
RYDAPT kapseli, pehmeä 25 mg
Vaikuttavat aineet ja niiden määrät
Yksi pehmeä kapseli sisältää 25 mg midostauriinia.
Apuaineet, joiden vaikutus tunnetaan
Yksi pehmeä kapseli sisältää noin 83 mg vedetöntä etanolia ja 415 mg makrogoliglyserolihydroksistearaattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Pehmeä kapseli (kapseli).
Kliiniset tiedot
Käyttöaiheet
Rydapt on tarkoitettu käytettäväksi:
- äskettäin todetun akuutin myelooisen leukemian (AML) hoitoon aikuispotilaille, jotka ovat FLT3-mutaatiopositiivisia (ks. kohta Annostus ja antotapa). Rydaptia käytetään ensin yhdessä tavanomaisen daunorubisiinilla ja sytarabiinilla toteutettavan induktiohoidon ja suuriannoksisella sytarabiinilla toteutettavan konsolidaatiosolunsalpaajahoidon kanssa, ja sen jälkeen täydellisen vasteen saavuttaneille potilaille ylläpitohoitona, jossa Rydapt on ainoa lääke;
- ainoana lääkkeenä aggressiivista systeemistä mastosytoosia (ASM), systeemistä mastosytoosia, johon liittyy hematologinen maligniteetti (SM‑AHN), tai syöttösoluleukemiaa (MCL) sairastavien aikuispotilaiden hoitoon.
Ehto
Ainoastaan syöpälääkkeiden antoon perehtyneen lääkärin tulee aloittaa hoito.
Annostus ja antotapa
Rydapt-hoidon aloittamisesta vastaa syöpähoitojen toteuttamiseen perehtynyt lääkäri.
Ennen midostauriinin käyttöä AML-potilaiden FLT3-mutaatiostatus (internal tandem duplication [ITD] tai tyrosine kinase domain [TKD]) on vahvistettava validoidulla tutkimuksella.
Annostus
Rydapt otetaan suun kautta kahdesti vuorokaudessa noin 12 tunnin välein. Kapselit otetaan ruoan kanssa (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Ennaltaehkäisevää pahoinvointilääkitystä annetaan paikallisen hoitokäytännön mukaisesti potilaan sietokyvyn mukaan.
AML
Suositeltava Rydapt-annos on 50 mg suun kautta kahdesti vuorokaudessa.
Induktio- ja konsolidaatiohoidossa Rydapt annetaan solunsalpaajahoitojaksojen päivinä 8–21. Tämän jälkeen sitä käytetään täydellisen vasteen saavuttaneille potilaille joka päivä ylläpitohoitona ainoana lääkkeenä relapsiin saakka enintään 12 hoitojakson ajan siten, että kunkin hoitojakson pituus on 28 vuorokautta (ks. kohta Käyttöaiheet). Potilailla, jotka saavat hematopoieettisen kantasolusiirron, Rydapt-hoito on lopetettava 48 tuntia ennen siirron esihoitoa.
Annosmuutokset AML:n hoidossa
Taulukossa 1 esitetään Rydapt-valmisteen annosmuutossuositukset AML:n hoidossa.
Taulukko 1 Rydapt-hoidon tauottamista, annoksen pienentämistä ja hoidon lopettamista koskevat suositukset AML:n hoidossa
Vaihe | Kriteerit | Rydapt-valmisteen annostelu |
Induktio-, konsolidaatio- ja ylläpitohoito | Asteen 3/4 keuhkoinfiltraatit | Rydapt-hoito tauotetaan hoitojakson loppuun saakka. Rydapt-hoito aloitetaan uudestaan samalla annoksella, kun infiltraatit korjautuvat asteelle ≤ 1. |
Muut asteen 3/4 ei-hematologiset toksisuudet | Rydapt-hoito tauotetaan, kunnes toksisuus, jonka katsotaan ainakin mahdollisesti liittyvän Rydapt-hoitoon, on korjaantunut asteelle ≤ 2. Tämän jälkeen Rydapt aloitetaan uudestaan. | |
QTc-aika > 470 msek, ≤ 500 msek | Rydapt-annostus pienennetään tasolle 50 mg kerran vuorokaudessa loppujakson ajaksi. Rydapt aloitetaan uudestaan aloitusannoksella seuraavassa hoitojaksossa, jos QTc-aika kohenee tasolle ≤ 470 msek kyseisen jakson alussa. Muussa tapauksessa jatketaan annostuksella 50 mg Rydaptia kerran vuorokaudessa. | |
QTc-aika > 500 msek | Rydapt-hoito tauotetaan tai keskeytetään loppujakson ajaksi. Jos QTc kohenee tasolle ≤ 470 msek juuri ennen seuraavaa jaksoa, Rydapt aloitetaan uudestaan aloitusannoksella. Jos QTc-aika ei kohene seuraavan hoitojakson alkuun mennessä, Rydaptia ei anneta kyseisen jakson aikana. Rydapt-hoito voidaan tauottaa niin moneksi jaksoksi kuin tarpeen, kunnes QTc kohenee. | |
Vain ylläpitohoito | Asteen 4 neutropenia (absoluuttinen neutrofiilien määrä < 0,5 x 109/l) | Rydapt-hoito tauotetaan, kunnes absoluuttinen neutrofiilien määrä on ≥ 1,0 x 109/l. Tämän jälkeen jatketaan annoksella 50 mg kahdesti vuorokaudessa. Jos neutropenia (absoluuttinen neutrofiilien määrä < 1,0 x 109/l) jatkuu > 2 viikkoa ja sen epäillään liittyvän Rydapt-hoitoon, Rydapt-hoito lopetetaan. |
Pitkittynyt asteen 1/2 toksisuus | Pitkittynyt asteen 1 tai 2 toksisuus, joka on potilaan mielestä sietämätön, voi johtaa jopa 28 vrk pituiseen hoidon keskeytykseen. | |
ANC: Absoluuttinen neutrofiilimäärä | ||
ASM, SM-AHN ja MCL
Rydapt-valmisteen suositeltava aloitusannos on 100 mg suun kautta kahdesti vuorokaudessa.
Hoitoa jatketaan niin pitkään kuin siitä on havaittavaa kliinistä hyötyä tai kunnes ilmenee sellaista toksisuutta joka voi olla hoidon esteenä.
Annosmuutokset ASM:n, SM-AHN:n ja MCL:n hoidossa
Taulukossa 2 esitetään Rydapt-valmisteen annosmuutossuositukset ASM:n, SM-AHN:n ja MCL:n hoidossa.
