
ALTUVOCT injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU, 4000 IU
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
ALTUVOCT 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 250 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 83 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 500 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 167 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 750 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 750 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 250 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 1 000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 1 000 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 333 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 2 000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 2 000 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 667 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 3 000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 3 000 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 1 000 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
ALTUVOCT 4 000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 4 000 IU efanesoktokogi alfaa. Käyttökuntoon saattamisen jälkeen ALTUVOCT sisältää noin 1 333 IU/ml ihmisen hyytymistekijä VIII:aa, efanesoktokogi alfaa.
Teho (IU) on määritetty käyttämällä aktivoituun partiaaliseen tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymistestiä, jossa käytettiin Actin-FSL-reagenssia.
Efanesoktokogi alfa (ihmisen hyytymistekijä VIII [rDNA]) on proteiini, joka sisältää 2 829 aminohappoa.
Efanesoktokogi alfa valmistetaan yhdistelmä-DNA-tekniikalla ihmisalkion munuaisten (HEK) solulinjassa. Valmistusprosessissa ei käytetä lainkaan ihmisestä tai eläimestä peräisin olevia raaka-aineita.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
A‑hemofiliaa (synnynnäinen hyytymistekijä VIII:n puute) sairastavien potilaiden verenvuotojen hoito ja ennaltaehkäisy.
ALTUVOCT-valmistetta voidaan käyttää kaikille ikäryhmille.
Ehto
Hoito tulee aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää toteuttaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Saatuaan asianmukaisen opastuksen oikean injektiotekniikan käyttöön (ks. kohta Käyttö- ja käsittelyohjeet ja pakkausseloste) potilas voi pistää ALTUVOCT-valmisteen itse, tai potilasta hoitava henkilö voi pistää valmisteen, jos lääkäri katsoo sen asianmukaiseksi.
Hoidon valvonta
Yksittäisten potilaiden vaste tekijä VIII:lle saattaa vaihdella, mikä ilmenee puoliintumisaikojen ja saantojen eroina. Painoon perustuvia annoksia on ehkä säädettävä alipainoisten tai ylipainoisten potilaiden kohdalla. Rutiininomaisessa estohoidossa tekijä VIII:n pitoisuuksia ei yleensä tarvitse seurata annoksen säätämistarkoituksessa. Suurten leikkausten tai hengenvaarallisten verenvuotojen tapauksessa tekijä VIII:n pitoisuudet on määritettävä, jotta annos ja toistettujen injektioiden antotiheys voidaan määrittää.
Käytettäessä in vitro tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä tekijä VIII:n aktiivisuuden määrittämiseksi potilaiden verinäytteistä, plasman tekijä VIII:n aktiivisuuden tuloksiin saattavat vaikuttaa huomattavasti sekä aPTT-reagenssin tyyppi että määrityksessä käytettävä viitestandardi. Merkittäviä eroja voi olla myös aPTT-pohjaisen yksivaiheisen hyytymismäärityksen ja Euroopan farmakopean mukaisen kromogeenisen määrityksen tuloksissa. Tällä on merkitystä etenkin vaihdettaessa laboratoriota ja/tai määrityksessä käytettäviä reagensseja.
On suositeltavaa käyttää validoitua yksivaiheista hyytymismääritystä ALTUVOCT-valmisteen aikaansaaman plasman tekijä VIII:n aktiivisuuden määrittämiseen. Kliinisen kehityksen aikana käytettiin Actin-FSL:ään perustuvaa yksivaiheista hyytymismääritystä.
Kliinisten tutkimusnäytteiden vertailevan analyysin tuloksien perusteella kromogeenisen määrityksen tulos on jaettava luvulla 2,5, jotta saadaan likiarvo potilaan tekijä VIII:n aktiivisuudesta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lisäksi kenttätutkimuksessa, jossa vertailtiin erilaisia aPTT-reagensseja, tekijä VIII:n aktiivisuustasot todettiin noin 2,5-kertaisiksi, kun yksivaiheisessa hyytymismäärityksessä käytettiin Actin-FS-reagenssia Actin-FSL-reagenssin sijaan, ja 30 % pienemmiksi, kun käytettiin SynthASil-reagenssia.
Annostus
Korvaushoidon annos ja kesto riippuvat tekijä VIII:n puutoksen vaikeusasteesta, verenvuodon laajuudesta ja vuotokohdasta sekä potilaan kliinisestä tilasta.
Annettavien tekijä VIII ‑yksiköiden määrä ilmaistaan kansainvälisinä yksikköinä (International Units, IU), mikä perustuu tekijä VIII ‑valmisteita koskevaan voimassaolevaan WHO-standardiin. Tekijä VIII:n aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliin ihmisen plasmaan) tai mieluiten IU-yksikköinä (plasman tekijä VIII ‑pitoisuuden kansainvälisen standardin mukaan).
Yksi kansainvälinen yksikkö (IU) tekijä VIII ‑aktiivisuutta vastaa tekijä VIII:n määrää yhdessä millilitrassa normaalia ihmisen plasmaa.
Jos annos on 50 IU tekijä VIII:aa painokiloa kohti, in vivo odotettavissa oleva tekijä VIII:n saanto plasmassa, joka ilmaistaan yksikkönä IU/dl (tai % normaalista), arvioidaan seuraavalla laskukaavalla:
Arvioitu tekijä VIII:n lisäys (IU/dl tai % normaalista) = 50 IU/kg × 2 (IU/dl per IU/kg)
Tarvittaessa toteutettava hoito
ALTUVOCT-valmisteen annostus tarvittaessa toteutettavassa hoidossa, verenvuototapausten hallinnassa ja perioperatiivisessa hoidossa esitetään taulukossa 1.
Taulukko 1: ALTUVOCT-annostusohje verenvuototapausten hoitoa ja leikkauksia varten
| Verenvuodon määrä / kirurgisen toimenpiteen tyyppi | Suositeltu annos | Lisätietoja |
| Verenvuoto | ||
| Alkava hemartroosi, lihasverenvuoto tai suun verenvuoto | Kerta-annos 50 IU/kg | Jos kyseessä on lievä tai keskivaikea verenvuoto, joka ilmenee 2–3 vuorokautta estohoitoannoksen jälkeen, voidaan käyttää pienempää annosta 30 IU/kg. Lisäannosta 30–50 IU/kg voidaan harkita 2–3 vuorokauden kuluttua. |
| Laajempi hemartroosi, lihasverenvuoto tai hematooma | Kerta-annos 50 IU/kg | Lisäannoksia 30–50 IU/kg 2–3 vuorokauden välein voidaan harkita, kunnes verenvuoto lakkaa. |
| Hengenvaaralliset verenvuodot | Kerta-annos 50 IU/kg | Lisäannoksia 30–50 IU/kg 2–3 vuorokauden välein voidaan antaa, kunnes vaara on ohi. |
| Leikkaus | ||
| Pieni leikkaus, kuten hampaanpoisto | Kerta-annos 50 IU/kg | Lisäannosta 30–50 IU/kg voidaan harkita 2–3 vuorokauden kuluttua. |
| Suuri leikkaus | Kerta-annos 50 IU/kg | Lisäannoksia 30–50 IU/kg 2–3 vuorokauden välein voidaan antaa kliinisen tarpeen mukaan, kunnes haava on parantunut riittävästi. |
Kun verenvuodon hoitamisen jälkeen palataan estohoitoon (jos aiheellista), verenvuodon hoitamiseen annetun viimeisen 50 IU/kg annoksen jälkeen on suositeltavaa odottaa vähintään 72 tuntia ennen kuin hoitoa jatketaan estohoitoannoksella. Tämän jälkeen estohoitoa voidaan jatkaa tavalliseen tapaan potilaan normaalin antoaikataulun mukaisesti.
