
VOYDEYA tabletti, kalvopäällysteinen 50 mg+100 mg, 100 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Voydeya 50 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 50 mg danikopaania.
Voydeya 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg danikopaania.
Apuaine, jonka vaikutus tunnetaan
Yksi 50 mg:n tabletti sisältää 57,5 mg laktoosia laktoosimonohydraattina.
Yksi 100 mg:n tabletti sisältää 115 mg laktoosia laktoosimonohydraattina.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen.
Kliiniset tiedot
Käyttöaiheet
Voydeya on tarkoitettu ravulitsumabin tai ekulitsumabin lisänä paroksysmaalista nokturnaalista hemoglobinuriaa (PNH) sairastaville aikuisille, joilla on residuaalinen hemolyyttinen anemia (ks. kohta Farmakodynamiikka).
Ehto
Hoito on aloitettava hematologisiin sairauksiin perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Hoidon saa aloittaa vain veritautien hoitoon perehtynyt terveydenhuollon ammattilainen.
Annostus
Suositeltu aloitusannos on 150 mg kolmesti päivässä suun kautta, noin 8 tunnin välein (± 2 tuntia). Annos voidaan suurentaa 200 mg:aan kolmesti päivässä vähintään 4 viikkoa kestäneen hoidon jälkeen kliinisen vasteen perusteella.
Annosten väliin jääminen
Potilaita on neuvottava ottamaan unohtunut annos heti muistettaessa, jollei ole jo melkein seuraavan annoksen aika. Jälkimmäisessä tapauksessa unohtunut annos on jätettävä väliin ja lääkevalmisteen seuraava annos on otettava tavanomaisen aikaan. Potilaita on kiellettävä ottamasta 2 annosta samaan aikaan.
Hoidon lopettaminen
Hoidon lopettamisen jälkeen voi esiintyä alaniiniaminotransferaasin (ALAT) nousua (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), joten jos hoito lopetetaan, annosta on pienennettävä asteittain 6 päivän kuluessa seuraavasti:
- 100 mg:n annos: 100 mg kahdesti päivässä 3 päivän ajan, minkä jälkeen 100 mg kerran päivässä 3 päivän ajan.
- 150 mg:n annos: 100 mg kolmesti päivässä 3 päivän ajan, minkä jälkeen 50 mg kolmesti päivässä 3 päivän ajan.
- 200 mg:n annos: 100 mg kolmesti päivässä 3 päivän ajan, minkä jälkeen 100 mg kahdesti päivässä 3 päivän ajan.
Erityisryhmät
Iäkkäät potilaat
Annosta ei tarvitse muuttaa iäkkäille potilaille. Danikopaanin käytöstä ≥ 65 vuoden ikäisille potilaille on kuitenkin vain vähän kokemusta (ks. kohta Farmakodynamiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa lievää (arvioitu glomerulusten suodatusnopeus [eGFR] ≥ 60 – < 90 ml/min/1,73 m2) tai keskivaikeaa (eGFR ≥ 30 – < 60 ml/min/1,73 m2) munuaisten vajaatoimintaa sairastaville potilaille. Vaikeaa (eGFR < 30 ml/min/1,73 m2) munuaisten vajaatoimintaa sairastaville potilaille suositeltu aloitusannos on 100 mg kolmesti päivässä suun kautta, noin 8 tunnin välein (± 2 tuntia). Annos voidaan suurentaa 150 mg:aan kolmesti päivässä vähintään 4 viikkoa kestäneen hoidon jälkeen kliinisen vasteen perusteella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa lievää (Child-Pugh-luokka A) tai keskivaikeaa (Child-Pugh-luokka B) maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka). Vaikeaa (Child-Pugh-luokka C) maksan vajaatoimintaa sairastavia potilaita koskevia tutkimuksia ei ole tehty, eikä danikopaanin käyttöä siksi suositella tässä potilasryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Voydeya-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Tabletit tulee ottaa ruoan (aterian tai välipalan) kanssa (ks. kohta Farmakokinetiikka).
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Potilaat, joilla on aktiivinen Neisseria meningitidis -infektio hoitoa aloitettaessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Potilaat, joilla ei ole voimassa olevaa rokotussuojaa Neisseria meningitidis -infektiota vastaan, ellei potilas saa ennaltaehkäisevää hoitoa asianmukaisilla antibiooteilla 2 viikon ajan rokotuksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Yleistä tietoa
Danikopaania ei saa antaa monoterapiana, sillä tehoa ei ole varmistettu. Valmistetta saa määrätä ainoastaan ravulitsumabin tai ekulitsumabin lisäksi.
Vakavat infektiot
Meningokokki-infektiot
Potilaat, jotka saavat hoitoa komplementtia inaktivoivilla aineilla, saattavat olla tavallista alttiimpia meningokokki-infektioille (Neisseria meningitidis). Potilaiden rokotesuoja meningokokki-infektioita vastaan on oltava ajan tasalla voimassa olevien kansallisten rokotussuositusten mukaisesti ennen ensimmäisen danikopaaniannoksen saamista.
