle annetun efgartigimodi alfan kliinisestä tutkimuksesta yleistyneen myasthenia graviksen hoidosta (n = 168), kaikki injektiokohdan reaktiot olivat vaikeusasteeltaan lieviä tai kohtalaisia, eivätkä ne johtaneet hoidon keskeyttämiseen. 44,0 %:lle (n = 74) potilaista tuli injektiokohdan reaktio. Injektiokohdan reaktiot ilmenivät 24 tunnin sisällä annosta 78,4 %:lla (58/74) potilaista, ja ne hävisivät ilman hoitoa 85,1 %:lla (63/74) potilaista. Injektiokohdan reaktioiden esiintyvyys oli suurinta ensimmäisen hoitojakson aikana. Niitä raportoitiin 36,3 %:lla (61/168) potilaista ensimmäisen hoitojakson aikana, ja ne vähenivät 20,1 %:iin (30/149), 15,4 %:iin (18/117) ja 12,5 %:iin (10/80) potilaista toisen, kolmannen ja neljännen hoitojakson aikana. Yhdistetyssä tietoaineistossa kahdesta kliinisestä tutkimuksesta CIDP-potilailla, jotka saivat jatkuvasti efgartigimodi alfaa ihon alle, injektiokohdan reaktioiden ilmaantuvuus oli 26 % (61/235). Kolmen kuukauden aikaväleinä tehty analyysi osoitti, että niiden tutkittavien prosenttiosuus, joilla oli injektiokohdan reaktioita, oli suurin ensimmäisten kolmen hoitokuukauden aikana (73 [22,2 %] tutkittavaa) ja väheni myöhempi kolmen kuukauden aikavälien aikana (vaihteluväli 0–17 [6,8 %] tutkittavaa).
Infektiot
Laskimoon annetun efgartigimodi alfan lumekontrolloidussa ARGX‑113‑1704-tutkimuksessa yleistyneen myasthenia graviksen hoidosta yleisimmin ilmoitettuja haittavaikutuksia olivat infektiot, ja yleisimpiä ilmoitettuja infektioita olivat ylähengitystieinfektiot (10,7 %:lla [n = 9] efgartigimodi alfaa laskimoon saaneista potilaista ja 4,8 %:lla [n = 4] lumelääkettä saaneista potilaista) ja virtsatieinfektiot (9,5 %:lla [n = 8] efgartigimodi alfaa laskimoon saaneista potilaista ja 4,8 %:lla [n = 4] lumelääkettä saaneista potilaista). Nämä infektiot olivat vaikeusasteeltaan lieviä tai kohtalaisia efgartigimodi alfaa laskimoon saaneilla potilailla (≤ asteen 2 tasoisia Common Terminology Criteria for Adverse Events -luokituksen mukaisesti). Kaiken kaikkiaan hoitoon liittyviä infektioita raportoitiin 46,4 %:lla (n = 39) efgartigimodi alfaa laskimoon saaneista potilaista ja 37,3 %:lla (n = 31) lumelääkettä saaneista potilaista. Mediaaniaika hoidon aloittamisesta infektioiden ilmaantumiseen oli 6 viikkoa. Infektioiden ilmaantuvuus ei lisääntynyt myöhempien hoitojaksojen myötä. Hoito lopetettiin tai keskeytettiin tilapäisesti infektion vuoksi alle 2 %:lla potilaista. CIDP-potilailla tehdyn ARGX-113-1802-tutkimuksen lumekontrolloidussa osassa ihon alle annetun efgartigimodi alfan jatkuva anto ei ollut yhteydessä infektioiden ilmaantuvuuden lisääntymiseen (31,5 % [35/111] efgartigimodi alfaa ihon alle saaneessa ryhmässä ja 33,6 % [37/110] lumelääkeryhmässä) (ks. kohta Farmakodynamiikka).
Toimenpidepäänsärky
Toimenpidepäänsärkyä raportoitiin 4,8 %:lla efgartigimodi alfaa laskimoon saaneista potilaista ja 1,2 %:lla lumelääkettä saaneista potilaista. Toimenpidepäänsärky raportoitiin, kun päänsäryn katsottiin liittyvän ajallisesti efgartigimodi alfan laskimonsisäiseen infuusioon. Kaikki toimenpidepäänsäryt olivat lieviä tai kohtalaisia, lukuun ottamatta yhtä vaikea-asteisena raportoitua tapahtumaa (asteen 3 tasoinen).
Kaikki muut haittavaikutukset olivat lieviä tai kohtalaisia, lukuun ottamatta yhtä lihaskivun tapausta (asteen 3 tasoinen).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Efgartigimodi alfan yliannostukseen ei tiedetä liittyvän erityisiä merkkejä ja oireita. Yliannostustapauksissa mahdollisten haittavaikutusten ei odoteta eroavan suositellun annoksen käytön yhteydessä mahdollisesti havaittavista haittavaikutuksista. Potilasta on seurattava haittavaikutusten varalta, ja sopiva oireenmukainen tukihoito on aloitettava. Efgartigimodi alfan yliannostukseen ei ole olemassa spesifistä vastalääkettä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, selektiiviset immunosuppressantit, ATC‑koodi: L04AA58
Vaikutusmekanismi
Efgartigimodi alfa on humaani IgG1-vasta-ainefragmentti, jonka affiniteettia neonataali-Fc-reseptoriin (FcRn) on lisätty suunnitellulla muokkauksella. Efgartigimodi alfa sitoutuu FcRn-reseptoriin, mikä johtaa verenkierrossa olevan IgG:n, myös patogeenisten IgG-autovasta-aineiden, määrän vähenemiseen. Efgartigimodi alfa ei vaikuta muiden immunoglobuliinien (IgA, IgD, IgE tai IgM) pitoisuuksiin eikä vähennä albumiinin pitoisuuksia.
IgG-autovasta-aineet ovat IgG-välitteisten autoimmuunisairauksien patogeneesin perimmäinen syy.