Taulukko 2 Rydapt-hoidon tauottamista, annoksen pienentämistä ja hoidon lopettamista koskevat suositukset ASM:n, SM-AHN:n ja MCL:n hoidossa
Kriteerit | Rydapt-valmisteen annostelu |
Absoluuttinen neutrofiilimäärä (ANC) < 1,0 x 109/l potilailla, joilla ei ole syöttösoluleukemiaa, kun haitan katsotaan liittyvän Rydapt-hoitoon, tai ANC alle 0,5 x 109/l potilailla, joiden ANC-lähtöarvo on 0,5–1,5 x 109/l, kun haitan katsotaan liittyvän Rydapt-hoitoon | Rydapt tauotetaan, kunnes ANC on ≥ 1,0 x 109/l. Tämän jälkeen Rydapt-hoitoa jatketaan annoksella 50 mg x 2. Jos potilas sietää tämän, annosta suurennetaan tasolle 100 mg x 2. Rydapt lopetetaan, jos ANC on alhainen > 21 vrk ajan ja tämän epäillään liittyvän Rydapt-hoitoon. |
Trombosyytit alle 50 x 109/l potilailla, joilla ei ole syöttösoluleukemiaa, kun haitan katsotaan liittyvän Rydapt-hoitoon, tai trombosyytit alle 25 x 109/l potilailla, joiden trombosyyttien lähtöarvo on 25–75 x 109/l, kun haitan katsotaan liittyvän Rydapt-hoitoon | Rydapt tauotetaan, kunnes trombosyyttimäärä on ≥ 50 x 109/l. Tämän jälkeen Rydapt-hoitoa jatketaan annoksella 50 mg x 2. Jos potilas sietää tämän, annosta suurennetaan tasolle 100 mg x 2. Rydapt lopetetaan, jos trombosyyttimäärä on pieni > 21 vrk ajan ja tämän epäillään liittyvän Rydapt-hoitoon. |
Hemoglobiini alle 8 g/dl potilailla, joilla ei ole syöttösoluleukemiaa, kun haitan katsotaan liittyvän Rydapt-hoitoon, tai henkeä uhkaava anemia potilailla, joiden hemoglobiinilähtöarvo on 8–10 g/dl, kun haitan katsotaan liittyvän Rydapt-hoitoon | Rydapt tauotetaan, kunnes hemoglobiini on ≥ 8 g/dl. Tämän jälkeen Rydapt-hoitoa jatketaan annoksella 50 mg x 2. Jos potilas sietää tämän, annosta suurennetaan tasolle 100 mg x 2. Rydapt lopetetaan, jos hemoglobiini on matala > 21 vrk ajan ja tämän epäillään liittyvän Rydapt-hoitoon. |
Asteen 3/4 pahoinvointi ja/tai oksentelu optimaalisesta pahoinvointilääkityksestä huolimatta | Rydapt tauotetaan 3 päivän (6 annoksen) ajaksi. Tämän jälkeen Rydapt-hoitoa jatketaan annoksella 50 mg x 2. Jos potilas sietää tämän, annosta suurennetaan vähitellen tasolle 100 mg x 2. |
Muut asteen 3/4 ei-hematologiset toksisuudet | Rydapt tauotetaan, kunnes tapahtuma on korjaantunut asteelle ≤ 2. Tämän jälkeen Rydapt-hoitoa jatketaan annoksella 50 mg x 2. Jos potilas sietää tämän, annosta suurennetaan tasolle 100 mg x 2. Rydapt-hoito lopetetaan, jos toksisuus ei korjaannu 21 vrk kuluessa asteelle ≤ 2 tai vaikea toksisuus uusiutuu pienennetyllä Rydapt‑annoksella. |
CTCAE ‑kriteerien mukaiset vaikeusasteluokat: aste 1 = lievät oireet; 2 = keskivaikeat oireet; 3 = vaikeat oireet; 4 = henkeä uhkaavat oireet. | |
Väliin jääneet annokset
Jos annos jää väliin, potilaan on otettava seuraava annos tavanomaisen aikataulun mukaiseen aikaan.
Jos potilas oksentaa, hänen ei pidä ottaa ylimääräistä Rydapt-annosta. Seuraava annos otetaan tavanomaisen aikataulun mukaan.
Erityisryhmät
Iäkkäät potilaat (≥ 65-vuotiaat)
Annostusta ei tarvitse muuttaa yli 65-vuotiaita potilaita hoidettaessa (ks. kohta Farmakokinetiikka). 60-vuotiailla ja sitä vanhemmilla potilailla Rydaptia tulee käyttää vain, jos intensiivinen induktiokemoterapia soveltuu potilaalle, hänen toimintakykynsä on riittävä, eikä hänellä ole merkittäviä liitännäissairauksia.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidosta on niukasti kliinistä kokemusta ja loppuvaiheen munuaistautia sairastavien potilaiden hoidosta ei ole tietoja. (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta (Child–Pugh-luokka A tai B) (ks. kohta Farmakokinetiikka). Midostauriinin ja sen aktiivisen metaboliitin CGP62221:n altistus on merkittävästi pienempi vaikeaa maksan vajaatoimintaa sairastavilla potilailla kuin potilailla, joiden maksan toiminta on normaalia (ks. kohta Farmakokinetiikka). Tehoon liittyvät tiedot ovat kuitenkin puutteelliset annosmuutosten tarpeen arvioimiseksi vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Akuutti promyelosyyttinen leukemia
Rydaptin käyttöä potilaille, joilla on akuutti promyelosyyttinen leukemia ei ole tutkittu, eikä käyttöä näin ollen suositella tämän potilaspopulaation hoidossa.
Pediatriset potilaat
Pediatristen potilaiden AML:n hoidossa Rydapt-valmistetta ei pidä käyttää yhdessä intensiivisen yhdistelmäsolunsalpaajahoidon (mukaan lukien antrasykliinien, fludarabiinin ja sytarabiinin) kanssa, sillä tähän liittyy hitaan hematologisen toipumisen riski (kuten pitkittyneen, vaikean neutropenian ja trombosytopenian riski) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Antotapa
Rydapt on tarkoitettu otettavaksi suun kautta.
Kapselit niellään kokonaisina vesilasillisen kera. Niitä ei saa avata, murskata eikä pureskella, jotta varmistetaan asianmukainen annostus, ja vältetään kapselin sisällön epämiellyttävä maku.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Samanaikainen vahvojen CYP3A4-indusorien kuten rifampisiinin, mäkikuisman (Hypericum perforatum), karbamatsepiinin, entsalutamidin tai fenytoiinin käyttö (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Neutropenia ja infektiot
Neutropeniaa on esiintynyt potilailla, jotka ovat saaneet Rydapt-valmistetta ainoana lääkkeenä tai yhdessä solunsalpaajahoidon kanssa (ks. kohta Haittavaikutukset). Vaikea neutropenia (absoluuttinen neutrofiilien määrä < 0,5 x 109/l) yleensä korjautui, kun Rydapt-lääkitys tauotettiin tilanteen korjautumiseen asti tai kun ASM:n, SM-AHN:n ja MCL:n tutkimuksissa hoito lopetettiin. Valkosolujen määriä on seurattava säännöllisesti etenkin hoidon aloitusvaiheessa.
Jos potilaalle kehittyy selittämätön, vaikea neutropenia, Rydapt-hoito tauotetaan, kunnes absoluuttinen neutrofiilien määrä on ≥ 1,0 x 109/l, kuten taulukoissa 1 ja 2 suositellaan. Rydapt-hoito on lopetettava, jos potilaalle kehittyy toistuva tai pitkittynyt vaikea neutropenia, jonka epäillään liittyvän Rydapt-hoitoon (ks. kohta Annostus ja antotapa).
Aktiiviset, vakavat infektiot on saatava hallintaan ennen Rydapt-monoterapian aloittamista. Potilaita on seurattava infektioiden oireiden ja löydösten, myös laitteisiin liittyvien infektioiden varalta. Jos potilaalla todetaan infektio, asianmukainen hoito on aloitettava ripeästi ja Rydapt-hoito on lopetettava tarvittaessa.
Sydämen toimintahäiriö
Potilaat, joilla oli oireinen kongestiivinen sydämen vajaatoiminta, suljettiin pois kliinisistä tutkimuksista. ASM:ia, SM-AHN:ia tai MCL:aa sairastavilla potilailla tehdyissä tutkimuksissa esiintyi sydämen toimintahäiriöitä, kuten kongestiivista sydämen vajaatoimintaa (myös kuolemaan johtaneita tapauksia) ja vasemman kammion ejektiofraktion ohimenevää pienenemistä. Satunnaistetussa AML-potilailla tehdyssä tutkimuksessa Rydapt + solunsalpaajahoito ‑ryhmän ja lumelääke + solunsalpaajahoito ‑ryhmän välillä ei todettu eroja kongestiivisen sydämen vajaatoiminnan suhteen. Riskiryhmään kuuluvilla potilailla Rydapt-valmistetta on käytettävä varoen ja potilaan vointia on seurattava tarkoin arvioimalla vasemman kammion ejektiofraktio, kun se on kliinisesti aiheellista (lähtötilanteessa ja hoidon aikana).
