
WINREVAIR injektiokuiva-aine ja liuotin, liuosta varten 45 mg, 60 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Winrevair 45 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 45 mg sotaterseptia (sotatercept). Käyttökuntoon saattamisen jälkeen yksi millilitra liuosta sisältää 50 mg sotaterseptia.
Winrevair 60 mg injektiokuiva-aine ja liuotin, liuosta varten
Yksi injektiopullo sisältää 60 mg sotaterseptia (sotatercept). Käyttökuntoon saattamisen jälkeen yksi millilitra liuosta sisältää 50 mg sotaterseptia.
Sotatersepti on rekombinantti homodimeerinen fuusioproteiini, joka koostuu ihmisen tyypin IIA aktiviinireseptorin (ActRIIA) solunulkoisesta domeenista ja siihen liittyneestä ihmisen IgG1:n Fc‑domeenista, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten (injektiokuiva-aine).
Kliiniset tiedot
Käyttöaiheet
Winrevair yhdistelmänä keuhkovaltimoverenpainetaudin muiden hoitojen kanssa on tarkoitettu keuhkovaltimoverenpainetaudin hoitoon aikuisille potilaille, joiden WHO-toimintakykyluokka on II, III tai IV (ks. kohta Farmakodynamiikka).
Ehto
Hoidon saa aloittaa vain keuhkovaltimoverenpainetaudin diagnosointiin ja hoitoon perehtyneen lääkärin määräyksestä ja toteuttaa hänen valvonnassaan.
Annostus ja antotapa
Winrevair-hoidon saa aloittaa vain keuhkovaltimoverenpainetaudin diagnosointiin ja hoitoon perehtyneen lääkärin määräyksestä ja toteuttaa hänen valvonnassaan.
Annostus
Winrevair-valmistetta annetaan 3 viikon välein kertainjektiona ihon alle potilaan painon mukaan.
Suositeltu aloitusannos
Hemoglobiiniarvo (Hb) ja verihiutaleiden määrä on määritettävä ennen ensimmäisen annoksen antamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoidon aloittaminen on vasta‑aiheista, jos verihiutaleiden määrä on jatkuvasti < 50 x 109/l (ks. kohta Vasta-aiheet).
Hoito aloitetaan antamalla 0,3 mg/kg kerta-annoksena (ks. taulukko 1).
Taulukko 1: Injektionesteen määrä, kun annos on 0,3 mg/kg
| Potilaan paino (kg) | Injektionesteen määrä (ml)* | Injektiosetin tyyppi |
| 30,0–40,8 | 0,2 | Injektiosetti, jossa on yksi 45 mg:n injektiopullo |
| 40,9–57,4 | 0,3 | |
| 57,5–74,1 | 0,4 | |
| 74,2–90,8 | 0,5 | |
| 90,9–107,4 | 0,6 | |
| 107,5–124,1 | 0,7 | |
| 124,2–140,8 | 0,8 | |
| 140,9–157,4 | 0,9 | |
| 157,5–174,1 | 1,0 | Injektiosetti, jossa on yksi 60 mg:n injektiopullo |
| 174,2–180,0 | 1,1 |
*Käyttökuntoon saatetun liuoksen pitoisuus on 50 mg/ml (ks. kohta Käyttö- ja käsittelyohjeet)
Suositeltu tavoiteannos
Kolme viikkoa aloitusannoksen (kerta-annos, 0,3 mg/kg) antamisen jälkeen annos tulee suurentaa suositellulle tavoitetasolle 0,7 mg/kg, kun on varmistettu, että hemoglobiiniarvo (Hb) ja verihiutaleiden määrä ovat hyväksyttävät (ks. kohta Annostus ja antotapa ”Annoksen muuttaminen hemoglobiiniarvon suurenemisen tai verihiutaleiden määrän pienenemisen vuoksi”). Hoitoa tulee jatkaa annoksella 0,7 mg/kg 3 viikon välein, ellei annoksen muuttaminen ole tarpeen.
Taulukko 2: Injektionesteen määrä, kun annos on 0,7 mg/kg
| Potilaan paino (kg) | Injektionesteen määrä (ml)* | Injektiosetin tyyppi |
| 30,0–31,7 | 0,4 | Injektiosetti, jossa on yksi 45 mg:n injektiopullo |
| 31,8–38,9 | 0,5 | |
| 39,0–46,0 | 0,6 | |
| 46,1–53,2 | 0,7 | |
| 53,3–60,3 | 0,8 | |
| 60,4–67,4 | 0,9 | |
| 67,5–74,6 | 1,0 | Injektiosetti, jossa on yksi 60 mg:n injektiopullo |
| 74,7–81,7 | 1,1 | |
| 81,8–88,9 | 1,2 | |
| 89,0–96,0 | 1,3 | Injektiosetti, jossa on kaksi 45 mg:n injektiopulloa |
| 96,1–103,2 | 1,4 | |
| 103,3–110,3 | 1,5 | |
| 110,4–117,4 | 1,6 | |
| 117,5–124,6 | 1,7 | |
| 124,7–131,7 | 1,8 | |
| 131,8–138,9 | 1,9 | Injektiosetti, jossa on kaksi 60 mg:n injektiopulloa |
| 139,0–146,0 | 2,0 | |
| 146,1–153,2 | 2,1 | |
| 153,3–160,3 | 2,2 | |
| 160,4–167,4 | 2,3 | |
| vähintään 167,5 | 2,4 |
*Käyttökuntoon saatetun liuoksen pitoisuus on 50 mg/ml (ks. kohta Käyttö- ja käsittelyohjeet)
Annoksen muuttaminen hemoglobiiniarvon suurenemisen tai verihiutaleiden määrän pienenemisen vuoksi
Hemoglobiiniarvoa ja verihiutaleiden määrää on seurattava, kunnes viisi ensimmäistä annosta on annettu, tai kauemmin, jos arvot vaihtelevat. Tämän jälkeen hemoglobiiniarvo ja verihiutaleiden määrä on tutkittava 3–6 kuukauden välein, ja annosta on muutettava tarvittaessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Hoitoa on lykättävä 3 viikolla (lykkäämällä antamista yhden annosvälin verran), jos jokin seuraavista ilmenee:
- hemoglobiiniarvo on suurentunut > 1,24 mmol/l (20 g/l) edellisen annoksen saamisen jälkeen ja on viitealueen ylärajan yläpuolella
- hemoglobiiniarvo on suurentunut > 2,48 mmol/l (40 g/l) lähtötilanteesta
- hemoglobiiniarvo on suurentunut > 1,24 mmol/l (20 g/l) viitealueen ylärajan yläpuolelle
- verihiutaleiden määrä on pienentynyt tasolle < 50 x 109/l.
Hemoglobiiniarvo ja verihiutaleiden määrä on määritettävä uudelleen ennen hoidon uudelleenaloittamista.
Jos hoito lykkääntyy yli 9 viikolla, hoito tulee aloittaa uudelleen annoksella 0,3 mg/kg, ja annos tulee suurentaa tasolle 0,7 mg/kg, kun on varmistettu, että hemoglobiiniarvo ja verihiutaleiden määrä ovat hyväksyttävät.
Jos hoito lykkääntyy yli 9 viikolla, koska verihiutaleiden määrä on jatkuvasti < 50 x 109/l, lääkärin on arvioitava uudelleen hoidon hyöty-riskisuhde kyseisen potilaan kohdalla ennen hoidon uudelleenaloittamista.
Annoksen jääminen väliin
Jos annos jää väliin, se on annettava mahdollisimman pian. Jos väliin jäänyttä annosta ei oteta 3 päivän sisällä suunnitellusta ajankohdasta, aikataulua on muutettava, jotta 3 viikon annosväli säilyy.
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäillä (vähintään 65‑vuotiailla) potilailla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen munuaisten vajaatoiminnan vuoksi ei ole tarpeen (ks. kohta Farmakokinetiikka). Saatavilla on vain tähän tietoa sotaterseptin käytöstä keuhkovaltimoverenpainetautia sairastaville potilaille, joilla on vaikea munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] < 30 ml/min/1,73 m2).
Maksan vajaatoiminta
Annoksen muuttaminen maksan vajaatoiminnan (Child–Pughin luokat A–C) vuoksi ei ole tarpeen. Sotaterseptia ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Winrevair-valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Winrevair on tarkoitettu vain yhtä käyttökertaa varten.
Valmiste on saatettava käyttökuntoon ennen käyttöä. Käyttökuntoon saatettu lääkevalmiste on kirkas tai opaalinhohtoinen ja väritön tai heikosti ruskehtavankeltainen liuos.
Winrevair annetaan injektiona ihon alle vatsaan (vähintään 5 cm:n päähän navasta), olkavarteen tai reiden yläosaan. Sitä ei saa pistää kohtaan, jossa on arpia, aristusta tai mustelmia. Samaa pistoskohtaa ei saa käyttää kahdella peräkkäisellä kerralla.
Winrevair injektiokuiva-aine ja liuotin, liuosta varten on tarkoitettu käytettäväksi terveydenhuollon ammattilaisen ohjauksessa. Potilaat ja heitä hoitavat henkilöt voivat pistää lääkevalmistetta, jos se katsotaan tarkoituksenmukaiseksi ja jos he saavat terveydenhuollon ammattilaiselta koulutuksen siitä, miten Winrevair injektiokuiva-aine ja liuotin, liuosta varten saatetaan käyttökuntoon, valmistellaan, mitataan ja annetaan injektiona. Myöhemmällä käynnillä pian koulutuksen jälkeen terveydenhuollon ammattilaisen on varmistettava, että potilas tai häntä hoitava henkilö osaa suorittaa nämä vaiheet oikein. Terveydenhuollon ammattilaisen on myös harkittava potilaan tai häntä hoitavan henkilön käyttämän antotekniikan tarkistamista, jos annosta muutetaan, potilas tarvitsee toisenlaisen injektiosetin tai potilaalle kehittyy erytrosytoosi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) tai milloin tahansa terveydenhuollon ammattilainen katsoo sen tarpeelliseksi.
Katso kohdasta Käyttö- ja käsittelyohjeet yksityiskohtaiset ohjeet Winrevair-valmisteen asianmukaisesta valmistelusta ja antamisesta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Verihiutaleiden määrä jatkuvasti < 50 x 109/l ennen hoidon aloittamista.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Erytrosytoosi
Potilailla on sotaterseptihoidon aikana havaittu suurentuneita hemoglobiiniarvoja. Vaikea erytrosytoosi saattaa suurentaa tromboembolisten tapahtumien ja hyperviskositeettioireyhtymän riskiä. Varovaisuutta on noudatettava, jos potilaalla on erytrosytoosi ja suurentunut tromboembolisten tapahtumien riski. Hemoglobiiniarvo on määritettävä ennen jokaisen annoksen antamista, kunnes viisi ensimmäistä annosta on annettu, tai kauemmin, jos arvot vaihtelevat. Sen jälkeen arvo on määritettävä 3–6 kuukauden välein, jotta voidaan arvioida, onko annoksen muuttaminen tarpeen (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Jos potilaalle kehittyy erytrosytoosi, terveydenhuollon ammattilaisen on harkittava, pitääkö potilaan tai häntä hoitavan henkilön lääkkeenantotekniikkaa arvioida uudelleen.