Myasthenia graviksessa ne heikentävät neuromuskulaarista transmissiota sitoutumalla asetyylikoliinireseptoreihin (AChR), lihasspesifiseen tyrosiinikinaasiin (MuSK) tai pienitiheyksiseen lipoproteiinireseptoriin liittyvään proteiini 4:ään (LRP4).
CIDP:ssä useasta lähteestä peräisin oleva näyttö viittaa IgG-autovasta-aineiden keskeiseen rooliin tämän sairauden patogeneesissä. Tämä näyttö sisältää autoreaktiivisten IgG-vasta-aineiden osoittamisen myelinoitujen hermojen komponentteja vasten, passiivisen CIDP:n oireiden siirtymisen eläinmalleihin CIDP-potilaiden seerumia tai IgG-vasta-aineita käyttäen sekä plasmanvaihdon ja immunoadsorption terapeuttisen vaikutuksen CIDP-potilaiden hoidossa.
Farmakodynaamiset vaikutukset
Laskimoon annettava lääkemuoto
Kaksoissokkoutetussa, lumekontrolloidussa yleistynyttä myasthenia gravista sairastavilla potilailla tehdyssä ARGX‑113‑1704-tutkimuksessa efgartigimodi alfa 10 mg/kg kerran viikossa 4 viikon ajan annettuna pienensi seerumin IgG-pitoisuuksia ja AChR-autovasta-ainepitoisuuksia (AChR‑Ab). Suurin keskimääräinen prosentuaalinen IgG-kokonaispitoisuuden lasku lähtötasoon verrattuna oli 61 % viikon kuluttua ensimmäisen hoitojakson viimeisestä infuusiosta. Pitoisuus palasi lähtötasoon 9 viikon kuluttua viimeisestä infuusiosta. Samanlaisia vaikutuksia havaittiin myös kaikilla IgG:n alatyypeillä. AChR‑Ab-pitoisuuden pieneneminen toteutui ajallisesti vastaavanlaisella tavalla. Pitoisuuden keskimääräinen prosentuaalinen lasku oli suurimmillaan (58 %) viikon kuluttua viimeisestä infuusiosta ja palautui lähtötasolle 7 viikon kuluttua viimeisestä infuusiosta. Samanlaisia muutoksia havaittiin tutkimuksen toisen hoitojakson aikana.
Ihon alle annettava lääkemuoto
ARGX-113-2001-tutkimuksessa AChR‑Ab-pitoisuudet vähenivät samalla tavalla ajan myötä kuin IgG-kokonaispitoisuudet, ja ne olivat samanlaisia efgartigimodi alfaa ihon alle ja laskimoon saaneissa ryhmissä. Suurin AChR‑Ab-pitoisuuksien keskimääräinen prosentuaalinen väheneminen havaittiin viikon kuluttua viimeisestä antokerrasta ja oli 62,2 % efgartigimodi alfaa ihon alle saaneessa ryhmässä ja 59,6 % efgartigimodi alfaa laskimoon saaneessa ryhmässä. Sekä efgartigimodi alfaa ihon alle saaneessa ryhmässä että efgartigimodi alfaa laskimoon saaneessa ryhmässä IgG-kokonaispitoisuuden väheneminen ja AChR‑Ab-pitoisuuden väheneminen oli yhteydessä kliiniseen vasteeseen, kun sitä mitattiin MG‑ADL‑kokonaispistemäärän muutoksena lähtötasosta (ks. kuva 1).
Kuva 1. IgG-kokonaispitoisuuden ja AChR-Ab:n suhde MG‑ADL-kokonaispistemäärään AChR-Ab-seropositiivisten populaatiossa, joka sai efgartigimodi alfaa ihon alle (1A) tai efgartigimodi alfaa laskimoon (1B) (tutkimus ARGX-113-2001)
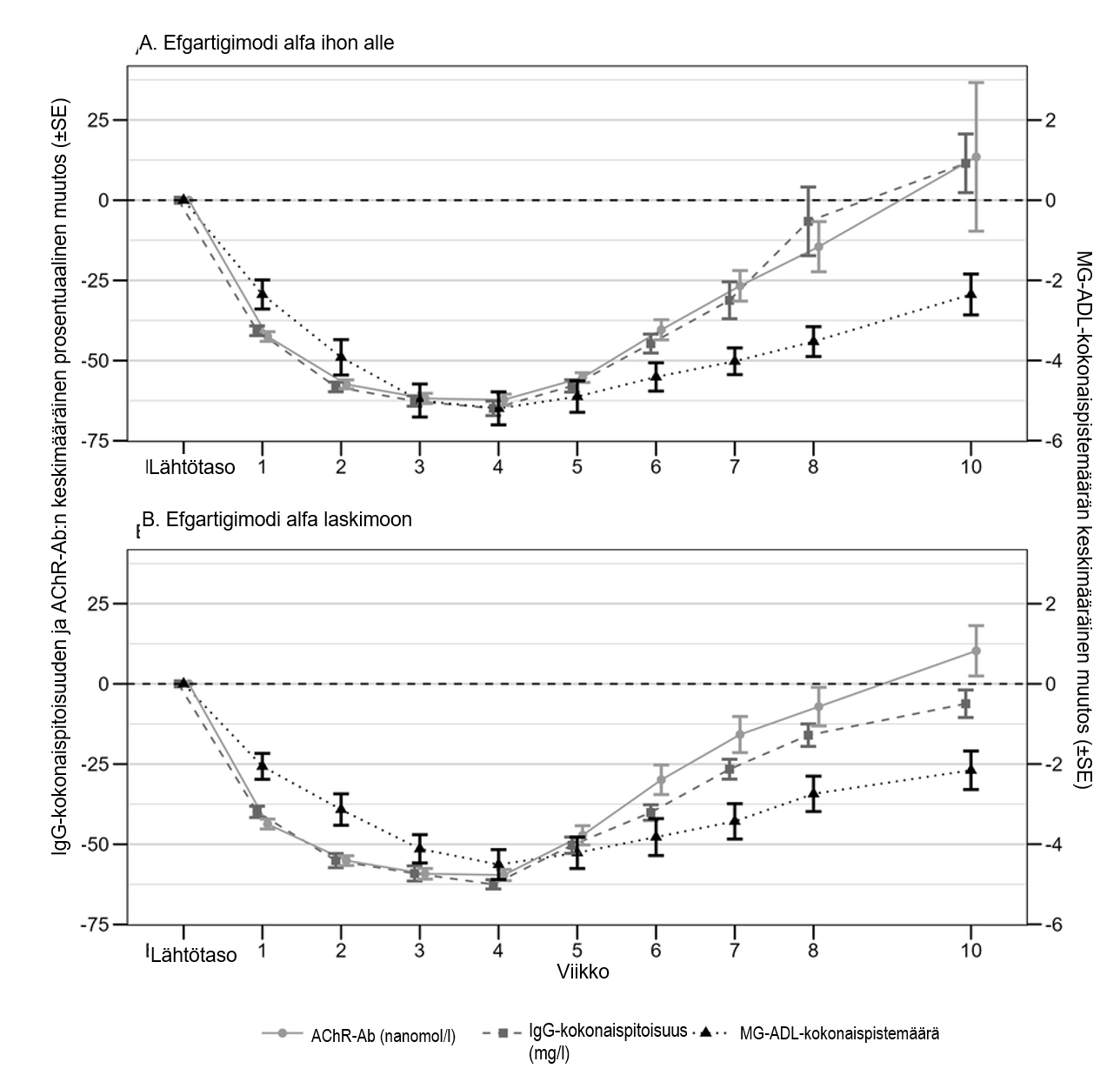
ARGX‑113‑1802-tutkimuksessa CIDP-potilaat saivat jatkuvasti kerran viikossa efgartigimodi alfaa ihon alle annoksella 1 000 mg. Keskimääräinen prosentuaalinen muutos lähtötasosta IgG-kokonaispitoisuuksissa säilyi viikolta 4 alkaen koko hoitojakson ajan (keskimääräinen väheneminen lähtötasosta oli 66,8–71,6 %).
