
TEVIMBRA infuusiokonsentraatti, liuosta varten 100 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi ml infuusiokonsentraattia liuosta varten sisältää 10 mg tislelitsumabia.
Yksi 10 ml:n injektiopullo sisältää 100 mg tislelitsumabia (100 mg/10 ml).
Tislelitsumabi on Fc‑muunnettu, humanisoitu immunoglobuliini G4:n (IgG4) monoklonaalinen vasta‑ainevariantti, joka tuotetaan rekombinanteissa kiinanhamsterin munasarjasoluissa.
Apuaine, jonka vaikutus tunnetaan
Yksi ml infuusiokonsentraattia liuosta varten sisältää 1,6 mg natriumia ja 0,2 mg polysorbaatti 20:tä (E432).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Ei‑pienisoluinen keuhkosyöpä
Tevimbra on tarkoitettu leikkaukseen soveltuvan ei-pienisoluisen keuhkosyövän hoitoon aikuispotilaille, joilla on suuri taudin uusiutumisriski (ks. valintakriteerit kohdasta Farmakodynamiikka). Tevimbra annetaan tällöin ensin yhdistelmänä platinapohjaisen solunsalpaajahoidon kanssa neoadjuvanttihoitona, minkä jälkeen valmisteen käyttöä jatketaan adjuvanttihoidossa monoterapiana.
Tevimbra, yhdessä pemetreksedin ja platinaa sisältävän solunsalpaajahoidon kanssa, on tarkoitettu ei‑levyepiteeliperäisen ei‑pienisoluisen keuhkosyövän ensilinjan hoitoon aikuispotilaille, joiden kasvaimissa kasvainsolujen PD-L1-ekspressio on vähintään 50 %, eikä niissä ole EGFR‑mutaatioita tai ALK‑positiivisia mutaatioita, ja joilla on
- paikallisesti edennyt ei‑pienisoluinen keuhkosyöpä ja joille leikkaus tai platinapohjainen kemosädehoito ei sovellu tai
- metastasoitunut ei‑pienisoluinen keuhkosyöpä.
Tevimbra, yhdessä karboplatiinin ja joko paklitakselin tai nab‑paklitakselin kanssa, on tarkoitettu levyepiteeliperäisen ei‑pienisoluisen keuhkosyövän ensilinjan hoitoon aikuispotilaille, joilla on
- paikallisesti edennyt ei‑pienisoluinen keuhkosyöpä ja joille leikkaus tai platinapohjainen kemosädehoito ei sovellu tai
- metastasoitunut ei‑pienisoluinen keuhkosyöpä.
Tevimbra on tarkoitettu monoterapiana paikallisesti edenneen tai metastasoituneen ei‑pienisoluisen keuhkosyövän hoitoon aikuispotilaille aiemman platinapohjaisen hoidon jälkeen. Potilaiden, joilla on ei‑pienisoluinen keuhkosyöpä, jossa on EGFR‑mutaatio tai ALK‑positiivisuus, on täytynyt saada myös kohdennettua hoitoa ennen tislelitsumabin antoa.
Pienisoluinen keuhkosyöpä
Tevimbra, yhdessä etoposidin ja platinasolunsalpaajahoidon kanssa, on tarkoitettu ensisilinjan hoidoksi aikuispotilaille, joilla on levinnyt pienisoluinen keuhkosyöpä (ES-SCLC).
Mahalaukun tai gastroesofageaalisen junktion adenokarsinooma
Tevimbra, yhdessä platina- ja fluoropyrimidiinipohjaisen solunsalpaajahoidon kanssa, on tarkoitettu ensilinjan hoidoksi aikuispotilaille, joilla on HER-2-negatiivinen paikallisesti edennyt, leikattavaksi soveltumaton tai metastasoitunut mahalaukun tai gastroesofageaalisen junktion adenokarsinooma, ja joiden kasvaimet ilmentävät PD-L1:tä siten, että kasvainalueen positiivisuuspistemäärä (TAP-pistemäärä) on vähintään 5 % (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinooma
Tevimbra, yhdessä platinapohjaisen solunsalpaajahoidon kanssa, on tarkoitettu leikattavaksi soveltumattoman, paikallisesti edenneen tai metastasoituneen ruokatorven levyepiteelikarsinooman ensilinjan hoitoon aikuispotilaille, joiden kasvaimet ilmentävät PD-L1:tä siten, että TAP-pistemäärä on vähintään 5 % (ks. kohta Farmakodynamiikka).
Tevimbra on tarkoitettu monoterapiana leikattavaksi soveltumattoman, paikallisesti edenneen tai metastasoituneen ruokatorven levyepiteelikarsinooman hoitoon aikuispotilaille aiemman platinapohjaisen solunsalpaajahoidon jälkeen.
Nenänielukarsinooma
Tevimbra, yhdessä gemsitabiinin ja sisplatiinin kanssa, on tarkoitettu ensilinjan hoidoksi aikuispotilaille, joilla on uusiutunut tai metastasoitunut nenänielukarsinooma tai nenänielukarsinooma, jota ei voida hoitaa kuratiivisella leikkauksella eikä sädehoidolla.
Ehto
Valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sitä.
Annostus ja antotapa
Tevimbra‑hoidon aloittaa ja hoitoa valvoo lääkäri, jolla on kokemusta syövän hoidosta.
PD-L1-testaus
Jos käyttöaiheessa on niin täsmennetty, kasvaimen PD-L1-ekspression perusteella tehtävä potilaiden valinta Tevimbra-hoitoon on suoritettava CE-merkityllä IVD-laitteella, jolla on vastaava käyttötarkoitus. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, tällöin on käytettävä vaihtoehtoista validoitua testiä (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus
Tevimbra-monoterapia
Suositeltu Tevimbra‑annos on joko 200 mg 3 viikon välein tai 400 mg 6 viikon välein infuusiona laskimoon. Leikkaushoitoon soveltuvan ei-pienisoluisen keuhkosyövän adjuvanttihoitovaiheessa suositeltu Tevimbra-annos on 400 mg infuusiona laskimoon 6 viikon välein.
Tevimbra-yhdistelmähoito
Suositeltu Tevimbra‑annos on joko 200 mg 3 viikon välein tai 400 mg 6 viikon välein infuusiona laskimoon yhdistelmähoitona solunsalpaajahoidon kanssa.
Kun Tevimbra-valmistetta ja solunsalpaajahoitoa annetaan samana päivänä, Tevimbra-valmiste on annettava ennen solunsalpaajahoitoa. Solunsalpaajahoidon annostus ja solunsalpaajahoidon haittavaikutuksia estävänä esilääkityksenä käytettäviä kortikosteroideja koskevat suositukset on tarkistettava solunsalpaajahoitovalmisteen valmisteyhteenvedosta.
Hoidon kesto
Tevimbra‑hoitoa jatketaan, kunnes tauti etenee tai ilmenee liiallista toksisuutta (ks. kohta Farmakodynamiikka).
Leikkaushoitoon soveltuvan ei-pienisoluisen keuhkosyövän neoadjuvantti- ja adjuvanttihoito toteutetaan seuraavasti: potilaat saavat yhdistelmänä solunsalpaajahoidon kanssa Tevimbra-neoadjuvanttihoitoa (200 mg 3 viikon välein) 3 tai 4 hoitojakson ajan tai kunnes taudin eteneminen sulkee pois parantavan leikkaushoidon tai ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä. Tämän jälkeen annetaan adjuvanttihoitona Tevimbra-monoterapiaa (400 mg 6 viikon välein) enintään 8 hoitojakson ajan tai kunnes tauti uusiutuu, metastasoituu tai ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä.
Annoksen viivästyminen tai hoidon lopettaminen (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet)
Tevimbra‑annoksen pienentämistä ei suositella monoterapiassa eikä yhdistelmähoidossa. Tevimbra‑hoito on tauotettava tai lopetettava turvallisuuden ja siedettävyyden perusteella taulukossa 1 kuvatulla tavalla.
Tarkemmat ohjeet immuunivälitteisten haittavaikutusten hoitoon on kuvattu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Taulukko 1 Suositellut Tevimbra‑hoidon muutokset
| Immuunivälitteinen haittavaikutus | Vaikeusaste1 | Tevimbra‑hoidon muutos |
| Pneumoniitti | Aste 2 | Tauotetaan.2,3 |
| Uusiutunut aste 2; aste 3 tai 4 | Lopetetaan pysyvästi.3 | |
| Hepatiitti | ALAT tai ASAT > 3–8 x ULN tai kokonaisbilirubiini > 1,5–3 x ULN | Tauotetaan.2,3 |
| ALAT tai ASAT > 8 x ULN tai kokonaisbilirubiini > 3 x ULN | Lopetetaan pysyvästi.3 | |
| Ihottuma | Aste 3 | Tauotetaan.2,3 |
| Aste 4 | Lopetetaan pysyvästi.3 | |
| Vaikeat ihoreaktiot | Epäilty vaikea ihoreaktio, mukaan lukien Stevens–Johnsonin oireyhtymä ja toksinen epidermaalinen nekrolyysi | Tauotetaan.2,3 Jos Stevens–Johnsonin oireyhtymää tai toksista epidermaalista nekrolyysiä epäillään, hoitoa ei saa aloittaa uudelleen ennen kuin oireyhtymä tai nekrolyysi on suljettu pois asianmukaisten erikoislääkärien konsultoinnin jälkeen. |
| Vahvistettu vaikea ihoreaktio, mukaan lukien Stevens–Johnsonin oireyhtymä ja toksinen epidermaalinen nekrolyysi | Lopetetaan pysyvästi. | |
| Koliitti | Aste 2 tai 3 | Tauotetaan.2,3 |
| Uusiutunut aste 3; aste 4 | Lopetetaan pysyvästi.3 | |
| Myosiitti/rabdomyolyysi | Aste 2 tai 3 | Tauotetaan.2,3 |
| Uusiutunut aste 3; aste 4 | Lopetetaan pysyvästi.3 | |
| Kilpirauhasen vajaatoiminta | Aste 2, 3 tai 4 | Kilpirauhasen vajaatoimintaa voidaan hoitaa korvaushoidolla keskeyttämättä syöpähoitoa. |
| Kilpirauhasen liikatoiminta | Aste 3 tai 4 | Tauotetaan.2 Jos asteen 3 tai 4 haitta on lievittynyt asteeseen ≤ 2 ja on hallinnassa kilpirauhasen liikatoiminnan estohoidolla, Tevimbra‑hoidon jatkamista voidaan tarvittaessa harkita kortikosteroidihoidon asteittaisen purkamisen jälkeen. Muussa tapauksessa hoito on lopetettava. |
| Lisämunuaisten vajaatoiminta | Aste 2 | Hoidon tauottamista on harkittava, kunnes haitta on saatu hallintaan hormonikorvaushoidolla. |
| Aste 3 tai 4 | Tauotetaan.3 Jos asteen 3 tai 4 haitta on lievittynyt asteeseen ≤ 2 ja on hallinnassa hormonikorvaushoidolla, Tevimbra‑hoidon jatkamista voidaan tarvittaessa harkita kortikosteroidihoidon asteittaisen purkamisen jälkeen. Muussa tapauksessa hoito on lopetettava.3 | |
| Hypofysiitti | Aste 2 | Hoidon tauottamista on harkittava, kunnes haitta on saatu hallintaan hormonikorvaushoidolla. |
| Aste 3 tai 4 | Tauotetaan.2,3 Jos asteen 3 tai 4 haitta on lievittynyt asteeseen ≤ 2 ja on hallinnassa hormonikorvaushoidolla, Tevimbra‑hoidon jatkamista voidaan tarvittaessa harkita kortikosteroidihoidon asteittaisen purkamisen jälkeen. Muussa tapauksessa hoito on lopetettava.3 | |
| Tyypin 1 diabetes | Tyypin 1 diabetes, johon liittyy asteen ≥ 3 hyperglykemia (glukoosi >250 mg/dl tai > 13,9 mmol/l) tai johon liittyy ketoasidoosi | Tauotetaan. Jos asteen 3 tai 4 haitta on lievittynyt insuliinihoidolla asteeseen ≤ 2, Tevimbra‑hoidon jatkamista voidaan tarvittaessa harkita, kun diabetes on saatu hyvään hoitotasapainoon. Muussa tapauksessa hoito on lopetettava. |
| Nefriitti ja siihen liittyvä munuaisten toimintahäiriö | Aste 2 (kreatiniini > 1,5–3 x lähtöarvo tai > 1,5–3 x ULN) | Tauotetaan.2,3 |
| Aste 3 (kreatiniini > 3 x lähtöarvo tai > 3–6 x ULN) tai aste 4 (kreatiniini > 6 x ULN) | Lopetetaan pysyvästi.3 | |
| Myokardiitti | Aste 2, 3 tai 4 | Lopetetaan pysyvästi.3 |
| Neurologiset toksisuudet | Aste 2 | Tauotetaan.2,3 |
| Aste 3 tai 4 | Lopetetaan pysyvästi.3 | |
| Pankreatiitti | Asteen 3 pankreatiitti tai asteen 3 tai 4 seerumin amylaasi‑ tai lipaasiarvon suureneminen (> 2 x ULN) | Tauotetaan.2,3 |
| Aste 4 | Lopetetaan pysyvästi.3 | |
| Muut immuunivälitteiset haittavaikutukset | Aste 3 | Tauotetaan.2,3 |
| Uusiutunut aste 3; aste 4 | Lopetetaan pysyvästi.3 | |
| Muut haittavaikutukset | ||
| Infuusioreaktiot | Aste 1 | Esilääkitystä myöhempien infuusioreaktioiden estämiseksi on harkittava. Infuusionopeutta hidastetaan 50 %:lla. |
| Aste 2 | Infuusio keskeytetään. Infuusiota jatketaan, jos haitta häviää tai lievittyy asteeseen 1, ja infuusionopeutta hidastetaan 50 %:lla. | |
| Aste 3 tai 4 | Lopetetaan pysyvästi. | |
ALAT = alaniiniaminotransferaasi, ASAT = aspartaattiaminotransferaasi, ULN = viitealueen yläraja 1 Toksisuusasteet perustuvat NCI‑CTCAE v4.0 ‑kriteereihin (National Cancer Institute Common Terminology Criteria for Adverse Events, versio 4.0). Hypofysiitin aste perustuu NCI‑CTCAE v5.0 ‑kriteereihin. 2 Hoitoa jatketaan, jos haitta on korjautunut kokonaan tai osittain (asteeseen 0–1) sen jälkeen, kun kortikosteroidihoito on asteittain purettu vähintään 1 kuukauden aikana. Hoito lopetetaan pysyvästi, jos haitta ei ole korjautunut kokonaan eikä osittain 12 viikon kuluessa kortikosteroidihoidon aloittamisesta tai jos prednisoniannosta ei voida pienentää tasolle ≤ 10 mg/vrk (tai vastaavalle tasolle) 12 viikon kuluessa kortikosteroidihoidon aloittamisesta. 3 1–2 mg/kg/vrk aloitusannos prednisonia tai vastaava hoito ja sen jälkeen hoidon asteittainen purku tasolle ≤ 10 mg/vrk (tai vastaavalle tasolle) vähintään 1 kuukauden aikana on suositeltavaa lukuun ottamatta pneumoniittia, johon suositeltava aloitusannos on 2–4 mg/kg/vrk. | ||
Erityisryhmät
Pediatriset potilaat
Tevimbra‑valmisteen turvallisuutta ja tehoa alle 18‑vuotiaiden potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Iäkkäät
≥ 65‑vuotiaiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Haittavaikutukset).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavista potilaista on liian vähän tietoa annostelusuositusten antamiseksi kyseiselle populaatiolle (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Vaikeaa maksan vajaatoimintaa sairastavista potilaista on liian vähän tietoa annostelusuositusten antamiseksi kyseiselle populaatiolle (ks. kohta Farmakokinetiikka).
Antotapa
Tevimbra on tarkoitettu annettavaksi vain laskimoon. Tevimbra on annettava infuusiona, eikä sitä saa antaa laskimoon nopeana injektiona / kertaluonteisena bolusinjektiona. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Ensimmäinen 200 mg:n infuusio on annettava 60 minuutin kestoisena. Jos potilas sietää sen hyvin, seuraavat infuusiot voidaan antaa 30 minuutin kestoisina. Infuusio on annettava infuusioletkulla, jossa on steriili, pyrogeeniton, heikosti proteiinia sitova 0,2 tai 0,22 mikrometrin kiinteä (in‑line) tai irrallinen (add‑on) suodatin.
Tevimbran 400 mg:n aloitusannoksen infuusio on annettava 120 minuutin kestoisena (90 minuutin kestoisena, kun sitä käytetään jatkohoitona 3 viikon välein annetun 200 mg:n annoksen jälkeen). Jos potilas sietää sen hyvin, toinen infuusio voidaan antaa 60 minuutin kestoisena. Jos potilas sietää toisen infuusion hyvin, myöhemmät infuusiot voidaan antaa 30 minuutin kestoisina.
Muita lääkevalmisteita ei saa sekoittaa samassa infuusioletkussa eikä antaa samalla infuusioletkulla.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
PD-L1-statuksen arviointi
Kasvaimen PD-L1-statusta arvioitaessa on tärkeää valita hyvin validoitu menetelmä väärien negatiivisten ja väärien positiivisten määritysten minimoimiseksi.
Potilaskortti
Tevimbra‑hoitoa saaville potilaille on annettava potilaskortti, jossa kerrotaan immuunivälitteisten haittavaikutusten riskeistä Tevimbra‑hoidon aikana (ks. myös pakkausseloste).
Lääkkeen määrääjän on keskusteltava potilaan kanssa Tevimbra‑hoidon aikaisten immuunivälitteisten haittavaikutusten riskeistä.
Immuunivälitteiset haittavaikutukset
Tislelitsumabihoidon aikana on ilmoitettu immuunivälitteisiä haittavaikutuksia, myös kuolemaan johtaneita tapauksia (ks. kohta Haittavaikutukset). Valtaosa tapauksista lievittyi, kun tislelitsumabihoito keskeytettiin ja potilaalle annettiin kortikosteroideja ja/tai elintoimintoja tukevaa hoitoa. Immuunivälitteisiä haittavaikutuksia on ilmoitettu myös viimeisen tislelitsumabiannoksen jälkeen. Immuunivälitteiset haittavaikutukset voivat vaikuttaa samanaikaisesti useaan elinjärjestelmään.
Jos immuunivälitteistä haittavaikutusta epäillään, syyn vahvistamiseksi tai muiden syiden (myös infektion) poissulkemiseksi on tehtävä riittävä arviointi. Tislelitsumabihoito on tauotettava ja potilaalle on annettava kortikosteroideja haittavaikutuksen vaikeusasteen perusteella (ks. kohta Annostus ja antotapa). Kliinisistä tutkimuksista saatujen suppeiden tietojen perusteella muiden systeemisten immunosuppressanttien antoa voidaan harkita, jos immuunivälitteistä haittavaikutusta ei saada hallintaan kortikosteroideilla (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Jos haitta korjautuu asteeseen ≤ 1, aloitetaan kortikosteroidihoidon asteittainen purku ja sitä jatketaan vähintään 1 kuukauden ajan.
Havainnoivista tutkimuksista peräisin olevien tietojen perusteella immuunivälitteisten haittavaikutusten riski immuunitarkistuspisteen estäjähoidon jälkeen voi olla koholla potilailla, joilla on olemassa oleva autoimmuunisairaus (AID), verrattuna riskiin potilailla, joilla ei ole olemassa olevaa autoimmuunisairautta. Tämän lisäksi taustalla olevan autoimmuunisairauden pahenemisvaiheita ilmeni usein, mutta suurin osa niistä oli lieviä ja hoidettavissa.
Immuunivälitteinen pneumoniitti
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteistä pneumoniittia, myös kuolemaan johtaneita tapauksia. Potilaita on seurattava pneumoniitin oireiden ja löydösten varalta. Jos pneumoniittia epäillään, potilaalle on tehtävä kuvantamistutkimus ja infektioperäiset ja tautiin liittyvät syyt on suljettava pois.
Immuunivälitteisen pneumoniitin yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Immuunivälitteinen hepatiitti
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteistä hepatiittia, myös kuolemaan johtaneita tapauksia. Potilaita on seurattava hepatiitin oireiden ja löydösten ja maksatoiminnan muutosten varalta. Maksan toimintakokeet on tehtävä lähtötilanteessa ja määräajoin hoidon aikana.
Immuunivälitteisen hepatiitin yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Immuunivälitteiset ihoreaktiot
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteistä ihottumaa tai dermatiittia. Potilaita on seurattava epäiltyjen ihoreaktioiden varalta, ja muut syyt on suljettava pois. Tislelitsumabihoito on tauotettava tai lopetettava pysyvästi ihoon kohdistuvan haittavaikutuksen vaikeusasteen perusteella taulukon 1 suositusten mukaisesti (ks. kohta Annostus ja antotapa).
Tislelitsumabia saaneilla potilailla on ilmoitettu vaikeita ihoreaktioita, kuten monimuotoinen punavihoittuma (erythema multiforme (EM)), Stevens–Johnsonin oireyhtymä (SJS) ja toksinen epidermaalinen nekrolyysi (TEN), joista jotkut ovat johtaneet kuolemaan (katso kohta Haittavaikutukset). Potilaita on seurattava vaikeiden ihoreaktioiden oireiden ja löydösten varalta (esim. ennakko‑oireena kuume, flunssankaltaiset oireet, limakalvomuutokset tai etenevä ihottuma), ja muut syyt on suljettava pois. Jos vaikeaa ihoreaktiota epäillään, tislelitsumabihoito on tauotettava ja potilas on ohjattava erikoissairaanhoitoon arviointia ja hoitoa varten. Jos vaikea ihoreaktio vahvistetaan, tislelitsumabihoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Immuunivälitteinen koliitti
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteistä koliittia, johon on usein liittynyt ripuli. Potilaita on seurattava koliitin oireiden ja löydösten varalta. Infektioperäiset ja tautiin liittyvät syyt on suljettava pois.
Immuunivälitteisen koliitin yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Immuunivälitteiset endokrinopatiat
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteisiä endokrinopatioita, mukaan lukien kilpirauhasen häiriöitä, lisämunuaisten vajaatoimintaa, hypofysiittia ja tyypin 1 diabetesta. Nämä voivat vaatia elintoimintoja tukevaa hoitoa umpierityshäiriön tyypistä riippuen. Immuunivälitteiset endokrinopatiat voivat vaatia pitkäaikaista hormonikorvaushoitoa.