Estohoito
Rutiininomaiseen estohoitoon aikuisille ja lapsille suositeltu annostus on 50 IU/kg ALTUVOCT-valmistetta kerran viikossa.
Erityisryhmät
Iäkkäät
Valmisteen käytöstä ≥ 65-vuotiaille potilaille on vain vähän kokemusta. Annossuositukset ovat samat kuin < 65-vuotiaille potilaille.
Pediatriset potilaat
Annossuositukset ovat samat kuin aikuisille.
Antotapa
Laskimoon.
Koko ALTUVOCT-annos on injisoitava laskimoon 1–10 minuutin aikana. Antonopeus on määritettävä potilaskohtaisesti.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
Allergistyyppisiä yliherkkyysreaktioita, myös anafylaktisia reaktioita, on havaittu ALTUVOCT‑valmistetta käytettäessä. Potilaita on neuvottava lopettamaan lääkevalmisteen käyttö välittömästi ja ottamaan yhteyttä lääkäriin, jos yliherkkyysoireita ilmaantuu. Potilaille on kerrottava varhaisista yliherkkyysreaktioiden oireista, joita ovat esim. paukamat, yleistynyt nokkosihottuma, kutina, pahoinvointi, oksentelu, puristuksen tunne rinnassa, hengityksen vinkuminen, verenpaineen lasku ja anafylaksia.
Sokkitapauksissa potilasta on hoidettava yleisten sokinhoito-ohjeiden mukaisesti.
Vasta-aineet eli inhibiittorit
Tekijä VIII:aa neutraloivien vasta-aineiden (inhibiittorien) muodostuminen on tunnettu komplikaatio A‑hemofiliapotilaiden hoidossa. Nämä inhibiittorit ovat yleensä IgG-immunoglobuliineja, jotka estävät tekijä VIII ‑hyytymistoiminnan aktivoitumisen ja joiden määrä ilmaistaan Bethesda-yksikköinä (Bethesda Units, BU) millilitrassa plasmaa käyttämällä muunneltua määritystä. Inhibiittorien muodostumisen riski riippuu taudin vaikeusasteesta ja tekijä VIII ‑altistuksesta. Riski on suurin 50 ensimmäisen altistuspäivän aikana, mutta jatkuu läpi elämän, vaikkakin melko harvinaisena.
Inhibiittorien muodostumisen kliininen merkitys riippuu inhibiittorititteristä. Matalan titterin inhibiittoreihin liittyy pienempi riittämättömän kliinisen vasteen riski kuin korkean titterin inhibiittoreihin.
Yleisesti ottaen kaikkia hyytymistekijä VIII ‑valmisteilla hoidettavia potilaita on seurattava huolellisesti inhibiittorien muodostumisen varalta asianmukaisen kliinisen havainnoinnin ja laboratoriokokeiden avulla. Jos odotettua tekijä VIII:n aktiivisuutta plasmassa ei saavuteta tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, on potilaalta testattava tekijä VIII:n inhibiittorien esiintyminen. Jos potilaan inhibiittoripitoisuus on korkea, tekijä VIII ‑hoito ei ehkä ole tehokasta ja muita hoitovaihtoehtoja on syytä harkita. Näiden potilaiden hoidon on tapahduttava sellaisten lääkäreiden valvonnassa, joilla on kokemusta sekä hemofilian tekijä että VIII:n inhibiittoreiden hoidosta.
Laboratoriotestien valvonta
Jos käytetään kromogeenista määritystä tai yksivaiheista hyytymismääritystä, jossa käytetään Actin-FS-reagenssia, potilaan tekijä VIII:n aktiivisuustason likiarvo lasketaan jakamalla tulos luvulla 2,5 (ks. kohta Annostus ja antotapa). On syytä ottaa huomioon, että tällä muunnoskertoimella saadaan vain arvio (kromogeenisen määrityksen / yksivaiheisen hyytymismäärityksen keskimääräinen Actin-FSL-suhde: 2,53; keskihajonta: 1,54; Q1: 1,98; Q3: 2,96; N = 3 353).
Sydän- ja verisuonitapahtumat
Potilailla, joilla on ennestään kardiovaskulaarisia riskitekijöitä, korvaushoito tekijä VIII ‑valmisteilla voi lisätä kardiovaskulaarista riskiä.
Katetreihin liittyvät komplikaatiot
Jos hoito edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrointikohdan tromboosi.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat sekä aikuisia että lapsia.
Yhteisvaikutukset
Yhteisvaikutuksia ihmisen hyytymistekijä VIII (rDNA) ‑valmisteiden ja muiden lääkevalmisteiden välillä ei ole raportoitu.
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Tekijä VIII ‑valmisteilla ei ole tehty lisääntymistä koskevia eläinkokeita. A‑hemofilia on naisilla harvinainen, joten kokemusta tekijä VIII:n käytöstä raskauden ja imetyksen aikana ei ole saatavilla. Tämän vuoksi tekijä VIII ‑valmistetta saa käyttää raskauden ja imetyksen aikana vain, jos se on selvästi aiheellista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ALTUVOCT-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Harvinaisissa tapauksissa on havaittu yliherkkyyttä tai allergisia reaktioita (näitä saattavat olla mm. angioedeema, injektiokohdan polte ja kirvely, vilunväristykset, punoitus, yleistynyt nokkosihottuma, päänsärky, paukamat, matala verenpaine, letargia, pahoinvointi, levottomuus, takykardia, puristuksen tunne rinnassa, pistely, oksentelu, hengityksen vinkuminen), jotka voivat joissakin tapauksissa kehittyä vaikeaksi anafylaksiaksi (mukaan lukien sokki).
Tekijä VIII ‑valmisteita, myös ALTUVOCT-valmistetta, saaville A‑hemofiliapotilaille voi kehittyä neutraloivia vasta-aineita (inhibiittoreita) (ks. kohta Farmakodynamiikka). Mikäli tällaisia inhibiittoreita ilmaantuu, se näkyy riittämättömänä kliinisenä vasteena hoidolle. Tällaisissa tapauksissa on suositeltavaa ottaa yhteyttä hemofiliaan erikoistuneeseen hoitoyksikköön.
Haittavaikutustaulukko
Seuraava taulukko 2 on MedDRA-elinjärjestelmäluokituksen mukainen (elinjärjestelmäluokka ja suositeltu termi). Haittavaikutusten yleisyydet perustuvat faasin 3 kliinisistä tutkimuksista saatuihin raportteihin yhteensä 277:stä vaikeaa A‑hemofiliaa sairastavasta tutkittavasta, jotka olivat saaneet aiemmin hoitoa (PTP). Näistä potilaista 161 (58,2 %) oli aikuisia (18-vuotiaita tai vanhempia), 37 (13,4 %) oli nuoria (12 < 18-vuotiaita) ja 79 (28,5 %) oli alle 12-vuotiaita lapsia.