Kliininen teho ja turvallisuus
Yleistynyt myasthenia gravis
Laskimoon annettava lääkemuoto
Efgartigimodi alfan tehoa aikuisten yleistyneen myasthenia graviksen (gMG) hoidossa tutkittiin 26 viikkoa kestäneessä satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (ARGX‑113‑1704).
Tässä tutkimuksessa potilaiden oli täytettävä seuraavat pääkriteerit seulonnassa:
- Myasthenia Gravis Foundation of American (MGFA) kliininen luokitus: luokka II, III tai IV
- potilaalla AChR-vasta-aineiden positiiviset tai negatiiviset serologiset testitulokset
- MG-Activity of Daily Living (MG‑ADL) -asteikon kokonaispistemäärä ≥ 5
- potilas sai ennen seulontaa vakaina annoksina myasthenia graviksen hoitoa, johon kuului asetyylikoliiniesteraasin (AChE) estäjiä, steroideja tai ei-steroidaalista immunosuppressiivista hoitoa (NSIST) joko yhdistelmähoitona tai yksilääkehoitona [NSIST-hoitoja olivat mm. atsatiopriini, metotreksaatti, siklosporiini, takrolimuusi, mykofenolaattimofetiili ja syklofosfamidi]
- IgG-pitoisuus vähintään 6 g/l.
Tutkimuksista suljettiin pois potilaat, joilla oli MGFA:n luokan V yleistynyt myasthenia gravis; potilaat, joilla ei todettu kliinistä vastetta plasmanvaihdolle; potilaat, jotka olivat saaneet plasmanvaihtoa, IVIg-hoitoa yhtä kuukautta ja monoklonaalisia vasta-aineita kuusi kuukautta ennen hoidon aloittamista; potilaat, joilla oli aktiivinen (akuutti tai krooninen) hepatiitti B-infektio, hepatiitti C-seropositiivisuus tai AIDS-diagnoosi.
Tutkimukseen otettiin yhteensä 167 potilasta, jotka satunnaistettiin saamaan joko efgartigimodi alfaa laskimoon (n = 84) tai lumelääkettä (n = 83). Lähtötason ominaisuudet olivat samanlaiset eri hoitoryhmissä, mukaan lukien mediaani-ikä diagnoosin saamisen hetkellä [45 (19 ‑ 81) vuotta], sukupuoli [useimmat olivat naisia; 75 % efgartigimodi alfaa saaneista ja 66 % lumelääkettä saaneista], rotu [useimmat potilaat olivat valkoihoisia, 84,4 %] ja mediaaniaika diagnoosin saamisesta [8,2 vuotta (efgartigimodi alfa) ja 6,9 vuotta (lumelääke)].
Suurimmalla osalla potilaista (77 %:lla kummassakin ryhmässä) todettiin AChR-vasta-aineita (AChR‑Ab), ja 23 %:lla potilaista oli negatiiviset AChR-Ab-tulokset.
Tutkimuksen aikana yli 80 % kummankin ryhmän potilaista sai AChE:n estäjiä, yli 70 % kummankin hoitoryhmän potilaista sai steroideja ja noin 60 % kummankin hoitoryhmän potilaista sai vakaina annoksina ei-steroidaalisia immunosuppressiivisia hoitoja (NSIST). Tutkimuksen alussa noin 30 %:lla kummankaan hoitoryhmän potilaista ei ollut aiempaa NSIST-altistusta.
MG-ADL-kokonaispistemäärän mediaani oli 9,0 molemmissa hoitoryhmissä, ja Quantitative Myasthenia Gravis (QMG) -kokonaispistemäärän mediaani oli 17 efgartigimodi alfa -ryhmässä ja 16 lumelääkeryhmässä.
Potilaat saivat efgartigimodi alfaa laskimoon 10 mg/kg kerran viikossa 4 viikon ajan enintään 3 hoitojakson verran.