QTc-ajan pitenemistä havaittiin midostauriinihoitoa saaneilla potilailla tavallista yleisemmin (ks. kohta Haittavaikutukset). Tälle havainnolle ei kuitenkaan löydetty selittävää mekanismia. Varovaisuus on tarpeen potilailla, joilla on QTc-ajan pitenemisen riski (esim. samanaikaisten lääkehoitojen ja/tai elektrolyyttitasapainon häiriöiden vuoksi). QT-ajan arviointia EKG:llä on harkittava, jos Rydaptia käytetään samanaikaisesti QT-aikaa mahdollisesti pidentävien lääkevalmisteiden kanssa.
Keuhkotoksisuus
Potilailla, jotka ovat saaneet Rydapt-valmistetta ainoana lääkkeenä tai yhdessä solunsalpaajahoidon kanssa, on esiintynyt interstitiaalista keuhkosairautta ja pneumoniittia. Jotkin tapaukset ovat johtaneet kuolemaan. Potilaita on seurattava interstitiaaliseen keuhkosairauteen tai pneumoniittiin viittaavien keuhko-oireiden varalta, ja Rydapt-hoito on lopetettava, jos potilaalla todetaan interstitiaaliseen keuhkosairauteen tai pneumoniittiin ilman tarttuvaa etiologiaa viittaavia asteen ≥ 3 keuhko-oireita (NCI:n CTCAE-kriteerit).
Alkio- ja sikiötoksisuus ja imetys
Raskaana oleville naisille on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä. Naisia, jotka voivat tulla raskaaksi, on kehotettava tekemään raskaustesti Rydapt-hoidon aloittamista edeltävien 7 vrk aikana ja käyttämään tehokasta ehkäisyä Rydapt-hoidon aikana ja vähintään 4 kuukauden ajan hoidon lopettamisen jälkeen.
Rydapt saattaa aiheuttaa vakavia haittavaikutuksia rintaruokittavalle lapselle, joten naisten on lopetettava rintaruokinta Rydapt-hoidon ajaksi ja vähintään 4 kuukaudeksi hoidon lopettamisen jälkeen (ks. kohta Raskaus ja imetys).
Pediatriset potilaat
Pediatristen potilaiden AML:n hoidossa Rydapt-valmistetta ei pidä käyttää yhdessä intensiivisen yhdistelmäsolunsalpaajahoidon (mukaan lukien antrasykliinien, fludarabiinin ja sytarabiinin) kanssa, sillä tähän liittyy hitaan hematologisen toipumisen riski (kuten pitkittyneen, vaikean neutropenian ja trombosytopenian riski) (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Vaikea munuaisten vajaatoiminta
Varovaisuutta on noudatettava, jos midostauriinin antoa harkitaan potilaille, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaistauti, ja näitä potilaita on seurattava tarkasti toksisuuden varalta (ks. kohta Farmakokinetiikka).
Yhteisvaikutukset
Varovaisuus on tarpeen, jos midostauriinia saavalle potilaalle määrätään samanaikaisesti CYP3A4-toimintaa voimakkaasti estäviä lääkevalmisteita kuten sienilääkkeitä (esim. ketokonatsoli), tiettyjä viruslääkkeitä (esim. ritonaviiri), makrolidiantibiootteja (esim. klaritromysiini) tai nefatsodonia, sillä ne saattavat suurentaa plasman midostauriinipitoisuuksia, etenkin midostauriinihoitoa (uudelleen) aloitettaessa (ks. kohta Yhteisvaikutukset). Tällöin on harkittava muita lääkevalmisteita, jotka eivät estä voimakkaasti CYP3A4-toimintaa. Jos muita tyydyttäviä hoitovaihtoehtoja ei ole, potilaiden vointia on seurattava tarkoin midostauriiniin liittyvän toksisuuden varalta.
Apuaineet
Tämä lääkevalmiste sisältää makrogoliglyserolihydroksistearaattia, joka saattaa aiheuttaa vatsavaivoja ja ripulia.
Tämä lääkevalmiste sisältää 666 mg alkoholia (etanolia) per 200 mg:n annos (enimmäisvuorokausiannos), joka vastaa 14 tilavuusprosenttia vedetöntä etanolia. Alkoholimäärä 200 mg:ssa tätä lääkevalmistetta vastaa 17 ml:aa olutta tai 7 ml:aa viiniä. Tämän lääkevalmisteen sisältämä pieni määrä alkoholia ei aiheuta havaittavia vaikutuksia. Alkoholi saattaa olla haitallista potilaille, joilla on alkoholiin liittyviä ongelmia, epilepsia tai maksavaivoja, tai raskauden tai rintaruokinnan aikana.
Yhteisvaikutukset
Midostauriinilla on runsas pääasiassa CYP3A4-entsyymivälitteinen maksametabolia. Monet samanaikaisesti käytettävät lääkevalmisteet joko kiihdyttävät tai estävät CYP3A4-entsyymitoimintaa.
Muiden lääkevalmisteiden vaikutus Rydapt-valmisteeseen
Lääkevalmisteet tai aineet, joiden tiedetään vaikuttavan CYP3A4-toimintaan, saattavat vaikuttaa midostauriinin pitoisuuksiin plasmassa ja siten Rydapt-valmisteen turvallisuuteen ja/tai tehoon.
Voimakkaat CYP3A4:n induktorit
Rydapt-valmisteen ja voimakkaiden CYP3A4:n induktorien (esim. karbamatsepiini, rifampisiini, entsalutamidi, fenytoiini, mäkikuisma [Hypericum perforatum]) samanaikainen käyttö on vasta-aiheista (ks. kohta Vasta-aiheet). Voimakkaat CYP3A4:n induktorit pienentävät altistusta midostauriinille ja sen aktiivisille metaboliiteille (CGP52421 ja CGP62221). Kun terveillä tutkittavilla tehdyssä tutkimuksessa voimakkaan CYP3A4:n induktorin rifampisiinin (annostus 600 mg vuorokaudessa) suhteen vakaassa tilassa oleville tutkittaville annettiin 50 mg kerta-annos midostauriinia, pieneni midostauriinin Cmax-arvo keskimäärin 73 % ja AUCinf-arvo keskimäärin 96 %. Sama ilmiö toistui CGP62221:n kohdalla. CGP52421:n AUClast-arvon keskiarvo pieneni 60 %.
Voimakkaat CYP3A4:n estäjät
Voimakkaat CYP3A4:n estäjät saattavat suurentaa veren midostauriinipitoisuuksia. Tutkimuksessa, johon osallistui 36 tervettä tutkittavaa, ketokonatsolin (voimakas CYP3A4:n estäjä) suhteen vakaassa tilassa oleville tutkittaville annettiin 50 mg kerta-annos midostauriinia. Midostauriinialtistus suureni merkitsevästi (Cmax-arvo suureni 1,8-kertaiseksi ja AUCinf-arvo 10-kertaiseksi) ja CGP62221:n AUCinf-arvo suureni 3,5-kertaiseksi, kun taas aktiivisten metaboliittien (CGP62221 ja CGP52421) Cmax-arvo pieneni puoleen (ks. kohta Farmakokinetiikka). Potilaspopulaation osajoukossa (N = 7) jossa potilaat olivat vakaassa tilassa sekä midostauriinin (50 mg kahdesti vuorokaudessa) että itrakonatsolin (voimakas CYP3A4:n estäjä) suhteen, vakaan tilan midostauriinialtistus (Cmin) suureni 2,09-kertaiseksi. CGP52421:n Cmin suureni 1,3-kertaiseksi, kun taas CGP62221-altistukseen ei havaittu merkittävää vaikutusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Rydapt-valmisteen vaikutus muihin lääkevalmisteisiin
CYP-entsyymien substraatit
Bupropionin (CYP2B6:n substraatti) kerta-annos yhdessä toistuvien midostauriiniannosten (50 mg kahdesti vuorokaudessa) kanssa laski vakaassa tilassa bupropionin AUCinf -arvoa 48 % ja AUClast -arvoa 49 % ja Cmax -arvoa 55 % verrattuna pelkkään bupropioniin. Tämä osoittaa, että midostauriini on CYP2B6:n lievä indusoija. Lääkevalmisteiden, joilla on kapea terapeuttinen leveys ja jotka ovat CYP2B6:n substraatteja (esim. bupropioni tai efavirentsi), samanaikaisessa käytössä midostauriinin kanssa tulee noudattaa varovaisuutta ja annoksen säätäminen voi olla tarpeen optimaalisen altistuksen ylläpitämiseksi.