Jos potilaan hoito aloitetaan ennen kuin meningokokkirokotteen saamisesta on kulunut 2 viikkoa, potilaan on saatava asianmukaista ennaltaehkäisevää antibioottihoitoa 2 viikon ajan rokotuksen jälkeen. Potilaat on rokotettava seroryhmiä A, C, Y ja W135 vastaan yleisesti patogeenisten seroryhmien aiheuttamien meningokokki-infektioiden ehkäisemiseksi. Lisäksi suositellaan rokottamista seroryhmää B vastaan, mikäli saatavilla. Bakteerilääkkeiden asianmukaista käyttöä koskevat viralliset suositukset on huomioitava.
Kaikkia danikopaanihoitoa saavia potilaita on seurattava meningokokki-infektion ja -sepsiksen varhaisten merkkien varalta. Infektiota epäiltäessä potilas on tutkittava välittömästi ja hoito asianmukaisilla antibiooteilla aloitettava. Potilaille on kerrottava näistä merkeistä ja oireista, ja heitä on neuvottava hakeutumaan välittömästi lääkärinhoitoon, jos niitä esiintyy.
Muut vakavat infektiot
Danikopaania on annettava varoen potilaille, joilla on aktiivisia systeemisiä infektioita. Danikopaani estää selektiivisesti komplementin oikotietä aktivoitumasta, joten potilaat saattavat olla tavallista alttiimpia vakaville infektioille (joiden aiheuttaja on jokin muu kuin Neisseria meningitidis). Ennen danikopaanihoidon lisäämistä ravulitsumabi- tai ekulitsumabihoitoon on suositeltavaa, että potilaille aloitetaan voimassa olevien rokotusohjeiden mukaiset rokotukset.
Vaikea munuaisten vajaatoiminta
Jos vaikeaa munuaisten vajaatoimintaa sairastavan potilaan annos suurennetaan 150 mg:aan kolmesti päivässä, potilasta on seurattava haittatapahtumien varalta danikopaanihoidon aikana näillä potilailla odotettavissa olevan suuremman altistuksen vuoksi.
Pieni kehonpaino
Alle 60 kg painavia potilaita on seurattava haittatapahtumien varalta danikopaanihoidon aikana näillä potilailla odotettavissa olevan suuremman altistuksen vuoksi.
Maksaentsyymien nousu
Kliinisissä tutkimuksissa on havaittu alaniiniaminotransferaasin (ALAT) nousua (ks. kohta Haittavaikutukset). Maksaentsyymien mittaaminen ennen hoidon aloittamista on suositeltavaa. Hoidon aloittamisen jälkeen suositellaan tavanomaista, PNH:n hoitosuositusten mukaista laboratorioseurantaa. Hoidon keskeyttämistä tai lopettamista on harkittava, jos maksaentsyymien nousu on kliinisesti merkittävää tai potilaalle ilmaantuu oireita. Danikopaania ei suositella vaikeaa maksan vajatoimintaa sairastaville potilaille (ks. kohta Annostus ja antotapa).
Hoidon lopettaminen
Käytettäessä annoksia, jotka olivat suurempia kuin 200 mg kolmesti päivässä, terveillä henkilöillä esiintyi ALAT-arvojen nousua, kun hoito lopetettiin ilman asteittaista annoksen pienentämistä (ks. kohta Yliannostus). Hoidon lopettamisen yhteydessä annosta on pienennettävä asteittain 6 päivän kuluessa (ks. kohta Annostus ja antotapa).
Apuaineet, joiden vaikutus tunnetaan
Laktoosi
Tämä lääkevalmiste sisältää laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per kalvopäällysteinen tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Danikopaanin vaikutus muihin lääkevalmisteisiin
P-gp:n substraatit
P-gp:n substraatti feksofenadiinin 180 mg:n kerta-annoksen anto suun kautta yhdessä 150 mg:n annoksina kolmesti päivässä annetun danikopaanihoidon kanssa suurensi feksofenadiinin Cmax-arvoa 1,42-kertaisesti ja AUC0-inf-arvoa 1,62-kertaisesti.
Tulokset viittaavat siihen, että danikopaani on P-gp:n heikko estäjä. Varovaisuus saattaa olla tarpeen samanaikaisessa käytössä sellaisten lääkevalmisteiden kanssa, joiden tiedetään olevan P-gp:n substraatteja (kuten dabigatraani, digoksiini, edoksabaani, feksofenadiini, takrolimuusi).
BCRP:n substraatit
BCRP:n substraatti rosuvastatiinin 20 mg:n kerta-annoksen anto suun kautta yhdessä 200 mg:n annoksina kolmesti päivässä annetun danikopaanihoidon kanssa suurensi rosuvastatiinin Cmax-arvoa 3,29-kertaisesti ja AUC0-inf-arvoa 2,25-kertaisesti. Tulokset viittaavat siihen, että danikopaani on BCRP:n estäjä. Varovaisuus saattaa olla tarpeen samanaikaisessa käytössä sellaisten lääkevalmisteiden kanssa, joiden tiedetään olevan BCRP:n substraatteja (kuten rosuvastatiini ja sulfasalatsiini).