Efgartigimodi alfan tehoa mitattiin käyttämällä Myasthenia Gravis-Specific Activities of Daily Living -asteikkoa (MG-ADL), jossa arvioidaan yleistyneen myasthenia graviksen vaikutusta päivittäisiin toimintoihin. Kokonaispistemäärän vaihteluväli on 0 ‑ 24, ja korkeammat pisteet viittaavat suurempaan toiminnanvajaukseen. Tässä tutkimuksessa MG-ADL-vasteen saavuttavalla potilaalla MG‑ADL-kokonaispistemäärän oli oltava ≥ 2 pistettä pienempi hoitojakson lähtötasoon verrattuna vähintään 4 peräkkäisen viikon ajan siten, että ensimmäinen pienempi pistemäärä havaittiin viimeistään 1 viikon kuluttua hoitojakson viimeisestä infuusiosta.
Efgartigimodi alfan tehoa mitattiin myös QMG‑kokonaispistemäärällä. Kyseessä on luokitusjärjestelmä, joka arvioi lihasheikkoutta kokonaispistemäärällä 0 ‑ 39 siten, että korkeammat pistemäärät tarkoittavat vaikea-asteisempaa toiminnanvajausta. Tässä tutkimuksessa QMG-vasteen saavuttavalla potilaalla QMG-kokonaispistemäärän oli oltava ≥ 3 pistettä pienempi hoitojakson lähtötasoon verrattuna vähintään 4 peräkkäisen viikon ajan siten, että ensimmäinen pienempi pistemäärä havaittiin viimeistään 1 viikon kuluttua hoitojakson viimeisestä infuusiosta.
Ensisijaisena tehon päätetapahtumana oli MG‑ADL-vasteen saavuttaneiden prosentuaalisen osuuden vertailu ensimmäisen hoitojakson (C1) aikana AChR‑Ab-seropositiivisen populaation eri hoitoryhmien välillä.
Keskeisenä toissijaisena päätetapahtumana oli ensimmäisen hoitojakson aikana QMG-vasteen saavuttaneiden prosentuaalisen osuuden vertailu AChR‑Ab-seropositiivisten potilaiden molempien hoitoryhmien välillä.
Taulukko 2. MG-ADL- ja QMG-vasteen saavuttaneiden osuudet ensimmäisen hoitojakson aikana AChR-Ab-seropositiivisessa populaatiossa (mITT-analyysijoukko)
| Populaatio | Efgartigimodi alfa n/N (%) | Lumelääke n/N (%) | P-arvo | Efgartigimodi alfan ja lumelääkkeen ero (95 %:n luottamusväli) | |
| MG-ADL | AChR‑Ab-seropositiivinen | 44/65 (67,7) | 19/64 (29,7) | < 0,0001 | 38,0 (22,1; 54,0) |
| QMG | AChR‑Ab-seropositiivinen | 41/65 (63,1) | 9/64 (14,1) | < 0,0001 | 49,0 (34,5; 63,5) |
AChR-Ab = asetyylikoliinireseptorivasta-aine; MG-ADL = Myasthenia Gravis Activities of Daily Living ‑asteikko (myasthenia graviksen vaikutukset päivittäisiin toimintoihin); QMG = Quantitative Myasthenia Gravis (kvantitatiivinen myasthenia gravis -asteikko); mITT = modifioitu hoitoaikeen mukainen; n = niiden potilaiden lukumäärä, joista on ilmoitettu havaintoarvo; N = potilaiden lukumäärä analyysijoukossa;
Logistinen regressiomalli, joka on ositettu AChR-Ab-statuksen (jos tiedossa), japanilaisuuden/ei-japanilaisuuden ja standardihoidon mukaan, lähtötilanteen MG-ADL kovariaattina/QMG kovariaatteina
Kaksipuolinen tarkka p-arvo
Analyysit osoittavat, että toisen hoitojakson aikana MG-ADL-vasteen saavuttaneiden osuus oli samanlainen kuin ensimmäisen hoitojakson aikana (ks. taulukko 3).
Taulukko 3. MG-ADL- ja QMG-vasteen saavuttaneiden osuudet toisen hoitojakson aikana AChR‑Ab-seropositiivisessa populaatiossa (mITT-analyysijoukko)
|
AChR-Ab = asetyylikoliinireseptorivasta-aine; MG-ADL = Myasthenia Gravis Activities of Daily Living ‑asteikko (myasthenia graviksen vaikutukset päivittäisiin toimintoihin); QMG = Quantitative Myasthenia Gravis (kvantitatiivinen myasthenia gravis -asteikko); mITT = modifioitu hoitoaikeen mukainen; n = niiden potilaiden lukumäärä, joista on ilmoitettu havaintoarvo; N = potilaiden lukumäärä analyysijoukossa.
Eksploratiiviset tiedot osoittavat, että AChR‑Ab-seropositiivisista MG‑ADL-vasteen saavuttaneista efgartigimodi alfa -hoitoa laskimoon saaneista 37/44 potilaalla (84 %:lla) havaittiin vasteen alkaminen 2 viikon kuluessa ensimmäisestä infuusiosta.
Kaksoissokkoutetussa lumekontrolloidussa tutkimuksessa (ARGX‑113‑1704) aikaisin mahdollinen aika seuraavan hoitojakson aloittamiseen oli 8 viikkoa ensimmäisen hoitojakson ensimmäisen infuusion jälkeen. Kokonaispopulaatiossa keskimääräinen aika toiseen hoitojaksoon efgartigimodi alfaa laskimoon saaneessa ryhmässä oli 13 viikkoa (keskihajonta 5,5 viikkoa) ja mediaaniaika oli 10 viikkoa (8 ‑ 26 viikkoa) ensimmäisen hoitojakson ensimmäisestä infuusiosta. Avoimessa jatkotutkimuksessa (ARGX‑113‑1705) aikaisin mahdollinen aika seuraavien hoitojaksojen aloittamiseen oli 7 viikkoa.
Hoitovasteen saavuttaneilla potilailla kliinisen kohentumisen kesto oli 5 viikkoa 5/44 potilaalla (11 %:lla), 6 ‑ 7 viikkoa 14/44 potilaalla (32 %:lla), 8 ‑ 11 viikkoa 10/44 potilaalla (23 %:lla) ja 12 viikkoa tai enemmän 15/44 potilaalla (34 %:lla).