Immuunivälitteisten endokrinopatioiden yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Kilpirauhasen häiriöt
Tislelitsumabia saaneilla potilailla on ilmoitettu kilpirauhasen häiriöitä, mukaan lukien tyreoidiittia sekä kilpirauhasen vajaatoimintaa ja liikatoimintaa. Potilaita on seurattava kilpirauhasen toiminnan muutosten varalta ja kilpirauhasen häiriöiden kliinisten oireiden ja löydösten varalta (hoidon alkaessa, säännöllisesti hoidon aikana ja lisäksi tarvittaessa kliinisen arvioinnin perusteella). Kilpirauhasen vajaatoimintaa voidaan hoitaa korvaushoidolla keskeyttämättä syöpähoitoa ja käyttämättä kortikosteroideja. Kilpirauhasen liikatoimintaa voidaan hoitaa oireenmukaisesti (ks. kohta Annostus ja antotapa).
Lisämunuaisten vajaatoiminta
Tislelitsumabia saaneilla potilailla on ilmoitettu lisämunuaisten vajaatoimintaa. Potilaita on seurattava lisämunuaisten vajaatoiminnan oireiden ja löydösten varalta. Lisämunuaisten toiminnan ja hormonipitoisuuksien seurantaa on harkittava. Kortikosteroideja ja hormonikorvaushoitoa on annettava kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Hypofysiitti
Tislelitsumabia saaneilla potilailla on ilmoitettu hypofysiittia. Potilaita on seurattava hypofysiitin / aivolisäkkeen vajaatoiminnan oireiden ja löydösten varalta. Aivolisäkkeen toiminnan ja hormonipitoisuuksien seurantaa on harkittava. Kortikosteroideja ja hormonikorvaushoitoa on annettava kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
Tyypin 1 diabetes
Tislelitsumabia saaneilla potilailla on ilmoitettu tyypin 1 diabetesta, mukaan lukien diabeettista ketoasidoosia. Potilaita on seurattava hyperglykemian ja muiden diabeteksen oireiden ja löydösten varalta. Tyypin 1 diabetesta on hoidettava insuliinilla. Jos potilaalla on vaikea (asteen ≥ 3) hyperglykemia tai ketoasidoosi, tislelitsumabihoito on tauotettava ja potilaalle on annettava antihyperglykeemistä hoitoa (ks. kohta Annostus ja antotapa). Tislelitsumabihoitoa voidaan jatkaa, kun diabetes on saatu hyvään hoitotasapainoon.
Immuunivälitteinen nefriitti ja siihen liittyvä munuaisten toimintahäiriö
Tislelitsumabia saaneilla potilailla on ilmoitettu immuunivälitteistä nefriittiä ja siihen liittyvää munuaisten toimintahäiriötä. Potilaita on seurattava munuaistoiminnan muutosten (seerumin kreatiniinipitoisuuden suurenemisen) varalta, ja muut munuaisten toimintahäiriön syyt on suljettava pois.
Immuunivälitteisessä nefriitissä ja siihen liittyvässä munuaisten toimintahäiriön yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Muut immuunivälitteiset haittavaikutukset
Tislelitsumabia saaneilla on ilmoitettu myös seuraavia kliinisesti merkittäviä immuunivälitteisiä haittavaikutuksia: myosiitti, myokardiitti, artriitti, polymyalgia rheumatica, perikardiitti, ei-infektiivinen virtsarakkotulehdus, immuunitrombosytopenia, enkefaliitti, myasthenia gravis, Sjögrenin oireyhtymä ja Guillain–Barrén oireyhtymä (ks. kohta Haittavaikutukset).
Kyseisten muiden immuunivälitteisten haittavaikutusten yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Kiinteän elinsiirteen hyljintä
Kiinteiden elinsiirteiden hyljintää on ilmoitettu PD‑1:n estäjillä hoidetuilla potilailla markkinoilletulon jälkeen. Tislelitsumabihoito voi suurentaa kiinteän elinsiirteen hyljintäriskiä. Tislelitsumabihoidon hyöty verrattuna mahdollisen elinsiirteen hyljinnän riskiin on otettava huomioon kyseisillä potilailla.
Hemofagosyyttinen lymfohistiosytoosi
Hemofagosyyttistä lymfohistiosytoosia (Haemophagocytic lymphohistiocytosis, HLH) on ilmoitettu tislelitsumabia saaneilla potilailla (ks. kohta Haittavaikutukset). HLH on hengenvaarallinen oireyhtymä, jonka tunnuspiirteitä ovat kuume, ihottuma, lymfadenopatia, hepato- ja/tai splenomegalia sekä sytopeniat. Potilaita on seurattava HLH:n kliinisten oireiden ja löydösten varalta. Jos HLH:ia epäillään, tislelitsumabihoito on keskeytettävä HLH:n diagnostiikan tekemiseksi ja hoidon aloittamiseksi. Jos HLH vahvistuu, tislelitsumabihoidon anto on keskeytettävä.
Infuusioreaktiot
Tislelitsumabia saaneilla potilailla on ilmoitettu vaikeita infuusioreaktioita (≥ aste 3) (ks. kohta Haittavaikutukset). Anafylaksiatapauksia, mukaan lukien anafylaktinen reaktio ja anafylaktinen sokki, on raportoitu markkinoille tulon jälkeen. Potilaita on seurattava infuusioreaktioiden oireiden ja löydösten varalta.
Infuusioreaktioiden yhteydessä on tehtävä taulukossa 1 suositellut hoitomuutokset (ks. kohta Annostus ja antotapa).
Potilaat, jotka suljettiin pois kliinisistä tutkimuksista
Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli jokin seuraavista tiloista: ECOG‑toimintakykyluokka lähtötilanteessa ≥ 2; aktiiviset aivo‑ tai leptomeningeaaliset metastaasit; aktiivinen autoimmuunitauti tai taustalla autoimmuunitauti, joka voi uusiutua; mikä tahansa tila, joka oli vaatinut systeemistä hoitoa joko kortikosteroideilla (> 10 mg/vrk prednisonia tai vastaava hoito) tai muilla immunosuppressanteilla tutkimushoitoa edeltävien 14 vuorokauden aikana; aktiivinen tai hoitamaton HIV; hoitamaton hepatiitti B tai hepatiitti C; taustalla interstitiaalinen keuhkosairaus; elävän rokotteen anto tutkimushoitoa edeltävien 14 vuorokauden aikana; infektio, joka oli vaatinut systeemistä hoitoa tutkimushoitoa edeltävien 14 vuorokauden aikana; taustalla vaikea yliherkkyys jollekin muulle monoklonaaliselle vasta‑aineelle. Tietojen puutteen takia tislelitsumabia on käytettävä varoen kyseisissä populaatioissa mahdollisten hyötyjen ja riskien huolellisen yksilöllisen harkinnan jälkeen.
Potilaat, joilla on ruokavalion natriumrajoitus
Yksi ml tätä lääkevalmistetta sisältää 0,069 mmol (eli 1,6 mg) natriumia. Tämä lääkevalmiste sisältää 16 mg natriumia per 10 ml:n injektiopullo, joka vastaa 0,8 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Tevimbra laimennetaan natriumkloridi 9 mg/ml (0,9 %) infuusioliuokseen. Tämä on otettava huomioon niiden potilaiden kohdalla, joilla on ruokavalion natriumrajoitus (ks. kohta Käyttö- ja käsittelyohjeet).
Polysorbaatti 20 (E432)
Tämä lääkevalmiste sisältää 0,2 mg polysorbaatti 20:tä per ml konsentraattia, joka vastaa 4 mg:aa kahdessa 10 ml:n injektiopullossa, jotka on tarkoitettu yhteen Tevimbra-valmisteen infuusioon. Polysorbaatit saattavat aiheuttaa allergisia reaktioita. Potilaiden todetut allergiat on otettava huomioon.
Yhteisvaikutukset
Tislelitsumabi on humanisoitu monoklonaalinen vasta‑aine, joka poistuu verenkierrosta kataboloitumalla. Varsinaisia muodollisia farmakokineettisiä yhteisvaikutustutkimuksia ei ole tehty. Monoklonaaliset vasta‑aineet eivät metaboloidu sytokromi P450 ‑entsyymien (CYP‑entsyymien) eivätkä muiden lääkeainemetaboliaan osallistuvien entsyymien välityksellä. Muiden samanaikaisesti käytettävien lääkevalmisteiden inhiboiva tai indusoiva vaikutus näiden entsyymien toimintaan ei siis todennäköisesti vaikuta tislelitsumabin farmakokinetiikkaan.
Systeemisten kortikosteroidien ja muiden immunosuppressanttien käyttöä on vältettävä lähtötilanteessa ennen tislelitsumabihoidon aloittamista lukuun ottamatta systeemisten kortikosteroidien pieniä annoksia (10 mg/vrk prednisonia tai vastaava hoito), sillä ne saattavat häiritä tislelitsumabin farmakodynaamista aktiivisuutta ja tehoa. Systeemisiä kortikosteroideja ja muita immunosuppressantteja voidaan kuitenkin käyttää tislelitsumabihoidon aloittamisen jälkeen immuunivälitteisten haittavaikutusten hoitoon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kortikosteroideja voidaan käyttää myös esilääkityksenä, kun tislelitsumabia käytetään yhdessä solunsalpaajahoidon kanssa, antiemeettisenä estohoitona ja/tai lievittämään solunsalpaajahoitoon liittyviä haittavaikutuksia.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy
Tislelitsumabia ei pidä käyttää sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä tehokasta ehkäisyä, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa tislelitsumabilla. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä (menetelmät, joihin liittyvä raskausprosentti on alle 1 %) tislelitsumabihoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Tietoja tislelitsumabin käytöstä raskaana oleville naisille ei ole saatavilla. Vaikutusmekanismin perusteella tislelitsumabin anto raskaana olevalle naiselle voi aiheuttaa haittaa sikiölle.
Eläimillä ei ole tutkittu tislelitsumabin vaikutusta lisääntymiseen. Hiiren tiineysmalleissa on kuitenkin osoitettu, että PD‑1/PD‑L1‑signalointireitin estyminen häiritsee toleranssia sikiötä kohtaan, mikä lisää sikiökuolemia.
Ihmisen G4‑immunoglobuliinien (IgG4) tiedetään läpäisevän istukan, ja koska tislelitsumabi on IgG4‑variantti, tislelitsumabi voi kulkeutua äidistä kehittyvään sikiöön. Naisille on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Tislelitsumabia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa tislelitsumabilla.
Imetys
Ei tiedetä, erittyykö tislelitsumabi ihmisen rintamaitoon. Sen vaikutuksia rintaruokittuihin vastasyntyneisiin/imeväisiin ja maidontuotantoon ei myöskään tunneta.
Tevimbra‑valmisteen aiheuttamien, rintaruokittuihin vastasyntyneisiin/imeväisiin kohdistuvien vakavien haittavaikutusten riskin vuoksi naisia on ohjeistettava olemaan imettämättä Tevimbra‑hoidon aikana ja vähintään 4 kuukauden ajan viimeisen Tevimbra‑annoksen jälkeen.
Hedelmällisyys
Tislelitsumabin mahdollisista vaikutuksista hedelmällisyyteen ei ole saatavilla kliinisiä tietoja. Tislelitsumabilla ei ole tehty lisääntymis‑ eikä kehitystoksisuustutkimuksia. Kolme kuukautta kestäneessä, toistuvaisannoksilla tehdyssä toksisuustutkimuksessa ei havaittu merkittäviä vaikutuksia uros‑ eikä naaraspuolisten jaavanmakakien lisääntymiselimiin, kun tislelitsumabia annettiin 3, 10 tai 30 mg/kg 2 viikon välein 13 viikon ajan (7 antokertaa) (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tevimbra‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Joillain potilailla on ilmoitettu väsymystä tislelitsumabin annon jälkeen (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tislelitsumabimonoterapian turvallisuus perustuu yhdistettyihin tietoihin 1 952 potilaasta, joilla oli erityyppisiä kasvaimia ja jotka saivat 200 mg tislelitsumabia 3 viikon välein. Yleisimmät haittavaikutukset (≥ 20 %) olivat anemia (27,7 %), ASAT-arvon nousu (24,7 %), väsymys (24,6 %) ja ALAT-arvon nousu (22,0 %). Yleisimmät asteen 3/4 haittavaikutukset (≥ 2 %) olivat anemia (4,8 %), ASAT-arvon nousu (3,7 %), keuhkokuume (3,6 %), hyponatremia (2,9 %), veren bilirubiinipitoisuuden nousu (2,8 %), hypertensio (2,4 %) ja väsymys (2,1 %). Potilaista 1,0 %:lle kehittyi haittavaikutus, joka johti kuolemaan. Kuolemaan johtaneet haittavaikutukset olivat keuhkokuume (0,61 %), pneumoniitti (0,10 %), hepatiitti (0,10 %), trombosytopenia (0,05 %), hengenahdistus (0,05 %) ja ruokahalun heikentyminen (0,05 %). 1 952 potilaasta 40,7 % altistui tislelitsumabille yli 6 kuukauden ajan ja 24,7 % yli 12 kuukauden ajan.
Tislelitsumabin turvallisuus samanaikaisessa käytössä solunsalpaajahoidon kanssa perustuu tietoihin 1 950 potilaasta, joilla oli erilaisia kasvaintyyppejä ja jotka saivat 200 mg tislelitsumabia 3 viikon välein, paitsi tutkimuksessa BGB A317-315, jossa potilaat saivat myös tislelitsumabia 400 mg:n annoksella 6 viikon välein adjuvanttihoitona neoadjuvanttihoidon ja leikkauksen jälkeen. Yleisimmät haittavaikutukset (≥ 20 %) olivat neutropenia (71,6 %), anemia (67,2 %), trombosytopenia (48,7 %), pahoinvointi (43,3 %), väsymys (40,8 %), ruokahalun heikkeneminen (40,1 %), ALAT-arvon nousu (30,6 %), ASAT‑arvon nousu (30,3 %), ihottuma (21,4 %) ja ripuli (20,3 %). Yleisimmät asteen 3/4 haittavaikutukset (≥ 2 %) olivat neutropenia (45,2 %), anemia (14,5 %), trombosytopenia (14,1 %), hyponatremia (4,6 %), hypokalemia (4,5 %), väsymys (4,2 %), keuhkokuume (4,0 %), lymfopenia (3,1 %), ihottuma (2,9 %), ruokahalun heikkeneminen (2,6 %), ASAT-arvon nousu (2,2 %) ja ALAT-arvon nousu (2,1 %). Potilaista 1,3 %:lle kehittyi haittavaikutus, joka johti kuolemaan. Kuolemaan johtaneet haittavaikutukset olivat keuhkokuume (0,50 %), pneumoniitti (0,30 %), hengenahdistus (0,20 %), myokardiitti (0,20 %), hepatiitti (0,05 %), trombosytopenia (0,05 %), koliitti (0,05 %), hypokalemia (0,05 %) ja myosiitti (0,05 %). 1 950 potilaasta 56,5 % altistui tislelitsumabille vähintään 6 kuukauden ajan ja 31,9 % vähintään 12 kuukauden ajan.
Haittavaikutustaulukko
Taulukossa 2 esitetään haittavaikutukset, joita on ilmoitettu Tevimbra‑monoterapiaa saaneiden potilaiden (N = 1 952) ja yhdessä solunsalpaajahoidon kanssa (N = 1 950) yhdistetyssä tietoaineistossa. Haittavaikutukset esitetään MedDRA‑elinjärjestelmäluokittain. Haittavaikutukset on esitetty kussakin elinjärjestelmäluokassa haittavaikutuksen yleisyyden mukaan alenevassa järjestyksessä. Haittavaikutusten yleisyydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Kussakin yleisyysluokassa haittavaikutukset on esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2 Haittavaikutukset Tevimbra‑monoterapiassa (N = 1 952) ja Tevimbra‑valmisteen ja solunsalpaajahoidon yhdistelmää saaneilla (N = 1 950)
Tislelitsumabin monoterapia N = 1 952 | Tislelitsumabi + solunsalpaajahoito N = 1 950 | |
Haittavaikutukset | Yleisyys (kaikki asteet) | Yleisyys (kaikki asteet) |
Infektiot | ||
Keuhkokuume1 | Yleinen* | Hyvin yleinen* |
Veri ja imukudos | ||
Anemia2 | Hyvin yleinen | Hyvin yleinen |
Trombosytopenia3 | Hyvin yleinen* | Hyvin yleinen |
Neutropenia4 | Yleinen | Hyvin yleinen |
Lymfopenia5 | Yleinen | Hyvin yleinen |
Hemofagosyyttinen lymfohistiosytoosi | Tuntematon | Harvinainen |
Immuunijärjestelmä | ||
Sjögrenin oireyhtymä | # | Melko harvinainen |
Umpieritys | ||
Kilpirauhasen vajaatoiminta6 | Hyvin yleinen | Hyvin yleinen |
Kilpirauhasen liikatoiminta7 | Yleinen | Yleinen |
Tyreoidiitti8 | Yleinen | Melko harvinainen |
Lisämunuaisten vajaatoiminta9 | Melko harvinainen | Melko harvinainen |
Hypofysiitti10 | Melko harvinainen | Melko harvinainen |
Aineenvaihdunta ja ravitsemus | ||
Hyperglykemia11 | Yleinen | Hyvin yleinen |
Hyponatremia12 | Yleinen | Hyvin yleinen |
Hypokalemia13 | Yleinen | Hyvin yleinen* |
Diabetes14 | Melko harvinainen | Yleinen |
Hermosto | ||
Guillain–Barrén oireyhtymä | Harvinainen | Harvinainen |
Enkefaliitti15 | # | Harvinainen |
Myasthenia gravis | # | Harvinainen |
Silmät | ||
Uveiitti16 | Melko harvinainen | Melko harvinainen |
Sydän | ||
Myokardiitti17 | Melko harvinainen | Yleinen* |
Perikardiitti | Melko harvinainen | Harvinainen |
Verisuonisto | ||
Hypertensio18 | Yleinen | Yleinen |
Hengityselimet, rintakehä ja välikarsina | ||
Yskä | Hyvin yleinen | Hyvin yleinen |
Hengenahdistus | Yleinen* | Yleinen* |
Pneumoniitti19 | Yleinen* | Yleinen* |
Ruoansulatuselimistö | ||
Pahoinvointi | Hyvin yleinen | Hyvin yleinen |
Ripuli20 | Hyvin yleinen | Hyvin yleinen |
Stomatiitti21 | Yleinen | Yleinen |
Pankreatiitti22 | Melko harvinainen | Yleinen |
Koliitti23 | Melko harvinainen | Yleinen |
Keliakia | Harvinainen | # |
Maksa ja sappi | ||
Hepatiitti24 | Yleinen* | Yleinen* |
Iho ja ihonalainen kudos | ||
Ihottuma25 | Hyvin yleinen | Hyvin yleinen |
Kutina | Hyvin yleinen | Hyvin yleinen |
Vitiligo26 | Melko harvinainen | Melko harvinainen |
Erythema multiforme | Melko harvinainen | Harvinainen |
Stevens–Johnsonin oireyhtymä | Harvinainen | # |
Toksinen epidermaalinen nekrolyysi27 | Tuntematon* | Tuntematon* |
Luusto, lihakset ja sidekudos | ||
Nivelkipu | Yleinen | Hyvin yleinen |
Lihaskipu | Yleinen | Yleinen |
Myosiitti28 | Melko harvinainen | Melko harvinainen* |
Artriitti29 | Melko harvinainen | Yleinen |
Munuaiset ja virtsatiet | ||
Nefriitti30 | Melko harvinainen | Melko harvinainen |
Ei-infektiivinen virtsarakkotulehdus31 | Harvinainen | # |
Yleisoireet ja antopaikassa todettavat haitat | ||
Väsymys32 | Hyvin yleinen | Hyvin yleinen |
Kuume33 | Hyvin yleinen | Hyvin yleinen |
Ruokahalun heikentyminen | Hyvin yleinen* | Hyvin yleinen |
Tutkimukset | ||
ASAT‑arvon suureneminen | Hyvin yleinen | Hyvin yleinen |
ALAT‑arvon suureneminen | Hyvin yleinen | Hyvin yleinen |
Veren bilirubiinipitoisuuden suureneminen34 | Hyvin yleinen | Hyvin yleinen |
Veren AFOS‑pitoisuuden suureneminen | Yleinen | Yleinen |
Veren kreatiniiniarvon suureneminen | Yleinen | Hyvin yleinen |
Vammat, myrkytykset ja hoitokomplikaatiot | ||
Infuusioreaktio35 | Yleinen | Yleinen |
1 Keuhkokuume sisältää seuraavat haittavaikutustermit: keuhkokuume, alahengitystieinfektio, alahengitysteiden bakteeri‑infektio, bakteeriperäinen keuhkokuume, sieniperäinen keuhkokuume, ja pneumocystis jirovecii ‑keuhkokuume, brokopulmonaalinen aspergilloosi, hiivaperäinen keuhkokuume, mykoplasmaperäinen keuhkokuume, stafylokokkiperäinen keuhkokuume ja virusperäinen keuhkokuume. 2 Anemia sisältää seuraavat haittavaikutustermit: anemia ja hemoglobiiniarvon pieneneminen. 3 Trombosytopenia sisältää seuraavat haittavaikutustermit: trombosytopenia, trombosyyttiarvon pieneneminen ja immuunitrombosytopenia. 4 Neutropenia sisältää seuraavat haittavaikutustermit: neutropenia ja neutrofiiliarvon pieneneminen. 5 Lymfopenia sisältää seuraavat haittavaikutustermit: lymfopenia, lymfosyyttiarvon pieneneminen ja lymfosyyttien prosenttiosuuden pieneneminen. 6 Kilpirauhasen vajaatoiminta sisältää seuraavat haittavaikutustermit: kilpirauhasen vajaatoiminta, kilpirauhasen vasta-aineiden pitoisuuden suureneminen, immuunivälitteinen kilpirauhasen vajaatoiminta, kilpirauhashormonien pitoisuuksien pieneneminen, vapaan tyroksiinin pitoisuuden pieneneminen, vapaan trijodityroniinin pitoisuuden pieneneminen, trijodityroniinin pitoisuuden pieneneminen, primaarinen kilpirauhasen vajaatoiminta, sentraalinen kilpirauhasen vajaatoiminta ja tyroksiinipitoisuuden pieneneminen. 7 Kilpirauhasen liikatoiminta sisältää seuraavat haittavaikutustermit: veren TSH‑pitoisuuden pieneneminen, kilpirauhasen liikatoiminta, vapaan tyroksiinin pitoisuuden suureneminen, tyroksiinipitoisuuden suureneminen, vapaan trijodityroniinin pitoisuuden suureneminen ja trijodityroniinin pitoisuuden suureneminen. 8 Tyreoidiitti sisältää seuraavat haittavaikutustermit: tyreoidiitti, autoimmuunityreoidiitti, immuunivälitteinen tyreoidiitti, hiljainen tyreoidiitti ja subakuutti tyreoidiitti. 9 Lisämunuaisten vajaatoiminta sisältää seuraavat haittavaikutustermit: Addisonin tauti, lisämunuaisten vajaatoiminta, glukokortikoidivaje, immuunivälitteinen lisämunuaisen vajaatoiminta, primaarinen lisämunuaisten vajaatoiminta ja sekundaarinen lisämunuaiskuoren vajaatoiminta. 10 Hypofysiitti sisältää seuraavat haittavaikutustermit: hypofysiitti ja aivolisäkkeen vajaatoiminta. 11 Hyperglykemia sisältää seuraavat haittavaikutustermit: hyperglykemia ja veren glukoosipitoisuuden suureneminen. 12 Hyponatremia sisältää seuraavat haittavaikutustermit: hyponatremia ja veren natriumpitoisuuden pieneneminen. 13 Hypokalemia sisältää seuraavat haittavaikutustermit: hypokalemia ja veren kaliumpitoisuuden pieneneminen. 14 Diabetes sisältää seuraavat haittavaikutustermit: diabetes, diabeettinen ketoasidoosi, ketoasidoosi, tyypin 1 diabetes ja latentti autoimmuunidiabetes aikuisilla. 15 Enkefaliitti sisältää seuraavan haittavaikutustermin: immuunivälitteinen enkefaliitti. 16 Uveiitti sisältää seuraavat haittavaikutustermit: korioretiniitti, iridosykliitti, uveiitti ja iriitti. 17 Myokardiitti sisältää seuraavat haittavaikutustermit: myokardiitti, immuunivälitteinen myokardiitti ja autoimmuunimyokardiitti. 18 Hypertensio sisältää seuraavat haittavaikutustermit: hypertensio, verenpaineen kohoaminen ja essentiaalinen hypertensio. 19 Pneumoniitti sisältää seuraavat haittavaikutustermit: pneumoniitti, immuunivälitteinen keuhkosairaus, interstitiaalinen keuhkosairaus ja organisoituva keuhkokuume. 20 Ripuli sisältää seuraavat haittavaikutustermit: ripuli ja tihentynyt suolen toiminta. 21 Stomatiitti sisältää seuraavat haittavaikutustermit: stomatiitti, suun haavaumat, suun limakalvon eroosio ja aftahaavaumat. 22 Pankreatiitti sisältää seuraavat haittavaikutustermit: amylaasiarvon suureneminen, lipaasiarvon suureneminen, pankreatiitti ja akuutti pankreatiitti. 23 Koliitti sisältää seuraavat haittavaikutustermit: autoimmuunikoliitti, koliitti, haavainen koliitti ja immuunivälitteinen enterokoliitti. 24 Hepatiitti sisältää seuraavat haittavaikutustermit: hepatiitti, lääkkeen aiheuttama maksavaurio, maksatoksisuus, poikkeava maksatoiminta, immuunivälitteinen hepatiitti, maksavaurio ja autoimmuunihepatiitti. 25 Ihottuma sisältää seuraavat haittavaikutustermit: ihottuma, makulopapulaarinen ihottuma, ekseema, punoittava ihottuma, dermatiitti, akuutti kuumeinen neutrofiilinen dermatoosi, autoimmuunidermatiitti, allerginen dermatiitti, papulaarinen ihottuma, nokkosihottuma, punoitus, ihon kesiminen, lääkeaineihottuma, makulaarinen ihottuma, psoriaasi, pustulaarinen ihottuma, aknea muistuttava dermatiitti, kutiava ihottuma, likenoidi keratoosi, käsien dermatiitti, immuunivälitteinen dermatiitti, follikulaarinen ihottuma, kyhmyruusu ja pemfigoidi. 26 Vitiligo sisältää seuraavat haittavaikutustermit: ihon hypopigmentaatio, ihon depigmentaatio ja ja vitiligo. 27 Markkinoille tulon jälkeinen kokemus. 28 Myosiitti sisältää seuraavat haittavaikutustermit: myosiitti, rabdomyolyysi ja immuunivälitteinen myosiitti. 29 Artriitti sisältää seuraavat haittavaikutustermit: artriitti, polyartriitti ja immuunivälitteinen artriitti. 30 Nefriitti sisältää seuraavat haittavaikutustermit: nefriitti, fokaalinen segmentaalinen glomeruloskleroosi, membranoosinen glomerulusnefriitti, immuunivälitteinen munuaisten toimintahäiriö, tubulointerstitiaalinen nefriitti ja immuunivälitteinen nefriitti. 31 Ei-infektiivinen virtsarakkotulehdus sisältää seuraavat haittavaikutustermit: ei-infektiivinen virtsarakkotulehdus ja immuunivälitteinen virtsarakkotulehdus. Immuunivälitteisen virtsarakkotulehduksen tapauksia on raportoitu markkinoille tulon jälkeen. 32 Väsymys sisältää seuraavat haittavaikutustermit: väsymys, astenia, fyysisen toimintakyvyn heikkeneminen, huonovointisuus ja letargia. 33 Kuume sisältää seuraavat haittavaikutustermit: lämpötilan nousu ja kuume. 34 Veren bilirubiinipitoisuuden suureneminen sisältää seuraavat haittavaikutustermit: veren bilirubiinipitoisuuden suureneminen, konjugoituneen bilirubiinin pitoisuuden suureneminen, konjugoitumattoman bilirubiinin pitoisuuden suureneminen ja hyperbilirubinemia. 35 Infuusioreaktio sisältää seuraavat haittavaikutustermit: anafylaktinen reaktio, vilunväristykset, sarveiskalvon edeema, allerginen dermatiitti, lääkeihottuma, lääkeyliherkkyys, kasvojen edeema, ikenien turvotus, yliherkkyys, kurkun obstruktio, kurkun turvotus, huulten edeema, huulten turvotus, suun turvotus, allerginen kutina, ihottuma, erytematoottinen ihottuma, makulaarinen ihottuma, kutiseva ihottuma, allerginen nuha, kasvojen turvotus, kielen edeema, tyypin I yliherkkyys, urtikaria, infuusioreaktio ja infuusioon liittyvä yliherkkyysreaktio. * Sisältää kuolemaan johtaneet tapaukset # Ei raportoitu tässä yhdistetyssä tietojoukossa | ||
Valikoitujen haittavaikutusten kuvaus
Seuraavat tiedot kuvaavat tislelitsumabimonoterapian merkittäviä haittavaikutuksia kliinisissä tutkimuksissa. Tiedot tislelitsumabin merkittävistä haittavaikutuksista solunsalpaajia sisältävässä yhdistelmähoidossa esitetään, jos tislelitsumabimonoterapiaan verrattuna havaittiin kliinisesti merkittäviä eroja.