In vitro -tietojen perusteella midostauriini ja sen metaboliitit CGP52421 ja CGP62221 ovat CYP1A2:n ja CYP2E1:n estäjiä ja CYP1A2:n indusoijia. Tämän vuoksi lääkevalmisteiden, joilla on kapea terapeuttinen leveys ja jotka ovat CYP1A2:n (esim. titsanidiini) ja CYP2E1:n (esim. klooritsoksatsoni) substraatteja, samanaikaisessa käytössä midostauriinin kanssa tulee noudattaa varovaisuutta ja annoksen säätäminen voi olla tarpeen optimaalisen altistuksen ylläpitämiseksi.
Kuljettajaproteiinien substraatit
Terveillä tutkittavilla rosuvastatiinin kerta-annos (BCPR:n substraatti) yhdessä midostauriinin kerta-annoksen (100 mg) kanssa nosti rosuvastatiinin AUCinf -arvoa 37 % ja AUClast -arvoa 48 % ja Cmax -arvo noin tuplaantui (2,01-kertaisesti) verrattuna pelkkään rosuvastatiiniin. Tämä osoittaa, että midostauriinilla on lievä estovaikutus BCRP:n substraatteihin. Lääkevalmisteiden, joilla on kapea terapeuttinen leveys ja jotka ovat BCRP-kuljettajaproteiinin substraatteja (esim. rosuvastatiini tai atorvastatiini) samanaikaisessa käytössä midostauriinin kanssa tulee noudattaa varovaisuutta ja annoksen säätäminen voi olla tarpeen optimaalisen altistuksen ylläpitämiseksi.
Hormonaalinen raskaudenehkäisy
Toistuvien midostauriiniannosten (50 mg kahdesti vuorokaudessa) ja etinyyliestradiolia ja levonorgestreelia sisältävien suun kautta otettavien raskaudenehkäisyvalmisteiden välillä ei havaittu vakaassa tilassa kliinisesti merkitseviä farmakokineettisiä yhteisvaikutuksia terveillä naisilla. Tämän vuoksi midostauriinin yhtäaikaisen käytön ei odoteta vaarantavan tämän yhdistelmän ehkäisytehoa.
Yhteisvaikutukset ruoan kanssa
Terveillä koehenkilöillä midostauriinin imeytyminen (AUC) lisääntyi keskimäärin 22 %, kun Rydapt otettiin tavanomaisen aterian kanssa, ja keskimäärin 59 %, kun se otettiin runsasrasvaisen aterian kanssa. Midostauriinin ottaminen tavanomaisen aterian kanssa pienensi lääkkeen huippupitoisuutta (Cmax) 20 % ja sen ottaminen runsasrasvaisen aterian kanssa 27 % verrattuna tilanteeseen, jossa lääke otettiin tyhjään mahaan (ks. kohta Farmakokinetiikka).
On suositeltavaa ottaa Rydapt ruoan kanssa.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Naisille, jotka voivat tulla raskaaksi, on kerrottava midostauriinin aiheuttavan eläintutkimuksissa haittaa kehittyvälle sikiölle. Seksuaalisesti aktiivisia naisia, jotka voivat tulla raskaaksi, kehotetaan tekemään raskaustesti 7 päivän sisällä ennen Rydapt-hoidon aloittamista ja käyttämään tehokasta ehkäisyä (menetelmää, jonka raskausprosentti on alle 1 %) Rydapt-hoidon aikana ja vähintään 4 kuukauden ajan sen päättymisen jälkeen.
Raskaus
Midostauriinin anto raskaana olevalle naiselle voi aiheuttaa haittaa sikiölle. Raskaana olevilla naisilla ei ole tehty riittäviä ja kontrolloituja tutkimuksia. Rotalla ja kanilla tehdyissä lisääntymistutkimuksissa todettiin, että midostauriini aiheutti sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Rydapt-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä. Raskaana oleville naisille on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Imetys
Ei tiedetä, erittyvätkö midostauriini tai sen aktiiviset metaboliitit ihmisen rintamaitoon. Eläinkokeista saatujen tietojen mukaan midostauriini ja sen aktiiviset metaboliitit erittyvät imettävien rottien maitoon. Imetys on lopetettava Rydapt-hoidon ajaksi ja vähintään 4 kuukaudeksi hoidon päättymisen jälkeen.
Hedelmällisyys
Rydapt-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläinkokeissa on havaittu hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Rydapt-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Heite- ja kiertohuimausta on ilmoitettu Rydapt-valmistetta ottavilla potilailla ja tämä on otettava huomioon, kun arvioidaan potilaan kykyä ajaa tai käyttää koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
AML
Rydapt-valmisteen (50 mg kahdesti vuorokaudessa) turvallisuusarviointi äskettäin todettua FLT3-mutaatiopositiivista AML:aa sairastavilla potilailla perustuu satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun vaiheen III tutkimukseen, johon osallistui 717 potilasta. Altistuksen kokonaiskeston mediaani oli 42 vrk (vaihteluväli 2–576 vrk) Rydapt-valmistetta ja tavanomaista solunsalpaajahoitoa saaneessa ryhmässä ja 34 vrk (vaihteluväli 1–465 vrk) lumelääkettä ja tavanomaista solunsalpaajahoitoa saaneessa ryhmässä. Ylläpitovaiheeseen osallistuneilla 205 potilaalla (120 potilasta Rydapt-ryhmässä ja 85 lumelääkeryhmässä) altistuksen mediaanikesto oli ylläpitovaiheessa molemmissa ryhmissä 11 kk (16–520 vrk Rydapt-valmistetta saaneiden potilaiden ryhmässä ja 22–381 vrk lumelääkeryhmässä).
Yleisimpiä Rydapt-valmistetta saaneessa ryhmässä esiintyneitä haittavaikutuksia olivat kuumeinen neutropenia (83,4 %), pahoinvointi (83,4 %), eksfoliatiivinen dermatiitti (61,6 %), oksentelu (60,7 %), päänsärky (45,9 %), petekiat (35,8 %) ja kuume (34,5 %). Yleisimpiä asteen 3/4 haittavaikutuksia olivat kuumeinen neutropenia (83,5 %), lymfopenia (20,0 %), laitteeseen liittyvät infektiot (15,7 %), eksfoliatiivinen dermatiitti (13,6 %), hyperglykemia (7,0 %) ja pahoinvointi (5,8 %). Yleisimmät laboratoriopoikkeavuudet olivat hemoglobiinipitoisuuden pieneneminen (97,3 %), absoluuttisen neutrofiilimäärän pieneneminen (86,7 %), ALAT-arvon suureneminen (84,2 %), ASAT-arvon suureneminen (73,9 %) ja hypokalemia (61,7 %). Yleisimmät asteen 3/4 laboratoriopoikkeavuudet olivat absoluuttisen neutrofiilimäärän pieneneminen (85,8 %), hemoglobiinipitoisuuden pieneneminen (78,5 %), ALAT-arvon suureneminen (19,4 %) ja hypokalemia (13,9 %).
Vakavia haittavaikutuksia esiintyi yhtä usein Rydapt-valmistetta saaneiden potilaiden ryhmässä kuin lumelääkeryhmässä. Yleisin vakava haittavaikutus molemmissa ryhmissä oli kuumeinen neutropenia (16 %).
Rydapt-ryhmässä 3,1 % potilaista ja lumelääkeryhmässä 1,3 % potilaista lopetti hoidon minkä tahansa haittavaikutuksen vuoksi. Rydapt-ryhmässä yleisin hoidon lopettamiseen johtanut asteen 3/4 haittavaikutus oli eksfoliatiivinen dermatiitti (1,2 %).