Ihon alle annettava lääkemuoto
10 viikon pituinen satunnaistettu, avoin, rinnakkaisryhmissä toteutettu monikeskustutkimus (ARGX‑113‑2001) tehtiin gMG-aikuispotilailla ihon alle annettavan efgartigimodi alfan ja laskimoon annettavan efgartigimodi alfan farmakodynaamisen vaikutuksen yhdenveroisuuden arvioimiseksi. Pääasialliset mukaanotto- ja poissulkukriteerit olivat samat kuin ARGX‑113‑1704-tutkimuksessa.
Yhteensä 110 potilasta satunnaistettiin ja sai yhden kerran viikossa annetun 4 viikon hoitojakson verran joko 1 000 mg efgartigimodi alfaa ihon alle (n = 55) tai 10 mg/kg efgartigimodi alfaa laskimoon (n = 55). Suurin osa potilaista oli AChR‑vasta-aine (AChR‑Ab) -positiivisia: 45 potilasta (82 %) efgartigimodi alfaa ihon alle saaneessa ryhmässä ja 46 potilasta (84 %) efgartigimodi alfaa laskimoon saaneessa ryhmässä. Kaikki potilaat saivat ennen seulontaa vakaalla annoksella myasthenia graviksen hoitoa, joka sisälsi AChE:n estäjiä, steroideja tai NSIST-hoitoa joko yhdistelmänä tai yksinään.
Lähtötason ominaisuudet olivat samanlaisia hoitoryhmien välillä.
Tutkimuksen aikana yli 80 % kummankin ryhmän potilaista sai AChE:n estäjiä, yli 60 % kummankin ryhmän potilaista sai steroideja, ja noin 40 % kummankin hoitoryhmän potilaista sai NSIST-hoitoa vakailla annoksilla. Tutkimuksessa aloittaessaan noin 56 % potilaista kummassakin hoitoryhmässä ei ollut aiemmin saanut NSIST-hoitoa.
Ensisijaisena päätetapahtumana oli hoitoryhmien vertailu IgG-kokonaispitoisuuksien prosentuaalisessa vähenemisessä lähtötasolta päivään 29 mennessä kokonaispopulaatiossa. AChR-Ab-seropositiivisen populaation tulokset osoittavat ihon alle annettavan efgartigimodi alfan ja laskimoon annettavan efgartigimodi alfan yhdenveroisuuden (ks. taulukko 4).
Taulukko 4. ANCOVA-analyysi IgG-kokonaispitoisuuden prosentuaalisesta muutoksesta lähtötasolta päivään 29 mennessä AChR-Ab-seropositiivisessa populaatiossa (mITT-analyysijoukko)
| Efgartigimodi alfa SC | Efgartigimodi alfa IV | Ero Efgartigimodi alfa SC-Efgartigimodi alfa IV | ||||||
| N | LS-keskiarvo | (95 %:n luottamusväli) | N | LS-keskiarvo | (95 %:n luottamusväli) | Keskiarvon eron LS | (95 %:n luottamusväli) | p-arvo |
| 41 | ‑66,9 | ‑69,78, ‑64,02 | 43 | ‑62,4 | ‑65,22, ‑59,59 | ‑4,5 | ‑8,53, ‑0,46 | < 0,0001 |
AChR-Ab = asetyylikoliinireseptorivasta-aine; ANCOVA = kovarianssianalyysi; SC = ihon alle; IV = laskimoon; LS = pienimmät neliösummat; mITT = modifioitu hoitoaikeen mukainen analyysijoukko; N = potilaiden lukumäärä per ryhmä, jotka sisällytettiin mukaan ANCOVA-analyysiin.
Toissijaisina tehon päätetapahtumina olivat vertailut hoitoryhmien välillä MG-ADL- ja QMG-vasteen saaneiden prosenttiosuudessa tutkimuksessa ARGX‑113‑1704 määritellyllä tavalla. AChR‑Ab-seropositiivisen populaation tulokset on esitetty taulukossa 5.
Taulukko 5. MG-ADL- ja QMG-vasteen saaneet päivän 29 kohdalla AChR-Ab-seropositiivisessa populaatiossa (mITT-analyysijoukko)
| Efgartigimodi alfa SC n/N (%) | Efgartigimodi alfa IV n/N (%) | Ero Efgartigimodi alfa SC-Efgartigimodi alfa IV (95 %:n luottamusväli) | |
| MG-ADL | 32/45 (71,1) | 33/46 (71,7) | ‑0,6 (‑19,2–17,9) |
| QMG | 31/45 (68,9) | 24/45 (53,3) | 15,6 (‑4,3–35,4) |
AChR-Ab = asetyylikoliinireseptorivasta-aine; MG‑ADL = Myasthenia Gravis Activities of Daily Living ‑asteikko (myasthenia graviksen vaikutukset päivittäisiin toimintoihin); QMG = Quantitative Myasthenia Gravis (kvantitatiivinen myasthenia gravis -asteikko); SC = ihon alle; IV = laskimoon; mITT = modifioitu hoitoaikeen mukainen; n = potilaiden lukumäärä, joilta havaintoarvo on raportoitu; N = potilaiden lukumäärä analyysijoukossa.
AChR‑Ab-seropositiivisista MG‑ADL-vasteen saaneista potilaista saatujen eksploratiivisten tietojen perusteella vasteen havaittiin alkavan 2 viikon sisällä ensimmäisestä antokerrasta 28/32 (88 %) potilaalla, jotka saivat efgartigimodi alfaa ihon alle, ja 27/33 (82 %) potilaalla, jotka saivat efgartigimodi alfaa laskimoon.
Krooninen inflammatorinen demyelinoiva polyneuropatia
Ihon alle annettavan efgartigimodi alfan tehoa CIDP:tä sairastavien aikuisten hoidossa tutkittiin prospektiivisessa monikeskustutkimuksessa ARGX‑113‑1802, joka tehtiin kahdessa hoitovaiheessa: avoin vaihe A ja satunnaistettua hoidon lopetusta käyttävä, kaksoissokkoutettu ja lumekontrolloitu vaihe B.