Immuunivälitteinen pneumoniitti
Immuunivälitteistä pneumoniittia esiintyi 5,1 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (1,3 %), asteen 2 (2,1 %), asteen 3 (1,3 %), asteen 4 (0,3 %) ja asteen 5 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 4,1 kk (vaihteluväli: 1,0 vrk – 55,0 kk) ja ilmaantumisesta korjautumiseen kuluneen ajan mediaani 2,8 kk (vaihteluväli: 7,0 vrk – 33,7 kk). Tislelitsumabihoito lopetettiin pysyvästi 1,8 %:lla potilaista ja tauotettiin 1,9 %:lla potilaista. Pneumoniitti korjautui 47,0 %:lla potilaista.
Tislelitsumabia monoterapiana saaneilla potilailla pneumoniittia esiintyi enemmän aiemmin rintakehän sädehoitoa saaneilla potilailla (8,4 %) verrattuna potilaisiin, jotka eivät olleet saaneet rintakehän sädehoitoa (3,6 %).
Pneumoniittia esiintyi 11,2 %:lla ei-pienisoluista keuhkosyöpää sairastavista potilaista, jotka saivat tislelitsumabihoitoa yhdistelmänä solunsalpaajahoidon kanssa. Ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka saivat tislelitsumabimonoterapiaa, pneumoniittia esiintyi 8,3 %:lla potilaista.
Immuunivälitteinen hepatiitti
Immuunivälitteistä hepatiittia esiintyi 1,2 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,1 %), asteen 2 (0,2 %), asteen 3 (0,6 %) ja asteen 4 (0,3 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 22,0 vrk (vaihteluväli: 1,0 vrk – 4,1 kk) ja ilmaantumisesta korjautumiseen kuluneen ajan mediaani 1,1 kk (vaihteluväli: 6,0 vrk – 6,6 kk). Immuunivälitteisen hepatiitin takia tislelitsumabihoito lopetettiin pysyvästi 0,3 %:lla potilaista ja tauotettiin 0,8 %:lla potilaista. Hepatiitti korjautui 60,9 %:lla potilaista.
Immuunivälitteiset ihoreaktiot
Immuunivälitteisiä ihoreaktioita esiintyi 12,6 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (7,7 %), asteen 2 (3,7 %), asteen 3 (1,0 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 1,5 kk (vaihteluväli: 1,0 vrk – 36,1 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 1,1 kk (vaihteluväli: 1,0 vrk – 36,7 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja tauotettiin 1,3 %:lla potilaista. Ihoreaktiot korjautuivat 72,0 %:lla potilaista.
SJS- ja TEN-tapauksia on raportoitu markkinoille tulon jälkeen. Jotkin näistä ovat johtaneet kuolemaan (katso kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Immuunivälitteinen koliitti
Immuunivälitteistä koliittia esiintyi 0,6 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 2 (0,4 %) ja asteen 3 (0,2 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 6,0 kk (vaihteluväli: 6,0 vrk – 26,5 kk) ja ilmaantumisesta korjautumiseen kuluneen ajan mediaani 28,0 vrk (vaihteluväli: 9,0 vrk – 26,7 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja tauotettiin 0,4 %:lla potilaista. Koliitti korjautui 81,8 %:lla potilaista.
Immuunivälitteinen myosiitti/rabdomyolyysi
Immuunivälitteistä myosiittia/rabdomyolyysia esiintyi 0,8 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,3 %), asteen 2 (0,3 %), asteen 3 (0,2 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 1,5 kk (vaihteluväli: 15,0 vrk – 39,3 kk) ja ilmaantumisesta korjautumiseen kuluneen ajan mediaani 1,2 kk (vaihteluväli: 5,0 vrk – 5,2 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,2 %:lla potilaista ja tauotettiin 0,5 %:lla potilaista. Myosiitti/rabdomyolyysi korjautui 75,0 %:lla potilaista.
Immuunivälitteiset endokrinopatiat
Kilpirauhasen häiriöt
Kilpirauhasen vajaatoiminta:
Kilpirauhasen vajaatoimintaa esiintyi 13,8 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (6,4 %), asteen 2 (7,3 %), asteen 3 (0,1 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 4,0 kk (vaihteluväli: 1,0 vrk – 29,9 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani 2,1 kk (vaihteluväli: 2,0 vrk – 27,0 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja hoito tauotettiin 0,6 %:lla potilaista. Kilpirauhasen vajaatoiminta korjautui 36,4 %:lla potilaista.
Kilpirauhasen liikatoiminta:
Kilpirauhasen liikatoimintaa esiintyi 5,1 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (4,4 %) ja asteen 2 (0,7 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 2,1 kk (vaihteluväli: 6,0 vrk – 39,4 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 1,4 kk (vaihteluväli: 8,0 vrk – 22,1 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja tauotettiin 0,3 %:lla potilaista. Kilpirauhasen liikatoiminta korjautui 77,0 %:lla potilaista.
Tyreoidiitti:
Tyreoidiittia esiintyi 1,1 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,5 %) ja asteen 2 (0,6 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 2,0 kk (vaihteluväli: 14,0 vrk – 20,7 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 2,0 kuukautta (vaihteluväli: 20,0 vrk – 15,3 kk). Tislelitsumabihoitoa ei lopetettu pysyvästi yhdelläkään potilaalla. Hoito tauotettiin 0,2 %:lla potilaista. Tyreoidiitti korjautui 38,1 %:lla potilaista.
Lisämunuaisten vajaatoiminta
Lisämunuaisten vajaatoimintaa esiintyi 0,5 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 2 (0,3 %), asteen 3 (0,2 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 10,3 kk (vaihteluväli: 1,4–16,9 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 1,9 kuukautta (vaihteluväli: 30,0 vrk – 13,6 kk). Tislelitsumabihoitoa ei lopetettu pysyvästi yhdelläkään potilaalla. Hoito tauotettiin 0,4 %:lla potilaista. Lisämunuaisten vajaatoiminta korjautui 30,0 %:lla potilaista.
Hypofysiitti
Hypofysiittia (aste 2) esiintyi 0,3 %:lla tislelitsumabimonoterapiaa saaneista potilaista.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 9,0 kk (vaihteluväli: 22,0 vrk–16,2 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 2,3 kuukautta (vain yksi korjautumisen tapahtuma). Tislelitsumabihoitoa ei lopetettu pysyvästi eikä tauotettu yhdelläkään potilaalla. Hypofysiitti korjautui 20,0 %:lla potilaista.
Tyypin 1 diabetes
Tyypin 1 diabetesta esiintyi 0,6 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,1 %), asteen 2 (0,3 %), asteen 3 (0,2 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 6,5 kk (vaihteluväli: 1,1–36,1 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 22,0 vuorokautta (vaihteluväli: 5,0 vrk – 3,6 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,2 %:lla potilaista ja tauotettiin 0,2 %:lla potilaista. Tyypin 1 diabetes korjautui 8,3 %:lla potilaista.
Immuunivälitteinen nefriitti ja munuaisten toimintahäiriö
Immuunivälitteistä nefriittiä ja munuaisten toimintahäiriötä esiintyi 0,2 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,1 %), asteen 2 (0,1 %) ja asteen 3 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 1,5 kk (vaihteluväli: 15,0 vrk – 12,1 kk). Ilmaantumisesta korjautumiseen kuluneen ajan mediaani oli 9,0 vuorokautta (sama kahdella korjautumisen tapahtumalla). Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja tauotettiin 0,1 %:lla potilaista. Immuunivälitteinen nefriitti ja munuaisten toimintahäiriö korjautuivat 50,0 %:lla potilaista.
Immuunivälitteinen myokardiitti
Immuunivälitteistä myokardiittia esiintyi 0,8 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 1 (0,4 %), asteen 2 (0,2 %), asteen 3 (0,2 %) ja asteen 4 (0,1 %) tapahtumat.
Ensimmäisestä annoksesta tapahtuman ilmaantumiseen kuluneen ajan mediaani oli 1,6 kk (vaihteluväli: 14,0 vrk – 33,6 kk) ja ilmaantumisesta korjautumiseen kuluneen ajan mediaani 1,2 kk (vaihteluväli: 4,0 vrk – 15,6 kk). Tislelitsumabihoito lopetettiin pysyvästi 0,4 %:lla potilaista ja tauotettiin 0,4 %:lla potilaista. Myokardiitti korjautui 60,0 %:lla potilaista.
Myokardiittia esiintyi 1,2 %:lla tislelitsumabihoitoa yhdistelmänä solunsalpaajahoidon kanssa saaneista potilaista, mukaan lukien asteen 5 (0,2 %) tapahtumat.
Immuunijärjestelmän tarkistuspisteen estäjien luokkavaikutukset
Muilla immuunijärjestelmän tarkistuspisteen estäjillä annetun hoidon aikana on havaittu seuraavia haittavaikutuksia, joita voi ilmaantua myös tislelitsumabihoidon aikana: haiman eksokriininen vajaatoiminta.
Infuusioreaktiot
Infuusioreaktioita esiintyi 3,0 %:lla tislelitsumabimonoterapiaa saaneista potilaista, mukaan lukien asteen 3 (0,1 %) tapahtumat. Tislelitsumabihoito lopetettiin pysyvästi 0,1 %:lla potilaista ja tauotettiin 0,1 %:lla potilaista.
Anafylaksiatapauksia, mukaan lukien anafylaktinen reaktio ja anafylaktinen sokki, on raportoitu markkinoille tulon jälkeen.
Laboratorioarvojen poikkeavuudet
Tislelitsumabimonoterapiaa saaneilla potilailla lähtötilanteen laboratorioarvojen muuttumista asteen 3 tai 4 poikkeamiksi esiintyi seuraavilla prosenttiosuuksilla: hemoglobiiniarvon suureneminen 0,1 %:lla, hemoglobiiniarvon pieneneminen 4,4 %:lla, leukosyyttiarvon pieneneminen 0,9 %:lla, lymfosyyttiarvon pieneneminen 8,9 %:lla, lymfosyyttiarvon suureneminen 0,2 %:lla, neutrofiiliarvon pieneneminen 2,1 %:lla, trombosyyttiarvon pieneneminen 1,3 %:lla, ALAT‑arvon suureneminen 2,6 %:lla, albumiiniarvon pieneneminen 0,3 %:lla, AFOS‑arvon suureneminen 2,7 %:lla, ASAT‑arvon suureneminen 4,8 %:lla, bilirubiiniarvon suureneminen 2,8 %:lla, kreatiinikinaasiarvon suureneminen 1,9 %:lla, kreatiniiniarvon suureneminen 1,2 %:lla, glukoosiarvon suureneminen 4,4 %:lla, glukoosiarvon pieneneminen 0,5 %:lla, kaliumarvon suureneminen 0,9 %:lla, kaliumarvon pieneneminen 2,9 %:lla, natriumarvon suureneminen 0,1 %:lla ja natriumarvon pieneneminen 6,5 %:lla.
Tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneilla potilailla lähtötilanteen laboratorioarvojen muuttumista asteen 3 tai 4 poikkeamiksi esiintyi seuraavilla prosenttiosuuksilla: hemoglobiiniarvon pieneneminen 14,2 %:lla, leukosyyttiarvon pieneneminen 23,3 %:lla, lymfosyyttiarvon pieneneminen 17,9 %:lla, lymfosyyttiarvon suureneminen 0,1 %:lla, neutrofiiliarvon pieneneminen 47,2 %:lla, trombosyyttiarvon pieneneminen 14,1 %:lla, ALAT‑arvon suureneminen 3,5 %:lla, albumiiniarvon pieneneminen 0,5 %:lla, AFOS‑arvon suureneminen 0,8 %:lla, ASAT‑arvon suureneminen 3,1 %:lla, bilirubiiniarvon suureneminen 2,0 %:lla, kreatiinikinaasiarvon suureneminen 2,3 %:lla, kreatiniiniarvon suureneminen 1,8 %:lla, glukoosiarvon pieneneminen 0,5 %:lla, glukoosiarvon suureneminen 1,2 %:lla, kaliumarvon suureneminen 1,3 %:lla, kaliumarvon pieneneminen 7,6 %:lla, natriumarvon suureneminen 0,3 %:lla ja natriumarvon pieneneminen 11,5 %:lla.
Immunogeenisuus
Lääkkeelle kehittyviä vasta‑aineita voitiin arvioida 3 614 potilaalla. 21,1 %:lla potilaista todettiin hoidon aikana lääkkeelle kehittyneitä vasta‑aineita ja 0,9 %:lla neutraloivia vasta‑aineita. Populaatiofarmakokinetiikan analryysi osoitti, että lääkkeelle kehittyneiden vasta‑aineiden status oli tilastollisesti merkitsevä kovariaatti puhdistuman suhteen; hoidon aikana tislelitsumabille kehittyneillä vasta‑aineilla ei kuitenkaan näytä olevan kliinisesti merkittävää vaikutusta farmakokinetiikkaan eikä tehoon.
200 mg:n annoksen 3 viikon välein monoterapiana tai yhdistelmähoitona (mukaan lukien adjuvanttihoito 400 mg 6 viikon välein leikkaushoitoon soveltuvan ei-pienisoluisen keuhkosyövän hoitoon) saaneilla potilailla, joilla lääkkeelle kehittyviä vasta‑aineita voitiin arvioida, havaittiin seuraavanlaisia haittatapahtumaprosentteja (vasta‑ainepositiivinen populaatio vs. vasta‑ainenegatiivinen populaatio): asteen ≥ 3 haittatapahtumat 52,5 % vs. 42,1 %, vakavat haittatapahtumat 39,0 % vs. 31,8 %, tislelitsumabihoidon lopettamiseen johtaneet haittatapahtumat 12,3 % vs. 11,4 % (monoterapia); vähintään asteen 3 haittatapahtumat 80,0 % ja 78,6 %, vakavat haittatapahtumat 43,3 % ja 41,0 %, tislelitsumabihoidon lopettamiseen johtaneet haittatapahtumat 13,6 % ja 13,5 % (yhdistelmähoito). Potilailla, joille kehittyi hoidon aikana vasta‑aineita, oli yleensä kokonaisuudessaan huonompi terveydentila ja huonommat tautiominaisuudet lähtötilanteessa, mikä voi sekoittaa turvallisuusanalyysin tulkintaa. Saatavilla olevat tiedot eivät mahdollista selvien johtopäätösten tekoa mahdollisista haittavaikutusmaisista kaavoista.
Iäkkäät
Yleisesti ottaen tislelitsumabimonoterapian ja solunsalpaajahoidon kanssa yhdessä käytön turvallisuudessa ei havaittu eroja < 65‑vuotiaiden ja 65–74‑vuotiaiden potilaiden välillä. 75 vuotta täyttäneistä potilaista on liian vähän tietoa johtopäätösten tekemiseksi.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tislelitsumabin yliannostuksesta ei ole tietoa. Yliannostustapauksissa potilaita on seurattava tarkoin haittavaikutusten oireiden ja löydösten varalta, ja asianmukainen oireenmukainen hoito on aloitettava välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset aineet, monoklonaaliset vasta‑aineet ja vasta‑ainekonjugoidut lääkkeet, ATC‑koodi: L01FF09
Vaikutusmekanismi
Tislelitsumabi on humanisoitu immunoglobuliini G4:n (IgG4) monoklonaalinen vasta‑ainevariantti PD‑1:tä vastaan. Se sitoutuu ihmisen PD‑1:n solunulkoiseen domeeniin ja estää kilpailevasti sekä PD‑L1:n että PD‑L2:n sitoutumista. Soluilla tehdyissä in vitro ‑määrityksissä tislelitsumabi estää PD‑1‑välitteistä negatiivista signalointia ja vahvistaa T‑solujen toiminnallista aktiivisuutta.
Kliininen teho ja turvallisuus
Tislelitsumabin tehon ja turvallisuuden altistus-vastesuhteiden mallinnuksen ja simulaation perusteella 3 viikon välein annetun 200 mg:n annoksen ja 6 viikon välein annetun 400 mg:n annoksen tehossa tai turvallisuudessa ei ole kliinisesti merkittäviä eroja.