Vaikea trombosytopenia
Joillakin sotaterseptia saaneilla potilailla on havaittu verihiutaleiden määrän pienenemistä, myös vaikeaa trombosytopeniaa (verihiutaleiden määrä < 50 x 109/l). Trombosytopeniaa ilmoitettiin useammin potilailla, jotka saivat myös prostasykliini-infuusiohoitoa (7,7–24,5 %), verrattuna potilaisiin, jotka eivät saaneet prostasykliini-infuusiohoitoa (0,0–3,7 %) (ks. kohta Haittavaikutukset). Vaikea-asteinen trombosytopenia saattaa suurentaa verenvuototapahtumien riskiä. Verihiutaleiden määrä on määritettävä ennen jokaisen annoksen antamista viiden ensimmäisen annoksen kohdalla, tai kauemmin, jos arvot vaihtelevat, sekä sen jälkeen 3–6 kuukauden välein, jotta voidaan arvioida, onko annoksen muuttaminen tarpeen (ks. kohta Annostus ja antotapa).
Vakava verenvuoto
Kliinisissä tutkimuksissa sotaterseptihoidon aikana on havaittu vakavia verenvuototapahtumia (mukaan lukien ruoansulatuskanavan verenvuoto ja kallonsisäinen verenvuoto) 4,3–7,0 %:lla potilaista (ks. kohta Haittavaikutukset).
Potilaita, joilla ilmeni vakavia verenvuototapahtumia, koski todennäköisemmin jokin seuraavista: he saivat prostasykliinitaustahoitoa ja/tai antitromboottisia lääkkeitä, heidän trombosyyttiarvonsa olivat matalat tai he olivat vähintään 65‑vuotiaita. Potilaille on kerrottava verenhukan mahdollisista merkeistä ja oireista. Lääkärin on arvioitava ja hoidettava verenvuototapahtumia asianmukaisesti. Sotaterseptia ei pidä antaa, jos potilaalla on parhaillaan vakava verenvuototapahtuma.
Kliinisten tietojen rajoitukset
Kliinisissä tutkimuksissa ei ollut mukana tutkittavia, joilla oli immuunikatovirukseen (HIV) liittyvä keuhkovaltimoverenpainetauti, portahypertensioon liittyvä keuhkoverenpainetauti, skistosomiaasiin liittyvä keuhkovaltimoverenpainetauti tai keuhkojen veno-okklusiiviseen tautiin liittyvä keuhkovaltimoverenpainetauti.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää natriumia alle 1 mmol (23 mg) per annos, eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää 0,20 mg polysorbaatti 80:tä per ml käyttökuntoon saatettua liuosta. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on suositeltavaa tehdä raskaustesti ennen hoidon aloittamista. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen saamisen jälkeen, jos hoito lopetetaan (ks. kohta Prekliiniset tiedot turvallisuudesta).
Raskaus
Sotaterseptin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (enemmän alkionmenetyksiä kiinnittymisen jälkeen, sikiöiden painon pienenemistä ja luutumisen hidastumista) (ks. kohta Prekliiniset tiedot turvallisuudesta).
Winrevair-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö sotatersepti ja/tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
Imetys on lopetettava hoidon ajaksi ja siihen asti, kunnes viimeisen annoksen saamisesta on kulunut 4 kuukautta.
Hedelmällisyys
Eläimillä tehtyjen havaintojen perusteella sotatersepti saattaa heikentää naisten ja miesten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Sotaterseptilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Joko STELLAR-, ZENITH- tai HYPERION-tutkimuksessa yleisimmin raportoituja haittavaikutuksia (suurin esiintymistiheys tutkimuksissa) olivat nenäverenvuoto (45,3 %), päänsärky (26,7 %), telangiektasia (26,3 %), ripuli (25,6 %), hemoglobiiniarvon suureneminen (15,1 %), trombosytopenia (15,1 %), heitehuimaus (14,7 %), selkäkipu (14 %), ihottuma (12,3 %) ruoansulatuskanavan verenvuoto (11,6 %) ja ienverenvuoto (10,5 %).
Yleisimmin raportoituja vakavia haittavaikutuksia olivat trombosytopenia (< 1,2 %), nenäverenvuoto (< 1,2 %) ja heitehuimaus (< 1,2 %).
Haittavaikutuksia, jotka yleisimmin johtivat hoidon keskeyttämiseen, olivat nenäverenvuoto ja telangiektasia.
Haittavaikutustaulukko
Sotaterseptin turvallisuutta on arvioitu vaiheen 3 lumekontrolloiduissa STELLAR-, ZENITH- ja HYPERION-tutkimuksissa. STELLAR-tutkimukseen osallistui 163 potilasta, ZENITH-tutkimukseen 86 potilasta ja HYPERION-tutkimukseen 160 potilasta, joilla oli keuhkovaltimoverenpainetauti ja joille annettiin sotaterseptia (ks. kohta Farmakodynamiikka). Sotaterseptihoidon keston mediaani oli STELLAR-tutkimuksessa 313 päivää, ZENITH-tutkimuksessa 434,5 päivää ja HYPERION-tutkimuksessa 442,5 päivää.
Taulukossa 3 on esitetty lumekontrolloiduissa kliinisissä tutkimuksissa ja valmisteen markkinoilletulon jälkeen sotaterseptin käytön yhteydessä ilmoitetut haittavaikutukset. Ne on lueteltu MedDRA-elinjärjestelmäluokituksen ja esiintymistiheyden mukaan. Esiintymistiheydet määritellään seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva, markkinoilletulon jälkeen saatu tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3: Haittavaikutukset
Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
Infektiot | Yleinen | Virtsatieinfektiot |
Veri ja imukudos | Hyvin yleinen | Trombosytopenia1,2 |
Hermosto | Hyvin yleinen | Heitehuimaus |
Sydän | Tuntematon | Perikardiumeffuusio1 |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Nenäverenvuoto |
Melko harvinainen | Intrapulmonaarinen oikovirtaus3 | |
Ruoansulatuselimistö | Hyvin yleinen | Ripuli |
| Yleinen | Paksusuolen angiektasia |
Iho ja ihonalainen kudos | Hyvin yleinen | Telangiektasia1 |
Yleinen | Punoitus | |
Luusto, lihakset ja sidekudos | Hyvin yleinen | Selkäkipu4 |
Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Pistoskohdan kutina |
Tutkimukset | Yleinen | Kohonnut verenpaine1,6 |
1 Ks. valikoitujen haittavaikutusten kuvaus
2 Sisältää seuraavat: ”trombosytopenia” ja ”verihiutaleiden määrän pieneneminen”
3 Ks. ”pitkän aikavälin turvallisuustiedot”
4 Esiintymistiheysluokitus perustuu ZENITH-tutkimukseen
5 Sisältää seuraavat: ”ruoansulatuskanavan verenvuoto”, ”ylemmän ruoansulatuskanavan verenvuoto”, ”verioksennus”, ”alemman ruoansulatuskanavan verenvuoto”, ”hematoketsia”, ”peräsuolen verenvuoto”, ”meleena” ja ”hemorraginen gastriitti”
6 Sisältää seuraavat: ”hypertensio”, ”kohonnut diastolinen verenpaine” ja ”verenpaineen kohoaminen”
Valikoitujen haittavaikutusten kuvaus
Hemoglobiiniarvon suureneminen
STELLAR-tutkimuksessa hemoglobiiniarvon suurenemista (”suurentunut hemoglobiiniarvo” ja ”polysytemia”) ilmoitettiin 8,6 %:lla sotaterseptia saaneista potilaista. Laboratoriodatan perusteella hemoglobiiniarvon kohtalaista suurenemista (> 1,24 mmol/l [20 g/l] viitealueen ylärajan yläpuolelle) ilmeni 15,3 %:lla sotaterseptia saaneista potilaista.
ZENITH-tutkimuksessa hemoglobiiniarvon suurenemista ilmoitettiin 15,1 %:lla sotaterseptia saaneista potilaista. Laboratoriodatan perusteella hemoglobiiniarvon kohtalaista suurenemista ilmeni 7,1 %:lla sotaterseptia saaneista potilaista.
HYPERION-tutkimuksessa hemoglobiiniarvon suurenemista ilmoitettiin 11,3 %:lla sotaterseptia saaneista potilaista. Laboratoriodatan perusteella hemoglobiiniarvon kohtalaista suurenemista ilmeni 5,7 %:lla sotaterseptia saaneista potilaista.
Hemoglobiiniarvojen suurenemista hoidettiin annosta muuttamalla (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Trombosytopenia
STELLAR-tutkimuksessa trombosytopeniaa (”trombosytopenia” ja ”pienentynyt verihiutaleiden määrä”) ilmoitettiin 10,4 %:lla sotaterseptia saaneista potilaista. Verihiutaleiden määrän vaikea-asteista pienenemistä niin, että määrä oli < 50 x 109/l, ilmeni 3,1 %:lla sotaterseptia saaneista potilaista. Trombosytopeniaa ilmoitettiin useammin potilailla, jotka saivat myös prostasykliini-infuusiohoitoa (21,5 %), kuin potilailla, jotka eivät saaneet prostasykliini-infuusiohoitoa (3,1 %).
ZENITH-tutkimuksessa trombosytopeniaa ilmoitettiin 15,1 %:lla sotaterseptia saaneista potilaista. Verihiutaleiden määrän vaikea-asteista pienenemistä niin, että määrä oli < 50 x 109/l, ilmeni 6,0 %:lla sotaterseptia saaneista potilaista. Trombosytopeniaa ilmoitettiin vain potilailla, jotka saivat myös prostasykliini-infuusiohoitoa (24,5 %).
HYPERION-tutkimuksessa trombosytopeniaa ilmoitettiin 4,4 %:lla sotaterseptia saaneista potilaista. Verihiutaleiden määrän vaikea-asteista pienenemistä niin, että määrä oli < 50 x 109/l, ei ilmennyt sotaterseptia saaneilla potilailla. Trombosytopeniaa ilmoitettiin useammin potilailla, jotka saivat myös prostasykliini-infuusiohoitoa (7,7 %), kuin potilailla, jotka eivät saaneet prostasykliini-infuusiohoitoa (3,7 %).
Trombosytopenia hoidettiin annosta muuttamalla (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Telangiektasia
STELLAR-tutkimuksessa telangiektasiaa todettiin 16,6 %:lla sotaterseptia saaneista potilaista. Mediaaniaika tapahtuman ilmaantumiseen oli 18,6 viikkoa. Telangiaektasian vuoksi hoidon lopettaneiden osuus oli sotaterseptiryhmässä 1 %.
ZENITH-tutkimuksessa telangiektasiaa todettiin 25,6 %:lla sotaterseptia saaneista potilaista. Mediaaniaika tapahtuman ilmaantumiseen oli 12,8 viikkoa. Sotaterseptiryhmässä yksikään potilas ei lopettanut hoitoa telangiektasian vuoksi.