Potilaat olivat joko käyttäneet CIDP-hoitoa tutkimuksessa aloittamista edeltävien 6 kuukauden aikana tai olleet ilman tätä hoitoa. Potilaat, jotka olivat saaneet aikaisempaa CIDP-hoitoa sekä potilaat, jotka eivät saaneet CIDP-hoitoa, ja joilla ei ollut dokumentoitua näyttöä CIDP:n viimeaikaisesta heikkenemisestä, aloittivat hoidottoman alkuseurantajakson. Potilaat, joilla todettiin näyttöä kliinisesti merkittävästä heikkenemisestä, aloittivat tutkimuksen vaiheessa A. Potilaat, jotka olivat olleet ilman CIDP-hoitoa ja joilla oli viimeaikaista näyttöä CIDP:n heikkenemisestä, jättivät väliin alkuseurantajakson ja aloittivat suoraan vaiheessa A.
Yhteensä 322 potilasta otettiin mukaan vaiheeseen A. Potilaat saivat enintään 12 kertaviikkoista injektiota efgartigimodi alfaa ihon alle 1 000 mg:n annoksella, kunnes heillä todettiin näyttöä kliinisestä kohentumisesta kahdella peräkkäisellä tutkimuskäynnillä. Tämän jälkeen potilaat, joilla todettiin näyttöä kliinisestä kohentumisesta, aloittivat tutkimuksen vaiheessa B, ja heidät satunnaistettiin saamaan viikoittain joko efgartigimodi alfaa ihon alle (111 potilasta) tai lumelääkettä (110 potilasta). Näyttö kliinisestä kohentumisesta määriteltiin kliiniseksi kohentumiseksi muokatulla Inflammatory Neuropathy Cause and Treatment (aINCAT) -mittarilla tai kohentumiseksi Inflammatory Rasch-built Overall Disability Scale (I‑RODS) -asteikolla/otevoimassa potilailla, joiden tulokset näillä mittareilla heikkenivät vain ennen vaihetta A.
Vaiheessa A potilaiden mediaani-ikä oli 54 vuotta (vaihteluväli 20–82 vuotta), mediaaniaika CIDP-diagnoosista oli 2,8 vuotta ja INCAT-pistemäärän mediaani oli 4,0. 65 % potilaista oli miehiä ja 66 % oli valkoihoisia. Vaiheessa B potilaiden mediaani-ikä oli 55 vuotta (vaihteluväli 20–82 vuotta), mediaaniaika CIDP-diagnoosista oli 2,2 vuotta ja INCAT-pistemäärän mediaani oli 3,0. 64 % potilaista oli miehiä ja 65 % oli valkoihoisia. Vaiheen B lähtötason ominaisuudet olivat samanlaiset hoitoryhmien välillä.
Vaiheen A ensisijaisena päätetapahtumana oli vasteen saaneiden prosenttiosuus, määritelmänä ne potilaat, jotka saavuttivat vahvistetun näytön kliinisestä kohentumisesta. 66,5 % potilaista saavutti ensisijaisen päätetapahtuman. Tarkemmat tiedot ovat taulukossa 6.
Vaiheen A toissijaisena päätetapahtumana oli aika ensimmäiseen vahvistettuun näyttöön kliinisestä kohentumisesta. Viikko 4 oli aikaisin ajankohta, jolloin kliinistä kohentumista koskevan näytön kriteerit voivat täyttyä. Kyseisessä ajankohdassa jopa 40 % potilaista saavutti näytön kliinisestä kohentumisesta. Ennalta määritetyn lisäanalyysin perusteella 25 %:lla potilaista todettiin kliinisesti merkittävä kohentuminen 9 vuorokauden kulutta vähintään yhdessä kolmesta parametrista (aINCAT, I‑RODS tai otevoima).
Suurin osa potilaista saavutti vahvistetun näytön kliinisestä kohentumisesta kaikissa aiemmin CIDP-lääkitystä saaneiden ryhmissä.
Taulukko 6. Näyttö kliinisestä kohentumisesta CIDP-potilailla tutkimuksen ARGX-113-1802 vaiheessa A
| Vasteen saaneet kliinistä kohentumista koskevan näytön perusteella ja aika ensimmäiseen vahvistettuun näyttöön kliinisestä kohentumisesta | Vaihe A |