Raskaus ja imetys
Raskaus
Danikopaanin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa terapeuttisesti oleellisella annostuksella ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Voydeya-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet danikopaanin ja/tai sen metaboliittien erittyvän maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Voydeya-valmistetta ei pidä käyttää imetyksen aikana, eikä imetystä pidä aloittaa ennen kuin hoidon lopettamisesta on kulunut 3 päivää.
Hedelmällisyys
Danikopaanin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoja saatavilla. Eläimillä tehdyissä tutkimuksissa on havaittu mahdollisia vaikutuksia urosten hedelmällisyyteen ja lisääntymistoimintoihin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Voydeya-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset ovat kuume (28,1 %), päänsärky (25,0 %) ja maksaentsyymien nousu (11,5 %).
Haittavaikutusten taulukkomuotoinen luettelo
Taulukko 1 sisältää danikopaanin kliinisisissä tutkimuksissa raportoidut haittavaikutukset. Haittavaikutukset on lueteltu elinjärjestelmäluokittain, Preferred Terms -termejä käyttäen ja seuraavien MedDRA-yleisyysluokkien mukaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100). Haittavaikutukset on kussakin yleisyysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutusten taulukkomuotoinen luettelo
| MedDRA-elinjärjestelmäluokka | Hyvin yleinen (≥ 1/10) | Yleinen (≥ 1/100, < 1/10) |
| Hermosto | Päänsärky | |
| Verisuonisto | Hypertensio | |
| Ruoansulatuselimistö | Oksentelu | |
| Maksa ja sappi | Maksaentsyymien nousua | |
| Luusto, lihakset ja sidekudos | Raajakipu | |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume |
a Maksaentsyymien nousu käsittää seuraavat Preferred Terms -termit: alaniiniaminotransferaasin nousu, maksan toiminnan poikkeavuudet, maksaentsyymien nousu ja transaminaasien nousu.
Valittujen haittavaikutusten kuvaus
Maksaentsyymien nousu
Tutkimuksen ALXN2040-PNH-301 12 viikon satunnaistetun kontrollijakson aikana
ALAT-arvon nousuun liittyviä laboratorioarvojen poikkeavuuksia havaittiin 14,0 %:lla danikopaania saaneista potilaista. Danikopaaniryhmässä ALAT nousi > 3 – ≤ 5 kertaa viitealueen ylärajaa suuremmaksi 8,8 %:lla potilaista ja > 5 – ≤ 10 kertaa viitealueen ylärajaa suuremmaksi 5,3 %:lla potilaista. Kaikki potilaat olivat oireettomia, ja arvojen nousu oli kaikilla potilailla ohimenevä. Osa nousuista liittyi hemolyysiin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Terveille tutkittaville on annettu enimmillään 1 200 mg:n kerta-annoksia ja enimmillään 800 mg:n annoksia kahdesti päivässä. Hoidon lopettamisen jälkeistä ALAT-arvon nousua, kun hoitoa ei lopetettu asteittain, esiintyi 2 tutkittavalla, jotka saivat 500 mg ja 800 mg kahdesti päivässä 14 päivän ajan. Kaikki poikkeavat ALAT-löydökset olivat itsestään ohimeneviä, eikä niihin liittynyt maksan toiminnan poikkeavuuksia.
Yliannostus saattaa aiheuttaa aminotransferaasien ja muiden maksa-arvojen nousua. Yleistä elintoimintoja tukeva hoitoa suositellaan. Ei tiedetä, poistuuko danikopaani dialyysissa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, komplementin estäjät, ATC-koodi: L04AJ09
Vaikutusmekanismi
Danikopaani sitoutuu palautuvasti komplementtitekijään D (FD) ja estää selektiivisesti sen toimintaa. FD:tä estämällä danikopaani estää selektiivisesti komplementin oikotietä aktivoitumasta, jolloin useiden efektorien – mukaan lukien C3-fragmenttien – tuotanto estyy oikotien aktivoitumisen jälkeen. Kaksi muuta komplementtitietä (klassinen tie ja lektiinitie) pysyvät aktiivisina. Danikonipaanin komplementin oikotietä salpaava vaikutus estää C3-fragmentteja kertymästä PNH-punasoluihin. Tämä kertyminen on keskeinen syy ekstravaskulaariseen hemolyysiin, joka voi muuttua kliinisesti merkittäväksi pienellä osalla komplementti C5:n estohoitoa saavista PNH-potilaista. Komplementti C5:n eston ylläpitäminen ehkäisee PNH:n taustalla olevan komplementin aktivoitumisen henkeä uhkaavia patofysiologisia seurauksia.
Farmakodynaamiset vaikutukset
Kliinisessä tutkimuksessa ravulitsumabi- tai ekulitsumabihoitoa saavilla PNH-potilailla, joilla esiintyi kliinisesti merkittävää ekstravaskulaarista hemolyysia, danikopaani esti odotetusti komplementin oikotien aktiivisuutta, pienensi plasman Bb-pitoisuutta (FD:n aikaansaama komplementtitekijä B:n pilkkoutumistuote) ja vähensi C3-fragmenttien kertymistä kiertäviin PNH-punasoluihin.