HYPERION-tutkimuksessa telangiektasiaa todettiin 26,3 %:lla sotaterseptia saaneista potilaista. Mediaaniaika tapahtuman ilmaantumiseen oli 19,6 viikkoa. Telangiaektasian vuoksi hoidon lopettaneiden osuus oli sotaterseptiryhmässä 0,6 %.
Kohonnut verenpaine
Kohonnutta verenpainetta ilmoitettiin STELLAR-tutkimuksessa 4,3 %:lla sotaterseptia saaneista potilaista. Sotaterseptia saaneilla potilailla systolisen verenpaineen keskiarvo oli kohonnut lähtötilanteesta 2,2 mmHg ja diastolisen verenpaineen keskiarvo oli kohonnut 4,9 mmHg 24 viikon kohdalla.
Kohonnutta verenpainetta ilmoitettiin ZENITH-tutkimuksessa 2,3 %:lla sotaterseptia saaneista potilaista. Sotaterseptia saaneilla potilailla systolisen verenpaineen keskiarvo oli kohonnut lähtötilanteesta 3,1 mmHg ja diastolisen verenpaineen keskiarvo oli kohonnut 5,1 mmHg 24 viikon kohdalla.
Kohonnutta verenpainetta ilmoitettiin HYPERION-tutkimuksessa 6,9 %:lla sotaterseptia saaneista potilaista. Sotaterseptia saaneilla potilailla systolisen verenpaineen keskiarvo oli kohonnut lähtötilanteesta 3,6 mmHg ja diastolisen verenpaineen keskiarvo oli kohonnut 4,5 mmHg 24 viikon kohdalla.
Perikardiumeffuusio
Sotaterseptihoitoa saaneilla potilailla on ilmoitettu uutta tai pahenevaa perikardiumeffuusiota (myös sydäntamponaatiota) keuhkovaltimoverenpainetautiin liittyvän hemodynamiikan paranemisesta tai vakiintumisesta huolimatta. Useimmat tapauksista ilmoitettiin potilailla, joilla oli sidekudossairauteen liittyvä keuhkovaltimoverenpainetauti, aiempaa perikardiumeffuusiota tai kummatkin näistä. Useimmat potilaat saivat myös prostasykliinianalogeja.
Iäkkäät
Verenvuototapahtumia lukuun ottamatta (erityisen kliinisen seurannan kohteena olleiden haittatapahtumien kollektiivinen ryhmä) turvallisuudessa ei havaittu eroja < 65‑vuotiaiden ja ≥ 65‑vuotiaiden potilaiden alaryhmien välillä.
STELLAR-tutkimuksessa verenvuototapahtumia ilmeni yleisemmin sotaterseptia saaneiden vanhempien potilaiden alaryhmässä (52 %:lla vs. 31,9 %:lla alle 65-vuotiaista potilaista), mutta ikäryhmien välillä ei ollut merkittävää eroa minkään tietyn verenvuototapahtuman suhteen. Vakavia verenvuotoja ilmeni 3,6 %:lla < 65-vuotiaista ja 8,0 %:lla ≥ 65-vuotiaista sotaterseptia saaneista potilaista.
ZENITH-tutkimuksessa verenvuototapahtumia ilmeni yleisemmin sotaterseptia saaneiden vanhempien potilaiden alaryhmässä (73,3 %:lla vs. 60,7 %:lla alle 65-vuotiaista potilaista). Vakavia verenvuotoja ilmeni 3,6 %:lla < 65-vuotiaista ja 13,3 %:lla ≥ 65-vuotiaista sotaterseptia saaneista potilaista.
HYPERION-tutkimuksessa verenvuototapahtumia ilmeni yleisemmin sotaterseptia saaneiden vanhempien potilaiden alaryhmässä (55,9 %:lla vs. 38 %:lla alle 65‑vuotiaista potilaista). Vakavia verenvuotoja ilmeni 2,2 %:lla < 65‑vuotiaista ja 13,2 %:lla ≥ 65‑vuotiaista sotaterseptia saaneista potilaista.
Pitkän aikavälin turvallisuustiedot
Yhdistettyjä pitkän aikavälin turvallisuustietoja on saatavilla 431 potilaasta, jotka osallistuivat vaiheen 2 ja vaiheen 3 kliinisiin tutkimuksiin (PULSAR, SPECTRA ja STELLAR). Suurin osa näistä potilaista jatkoi SOTERIA-tutkimukseen, joka on sotaterseptin pitkän aikavälin turvallisuutta ja tehoa koskeva meneillään oleva avoin seurantatutkimus. Altistuksen keston keskiarvo oli 173 viikkoa, ja altistuksen enimmäiskesto oli 355 viikkoa. Turvallisuusprofiili oli yleisesti samanlainen kuin STELLAR-avaintutkimuksessa. Oikealta vasemmalle suuntautuvaa intrapulmonaarista oikovirtausta on raportoitu osallistujilla, joille kehittyi paheneva hypoksemia keuhkovaltimoverenpainetautiin liittyvän hemodynamiikan paranemisesta huolimatta.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveillä vapaaehtoisilla toteutetussa vaiheen 1 tutkimuksessa yhdellä osallistujalla, joka sai sotaterseptia annoksella 1 mg/kg, ilmeni hemoglobiiniarvon suurenemista, johon liittyi oireinen kohonnut verenpaine. Nämä korjaantuivat flebotomialla.
Jos keuhkovaltimoverenpainetautia sairastavalla potilaalla ilmenee yliannostus, hemoglobiiniarvoa on seurattava tarkasti suurenemisen ja verenpaineen kohoamisen varalta, ja tarvittaessa on annettava tukihoitoa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Sotatersepti ei poistu verestä hemodialyysilla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: verenpainelääkkeet, keuhkoverenpainetaudin lääkkeet. ATC-koodi: C02KX06
Vaikutusmekanismi
Sotatersepti on aktiviinin signaalinvälityksen estäjä. Se sitoutuu erittäin selektiivisesti aktiviini A:han, joka on transformoiva kasvutekijä β (TGF‑β) ‑ligandien suurperheeseen kuuluva dimeerinen glykoproteiini. Sitoutumalla tyypin IIa aktiviinireseptoriin (ActRIIA) aktiviini A säätelee keskeisiä tulehdukseen, soluproliferaatioon, apoptoosiin ja kudosten homeostaasiin liittyviä signaaleja.
Aktiviini A:n pitoisuudet ovat suurentuneet keuhkovaltimoverenpainetautia sairastavilla potilailla. Aktiviinin sitoutuminen ActRIIA-reseptoriin edistää proliferatiivista signaalinvälitystä ja vähentää antiproliferatiivista luun morfogeneettisen proteiinin tyypin II reseptorin (BMPRII) signaalinvälitystä. Keuhkovaltimoverenpainetaudin taustalla oleva ActRIIA-BMPRII-signaalien epätasapaino saa aikaan verisuonisolujen hyperproliferaatiota, mikä aiheuttaa keuhkovaltimoiden seinämien patologista remodellaatiota ja valtimon luumenin ahtautumista, suurentaa keuhkoverenkierron vastusta ja johtaa keuhkovaltimopaineen suurenemiseen ja oikean kammion toimintahäiriöön.
Sotatersepti muodostuu ligandia sitovasta rekombinantista homodimeerisestä tyypin IIA aktiviinireseptorin ja Fc‑domeenin (ActRIIA‑Fc) fuusioproteiinista, joka sitoo liiallista aktiviini A:ta ja muita ActRIIA-reseptorin ligandeja ja estää siten aktiviinin signaalinvälityksen. Näin sotatersepti palauttaa proliferaatiota edistävän (ActRIIA/Smad2/3-välitteisen) ja antiproliferatiivisen (BMPRII/Smad1/5/8-välitteisen) signaalinvälityksen tasapainoa ja vaikuttaa siten verisuoniproliferaatioon.
Farmakodynaamiset vaikutukset
Vaiheen 2 kliinisessä tutkimuksessa (PULSAR) arvioitiin keuhkoverenkierron vastusta keuhkovaltimoverenpainetautia sairastavilla potilailla 24 viikon pituisen sotaterseptihoidon jälkeen. Keuhkoverenkierron vastus pieneni lähtötilanteesta merkittävästi enemmän ryhmissä, joissa tutkittavat saivat sotaterseptia annoksella 0,7 mg/kg tai 0,3 mg/kg, kuin lumeryhmässä. Lumeryhmän tietojen suhteen korjatun pienimmän neliösumman keskiarvon ero lähtötilanteeseen nähden oli ‑269,4 dyn*s/cm5 (95 %:n luottamusväli: ‑365,8, ‑173,0) sotaterseptia annoksella 0,7 mg/kg saaneiden ryhmässä ja ‑151,1 dyn*s/cm5 (95 %:n luottamusväli: ‑249,6, ‑52,6) sotaterseptia annoksella 0,3 mg/kg saaneiden ryhmässä.
Keuhkovaltimoverenpainetaudin rottamalleissa sotaterseptianalogi vähensi proinflammatoristen markkerien ilmentymistä keuhkovaltimoiden seinämissä, vähensi leukosyyttien kertymistä, esti endoteeli- ja sileälihassolujen proliferaatiota ja edisti apoptoosia vaurioituneissa verisuonissa. Näihin solumuutoksiin liittyi verisuonten seinämien ohenemista, valtimoiden ja oikean kammion patologisten muutosten kumoutumista ja hemodynamiikan paranemista.
Immunogeenisuus
Lääkevasta-aineita havaittiin STELLAR-tutkimuksessa 27 %:lla, ZENITH-tutkimuksessa 43 %:lla ja HYPERION-tutkimuksessa 40 %:lla potilaista. Lääkevasta-aineiden vaikutuksista farmakokinetiikkaan, tehoon tai turvallisuuteen ei havaittu näyttöä.
Kliininen teho ja turvallisuus
STELLAR
Sotaterseptin tehoa arvioitiin keuhkovaltimoverenpainetautia sairastavilla aikuisilla potilailla STELLAR-avaintutkimuksessa. STELLAR oli kaksoissokkoutettu, lumekontrolloitu, rinnakkaisryhmillä toteutettu kliininen monikeskustutkimus, johon osallistui 323 keuhkovaltimoverenpainetautia (WHO-toimintakykyluokka II tai III) sairastavaa potilasta. Potilaat satunnaistettiin suhteessa 1:1 saamaan ihon alle 3 viikon välein joko sotaterseptia (aloitusannos 0,3 mg/kg, suurennettiin tavoiteannokseksi 0,7 mg/kg) (n = 163) tai lumelääkettä (n = 160). Pitkän aikavälin kaksoissokkoutetun hoitojakson aikana potilaat jatkoivat siinä ryhmässä, johon heidät oli satunnaistettu, kunnes kaikki potilaat olivat suorittaneet loppuun viikon 24.