| Efgartigimodi alfa SC (N = 322) | |
Vaste, näyttö kliinisestä kohentumisesta (potilaat, joilla oli vahvistettu kliininen kohentuminen) n/N (%) (95 %:n luottamusväli) | 214/322 (66,5 %) (61,0; 71,6) |
Aika ensimmäiseen vahvistettuun näyttöön kliinisestä kohentumisesta, vuorokautta mediaani (95 %:n luottamusväli) | 43,0 (31,0; 51,0) |
n = potilaiden määrä, joilta havainto raportoitiin; N = potilaiden määrä analyysijoukossa
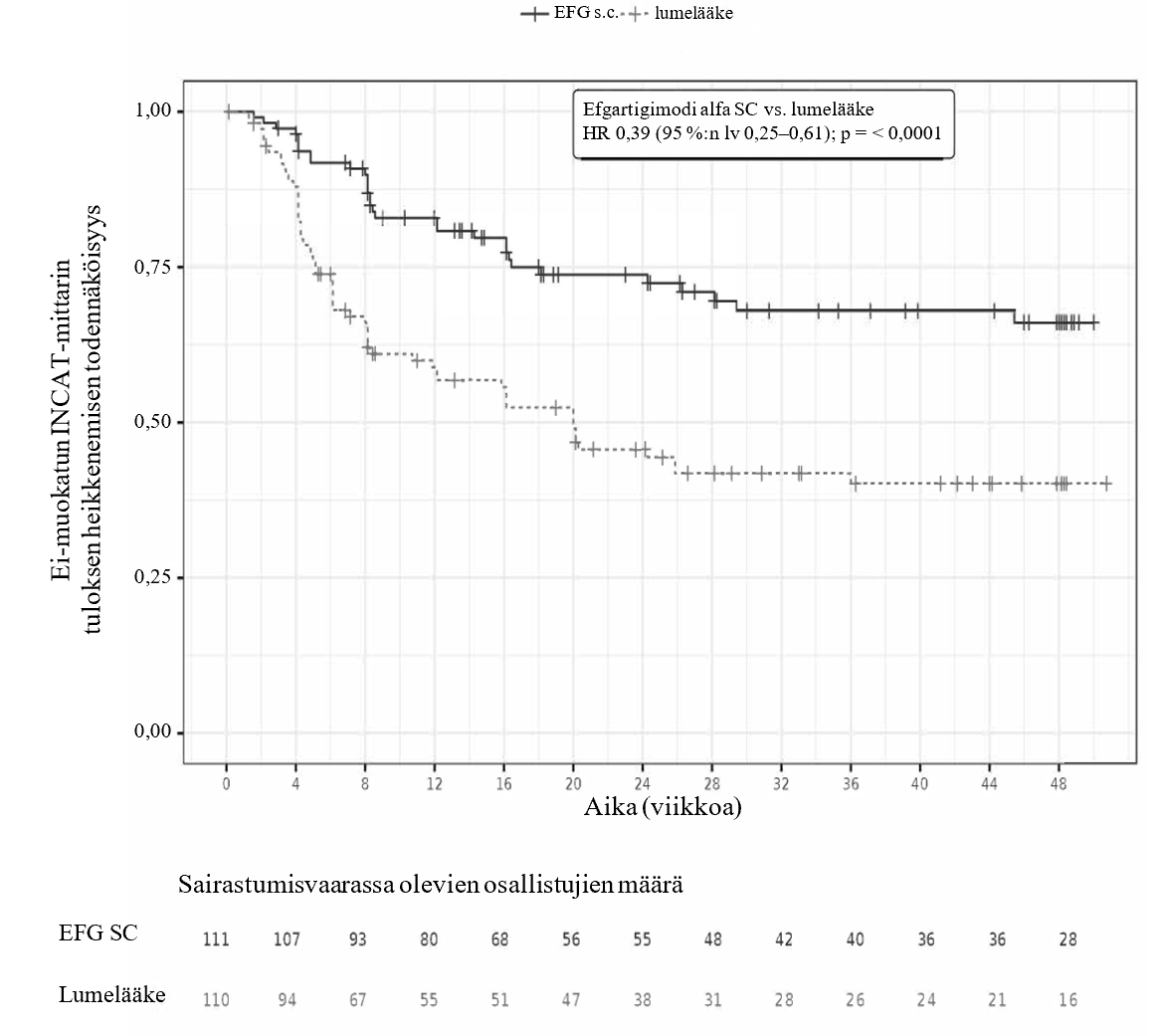
Vaiheessa B ensisijaisen päätetapahtuman määritelmänä oli aika ensimmäiseen näyttöön kliinisestä heikkenemisestä (1 pisteen lisäys aINCAT-mittarissa vaiheen B lähtötasoon verrattuna, joka vahvistettiin seuraavalla käynnillä ensimmäisen 1 pisteen lisäyksen jälkeen aINCAT-mittarissa, tai ≥ 2 pisteen lisäys aINCAT-mittarissa vaiheen B lähtötasoon verrattuna). Efgartigimodi alfaa ihon alle saaneet potilaat välttivät relapsit (kliinisen heikkenemisen) merkitsevästi pidemmän aikaa kuin lumelääkettä saaneet potilaat. Tämän osoittaa riskitiheyksien suhde 0,394 [95 %:n luottamusväli (0,253; 0,614)]. 31/111 (27,9 %) potilaista, jotka saivat tutkimuksen vaiheessa B efgartigimodi alfaa ihon alle, sai relapsin; lumelääkettä saaneista relapsin sai 59/110 (53,6 %). Tulokset esitetään taulukossa 7 ja kuvassa 2.
Taulukko 7. Ensimmäinen näyttö kliinisestä heikkenemisestä CIDP-potilailla tutkimuksen ARGX-113-1802 vaiheessa B
| Aika ensimmäiseen aINCAT-pistemäärän nousuun (kliininen heikkeneminen) | Vaihe B | |
Efgartigimodi alfa SC (N = 111) | Lumelääke (N = 110) | |
| Riskitiheyksien suhde (95 %:n luottamusväli) | 0,394 (0,253; 0,614) p-arvo < 0,0001 | |
| Mediaaniaika vuorokausina (95 %:n luottamusväli) | EL (EL; EL) | 140,0 (75,0; EL) |
EL = ei laskettu; N = potilaiden määrä analyysijoukossa; aINCAT = muokattu Inflammatory Neuropathy Cause and Treatment -arviointiasteikko
Kuva 2. Aika ensimmäiseen aINCAT-pistemäärän heikkenemiseen (Kaplan-Meierin kuvaaja) CIDP-potilailla tutkimuksen ARGX-113-1802 vaiheessa B
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Vyvgart‑valmisteen käytöstä myasthenia graviksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Vyvgart‑valmisteen käytöstä kroonisen inflammatorisen demyelinoivan polyneuropatian hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Populaatiofarmakokineettisen analyysin perusteella ihon alle annettavan efgartigimodi alfan 1 000 mg:n annoksen arvioitu biologinen hyötyosuus on 77 %.
Keskimääräinen matalin pitoisuus (Ctrough) 4 viikon pituisen kertaviikkoisen annon jälkeen oli efgartigimodi alfa 1 000 mg ihon alle -hoidon osalta 22,0 mikrog/ml (37 % CV) ja efgartigimodi alfa 10 mg/kg laskimoon -hoidon osalta 14,9 mikrog/ml (43 % CV). Efgartigimodi alfan AUC0-168h-arvo yhden hoitojakson annon jälkeen oli samanlainen 1 000 mg ihon alle -hoidon ja 10 mg/kg laskimoon -hoidon yhteydessä.
Jatkuvasti ihon alle annettavaa efgartigimodi alfaa 1 000 mg kerran viikossa saaneilla potilailla keskimääräinen matalin pitoisuus vaihteli välillä 14,9–20,1 mikrog/ml.
Jakautuminen
Terveillä tutkittavilla ja potilailla tehdyn populaatiofarmakokineettisen analyysin perusteella jakautumistilavuus on 18 l.