Sydämen sähköfysiologia
Danikopaanin suun kautta annetut 400 mg:n, 800 mg:n ja 1 200 mg:n kerta-annokset eivät pidentäneet QTc-aikaa. EKG-tutkimuksissa ei havaittu johdonmukaisia huolta aiheuttavia poikkeavuuksia intervallien tai aaltomuotojen suhteen.
Kliininen teho ja turvallisuus
Danikopaanin tehoa ja turvallisuutta aikuisilla PNH-potilailla, joilla esiintyi kliinisesti merkittävää ekstravaskulaarista hemolyysia, arvioitiin faasin 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (ALXN2040-PNH-301). Tutkimukseen otettiin 86 aneemista PNH-potilasta (hemoglobiini ≤ 95 g/l [5,9 mmol/l]), jotka olivat saaneet ravulitsumabi- tai ekulitsumabihoitoa vakaalla annoksella vähintään edeltävän 6 kuukauden ajan ja joiden absoluuttinen retikulosyyttimäärä oli ≥ 120 × 109/l, ilman verensiirtoja tai verensiirroista huolimatta.
Danikopaania annettiin kohdassa Annostus ja antotapa olevien annostussuositusten mukaisesti (150 mg kolmesti päivässä tai enintään 200 mg kolmesti päivässä kliinisen vasteen perusteella).
Potilaiden rokotushistoria selvitettiin, ja potilaat oli pitänyt rokottaa meningokokki-infektioita vastaan ennen danikopaanihoidon aloittamista. Jos rokotteen saamista tutkimusta edeltävän 3 vuoden aikana ei kyetty vahvistamaan, potilas oli rokotettava danikopaanihoidon alkaessa.
Potilaat satunnaistettiin suhteessa 2:1 saamaan danikopaania tai lumelääkettä kolmesti päivässä 12 viikon ajan ravulitsumabi- tai ekulitsumabitaustahoitonsa lisäksi. Viikon 12 jälkeen kaikki potilaat saivat danikopaania ravulitsumabi- tai ekulitsumabitaustahoidon lisänä viikkoon 24 asti. Hoitojaksojen jälkeen (viikolla 24) potilaille tarjottiin mahdollisuus osallistua pitkäaikaiseen jatkovaiheeseen, jossa danikopaanin antoa jatkettiin ravulitsumabi- tai ekulitsumabitaustahoidon lisänä.
Demografiset ja lähtötilanteen ominaisuudet olivat pääosin tasapainossa hoitoryhmien välillä. PNH-sairaushistoria oli samankaltainen danikopaaniryhmän ja lumelääkeryhmän välillä. Keskimääräinen ikä lähtötilanteessa oli 52,8 vuotta, ja suurin osa potilaista oli naisia (62,8 %). Lähtötilanteen keskimääräinen hemoglobiinipitoisuus oli 77,5 g/l [4,81 mmol/l] ja keskimääräinen retikulosyyttimäärä oli 239,40 × 109/l. Potilaita, joille oli siirretty punasolutiivistettä/kokoverta ensimmäistä annosta edeltävän 24 viikon aikana, oli 76 (88,4 %), ja verensiirtojen keskimääräinen lukumäärä oli 2,6. Keskimääräinen LDH-taso oli 298,13 U/l ja keskimääräiset FACIT-Fatigue-pisteet olivat 33,24. Tutkimukseen otetuista potilaista 51 (59,3 %) sai ravulitsumabia ja 35 (40,7 %) ekulitsumabia.
Ensisijainen päätetapahtuma oli hemoglobiinipitoisuuden muutos lähtötilanteesta viikon 12 loppuun. Toissijaiset päätetapahtumat olivat niiden potilaiden osuus, joiden hemoglobiinipitoisuus viikon 12 kohdalla oli noussut ≥ 20 g/l [1,2 mmol/l] ilman verensiirtoja, niiden potilaiden osuus, jotka eivät olleet tarvinneet verensiirtoja viikon 12 loppuun mennessä, väsymystä mittaavien FACIT-Fatigue-pisteiden muutos lähtötilanteesta viikon 12 kohdalla sekä absoluuttisen retikulosyyttimäärän muutos lähtötilanteesta viikon 12 kohdalla. Verensiirtoja tarvitsemattomiksi katsottiin vain ne potilaat, jotka eivät saaneet verensiirtoja eivätkä täyttäneet tutkimussuunnitelmassa määritettyjä verensiirron suosituskriteerejä lähtötilanteen ja 12 viikon pituisen hoitojakson 1 päättymisen välillä.
Tehoanalyysissa käytetty ensisijainen näyttö perustui etukäteen määritettyyn analyysiin, joka tehtiin, kun ensimmäiset 63 satunnaistettua osallistujaa olivat päättäneet 12 viikon pituisen hoitojakson 1 tai lopettaneet tutkimukseen osallistumisen sitä ennen.