Haittavaikutuksia (esitetty yhteenvetona taulukossa 2) raportoitiin kliinisissä tutkimuksissa 111 (40,1 %) tutkittavalla 277:stä tutkittavasta, jotka saivat rutiininomaista estohoitoa tai tarvittaessa annettavaa hoitoa, sekä myyntiintulon jälkeisessä seurannassa.
Esiintyvyys on arvioitu seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on lueteltu kussakin esiintyvyysluokassa vakavuuden mukaan alanevassa järjestyksessä.
Taulukko 2: ALTUVOCT-valmisteesta raportoidut haittavaikutukset
| MedDRA-elinjärjestelmäluokka | Haittavaikutukset | Esiintyvyysluokka |
| Veri ja imukudos | Hyytymistekijä VIII:n inhibitio1 | Hyvin yleinen (PUP)2 Melko harvinainen (PTP)3 |
| Immuunijärjestelmä | Yliherkkyys, anafylaktiset reaktiot1 | Tuntematon |
| Hermosto | Päänsärky4 | Hyvin yleinen |
| Ruoansulatuselimistö | Oksentelu | Yleinen |
| Iho ja ihonalainen kudos | Ekseema | Yleinen |
| Ihottuma5 | Yleinen | |
| Nokkosihottuma6 | Yleinen | |
| Luusto, lihakset ja sidekudos | Nivelkipu | Hyvin yleinen |
| Raajakipu | Yleinen | |
| Selkäkipu | Yleinen | |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume | Yleinen |
| Injektiokohdan reaktio7 | Melko harvinainen |
1 Raportoitu myyntiintulon jälkeen.
2 PUP = aiemmin hoitamattomat potilaat (Previously-Untreated Patients). Esiintyvyys perustuu muilla hyytymistekijä VIII ‑valmisteilla tehtyihin tutkimuksiin, joissa oli mukana aiemmin hoitamattomia potilaita, joilla oli vaikea A‑hemofilia.
3 PTP = aiemmin hoidetut potilaat (Previously-Treated Patients). Esiintyvyys perustuu kaikilla hyytymistekijä VIII ‑valmisteilla tehtyihin tutkimuksiin, joissa oli mukana aiemmin hoidettuja potilaita, joilla oli vaikea A-hemofilia.
4 Päänsärky, mukaan lukien migreeni.
5 Ihottuma, mukaan lukien makulopapulaarinen ihottuma.
6 Nokkosihottuma, mukaan lukien papulaarinen nokkosihottuma.
7 Injektiokohdan reaktio, mukaan lukien injektiokohdan hematooma ja injektiokohdan dermatiitti.
Pediatriset potilaat
Haittavaikutuksissa ei todettu ikään liittyviä eroja pediatristen potilaiden ja aikuispotilaiden välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
[Swedish]
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Yliannostus
Ihmisen hyytymistekijä VIII (rDNA) ‑valmisteiden yliannostusoireita ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: veren hyytymistekijät, hyytymistekijä VIII, ATC-koodi: B02BD02.
Vaikutusmekanismi
Efanesoktokogi alfa on tekijä VIII ‑korvaushoito. Aktivoitunut tekijä VIII toimii aktivoituneen tekijä IX:n kofaktorina ja nopeuttaa tekijä X:n muuttumista aktivoituneeksi tekijä X:ksi. Aktivoitunut tekijä X muuttaa protrombiinin trombiiniksi. Trombiini muuttaa tämän jälkeen fibrinogeenin fibriiniksi, jolloin hyytymä muodostuu. A‑hemofilia on sukupuolisidonnainen perinnöllinen veren hyytymissairaus, joka johtuu toimivan tekijä VIII:C:n pienentyneistä pitoisuuksista ja johtaa verenvuotoihin nivelissä, lihaksissa tai sisäelimissä joko spontaanisti tai tapaturman tai leikkausvamman seurauksena. Korvaushoidon ansiosta tekijä VIII:n pitoisuus plasmassa suurenee, jolloin hyytymistekijän puutos ja vuototaipumus hetkellisesti korjaantuvat.
On huomioitava, että vuositasolla lasketut verenvuotomäärät (annualized bleeding rate, ABR) eivät ole vertailukelpoisia eri hyytymistekijäkonsentraattien välillä eivätkä eri kliinisten tutkimusten välillä.
ALTUVOCT (efanesoktokogi alfa) eli rekombinantti hyytymistekijä VIII Fc – von Willebrand ‑tekijä XTEN on rekombinantti fuusioproteiini, joka korvaa tilapäisesti tehokkaaseen hemostaasiin tarvittavan hyytymistekijä VIII:n.
Efanesoktokogi alfa on FVIII-proteiini, joka on suunniteltu olemaan sitoutumatta endogeeniseen VWF:ään ja estämään näin FVIII:n ja VWF:n interaktioista johtuvan puoliintumisaikarajoituksen vaikutuksen. Alue, jolla on interaktioita FVIII:n kanssa, on VWF:n D’D3-domeeni. Kun VWF:n D’D3-domeeni liitetään rFVIII-Fc-fuusioproteiiniin, se antaa FVIII:lle suojaa ja vakautta, estää FVIII:n ja endogeenisen VWF:n interaktiot ja poistaa siten VWF:n puhdistuman FVIII:n puoliintumisajalle asettamat rajoitukset.
Ihmisen immunoglobuliini G1:n (IgG1) Fc-osa sitoutuu vastasyntyneen Fc-reseptoriin (FcRn). FcRn on osa luonnollisesti esiintyvää reittiä, joka hidastaa immunoglobuliinien lysosomaalista hajoamista kuljettamalla näitä proteiineja takaisin verenkiertoon, mikä johtaa fuusioproteiinien pidempään puoliintumisaikaan plasmassa.
Efanesoktokogi alfa sisältää kahta XTEN-polypeptidiä, jotka lisäävät sen farmakokinetiikkaa entisestään. FVIII:n luonnollinen B-domeeni (viittä aminohappoa lukuun ottamatta) korvautuu ensimmäisellä XTEN-polypeptidillä, joka asettuu FVIII:n N745- ja E1649-aminohappojäänteiden väliin. Toinen XTEN-polypeptidi asettuu D’D3-domeenin ja Fc:n väliin.
Kliininen teho ja turvallisuus
ALTUVOCT-valmisteen turvallisuutta, tehoa ja farmakokinetiikkaa on arvioitu kahdessa prospektiivisessa, avoimessa faasin 3 kliinisessä monikeskustutkimuksessa (toinen tutkimus tehtiin aikuisille ja nuorille [XTEND‑1] ja toinen oli pediatrinen tutkimus < 12-vuotiaille lapsille [XTEND‑Kids, ks. Pediatriset potilaat]) aiemmin hoidetuille potilaille, joilla oli vaikea A‑hemofilia (endogeeninen FVIII-aktiivisuus < 1 % tai dokumentoitu vaikeaan A‑hemofiliaan yhdistetty geneettinen mutaatio). Myös ALTUVOCT-valmisteen pitkäaikaisturvallisuutta ja ‑tehoa arvioidaan parhaillaan pitkäkestoisessa jatkotutkimuksessa.
Kaikissa tutkimuksissa arvioitiin rutiininomaisen estohoidon tehoa viikoittaisella annoksella 50 IU/kg ja määritettiin valmisteen hemostaattinen teho verenvuototapahtumien hoidossa ja perioperatiivisessa hoidossa suurten ja pienten kirurgisten toimenpiteiden yhteydessä. Lisäksi ALTUVOCT-valmisteella toteutetun estohoidon tehoa verrattuna aiempaan tekijä VIII ‑estohoitoon arvioitiin potilaskohtaisten vertailujen avulla tutkittavilla, jotka olivat osallistuneet prospektiiviseen havainnointitutkimukseen (OBS16221) ennen XTEND‑1-tutkimukseen kirjautumista.