Turvallisuusprofiili ylläpitovaiheessa
Taulukossa 3 esitetään haittavaikutusten ilmaantuvuus koko tutkimuksen aikana. Kun ylläpitovaiheen (Rydapt tai lumelääke monoterapiana) tiedot arvioitiin erikseen, haittavaikutusten tyypissä ja vaikeusasteessa todettiin eroja. Ylläpitovaiheessa haittavaikutusten kokonaisilmaantuvuus oli yleisesti pienempi kuin induktio- ja konsolidaatiovaiheessa. Haittavaikutusten ilmaantuvuudet olivat ylläpitovaiheessa kuitenkin suurempia Rydapt-ryhmässä kuin lumeryhmässä. Seuraavia haittavaikutuksia esiintyi ylläpitohoidon aikana useammin midostauriiniryhmässä kuin lumeryhmässä: pahoinvointi (46,4 % vs. 17,9 %), hyperglykemia (20,0 % vs. 12,5 %) oksentelu (19 % vs. 5,4 %) ja QT-ajan piteneminen (11,9 % vs. 5,4 %).
Useimmat ilmoitetuista hematologisista poikkeavuuksista ilmenivät induktio- ja konsolidaatiovaiheissa, kun potilaat saivat Rydapt- tai lumelääkehoitoa yhdessä solunsalpaajahoidon kanssa. Ylläpitovaiheessa yleisimpiä Rydapt-ryhmässä ilmoitettuja asteen 3/4 hematologisia poikkeavuuksia olivat absoluuttisen neutrofiilien määrän lasku (20,8 % Rydapt- ja 18,8 % lumelääkeryhmässä) ja leukopenia (7,5 % Rydapt- ja 5,9 % lumelääkeryhmässä).
Ylläpitovaiheessa ilmoitetut haittavaikutukset johtivat hoidon lopettamiseen Rydapt-ryhmässä 1,2 %:lla potilaista ja lumelääkeryhmässä ei yhdelläkään.
ASM, SM-AHN ja MCL
Rydapt-monoterapian (100 mg kahdesti vuorokaudessa) turvallisuutta ASM:ia, SM-AHN:ia ja MCL:aa sairastavilla potilailla arvioitiin 142 potilaalla kahdessa yksihaaraisessa, avoimessa monikeskustutkimuksessa. Rydapt-altistuksen mediaanikesto oli 11,4 kk (vaihteluväli 0‑81 kk).
Yleisimpiä haittavaikutuksia olivat pahoinvointi (82 %), oksentelu (68 %), ripuli (51 %), ääreisosien turvotus (35 %) ja uupumus (31 %). Yleisimpiä asteen 3/4 haittavaikutuksia olivat uupumus (8,5 %), sepsis (7,7 %), keuhkokuume (7 %), kuumeinen neutropenia (7 %) ja ripuli (6,3 %). Yleisimpiä ei-hematologisia laboratorioarvojen poikkeavuuksia olivat hyperglykemia (93,7 %), kokonaisbilirubiinipitoisuuden suureneminen (40,1 %), lipaasipitoisuuden suureneminen (39,4 %), aspartaattiaminotransferaasi (ASAT) -arvon nousu (33,8 %) ja alaniiniaminotransferaasi (ALAT) -arvon nousu (33,1 %). Yleisimpiä hematologisten laboratorioarvojen poikkeavuuksia olivat absoluuttisen lymfosyyttimäärän lasku (73,2 %) ja absoluuttisen neutrofiilimäärän lasku (58,5 %). Yleisimpiä asteen 3/4 laboratorioarvojen poikkeavuuksia olivat absoluuttisen lymfosyyttimäärän lasku (45,8 %), absoluuttisen neutrofiilimäärän lasku (26,8 %), hyperglykemia (19 %) ja lipaasipitoisuuden suureneminen (17,6 %).
Haittavaikutukset johtivat annosmuutoksiin (hoidon tauottamiseen tai annoksen muuttamiseen) 31 %:lla potilaista. Yleisimpiä annosmuutokseen johtaneita haittavaikutuksia (ilmaantuvuus ≥ 5 %) olivat pahoinvointi ja oksentelu.
Hoidon lopettamiseen johtaneita haittavaikutuksia ilmeni 9,2 %:lla potilaista. Yleisimpiä (ilmaantuvuus ≥ 1 %) olivat kuumeinen neutropenia, pahoinvointi, oksentelu ja pleuraeffuusio.
Haittavaikutustaulukot
Haittavaikutukset luetellaan MedDRA-elinjärjestelmäluokan mukaisesti. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa yleisyysjärjestyksessä yleisimmistä alkaen, perustuen seuraaviin määritelmiin (CIOMS III): hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
AML
Taulukossa 3 esitettävät ilmoitettujen haittavaikutusten yleisyysluokat perustuvat vaiheen III tutkimukseen, jonka potilailla oli äskettäin todettu FLT3-mutaatiopositiivinen AML sekä myyntiluvan myöntämisen jälkeisen vaiheen kokemuksiin.
Taulukko 3 AML:ssa havaitut haittavaikutukset
Haittavaikutus | Kaikki asteet | Asteet 3/4 | Yleisyysluokka |
Rydapt + kemoterapia n = 2291 % | Rydapt + kemoterapia n = 3451 % | ||
Infektiot | |||
Laitteeseen liittyvä infektio | 24 | 15,7 | Hyvin yleinen |
Ylähengitystieinfektio | 5,2 | 0,6 | Yleinen |
Neutropeeninen sepsis | 0,9 | 3,5 | Melko harvinainen |
Veri ja imukudos | |||
Kuumeinen neutropenia | 83,4 | 83,5 | Hyvin yleinen |
Petekiat | 35,8 | 1,2 | Hyvin yleinen |
Lymfopenia | 16,6 | 20 | Hyvin yleinen |
Immuunijärjestelmä | |||
Yliherkkyys | 15,7 | 0,6 | Hyvin yleinen |
Aineenvaihdunta ja ravitsemus | |||
Hyperurikemia | 8,3 | 0,6 | Yleinen |
Psyykkiset häiriöt | |||
Unettomuus | 12,2 | 0 | Hyvin yleinen |
Hermosto | |||
Päänsärky | 45,9 | 2,6 | Hyvin yleinen |
Pyörtyminen | 5,2 | 4,6 | Yleinen |
Vapina | 3,9 | 0 | Yleinen |
Silmät | |||
Silmäluomien turvotus | 3,1 | 0 | Yleinen |
Sydän | |||
Hypotensio | 14,4 | 5,5 | Hyvin yleinen |
Sinustakykardia | 9,6 | 1,2 | Yleinen |
Hypertensio | 7,9 | 2,3 | Yleinen |
Perikardiumeffuusio | 3,5 | 0,6 | Yleinen |
Hengityselimet, rintakehä ja välikarsina | |||
Nenäverenvuoto | 27,5 | 2,6 | Hyvin yleinen |
Kurkunpään kipu | 11,8 | 0,6 | Hyvin yleinen |
Interstitiaalinen keuhkosairaus / pneumoniitti2 | 11,4 | 4,9 | Hyvin yleinen |
Hengenahdistus | 10,9 | 5,5 | Hyvin yleinen Yleinen |
Pleuraeffuusio | 5,7 | 0,9 | |
Nenänielutulehdus | 8,7 | 0 | Yleinen |
Äkillinen hengitysvajausoireyhtymä | 2,2 | 2,3 | Yleinen |
Ruoansulatuselimistö | |||
Pahoinvointi | 83,4 | 5,8 | Hyvin yleinen |
Oksentelu | 60,7 | 2,9 | Hyvin yleinen |
Suutulehdus | 21,8 | 3,5 | Hyvin yleinen |
Ylävatsakipu | 16,6 | 0 | Hyvin yleinen |
Peräpukamat | 15,3 | 1,4 | Hyvin yleinen |
Epämukavat tuntemukset peräaukon ja peräsuolen alueella | 7 | 0,9 | Yleinen |
Epämukava tunne vatsassa | 3,5 | 0 | Yleinen |
Iho ja ihonalainen kudos | |||
Eksfoliatiiivinen dermatiitti | 61,6 | 13,6 | Hyvin yleinen |
Voimakas hikoilu | 14,4 | 0 | Hyvin yleinen |
Kuiva iho | 7 | 0 | Yleinen |
Sarveiskalvotulehdus | 6,6 | 0,3 | Yleinen |
Akuutti kuumeinen neutrofiilinen dermatoosi3 | - | - | Tuntematon |