Ravulitsumabi- tai ekulitsumabihoidon lisänä annettu danikopaani oli ravulitsumabi- tai ekulitsumabihoidon lisänä annettua lumelääkettä parempi ensisijaisen päätetapahtuman osalta. Danikopaanihoito sai aikaan tilastollisesti merkitsevän hemoglobiinipitoisuuden nousun lähtötilanteesta viikolle 12. Hemoglobiiniarvon pienimmän neliösumman keskiarvon muutos lähtötilanteesta oli danikopaaniryhmässä 29,4 g/l [1,82 mmol/l] ja lumelääkeryhmässä 5,0 g/l [0,31 mmol/l]. Hoitoryhmien välinen ero oli 24,4 g/l [1,51 mmol/l] (95 %:n CI: 1,69 [1,05], 3,20 [1,99]); p < 0,0001). Danikopaanihoidolla saavutettiin tilastollisesti merkitsevää parannusta lumelääkkeeseen verrattuna myös kaikkien neljän toissijaisen päätetapahtuman suhteen: niiden potilaiden osuus, joiden hemoglobiinipitoisuus nousi ≥ 20 g/l [1,2 mmol/l] ilman verensiirtoja (59,5 % vs. 0 %, hoitoero: 46,9 [95 %:n CI: 29,2; 64,7]; p < 0,0001), verensiirtoja tarvitsemattomien potilaiden osuus (83,3 % vs. 38,1 %, hoitoero: 41,7 [95 %:n CI: 22,7; 60,8]; p = 0,0004), FACIT-Fatigue-pisteiden muutos (7,97 vs. 1,85, hoitoero: 6,12 [95 %:n CI: 2,33; 9,91]; p = 0,0021) ja absoluuttisen retikulosyyttimäärän muutos (-83,8 vs. 3,5, hoitoero: -87,2 [95 %:n CI: ‑117,7; ‑56,7]; p < 0,0001).
Kaikkiin satunnaistettuihin potilaisiin (N = 86) perustuvat täydentävät tulokset viikolta 12 vastaavat ensisijaisesta tehoanalyysista (N = 63) saatuja tuloksia. Ravulitsumabi- tai ekulitsumabihoidon lisänä annettu danikopaani oli ravulitsumabi- tai ekulitsumabihoidon lisänä annettua lumelääkettä parempi ensisijaisen päätetapahtuman suhteen. Danikopaanihoito sai aikaan tilastollisesti merkitsevän hemoglobiinipitoisuuden nousun lähtötilanteesta viikolle 12 (ks. taulukko 2 ja kuva 1).
Danikopaanihoidolla saavutettiin tilastollisesti merkitsevää parannusta lumelääkkeeseen verrattuna myös kaikkien neljän toissijaisen päätetapahtuman suhteen (ks. taulukko 2).
12 viikon pituisen hoitojakson 1 aikana annos suurennettiin 14:lle danikopaania lisälääkkeenä saaneelle potilaalle 57:stä (24,6 %) 150 mg:sta kolmesti päivässä 200 mg:aan kolmesti päivässä. Neljä potilasta (joista kaksi oli satunnaistettu saamaan danikopaania ja kaksi lumelääkettä) keskeytti hoidon hoitojakson 1 aikana. Kukaan potilaista ei lopettanut hoitoa hemolyysin takia.
Taulukko 2: Ensisijaisten ja toissijaisten päätetapahtumien analyysit viikon 12 kohdalla (kaikki satunnaistetut potilaat)
Danikopaani (ravulitsumabin tai ekulitsumabin lisänä) N = 57 | Lumelääke (ravulitsumabin tai ekulitsumabin lisänä) N = 29 | |
| Hemoglobiinipitoisuuden muutos (ensisijainen päätetapahtuma) | ||
| Keskimääräinen muutos lähtötilanteesta viikolle 12 (g/l [mmol/l]) | 2,81 [1,74] | 0,46 [0,29] |
| Hoitoero* (95 %:n CI) | 2,35 [1,46] (1,63 [1,01], 3,06 [1,90]) | |
| Niiden potilaiden osuus, joiden hemoglobiinipitoisuus nousi ≥ 20 g/l [1,2 mmol/l] ilman verensiirtoja | ||
| Viikon 12 kohdalla (%) | 54,4 | 0 |
| Hoitoero** (95 %:n CI) | 47,5 (32,6; 62,4) | |
| Verensiirtoja tarvitsemattomien potilaiden osuus | ||
| 12 viikon pituisen hoitojakson aikana (%) | 78,9 | 27,6 |
| Hoitoero** (95 %:n CI) | 48,4 (31,8; 64,9) | |
| FACIT-Fatigue-pisteiden muutos | ||
| Keskimääräinen muutos lähtötilanteesta viikolle 12 | 8,10 | 2,38 |
| Hoitoero* (95 %:n CI) | 5,72 (2,62; 8,83) | |
| Absoluuttisen retikulosyyttimäärän muutos | ||
| Keskimääräinen muutos lähtötilanteesta viikolle 12 (109/l) | -92,5 | -0,8 |
| Hoitoero* (95 %:n CI) | -91,7 (-120,1; -63,4) | |
* Perustuu toistettujen mittausten sekamalliin.
** Osuuksien ero ja sen 95 %:n luottamusväli on laskettu Miettisen ja Nurmisen menetelmällä ja korjattu ositustekijöiden mukaan.