Tähän tutkimukseen osallistuneet tutkittavat olivat aikuisia, joiden mediaani-ikä oli 48,0 vuotta (vaihteluväli: 18–82 vuotta), ja heistä 16,7 % oli ≥ 65‑vuotiaita. Painon mediaani oli 68,2 kg (vaihteluväli: 38,0–141,3 kg), 89,2 % tutkittavista oli valkoihoisia, 79,3 % oli taustaltaan muita kuin espanjankielisiä tai latinalaisamerikkalaisia, ja 79,3 % oli naisia. Potilaiden keuhkovaltimoverenpainetauti oli etiologialtaan yleisimmin idiopaattinen primaarinen keuhkovaltimoverenpainetauti (58,5 %), periytyvä primaarinen keuhkovaltimoverenpainetauti (18,3 %) tai sidekudossairauteen liittyvä keuhkovaltimoverenpainetauti (14,9 %), keuhkovaltimoverenpainetauti, joka liittyi yksinkertaiseen synnynnäiseen sydänvikaan, jonka yhteydessä oli korjattu systeemisen verenkierron ja keuhkoverenkierron välisiä oikovirtauksia (5 %) tai lääkkeisiin tai toksiineihin liittyvä keuhkovaltimoverenpainetauti (3,4 %). Keuhkovaltimoverenpainetautidiagnoosista seulontaan kuluneen ajan keskiarvo oli 8,76 vuotta.
Suurin osa tutkittavista sai keuhkovaltimoverenpainetaudin taustahoitona kolmea valmistetta (61,3 %) tai kahta valmistetta (34,7 %), ja yli kolmasosa (39,9 %) tutkittavista sai prostasykliini-infuusiovalmistetta. Tutkittavia, joiden WHO-toimintakykyluokka oli II, oli 48,6 %, ja tutkittavia, joiden toimintakykyluokka oli III, oli 51,4 %. STELLAR-tutkimuksesta suljettiin pois potilaat, joilla oli todettu HIV-infektioon liittyvä keuhkovaltimoverenpainetauti, keuhkovaltimoverenpainetauti, johon liittyi portahypertensio, skistosomiaasiin liittyvä keuhkovaltimoverenpainetauti tai keuhkojen veno-okklusiivinen tauti.
Ensisijainen tehoa koskeva päätetapahtuma oli 6 minuutin kävelytestin (6MWD) tuloksen muutos lähtötilanteesta viikon 24 kohdalla. Sotaterseptihoitoryhmässä lumeryhmän tietojen suhteen korjattu 6MWD-testituloksen muutos lähtötilanteesta oli viikon 24 kohdalla 40,8 metriä (95 %:n luottamusväli: 27,5, 54,1; p < 0,001). Viikon 24 kohdalla todettujen lumeryhmän tietojen suhteen korjattujen 6MWD-testitulosten muutosten mediaani arvioitiin myös alaryhmittäin. Hoitovaikutus oli johdonmukainen sukupuoleen, keuhkovaltimoverenpainetaudin diagnostiseen ryhmään, lähtötilanteessa käytettyyn taustahoitoon, lähtötilanteessa käytettyyn prostasykliini-infuusiohoitoon, WHO-toimintakykyluokkaan ja lähtötilanteen keuhkoverenkierron vastukseen perustuvien alaryhmien välillä.
Toissijaisiin päätetapahtumiin kuuluivat paremmat tulokset usean osa-alueen paranemisen osalta (multicomponent improvement, MCI), keuhkoverenkierron vastuksen pieneneminen, B‑tyypin natriureettisen N‑terminaalisen propeptidin (NT‑proBNP) pitoisuuden pieneneminen, WHO-toimintakykyluokan paraneminen sekä pidempi aika kuolemaan tai ensimmäiseen kliinisen tilan pahenemistapahtumaan.
Usean osa-alueen paraneminen oli ennalta määritelty päätetapahtuma, jonka mittarina oli niiden potilaiden osuus, joiden osalta oli viikon 24 kohdalla saavutettu lähtötilanteeseen verrattuna kaikki seuraavat kolme kriteeriä: 6MWD-testituloksen paraneminen (piteneminen ≥ 30 m), NT‑proBNP-pitoisuuden paraneminen (NT‑proBNP-pitoisuuden pieneneminen ≥ 30 % tai NT‑proBNP-pitoisuuden pysyminen tasolla < 300 ng/l tai tämän tason saavuttaminen) ja WHO-toimintakykyluokan paraneminen tai WHO-toimintakykyluokan II säilyminen.
Taudin etenemisen mittarina oli aika kuolemaan tai ensimmäiseen kliinisen tilan pahenemistapahtumaan. Kliinisen tilan pahenemistapahtumia olivat joutuminen keuhkon- ja/tai sydämensiirtojonoon tilan pahenemisen vuoksi, tarve aloittaa oirelääkitys hyväksytyllä keuhkovaltimoverenpainetaudin hoitoon käytettävällä valmisteella tai tarve suurentaa prostasykliini-infuusion annosta ≥ 10 %, eteisseptostomian tarve, sairaalahoito (≥ 24 tuntia) keuhkovaltimoverenpainetaudin pahenemisen vuoksi ja keuhkovaltimoverenpainetaudin paheneminen (WHO-toimintakykyluokan huononeminen ja 6MWD-testituloksen pieneneminen ≥ 15 % riippumatta siitä, ilmenivätkö nämä tapahtumat samaan aikaan tai eri aikoina). Tietoja kliinisen tilan pahenemistapahtumista ja kuolemista kerättiin, kunnes viimeinen potilas oli ollut viikon 24 vastaanottokäynnillä (tiedot tiedonkeruun päättymispäivään asti; altistuksen keston mediaani 33,6 viikkoa).
Viikon 24 kohdalla usean osa-alueen paranemista havaittiin 38,9 %:lla sotaterseptihoitoa saaneista potilaista vs. 10,1 %:lla lumeryhmästä (p < 0,001). Keuhkoverenkierron vastuksen osalta sotatersepti- ja lumehoidon välisen eron mediaani oli -234,6 dyn*s/cm5 (95 %:n luottamusväli: -288,4, -180,8; p < 0,001). NT-proBNP-pitoisuuden osalta sotatersepti- ja lumehoidon välisen eron mediaani oli -441,6 pg/ml (95 %:n luottamusväli: -573,5, -309,6; p < 0,001). WHO-toimintakykyluokka parani lähtötilanteesta 29 %:lla sotaterseptiryhmän potilaista ja 13,8 %:lla lumeryhmän potilaista (p < 0,001).
Sotaterseptihoito vähensi kuolemantapauksia ja kliinisen tilan pahenemistapahtumia 82 % (riskitiheyksien suhde [HR]: 0,182, 95 %:n luottamusväli: 0,075, 0,441; p < 0,001) lumelääkkeeseen verrattuna (ks. taulukko 4). Sotaterseptin hoitovaikutus lumelääkkeeseen verrattuna alkoi viikkoon 10 mennessä ja jatkui tutkimuksen keston ajan.
Taulukko 4: Kuolema tai kliinisen tilan pahenemistapahtumat STELLAR-tutkimuksessa
| Sotatersepti | Lumelääke |
Niiden tutkittavien kokonaismäärä, jotka kuolivat tai joilla ilmeni ainakin yksi kliinisen tilan pahenemistapahtuma, n (%) | 7 (4,3) | 29 (18,1) |
Kuoleman tai kliinisen tilan ensimmäisen pahenemistapahtuman arviointi*, n (%) |
|
|
Kuolema | 2 (1,2) | 6 (3,8) |
Joutuminen keuhkon- ja/tai sydämensiirtojonoon tilan pahenemisen vuoksi | 1 (0,6) | 1 (0,6) |
Eteisseptostomian tarve | 0 (0,0) | 0 (0,0) |
Keuhkovaltimoverenpainetautiin liittyvä sairaalahoito (≥ 24 tuntia) | 0 (0,0) | 8 (5,0) |
Keuhkovaltimoverenpainetaudin paheneminen† | 4 (2,5) | 15 (9,4) |
* Tutkittavalta voi olla kirjattuna useampia kuin yksi arviointi ensimmäisestä kliinisen tilan pahenemistapahtumasta. Tutkittavia, joilta oli kirjattuna useampia kuin yksi arviointi ensimmäisestä kliinisen tilan pahenemistapahtumasta, oli lumelääkettä saaneista 2 ja sotaterseptia saaneista ei yhtään. Analyysista suljettiin pois komponentti ”tarve aloittaa oirelääkitys hyväksytyllä keuhkovaltimoverenpainetaudin hoitoon käytettävällä valmisteella tai tarve suurentaa prostasykliini-infuusion annosta vähintään 10 %”.
† Keuhkovaltimoverenpainetaudin paheneminen määritellään molempien seuraavien tapahtumien ilmenemisenä samaan aikaan, vaikka ne olisivat alkaneet eri aikoina, verrattuna lähtötilanteeseen: (a) WHO-toimintakykyluokan huononeminen (esim. luokasta II luokkaan III, luokasta III luokkaan IV tai luokasta II luokkaan IV); ja (b) 6MWD-testituloksen pieneneminen ≥ 15 % (vahvistettuna kahdella 6MWT-testillä, joiden välillä on vähintään 4 tuntia ja enintään 1 viikko).
N = tutkittavien määrä koko analyysijoukossa; n = tutkittavien määrä kategoriassa. Prosentuaaliset osuudet on laskettu seuraavasti: (n/N) * 100.
ZENITH
Sotaterseptin tehoa arvioitiin ZENITH-tutkimuksessa keuhkovaltimoverenpainetautia sairastavilla aikuisilla potilailla, joiden WHO-toimintakykyluokka oli III tai IV ja joilla kuolleisuusriski oli suuri. ZENITH oli kaksoissokkoutettu, lumekontrolloitu, rinnakkaisryhmillä toteutettu kliininen monikeskustutkimus, johon osallistui 172 potilasta. Potilaat satunnaistettiin suhteessa 1:1 saamaan ihon alle 3 viikon välein joko sotaterseptia (aloitusannos 0,3 mg/kg, suurennettiin tavoiteannokseksi 0,7 mg/kg) (n = 86) tai lumelääkettä (n = 86). Potilaat, joilla ei ilmennyt ensisijaisen yhdistelmäpäätetapahtuman tapahtumaa, jatkoivat kaksoissokkoutettua lumekontrolloitua hoitojaksoa. Potilailla, joilla ilmeni keuhkovaltimoverenpainetaudin pahenemiseen liittyvä, vähintään 24 tuntia kestänyt sairaalahoitotapahtuma, oli sen sijaan mahdollisuus osallistua SOTERIA-tutkimukseen, joka on avoin pitkän aikavälin seurantatutkimus.