Luusto, lihakset ja sidekudos | |||
Selkäkipu | 21,8 | 1,4 | Hyvin yleinen |
Nivelkipu | 14 | 0,3 | Hyvin yleinen |
Luustokipu | 9,6 | 1,4 | Yleinen |
Raajakipu | 9,6 | 1,4 | Yleinen |
Niskakipu | 7,9 | 0,6 | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | |||
Kuume | 34,5 | 3,2 | Hyvin yleinen |
Katetriin liittyvä tromboosi | 3,5 | 2 | Yleinen |
Tutkimukset | |||
Hemoglobiinipitoisuuden pieneneminen* | 97,3 | 78,5 | Hyvin yleinen |
Absoluuttisen neutrofiilimäärän lasku* | 86,7 | 85,8 | Hyvin yleinen |
ALAT-arvojen suureneminen* | 84,2 | 19,4 | Hyvin yleinen |
ASAT-arvon suureneminen* | 73,9 | 6,4 | Hyvin yleinen |
Hypokalemia* | 61,7 | 13,9 | Hyvin yleinen |
Hyperglykemia | 20,1 | 7 | Hyvin yleinen |
Hyponatremia* | 20 | 1,2 | Hyvin yleinen |
QT-ajan pidentyminen EKG:ssä3 | 19,7 | 5,8 | Hyvin yleinen |
Pidentynyt aktivoitu partiaalinen tromboplastiiniaika (APTT) | 12,7 | 2,6 | Hyvin yleinen |
Hyperkalsemia* | 6,7 | 0,6 | Yleinen |
Painonnousu | 6,6 | 0,6 | Yleinen |
1Pohjois-Amerikan tutkimuskeskuksissa kerättiin 13:n ennalta määritetyn haittatapahtuman osalta kaiken asteisten tapahtumien tiedot. Kaikkien muiden haittatapahtumien osalta kerättiin vain asteiden 3 ja 4 tiedot. Tästä syystä kaikkien asteiden haittatapahtumat esitetään yhteenvetona vain muiden kuin pohjoisamerikkalaisten keskusten potilaiden osalta, kun taas asteen 3 ja 4 haitat esitetään yhteenvetona kaikkien tutkimuskeskusten potilaiden osalta. 2Tämä haittavaikutus sisällytettiin, kun se tunnistettiin Rydapt-valmisteen myyntiluvan myöntämisen jälkeisessä vaiheessa spontaanien tapaustutkimusten ja kirjallisuudessa esitettyjen tapauksien kautta. Vaiheen III tutkimuksessa ei raportoitu interstitiaalista keuhkosairautta. 3Nämä haittavaikutukset sisällytettiin, kun ne tunnistettiin myyntiluvan myöntämisen jälkeisessä vaiheessa. * Esiintymistiheys perustuu laboratorioarvoihin. | |||
ASM, SM-AHN ja MCL
Taulukossa 4 esitettävät haittavaikutusten yleisyysluokat perustuvat yhdistettyihin tietoihin kahdesta tutkimuksesta, joiden potilailla oli ASM, SM-AHN tai MCL.
Taulukko 4 ASM:ssa, SM-AHN:ssa ja MCL:ssa havaitut haittavaikutukset
Haittavaikutus | Rydapt (100 mg x 2) N = 142 | Yleisyysluokka | |
Kaikki asteet % | Asteet 3/4 % | ||
Infektiot | |||
Virtsatieinfektio | 13 | 2,8 | Hyvin yleinen |
Ylähengitystieinfektio | 11 | 1,4 | Hyvin yleinen |
Keuhkokuume | 8,5 | 7,0 | Yleinen |
Sepsis | 7,7 | 7,7 | Yleinen |
Keuhkoputkitulehdus | 5,6 | 0 | Yleinen |
Suun herpes | 4,9 | 0 | Yleinen |
Virtsarakkotulehdus | 4,2 | 0 | Yleinen |
Poskiontelotulehdus | 4,2 | 0,7 | Yleinen |
Ruusu | 3,5 | 1,4 | Yleinen |
Vyöruusu | 3,5 | 0,7 | Yleinen |
Veri ja imukudos | |||
Kuumeinen neutropenia | 7,7 | 7,0 | Yleinen |
Immuunijärjestelmä | |||
Yliherkkyys | 2,1 | 0 | Yleinen |
Anafylaktinen sokki | 0,7 | 0,7 | Melko harvinainen |
Hermosto | |||
Päänsärky | 26 | 1,4 | Hyvin yleinen |
Heitehuimaus | 13 | 0 | Hyvin yleinen |
Tarkkaavuuden häiriö | 7 | 0 | Yleinen |
Vapina | 6,3 | 0 | Yleinen |
Kuulo ja tasapainoelin | |||
Kiertohuimaus | 4,9 | 0 | Yleinen |
Verisuoniston häiriöt | |||
Hypotensio | 9,2 | 2,1 | Yleinen |
Hematooma | 6,3 | 0,7 | Yleinen |
Hengityselimet, rintakehä ja välikarsina | |||
Hengenahdistus | 18 | 5,6 | Hyvin yleinen |
Yskä | 16 | 0,7 | Hyvin yleinen |
Pleuraeffuusio | 13 | 4,2 | Hyvin yleinen |
Nenäverenvuoto | 12 | 2,8 | Hyvin yleinen |
Suunielun kipu | 4,2 | 0 | Yleinen |
Interstitiaalinen keuhkosairaus / pneumoniitti1 | 2,1 | 0 | Yleinen |
Ruoansulatuselimistö | |||
Pahoinvointi | 82 | 5,6 | Hyvin yleinen |
Oksentelu | 68 | 5,6 | Hyvin yleinen |
Ripuli | 51 | 6,3 | Hyvin yleinen |
Ummetus | 29 | 0,7 | Hyvin yleinen |
Dyspepsia | 5,6 | 0 | Yleinen |
Ruoansulatuskanavan verenvuoto | 4,2 | 3,5 | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | |||
Ääreisosien turvotus | 35 | 3,5 | Hyvin yleinen |
Uupumus | 31 | 8,5 | Hyvin yleinen |
Kuume | 27 | 4,2 | Hyvin yleinen |
Voimattomuus | 4,9 | 0,7 | Yleinen |
Vilunväristykset | 4,9 | 0 | Yleinen |
Turvotus | 4,2 | 0,7 | Yleinen |
Tutkimukset | |||
Hyperglykemia (muu kuin paastoarvo)* | 93,7 | 19,0 | Hyvin yleinen |
Absoluuttisen lymfosyyttimäärän lasku* | 73,2 | 45,8 | Hyvin yleinen |
Absoluuttisen neutrofiilimäärän lasku* | 58,5 | 26,8 | Hyvin yleinen |
Veren kokonaisbilirubiinipitoisuuden suureneminen* | 40,1 | 4,9 | Hyvin yleinen |
Lipaasipitoisuuden suureneminen* | 39,4 | 17,6 | Hyvin yleinen |
ASAT-arvojen suureneminen* | 33,8 | 2,8 | Hyvin yleinen |
ALAT-arvon suureneminen* | 33,1 | 3,5 | Hyvin yleinen |
Amylaasiarvon suureneminen* | 20,4 | 7,0 | Hyvin yleinen |
QT-ajan pidentyminen EKG:ssä1 | 10,6 | 0,7 | Hyvin yleinen |
Painonnousu | 5,6 | 2,8 | Yleinen |
Vammat ja myrkytykset | |||
Kontuusio | 6,3 | 0 | Yleinen |
Kaatuminen | 4,2 | 0,7 | Yleinen |
* Esiintymistiheys perustuu laboratorioarvoihin. 1 Nämä haittavaikutukset sisällytettiin, kun ne tunnistettiin myyntiluvan myöntämisen jälkeisessä vaiheessa. | |||
Tiettyjen haittavaikutusten kuvaus
Ruoansulatuselimistö
Pahoinvointia, oksentelua ja ripulia havaittiin AML-, ASM-, SM‑AHN- ja MCL-potilailla. ASM-, SM‑AHN- ja MCL-potilailla nämä tapahtumat johtivat annosmuutoksiin tai hoidon tauottamiseen 26 %:lla potilaista ja hoidon lopettamiseen 4,2 %:lla potilaista. Useimmat tapahtumat ilmenivät ensimmäisten 6 hoitokuukauden aikana, ja ne pystyttiin hoitamaan profylaktisella tukilääkityksellä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ihmisellä tapahtuneista yliannostuksista on olemassa hyvin niukasti tietoa. Enintään 600 mg kerta-annosten välitön siedettävyys on ollut hyväksyttävä. Havaittuja haittavaikutuksia olivat ripuli, vatsakipu ja oksentelu.