Lyhenteet: CI = luottamusväli; FACIT = Functional Assessment of Chronic Illness Therapy;
Kuva 1: Hemoglobiinipitoisuuden keskimääräinen muutos lähtötilanteesta viikolle 12 (kaikki satunnaistetut potilaat)
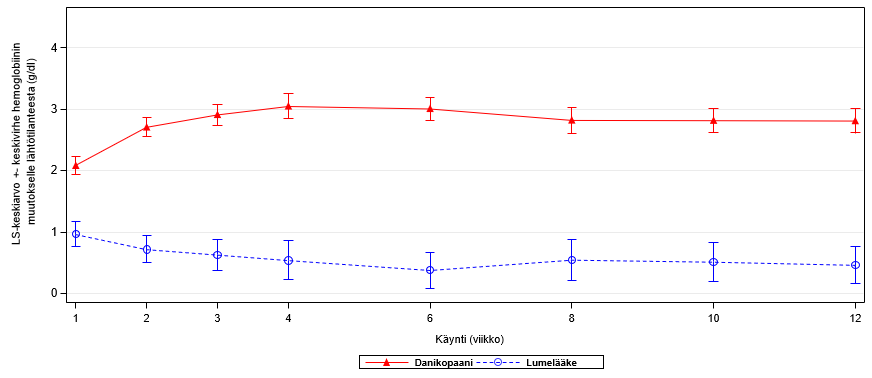
Tulokset viikon 24 kohdalla olivat yhdenmukaisia viikon 12 kohdalla saatujen tulosten kanssa, mikä tuki tehon säilymistä. Danikopaania 24 viikon ajan saaneilla 55 PNH-potilaalla hemoglobiinipitoisuuden lähtötilanteen jälkeisen muutoksen pienimmän neliösumman keskiarvo 24 viikon kohdalla oli 29,5 g/l [1,83 mmol/l] (95 %:n CI: 2,42 [1,50], 3,48 [2,16]). Potilaista 69,1 % säilytti verensiirroista riippumattomuutensa viikon 24 loppuun asti, ja 41,8 %:lla hemoglobiinipitoisuus oli viikon 24 kohdalla noussut ≥ 20 g/l [1,2 mmol/l] ilman verensiirtoja. Lisäksi näiden potilaiden FACIT-Fatigue-pisteissä havaittiin johdonmukaista parannusta, joka säilyi viikon 24 loppuun asti; keskimääräinen muutos lähtötilanteesta oli 6,19 (95 %:n CI: 4,10; 8,29).
Kaikkiaan 80 potilasta osallistui pitkäaikaiseen jatkovaiheeseen, jonka aikana kaikki potilaat saivat danikopaania. Tehoa koskevat tulokset viikolle 72 asti ovat yhdenmukaisia viikon 12 ja viikon 24 kohdalla saatujen tulosten kanssa. Danikopaania 72 viikon ajan saaneilla potilailla (N = 38) hemoglobiinipitoisuuden keskimääräinen muutos lähtötilanteesta viikolle 72 oli 28,1 g/l [1,74 mmol/l].
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset
Voydeya-valmisteen käytöstä PNH:n hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Suun kautta annettu danikopaani imeytyy nopeasti, ja suurin havaittava pitoisuus saavutetaan noin 3 tunnin jälkeen annosta. Annosalueella 200–800 mg Cmax suureni vähemmän kuin suhteessa annokseen, mikä johtuu todennäköisesti liukoisuuden rajoittamasta imeytymisestä. Danikopaanin anto runsasrasvaisen aterian kanssa suurensi AUC-arvoa noin 25 % ja Cmax-arvoa noin 93 % verrattuna paastotilassa antoon. Tmax-arvon mediaani oli samankaltainen danikopaanin ravitussa tilassa annon ja paastotilassa annon jälkeen, noin 3,0 tuntia vs. 2,5 tuntia (ks. kohta Annostus ja antotapa).
Danikopaanin läpäisevyys on suuri ja se on P‑gp:n substraatti in vitro, mutta sen effluksisuhde on pieni. P‑gp-uloskuljetus ei ilmeisesti vaikuta suun kautta otetun danikopaanin altistukseen ruoansulatuskanavassa. Danikopaani ei ole BCRP:n, OATP1B1:n eikä OATP1B3:n substraatti.
Jakautuminen
Danikopaani sitoutuu voimakkaasti ihmisen plasman proteiineihin (91,5–94,3 %), ja jakautuu pääasiassa plasmaan, ja sen kokovereen ja plasmaan jakautumisen AUC0‑∞-keskiarvojen suhde on 0,545. Danikopaanin pitoisuus plasmassa vaikutti laskevan kaksivaiheisesti huippupitoisuuden saavuttamisen (Tmax) jälkeen. Populaatiofarmakokineettisen mallin avulla arvioitu näennäinen jakautumistilavuus 75 kg:n painoiselle henkilölle suun kautta annossa oli 168 litraa keskustilan osalta (Vc/F) ja 234 litraa perifeerisen tilan osalta (Vp/F) (yhteensä 402 litraa), mikä viittaa danikopaanin kohtalaiseen perifeerisiin kudoksiin jakautumiseen.