ZENITH-tutkimukseen osallistuneet tutkittavat olivat aikuisia, joiden mediaani-ikä oli 57,5 vuotta (vaihteluväli: 18–75 vuotta), ja heistä 29,1 % oli ≥ 65-vuotiaita. 86,6 % tutkittavista oli valkoihoisia, 87,8 % oli taustaltaan muita kuin espanjankielisiä tai latinalaisamerikkalaisia, ja 76,7 % oli naisia. Tutkimuksen osallistujien keuhkovaltimoverenpainetauti oli etiologialtaan idiopaattinen primaarinen keuhkovaltimoverenpainetauti (50,0 %), sidekudossairauteen liittyvä keuhkovaltimoverenpainetauti (27,9 %), periytyvä primaarinen keuhkovaltimoverenpainetauti (10,5 %), lääkkeisiin tai toksiineihin liittyvä keuhkovaltimoverenpainetauti (6,4 %) tai keuhkovaltimoverenpainetauti, joka liittyi korjattuihin synnynnäisiin oikovirtauksiin (5,2 %). Keuhkovaltimoverenpainetautidiagnoosista seulontaan kuluneen ajan keskiarvo oli 7,68 vuotta. Tutkittavat saivat keuhkovaltimoverenpainetaudin taustahoitona kolmea valmistetta (72,1 %) tai kahta valmistetta (27,9 %), ja 59,3 % tutkittavista sai prostasykliini-infuusiovalmistetta. Tutkittavia, joiden WHO-toimintakykyluokka oli III, oli 74,4 %, ja tutkittavia, joiden WHO-toimintakykyluokka oli IV, oli 25,6 %. REVEAL Lite 2 -riskipistemäärä oli 2,3 %:lla tutkittavista < 9, 67,4 %:lla tutkittavista 9–10 ja 30,2 %:lla tutkittavista ≥ 11. ZENITH-tutkimuksesta suljettiin pois potilaat, joilla oli todettu HIV-infektioon liittyvä keuhkovaltimoverenpainetauti, keuhkovaltimoverenpainetauti, johon liittyi portahypertensio, keuhkojen veno-okklusiivinen tauti, keuhkojen kapillaarinen hemangiomatoosi tai ilmeisiä merkkejä hiussuoni- ja/tai laskimoaffisiosta.
Ensisijainen tehoa koskeva päätetapahtuma oli aika ensimmäiseen tapahtumaan, joka oli mistä tahansa syystä johtunut kuolema, keuhkonsiirto tai keuhkovaltimoverenpainetaudin pahenemiseen liittyvä vähintään 24 tuntia kestänyt sairaalahoito. Sotaterseptihoito pienensi ensisijaisen yhdistelmäpäätetapahtuman ensimmäisen tapahtuman esiintyvyyttä 76 % (riskitiheyksien suhde [HR]: 0,24; 95 %:n luottamusväli: 0,13, 0,43; p < 0,0001) (ks. taulukko 5). Ensisijaisen yhdistelmäpäätetapahtuman tapahtumia ilmeni pienemmällä määrällä tutkittavia sotaterseptihoitoryhmässä (15:llä [17,4 %]) kuin lumeryhmässä (47:llä [54,7 %]) tiedonkeruun päättymispäivään mennessä.
Mediaaniaika ensimmäiseen tapahtumaan oli 9,6 kuukautta (95 %:n luottamusväli: 6,2, 14,8) lumeryhmässä. Kaplan–Meier-käyrät alkoivat erkaantua toisistaan noin viikon 5 kohdalla, ja ero suureni jäljellä olevan tutkimusjakson ajan (ks. kuva 1). Hoitovaikutus oli johdonmukainen eri alaryhmissä, jotka perustuivat ikään, sukupuoleen, keuhkovaltimoverenpainetaudin alatyyppiin (sidekudossairauteen liittyvä vs. sidekudossairauteen liittymätön), WHO-toimintakykyluokkaan, lähtötilanteessa käytettyyn taustahoitoon (kaksi vs. kolme valmistetta), lähtötilanteessa käytettyyn prostasykliini-infuusiohoitoon, lähtötilanteen keuhkoverenkierron vastukseen ja lähtötilanteen eGFR-arvoon.
Taulukko 5: Ensisijaisen päätetapahtuman komponentit ZENITH-tutkimuksessa
| Sotatersepti (N = 86) n (%) | Lumelääke (N = 86) n (%) | Riskitiheyksien suhde (95 %:n luottamusväli) p‑arvoa |
Niiden tutkittavien määrä (%), joilla ilmeni ≥ 1 ensisijainen tapahtuma ZENITH-tutkimuksen aikana tai sen jälkeen Ensisijaisen yhdistelmäpäätetapahtuman ensimmäinen tapahtuma, komponenttien mukaanb | 15 (17,4)
6 (7,0) | 47 (54,7)
3 (3,5) | 0,24 (0,13, 0,43)
|
Tutkittavat, joilla ilmeni mikä tahansa yhdistelmäpäätetapahtuman komponenttien tapahtumad |
7 (8,1) |
13 (15,1) |
|
a Ensisijaisen yhdistelmäpäätetapahtuman analyysi sisältää ensimmäiset vahvistetut sairastuvuus- ja kuolleisuustapahtumat tiedonkeruun päättymispäivään mennessä. Analyysiin sisältyvät kaikki kuolemantapaukset ennen tiedonkeruun päättymispäivää riippumatta siitä, olivatko ne vahvistettuja ja ilmenivätkö ne ZENITH-tutkimuksen aikana tai sen jälkeen.
b Jos tutkittavalla ilmeni > 1 komponentti, vain ensimmäisenä ilmennyt komponentti otettiin huomioon.
c Sisältää kaikki tiedonkeruun päättymispäivään mennessä tapahtuneet kuolemat lukuun ottamatta niitä, jotka tapahtuivat keuhkonsiirron tai SOTERIA-tutkimuksessa aloittamisen jälkeen.
d Ensisijaisen yhdistelmäpäätetapahtuman kukin komponentti on esitetty itsenäisenä tuloksena. Jos tutkittavalla todettiin useita ensisijaisen päätetapahtuman määritelmän täyttäviä tapahtumia, kyseisen tutkittavan tietoja on useammalla kuin yhdellä rivillä.
e Vastaa ZENITH-tutkimuksen ensimmäistä toissijaista päätetapahtumaa.
Kuva 1: ZENITH-tutkimuksessa todettu aika ensimmäiseen tapahtumaan, joka oli mistä tahansa syystä johtunut kuolema, keuhkonsiirto tai keuhkovaltimoverenpainetaudin pahenemiseen liittyvä vähintään 24 tuntia kestänyt sairaalahoito – Kaplan–Meier-kuvaaja
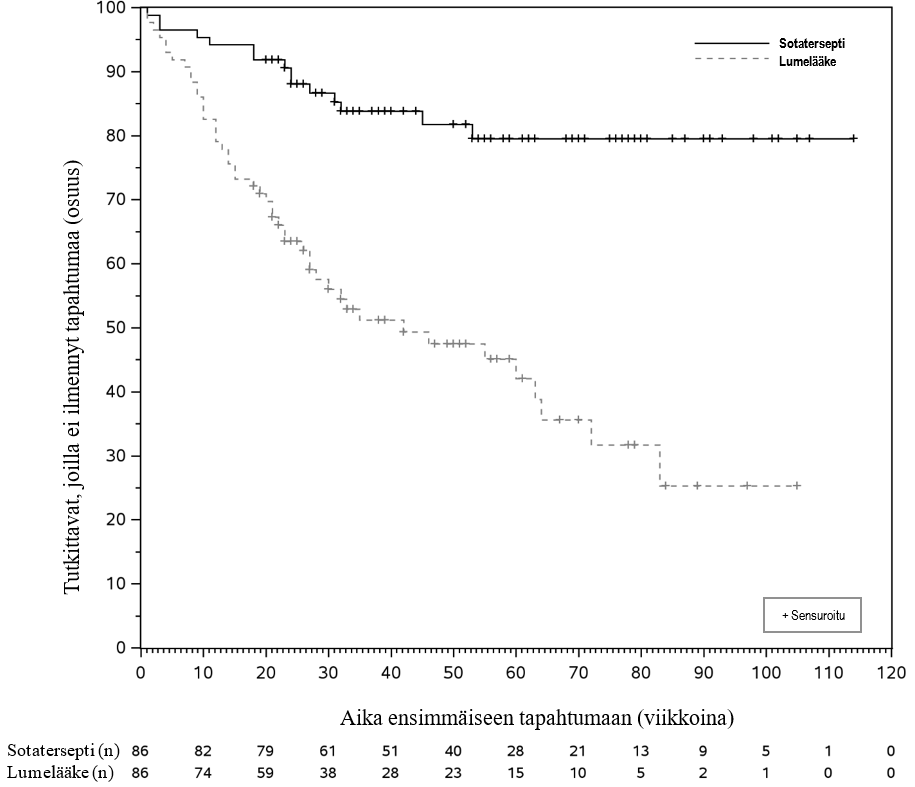
n = niiden tutkittavien määrä, joita riski koskee
Ensisijaista päätetapahtumaa koskevien tulosten perusteella tutkimus lopetettiin hyvän tehon vuoksi välianalyysin kohdalla. Hierarkkisessa testausmenettelyssä ensimmäisen toissijaisen päätetapahtuman eli kokonaiselossaolon primaarianalyysi sisälsi kaikki tiedonkeruun päättymispäivään mennessä tapahtuneet kuolemat lukuun ottamatta niitä, jotka tapahtuivat keuhkonsiirron tai pitkän aikavälin seurantatutkimuksessa aloittamisen jälkeen (ks. taulukko 5). Piste-estimaatti kokonaiselossaoloajan riskitiheyksien suhteelle oli sotaterseptihoitoryhmässä parempi kuin lumeryhmässä (riskitiheyksien suhde: 0,42; 95 %:n luottamusväli: 0,17, 1,07; p = 0,0313), mutta välianalyysin kohdalla tilastollisen merkitsevyyden rajaa (p < 0,0021) ei ollut saavutettu.
Muita toissijaisia päätetapahtumia olivat pidentynyt elossaoloaika ilman elinsiirtoa, NT proBNP-pitoisuuden pieneneminen, keuhkovaltimopaineen keskiarvon pieneneminen, keuhkoverenkierron vastuksen pieneneminen, 6MWD-testituloksen paraneminen, sydämen minuuttitilavuuden suureneminen ja WHO-toimintakykyluokan paraneminen.
Piste-estimaatti elossaoloajalle ilman elinsiirtoa oli sotaterseptihoitoryhmässä parempi kuin lumeryhmässä (riskitiheyksien suhde: 0,34; 95 %:n luottamusväli: 0,15, 0,78). Viikon 24 kohdalla sotatersepti- ja lumehoidon välisen eron mediaani oli NT proBNP-pitoisuuden osalta -2 339,1 pg/ml (95 %:n luottamusväli: -3 378,7, -1 299,4). Keuhkovaltimopaineen keskiarvon osalta sotatersepti- ja lumehoidon välisen eron mediaani oli -21,2 mmHg (95 %:n luottamusväli: -27,8, -14,6). Keuhkoverenkierron vastuksen osalta sotatersepti- ja lumehoidon välisen eron mediaani oli -339,6 dyn*s/cm5 (95 %:n luottamusväli: -511,1, -168,1). 6MWD-testitulosten osalta sotatersepti- ja lumehoidon välisen eron mediaani oli 63,0 m (95 %:n luottamusväli: 23,2, 102,7). Sydämen minuuttitilavuuden osalta sotatersepti- ja lumehoidon välisen eron mediaani oli 0,5 l/min (95 %:n luottamusväli: -0,2, 1,2). WHO-toimintakykyluokka parani lähtötilanteesta 55,8 %:lla sotaterseptiryhmän potilaista ja 27,9 %:lla lumeryhmän potilaista.