Ei‑pienisoluinen keuhkosyöpä
Leikkaushoitoon soveltuvan ei-pienisoluisen keuhkosyövän neoadjuvantti- ja adjuvanttihoito: BGB-A317-315
BGB-A317-315 oli vaiheen 3 satunnaistettu, lumekontrolloitu, kaksoissokkoutettu tutkimus. Tutkimuksessa arvioitiin tislelitsumabin tehoa ja turvallisuutta, kun sitä annettiin yhdistelmänä kahden valmisteen platinapohjaisen solunsalpaajahoidon kanssa neoadjuvanttihoitona ja valmisteen käyttöä jatkettiin sen jälkeen adjuvanttihoidossa. Tätä verrattiin neoadjuvanttihoitoon lumelääkkeellä ja kahden valmisteen platinapohjaisella solunsalpaajahoidolla, jota seurasi lumelääkeadjuvanttihoito. Tutkittavilla potilailla oli leikkaushoitoon soveltuva levinneisyysasteen II tai IIIA ei-pienisoluinen keuhkosyöpä.
Tutkimukseen otettiin mukaan potilaita, joilla oli histologisesti vahvistettu levinneisyysasteen II tai IIIA (AJCC 8. painos) ei-pienisoluinen keuhkosyöpä, ECOG-toimintakykyluokka 0 tai 1, ei tunnettuja EGFR-mutaatioita eikä ALK-geenin translokaatioita sekä vahvistettu soveltuvuus R0-resektioon parantavassa tarkoituksessa. Levinneisyysasteen IIIB potilaita ei otettu mukaan tutkimukseen.
Seuraavilla valintakriteereillä määritettiin potilaat, joilla oli suuri uusiutumisen riski ja jotka otettiin mukaan terapeuttisen käyttöaiheen ryhmään ja vastaavat potilasjoukkoa, joilla on levinneisyysasteen II – IIIA tauti AJCC:n levinneisyysasteen luokitusjärjestelmän 8. painoksen mukaisesti:
- kasvaimen koko > 4 cm tai minkä tahansa kokoinen kasvain, johon liittyy N1- tai N2-status
- rintakehän rakenteisiin tunkeutuva kasvain (tunkeutuu suoraan viskeraaliseen keuhkopussiin, parietaaliseen keuhkopussiin, rintakehän seinämään, pääkeuhkoputkeen, palleahermoon, mediastinaaliseen keuhkopussiin tai parietaaliseen perikardiumiin)
- > 4 cm:n kokoinen kasvain, joka aiheuttaa absorptioatelektaasin, joka ulottuu hiluksen seutuun, vaikuttaa keuhkojen joihinkin osiin tai koko keuhkoihin tai vaikuttaa päärungon keuhkoputkiin riippumatta etäisyydestä harjuun, tai joka tunkeutuu viskeraaliseen keuhkopussiin (PL1 tai PL2) N0-statuksen osalta
- kasvain, jossa on erillisiä kyhmyjä samassa lohkossa kuin missä primaari keuhkosyöpä on.
Yhteensä 453 potilasta satunnaistettiin (suhteessa 1:1) saamaan hoitona
- tislelitsumabiryhmä: neoadjuvanttihoitona tislelitsumabia 200 mg päivänä 1 yhdessä joko sisplatiinin 75 mg/m2 tai karboplatiinin AUC 5 mg/ml/min ja pemetreksedin 500 mg/m2 tai paklitakselin 175 mg/m2 kanssa kunkin 21 vuorokauden pituisen hoitojakson päivänä 1, 3–4 hoitojakson ajan. Leikkauksen jälkeen annettiin adjuvanttihoitona tislelitsumabia 400 mg 6 viikon välein enintään 8 hoitojakson ajan.
- lumelääkeryhmä: neoadjuvanttihoitona lumelääkettä päivänä 1 yhdessä joko sisplatiinin 75 mg/m2 tai karboplatiinin AUC 5 mg/ml/min ja pemetreksedin 500 mg/m2 tai paklitakselin 175 mg/m2 kanssa kunkin 21 vuorokauden pituisen hoitojakson päivänä 1, 3–4 hoitojakson ajan. Leikkauksen jälkeen annettiin adjuvanttihoitona lumelääkettä 6 viikon välein enintään 8 hoitojakson ajan.
Potilaat, joilla oli ei-levyepiteeliperäinen histologia, saivat pemetreksediä, ja potilaat, joilla oli levyepiteeliperäinen histologia, saivat paklitakselia. Tutkijat tekivät valinnan sisplatiinin tai karboplatiinin välillä kaikkien potilaiden kohdalla. Jos se oli aiheellista, potilaat saivat leikkauksen jälkeen adjuvanttisädehoitoa ennen tislelitsumabi- tai lumelääkeadjuvanttihoitoa. Tislelitsumabi- ja solunsalpaajahoito jatkui hoidon päättymiseen, taudin etenemiseen, haittatapahtumaan, jota ei voida hyväksyä, kuolemaan tai potilaan ja/tai tutkijan tekemään tutkimushoidon lopetuspäätökseen saakka.
Kaksi ensisijaista päätetapahtumaa olivat tapahtumavapaa elossaolo (EFS) sokkoutetun riippumattoman keskitetyn arvioinnin (BICR) perusteella ja merkittävän patologisen vasteen (MPR) osuus sokkoutetun riippumattoman patologisen arvioinnin (BIPR) perusteella. Toissijaisia tehon päätetapahtumia olivat patologisen täydellisen vasteen (pCR) osuus BIPR-arvioinnin perusteella ja kokonaiselossaolo (OS).
Demografiset ominaisuudet ja lähtötilanteen ominaisuudet olivat yleisesti ottaen samanlaiset kahden hoitoryhmän välillä. Lähtötilanteen ominaisuudet kaikkien 453 satunnaistetun potilaan osalta olivat seuraavanlaiset: iän mediaani 62 vuotta (vaihteluväli 30–80 vuotta), 40 % potilaista oli ≥ 65-vuotiaita, 3,3 % potilaista oli ≥ 75-vuotiaita, 90,5 % potilaista oli miehiä, 100 % oli aasialaisia (kaikki otettiin tutkimukseen Kiinassa), 65,3 %:lla oli ECOG-toimintakykyluokka 0, 84,5 % tupakoi tällä hetkellä tai oli tupakoinut aiemmin, 78,1 %:lla oli diagnosoitu levyepiteeliperäinen histologia, 58,5 %:lla oli levinneisyysasteen IIIA tauti ja 57,8 %:lla oli ≥ 1%:n PD-L1-ekspressio.
Parantava leikkaus oli tehty 84,1 %:lle potilaista, jotka olivat tislelitsumabia ja platinaa sisältävän solunsalpaajahoidon ryhmässä, ja 76,2 %:lle potilaista, jotka olivat platinaa sisältävän solunsalpaajahoidon ryhmässä.
Tutkimus osoitti, että MPR, EFS, pCR ja OS olivat tilastollisesti merkitsevästi paremmat potilailla, jotka satunnaistettiin saamaan tislelitsumabia verrattuna lumelääkettä saaneisiin potilaisiin.
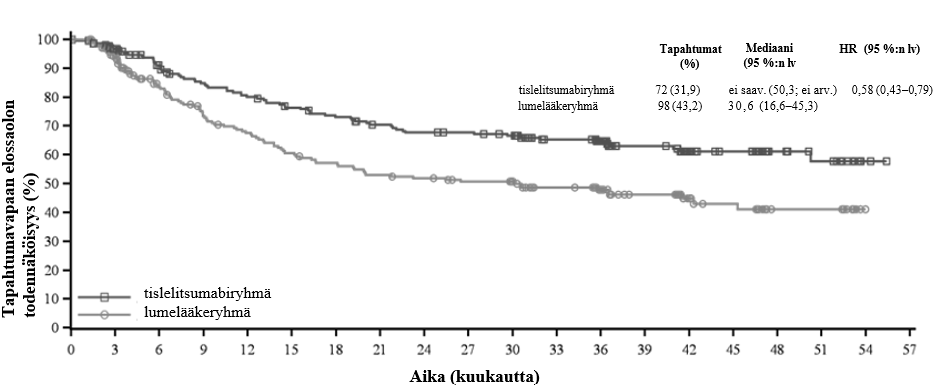
Ennalta määritetyssä tapahtumavapaan elossaolon (EFS) välianalyysissä (tiedonkeruun katkaisupäivä 21.8.2023) EFS:n riskitiheyksien suhde (HR) oli 0,56 (95 %:n lv: 0,40–0,79, yksisuuntainen p-arvo 0,0003) ja OS:n seurannan mediaaniajat käänteiseen Kaplan-Meierin menetelmään perustuen olivat 24,6 kuukautta tislelitsumabiryhmässä ja 22,7 kuukautta lumelääkeryhmässä.
Taulukossa 3, kuvassa 1 ja kuvassa 2 on esitetty yhteenveto tehoa koskevista tuloksista.
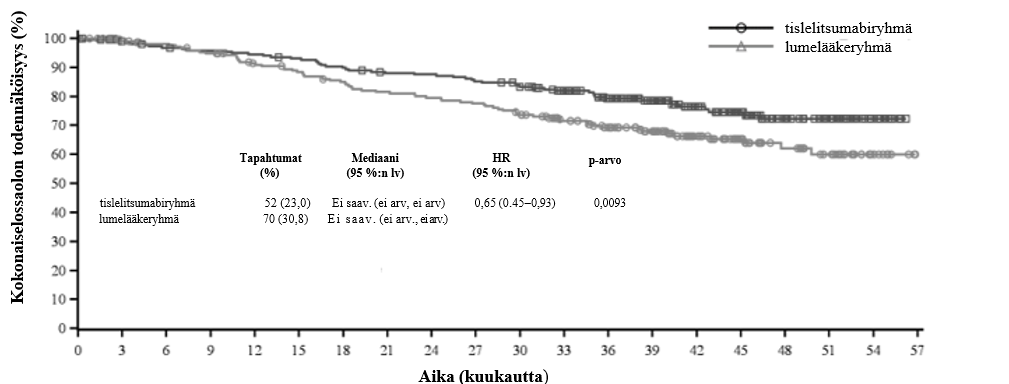
Ennalta määritetyssä loppuanalyysissä (tiedonkeruun katkaisupäivä 7.3.2025) elossaolon seurannan mediaaniajat käänteiseen Kaplan-Meierin menetelmään perustuen olivat 43,3 kuukautta (95 %:n lv: 41,2–44,6) tislelitsumabiryhmässä ja 41,6 kuukautta (95 %:n lv: 39,9–43,8) lumelääkeryhmässä.
Taulukko 3 Tehotulokset tutkimuksessa BGB-A317-3151
Tislelitsumabiryhmä (N=226) | Lumelääkeryhmä (N=227) | |
| Tapahtumavapaa elossaolo | ||
| Tapahtumia, n (%) | 72 (31,9) | 98 (43,2) |
| Mediaani (kuukautta) (95 %:n lv) | ei saav. (50,3; ei arv.) | 30,6 (16,6–45,3) |
| Riskitiheyksien suhde (95 %:n lv)a | 0,58 (0,43–0,79) | |
| Merkittävä patologinen vaste | ||
| n (%) | 127 (56,2) | 34 (15) |
| 95 %:n lvc | (49,5–62,8) | (10,6–20,3) |
| Ero, % (95 %:n lv)d | 41,1 (33,2–49,1) | |
| p-arvoe | < 0,0001 | |
| Kokonaiselossaolo | ||
| Kuolemia, n (%) | 52 (23,0) | 70 (30,8) |
| Mediaani (kuukautta) (95 %:n lv) | ei saav. (ei arv., ei arv.) | ei saav. (ei arv., ei arv.) |
| Riskitiheyksien suhde (95 %:n lv)a | 0,65 (0,45–0,93) | |
| p-arvob | 0,0093 | |
HR= riskitiheyksien suhde; lv = luottamusväli; ei arv. = ei arvioitavissa; ei saav. = ei saavutettu Potilaiden, joille ei ollut tehty leikkaus tai joilla ei ollut patologisia tuloksia, katsottiin jääneen ilman vastetta. 1 Ennalta määritetty merkittävän patologisen vasteen analyysi perustui tietoihin, joiden keruun katkaisupäivä oli 20.2.2023, ja ennalta määritetty tapahtumavapaan elossaolon ja kokonaiselossaolon analyysi perustui tietoihin, joiden keruun katkaisupäivä oli 7.3.2025. a Riskitiheyksien suhde ja 95 %:n luottamusvälit arvioitiin ositetun Coxin regressiomallin perusteella, joka oli ositettu histologian, taudin vaiheen ja PD-L1-ekspression osalta (IRT-järjestelmästä [Interactive response technology]). b P-arvo laskettiin log-rank-testillä, joka oli ositettu histologian, taudin vaiheen ja PD-L1-ekspression osalta IRT:stä. c 95 %:n luottamusväli arvioitiin Clopper-Pearsonin-menetelmällä. d Mantel-Haenszelin yleinen riskiero arvioitiin 95 %:n luottamusväleineen, jotka oli konstruoitu normaaliapproksimaatiolla, ja Saton varianssiestimaattorilla, joka oli ositettu histologian, taudin vaiheen ja PD-L1-ekspression osalta IRT:stä. e P-arvo saatiin Cochran-Mantel-Haenszelin menetelmällä, joka oli ositettu histologian, taudin vaiheen ja PD-L1-ekspression osalta IRT:stä. | ||
Kuva 1 Tapahtumavapaan elossaolon Kaplan-Meier-käyrä tutkimuksessa BGB-A317-315
| Riskille alttiina | ||||||||||||||||||||
| tislelitsumabiryhmä | 226 | 196 | 176 | 161 | 152 | 143 | 136 | 128 | 123 | 121 | 117 | 101 | 92 | 69 | 49 | 39 | 21 | 17 | 2 | 0 |
| lumelääkeryhmä | 227 | 187 | 149 | 128 | 117 | 105 | 98 | 91 | 88 | 83 | 79 | 69 | 59 | 47 | 29 | 22 | 11 | 11 | 0 | 0 |
Kuva 2 Kokonaiselossaolon Kaplan-Meier-käyrä tutkimuksessa BGB-A317-315
| Riskille alttiina | ||||||||||||||||||||
| tislelitsumabiryhmä | 226 | 218 | 212 | 209 | 206 | 202 | 195 | 189 | 188 | 183 | 176 | 163 | 143 | 121 | 91 | 69 | 47 | 36 | 15 | 0 |
| lumelääkeryhmä | 227 | 214 | 207 | 199 | 186 | 180 | 172 | 165 | 161 | 157 | 148 | 131 | 117 | 98 | 73 | 51 | 34 | 26 | 9 | 0 |
Alaryhmäanalyysi tehtiin tutkimuksessa BGB-A317-315 potilailla, joiden PD‑L1-ekspressio oli ≥ 1 % (tislelitsumabiryhmä [n=130; 58 %] vs. lumelääkeryhmä [n=132; 58 %]) ja joiden PD‑L1-ekspressio oli < 1 % (josta on poissuljettu ei arvioitavissa olevat/määrittämättömät arvot) (tislelitsumabiryhmä [n=89; 39 %] vs. lumelääkeryhmä [n=84; 37 %]). Tapahtumavapaan elossaolon riskitiheyksien suhde oli 0,53 (95 %:n lv: 0,35–0,79) potilailla, joiden PD‑L1-ekspressio oli ≥ 1 % ja 0,70 (95 %:n lv: 0,43–1,14) potilailla, joiden PD‑L1-ekspressio oli < 1 %. Kokonaiselossaolon riskitiheyksien suhde oli 0,61 (95 %:n lv: 0,38–0,98) potilailla, joiden PD‑L1-ekspressio oli ≥ 1 % ja 0,91 (95 %:n lv: 0,50–1,64) potilailla, joiden PD‑L1-ekspressio oli < 1 %.
Ei‑levyepiteeliperäisen ei‑pienisoluisen keuhkosyövän ensilinjan hoito: BGB‑A317‑304
BGB‑A317‑304 oli satunnaistettu, avoin vaiheen III monikeskustutkimus, jossa arvioitiin tislelitsumabin tehoa ja turvallisuutta yhdessä pemetreksedin ja platinan kanssa käytettynä verrattuna pelkkään platinan ja pemetreksedin yhdistelmään ensilinjan hoidossa aiempaa solunsalpaajahoitoa saamattomilla potilailla, joilla oli paikallisesti edennyt ei‑levyepiteeliperäinen ei‑pienisoluinen keuhkosyöpä ja joille leikkaus tai platinapohjainen kemosädehoito ei soveltunut tai joilla oli metastasoitunut ei‑levyepiteeliperäinen ei‑pienisoluinen keuhkosyöpä.
Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivisia aivojen tai leptomeningeaalisia metastaaseja, saatavissa olevalle kohdennetulle estäjähoidolle herkkä, tiedossa oleva EGFR‑mutaatio tai ALK‑translokaatio, aktiivinen autoimmuunitauti tai mikä tahansa tila, joka vaati systeemistä hoitoa joko kortikosteroideilla (> 10 mg/vrk prednisonia tai vastaava hoito) tai muilla immunosuppressanteilla.
Yhteensä 334 potilasta satunnaistettiin (2:1) saamaan tislelitsumabia (200 mg) yhdessä pemetreksedin (500 mg/m2) ja karboplatiinin (AUC 5 mg/ml/min) tai sisplatiinin (75 mg/m2) kanssa (T + PP ‑ryhmä, N = 223) tai pemetreksediä (500 mg/m2) ja karboplatiinia (AUC 5 mg/ml/min) tai sisplatiinia (75 mg/m2) (PP‑ryhmä, N = 111). Platinavalmisteen (sisplatiini tai karboplatiini) valinnasta päätti tutkija.
Hoitoa annettiin 3 viikon hoitojaksoina. Kun solunsalpaajahoitoa tai tislelitsumabin ja solunsalpaajahoidon yhdistelmää oli annettu tutkijan harkinnan mukaan 4, 5 tai 6 hoitojakson ajan, T + PP ‑ryhmän potilaille annettiin 200 mg tislelitsumabia ja 500 mg/m2 pemetreksediä 3 viikon hoitojaksoina, kunnes tauti eteni tai ilmeni liiallista toksisuutta. PP‑ryhmän potilaille annettiin vastaavasti pelkästään 500 mg/m2 pemetreksediä, kunnes tauti eteni tai ilmeni liiallista toksisuutta. Potilaille, joilla riippumaton arviointitoimikunta vahvisti taudin etenemisen, annettiin mahdollisuus siirtyä saamaan tislelitsumabimonoterapiaa 3 viikon hoitojaksoina.
Satunnaistamisen osittamistekijöitä olivat kasvainsolujen PD‑L1:n ilmentäminen (< 1 % vs. 1–49 % vs. ≥ 50 %) ja taudin aste (IIIB vs. IV) AJCC:n (American Joint Committee on Cancer) kriteerien mukaisesti luokiteltuna (Cancer Staging Manual, 7. laitos). PD‑L1:n ilmentäminen arvioitiin keskuslaboratoriossa Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin PD‑L1:tä ilmentävät kasvainsolut. Kasvaimet arvioitiin 6 viikon välein ensimmäisten 6 kuukauden ajan, 9 viikon välein seuraavien 6 kuukauden ajan ja tämän jälkeen 12 viikon välein.
Tutkimuksen BGB-A317-304 potilaiden lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 61 vuotta (vaihteluväli: 25–75), 29 % oli 65 vuotta täyttäneitä tai vanhempia, 74 % oli miehiä, 100 % oli aasialaisia (kaikki Kiinasta), 23,4 %:lla ECOG‑toimintakykyluokka oli 0 ja 76,6 %:lla 1, 18,3 %:lla taudin aste oli IIIB, 26,6 %:lla ALK‑uudelleenjärjestymän status ei ollut tiedossa ja 73,4 %:lla status oli negatiivinen, 36,2 % ei ollut koskaan tupakoinut ja 5,4 %:lla oli aivometastaaseja. Ikää, sukupuolta, ECOG‑toimintakykyluokkaa, taudin astetta, tupakointistatusta, kasvainsolujen PD‑L1:n ilmentämisosuutta ja aiempia syöpähoitoja koskevat ominaisuudet olivat ryhmissä samankaltaiset.
Ensisijainen tehon päätetapahtuma oli etenemättömyysaika (PFS), jonka arvioi riippumaton arviointitoimikunta RECIST v1.1 ‑kriteerien mukaan hoitoaikeen mukaisessa (ITT) analyysissä. Toissijaisia tehon päätetapahtumia olivat kokonaiselossaolo (OS), objektiivinen vasteprosentti (ORR) ja vasteen kesto (DoR) riippumattoman arviointitoimikunnan ja tutkijan arvioimina.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin välianalyysissä (tiedonkeruun katkaisupäivä 23.1.2020). Analyysi osoitti, että PFS parani T + PP ‑ryhmässä tilastollisesti merkitsevästi verrattuna PP‑ryhmään. Stratifioitu riskitiheyssuhde oli 0,65 (95 %:n lv: 0,47–0,91; p = 0,0054). PFS:n mediaani oli T + PP ‑ryhmässä 9,7 kk ja PP‑ryhmässä 7,6 kk. Käänteisellä Kaplan-Meierin menetelmällä lasketut kokonaiselossaolon seuranta-ajan mediaanit olivat 9,9 kuukautta T + PP -ryhmässä ja 9,7 kuukautta PP-ryhmässä.
Tehotulokset lopullisesta analyysistä (tiedonkeruun katkaisupäivä 26.10.2020) olivat yhdenmukaisia välianalyysin tulosten kanssa. Loppuanalyysin kohdalla käänteisellä Kaplan-Meierin menetelmällä lasketut kokonaiselossaolon seuranta-ajan mediaanit olivat 18,4 kuukautta T + PP -ryhmässä ja 18,0 kuukautta PP-ryhmässä.