Kliininen teho rutiininomaisessa estohoidossa aikuisilla/nuorilla
Loppuun suoritettuun, aikuisille ja nuorille tehtyyn tutkimukseen (XTEND‑1) otettiin kaikkiaan 159 aiemmin hoidettua potilasta (158 miestä ja 1 nainen), joilla oli vaikea A‑hemofilia. Tutkittavien ikä oli 12–72 vuotta, ja heistä 25 oli 12–17-vuotiaita nuoria. Kaikki 159 tutkimukseen otettua tutkittavaa saivat vähintään yhden ALTUVOCT-annoksen ja olivat tehon suhteen arviointikelpoisia. Tutkimuksen suoritti loppuun yhteensä 149 tutkittavaa (93,7 %).
Viikoittaisen ALTUVOCT-annoksen 50 IU/kg tehoa rutiininomaisessa estohoidossa arvioitiin vuositasolla laskettuina keskimääräisinä verenvuotomäärinä (ABR) (taulukko 3) ja vertaamalla tutkimuksessa annetun estohoidon aikaista ABR-määrää ennen tutkimusta saadun tekijä VIII ‑estohoidon aikaiseen ABR-määrään (taulukko 4). Kaikkiaan 133 aikuista ja nuorta, jotka olivat saaneet tekijä VIII ‑estohoitoa ennen tutkimukseen osallistumista, määrättiin saamaan ALTUVOCT-valmistetta rutiininomaisena estohoitona annoksella 50 IU/kg kerran viikossa 52 viikon ajan (haara A). Lisäksi 26 tutkittavaa, jotka olivat saaneet ajoittaista (tarvittaessa toteutettavaa) tekijä VIII ‑estohoitoa ennen tutkimukseen osallistumista, saivat ajoittaista (tarvittaessa toteutettavaa) ALTUVOCT-hoitoa annoksella 50 IU/kg 26 viikon ajan ja sen jälkeen rutiininomaista estohoitoa annoksella 50 IU/kg kerran viikossa 26 viikon ajan (haara B). Kaikkiaan 115 tutkittavaa sai hoitoa vähintään 50 altistuspäivän verran haarassa A, ja 17 tutkittavaa sai rutiininomaista estohoitoa vähintään 25 altistuspäivän verran haarassa B.
Taulukko 3: Yhteenveto vuositasolla lasketuista verenvuotomääristä (ABR) ALTUVOCT-estohoidon yhteydessä, tarvittaessa toteutettavan ALTUVOCT-hoidon yhteydessä ja ALTUVOCT-estohoitoon siirtymisen jälkeen ≥ 12-vuotiailla tutkittavilla
Päätetapahtuma1 | Haara A Estohoito2 | Haara B Tarvittaessa toteutettava hoito3 | Haara B Estohoito3 |
N = 133 | N = 26 | N = 26 | |
Verenvuodot | |||
ABR-keskiarvo (95 %:n lv)4 | 0,71 (0,52; 0,97) | 21,41 (18,81; 24,37) | 0,70 (0,33; 1,49) |
ABR-mediaani (IQR) | 0,00 (0,00; 1,04) | 21,13 (15,12; 27,13) | 0,00 (0,00; 0,00) |
Tutkittavat, joilla ei ollut yhtään verenvuotoa, % | 64,7 | 0 | 76,9 |
Spontaanit verenvuodot | |||
ABR-keskiarvo (95 %:n lv)4 | 0,27 (0,18; 0,41) | 15,83 (12,27; 20,43) | 0,44 (0,16; 1,20) |
ABR-mediaani (IQR) | 0,00 (0,00; 0,00) | 16,69 (8,64; 23,76) | 0,00 (0,00; 0,00) |
Tutkittavat, joilla ei ollut yhtään verenvuotoa, % | 80,5 | 3,8 | 84,6 |
Nivelten verenvuodot | |||
ABR-keskiarvo (95 %:n lv)4 | 0,51 (0,36; 0,72) | 17,48 (14,88; 20,54) | 0,62 (0,25; 1,52) |
ABR-mediaani (IQR) | 0,00 (0,00; 1,02) | 18,42 (10,80; 23,90) | 0,00 (0,00; 0,00) |
Tutkittavat, joilla ei ollut yhtään verenvuotoa, % | 72,2 | 0 | 80,8 |
1 Kaikki verenvuotopäätetapahtumien analyysit perustuvat hoidettuihin verenvuotoihin.
2 Tutkittavat määrättiin saamaan ALTUVOCT-estohoitoa 52 viikon ajan.
3 Tutkittavat määrättiin saamaan ALTUVOCT-hoitoa 26 viikon ajan.
4 Perustuu negatiiviseen binomimalliin.
ABR = vuositasolla laskettu verenvuotomäärä; lv = luottamusväli; IQR = kvartiiliväli, 25. persentiilin ja 75. persentiilin välinen alue.
Tutkimuksen aikaisten ABR-määrien ja tutkimusta edeltäneen estohoidon aikaisten ABR-määrien potilaskohtainen vertailu osoitti vuositasolla laskettujen verenvuotojen vähentyneen tilastollisesti merkitsevästi 77 %:lla rutiininomaisen ALTUVOCT-estohoidon aikana verrattuna tutkimusta edeltäneeseen tekijä VIII ‑estohoitoon (ks. taulukko 4).
Taulukko 4: Potilaskohtaiset vuositasolla lasketut verenvuotomäärät (ABR) ALTUVOCT-estohoidon aikana verrattuna tutkimusta edeltäneeseen tekijä VIII -estohoitoon ≥ 12-vuotiailla tutkittavilla
Päätetapahtuma | Tutkimuksen aikainen ALTUVOCT-estohoito 50 IU/kg kerran viikossa (N = 78) | Tutkimusta edeltänyt tavanomainen tekijä VIII -estohoito2 (N = 78) |
Havainnointijakson mediaani (viikkoa) (IQR) | 50,09 (49,07; 51,18) | 50,15 (43,86; 52,10) |
Verenvuodot | ||
ABR-keskiarvo (95 %:n lv)1 | 0,69 (0,43; 1,11) | 2,96 (2,00; 4,37) |
ABR-määrän väheneminen, % (95 %:n lv) p-arvo | 77 (58; 87) < 0,0001 | |
Tutkittavat, joilla ei ollut yhtään verenvuotoa, % | 64,1 | 42,3 |
ABR-mediaani (IQR) | 0,00 (0,00; 1,04) | 1,06 (0,00; 3,74) |
1 Perustuu negatiiviseen binomimalliin.
2 Prospektiivinen havainnointitutkimus (OBS16221).
ABR = vuositasolla laskettu verenvuotomäärä; lv = luottamusväli; IQR = kvartiiliväli, 25. persentiilin ja 75. persentiilin välinen alue.