Biotransformaatio
Suun kautta annettu danikopaani metaboloituu laajasti (96 %) hapettumalla, pelkistymällä ja hydrolysoitumalla. Amidihydrolyysin havaittiin olevan pääasiallinen eliminaatioreitti. CYP-välitteinen metabolia on minimaalista.
Eliminaatio
Suun kautta annetusta annoksesta pääosa eliminoituu ulosteen mukana (noin 69 % annoksesta, verrattuna 25 %:iin virtsan mukana). Populaatiofarmakokineettisessä analyysissa PNH-potilailla, joilla esiintyi kliinisesti merkittävää ekstravaskulaarista hemolyysiä, keskimääräinen t½ oli arviolta 7,91 tuntia.
Erityisryhmät
Populaatiofarmakokineettisessä analyysissa ei havaittu kliinisesti merkittäviä eroja danikopaanin farmakokinetiikassa sukupuolen, iän tai rodun perusteella.
Munuaisten vajaatoiminta
Kun vaikeaa munuaisten vajaatoimintaa sairastaville tutkittaville (eGFR < 30 ml/min/1,73 m2) annettiin 200 mg danikopaania suun kautta, danikopaanialtistus oli noin 50 % suurempi kuin tutkittavilla, joiden munuaistoiminta oli normaali. Munuaisten kautta erittyminen ei ole danikopaanin pääasiallinen eliminaatioreitti edes henkilöillä, joiden munuaistoiminta on normaali (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Keskivaikeaa maksan vajaatoimintaa sairastavien tutkittavien (Child-Pugh-luokka B) danikopaanialtistus ei eronnut merkittävästi tutkittavista, joiden maksan toiminta oli normaali (ks. kohta Annostus ja antotapa). Tutkimuksia ei ole tehty vaikeaa maksan vajatoimintaa sairastavilla potilailla (Child-Pugh-luokka C).
Prekliiniset tiedot turvallisuudesta
Rotilla (lajit, jotka eivät ole farmakologisesti herkkiä danikopaanille) tehdyssä 6 kuukautta kestäneessä toksisuustutkimuksessa havaittiin maksan, kilpirauhasen ja lisämunuaisen hypertrofiaa annoksilla 1000 mg/kg/vrk (noin 26 kertaa enemmän kuin ihmisen altistus annoksella 200 mg kolme kertaa vuorokaudessa AUC-arvon perusteella).
Koirilla tehdyssä 9 kuukauden pituisessa toksisuustutkimuksessa koirat eivät sietäneet annosta 150 mg/kg/vrk. Maksassa todettiin kohde-elimeen kohdistuvia vaikutuksia, jotka vastasivat hepatobiliaarista kolestaasia ja joita olivat mm. sapenjohdinten hypertrofia/hyperplasia ja sapen pigmenttiä vastaavan pigmentin kertyminen Kupfferin soluihin ja hepatosyytteihin. ASAT-, ALAT-, AFOS-, GGT- ja kokonaisbilirubiiniarvojen nousut korreloivat maksan histologisten löydösten kanssa. Sapenjohdinten hypertrofiaa/hyperplasiaa todettiin uroksilla, joiden saamat annokset olivat vähintään 75 mg/kg/vrk (AUC-arvon perusteella noin 5‑kertainen altistus verrattuna ihmisen altistukseen annoksella 200 mg kolmesti päivässä). Annoksella 75 mg/kg/vrk todetut löydökset olivat kuitenkin lievempiä ja heikompia, eikä niihin liittynyt vastaavia kliinis-patologisia löydöksiä.
Genotoksisuus/karsinogeenisuus
Danikopaani ei ollut genotoksinen bakteereilla tehdyssä takaisinmutaatiotestissä (Amesin testissä), ihmisen perifeerisen veren lymfosyyteillä tehdyssä mikrotumatestissä in vitro eikä rotilla tehdyssä mikrotumatestissä in vivo.
Danikopaani ei ollut karsinogeeninen TgRasH2-hiirillä tehdyssä 6 kuukauden pituisessa karsinogeenisuustutkimuksessa eikä rotilla tehdyssä 2 vuoden pituisessa karsinogeenisuustutkimuksessa. Rotilla tehdyssä tutkimuksessa suurimmalla 500 mg/kg/vrk:n annoksella todettu kohdun limakalvon epiteelin kasvainten ilmaantuvuus oli kuitenkin suurempi kuin verrokkieläimillä, joskin rottakannalla voi olla suuri kohdun limakalvon syövän taustailmaantuvuus. Tämän löydöksen kliinistä merkitystä ei tunneta.
Lisääntymis-/kehitystoksisuus
Kaniineilla tehdyssä, hedelmällisyyttä ja varhaista alkionkehitystä selvittäneessä tutkimuksessa urosten ja naaraiden lisääntymistoimintojen heikkenemistä todettiin annoksella 500 mg/kg/vrk, jonka siedettävyys on ollut huono. Uroksiin ja naaraisiin kohdistuvan lisääntymistoksisuuden osalta NOAEL-annoksen katsottiin olevan 250 mg/kg/vrk (7,2- ja 8,8-kertainen altistus verrattuna ihmisen altistukseen).