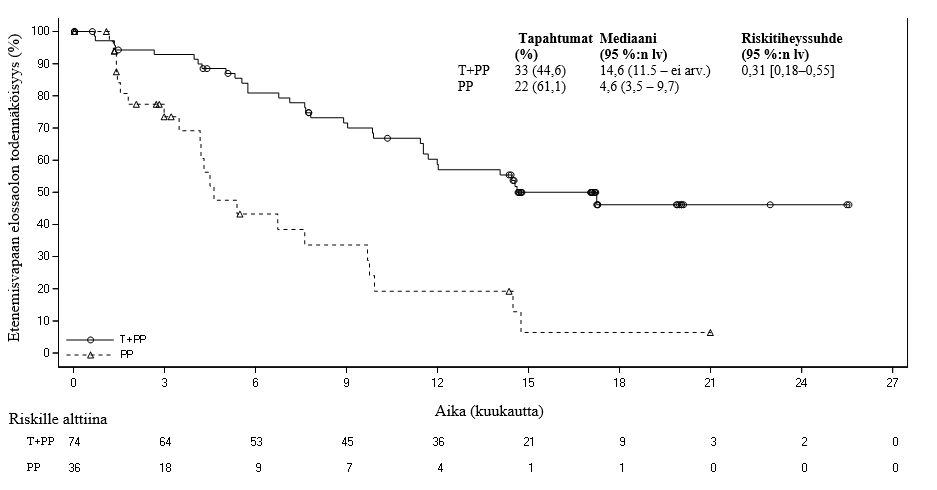
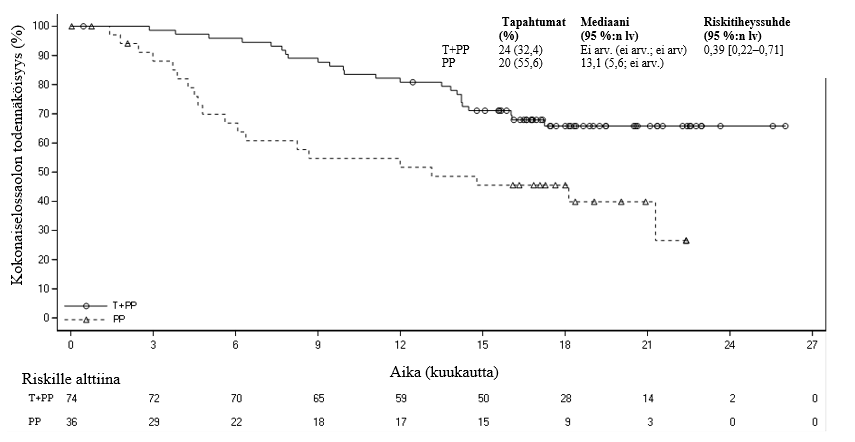
334 tutkimuksen BGB-A317-304 potilaasta 110:llä (33 %) potilaalla kasvainsolujen PD-L1:n ilmentäminen oli vähintään 50 %. Näistä potilaista 74 potilasta oli tislelitsumabi ja solunsalpaajahoito -ryhmässä ja 36 potilasta lumelääke ja solunsalpaajahoito -ryhmässä. Lopullisen analyysin tehotulokset potilailla, joilla kasvainsolujen PD-L1:n ilmentäminen oli vähintään 50 %, esitetään taulukossa 4. Kaplan-Meierin käyrä PFS:n ja OS:n osalta esitetään kuvissa 3 ja 4.
Taulukko 4 BGB‑A317‑304‑tutkimuksen tehotulokset potilailla, joilla kasvainsolujen PD-L1:n ilmentäminen oli vähintään 50 %
| Päätetapahtuma | Tislelitsumabi + pemetreksedi + platina (n = 74) | Pemetreksedi + platina (n = 36) |
| PFS | ||
| Tapahtumia, n (%) | 33 (44,6) | 22 (61,1) |
| PFS, mediaani (kk) (95 %:n lv) | 14,6 (11,5; ei arv.) | 4,6 (3,5–9,7) |
| Stratifioitu riskitiheyssuhdea, b (95 %:n lv) | 0,31 (0,18–0,55) | |
| OS | ||
| Kuolemat, n (%) | 24 (32,4) | 20 (55,6) |
| OS, mediaani (kk) (95 %:n lv) | Ei arv. (ei arv., ei arv.) | 13,1 (5,6; ei arv.) |
| Stratifioitu riskitiheyssuhdea (95 %:n lv) | 0,39 (0,22–0,71) | |
| Paras kokonaisvaste, n (%)b | ||
| ORRb, n (%) | 52 (70,3) | 11 (30,6) |
| 95 %:n lvc | (58,5–80,3) | (16,3–48,1) |
| DoRb | ||
| DoR, mediaani (kk) (95 %:n lv) | Ei arv. (13,2; ei arv.) | 8,5 (3,3; ei arv.) |
PFS = etenemättömyysaika; lv = luottamusväli; OS = kokonaiselossaolo; ORR = objektiivinen vasteprosentti; DoR = vasteen kesto; ei arv. = ei arvioitavissa. Mediaanit laskettiin Kaplan-Meierin menetelmällä ja 95 %:n luottamusväleillä, jotka laskettiin Brookmeyer-Crowleyn menetelmällä. a Riskitiheyssuhde laskettiin käyttämällä stratifioitua Coxin mallia ja pemetreksedi + platina ‑ryhmää viiteryhmänä. Ositustekijä: taudin aste (IIIB vs. IV). b Etenemättömyysaika perustui riippumattoman arviointitoimikunnan arviointiin, ja objektiivinen vasteprosentti ja vasteen kesto perustuivat riippumattoman arviointitoimikunnan vahvistamaan vasteeseen. c 95 %:n lv laskettiin Clopper-Pearsonin menetelmällä. | ||
Kuva 3 Etenemättömyysajan Kaplan‑Meier-käyrä BGB‑A317‑304-tutkimuksesta potilailla, joilla PD-L1 ≥ 50 %
Kuva 4Kokonaiselossaolon Kaplan-Meier-käyrä BGB-A317-304-tutkimuksessa potilailla, joilla PD-L1 ≥ 50 %
Levyepiteeliperäisen ei‑pienisoluisen keuhkosyövän ensilinjan hoito: BGB‑A317‑307
BGB‑A317‑307 oli satunnaistettu, avoin vaiheen III monikeskustutkimus, jossa tislelitsumabin ja paklitakselin + karboplatiinin tai nab‑paklitakselin + karboplatiinin yhdistelmän tehoa ja turvallisuutta verrattiin pelkän paklitakselin ja karboplatiinin yhdistelmän tehoon ja turvallisuuteen ensilinjan hoidossa aiempaa solunsalpaajahoitoa saamattomilla potilailla, joilla oli paikallisesti edennyt levyepiteeliperäinen ei‑pienisoluinen keuhkosyöpä ja joille leikkaus tai platinapohjainen kemosädehoito ei soveltunut tai joilla oli metastasoitunut levyepiteeliperäinen ei‑pienisoluinen keuhkosyöpä.
Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivisia aivojen tai leptomeningeaalisia metastaaseja, saatavissa olevalle kohdennetulle estäjähoidolle herkkä, tiedossa oleva EGFR‑mutaatio tai ALK‑translokaatio, aktiivinen autoimmuunitauti tai mikä tahansa tila, joka vaati systeemistä hoitoa joko kortikosteroideilla (> 10 mg/vrk prednisonia tai vastaava hoito) tai muilla immunosuppressanteilla.
Yhteensä 360 potilasta satunnaistettiin (1:1:1) saamaan tislelitsumabia (200 mg) yhdessä paklitakselin (175 mg/m2) ja karboplatiinin (AUC 5 mg/ml/min) kanssa (T + PC ‑ryhmä, N = 120), tislelitsumabia (200 mg) yhdessä nab‑paklitakselin (100 mg/m2) ja karboplatiinin (AUC 5 mg/ml/min) kanssa (T + nPC ‑ryhmä, N = 119) tai paklitakselia (175 mg/m2) ja karboplatiinia (AUC 5 mg/ml/min) (PC‑ryhmä, N = 121).
Hoitoa annettiin 3 viikon hoitojaksoina, kunnes potilas oli saanut 4–6 hoitojaksoa solunsalpaajahoitoa tai tislelitsumabin ja solunsalpaajahoidon yhdistelmää tutkijan harkinnan mukaan. T + nPC‑ ja T + PC ‑ryhmien potilaille annettiin tislelitsumabia, kunnes tauti eteni tai ilmeni liiallista toksisuutta. PC‑ryhmän potilaille annettiin taudin edettyä mahdollisuus siirtyä saamaan tislelitsumabimonoterapiaa 3 viikon hoitojaksoina.
Satunnaistamisen osittamistekijöitä olivat kasvainsolujen PD‑L1:n ilmentäminen (< 1 % vs. 1–49 % vs. ≥ 50 %) ja kasvaimen aste (IIIB vs. IV AJCC:n kriteerien mukaisesti luokiteltuna (Cancer Staging Manual, 7. laitos). PD‑L1:n ilmentäminen arvioitiin keskuslaboratoriossa Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin PD‑L1:tä ilmentävät kasvainsolut. Kasvaimet arvioitiin 6 viikon välein ensimmäisten 6 kuukauden ajan, 9 viikon välein seuraavien 6 kuukauden ajan ja tämän jälkeen 12 viikon välein taudin etenemiseen asti.
Tutkimuspopulaation lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 62,0 vuotta (vaihteluväli: 34–74), 35,3 % oli 65 vuotta täyttäneitä tai vanhempia, 91,7 % oli miehiä, 100 % oli aasialaisia (kaikki Kiinasta), 23,6 %:lla ECOG‑toimintakykyluokka oli 0 ja 76,4 %:lla 1, 33,9 %:lla taudin aste oli lähtötilanteessa IIIB ja 66,1 %:lla IV, 16,4 % ei ollut koskaan tupakoinut, 38,3 %:lla kasvainsolujen PD‑L1:n ilmentäminen oli < 1 %, 25,3 %:lla 1–49 % ja 34,7 %:lla ≥ 50 %. Ikää, sukupuolta, ECOG‑toimintakykyluokkaa, taudin astetta, tupakointistatusta, PD‑L1:n ilmentämisosuutta ja aiempia syöpähoitoja koskevat ominaisuudet olivat ryhmissä samankaltaiset.
Ensisijainen tehon päätetapahtuma oli etenemättömyysaika (PFS), jonka arvioi riippumaton arviointitoimikunta RECIST v1.1 ‑kriteerien mukaan hoitoaikeen mukaisessa (ITT) analyysissä. Päätetapahtumaa oli tarkoitus arvioida sekventiaalisesti T + PC‑ryhmässä verrattuna PC‑ryhmään ja T + nPC ‑ryhmässä verrattuna PC‑ryhmään. Toissijaisia tehon päätetapahtumia olivat kokonaiselossaolo (OS), objektiivinen vasteprosentti (ORR) ja vasteen kesto (DoR) riippumattoman arviointitoimikunnan ja tutkijan arvioimina.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin välianalyysissä (tiedonkeruun katkaisupäivä 6.12.2019). Analyysi osoitti, että PFS parani tilastollisesti merkitsevästi T + PC ‑ryhmässä (tislelitsumabi yhdessä paklitakselin ja karboplatiinin kanssa) ja T + nPC ‑ryhmässä (tislelitsumabi yhdessä nab‑paklitakselin ja karboplatiinin kanssa) verrattuna PC‑ryhmään (pelkästään paklitakseli ja karboplatiini). Stratifioitu HR T+PC -ryhmä vs. PC-ryhmä -vertailussa oli 0,48 (95 %:n lv: 0,34–0,69; p < 0,0001). Stratifioitu HR T+nPC -ryhmä vs. PC-ryhmä -vertailussa oli 0,45 (95 %:n lv: 0,32–0,64; p < 0,0001). Etenemisvapaan elossaolon mediaani oli T + PC ‑ryhmässä 7,6 kk, T + nPC ‑ryhmässä 7,6 kk ja PC‑ryhmässä 5,4 kk. Käänteisellä Kaplan-Meierin menetelmällä lasketut kokonaiselossaolon seuranta-ajan mediaanit olivat 8,8 kuukautta T + PC -ryhmässä, 8,8 kuukautta T + nPC -ryhmässä ja 8 kuukautta PC-ryhmässä.
Lopullisessa analyysissä (tiedonkeruun katkaisupäivä 30.9.2020) tulokset olivat yhtenevät välianalyysin tuloksiin nähden. Loppuanalyysin kohdalla käänteisellä Kaplan-Meierin menetelmällä lasketut kokonaiselossaolon seuranta-ajan mediaanit olivat 18,8 kuukautta T + PC -ryhmässä, 18,9 kuukautta T + nPC -ryhmässä ja 18,1 kuukautta PC-ryhmässä.
Lopullisen analyysin tehotulokset esitetään taulukossa 5, kuvassa 5 ja kuvassa 6.
Taulukko 5 BGB‑A317‑307‑tutkimuksen tehotulokset
| Päätetapahtuma | Tislelitsumabi + paklitakseli + karboplatiini (N = 120) | Tislelitsumabi + nab‑paklitakseli + karboplatiini (N = 119) | Paklitakseli + karboplatiini (N = 121) |
| PFS | |||
| Tapahtumia, n (%) | 80 (66,7) | 79 (66,4) | 86 (71,1) |
| PFS, mediaani (kk) (95 %:n lv) | 7,7 (6,7–10,4) | 9,6 (7,4–10,8) | 5,5 (4,2–5,6) |
| Stratifioitu riskitiheyssuhdea (95 %:n lv) | 0,45 (0,33–0,62) | 0,43 (0,31–0,60) | ‑ |
| OS | |||
| Kuolemat, n (%) | 48 (40,0) | 47 (39,5) | 52 (43,0) |
| OS, mediaani (kk) (95 %:n lv) | 22,8 (19,1; ei arv.) | ei arv. (18,6; ei arv.) | 20,2 (16,0, ei arv.) |
| Strafitioitu riskitiheyssuhde (95 %:n lv) | 0,68 (0,45–1,01) | 0,752 (0,50–1,12) | ‑ |
| ORRb | |||
| ORR, n (%) | 74 (61,7) | 74 (62,2) | 45 (37,2) |
| 95 %:n lv | (52,4–70,4) | (52,8–70,9) | (28,6–46,4) |
| DoRb | |||
| DoR, mediaani (kk) (95 %:n lv) | 13,2 (7,85–18,79) | 10,4 (8,34–17,15) | 4,8 (4,04–5,72) |
PFS = etenemättömyysaika; lv = luottamusväli; OS = kokonaiselossaolo; ORR = objektiivinen vasteprosentti; DoR = vasteen kesto; ei arv. = ei arvioitavissa. a Osittamistekijät: taudin aste (IIIB vs. IV) ja PD‑L1:n ilmentäminen kasvainsoluissa (≥ 50 % vs. 1–49 % vs. < 1 %). b Etenemättömyysaika perustui riippumattoman arviointitoimikunnan arviointiin ja objektiivinen vasteprosentti /vasteen kesto perustui riippumattoman arviointitoimikunnan vahvistamaan vasteeseen. | |||
Kuva 5 Etenemättömyysajan Kaplan-Meier‑käyrä BGB‑A317‑307‑tutkimuksessa riippumattoman arviointitoimikunnan arvioimana
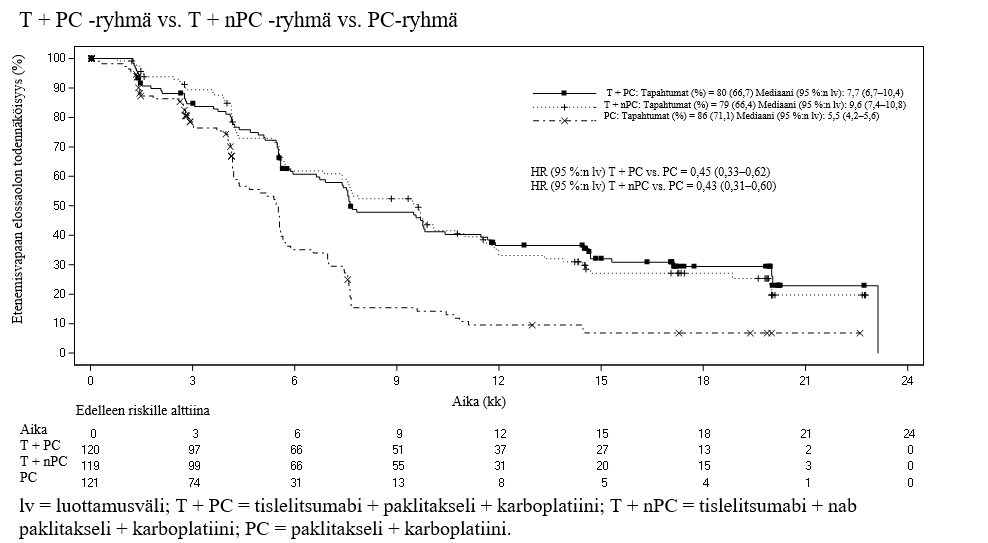
Kuva 6 Kokonaiselossaolon Kaplan-Meier-käyrä BGB-A317-307-tutkimuksessa
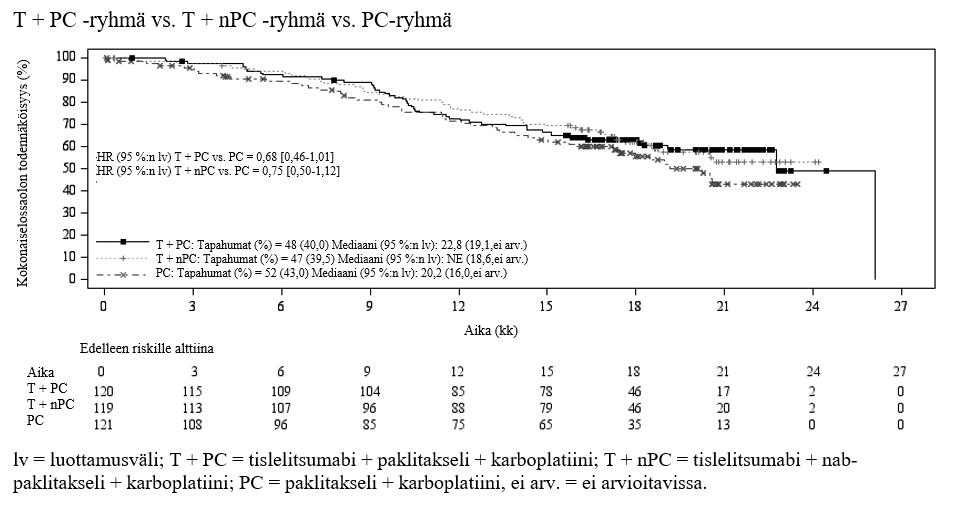
Alaryhmäanalyyseissä todettiin, että hoitovaikutus PFS‑aikaan oli yleisesti ottaen johdonmukainen erilaisissa merkittävimmissä demografisissa ja prognostisissa alaryhmissä, mukaan lukien PD‑L1:n ilmentäminen < 1 %, 1–49 % ja ≥ 50 % ja taudin asteet IIIB ja IV:
- T + PC ‑ryhmässä etenemättömyysajan HR oli 0,57 (95 %:n lv = 0,34–0,94) PD-L1:n < 1 %:n ilmentämisen osalta, 0,40 (95 %:n lv = 0,21–0,76) 1–49 %:n ilmentämisen osalta ja 0,44 (95 %:n lv = 0,26–0,75) ≥ 50 %:n ilmentämisen osalta.
- T + nPC ‑ryhmässä etenemättömyysajan HR oli 0,65 (95 %:n lv = 0,40–1,06) PD-L1:n < 1 %:n ilmentymisen osalta, 0,40 (95 %:n lv = 0,22–0,74) 1–49 %:n ilmentymisen osalta ja 0,33 (95 %:n lv = 0,18–0,59) ≥ 50 %:n ilmentymisen osalta.
Aiemmin hoidettu ei‑pienisoluinen keuhkosyöpä: BGB‑A317‑303
BGB‑A317‑303 oli satunnaistettu, avoin vaiheen III monikeskustutkimus, jossa arvioitiin tislelitsumabin tehoa ja turvallisuutta verrattuna dosetakseliin potilailla, joilla oli paikallisesti edennyt tai metastasoitunut ei‑pienisoluinen keuhkosyöpä (levyepiteeliperäinen tai ei‑levyepiteeliperäinen) ja joilla tauti oli edennyt aiemman platinapohjaisen hoito‑ohjelman aikana tai jälkeen.
Tutkimuksesta suljettiin pois potilaat, joilla oli tiedossa oleva EGFR‑mutaatio tai ALK‑uudelleenjärjestymä, aiempi hoito PD‑(L)1:n tai CTLA‑4:n estäjällä, aktiivinen autoimmuunitauti tai mikä tahansa tila, joka vaati systeemistä hoitoa joko kortikosteroideilla (> 10 mg/vrk prednisonia tai vastaava hoito) tai muilla immunosuppressiivisilla lääkkeillä.
Yhteensä 805 potilasta satunnaistettiin (suhteessa 2:1) saamaan 200 mg tislelitsumabia laskimoon 3 viikon välein (N = 535) tai 75 mg/m2 dosetakselia laskimoon 3 viikon välein (N = 270). Satunnaistamisen osittamistekijöitä olivat histologia (levyepiteeliperäinen vs. ei‑levyepiteeliperäinen), hoitolinja (toinen vs. kolmas linja) ja kasvainsolujen PD‑L1:n ilmentäminen (≥ 25 % vs. < 25 %). Dosetakselin ja tislelitsumabin antoa jatkettiin, kunnes tauti eteni (tutkijan arvio RECIST v1.1 ‑kriteerien mukaisesti) tai ilmeni liiallista toksisuutta. PD‑L1:n ilmentäminen arvioitiin keskuslaboratoriossa Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin PD‑L1:tä ilmentävät kasvainsolut. Kasvaimet arvioitiin 9 viikon välein 52 viikon ajan satunnaistamisen jälkeen ja tämän jälkeen 12 viikon välein. Elossaolostatusta seurattiin 3 kuukauden välein tutkimushoidon päättymisen jälkeen.
Tutkimuspopulaation lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 61 vuotta (vaihteluväli: 28–88), 32,4 % oli 65 vuotta täyttäneitä tai vanhempia, 3,2 % oli 75 vuotta täyttäneitä tai vanhempia, 77,3 % oli miehiä, 17,0 % oli valkoihoisia ja 79,9 % aasialaisia, 20,6 %:lla ECOG‑toimintakykyluokka oli 0 ja 79,4 %:lla 1, 85,5 %:lla oli metastasoitunut tauti, 30,3 % ei ollut koskaan tupakoinut, 46,0 %:lla tauti oli histologialtaan levyepiteeliperäinen ja 54,0 %:lla ei‑levyepiteeliperäinen, 65,8 %:lla oli villityypin EGFR‑status ja 34 %:lla tuntematon EGFR‑status, 46,1 %:lla oli villityypin ALK‑status ja 53,9 %:lla tuntematon ALK‑status, 7,1 %:lla oli aiemmin hoidettuja aivometastaaseja.
Potilaista 57,0 %:lla kasvainsolujen PD‑L1-ilmeneminen oli < 25 % ja 42,5 %:lla ≥ 25 %. Kaikki potilaat olivat saaneet aiemmin kahden platinavalmisteen yhdistelmähoitoa: 84,7 % potilaista oli saanut yhtä aiempaa hoitoa ja 15,3 % kahta aiempaa hoitoa.