HYPERION
Sotaterseptin tehoa arvioitiin HYPERION-tutkimuksessa aikuisilla potilailla, joilla oli vastadiagnosoitu keuhkovaltimoverenpainetauti (WHO-toimintakykyluokka II tai III, diagnoosi enintään 12 kuukautta ennen seulontaa) ja joilla oli keskisuuri tai suuri taudin etenemisen riski. HYPERION oli maailmanlaajuinen, kaksoissokkoutettu, lumekontrolloitu kliininen tutkimus, johon osallistui 320 potilasta. Potilaat satunnaistettiin suhteessa 1:1 saamaan ihon alle 3 viikon välein joko sotaterseptia (aloitusannos 0,3 mg/kg, suurennettiin tavoiteannokseksi 0,7 mg/kg) (n = 160) tai lumelääkettä (n = 160). HYPERION lopetettiin ennenaikaisesti ZENITH-tutkimuksen välianalyysistä saatujen positiivisten tulosten ja sotaterseptin kliinisestä tutkimusohjelmasta saatujen kokonaistietojen arvioinnin perusteella.
Tähän tutkimukseen osallistuneet tutkittavat olivat aikuisia, joiden mediaani-ikä oli 60,0 vuotta (vaihteluväli: 18–88 vuotta), ja heistä 40,6 % oli ≥ 65‑vuotiaita. 86,3 % tutkittavista oli valkoihoisia, 76,6 % oli taustaltaan muita kuin espanjankielisiä tai latinalaisamerikkalaisia ja 72,5 % oli naisia. Tutkimuksen osallistujien keuhkovaltimoverenpainetauti oli etiologialtaan idiopaattinen primaarinen keuhkovaltimoverenpainetauti (59,4 %), sidekudossairauteen liittyvä keuhkovaltimoverenpainetauti (30,3 %), periytyvä primaarinen keuhkovaltimoverenpainetauti (5,9 %), lääkkeisiin tai toksiineihin liittyvä keuhkovaltimoverenpainetauti (2,5 %) tai keuhkovaltimoverenpainetauti, joka liittyi yksinkertaiseen synnynnäiseen sydänvikaan, jonka yhteydessä oli korjattu systeemisen verenkierron ja keuhkoverenkierron välisiä oikovirtauksia (oikovirtausten korjauksesta kulunut vähintään 1 vuosi [1,9 %]). Keuhkovaltimoverenpainetautidiagnoosista kuluneen ajan keskiarvo oli 0,7 vuotta. Kaikki tutkittavat saivat keuhkovaltimoverenpainetaudin taustahoitona joko kahta valmistetta (72,2 %) tai kolmea valmistetta (27,8 %), ja 16,6 % tutkittavista sai prostasykliini-infuusiovalmistetta. Tutkittavia, joiden WHO-toimintakykyluokka oli II, oli 21,3 %, ja tutkittavia, joiden WHO-toimintakykyluokka oli III, oli 78,8 %. REVEAL Lite 2 ‑riskipistemäärä oli 20,6 %:lla tutkittavista < 6, 50,9 %:lla tutkittavista 6–7 ja 28,1 %:lla tutkittavista ≥ 8. COMPERA 2 ‑riskipistemäärä oli 1,9 %:lla tutkittavista matala, 64,1 %:lla tutkittavista lievästi kohonnut, 32,2 %:lla tutkittavista selvästi kohonnut ja 1,6 %:lla tutkittavista korkea, ja riskipistemäärä puuttui 0,3 %:lta tutkittavista. HYPERION-tutkimuksesta suljettiin pois potilaat, joilla oli todettu HIV-infektioon liittyvä keuhkovaltimoverenpainetauti, keuhkovaltimoverenpainetauti, johon liittyi portahypertensio, skistosomiaasiin liittyvä keuhkovaltimoverenpainetauti, keuhkojen veno-okklusiivinen tauti tai keuhkojen kapillaarinen hemangiomatoosi.
Ensisijainen tehoa koskeva päätetapahtuma oli aika kliinisen tilan pahenemiseen (TTCW), joka määriteltiin ajaksi kuolemaan tai ensimmäiseen vahvistettuun sairastuvuustapahtumaan. Tämä päätetapahtuma käsitti seuraavat tapahtumat: mistä tahansa syystä johtunut kuolema, keuhkovaltimoverenpainetaudin pahenemiseen liittyvä vähintään 24 tuntia kestänyt suunnittelematon sairaalahoito, eteisseptostomia, keuhkonsiirto ja 6MWD-testituloksen huononeminen lähtötilanteesta yhdistettynä ainakin yhteen seuraavista: WHO-toimintakykyluokan huononeminen, sydämen oikean puolen vajaatoiminnan pahenemisen merkit tai oireet, keuhkovaltimoverenpainetaudin uuden taustahoidon aloittaminen tai siirtyminen parenteraalisen antoreitin käyttöön.
Sotaterseptihoitoryhmässä kliinisen tilan ensimmäisen pahenemistapahtuman riski oli 76 % pienempi kuin lumeryhmässä (riskitiheyksien suhde: 0,24; 95 %:n luottamusväli: 0,14, 0,41; p < 0,0001) (ks. taulukko 6). Ensisijaisen päätetapahtuman tapahtumia ilmeni pienemmällä määrällä tutkittavia sotaterseptiryhmässä (17:llä [10,6 %]) kuin lumeryhmässä (59:llä [36,9 %]).
Mediaaniaika kliinisen tilan pahenemiseen oli 23,0 kuukautta (95 %:n luottamusväli: 17,3, ei saavutettu) lumeryhmässä. Kaplan–Meier-käyrät erkaantuivat toisistaan varhain. Erkaantuminen alkoi noin viikon 5 kohdalla, ja ero suureni jäljellä olevan tutkimusjakson ajan. (Ks. kuva 2.) Hoitovaikutus oli johdonmukainen eri alaryhmissä: ikä, sukupuoli, keuhkovaltimoverenpainetaudin alatyyppi (idiopaattinen primaarinen keuhkovaltimoverenpainetauti, sidekudossairauteen liittyvä keuhkovaltimoverenpainetauti), keuhkovaltimoverenpainetaudin taustahoito (kaksi vs. kolme valmistetta), WHO-toimintakykyluokka, prostasykliini-infuusiohoito vs. ei prostasykliini-infuusiohoitoa, keuhkoverenkierron vastus, eGFR, REVEAL Lite 2 -pistemäärä, lievästi kohonnut vs. selvästi kohonnut COMPERA 2 -riskipistemäärä.
Taulukko 6: Ensisijaisen päätetapahtuman komponentit HYPERION-tutkimuksessa
| Sotatersepti (N = 160) n (%) | Lumelääke (N = 160) n (%) | Riskitiheyksien suhde (95 %:n luottamusväli) p‑arvo |
Niiden tutkittavien määrä (%), joilla ilmeni ≥ 1 ensisijainen tapahtuma HYPERION-tutkimuksen aikana tai sen jälkeen* | 17 (10,6) | 59 (36,9) | 0,24 (0,14, 0,41) < 0,0001 |
Ensisijaisen yhdistelmäpäätetapahtuman ensimmäinen tapahtuma, komponenttien mukaan† |
|
|
|
Mistä tahansa syystä johtunut kuolema | 7 (4,4) | 5 (3,1) |
|
Keuhkovaltimoverenpainetautiin liittyvä > 24 tuntia kestänyt suunnittelematon sairaalahoito | 2 (1,3) | 12 (7,5) |
|
Eteisseptostomia | 0 | 0 |
|
Keuhkonsiirto | 0 | 0 |
|
Fyysisen suorituskyvyn heikkeneminen keuhkovaltimoverenpainetaudin vuoksi | 8 (5,0) | 46 (28,8) |
|
Tutkittavat, joilla ilmeni mikä tahansa yhdistelmäpäätetapahtuman komponenttien tapahtuma‡ |
|
|
|
Mistä tahansa syystä johtunut kuolema | 7 (4,4) | 6 (3,8) |
|
Keuhkovaltimoverenpainetautiin liittyvä ≥ 24 tuntia kestänyt suunnittelematon sairaalahoito | 3 (1,9) | 14 (8,8) |
|
Eteisseptostomia | 0 | 0 |
|
Keuhkonsiirto | 0 | 0 |
|
Fyysisen suorituskyvyn heikkeneminen keuhkovaltimoverenpainetaudin vuoksi§ | 8 (5,0) | 46 (28,8) |
|
* Ensisijaisen yhdistelmäpäätetapahtuman analyysi sisältää ensimmäiset vahvistetut kliinisen tilan pahenemistapahtumat. Analyysiin sisältyvät kaikki kuolemantapaukset riippumatta siitä, olivatko ne vahvistettuja ja ilmenivätkö ne HYPERION-tutkimuksen aikana vai sen jälkeen, lukuun ottamatta niitä, jotka ilmenivät SOTERIA-tutkimuksessa aloittamisen jälkeen.
† Jos tutkittavalla ilmeni > 1 komponentti, vain ensimmäisenä ilmennyt komponentti otettiin huomioon. Jos tutkittavalla ilmeni useita komponentteja samanaikaisesti ensimmäisenä tapahtumana, jokainen näistä komponenteista otettiin huomioon.
‡ Ensisijaisen yhdistelmäpäätetapahtuman kukin komponentti on esitetty itsenäisenä tuloksena. Jos tutkittavalla todettiin useita ensisijaisen päätetapahtuman määritelmän täyttäviä tapahtumia, kyseisen tutkittavan tietoja on useammalla kuin yhdellä rivillä.
§ Määritelmänä oli 6MWD-testituloksen pieneneminen seulontavaiheen tulosten keskiarvosta kahdessa peräkkäisessä testissä (joiden välillä on vähintään 4 tuntia) ja ainakin yksi seuraavista: WHO-toimintakykyluokan huononeminen, sydämen oikean puolen vajaatoiminnan pahenemisen merkit tai oireet tai keuhkovaltimoverenpainetaudin taustahoidon lisääminen tai muuttaminen.
Kuva 2: HYPERION-tutkimuksessa todettu aika ensimmäiseen kliinisen tilan pahenemistapahtumaan –Kaplan–Meier-kuvaaja
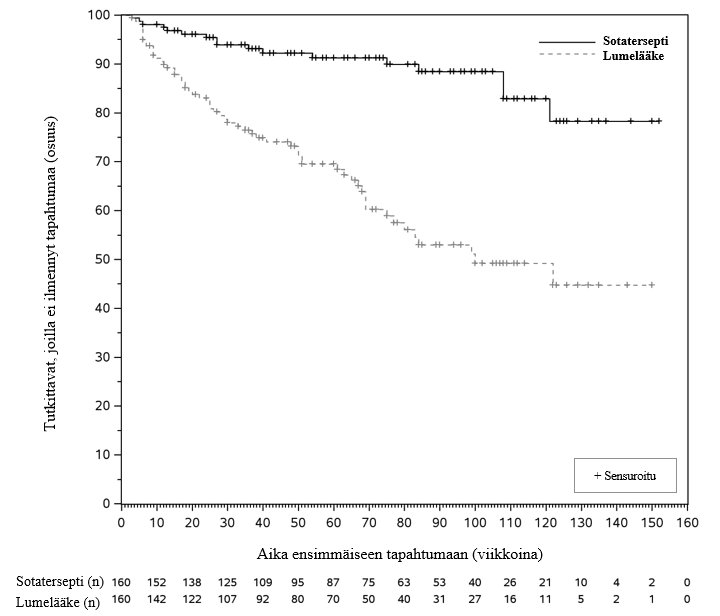
n = niiden tutkittavien määrä, joita riski koskee
Toissijaisiin päätetapahtumiin kuuluivat paremmat tulokset usean osa-alueen paranemisen (MCI-päätetapahtuma) osalta, NT‑proBNP-pitoisuuden pieneneminen, WHO-toimintakykyluokan paraneminen ja 6MWD-testituloksen paraneminen.