Potilaskohtainen vertailu (N = 26) ABR-määristä tarvittaessa toteutettavan ALTUVOCT-hoidon ensimmäisten 26 viikon aikana verrattuna ABR-määriin seuraavien 26 viikon aikana, jolloin tutkittavat saivat viikoittaista ALTUVOCT-estohoitoa (haara B), osoitti verenvuotojen vähentyneen kliinisesti merkittävästi 97 %:lla viikoittaista estohoitoa saaneilla tutkittavilla. Niiden tutkittavien määrä, joilla ei esiintynyt yhtään verenvuotoa, suureni 0:sta 76,9 %:iin.
Teho verenvuotojen hallinnassa
Aikuisilla ja nuorilla tehdyssä tutkimuksessa (XTEND‑1) kaikkiaan 362 verenvuototapahtumaa hoidettiin ALTUVOCT-valmisteella. Useimmat niistä ilmenivät tarvittaessa toteutettavaa hoitoa saaneessa haarassa B. Useimmat verenvuototapahtumat olivat nivelten verenvuotoja. Tutkittavat arvioivat ensimmäisellä injektiolla saavutettua vastetta vähintään 8 tuntia hoidon jälkeen. Vaste arvioitiin 4 pisteen asteikolla erinomaiseksi, hyväksi, kohtalaiseksi tai vasteen puuttumiseksi. Yhteenveto hoidon tehosta ≥ 12-vuotiaiden tutkittavien verenvuototapahtumien hallinnassa esitetään taulukossa 5. Verenvuototapahtumien hallinta oli samankaltaista kaikissa hoitohaaroissa.
Taulukko 5: Yhteenveto hoidon tehosta ≥ 12-vuotiaiden tutkittavien verenvuototapahtumien hallinnassa
Verenvuototapahtumien lukumäärä | (N = 362) | |
Verenvuototapahtuman hoitoon annettujen injektioiden lukumäärä, N (%) | 1 injektio 2 injektiota > 2 injektiota | 350 (96,7) 11 (3,0) 1 (0,3) |
Verenvuototapahtuman hoitoon annetun kokonaisannoksen mediaani (IU/kg) (IQR) | 50,93 (50,00; 51,85) | |
Arviointikelpoisten injektioiden lukumäärä | (N = 332) | |
Verenvuototapahtuman hoitovaste, N (%) | Erinomainen tai hyvä Kohtalainen Ei vastetta | 315 (94,9) 14 (4,2) 3 (0,9) |
Perioperatiivinen verenvuotojen hoito
Perioperatiivista hemostaasia arvioitiin 49 suuren leikkauksen yhteydessä 41 tutkittavalla (32 aikuista ja 9 nuorta ja lasta) faasin 3 tutkimuksissa. Näistä 49 suuresta leikkauksesta 48 leikkauksessa tarvittiin yksi preoperatiivinen annos hemostaasin ylläpitämiseksi leikkauksen aikana. Yhden rutiininomaisen estohoidon aikana tehdyn leikkauksen yhteydessä ei annettu preoperatiivista kyllästysannosta leikkauspäivänä eikä ennen leikkausta. Annoksen mediaani yhtä preoperatiivista injektiota kohti oli 50 IU/kg (vaihteluväli 12,7–84,7). Perioperatiivisessa vaiheessa (leikkausta edeltävästä päivästä 14. leikkauksen jälkeiseen päivään) injektioiden keskimääräinen (keskihajonta) kokonaisannos oli 171,85 (51,97) IU/kg ja lukumäärä 3,9 (1,4).
Suuren leikkauksen aikaisen hemostaattisen vasteen kliininen arviointi tehtiin 4 pisteen asteikolla (erinomainen, hyvä, kohtalainen tai huono / ei vastetta). ALTUVOCT-valmisteen hemostaattinen vaikutus arvioitiin erinomaiseksi tai hyväksi 48 leikkauksessa 49:stä (98 %). Arvioitu vaste ei ollut ”huono / ei vastetta” tai ”puuttuu” yhdenkään leikkauksen yhteydessä.
Arvioituja suuria leikkauksia olivat muun muassa suuret ortopediset toimenpiteet, kuten nivelten artroplastiat (polven, lonkan ja kyynärpään tekonivelleikkaukset), nivelten uusintaleikkaukset ja nilkan fuusioleikkaukset. Muita suuria leikkauksia olivat poskihampaan poisto, hampaan restaurointi ja hampaan poisto, ympärileikkaus, verisuonten epämuodostuman poisto, tyrän korjausleikkaus ja nenän/leuan korjausleikkaus. Lisäksi arvioitiin 25 pientä leikkausta. Kaikissa saatavilla olevissa tapauksissa hemostaasin raportoitiin olevan ”erinomainen”.
Immunogeenisuus
Immunogeenisuutta arvioitiin kliinisissä ALTUVOCT-tutkimuksissa aiemmin hoidetuilla aikuisilla ja lapsilla, joilla oli diagnosoitu vaikea A‑hemofilia. Kliinisissä tutkimuksissa ei todettu ALTUVOCT-inhibiittorien kehittymistä.
Faasin 3 kliinisissä tutkimuksissa (hoidon mediaanikesto 96,3 viikkoa) 4 arviointikelpoiselle potilaalle 276:sta (1,4 %) kehittyi ohimeneviä lääkevasta-aineita (ADA) hoidon aikana. Näyttöä lääkevasta-aineiden vaikutuksesta farmakokinetiikkaan, tehoon tai turvallisuuteen ei todettu.
Pediatriset potilaat
Rutiininomainen estohoito
Alle 12-vuotiaille lapsille rutiininomaisena estohoitona annetun viikoittaisen ALTUVOCT-annoksen 50 IU/kg tehoa arvioitiin vuositasolla laskettujen verenvuotomäärien (ABR) perusteella. Kaikkiaan 74 lasta (joista 38 oli < 6-vuotiaita ja 36 oli 6 – < 12-vuotiaita) määrättiin saamaan ALTUVOCT-valmistetta rutiininomaisena estohoitona annoksella 50 IU/kg laskimoon kerran viikossa 52 viikon ajan. Kaikilla 74 tutkittavalla, jotka saivat rutiininomaista estohoitoa, ABR-keskiarvo (95 %:n lv) oli 0,9 (0,6; 1,4) ja ABR-mediaani (Q1; Q3) oli 0 (0; 1,0) hoidettujen verenvuotojen osalta.
Herkkyysanalyysissä (N = 73), jonka ulkopuolelle jätettiin yksi tutkittava, joka ei saanut pitkään aikaan tutkimussuunnitelmassa määriteltyä viikoittaista estohoitoa, ABR-keskiarvo (95 %:n lv) oli 0,6 (0,4; 0,9) hoidetuille verenvuodoille [mediaani (Q1; Q3) 0 (0; 1,0)]. 47 lapsella (64,4 %) ei ollut hoitoa vaativia verenvuototapahtumia. Hoidettujen spontaanien verenvuotojen ABR-keskiarvo (95 %:n lv) oli 0,2 (0; 0,3) [mediaani (Q1; Q3) 0 (0; 0)]. Hoidettujen nivelen verenvuotojen ABR‑keskiarvo (95 %:n lv) oli 0,3 (0,2; 0,6) ja mediaani (Q1; Q3) oli 0 (0; 0).