Duaalisia ensisijaisia tehon päätetapahtumia olivat OS ITT‑analyysipopulaatiossa ja populaatiossa, jossa kasvainsolujen PD‑L1-ilmenemisosuus oli ≥ 25 %. Muita tehon päätetapahtumia olivat tutkijan arvioima PFS, ORR ja DoR.
BGB‑A317‑303‑tutkimuksessa saavutettiin molemmat duaaliset ensisijaiset päätetapahtumat eli OS ITT‑analyysipopulaatiossa ja PD‑L1 ≥ 25 % ‑analyysipopulaatiossa. Ennalta määritellyssä välianalyysissä (tiedonkeruun katkaisupäivä 10.8.2020) ITT‑populaatiossa havaittiin tilastollisesti merkitsevä OS:n piteneminen. Tulokset olivat parempia tislelitsumabiryhmässä (HR = 0,64; 95 %:n lv: 0,53–0,78; p < 0,0001). OS‑mediaani oli 17,2 kk tislelitsumabiryhmässä ja 11,9 kk dosetakseliryhmässä. Käänteisellä Kaplan-Meierin menetelmällä lasketut seuranta-ajan mediaanit olivat 19,5 kuukautta tislelitsumabiryhmässä ja 17,0 kuukautta dosetakseliryhmässä. Loppuanalyysin kohdalla (tiedonkeruun katkaisupäivä 15.7.2021) PD-L1 ≥ 25 % -analyysijoukossa havaittiin tilastollisesti merkitsevä OS:n piteneminen tislelitsumabiryhmän hyväksi (stratifioitu HR = 0,53; 95 %:n lv: 0,41–0,70; p < 0,0001). OS:n mediaani oli 19,3 kk tislelitsumabiryhmässä ja 11,5 kk dosetakseliryhmässä. Käänteisellä Kaplan-Meierin menetelmällä lasketut seuranta-ajan mediaanit loppuanalyysin kohdalla olivat 31,1 kuukautta tislelitsumabiryhmässä ja 27,9 kuukautta dosetakseliryhmässä.
Loppuanalyysi (tiedonkeruun katkaisupäivä 15.7.2021) osoitti, että ITT‑populaation tehotulokset olivat yhdenmukaiset välianalyysin tulosten kanssa.
Taulukossa 6 ja kuvassa 7 esitetään yhteenveto BGB‑A317‑303‑tutkimuksen lopullisen analyysin tehotuloksista (ITT‑analyysipopulaatio).
Taulukko 6 BGB‑A317‑303‑tutkimuksen tehotulokset
| Päätetapahtuma | Tislelitsumabi (N = 535) | Dosetakseli (N = 270) |
| OS | ||
| Kuolemat, n (%) | 365 (68,2) | 206 (76,3) |
| OS, mediaani (kk) (95 %:n lv) | 16,9 (15,24–19,09) | 11,9 (9,63–13,54) |
| Riskitiheyssuhde (95 %:n lv)a,b | 0,66 (0,56–0,79) | |
| PFS | ||
| Tapahtumia, n (%) | 451 (84,3) | 208 (77,0) |
| PFS, mediaani (kk) (95 %:n lv) | 4,2 (3,88–5,52) | 2,6 (2,17–3,78) |
| Riskitiheyssuhdea (95 %:n lv) | 0,63 (0,53–0,75) | |
| ORR (%) (95 %:n lv)c | 20,9 (17,56–24,63) | 3,7 (1,79–6,71) |
| DoRc | ||
| DoR, mediaani (kk) (95 %:n lv) | 14,7 (10,55–21,78) | 6,2 (4,11–8,31) |
OS = kokonaiselossaolo; lv = luottamusväli; PFS = etenemättömyysaika; ORR = objektiivinen vasteprosentti; DoR = vasteen kesto. Mediaanit laskettiin Kaplan-Meierin menetelmällä ja 95 %:n luottamusväleillä, jotka laskettiin Brookmeyer-Crowleyn menetelmällä. a Riskitiheyssuhde laskettiin käyttämällä stratifioitua Coxin mallia ja dosetakseliryhmää viiteryhmänä. b Osittamistekijöitä olivat histologia (levyepiteeliperäinen vs. ei‑levyepiteeliperäinen), hoitolinja (toinen vs. kolmas) ja kasvainsolujen PD‑L1:n ilmentäminen (PD‑L1‑osuus ≥ 25 % vs. < 25 %). c Tutkijan vahvistama. | ||
Kuva 7 Kokonaiselossaolon Kaplan-Meier‑käyrä BGB‑A317‑303‑tutkimuksessa (ITT‑analyysipopulaatio)
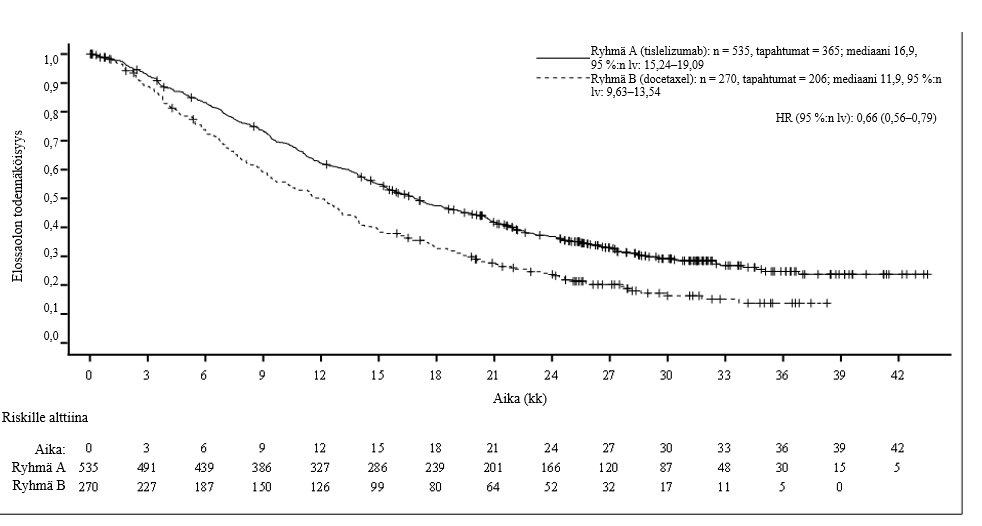
Ennalta määritellyt alaryhmäanalyysit osoittivat tislelitsumabin kannalta suotuisan, johdonmukaisesti hyödyllisen hoitovaikutuksen OS‑aikaan kaikissa merkittävimmissä demografisissa ja prognostisissa alaryhmissä.
Taulukossa 7 esitetään yhteenveto ennalta määriteltyjen alaryhmäanalyysien OS‑aikaa koskevista tehotuloksista kasvaimen PD‑L1:n ilmentämisen (< 25 % kasvainsoluista, ≥ 25 % kasvainsoluista) mukaan luokiteltuna.
Taulukko 7Kokonaiselossaoloaikaa koskevat tehotulokset kasvaimen PD‑L1:n ilmentämisen mukaan luokiteltuina (< 25 % kasvainsoluista, ≥ 25 % kasvainsoluista) BGB-A317-303-tutkimuksessa
| Tislelitsumabiryhmä | Dosetakseliryhmä | |
| N = 535 | N = 270 | |
| PD‑L1:n ilmentäminen kasvainsoluissa < 25 %, n | 307 | 152 |
| Tapahtumat, n (%) | 223 (72,6) | 117 (77,0) |
| OS:n mediaani (kuukautta) (95 % lv) | 15,2 (13,4–17,6) | 12,3 (9,3–14,3) |
| Riskitiheyksien suhde a (95 %:n lv) | 0,79 (0,64–0,99) | |
| PD‑L1:n ilmentäminen kasvainsoluissa ≥ 25 %, n | 227 | 115 |
| Tapahtumat, n (%) | 141 (62,1) | 86 (74,8) |
| OS:n mediaani (kuukautta) (95 %:n lv) | 19.3 (16,5–22,6) | 11,5 (8,2–13,5) |
| Riskitiheyksien suhde a (95 %:n lv) | 0,54 (0,41–0,71) | |
| a Riskitiheyksien suhde ja sen 95 %:n lv arvioitiin osittamattomasta Coxin mallista. | ||
Pienisoluinen keuhkosyöpä
Levinneen pienisoluisen keuhkosyövän ensilinjan hoito: BGB-A317-312
BGBA-317-312 on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa verrattiin tislelitsumabin tehoa ja turvallisuutta yhdessä sisplatiinin tai karboplatiinin sekä etoposidin kanssa lumelääkkeen ja sisplatiinin tai karboplatiinin sekä etoposidin yhdistelmän tehoon ensilinjan hoitona. Potilailla oli levinnyt pienisoluinen keuhkosyöpä (ES-SCLC).
Tutkimuksessa oli mukana potilaita, joilla oli histologisesti tai sytologisesti vahvistettu ES-SCLC:n diagnoosi, jotka eivät olleet aiemmin mitään systeemistä ES-SCLC:n hoitoa, ja joiden ECOG-toimintakykyluokka oli joko 0 tai 1.
Yhteensä 457 potilasta satunnaistettiin (suhteessa 1:1) saamaan:
- tislelitsumabi + solunsalpaajahoito -ryhmä: tislelitsumabia 200 mg ja karboplatiinia AUC 5 mg/ml/min tai sisplatiinia 75 mg/m2 päivänä 1 ja etoposidia 100 mg/m2 laskimoon kunkin 21 vuorokauden pituisen hoitojakson päivinä 1, 2 ja 3, enintään neljän hoitojakson ajan.
- lumelääke + solunsalpaajahoito -ryhmä: lumelääkettä ja karboplatiinia AUC 5 mg/ml/min tai sisplatiinia 75 mg/m2 päivänä 1 ja etoposidia 100 mg/m2 laskimoon kunkin 21 vuorokauden pituisen hoitojakson päivinä 1, 2 ja 3, enintään neljän hoitojakson ajan.
Platinalääkeaineen (sisplatiini tai karboplatiini) valinta perustui tutkijan harkintaan.
Tislelitsumabi 200 mg monoterapiaa tai lumelääkettä annettiin kolmen viikon välein taudin etenemiseen, kliinisen hyödyn häviämiseen tai kohtuuttomaan toksisuuteen saakka.
Satunnaistaminen ositettiin ECOG-toimintakykyluokan (0 tai 1), tutkijan valitseman solunsalpaajahoidon (karboplatiini tai sisplatiini) ja aivometastaasien (kyllä tai ei) osalta.
Ensisijaisena tehon päätetapahtumana oli kokonaiselossaolon hoitoaikeen mukaisessa analyysijoukossa. Toissijaisia tehon päätetapahtumia olivat tutkijan arvioima etenemisvapaa elossaolon (PFS), objektiivinen vasteen määrä (ORR) ja vasteen kesto (DoR) RECIST v1.1 -kriteerien mukaisesti.
Demografiset ominaisuudet ja lähtötilanteen ominaisuudet olivat yleisesti ottaen samanlaisia kahden hoitoryhmän välillä. Kaikkien 457 satunnaistettujen potilaiden lähtötilanteen ominaisuudet olivat seuraavat: mediaani-ikä 62 vuotta (vaihteluväli 31–78 vuotta), 37,2 % oli vähintään 65-vuotiaita; 81,4 % oli miehiä, 100 % oli aasialaisia (kaikki otettu mukaan tutkimukseen Kiinassa), 84,9 %lla oli ECOG-toimintakykyluokka 1; 1,1 %:lla oli sairaushistoriassa aivometastaaseja, 79 % sai karboplatiinia tutkijan valitsemana, 62,6 % tupakoi parhaillaan ja 89,3 %:lla oli vaiheen IV tauti AJCC 7th Edition -kriteerien mukaisesti.
Ennalta määritellyn loppuanalyysin (tietojen katkaisupäivä 19.4.2023) kohdalla BGB-A317-312-tutkimuksessa havaittiin tilastollisesti merkitsevä kokonaiselossaolon parantuminen tislelitsumabia ja solunsalpaajahoitoa saamaan satunnaistetuilla potilailla lumelääkettä ja solunsalpaajahoitoa saamaan satunnaistettujen ryhmään verrattuna. Ositettu riskitiheyksien suhde oli 0,75 (95 %:n lv: 0,61; 0,93); yksipuolinen p-arvo 0,004). Kokonaiselossaolon mediaani oli 15,5 kuukautta tislelitsumabia ja solunsalpaajahoitoa saaneiden ryhmässä ja 13,5 kuukautta lumelääkettä ja solunsalpaajahoitoa saaneiden ryhmässä.
Kuvaileva päivitetty analyysi (tietojen katkaisupäivä 29.12.2023) osoitti loppuanalyysin kanssa yhdenmukaisesti tehotulokset. Kokonaiselossaolon seurannan mediaaniaika käänteisen Kaplan-Meierin menetelmän perusteella oli 39,8 kuukautta (95 %:n lv: 36,2–41,4 kuukautta) tislelitsumabia ja solunsalpaajahoitoa saaneiden ryhmässä ja 36,4 kuukautta (95 %:n lv: 35,0–40,9 kuukautta) lumelääkettä ja solunsalpaajahoitoa saaneiden ryhmässä.
Päivitetyn analyysin tehotulokset on esitetty taulukossa 8 ja kuvassa 8. Tiedot potilaista, joilla oli aivometastaaseja, ovat liian vähäisiä johtopäätösten tekemiseksi kyseisessä potilasjoukossa.
Taulukko 8 Tehotulokset tutkimuksessa BGB-A317-312 – päivitetty analyysi
| Tislelitsumabi + solunsalpaajahoito (N = 227) | Lumelääke + solunsalpaajahoito (N = 230) | |
| Kokonaiselossolo | ||
| Kuolemat, n (%) | 175 (77,1) | 195 (84,8) |
| Mediaani (kuukautta) (95 %:n lv)a | 15,5 (13,5; 17,1) | 13,5 (12,1; 14,9) |
| Ositettu riskitiheyksien suhde (95 %:n lv)b | 0,78 (0,63; 0,95) | |
| Etenemisvapaa elossaolo | ||
| Tapahtumat, n (%) | 178 (78,4) | 207 (90,0) |
| Mediaani (kuukautta) (95 %:n lv)a | 4,7 (4,3; 5,5) | 4,3 (4,2; 4,4) |
| Ositettu riskitiheyksien suhde (95 %:n lv)b | 0,65 (0,53; 0,80) | |
| Vasteen kokonaismääräc (%) (95 %:n lv)d | 68,3 (61,8; 74,3) | 61,7 (55,1; 68,0) |
| Vasteen mediaanikesto (kuukautta)c (95% CI)a | 4,3 (4,1; 5,6) | 3,7 (3,0; 4,1) |
a Mediaani arvioitiin Kaplan-Meierin- menetelmällä 95 %:n luottamusväleineen käyttäen Brookmeyerin ja Crowleyn menetelmää log-log-muunnoksilla. b Riskitiheyksien suhde ja 95 %:n luottamusväli arvioitiin Coxin regressiomallilla, joka oli ositettu ECOG-toimintakykyluokan (1 vs 0) ja platinan (karboplatiini vs. sisplatiini) osalta käyttäen lumelääke + solunsalpaajahoito -yhdistelmää saaneita vertailuryhmänä. c Objektiiviset vasteet vahvistettiin RECIST v1.1 -kriteerien mukaisesti. d 95 %:n luottamusväli arvioitiin Clopper-Pearson-menetelmällä. | ||
Kuva 8 Kokonaiselossaolon Kaplan-Meier-käyrä tutkimuksessa BGB-A317-312
Mahalaukun tai gastroesofageaalisen junktion adenokarsinooma
Mahalaukun tai gastroesofageaalisen junktion adenokarsinooman ensilinjan hoito: BGB-A317-305
BGBA-317-305 on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa verrattiin tislelitsumabin tehoa yhdessä platina- ja fluoropyrimidiinipohjaisen solunsalpaajahoidon kanssa lumelääkkeen tehoon yhdessä platina- ja fluoropyrimidiinipohjaisen solunsalpaajahoidon kanssa ensilinjan hoitona. Potilailla oli paikallisesti edennyt, leikattavaksi soveltumaton tai metastasoitunut mahalaukun tai gastroesofageaalisen junktion adenokarsinooma.
Tutkimukseen otettiin mukaan vain potilaita, joilla oli histologisesti vahvistettu adenokarsinooma ja joita ei ollut hoidettu aiemmin systeemisellä hoidolla edenneen taudin takia. Potilaat olivat voineet saada aiemmin neoadjuvantti- tai adjuvanttihoitoa, mikäli se oli suoritettu loppuun eikä tauti ollut uusiutunut tai edennyt vähintään 6 kuukauteen.
Potilaat otettiin tutkimukseen kasvaimen PD‑L1‑ilmentymisen tasosta riippumatta. PD‑L1‑ilmentyminen arvioitiin keskuslaboratoriossa TAP (kasvainalueen positiivisuus) -pistemäärää käyttäen, jonka määritelmänä on kasvainalueen kasvainsolujen (kasvain ja mahdollinen desmoplastinen strooma) kattama kokonaisprosentti patologien visuaalisesti arvioimalla Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin minkä tahansa voimakkuuden PD‑L1-ilmentymä sekä kasvainsoluissa että kasvaimen sisältämissä immuunisoluissa.
Potilaat, joilla oli tyvisolusyöpä tai erilaistumaton tai muun histologisen tyypin mahalaukun tai gastroesofageaalisen junktion syöpä, sekä potilaat, joilla oli todettu HER-2-positiivisia kasvaimia, suljettiin pois tutkimuksesta.
Satunnaistaminen stratifioitiin maantieteellisen alueen mukaan (Kiina [mukaan lukien Taiwan] vs. Japani ja Etelä-Korea vs. muu mailma [ROW, mukaan lukien Yhdysvallat ja Eurooppa]), PD-L1-ilmentymisen (PD-L1:n TAP-pistemäärä vähintään 5 % vs. PD-L1:n TAP-pistemäärä alle 5 %), peritoneaalisen metastaasin esiintymisen mukaan (kyllä vs. ei) ja tutkijan valitseman solunsalpaajahoidon mukaan (ICC, oksaliplatiini ja kapesitabiini tai sisplatiini ja 5-FU).
Potilaat satunnaistettiin (1:1) saamaan joko 200 mg tislelitsumabia tai lumelääkettä 3 viikon välein yhdessä platina- ja fluoropyrimidiinipohjaisen solunsalpaajahoidon kanssa 21 vuorokauden hoitojaksona. Potilaat saivat tislelitsumabihoitoa (tai lumelääkettä), kunnes tauti eteni tai ilmeni liiallista toksisuutta. 24 kuukauden hoidon jälkeen tutkimushoitoa voitiin jatkaa pidempään kuin näiden kahden vuoden ajan, jos tutkija katsoi tämän olevan potilaan edun mukaista kliinisen hyöty-riski-arvion perusteella.
Solunsalpaajahoito koostui seuraavista:
- oksaliplatiinia 130 mg/m² laskimoon päivänä 1 ja kapesitabiinia 1 000 mg/ m² suun kautta kahdesti vuorokaudessa 14 peräkkäisen vuorokauden ajan, toistettuna 3 viikon välein. Oksaliplatiinia annettiin enintään 6 hoitojakson ajan ja kapesitabiinia annettiin ylläpitohoitona tutkijan harkinnan mukaan taudin etenemiseen tai kohtuuttomaan toksisuuteen asti.
tai
- sisplatiinia 80 mg/m² laskimoon päivänä 1 ja 5-FU-hoitoa 800 mg/m² jatkuvana laskimoinfuusiona 24 tunnin ajan päivinä 1–5, toistettuna 3 viikon välein. Sisplatiinia ja 5-FU-hoitoa annettiin enintään 6 hoitojakson ajan.
Ensisijainen tehon päätetapahtuma oli kokonaiselossaolo (OS) PD-L1-positiivisessa analyysijoukossa (PD-L1:n TAP-pistemäärä ≥ 5 %) ja hoitoaikeen mukaisessa populaatiossa (ITT, kaikki satunnaistetut potilaat). Toissijaisia tehon päätetapahtumia olivat etenemättömyysaika (PFS), objektiivinen vasteen kesto (ORR) ja vasteen kesto (DoR) tutkijan RECIST v1.1 ‑kriteerien mukaisesti arvioimina, sekä terveyteen liittyvä elämänlaatu (HRQoL).
Kasvaimet arvioitiin 6 viikon välein ensimmäisten 48 viikon ajan ja tämän jälkeen 9 viikon välein.
Yhteensä 997 potilasta satunnaistettiin saamaan tislelitsumabihoitoa yhdessä solunsalpaajahoidon kanssa (N = 501) tai saamaan lumelääkettä yhdessä solunsalpaajahoidon kanssa (N = 496). 997 potilaasta 546 potilaalla (54,8 %) oli PD-L1:n TAP-pistemäärä ≥ 5 % (tislelitsumabi yhdessä solunsalpaajahoidon kanssa: N = 274; lumelääke yhdessä solunsalpaajahoidon kanssa: N = 272). 931 potilasta (93,4 %) sai oksaliplatiini- ja kapesitabiinihoitoa (tislelitsumabi yhdessä solunsalpaajahoidon kanssa: N = 466, lumelääke yhdessä solunsalpaajahoidon kanssa: N = 465).
Potilaiden, joiden kasvaimet ilmensivät PD-L1:tä siten, että TAP-pistemäärä oli ≥ 5 %, lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 62 vuotta (vaihteluväli: 23–84), 39,2 % oli 65 vuotta täyttäneitä tai vanhempia, 72,2 % oli miehiä, 23,1 % oli valkoihoisia ja 73,8 % aasialaisia. 33,7 %:lla oli ECOG-toimintakykyluokka 0 ja 66,3 %:lla oli ECOG-toimintakykyluokka 1. Yhteensä 79,9 %:lla potilaista ensisijainen kasvain sijaitsi vatsassa. 98,5 %:lla oli lähtötilanteessa metastasoitunut tauti, 43,6 %:lla potilaista oli maksametastaasi ja 39,7 %:lla peritoneaalinen metastaasi.
Ennalta määritellyssä välianalyysissä BGB‑A317‑305‑tutkimuksessa havaittiin tilastollisesti merkitsevä kokonaiselossaolon kohentuminen tislelitsumabin ja solunsalpaajahoidon yhdistelmää saamaan satunnaistetuilla potilailla lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneisiin verrattuna niillä potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 %). Kokonaiselossaolon stratifioitu riskitiheyssuhde (HR) oli 0,74 (95 %:n lv: 0,59–0,94; yksisuuntainen p-arvo < 0,0056) ja kokonaiselossaolon mediaani 17,2 kuukautta tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä ja 12,6 kuukautta lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä. Tutkimuksessa todettiin myös tilastollisesti merkitsevä kohentuminen etenemättömyysajassa (FPS) potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 %. Etenemättömyysajan stratifioitu riskitiheyssuhde oli 0,67 (95 %:n lv: 0,55–0,83, yksisuuntainen p-arvo < 0,0001) ja etenemättömyysajan mediaani 7,2 kuukautta tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä ja 5,9 kuukautta lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä.