MCI-päätetapahtuman mittarina oli niiden tutkittavien osuus, joiden osalta oli viikon 24 kohdalla saavutettu lähtötilanteeseen verrattuna kaikki seuraavat tavoitteet: 6MWD-testituloksen paraneminen (kävelymatkan piteneminen ≥ 30 m), NT-proBNP-pitoisuuden paraneminen (NT-proBNP-pitoisuuden pieneneminen ≥ 30 % tai NT-proBNP-pitoisuuden pysyminen tasolla < 300 ng/l tai tämän tason saavuttaminen) ja WHO-toimintakykyluokan paraneminen tai WHO-toimintakykyluokan II säilyminen. Todettu tutkittavien osuus oli MCI-päätetapahtuman jokaisen yksittäisen komponentin osalta suurempi sotaterseptiryhmässä kuin lumeryhmässä. Niiden tutkittavien osuus, jotka täyttivät kaikki kolme MCI-päätetapahtuman kriteeriä, oli sotaterseptiryhmässä merkitsevästi suurempi (29,4 %) kuin lumeryhmässä (14,6 %) (p = 0,003). Viikon 24 kohdalla sotatersepti- ja lumehoidon välisen eron mediaani oli 6MWD-testitulosten osalta 21,4 m (95 %:n luottamusväli: 4,65, 38,24). NT-proBNP-pitoisuuden osalta sotatersepti- ja lumehoidon välisen eron mediaani oli -308,6 pg/ml (95 %:n luottamusväli: -434,84, -182,37). WHO-toimintakykyluokka parani lähtötilanteesta 55,2 %:lla sotaterseptiryhmän potilaista ja 38,9 %:lla lumeryhmän potilaista.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Winrevair-valmisteen käytöstä keuhkovaltimoverenpainetaudin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Kun keuhkovaltimoverenpainetautia sairastaville potilaille annettiin hoitoa vaiheen 2 ja vaiheen 3 PULSAR-, SPECTRA- ja STELLAR-tutkimuksissa annoksella 0,7 mg/kg 3 viikon välein, vakaan tilan AUC-arvon geometrinen keskiarvo (variaatiokerroin prosentteina [CV%]) oli 171,3 mikrog×d/ml (34,2 %) ja vakaan tilan huippupitoisuuden (Cmax) geometrinen keskiarvo (variaatiokerroin prosentteina [CV%]) oli 9,7 mikrog/ml (30 %). Sotaterseptin AUC ja Cmax suurenevat suhteessa annokseen. Vakaa tila saavutetaan noin 15 hoitoviikon kuluessa. Sotaterseptin AUC:n kumulaatiosuhde oli noin 2,2. Sotaterseptialtistus keuhkovaltimoverenpainetautia sairastavilla tutkittavilla vaiheen 3 ZENITH-tutkimuksessa vastasi edellä kuvattuja tietoja.
Imeytyminen
Ihon alle annettavan lääkemuodon absoluuttinen hyötyosuus on populaatiofarmakokineettisen analyysin perusteella noin 66 %. Sotaterseptin huippupitoisuuden saavuttamiseen kuluvan ajan (tmax) mediaani oli noin 7 päivää (vaihteluväli 2–8 päivää), kun valmistetta annettiin toistuvasti 4 viikon välein.
Jakautuminen
Sotaterseptin sentraalinen jakautumistilavuus (CV%) on noin 3,6 l (24,7 %). Perifeerinen jakautumistilavuus (CV%) on noin 1,7 l (73,3 %).
Biotransformaatio
Sotatersepti kataboloituu tavanomaisilla proteiinien hajoamisprosesseilla.
Eliminaatio
Sotaterseptin puhdistuma on noin 0,18 l/vrk. Terminaalisen puoliintumisajan geometrinen keskiarvo (CV%) on noin 21 vuorokautta (33,8 %).
Erityisryhmät
Ikä, sukupuoli ja etninen tausta
Sotaterseptin farmakokinetiikassa ei havaittu kliinisesti merkittäviä ikään (18–81 vuotta), sukupuoleen tai etniseen taustaan (82,9 % valkoihoisia, 3,1 % mustaihoisia, 7,1 % aasialaisia ja 6,9 % muita) perustuvia eroja.
Paino
Sotaterseptin puhdistuma ja sentraalinen jakautumistilavuus ovat sitä suuremmat, mitä suurempi on potilaan paino. Suositeltu painoon perustuva annostusohjelma johtaa yhdenmukaisiin sotaterseptialtistuksiin.
Munuaisten vajaatoiminta
Sotaterseptin farmakokinetiikka oli vastaavanlainen keuhkovaltimoverenpainetautia sairastavilla potilailla, joilla oli lievä tai kohtalainen munuaisten vajaatoiminta (eGFR 30–89 ml/min/1,73 m2), kuin potilailla, joiden munuaiset toimivat normaalisti (eGFR ≥ 90 ml/min/1,73 m2). Vaikea munuaisten vajaatoiminta (eGFR 15–30 ml/min/1,73 m2, n = 3) ei vaikuttanut sotaterseptin farmakokinetiikkaan. Sotaterseptin farmakokinetiikka oli vastaavanlainen myös potilailla, joilla oli keuhkovaltimoverenpainetautiin liittymätön loppuvaiheen munuaissairaus (eGFR < 15 ml/min/1,73 m2), kuin potilailla, joiden munuaiset toimivat normaalisti. Sotatersepti ei poistu verestä hemodialyysilla. Saatavilla on vain vähän tietoa sotaterseptin käytöstä keuhkovaltimoverenpainetautia sairastaville potilaille, joilla on vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2).
Maksan vajaatoiminta
Sotaterseptia ei ole tutkittu keuhkovaltimoverenpainetautia sairastavilla potilailla, joilla on maksan vajaatoiminta (Child–Pughin luokat A–C). Maksan vajaatoiminnan ei odoteta vaikuttavan sotaterseptin metaboliaan, sillä sotatersepti metaboloituu soluissa katabolisesti.
Prekliiniset tiedot turvallisuudesta
Sotaterseptilla ei ole tehty karsinogeenisuus- tai mutageenisuustutkimuksia.
Toistuvan altistuksen aiheuttama toksisuus
Pisimmät toksisuustutkimukset, joissa valmistetta annettiin ihon alle, kestivät rotilla 3 kuukautta ja apinoilla 9 kuukautta. Seuraavia haittavaikutuksia havaittiin rotilla: kiveksen viejätiehyiden / kivesten degeneraatio, lisämunuaisten kongestio/nekroosi, membranoproliferatiivinen munuaiskerästulehdus ja tubulointerstitiaalinen nefriitti. Munuaisten muutokset eivät korjaantuneet 1 kuukauden pituisen toipumisjakson jälkeen. Apinoilla havaittiin haittavaikutuksina muun muassa soluväliaineen suurentunutta määrää kortikomedullaarisen rajan alueella, glomerulusten pienentynyttä kokoa, munuaiskerästulehdusta ja tubulointerstitiaalista nefriittiä. Apinoilla todetut munuaismuutokset olivat osittain korjaantuneet 3 kuukauden toipumisjakson jälkeen. Rotilla ja apinoilla NOAEL-arvo (suurin annos, jolla ei saada merkitsevää vastetta) saavutettiin, kun sotaterseptialtistukset olivat ≤ 2-kertaisia verrattuna kliinisiin altistuksiin, joita todettiin suositellulla enimmäisannoksella ihmisillä. Muita löydöksiä, joita apinoilla esiintyi kliinisen altistuksen raja-arvoilla, olivat tulehdusinfiltraatit maksassa, pernan imukudosdepleetio ja tulehdusinfiltraatit aivokammion suonipunoksessa.
Lisääntymistoksisuus
Naaraiden hedelmällisyyttä koskeneessa tutkimuksessa havaittiin kiimakierron pitenemistä, tiinehtyvyyden heikkenemistä, ennen kiinnittymistä ja kiinnittymisen jälkeen tapahtuneiden alkionmenetysten määrän suurenemista ja elävien poikasten määrän pienenemistä poikueissa. Naaraiden hedelmällisyyteen liittyviä päätetapahtumia koskeva NOAEL-arvo saavutettiin, kun sotaterseptialtistus oli 2-kertainen verrattuna kliiniseen altistukseen (AUC-arvon perusteella), joka todettiin suositellulla enimmäisannoksella ihmisillä.
Uroksilla ilmeni korjaantumattomia histologisia muutoksia kivesten viejätiehyissä, kiveksissä ja lisäkiveksissä. Rotan kivesten histomorfologiset muutokset korreloivat pienentyneeseen hedelmällisyysindeksiin, joka korjaantui 13 viikon pituisen lääkkeettömän jakson aikana. Kivesten histologisiin muutoksiin liittyvää NOAEL-arvoa ei ole varmistettu, ja urosten hedelmällisyyden toiminnallisiin muutoksiin liittyvä NOAEL-arvo vastaa kaksinkertaista systeemistä altistusta ihmiselle suositellun enimmäisannoksen aiheuttamaan kliiniseen altistukseen nähden.
Alkion ja sikiön kehitykseen kohdistuvia toksisia vaikutuksia koskeneissa tutkimuksissa rotilla ja kaneilla ilmeni elävien sikiöiden määrän ja sikiöiden painon pienenemistä, luutumisen viivästymistä sekä sikiöiden resorption ja implantaation jälkeisten alkionmenetysten lisääntymistä. Ainoastaan rotilla havaittiin luuston muutoksia (ylimääräisten kylkiluiden lukumäärä suureni ja rintanikamien ja lannenikamien lukumäärissä tapahtui muutoksia). NOAEL-arvo saavutettiin, kun sotaterseptialtistus oli rotilla kaksinkertainen ja kaneilla 0,4-kertainen ihmiselle suositellun enimmäisannoksen aiheuttamaan kliiniseen altistukseen nähden.