Verenvuotojen hallinta
Valmisteen tehoa verenvuotojen hallinnassa arvioitiin pediatrisessa tutkimuksessa < 12-vuotiailla lapsilla lukuun ottamatta yhtä tutkittavaa, joka ei saanut pitkään aikaan tutkimussuunnitelmassa määriteltyä viikoittaista estohoitoa. ALTUVOCT-valmisteella hoidettiin yhteensä 43 verenvuototapahtumaa. Kaikista verenvuototapahtumista 95,3 % saatiin hallintaan yhdellä 50 IU/kg ALTUVOCT-injektiolla. Verenvuototapahtuman hoitamiseksi annetun kokonaisannoksen mediaani (Q1; Q3) oli 52,6 IU/kg (50,0–55,8).
Farmakokinetiikka
ALTUVOCT-valmisteen farmakokinetiikkaa arvioitiin faasin 3 XTEND‑1-tutkimuksessa, johon osallistui 159 aikuista ja nuorta, ja faasin 3 XTEND‑Kids-tutkimuksessa, johon osallistui 74 iältään < 12-vuotiasta lasta. Tutkittavat saivat viikoittain 50 IU/kg injektiona laskimoon. Alle 12-vuotiaiden lasten ryhmässä 37 tutkittavalta oli saatavana ALTUVOCT-kerta-annosten farmakokineettiset profiilit.
Efanesoktokogi alfan puoliintumisajan on osoitettu olevan noin 4-kertainen verrattuna tekijä VIII ‑valmisteisiin, joiden puoliintumisaika on tavanomainen, ja noin 2,5–3-kertainen verrattuna tekijä VIII ‑valmisteisiin, joiden puoliintumisaika on tavanomaista pidempi. ALTUVOCT-kerta-annoksen jälkeiset farmakokineettiset muuttujat esitetään taulukossa 6. Farmakokineettiset muuttujat perustuivat aPTT-pohjaisella yksivaiheisella hyytymismäärityksellä mitattuun tekijä VIII:n aktiivisuuteen plasmassa. Yhden 50 IU/kg:n annoksen jälkeen ALTUVOCT-valmisteella saavutettiin korkea ja pitkäkestoinen tekijä VIII:n aktiivisuus, ja sen puoliintumisaika oli pitkä kaikissa ikäkohorteissa. Pediatrisissa kohorteissa AUC-arvo pyrki suurenemaan ja puhdistuma pienenemään iän noustessa. Farmakokineettinen profiili vakaan tason vaiheessa (viikolla 26) oli verrattavissa ensimmäisen annoksen jälkeen todettuun farmakokineettiseen profiiliin.
Taulukko 6: ALTUVOCT-kerta-annoksen jälkeiset farmakokineettiset muuttujat iän mukaan (yksivaiheinen hyytymismääritys, kun käytössä on Actin-FSL-reagenssi)
Farmakokineettiset muuttujat Keskiarvo (SD) | Pediatrinen tutkimus | Aikuisille ja nuorille tehty tutkimus | ||
1 – < 6 vuotta | 6 – < 12 vuotta | 12 – < 18 vuotta | Aikuiset | |
N = 18 | N = 18 | N = 25 | N = 134 | |
AUC0-tau, IU*h/dl | 6 800 (1 120)b | 7 190 (1 450) | 8 350 (1 550) | 9 850 (2 010)a |
t½z, h | 38,0 (3,72) | 42,4 (3,70) | 44,6 (4,99) | 48,2 (9,31) |
CL, ml/h/kg | 0,742 (0,121) | 0,681 (0,139) | 0,582 (0,115) | 0,493 (0,121)a |
Vss, ml/kg | 36,6 (5,59) | 38,1 (6,80) | 34,9 (7,38) | 31,0 (7,32)a |
MRT, h | 49,6 (5,45) | 56,3 (5,10) | 60,0 (5,54) | 63,9 (10,2)a |
Cmax, IU/dl | 143 (57,8) | 113 (22,7) | 118 (24,9) | 133 (33,8) |
Inkrementaalinen saanto, IU/dl per IU/kg | 2,81 (1,1) | 2,24 (0,437) | 2,34 (0,490) | 2,64 (0,665) |
a Laskelma perustuu 128 profiiliin.
b N = 17
AUC0-tau = aktiivisuuden aikakäyrän alle jäävä alue annosvälin aikana, CL = puhdistuma, MRT = keskimääräinen elimistössä oloaika, SD = keskihajonta, t½z = terminaalinen puoliintumisaika, Vss = jakaantumistilavuus vakaan tason vaiheessa, Cmax = maksimiaktiivisuus
XTEND‑1-tutkimuksessa aikuisten viikoittaisella ALTUVOCT-estohoidolla saavutettu tekijä VIII:n aktiivisuus pysyi vakaan tason vaiheessa normaalina tai lähes normaalina (> 40 IU/dl) keskimäärin (keskihajonta) 4,1 (0,7) vuorokauden ajan. Tekijä VIII:n aktiivisuus säilyi suurempana kuin 10 IU/dl 83,5 %:lla aikuisista ja nuorista tutkittavista koko tutkimuksen ajan. Alle 12-vuotiaiden lasten viikoittaisella ALTUVOCT-hoidolla saavutettu tekijä VIII:n aktiivisuus pysyi vakaan tason vaiheessa normaalina tai lähes normaalina (> 40 IU/dl) 2–3 vuorokauden ajan ja tasolla > 10 IU/dl noin 7 vuorokauden ajan (ks. taulukko 7).
Taulukko 7: ALTUVOCT-valmisteen farmakokineettiset muuttujat vakaan tason vaiheessa iän mukaan (yksivaiheinen hyytymismääritys, kun käytössä on Actin-FSL-reagenssi)
Farmakokineettiset muuttujat Keskiarvo (SD) | Pediatrinen tutkimusa | Aikuisille ja nuorille tehty tutkimusa | ||
1 – < 6 vuotta | 6 – < 12 vuotta | 12 – < 18 vuotta | Aikuiset | |
N = 37 | N = 36 | N = 24 | N = 125 | |
Huippuarvo, IU/dl | 136 (48,9) (N = 35) | 131 (36,1) (N = 35) | 124 (31,2) | 150 (35,0) (N = 124) |
Inkrementaalinen saanto, IU/dl per IU/kg | 2,22 (0,83) (N = 35) | 2,10 (0,73) (N = 35) | 2,25 (0,61) (N = 22) | 2,64 (0,61) (N = 120) |
Aika tason 40 IU/dl saavuttamiseen, h | 68,0 (10,5)b | 80,6 (12,3)b | 81,5 (12,1)c | 98,1 (20,1)c |
Aika tason 20 IU/dl saavuttamiseen, h | 109 (14,0)b | 127 (14,5)b | 130 (15,7)c | 150 (27,7)c |
Aika tason 10 IU/dl saavuttamiseen, h | 150 (18,2)b | 173 (17,1)b | 179 (20,2)c | 201 (35,7)c |
Pienin arvo, IU/dl | 10,9 (19,7) (N = 36) | 16,5 (23,7) | 9,23 (4,77) (N = 22) | 18,0 (16,6) (N = 123) |
a Vakaan tason vaiheen huippuarvo, pienin arvo ja inkrementaalinen saanto laskettiin mittauksilla, jotka olivat saatavilla viikolla 52 / tutkimuksen lopussa farmakokineettisten näytteiden ottamiskäynnillä.
b Aika tekijä VIII:n aktiivisuuden saavuttamiseen ennustettiin pediatristen potilaiden populaatiofarmakokineettisestä mallista.
c Aika tekijä VIII:n aktiivisuuden saavuttamiseen ennustettiin aikuispotilaiden populaatiofarmakokineettisestä mallista.