Ennalta määritellyssä loppuanalyysissä BGB‑A317‑305‑tutkimuksessa havaittiin tilastollisesti merkitsevä kohentuminen kaikilla satunnaistetuilla potilailla. Kokonaiselossaolon stratifioitu riskitiheyssuhde (HR) oli 0,80 (95 %:n lv: 0,70–0,92; yksisuuntainen p-arvo < 0,0011) ja kokonaiselossaolon mediaani 15,0 kuukautta tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä ja 12,9 kuukautta lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä. Päivitetyt kokonaiselossaoloa koskevat tulokset potilaista, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 %, olivat kokonaiselossaolon ensisijaisen analyysin tulosten mukaisia.
Loppuanalyysin tehotulokset potilaista, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 %, esitetään taulukossa 9 ja kuvassa 9.
Taulukko 9 Tehotulokset tutkimuksessa BGB-A317-305 potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 % (loppuanalyysi)
| Tislelitsumabi + solunsalpaajahoito (n = 274) | Lumelääke + solunsalpaajahoito (n = 272) | |
| Patients with PD-L1 score ≥ 5% | ||
| Tutkimuksen seurannan mediaani (kuukautta)a | 32,5 | 32,2 |
| OS | ||
| Kuolemat, n (%) | 192 (70,1) | 219 (80,5) |
| Mediaanib (kk) (95 %:n lv) | 16,4 (13,6–19,1) | 12,8 (12,0–14,5) |
| Riskitiheyssuhdec (95 %:n lv) | 0,71 (0,58–0,86) | |
| p-arvoc,d | 0,0003e | |
| PFS | ||
| Taudin eteneminen tai kuolema, n (%) | 189 (69,0) | 216 (79,4) |
| Mediaanib (kk) (95 %:n lv) | 7,2 (5,8–8,4) | 5,9 (5,6–7,0) |
| Riskitiheyssuhdec (95 %:n lv) | 0,68 (0,56–0,83) | |
| ORR (%) (95 %:n lv) | 51,5 (45,4–57,5) | 42,6 (36,7–48,8) |
OS = kokonaiselossaolo; lv = luottamusväli; PFS = etenemättömyysaika; ORR = objektiivinen vasteprosentti. a Mediaaniarvoinen seuranta-aika arvioitiin käänteisellä Kaplan-Meierin menetelmällä. b Mediaanit arvioitiin Kaplan-Meierin menetelmällä 95 %:n luottamusväleillä Brookmeyerin ja Crowleyn menetelmää käyttäen. c Stratifioitu alueen (Itä-Aasia vs. Eurooppa, Yhdysvallat) ja peritoneaalisen metastaasin esiintymisen mukaan. d Yksipuolinen p-arvo stratifioidusta log rank-testistä. e Nominaalinen p-arvo. | ||
Kuva 9Kokonaiselossolon Kaplan-Meier-käyrä BGB-A317-305-tutkimuksessa potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5% (loppuanalyysi)
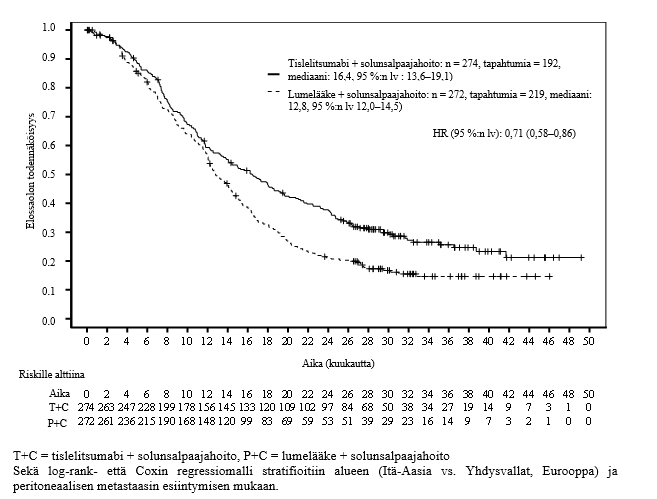
Ruokatorven levyepiteelikarsinooma
Ruokatorven levyepiteelikarsinooman ensilinjan hoito: BGB-A317-306
BGB-A317-306 on satunnaistettu, kaksoissokkoutettu, maailmanlaajuinen vaiheen III tutkimus, jossa verrattiin tislelitsumabin tehoa yhdessä platinapohjaisen solunsalpaajahoidon kanssa lumelääkkeen tehoon yhdessä platinapohjaisen solunsalpaajahoidon kanssa. Potilailla oli leikattavaksi soveltumaton, uusiutunut, paikallisesti edennyt tai metastasoitunut ruokatorven levyepiteelikarsinooma.
Tutkimukseen otettiin mukaan potilaita, joita ei ollut mahdollista hoitaa kemosädehoidolla eikä kuratiivisessa tarkoituksessa tehdyllä leikkauksella. Potilaat otettiin tutkimukseen kasvaimen PD‑L1‑ilmentymisen tasosta riippumatta. Jos saatavilla oli arkistoitu/tuore kasvainkudosnäyte, kasvaimen PD‑L1‑ilmentymisstatus tutkittiin retrospektiivisesti. PD‑L1‑ilmentyminen arvioitiin keskuslaboratoriossa TAP (kasvainalueen positiivisuus) -pistemäärää käyttäen, jonka määritelmänä on kasvainalueen kasvainsolujen (kasvain ja mahdollinen desmoplastinen strooma) kattama kokonaisprosentti Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin minkä tahansa voimakkuuden PD‑L1-ilmentymä sekä kasvainsoluissa että kasvaimen sisältämissä immuunisoluissa.
Potilaat, jotka olivat aiemmin saaneet systeemistä hoitoa edenneen tai metastasoituneen taudin hoitoon, suljettiin pois tutkimuksesta. Vähintään 6 kuukauden pituinen hoidoton väli edellytettiin, jos potilas oli saanut aiemmin neoadjuvantti/adjuvanttihoitoa platinapohjaisella solunsalpaajahoidolla.
Tutkimuksesta suljettiin pois potilaat, joilla oli merkkejä fisteleistä tai täydellinen ruokatorven tukos, joka ei ollut hoidettavissa.
Satunnaistaminen stratifioitiin maantieteellisen alueen mukaan (Aasia [pois lukien Japani] vs. Japani vs. muu mailma [ROW]), aiemman definitiivisen hoidon mukaan (kyllä vs. ei) ja tutkijan valitseman solunsalpaajahoidon mukaan (ICC, platina ja fluoropyrimidiini tai platina ja paklitakseli).
Potilaat satunnaistettiin (1:1) saamaan joko 200 mg tislelitsumabia tai lumelääkettä 3 viikon välein yhdessä tutkijan valitseman solunsalpaajahoidon (ICC) kanssa 21 vuorokauden hoitojaksona. Duplettisolunsalpaajahoito koostui seuraavista:
- platinaa (sisplatiinia [60–80 mg/m² laskimoon päivänä 1] tai oksaliplatiinia [130 mg/m² laskimoon päivänä 1]) ja fluoropyrimidiinia (5-FU [750–800 mg/ m² laskimoon päivinä 1–5] tai kapesitabiinia [1000 mg/ m² suun kautta kahdesti vuorokaudessa päivinä 1–14]), tai
- platinaa (sisplatiinia [60–80 mg/m² laskimoon päivänä 1 tai 2] tai oksaliplatiinia [130 mg/m² laskimoon päivänä 1 tai 2]) ja paklitakselia (175 mg/ m² laskimoon päivänä 1).
Potilaat saivat tislelitsumabihoitoa yhdessä solunsalpaajahoidon kanssa tai lumelääkettä yhdessä solunsalpaajahoidon kanssa, kunnes tauti eteni (tutkijan arvio RECIST v1.1 ‑kriteerien mukaisesti) tai ilmeni liiallista toksisuutta. 24 kuukauden hoidon jälkeen tutkimushoitoa voitiin jatkaa pidempään kuin näiden kahden vuoden ajan, jos tutkija katsoi tämän olevan potilaan edun mukaista kliinisen hyöty-riski-arvion perusteella.
Kasvaimet arvioitiin 6 viikon välein ensimmäisten 6 kuukauden ajan ja tämän jälkeen 9 viikon välein.
Ensisijainen tehon päätetapahtuma oli kokonaiselossaolo (OS) hoitoaikeen mukaisessa (ITT) populaatiossa. Toissijaisia tehon päätetapahtumia olivat etenemättömyysaika (PFS), objektiivinen vasteen kesto (ORR) ja vasteen kesto (DoR) tutkijan RECIST v1.1 ‑kriteerien mukaisesti arvioimina, kokonaiselossaolo PD-L1-positiivisten alaryhmässä (PD-L1:n TAP-pistemääärä ≥ 10 %) ja terveyteen liittyvä elämänlaatu (HRQoL).
Yhteensä 649 potilasta satunnaistettiin tislelitsumabihoitoon yhdessä solunsalpaajahoidon kanssa (N = 326) tai saamaan lumelääkettä yhdessä solunsalpaajahoidon kanssa (N = 323). 649 potilaasta 290 (44,7 %) potilasta sai platina + fluoropyrimidiini -hoitoa, 358 potilaalla oli PD-L1:n TAP-pistemäärä ≥ 5 %, 184 potilaalla oli PD-L1:n TAP-pistemäärä < 5 % ja 107 potilaalla oli tuntematon PD-L1-status.
Potilaiden, joiden kasvaimet ilmensivät PD-L1:tä siten, että TAP-pistemäärä oli ≥ 5 %, lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 63,0 vuotta (vaihteluväli: 40–84), 44,7 % oli 65 vuotta täyttäneitä tai vanhempia, 84,9 % oli miehiä, 20,9 % oli valkoihoisia ja 78,2 % aasialaisia. 87,7 %:lla oli tutkimukseen ottovaiheessa metastasoitunut tauti ja 12,3 %:lla oli paikallisesti edennyt tauti. Kaikilla potilailla oli histologisesti vahvistettu tyvisolusyöpä. Lähtötilanteen ECOG‑toimintakykyluokka oli 0 (29,9 %) tai 1 (70,1 %).
Välianalyysin tietojen katkaisupäivään (28.2.2022) mennessä BGB‑A317‑306‑tutkimuksessa havaittiin tilastollisesti merkitsevä kokonaiselossaolon kohentuminen kaikilla satunnaistetuilla potilailla. Stratifioitu kokonaiselossaolon riskitiheyssuhde (HR) oli 0,66 (95 %:n lv: 0,54–0,80; yksisuuntainen p-arvo < 0,0001) ja kokonaiselossaolon mediaani 17,2 kuukautta tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä ja 10,6 kuukautta lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä.
Päivitetyn analyysin, jossa oli 3 vuoden seuranta tietojen katkaisupiävään 24.11.2023 mennessä, tehotulokset olivat yhtenevät välianalyysin tulosten kanssa. Käänteisellä Kaplan-Meierin menetelmällä lasketut seurannan mediaaniajat olivat 44,2 kuukautta tislelitsumabin ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä ja 43,8 kuukautta lumelääkkeen ja solunsalpaajahoidon yhdistelmää saaneiden ryhmässä.
Tehotulokset kolmen vuoden seurannan kohdalla potilaista, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 %, esitetään taulukossa 10 ja kuvassa 10.
Taulukko 10 Tehotulokset tutkimuksessa BGB‑A317‑306 potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 % - 3 vuoden seuranta (tietojen katkaisupäivä 24.11.2023)
| Päätetapahtuma | Tislelitsumabi + solunsalpaajahoito (n = 172) | Lumelääke+ solunsalpaajahoito (n = 186) |
| OS | ||
| Kuolemat, n (%) | 128 (74,4) | 151 (81,2) |
| Mediaani (kk) (95 %:n lv) | 19,1 (16,1–24,1) | 10.0 (8,6–11,9) |
| Riskitiheyssuhde (95 %:n lv)a | 0,62 (0,49–0,79) | |
| p-arvob | < 0,0001 | |
| PFS | ||
| Tapahtumia, n (%) | 119 (69,2) | 153 (82,3) |
| Mediaani (kk) (95 %:n lv) | 8,2 (7,0–9,8) | 5,5 (4,3–6,4) |
| Riskitiheyssuhde (95 %:n lv)a | 0,50 (0,39–0,65) | |
| p-arvob | < 0,0001 | |
| ORR % (95 %:n lv)c | 64,0 (56,3–71,1) | 36,0 (29,1–43,4) |
OS = kokonaiselossaolo; lv = luottamusväli; PFS = etenemättömyysaika; ORR = objektiivinen vasteprosentti a Perustuu stratifioituun Coxin malliin. b Yksipuolinen nominaalinen p-arvo stratifioidusta log rank-testistä. c Tarkka Clopper-Person-kaksipuolinen luottamusväli. | ||
Kuva 10 Kokonaiselossaolon Kaplan-Meier-käyrä BGB‑A317‑306-tutkimuksessa potilailla, joiden PD-L1:n TAP-pistemäärä oli ≥ 5 % - 3 vuoden seuranta (tietojen katkaisupäivä 24.11.2023)
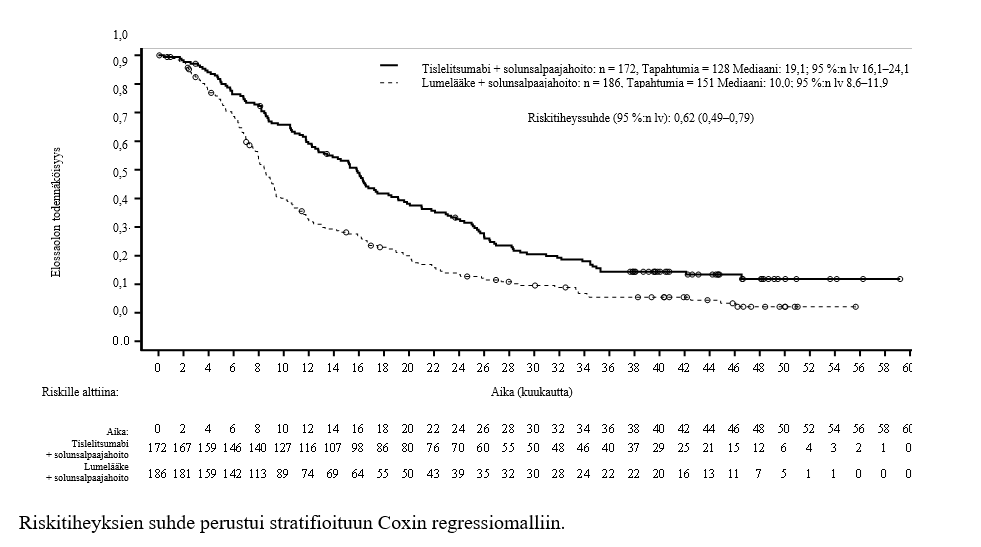
Aiemmin hoidettu ruokatorven levyepiteelikarsinooma: BGB‑A317‑302
BGB‑A317‑302 oli satunnaistettu, kontrolloitu, avoin, maailmanlaajuinen vaiheen III tutkimus, jossa tislelitsumabin tehoa verrattiin solunsalpaajahoidon tehoon. Potilailla oli leikattavaksi soveltumaton, uusiutunut, paikallisesti edennyt tai metastasoitunut ruokatorven levyepiteelikarsinooma, joka oli edennyt aiemman systeemisen hoidon aikana tai sen jälkeen. Potilaat otettiin tutkimukseen kasvaimen PD‑L1‑ilmentymisen tasosta riippumatta. Jos saatavilla oli arkistoitu/tuore kasvainkudosnäyte, kasvaimen PD‑L1‑ilmentymisstatus tutkittiin retrospektiivisesti. PD‑L1‑ilmentyminen arvioitiin keskuslaboratoriossa Ventana PD‑L1 (SP263) ‑määrityksellä, jossa värjäyksellä tunnistettiin PD‑L1-ilmentymä sekä kasvainsoluissa että kasvaimen sisältämissä immuunisoluissa.
Tutkimuksesta suljettiin pois potilaat, jotka olivat saaneet aiemmin hoitoa PD‑1:n/PD-L1:n estäjällä tai joilla kasvain oli invasoitunut ruokatorven karsinooman sijaintipaikan viereisiin elimiin (esim. aorttaan tai hengitysteihin).
Satunnaistaminen stratifioitiin maantieteellisen alueen mukaan (Aasia [pois lukien Japani] vs. Japani vs. Yhdysvallat/EU), ECOG‑toimintakykyluokan mukaan (0 vs. 1) ja tutkijan valitseman solunsalpaajahoitovaihtoehdon mukaan (paklitakseli vs. dosetakseli vs. irinotekaani). Tutkija valitsi solunsalpaajahoidon ennen satunnaistamista.
Potilaat satunnaistettiin (1:1) saamaan 200 mg tislelitsumabia laskimoon 3 viikon välein tai jotakin seuraavista solunsalpaajahoidoista laskimoon tutkijan valitsemana:
- paklitakselia 135–175 mg/m² päivänä 1 kolmen viikon välein (myös annoksina 80–100 mg/m2 viikoittain paikallisten ja/tai maakohtaisten tavanomaisen hoidon suositusten mukaisesti) tai
- dosetakselia 75 mg/m2 päivänä 1 kolmen viikon välein tai
- irinotekaania 125 mg/m2 päivinä 1 ja 8 kolmen viikon välein.
Potilaat saivat Tevimbra‑hoitoa tai yhtä tutkijan valitsemaa solunsalpaajahoitoa, kunnes tauti eteni (tutkijan arvio RECIST v1.1 ‑kriteerien mukaisesti) tai ilmeni liiallista toksisuutta.
Kasvaimet arvioitiin 6 viikon välein ensimmäisten 6 kuukauden ajan ja tämän jälkeen 9 viikon välein.
Ensisijainen tehon päätetapahtuma oli kokonaiselossaolo (OS) hoitoaikeen mukaisessa (ITT) populaatiossa. Toissijaisia tehon päätetapahtumia olivat OS PD‑L1‑positiivisessa analyysipopulaatiossa (PD‑L1‑osuus ≥ 10 % vCPS‑testillä [visually‑estimated Combined Positive Score, nyt tunnettu Tumour Area Positivity -pistemääränä eli TAP-pistemääränä] arvioituna [PD‑L1:n TAP-pistemäärä]), objektiivinen vasteprosentti (ORR), etenemättömyysaika (PFS) ja vasteen kesto (DoR) tutkijan RECIST v1.1 ‑kriteerien mukaisesti arvioimina.
Yhteensä 512 potilasta otettiin tutkimukseen ja satunnaistettiin tislelitsumabihoitoon (N = 256) tai tutkijan valitsemaan solunsalpaajahoitoon (N = 256; paklitakseli [n = 85], dosetakseli [n = 53] tai irinotekaani [n = 118]). Kyseisistä 512 potilaasta 142:lla (27,7 %) PD‑L1‑osuus oli ≥ 10 % ja 222:lla (43,4 %) < 10 %. Lähtötilanteen PD‑L1‑status ei ollut tiedossa 148 potilaalla (28,9 %).
Tutkimuspopulaation lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 63 vuotta (vaihteluväli: 35–86), 39,5 % oli 65 vuotta täyttäneitä tai vanhempia, 84 % oli miehiä, 19 % oli valkoihoisia ja 80 % aasialaisia, 25 %:lla ECOG‑toimintakykyluokka oli 0 ja 75 %:lla 1. Tutkimuspopulaatiosta 95 %:lla oli tutkimukseen ottovaiheessa metastasoitunut tauti. Kaikki potilaat olivat saaneet syöpään vähintään yhtä aiempaa solunsalpaajahoitoa, joka oli 97 %:lla potilaista platinapohjainen solunsalpaajayhdistelmähoito.
BGB‑A317‑302‑tutkimuksessa ennalta määritetyn loppuanalyysin kohdalla kokonaiselossaolo parani tislelitsumabiryhmään satunnaistetuilla potilailla tilastollisesti merkitsevästi verrattuna tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmään. Stratifioitu riskitiheyssuhde (HR) oli 0,70 (95 %:n lv: 0,57–0,85; yksisuuntainen p-arvo 0,0001) ja kokonaiselossaolon mediaani 8,6 kuukautta (95 %:n lv: 7,5–10,4) tislelitsumabiryhmässä ja 6,3 kuukautta (95 %:n lv: 5,3–7,0) tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmässä. Käänteisellä Kaplan–Meierin menetelmällä laskettu seuranta‑ajan mediaani oli tislelitsumabiryhmässä 20,8 kk ja tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmässä 21,1 kk.
Päivitetyn analyysin, jossa oli 24 kuukauden lisäseuranta ennalta määritetyn loppuanalyysin jälkeen, tehotulokset olivat yhtenevät loppuanalyysin tulosten kanssa. Käänteisellä Kaplan-Meierin menetelmällä lasketut seurannan mediaaniajat olivat 44,7 kuukautta tislelitsumabiryhmässä ja 44,0 kuukautta tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmässä.
Päivitetyn analyysin tehotulokset esitetään taulukossa 11 ja kuvassa 11.