Rotilla tehdyssä pre- ja postnataalista kehitystä koskeneessa tutkimuksessa ei havaittu sotaterseptiin liittyviä haittavaikutuksia ensimmäisen F‑polven (F1) poikasilla, joiden emojen arvioidut altistukset tiineyden aikana olivat enintään 2‑kertaisia verrattuna altistuksiin, joita todettiin suositellulla enimmäisannoksella ihmisillä. F1‑poikasilla, joiden emoille oli annettu valmistetta imetyksen aikana, poikasten painon lasku korreloi sukukypsyyden saavuttamisen viivästymisen kanssa. Poikasten kasvuun ja kehitykseen liittyvä NOAEL-arvo vastaa 0,6-kertaista systeemistä altistusta ihmiselle suositellun enimmäisannoksen aiheuttamaan kliiniseen altistukseen nähden.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Sitruunahappomonohydraatti (E330)
Natriumsitraatti (E331)
Polysorbaatti 80 (E433)
Sakkaroosi
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Käyttökuntoon saattamisen jälkeen
Valmisteen on osoitettu säilyvän käytön aikana biokemiallisesti ja biofysikaalisesti stabiilina 4 tuntia 30 °C:ssa.
Mikrobiologisista syistä lääkevalmiste on käytettävä käyttökuntoon saattamisen jälkeen välittömästi tai viimeistään 4 tunnin kuluttua.
Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja säilytysolosuhteet ennen käyttöä ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2–8 °C). Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
WINREVAIR injektiokuiva-aine ja liuotin, liuosta varten
45 mg (L:ei) 1 kpl (50 mg/ml, 1 liitin, ruisku, neula, 4 antiseptista pyyhettä) (7436,70 €), 2 x 1 kpl (50 mg/ml, 2 liitintä, ruisku, neula, 8 antiseptista pyyhettä) (14692,80 €)
60 mg (L:ei) 1 kpl (50 mg/ml, 1 liitin, ruisku, neula, 4 antiseptista pyyhettä) (9855,41 €)
PF-selosteen tieto
Winrevair 45 mg injektiokuiva-aine ja liuotin, liuosta varten
2 ml:n vetoinen, tyypin I lasista valmistettu injektiopullo, jossa on polymeeripinnoitettu bromibutyylikumitulppa ja alumiinisuljin limenvihreällä irtinapsautettavalla polypropeenikorkilla ja joka sisältää 45 mg sotaterseptia.
Esitäytetty ruisku (tyypin I lasista valmistettu sylinteriampulli, joka on suljettu bromibutyylikumitulpalla), jossa on 1 ml liuotinta.
Winrevair 60 mg injektiokuiva-aine ja liuotin, liuosta varten
2 ml:n vetoinen, tyypin I lasista valmistettu injektiopullo, jossa on polymeeripinnoitettu bromibutyylikumitulppa ja alumiinisuljin viininpunaisella irtinapsautettavalla polypropeenikorkilla ja joka sisältää 60 mg sotaterseptia.
Esitäytetty ruisku (tyypin I lasista valmistettu sylinteriampulli, joka on suljettu bromibutyylikumitulpalla), jossa on 1,3 ml liuotinta.
Winrevair injektiokuiva-aine ja liuotin, liuosta varten on saatavilla seuraavissa pakkauskoissa:
- injektiosetit, jotka sisältävät 1 injektiopullon, jossa on 45 mg kuiva-ainetta, 1 esitäytetyn ruiskun, jossa on 1,0 ml liuotinta, 1 annosteluruiskun, jonka mitta-asteikon tarkkuus on 0,1 ml, 1 injektiopulloliittimen (13 mm), 1 injektioneulan ja 4 antiseptistä pyyhettä
- injektiosetit, jotka sisältävät 2 injektiopulloa, joissa on 45 mg kuiva-ainetta, 2 esitäytettyä ruiskua, joissa on 1,0 ml liuotinta, 1 annosteluruiskun, jonka mitta-asteikon tarkkuus on 0,1 ml, 2 injektiopulloliitintä (13 mm), 1 injektioneulan ja 8 antiseptistä pyyhettä
- injektiosetit, jotka sisältävät 1 injektiopullon, jossa on 60 mg kuiva-ainetta, 1 esitäytetyn ruiskun, jossa on 1,3 ml liuotinta, 1 annosteluruiskun, jonka mitta-asteikon tarkkuus on 0,1 ml, 1 injektiopulloliittimen (13 mm), 1 injektioneulan ja 4 antiseptistä pyyhettä
- injektiosetit, jotka sisältävät 2 injektiopulloa, joissa on 60 mg kuiva-ainetta, 2 esitäytettyä ruiskua, joissa on 1,3 ml liuotinta, 1 annosteluruiskun, jonka mitta-asteikon tarkkuus on 0,1 ml, 2 injektiopulloliitintä (13 mm), 1 injektioneulan ja 8 antiseptistä pyyhettä.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kuiva-aine: valkoinen tai luonnonvalkoinen jauhe.
Liuotin: kirkas väritön injektionesteisiin käytettävä vesi.
Käyttö- ja käsittelyohjeet
Sopivan injektiosetin valinta
Jos potilaan paino edellyttää kahden 45 mg:n tai kahden 60 mg:n injektiopullon käyttöä, tulee käyttää yhtä 2 injektiopullon settiä, ei kahta 1 injektiopullon settiä. Näin potilaalle ei jouduta antamaan useita injektioita (ks. kohta Pakkaukset ja valmisteen kuvaus).
Käyttökuntoon saattamista ja antamista koskevat ohjeet
Winrevair injektiokuiva-aine ja liuotin, liuosta varten, on saatettava käyttökuntoon ennen käyttöä ja annettava kertainjektiona potilaan painon mukaan (ks. kohta Annostus ja antotapa).
Lue erillinen Käyttöohjeet-vihkonen, joka toimitetaan injektiosetin mukana. Siinä on yksityiskohtaiset vaiheittaiset ohjeet lääkevalmisteen valmistelusta ja antamisesta. Seuraavassa on yleiskatsaus käyttökuntoon saattamisesta ja lääkkeenannosta.
Käyttökuntoon saattaminen
- Ota setti jääkaapista ja odota 15 minuuttia, jotta esitäytetty ruisku (esitäytetyt ruiskut) ja lääkevalmiste lämpenevät huoneenlämpöisiksi ennen valmistelua.
- Tarkista injektiopullosta, ettei lääkevalmiste ole vanhentunut. Kuiva-aineen on oltava valkoista tai luonnonvalkoista, ja se saattaa olla kokonaisena tai rikkoutuneena kakkuna.
- Irrota kuiva-ainetta sisältävän injektiopullon kansi ja pyyhi kumitulppa antiseptisellä pyyhkeellä.
- Kiinnitä injektiopulloliitin injektiopulloon.
- Tarkasta silmämääräisesti, ettei esitäytetyssä ruiskussa ole vaurioita tai vuotoja ja että ruiskussa olevassa steriilissä vedessä ei ole näkyviä hiukkasia.
- Irrota esitäytetyn ruiskun korkki taittamalla ja kiinnitä ruisku injektiopulloliittimeen.
-
Ruiskuta kaikki steriili vesi injektiopulloon kiinnitetystä ruiskusta kuiva-ainetta sisältävään injektiopulloon:
- 45 mg:n injektiopullon mukana toimitettava esitäytetty ruisku sisältää 1,0 ml steriiliä vettä
- 60 mg:n injektiopullon mukana toimitettava esitäytetty ruisku sisältää 1,3 ml steriiliä vettä.
Käyttökuntoon saattamisen jälkeen 45 mg:n injektiopullosta saa enintään 0,9 ml:n lääkeannoksen ja 60 mg:n injektiopullosta enintään 1,2 ml:n lääkeannoksen. Lopullinen pitoisuus käyttökuntoon saattamisen jälkeen on 50 mg/ml.
- Pyörittele injektiopulloa varovasti lääkevalmisteen saattamiseksi käyttökuntoon. Älä ravista äläkä sekoita voimakkaasti.
- Anna injektiopullon seistä enintään 3 minuuttia, jotta kuplat häviävät.
- Tarkasta käyttökuntoon saatettu liuos silmämääräisesti. Kun valmiste on sekoitettu oikein, käyttökuntoon saatettu liuos on kirkasta tai opaalinhohtoista ja väritöntä tai heikosti ruskehtavankeltaista, eikä siinä pitäisi olla paakkuja tai kuiva-ainetta.
- Kierrä ruisku irti injektiopulloliittimestä ja hävitä tyhjä ruisku.
- Jos potilaalle on määrätty 2 injektiopullon injektiosetti, valmistele toinen injektiopullo toistamalla tässä kohdassa kuvatut vaiheet.
- Käytä käyttökuntoon saatettu liuos mahdollisimman pian, mutta viimeistään 4 tunnin kuluttua käyttökuntoon saattamisesta.
Annosteluruiskun valmistelu
- Tarkasta käyttökuntoon saatettu liuos silmämääräisesti ennen annosteluruiskun valmistelua. Käyttökuntoon saatetun liuoksen pitäisi olla kirkasta tai opaalinhohtoista ja väritöntä tai heikosti ruskehtavankeltaista, eikä siinä saa olla paakkuja tai kuiva-ainetta.
- Pyyhi injektiopulloliitin antiseptisella pyyhkeellä.
- Ota annosteluruisku pakkauksestaan ja kiinnitä ruisku injektiopulloliittimeen.
-
Käännä ruisku ja injektiopullo ylösalaisin, ja vedä ruiskuun potilaan painon mukaan laskettu tarvittava määrä injektionestettä.
- Jos annos edellyttää kahden injektiopullon käyttöä, vedä ruiskuun koko ensimmäisen injektiopullon sisältö ja lisää se hitaasti toiseen injektiopulloon, jotta saat varmasti oikean annoksen.
- Käännä ruisku ja injektiopullo ylösalaisin ja vedä ruiskuun tarvittava määrä lääkevalmistetta.
- Poista ylimääräinen lääkevalmiste tai ilma ruiskusta tarvittaessa painamalla mäntää.
- Irrota ruisku injektiopulloliittimestä ja kiinnitä neula.
Antaminen
Winrevair annetaan kertainjektiona ihon alle.
-
Valitse pistoskohta vatsasta (kohdan on oltava vähintään 5 cm:n päässä navasta), reiden yläosasta tai olkavarresta ja pyyhi pistoskohta antiseptisella pyyhkeellä. Valitse jokaiselle pistokselle uusi kohta, jossa ei ole arpia, aristusta eikä mustelmia.
- Jos potilas tai häntä hoitava henkilö antaa lääkkeen, hänet on koulutettava pistoksen antamiseen vain vatsaan tai reiden yläosaan (ks. Käyttöohjeet-vihkonen).
- Anna pistos ihon alle.
- Hävitä tyhjä ruisku. Älä käytä ruiskua uudelleen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Biologisten lääkevalmisteiden jäljitettävyyttä koskevat ohjeet, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Korvattavuus
WINREVAIR injektiokuiva-aine ja liuotin, liuosta varten
45 mg 1 kpl, 2 x 1 kpl
60 mg 1 kpl
- Ei korvausta.
ATC-koodi
C02KX06
Valmisteyhteenvedon muuttamispäivämäärä
07.05.2026
Yhteystiedot
 MSD FINLAND OY
MSD FINLAND OY Keilaniementie 1, PL 46
02151 Espoo
09 804 650
www.msd.fi
info@msd.fi