Huippuarvo = 15 minuuttia annoksen jälkeen vakaan tason vaiheessa; pienin arvo = annosta edeltävä tekijä VIII:n aktiivisuus vakaan tason vaiheessa; SD = keskihajonta
Prekliiniset tiedot turvallisuudesta
Rotilla ja apinoilla tehtyjen, toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten (mukaan lukien turvallisuusfarmakologiset mittaukset) ja veriyhteensopivuutta koskevan in vitro ‑tutkimuksen tulokset eivät viittaa erityiseen vaaraan ihmisille. Genotoksisuutta, karsinogeenisuutta, lisääntymistoksisuutta tai alkion- ja sikiönkehitystä koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Sakkaroosi
Kalsiumklorididihydraatti (E 509)
Histidiini
Arginiinihydrokloridi
Polysorbaatti 80 (E 433)
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Vain mukana toimitettua adapteria ja infuusiovälineistöä saa käyttää, koska hyytymistekijä VIII:n tarttuminen joidenkin injektiovälineiden sisäpintoihin voi aiheuttaa hoidon epäonnistumisen.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Kestoajan puitteissa lääkevalmistetta voidaan säilyttää huoneenlämmössä (enintään 30 °C:ssa) yhtäjaksoisesti enintään 6 kuukautta. Päivämäärä, jolloin lääkevalmiste otetaan pois jääkaapista, tulee merkitä pakkaukseen. Lääkevalmistetta ei saa siirtää enää takaisin jääkaappiin huoneenlämmössä säilyttämisen jälkeen. Valmistetta ei saa käyttää injektiopulloon painetun viimeisen käyttöpäivämäärän jälkeen tai 6 kuukauden kuluttua siitä, kun pakkaus on otettu jääkaapista, riippuen siitä, kumpi päivä on aikaisempi.
Käyttökuntoon saattamisen jälkeen
Lääkevalmiste on käytettävä heti käyttökuntoon saattamisen jälkeen. Ellei sitä käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ALTUVOCT injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 250 IU (infuusiovälineistö) (317,43 €)
500 IU (L:ei) 500 IU (infuusiovälineistö) (618,82 €)
1000 IU (L:ei) 1000 IU (infuusiovälineistö) (1196,65 €)
2000 IU (L:ei) 2000 IU (infuusiovälineistö) (2314,40 €)
3000 IU (L:ei) 3000 IU (infuusiovälineistö) (3381,30 €)
4000 IU (L:ei) 4000 IU (infuusiovälineistö) (4448,20 €)
PF-selosteen tieto
Jokainen pakkaus ALTUVOCT 250 IU, 500 IU, 750 IU, 1 000 IU, 2 000 IU, 3 000 IU ja 4 000 IU injektiokuiva-ainetta ja liuotinta, liuosta varten, sisältää seuraavat:
- kuiva-aine lasisessa (tyyppi I) injektiopullossa, jossa on klorobutyylikumitulppa
- steriili injektiopullon adapteri käyttökuntoon saattamista varten
- 3 ml liuotinta lasisessa esitäytetyssä ruiskussa, jossa on bromobutyylikumista valmistettu mäntätulppa
- männän varsi
- steriili infuusiovälineistö.
Valmisteen kuvaus:
Kuiva-aine: kylmäkuivattu, valkoinen tai luonnonvalkoinen jauhe tai kakku.
Liuotin: kirkas, väritön liuos
pH: 6,5–7,2
Osmolaalisuus: 586–688 mOsm/kg
Käyttö- ja käsittelyohjeet
ALTUVOCT annetaan laskimoon sen jälkeen, kun injektiokuiva-aine on saatettu käyttökuntoon ruiskun sisältämällä liuottimella. Injektiopulloa on pyöriteltävä varovasti, kunnes injektiokuiva-aine on kokonaan liuennut. Käyttökuntoon saatetun liuoksen on oltava kirkasta ja väritöntä tai hiukan opaalinhohtoista. Älä käytä liuosta, jos se on sameaa tai sisältää sakkaa.
Käytä aina aseptista tekniikkaa.
Lisätietoja käyttökuntoon saattamisesta
ALTUVOCT annetaan injektiona laskimoon sen jälkeen, kun injektiokuiva-aine on liuotettu esitäytetyn ruiskun sisältämällä liuottimella. ALTUVOCT-pakkaus sisältää seuraavat:
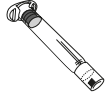 |  | 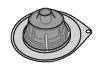 | 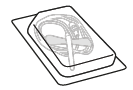 | |
| A. injektiokuiva-ainepullo | B. 3 ml liuotinta esitäytetyssä ruiskussa | C. männän varsi | D. injektiopullon adapteri | E. infuusiovälineistö |
Tarvitset myös steriilejä alkoholipyyhkeitä (F). Tämä laite ei sisälly ALTUVOCT-pakkaukseen.
Voit vetää liuosta useista injektiopulloista erilliseen isoon ruiskuun (G). Jos isoa ruiskua ei ole saatavilla, noudata vaiheita 6–8, joissa kuvataan liuoksen antaminen kustakin ruiskusta.
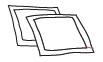 |  |
| F. alkoholipyyhkeitä | G. iso ruisku |
ALTUVOCT-valmistetta ei saa sekoittaa muiden injektio- tai infuusionesteiden kanssa.
Pese kädet ennen pakkauksen avaamista.
Käyttökuntoon saattaminen
| 1. | Valmistele injektiopullo | |||
| a. | Poista injektiopullon korkki Pitele injektiokuiva-ainepulloa (A) puhtaalla, tasaisella alustalla ja poista muovikorkki. | 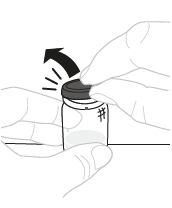 | ||
| b. | Puhdista injektiopullon yläosa Pyyhi injektiopullon yläosa alkoholipyyhkeellä. Puhdistuksen jälkeen varmista, ettei injektiopullon yläosa pääse kosketuksiin minkään kanssa. | 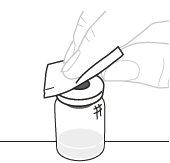 | ||
| c. | Avaa injektiopullon adapterin pakkaus Irrota paperinen suojus injektiopullon adapterin pakkauksesta (D). Älä koske injektiopullon adapteriin äläkä poista sitä pakkauksesta. | 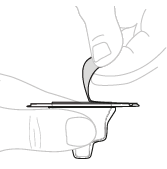 | ||
| d. | Kiinnitä injektiopullon adapteri Aseta injektiopullon adapterin pakkaus suoraan injektiopullon yläosan päälle. Paina tiukasti alaspäin, kunnes adapteri napsahtaa paikalleen. Piikki läpäisee injektiopullon tulpan. | 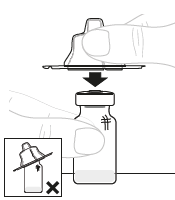 | ||
| 2. | Valmistele ruisku | |||
| a. | Kiinnitä männän varsi Kiinnitä männän varsi (C) 3 ml:n ruiskuun (B). Käännä männän vartta myötäpäivään, kunnes se on tiukasti paikoillaan. | 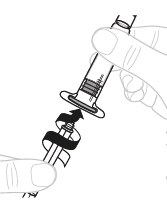 | ||
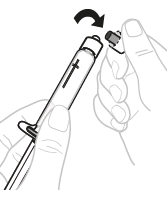
| b. | Irrota ruiskun korkki Napsauta 3 ml:n ruiskun valkoisen korkin yläosa irti katkoviivan kohdalta ja siirrä se syrjään.