Midostauriinille ei ole tiedossa spesifistä vastalääkettä. Yliannostustapauksissa potilaita on seurattava tarkoin haittavaikutusten oireiden tai löydösten varalta, ja asianmukainen oireenmukainen hoito tai tukihoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EX10
Vaikutusmekanismi
Midostauriini estää useita reseptorityrosiinikinaaseja, kuten FLT3- ja KIT-kinaaseja. Midostauriini estää FLT3-reseptorisignalointia ja saa aikaan solusyklin pysähtymisen ja apoptoosin leukemiasoluissa, jotka ilmentävät FLT3‑ITD- tai TKD-mutanttireseptoreja tai yli-ilmentävät FLT3-villityypin reseptoreja. In vitro -tietojen perusteella midostauriini estää D816V-mutatoituneiden KIT-reseptorien toimintaa potilailla saavutetuilla altistustasoilla (keskimääräinen saavutettu altistus yli IC50-arvon). In vitro -tietojen perusteella villityypin KIT-reseptorien toiminta estyy paljon vähemmässä määrin näillä pitoisuuksilla (keskimääräinen saavutettu altistus alle IC50-arvon). Midostauriini häiritsee poikkeavaa KIT D816V ‑välitteistä signalointia ja estää syöttösolujen proliferaatiota ja eloonjäämistä sekä histamiinin vapautumista.
Midostauriini estää myös useita muita reseptorityrosiinikinaaseja kuten PDGFR (verihiutalekasvutekijäreseptori) ja VEGFR2 (verisuonen endoteelikasvutekijäreseptori-2) kinaaseja sekä PKC-seriini-treoniinikinaasiperheen (proteiinikinaasi C -perhe) kinaaseja. Se sitoutuu näiden kinaasien katalyyttiseen alueeseen ja estää kyseisten kasvutekijöiden mitogeenista signalointia soluissa, mikä johtaa kasvun pysähtymiseen.
Midostauriinin käyttö yhdessä eri solunsalpaajien kanssa (sytarabiini, doksorubisiini, idarubisiini ja daunorubisiini) esti synergistisesti solukasvua FLT3‑ITD:tä ilmentävissä AML-solulinjoissa.
Farmakodynaamiset vaikutukset
Hiirimalleissa ja ihmisillä on tunnistettu kaksi päämetaboliittia, CGP62221 ja CGP52421. FLT3‑ITD-mutanttireseptoria ilmentävillä soluilla tehdyissä proliferaatiokokeissa CGP62221 osoittautui yhtä tehokkaaksi kuin kanta-aine, kun taas CGP52421-metaboliitin teho oli noin kymmenesosa kanta-aineen tehosta.
Sydämen elektrofysiologia
QT-täsmätutkimuksessa 192 terveellä henkilöllä, jotka saivat 75 mg annoksen kahdesti vuorokaudessa, midostauriinin ja CGP62221-metaboliitin ei todettu pidentävän QT-aikaa kliinisesti merkittävästi. Tutkimuksen kesto ei riittänyt pitkävaikutteisen CGP52421-metaboliitin mahdollisen QTc-aikaa pidentävän vaikutuksen arviointiin. QTcF-ajan muutosta lähtötilanteesta suhteessa midostauriinin ja molempien metaboliittien pitoisuuksiin arvioitiin tarkemmin vaiheen II tutkimuksessa, johon osallistui 116 ASM:ia, SM-AHN:ia tai MCL:aa sairastavaa potilasta. Mediaanisilla Cmin-huippupitoisuuksilla jotka saavutettiin annoksilla 100 mg kahdesti vuorokaudessa, ei midostauriinin eikä CGP62221- ja CGP52421-metaboliittien todettu aiheuttavan kliinisesti merkittävää QTcF-ajan pitenemistä, sillä näillä pitoisuuksilla saavutettujen ennakoitujen muutosten ylärajat olivat alle 10 ms (midostauriinilla 5,8 ms ja CGP62221- ja CGP52421-metaboliiteilla 2,4 ms ja 4,0 ms). 25,4 %:lla ASM:ia, SM-AHN:ia tai MCL:aa sairastavista potilaista QTcF-aika oli yli 450 ms ja 4,7 %:lla yli 480 ms vähintään yhdessä EKG-mittauksessa.
Kliininen teho ja turvallisuus
AML
Midostauriinin tehoa ja turvallisuutta tutkittiin satunnaistetussa, kaksoissokkoutetussa vaiheen III tutkimuksessa, johon osallistuneet 717 potilasta olivat 18–60-vuotiaita. Tutkimuksessa arvioitiin Rydapt-valmisteen ja tavanomaisen solunsalpaajahoidon yhdistelmähoitoa verrattuna lumelääkkeen ja tavanomaisen solunsalpaajahoidon yhdistelmään, sekä Rydapt-monoterapiaa ylläpitohoitona. Potilaat, joilla oli kliiniseen tutkimukseen liittyvässä määrityksessä FLT3-mutaatiopositiiviseksi todettu äskettäin diagnosoitu AML, satunnaistettiin (1:1) saamaan midostauriinia (50 mg kahdesti vuorokaudessa; n = 360) tai lumelääkettä (n = 357) sekventiaalisesti yhdessä tavanomaisen daunorubisiinilla (60 mg/m2 vuorokaudessa päivinä 1–3)/sytarabiinilla (200 mg/m2 vuorokaudessa päivinä 1–7) toteutetun induktiohoidon ja suuriannoksisen sytarabiinikonsolidaatiohoidon (3 g/m2 12 tunnin välein päivinä 1, 3, 5) kanssa, minkä jälkeen midostauriinia tai lumelääkettä annettiin yhtäjaksoisesti alkujaan määrätyn hoitoryhmän mukaan vielä enintään 12 hoitojakson ajan (28 vrk/hoitojakso). Tutkimukseen otetuilla potilailla sallittiin erilaisia AML:aan liittyviä sytogeneettisia poikkeavuuksia. Akuuttia promyelosyyttista leukemiaa (M3) tai hoitoon liittyvää AML:aa sairastavat potilaat suljettiin pois. Potilaat stratifioitiin FLT3-mutaatiostatuksen mukaan seuraaviin ryhmiin: TKD, ITD alleelisuhde < 0,7, tai ITD alleelisuhde ≥ 0,7.