Taulukko 11 BGB‑A317‑302‑tutkimuksen tehotulokset – päivitetty analyysi
| Päätetapahtuma | Tevimbra (N = 256) | Solunsalpaajahoito (N = 256) |
| OS | ||
| Kuolemat, n (%) | 233 (91,0) | 233 (91,0) |
| Mediaani (kk)a (95 %:n lv) | 8,6 (7,5–10,4) | 6,3 (5,3–7,0) |
| Riskitiheyssuhde (95 %:n lv)b | 0,71 (0,59–0,86) | |
| p‑arvoc | p = 0,0002 | |
| PFS, tutkijan arviod | ||
| Taudin eteneminen tai kuolema, n (%) | 229 (89,5) | 181 (70,7) |
| Mediaani (kk) (95 %:n lv) | 1,6 (1,4–2,7) | 2,1 (1,5–2,7) |
| Riskitiheyssuhde (95 %:n lv) | 0,82 (0,67–1,01) | |
| ORR, tutkijan vahvistamad | ||
| ORR (%) (95 %:n lv) | 15,2 (11,1–20,2) | 6,6 (3,9–10,4) |
| Vasteen mediaanikesto, tutkijan vahvistama (kk) (95 %:n lv) | 11,3 (6,5–14,4) | 6,3 (2,8–8,5) |
OS = kokonaiselossaolo; lv = luottamusväli; PFS = etenemättömyysaika; ORR = objektiivinen vasteprosentti. a Arvioitu Kaplan–Meierin menetelmällä. b Perustuu Coxin regressiomalliin, jossa hoito kovariaattina, ja stratifioitu lähtötilanteen ECOG‑toimintakykyluokan ja tutkijan valitseman solunsalpaajahoidon mukaan. c Nominaalinen yksisuuntainen p-arvo perustuu log‑rank‑testiin, joka stratifioitiin ECOG‑toimintakykyluokan ja tutkijan valitseman solunsalpaajahoidon mukaan. d Perustuu ad hoc ‑analyysiin. | ||
Kuva 11 OS (Kaplan–Meier‑käyrä) BGB‑A317‑302‑tutkimuksessa (ITT‑analyysipopulaatio) – päivitetty analyysi
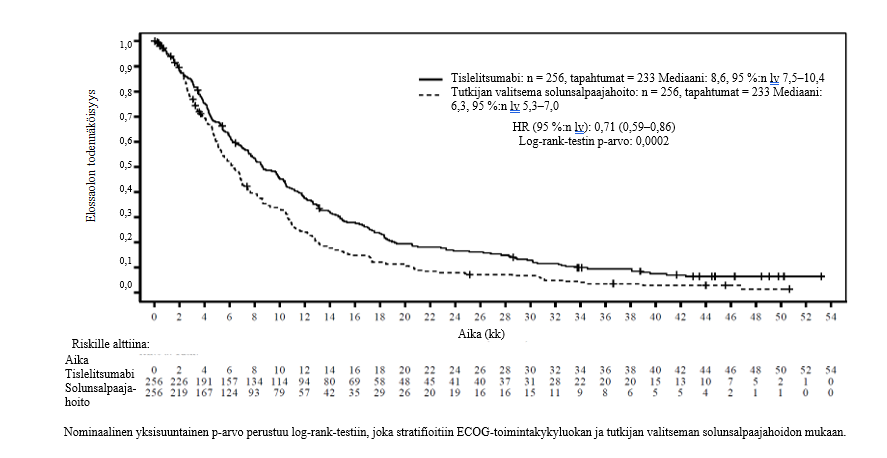
Teho PD‑L1‑alaryhmissä (päivitetty analyysi):
Päivitetyssä OS‑analyysissä PD‑L1‑positiivisten alaryhmässä (PD‑L1‑osuus ≥ 10 %) stratifioitu riskitiheyssuhde OS‑ajalle oli 0,54(95 %:n lv: 0,36–0,79). Elinajan mediaani oli tislelitsumabiryhmässä 10,2 kk (95 %:n lv: 8,5–14,5 kk) ja tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmässä 5,1 kk (95 %:n lv: 3,8–8,2 kk).
PD‑L1‑negatiivisten alaryhmässä (PD‑L1‑osuus < 10 %) stratifioitu HR OS‑ajalle oli 0,86 (95 %:n lv: 0,65–1,14): OS‑mediaani oli tislelitsumabiryhmässä 7,5 kk (95 %:n lv: 5,5–8,9 kk) ja tutkijan valitsemaa solunsalpaajahoitoa saaneiden ryhmässä 5,8 kk (95 %:n lv: 4,8–6,9 kk).
Nenänielukarsinooma
Uusiutuneen tai metastasoituneen nenänielukarsinooman ensilinjan hoito: BGB-A317-309
BGB‑A317‑309 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa verrattiin tislelitsumabin tehoa ja turvallisuutta yhdessä gemsitabiinin ja sisplatiinin kanssa lumelääkkeeseen yhdessä gemsitabiinin ja sisplatiinin kanssa ensilinjan hoitona. Potilailla oli uusiutunut tai metastasoitunut nenänielukarsinooma.
Potilaat eivät olleet saaneet hoitoa uusiutuneeseen tai metastasoituneeseen nenänielukarsinoomaan. Vähintään 6 kuukauden pituinen hoidoton väli edellytettiin, jos potilas oli saanut aiemmin neoadjuvanttisolunsalpaajahoitoa, adjuvanttisolunsalpaajahoitoa, sädehoitoa tai kemosädehoitoa ei-metastasoituneeseen tautiin kuratiivisella tarkoituksella. Tutkimuksesta suljettiin pois potilaat, joilla tauti oli uusiutunut paikallisesti siten, että se soveltui kuratiiviseen leikkaukseen tai sädehoitoon, sekä potilaat, jotka olivat saaneet aiemmin PD‑1:een tai PD‑L1:een kohdentuvia hoitoja.
Potilaat satunnaistettiin (1:1) saamaan joko tislelitsumabia 200 mg 3 viikon välein tai lumelääkettä yhdistelmänä päivänä 1 annettavan sisplatiinin 80 mg/m2 kanssa ja gemsitabiinin 1 g/m2 kanssa, joka annettiin jokaisen 21 vuorokauden hoitojakson päivinä 1 ja 8 4–6 hoitojakson ajan. Satunnaistetut potilaat stratifioitiin sukupuolen ja maksan metastaasistatuksen mukaan.
Tislelitsumabia tai lumelääkettä annettiin taudin etenemiseen tai kohtuuttomaan toksisuuteen saakka. Lumelääkeryhmän potilaille annettiin mahdollisuus siirtyä saamaan tislelitsumabimonoterapiaa riippumattoman arviointitoimikunnan vahvistaman taudin etenemisen jälkeen.
Ensisijainen tehon päätetapahtuma oli etenemättömyysaika (PFS), jonka arvioi riippumaton arviointitoimikunta RECIST v1.1 ‑kriteerien mukaan hoitoaikeen mukaisessa (ITT) analyysijoukossa. Toissijaisia tehon päätetapahtumia olivat kokonaiselossaolo (OS), tutkijan arvioima etenemättömyysaika, objektiivinen vasteprosentti (ORR) ja vasteen kesto (DoR) riippumattoman arviointitoimikunnan arvioimana.
Yhteensä 263 potilasta satunnaistettiin saamaan joko tislelitsumabia yhdessä gemsitabiinin ja sisplatiinin (N = 131) kanssa tai lumelääkettä yhdessä gemsitabiinin ja sisplatiinin (N = 132) kanssa.
Tutkimuspopulaation lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 50 vuotta (vaihteluväli: 23–74 vuotta), 91,6 % potilaista oli alle 65‑vuotiaita, 78,3 % potilaista oli miehiä, 63,1 %:lla ECOG-toimintakykyluokka oli 1, 100 % oli aasialaisia (Kiinasta, Thaimaasta ja Taiwanista) ja 46,7 % oli nykyisiä tai entisiä tupakoitsijoita. 95,1 %:lla tutkimuspopulaatiosta oli metastasoitunut tauti satunnaistamisen kohdalla. Nenänielukarsinooman histologisena alatyyppinä oli 86,3 %:lla ei‑keratinisoitunut, 6,5 %:lla keratinisoitunut levyepiteelikarsinooma ja 7,2 %:lla luokittelematon nenänielukarsinooma. Suurimmalla osalla (76 %) potilaista Epstein–Barrin viruksen (EBV) DNA:n määrä oli ≥ 500 IU/ml. Lähtötilanteen ominaisuudet olivat yleisesti ottaen samanlaisia kahden hoitoryhmän välillä.
Ennalta määritellyn välianalyysin (tietojen katkaisupäivä 26.3.2021) kohdalla BGB‑A317‑309-tutkimuksessa havaittiin tilastollisesti merkitsevä etenemättömyysajan parantuminen potilailla, jotka satunnaistettiin saamaan tislelitsumabia yhdessä gemsitabiinin ja sisplatiinin kanssa, verrattuna lumelääkettä yhdessä gemsitabiinin ja sisplatiinin kanssa saaneiden ryhmään. Stratifioitu riskitiheyssuhde oli 0,52 (95 %:n lv: 0,38–0,73; yksisuuntainen p-arvo < 0,0001), ja etenemättömyysajan mediaani oli 9,2 kuukautta tislelitsumabia ja solusalpaajahoitoa saaneiden ryhmässä ja 7,4 kuukautta lumelääkettä ja solunsalpaajahoitoa saaneiden ryhmässä.
Päivitetyn analyysin (tietojen katkaisupäivä 8.12.2023) tehotulokset olivat yhtenevät välianalyysin tulosten kanssa (taulukko 12 ja kuva 12). Analyysin ajankohtaan mennessä 52,3 % kontrolliryhmän potilaista oli siirtynyt saamaan tislelitsumabimonoterapiaa. Käänteisellä Kaplan–Meierin menetelmällä lasketut kokonaiselossaoloajan mediaanit seurannassa olivat 41,4 kuukautta tislelitsumabia ja solunsalpaajahoitoa saaneiden ryhmässä ja 40,8 kuukautta lumelääkettä ja solunsalpaajahoitoa saaneiden ryhmässä.
Tiedot vähintään 65‑vuotiaista nenänielukarsinoomapotilaista ovat liian vähäisiä johtopäätösten tekemiseksi kyseisessä potilasjoukossa.
Taulukko 12 Tehotulokset tutkimuksessa BGB-A317-309 (ITT-analyysijoukko) – päivitetty analyysi
| Päätetapahtuma | Tislelitsumabi + solunsalpaajahoito (N = 131) | Lumelääke + solunsalpaajahoito (N = 132) |
| PFS, riippumattoman arviointitoimikunnan arvio | ||
| Tapahtumia, n (%) | 95 (72,5) | 106 (80,3) |
| PFS, mediaani (kk) (95 %:n lv)a | 9,6 (7,6–11,6) | 7,4 (5,6–7,6) |
| Stratifioitu riskitiheyssuhde (95 %:n lv)b | 0,53 (0,39–0,71) | |
| OS | ||
| Kuolemat, n (%) | 55 (42,0) | 64 (48,5) |
| Mediaani (kk) (95 %:n lv)a | 45,3 (33,4; ei arv.) | 31,8 (25,0; ei arv.) |
| Stratifioitu riskitiheyssuhde (95 %:n lv)b | 0,73 (0,51–1,05) | |
Lyhenteet: ei arv. = ei arvioitavissa; OS = kokonaiselossaolo; lv = luottamusväli; PFS = etenemättömyysaika. a Mediaanit arvioitiin Kaplan–Meierin menetelmällä ja 95 %:n luottamusvälit arvioitiin Brookmeyerin ja Crowleyn menetelmällä. b Stratifioitu sukupuolen (mies vs. nainen) ja maksan metastaasistatuksen (kyllä vs. ei) mukaan. | ||
Kuva 12 Etenemättömyysajan Kaplan–Meier-käyrä tutkimuksessa BGB-A317-309 riippumattoman arviointitoimikunnan arvioimana (ITT-analyysijoukko) – päivitetty analyysi
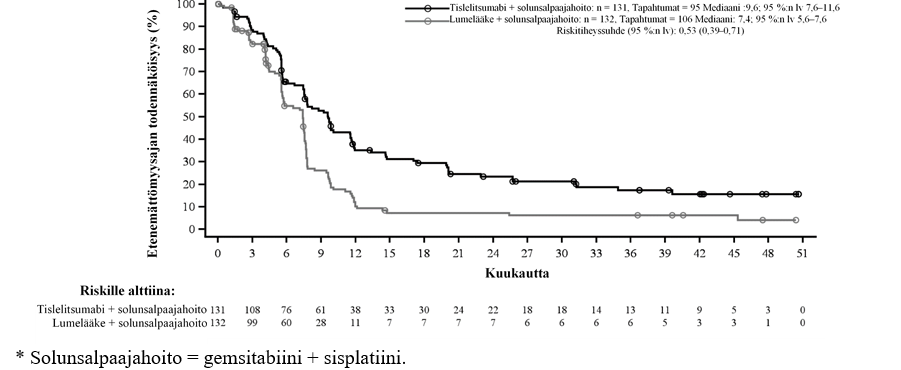
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tislelitsumabin käytöstä pahanlaatuisten kasvainten (lukuun ottamatta keskushermoston ja hematopoieettisten kudosten ja imukudosten kasvaimia) hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Tislelitsumabin farmakokinetiikkaa arvioitiin annosteltaessa Tevimbraa sekä monoterapiana että yhdessä solunsalpaajahoidon kanssa.
Tislelitsumabin farmakokinetiikan ominaisuuksia tutkittiin populaatiofarmakokinetiikan analyysissä. Pitoisuustietoja analysoitiin 2596 potilaalta, joilla oli pitkälle edennyt maligniteetti ja jotka saivat tislelitsumabia annoksina 0,5–10 mg/kg 2 viikon välein, 2,0 ja 5,0 mg kehon painokiloa kohti 3 viikon välein ja 200 mg 3 viikon välein.
90 %:n vakaan tilan tason saavuttamiseen kuluva aika on noin 84 vrk (12 viikkoa) annostuksella 200 mg 3 viikon välein, ja tislelitsumabin farmakokineettisen altistuksen vakaan tilan kumuloitumissuhde on noin kaksinkertainen.
Imeytyminen
Tislelitsumabi annetaan laskimoon, ja se on täten välittömästi ja täydellisesti biologisesti hyödynnettävissä.
Jakautuminen
Populaatiofarmakokinetiikan analyysin perusteella vakaan tilan jakautumistilavuus on 6,42 l, mikä on tyypillistä monoklonaalisille vasta‑aineille, joiden jakautuminen on suppeaa.
Biotransformaatio
Tislelitsumabi hajoaa todennäköisesti pieniksi peptideiksi ja aminohapoiksi kataboliareittien välityksellä.
Eliminaatio
Populaatiofarmakokinetiikan analyysin perusteella tislelitsumabin puhdistuma oli 0,153 l/vrk. Yksilöidenvälinen vaihtelu oli 26,3 %, terminaalisen puoliintumisajan geometrinen keskiarvo noin 23,8 vrk ja variaatiokerroin (CV) 31 %.
Lineaarisuus/ei‑lineaarisuus
Kun tislelitsumabin annostus oli 0,5–10 mg/kg 2 tai 3 viikon välein (mukaan lukien 200 mg 3 viikon välein ja 400 mg 6 viikon välein), tislelitsumabin farmakokinetiikan havaittiin olevan lineaarinen ja altistuksen suhteessa annokseen.
Erityisryhmät
Eri kovariaattien vaikutuksia tislelitsumabin farmakokinetiikkaan arvioitiin populaatiofarmakokinetiikan analyyseissä. Seuraavilla tekijöillä ei ollut kliinisesti merkittävää vaikutusta tislelitsumabialtistukseen: ikä (vaihteluväli 18–90 vuotta), paino (vaihteluväli 32–130 kg), sukupuoli, etninen tausta (valkoihoinen, aasialainen ja muut), lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma [CLCr] ≥ 30 ml/min), lievä tai keskivaikea maksan vajaatoiminta (kokonaisbilirubiiniarvo ≤ 3 x ULN ja mikä tahansa ASAT‑arvo) ja kasvaintaakka.
Munuaisten vajaatoiminta
Tislelitsumabia ei ole tutkittu nimenomaan munuaisten vajaatoimintapotilailla. Tislelitsumabia koskeneissa populaatiofarmakokinetiikan analyyseissä ei todettu kliinisesti merkittäviä eroja tislelitsumabin puhdistumassa, kun lievää munuaisten vajaatoimintaa (CLCr 60–89 ml/min, N = 1 046) ja keskivaikeaa munuaisten vajaatoimintaa (CLCr 30–59 ml/min, n = 320) sairastavia potilaita verrattiin tutkittaviin, joiden munuaistoiminta oli normaalia (CLCr ≥ 90 ml/min, n = 1 223). Lievä tai keskivaikea munuaisten vajaatoiminta ei vaikuttanut tislelitsumabialtistukseen (ks. kohta Annostus ja antotapa). Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden pienen määrän (n = 5) takia vaikean munuaisten vajaatoiminnan vaikutuksesta tislelitsumabin farmakokinetiikkaan ei voida tehdä lopullisia päätelmiä.
Maksan vajaatoiminta
Tislelitsumabia ei ole tutkittu nimenomaan maksan vajaatoimintapotilailla. Tislelitsumabia koskeneissa populaatiofarmakokinetiikan analyyseissä ei todettu kliinisesti merkittäviä eroja tislelitsumabin puhdistumassa, kun lievää maksan vajaatoimintaa (bilirubiini ≤ ULN ja ASAT > ULN tai bilirubiini > 1,0–1,5 x ULN ja mikä tahansa ASAT, n = 396) ja keskivaikeaa maksan vajaatoimintaa (bilirubiini > 1,5–3 x ULN ja mikä tahansa ASAT; n = 12) sairastavia potilaita verrattiin tutkittaviin, joiden maksatoiminta oli normaalia (bilirubiini ≤ ULN ja ASAT = ULN, n = 2 182) (ks. kohta Annostus ja antotapa). Vaikeaa maksan vajaatoimintaa (bilirubiini > 3 x ULN ja mikä tahansa ASAT, n = 2) sairastavien potilaiden pienen määrän takia vaikean maksan vajaatoiminnan vaikutusta tislelitsumabin farmakokinetiikkaan ei tunneta.
Prekliiniset tiedot turvallisuudesta
Kun jaavanmakakeille annettiin toistuvan altistuksen toksisuutta koskeneissa tutkimuksissa 3, 10, 30 tai 60 mg/kg annoksia laskimoon 2 viikon välein 13 viikon ajan (7 antokertaa), ilmeistä hoitoon liittyvää toksisuutta tai histopatologisia muutoksia ei havaittu annostuksen ollessa enimmillään 30 mg/kg 2 viikon välein. Altistus oli 4,3–6,6‑kertainen verrattuna 200 mg:n kliinisen annoksen tuottamaan altistukseen ihmisillä.
Tislelitsumabin kehitys‑ ja lisääntymistoksisuutta tai vaikutusta eläinten hedelmällisyyteen ei ole tutkittu.
Tislelitsumabin mahdollista karsinogeenisuutta tai genotoksisuutta ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraattidihydraatti
Sitruunahappomonohydraatti
L‑histidiinihydrokloridimonohydraatti
L‑histidiini
Trehaloosidihydraatti
Polysorbaatti 20 (E 432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Avattu injektiopullo
Injektiopullon avaamisen jälkeen lääkevalmiste on laimennettava ja infusoitava välittömästi (ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa).
Infuusionesteen valmistamisen jälkeen
Tevimbra ei sisällä säilöntäainetta. Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 10 päivän (240 tunnin) ajan 2–8 °C:n lämpötilassa. 10 päivän (240 tunnin) ajanjakso sisältää laimennetun liuoksen säilytyksen jääkaapissa (2–8 °C), huoneenlämpöiseksi (enintään 25 °C) palautumiseen vaadittavan ajan sekä infuusion antamiseen kuluvan ajan (enintään 4 tuntia).
Mikrobiologiselta kannalta valmiste on käytettävä välittömästi laimentamisen jälkeen.
Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja säilytysolosuhteet ovat käyttäjän vastuulla. Laimennettu liuos ei saa jäätyä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TEVIMBRA infuusiokonsentraatti, liuosta varten
100 mg (L:ei) 1 kpl (10 ml (10 mg/ml)) (2397,64 €)
PF-selosteen tieto
10 ml Tevimbra‑konsentraattia toimitetaan kirkkaassa tyypin 1 lasi‑injektiopullossa, jossa on harmaa, FluroTec‑päällysteinen klorobutyylitulppa ja irti napsautettava sinettikorkki.
Tevimbra on saatavana yksikköpakkauksissa, joissa on 1 injektiopullo, ja monipakkauksissa, joissa on 2 injektiopulloa (2 yhden injektiopullon pakkausta).
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön tai hieman kellertävä liuos.
Liuoksen pH on noin 6,5 ja osmolaliteetti noin 270–330 mOsm/kg.
Käyttö- ja käsittelyohjeet
Terveydenhuollon ammattilaisen on laimennettava infuusioneste aseptista tekniikkaa käyttäen.
Infuusionesteen valmistus
- Ota tarvittava määrä injektiopulloja jääkaapista. Älä ravista injektiopulloja.
- Tarkasta ennen antoa silmämääräisesti, että kummassakaan injektiopullossa ei ole hiukkasia eikä värimuutoksia. Konsentraatti on kirkasta tai hieman opalisoivaa, väritöntä tai hieman kellertävää liuosta. Älä käytä injektiopulloa, jos liuos on sameaa tai siinä näkyy hiukkasia tai värimuutoksia.
- Käännä injektiopullot varovasti ylösalaisin ravistamatta niitä. Vedä tarvittava määrä liuosta injektiopullo(i)sta ruiskuun ja siirrä liuos infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) NaCl‑injektionestettä. Laimennetun liuoksen lopullisen pitoisuuden on oltava 2–5 mg/ml. Sekoita laimennettua liuosta kääntelemällä pussia varovasti ylösalaisin vaahtoamisen ja liiallisten sekoitusvoimien estämiseksi.
Anto
- Anna laimennettu Tevimbra‑liuos infuusiona infuusioletkulla, jossa on steriili, pyrogeeniton, heikosti proteiinia sitova 0,2 tai 0,22 mikrometrin kiinteä (in‑line) tai irrallinen (add‑on) suodatin ja jonka pinta‑ala on noin 10 cm2.
- 200 mg kerran viikossa 3 viikon ajan -hoidon ensimmäinen infuusio on annettava 60 minuutin kestoisena. Jos potilas sietää sen hyvin, seuraavat infuusiot voidaan antaa 30 minuutin kestoisina.
Tevimbran 400 mg:n aloitusannoksen infuusio on annettava 120 minuutin kestoisena (90 minuutin kestoisena, kun sitä käytetään jatkohoitona 3 viikon välein annetun 200 mg:n annoksen jälkeen). Jos potilas sietää sen hyvin, toinen infuusio voidaan antaa 60 minuutin kestoisena. Jos potilas sietää toisen infuusion hyvin, myöhemmät infuusiot voidaan antaa 30 minuutin kestoisina.
- Muita lääkevalmisteita ei saa antaa samalla infuusioletkulla.
- Tevimbra‑valmistetta ei saa antaa laskimoon nopeana injektiona / kertaluonteisena bolusinjektiona.
- Infuusioletku on huuhdeltava infuusion lopuksi.
- Hävitä mahdollisesti käyttämättä jäänyt injektiopullon sisältö.
- Tevimbra‑injektiopullot on tarkoitettu vain yhtä käyttökertaa varten.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TEVIMBRA infuusiokonsentraatti, liuosta varten
100 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FF09
Valmisteyhteenvedon muuttamispäivämäärä
24.10.2025
Yhteystiedot
Gävlegatan 16
113 30 Stockholm
Sweden
beonemedicines.se
nordics@beonemed.com