|  | ||
| 3. | Liitä ruisku injektiopulloon | |||
| a. | Poista injektiopullon adapterin pakkaus Nosta pakkaus pois injektiopullon adapterin päältä ja hävitä se. | 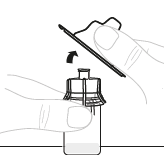 | ||
| b. | Liitä ruisku injektiopullon adapteriin Pitele injektiopullon adapteria sen alapäästä. Aseta ruiskun kärki injektiopullon adapterin yläosaan. Käännä ruiskua myötäpäivään, kunnes se on tiukasti paikoillaan. | 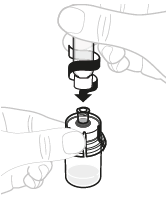 | ||
| 4. | Liuota kuiva-aine liuottimella | |||
| a. | Lisää liuotin injektiopulloon Paina männän vartta hitaasti alaspäin, kunnes kaikki liuotin on injektiopullossa. | 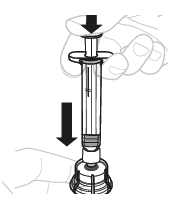 | ||
| b. | Liuota kuiva-aine Pidä peukalo männän varren päällä ja pyörittele injektiopulloa varovasti, kunnes kuiva-aine on liuennut. Ei saa ravistaa. | 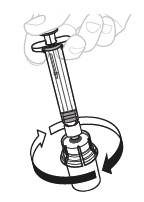 | ||
| c. | Tarkasta liuos Tarkasta liuos ennen antoa. Sen on oltava kirkasta ja väritöntä. Älä käytä liuosta, jos se on sameaa tai jos siinä näkyy hiukkasia. | |||
| 5. | Jos käytät useampia injektiopulloja | |||
| Jos annokseen tarvitaan useampia injektiopulloja, noudata seuraavia vaiheita (5a ja 5b). Muussa tapauksessa voit siirtyä vaiheeseen 6. | ||||
| a. | Toista vaiheet 1–4 Toista vaiheet 1–4 kaikkien injektiopullojen kohdalla, kunnes olet valmistellut riittävästi liuosta annosta varten. Poista 3 ml:n ruiskut kustakin injektiopullosta (ks. vaihe 6b), mutta jätä liuos injektiopulloihin. | 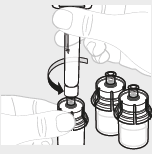 | ||
| b. | Käytä isoa ruiskua (G) Kunkin injektiopullon kohdalla liitä iso ruisku (G) injektiopullon adapteriin (ks. vaihe 3b) ja suorita vaihe 6 vetämällä liuos kustakin injektiopullosta isoon ruiskuun. Jos tarvitset vain osan injektiopullon sisällöstä, tarkista ruiskuun vedettävä määrä ruiskun mitta-asteikosta. | 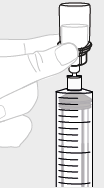 | ||
| 6. | Vedä liuos ruiskuun | |||
| a. | Vedä liuos injektiopullosta Varmista, että ruiskun kärki osoittaa ylöspäin. Vedä männän vartta hitaasti niin, että kaikki liuos virtaa ruiskuun. | 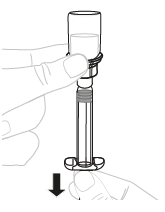 | ||
| b. | Irrota ruisku Pidä kiinni injektiopullon adapterista, kun irrotat ruiskun injektiopullosta. Irrota ruisku kääntämällä sitä vastapäivään. | 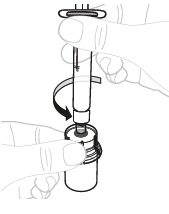 | ||
 Älä koske korkin sisäpuoleen tai ruiskun kärkeen.
Älä koske korkin sisäpuoleen tai ruiskun kärkeen.On suositeltavaa käyttää ALTUVOCT heti käyttökuntoon saattamisen jälkeen (ks. kohta Kestoaika).
Antaminen
| 7. | Valmistaudu antamaan pistos | ||
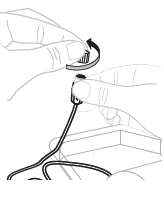
| a. | Irrota letkun korkki Avaa infuusiovälineistön (E) pakkaus (älä käytä infuusiovälineistöä, jos siinä on vaurioita). Irrota letkun korkki.
|  | |
| b. | Kiinnitä ruisku Kiinnitä käyttövalmis ruisku infuusiovälineistön letkun päähän kääntämällä ruiskua myötäpäivään. | 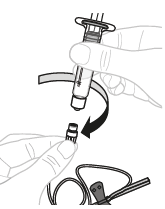 | |
| c. | Valmistele pistoskohta Käytä tarvittaessa kiristyssidettä. Pyyhi pistoskohta alkoholipyyhkeellä (F). | 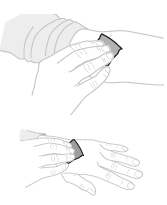 | |
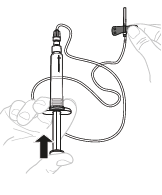
| d. | Poista ilma ruiskusta ja letkustosta Varmista, että ruiskun kärki osoittaa ylöspäin ja poista ilma painamalla männän vartta varovasti. Älä työnnä liuosta neulan läpi.
|  | |
| 8. | Pistä liuos | ||
| a. | Aseta neula Poista neulansuojus. Työnnä neula laskimoon ja poista kiristysside, jos käytit sellaista.
| ||
| b. | Pistä liuos Käyttövalmis liuos on pistettävä laskimoon 1–10 minuutin aikana. Antonopeus on määritettävä potilasmukavuuden perusteella. | ||
| 9. | Hävitä tarvikkeet turvallisesti | ||
| a. | Poista neula Poista neula. Käännä neulansuojus takaisin neulan päälle. Sen pitäisi napsahtaa paikoilleen. | 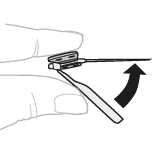 | |
| b. | Turvallinen hävittäminen Varmista, että kaikki pakkauksessa toimitetut tarvikkeet (pakkausmateriaaleja lukuun ottamatta) hävitetään turvallisesti lääkejätteelle tarkoitettuun säiliöön.
| ||
 Älä koske letkuston paljaaseen päähän.
Älä koske letkuston paljaaseen päähän. Ilman pistäminen laskimoon voi olla vaarallista.
Ilman pistäminen laskimoon voi olla vaarallista. Halutessasi voit kiinnittää neulan muovisiivekkeet paikalleen pistoskohtaan laastarilla, jotta neula ei pääse liikkumaan.
Halutessasi voit kiinnittää neulan muovisiivekkeet paikalleen pistoskohtaan laastarilla, jotta neula ei pääse liikkumaan. Tarvikkeita ei saa käyttää uudelleen.
Tarvikkeita ei saa käyttää uudelleen.Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ALTUVOCT injektiokuiva-aine ja liuotin, liuosta varten
250 IU 250 IU
500 IU 500 IU
1000 IU 1000 IU
2000 IU 2000 IU
3000 IU 3000 IU
4000 IU 4000 IU
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD02
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
Äyritie 18
01510 Vantaa
0201 558 840
www.sobi.fi
mail.fi@sobi.com