Biotransformaatio
Proteolyyttisten entsyymien odotetaan hajottavan efgartigimodi alfan pieniksi peptideiksi ja aminohapoiksi.
Eliminaatio
Terminaalinen puoliintumisaika on 80 ‑ 120 tuntia (3 ‑ 5 vuorokautta). Populaatiofarmakokineettisen analyysin perusteella puhdistuma on 0,128 l/h. Efgartigimodi alfan molekyylipaino on noin 54 kDa, joka on munuaisten suodattamien molekyylien koon rajalla.
Lineaarisuus/ei-lineaarisuus
Efgartigimodi alfan farmakokineettinen profiili on lineaarinen, annoksesta tai ajasta riippumatta, ja kertyminen on hyvin vähäistä.
Erityisryhmät
Ikä, sukupuoli, rotu ja paino
Ikä (19–84 vuotta), sukupuoli, rotu ja paino eivät vaikuttaneet efgartigimodi alfan farmakokinetiikkaan.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty erityisiä farmakokineettisiä tutkimuksia.
Munuaisten toiminnan merkkiaineena toimivan arvioidun glomerulusten suodatusnopeuden [eGFR] vaikutus populaatiofarmakokineettisen analyysin kovariaattina osoitti altistuksen lisääntyneen (11–21 %) potilailla, joilla oli lievä munuaisten vajaatoiminta (eGFR 60 ‑ 89 ml/min/1,73 m2). Erityistä annoksen muuttamista ei suositella potilaille, joilla on lievä munuaisten vajaatoiminta.
Kohtalaisen munuaisten vajaatoiminnan (eGFR-arvo 30 ‑ 59 ml/min/1,73 m2), ja vaikean munuaisten vajaatoiminnan (eGFR-arvo < 30 ml/min/1,73 m2) vaikutuksista efgartigimodi alfan farmakokineettisiin parametreihin ei ole olemassa riittävästi tietoja.
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavilla potilailla ei ole tehty erityisiä farmakokineettisiä tutkimuksia.
Kun maksan toiminnan merkkiaineiden vaikutusta käytettiin populaatiofarmakokineettisen analyysin kovariaatteina, efgartigimodi alfan farmakokinetiikkaan kohdistuvaa vaikutusta ei havaittu.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Rotilla ja kaneilla tehdyissä lisääntymistutkimuksissa efgartigimodi alfan laskimonsisäinen anto ei aiheuttanut haitallisia vaikutuksia hedelmällisyyteen ja tiineyteen, eikä teratogeenisia vaikutuksia havaittu annoksilla, jotka vastasivat 11-kertaista (rotat) ja 56-kertaista (kanit) ihmisen 10 mg/kg ‑tasoista altistusta AUC-arvon perusteella.
Karsinogeenisuus ja genotoksisuus
Efgartigimodi alfan karsinogeenisuutta ja genotoksisuutta ei ole tutkittu.
Hyaluronidaasia on löydetty useimmista ihmiskehon kudoksista. Rekombinanttia ihmisen hyaluronidaasia koskevat ei-kliiniset tiedot eivät viittaa erityiseen vaaraan ihmisille toistuvan altistuksen aiheuttamaa toksisuutta koskevissa konventionaalisissa tutkimuksissa, mukaan lukien turvallisuusfarmakologiset päätetapahtumat. Rekombinantti ihmisen hyaluronidaasilla tehdyissä lisääntymistoksikologiaa selvittäneissä tutkimuksissa havaittiin alkion ja sikiön toksisuutta hiirillä korkealla systeemisellä altistuksella, mutta ne eivät osoittaneet teratogeenistä potentiaalia.
Farmaseuttiset tiedot
Apuaineet
Rekombinantti ihmisen hyaluronidaasi (rHuPH20)
L-histidiini
L-histidiinihydrokloridimonohydraatti
L-metioniini
Polysorbaatti 20 (E432)
Natriumkloridi
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
18 kuukautta
Tarvittaessa avaamattomia injektiopulloja voidaan säilyttää huoneenlämmössä (enintään 30 °C) enintään 3 vuorokauden ajan. Huoneenlämmössä säilyttämisen jälkeen avaamattomat injektiopullot voidaan palauttaa jääkaappiin. Jos niitä säilytetään jääkaappisäilytyksen ulkopuolella ja ne palautetaan sen jälkeen jääkaappisäilytykseen, jääkaappisäilytyksen ulkopuolinen kokonaisaika ei saa olla yli 3 vuorokautta.
Mikrobiselta kannalta, ellei ruiskun valmistelumenetelmä poissulje mikrobikontaminaation riskiä, valmiste on käytettävä välittömästi. Jos valmistetta ei käytetä heti, käytönaikaiset säilytysajat ja säilytysolosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C ‑ 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Ei markkinoilla olevia pakkauksia.
PF-selosteen tieto
5,6 ml:n liuosta 6 ml:n tyypin I lasisessa injektiopullossa, jossa on kumitulppa, alumiinisinetti ja polypropyleenistä valmistettu repäisykorkki.
Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Kellertävä, kirkas tai opaalinhohtoinen, pH 6,0.
Käyttö- ja käsittelyohjeet
Vyvgart toimitetaan käyttövalmiina liuoksena kertakäyttöisessä injektiopullossa. Lääkevalmistetta ei tarvitse laimentaa.
Tarkista silmämääräisesti, että injektiopullon sisältö on kellertävää, kirkasta tai opaalinhohtoista liuosta ja että siinä ei ole hiukkasia. Jos näkyviä hiukkasia on havaittavissa, injektiopulloa ei saa käyttää.
Sen jälkeen kun injektiopullo on otettu pois jääkaapista, odota vähintään 15 minuutin ajan ennen injisoimista, jotta liuos lämpiää huoneenlämpöiseksi (ks. kohta Kestoaika).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VYVGART injektioneste, liuos
1000 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AA58
Valmisteyhteenvedon muuttamispäivämäärä
05.01.2026
Yhteystiedot
Industriepark-Zwijnaarde 7
9052 Gent
Belgium
0800 412838
medinfofi@argenx.com