Kaniineilla tehdyssä pre- ja postnataalista kehitystä selvittäneessä tutkimuksessa F1-uroksilla todettiin lisäkiveksen hännän siittiömäärän laskua (19 %, 20 % ja 18 %) kaikissa annosryhmissä (50, 125 ja 250 mg/kg/vrk), mutta se oli tilastollisesti merkitsevää vain pientä ja keskitason annosta saaneissa ryhmissä. Tämä ei vaikuttanut F1-sukupolven lisääntymiskykyyn.
Kaniineilla ei havaittu vaikutuksia varhaiseen alkioiden kehitykseen, sikiöiden kehitykseen tai poikasten syntymän jälkeiseen kehitykseen emon keskimääräisellä systeemisellä altistuksella, joka vastasi enimmillään noin 20-kertaista ihmisen altistusta. Rotilla ei havaittu vaikutuksia alkioiden ja sikiöiden kehitykseen emon altistuksella, joka vastasi noin 30-kertaista ihmisen altistusta 200 mg:n annoksella kolmesti päivässä.
Erittyminen maitoon
Danikopaania erittyi imettävien kaniinien maitoon, kun danikopaania annettiin suun kautta imetyspäivinä 4–10. Pitoisuus maidossa oli noin 5 kertaa emon plasmapitoisuutta suurempi annoksella 50 mg/kg/vrk ja noin 3,5 kertaa emon plasmapitoisuutta suurempi annoksella 250 mg/kg/vrk.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti, selluloosa, mikrokiteinen, kroskarmelloosinatrium, natriumlauryylisulfaatti, magnesiumstearaatti, piidioksidi, hydrofobinen, kolloidinen, hypromelloosiasetaattisuksinaatti
Kalvopäällyste
Polyvinyylialkoholi, titaanidioksidi (E171), makrogoli 4000, talkki
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
48 kuukautta suurtiheyspolyeteeni (HDPE) -purkissa
Purkin ensimmäisen avaamisen jälkeen: 48 vuorokautta
2 vuotta polyvinyylikloridi (PVC) / polyklooritrifluorietyleeni (PCTFE) / PVC -läpipainopakkauksissa
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VOYDEYA tabletti, kalvopäällysteinen
50 mg+100 mg (L:kyllä) 180 kpl (90x50mg+90x100mg) (6895,26 €)
100 mg (L:kyllä) 180 kpl (2x90) (9133,48 €)
PF-selosteen tieto
Purkki
HDPE-purkit, joissa on 90 kalvopäällysteistä tablettia, kuivatusaine ja lapsiturvallinen sinetti. Yhdessä pakkauksessa on 180 kalvopäällysteistä tablettia.
Seuraavat pakkauskoot ovat saatavilla:
-
Pakkaus, joka sisältää 1 purkin, jossa on 90 × 50 mg:n kalvopäällysteistä tablettia, ja 1 purkin, jossa on 90 × 100 mg:n kalvopäällysteistä tablettia.
-
Pakkaus, joka sisältää 2 purkkia, joissa on kussakin 90 × 100 mg:n kalvopäällysteistä tablettia.
Läpipainopakkaus
PVC/PCTFE/PVC-läpipainopakkaus. Yhdessä pakkauksessa on 168 kalvopäällysteistä tablettia.
Seuraavat pakkauskoot ovat saatavilla:
- Pakkaus, joka sisältää 4 läpipainopakkauskoteloa (lapsiturvallinen), jossa on kussakin 21 × 50 mg:n kalvopäällysteistä tablettia ja 21 × 100 mg:n kalvopäällysteistä tablettia.
- Pakkaus, joka sisältää 4 läpipainopakkauskoteloa (lapsiturvallinen), jossa on kussakin 42 × 100 mg:n kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Voydeya 50 mg kalvopäällysteiset tabletit
Valkoiset tai luonnonvalkoiset, pyöreät kalvopäällysteiset tabletit, joiden toiselle puolelle on kaiverrettu allekkain ”DCN” ja ”50” ja joiden toinen puoli on merkitsemätön. Tabletin koko on noin 8 mm.
Voydeya 100 mg kalvopäällysteiset tabletit
Valkoiset tai luonnonvalkoiset, pyöreät kalvopäällysteiset tabletit, joiden toiselle puolelle on kaiverrettu allekkain ”DCN” ja ”100” ja joiden toinen puoli on merkitsemätön. Tabletin koko on noin 10,3 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VOYDEYA tabletti, kalvopäällysteinen
50 mg+100 mg 180 kpl
100 mg 180 kpl
- Ei korvausta.
ATC-koodi
L04AJ09
Valmisteyhteenvedon muuttamispäivämäärä
12.01.2026
Yhteystiedot
Forskaren, Hagaplan 4
SE-113 68 Tukholma/Stockholm
Ruotsi/Sverige
+46 (0)8 557 727 50
alexion.nordics@alexion.com