Lähtötilanteen demografiset tiedot ja taudin ominaisuudet olivat yleisesti ottaen samaa luokkaa molemmissa hoitoryhmissä. Potilaiden mediaani-ikä oli 47 vuotta (vaihteluväli: 18–60 v), valtaosan ECOG-toimintakykyluokka oli 0 tai 1 (88,3 %), ja useimmilla potilailla oli de novo AML (95 %). Niistä potilaista, joiden etninen tausta oli ilmoitettu, 88,1 % oli valkoihoisia. Valtaosalla potilaista (77,4 %) oli FLT3-ITD-mutaatio. Heistä useimmilla (47,6 %) alleelisuhde oli pieni (< 0,7). 22,6 %:lla oli FLT3-TDK-mutaatio. Midostauriiniryhmässä 48 % potilaista ja lumeryhmässä 41 % potilaista oli miehiä.
Potilaat, joille tehtiin veren kantasolusiirto, lopettivat tutkimushoidon ennen kantasolusiirtoon valmistavan hoidon aloittamista. Kantasolusiirto tehtiin yhteensä 59,4 %:lle (214/360) midostauriinia ja tavanomaista solunsalpaajahoitoa saaneista potilaista ja 55,2 %:lle (197/357) lumelääkettä ja tavanomaista solunsalpaajahoitoa saaneista potilaista. Kaikkien potilaiden elossaoloa seurattiin.
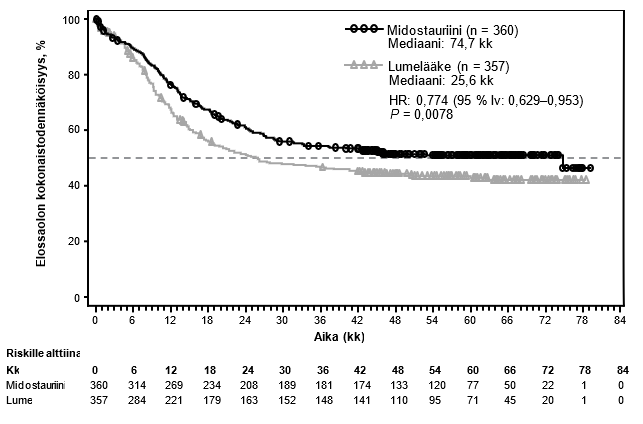
Tutkimuksen ensisijainen päätetapahtuma oli kokonaiselossaoloaika eli aika satunnaistamispäivästä kuolinpäivään kuolinsyystä riippumatta. Ensisijainen analyysi tehtiin, kun viimeisen potilaan satunnaistamisen jälkeinen seuranta-aika oli kestänyt vähintään noin 3,5 vuotta. Tutkimuksessa todettiin, että kokonaiselossaoloaika piteni tilastollisesti merkitsevästi ja kuoleman riski pieneni 23 % midostauriinin ja tavanomaisen solunsalpaajahoidon yhdistelmäryhmässä verrattuna lumelääkkeen ja tavanomaisen solunsalpaajahoidon yhdistelmäryhmään (ks. taulukko 6 ja kuva 1).
Kuva 1Kokonaiselossaoloajan Kaplan–Meier-käyrä, ei tietojen rajausta kantasolusiirtojen suhteen
Tärkein toissijainen päätetapahtuma oli tapahtumavapaa elossaoloaika (tapahtumaksi määriteltiin tilanne, jossa täydellistä remissiota ei saavutettu 60 päivän kuluessa tutkimussuunnitelman mukaisen hoidon aloittamisesta, relapsi tai mistä tahansa syystä johtuva kuolema). Tapahtumavapaa elossaoloaika oli midostauriinia ja tavanomaista solunsalpaajahoitoa saaneilla tilastollisesti merkitsevästi pidempi kuin lumelääkettä ja tavanomaista solunsalpaajahoitoa saaneilla (riskisuhde: 0,78 [95 % lv 0,66–0,93] p = 0,0024). Tapahtumavapaan elossaoloajan mediaani oli midostauriini + tavanomainen solunsalpaajahoito -ryhmässä 8,2 kk ja lume + tavanomainen solunsalpaajahoito ‑ryhmässä 3,0 kk; ks. taulukko 5.
Taulukko 5 Midostauriinin teho AML-potilaiden hoidossa
Tehoparametri | Midostauriini n = 360 | Lumelääke n = 357 | Riskisuhde* (95 % lv) | P-arvo¥ |
Kokonaiselossaoloaika1 | ||||
Kokonaiselossaoloajan mediaani kuukausina (95 % lv) | 74,7 (31,5–ei arvioitu) | 25,6 (18,6–42,9) | 0,77 (0,63–0,95) | 0,0078 |
Kaplan-Meier estimaatti 5 vuoden kohdalla (95 % lv) | 0,51 (0,45–0,56) | 0,43 (0,38–0,49) | ||
Tapahtumavapaa elossaoloaika (EFS)2 | ||||
EFS mediaani kuukausina, ottaen huomioon potilaat, jotka saavuttivat täydellisen remission 60 päivän kuluessa hoidon alusta (95 % lv) | 8,2 (5,4–10,7) | 3,0 (1,9–5,9) | 0,78 (0,66–0,93) | 0,0024 |
EFS mediaani kuukausina, ottaen huomioon potilaat, jotka saavuttivat täydellisen remission milloin tahansa induktiovaiheen aikana (95 % lv) | 10,2 (8,1–13,9) | 5,6 (2,9–6,7) | 0,73 (0,61–0,87) | 0,0001 |
Tauditon elossaoloaika | ||||
Taudittoman elossaoloajan mediaani kuukausina (95 % lv) | 26,7 (19,4–ei arvioitu) | 15,5 (11,3–23,5) | 0,71 (0,55–0,92) | 0,0051 |
Täydellinen remissio | ||||
60 päivän kuluessa hoidon alusta (%) | 212 (58,9) | 191 (53,5) | Ei arvioitu | 0,073§ |
milloin tahansa induktiovaiheen aikana (%) | 234 (65,0) | 207 (58,0) | Ei arvioitu | 0,027§ |
Relapsien kumulatiivinen ilmaantuvuus (CIR) | ||||
Mediaani (95 % lv) | Ei arvioitu (25,7–ei arvioitu) | 17,6 (12,7–46,3) | 0,68 (0,52–0,89) | 0,0023 |
1ensisijainen päätetapahtuma 2tärkein toissijainen päätetapahtuma *Riskisuhde arvioitiin Coxin regressiomallilla, ja tietojen stratifiointitekijänä käytettiin satunnaistamishetken FLT3-mutaatiostatusta. ¥1-tahoinen p-arvo laskettiin log-rank-testillä ja stratifiointitekijänä käytettiin satunnaistamishetken FLT3-mutaatiostatusta. §Ei merkitsevä | ||||
Midostauriinilla saavutettiin trendinomaisesti suurempi täydellisten remissioiden prosenttiosuus päivään 60 mennessä midostauriiniryhmässä (58,9 % vs. 53,5 %; p = 0,073), ja sama tulos säilyi, kun kaikki induktiovaiheessa saavutetut täydelliset remissiot otettiin huomioon (65,0 % vs. 58,0 %; p = 0,027). Täydellisen remission induktiovaiheessa saavuttaneilla potilaiden relapsien kumulatiivinen ilmaantuvuus 12 kuukauden kohdalla oli midostauriiniryhmässä 26 % ja lumelääkeryhmässä 41 %.
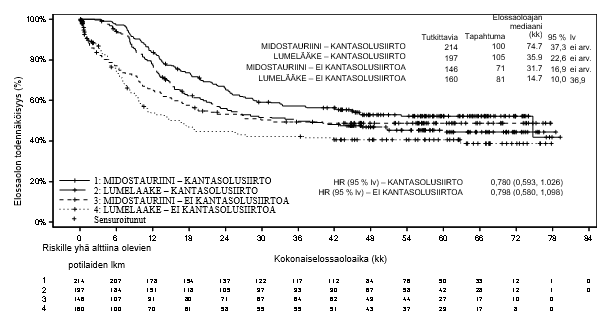
Kun tiedot rajattiin kantasolusiirron ajankohdan perusteella, kokonaiselossaoloajan ja tapahtumavapaan elossaoloajan herkkyysanalyysit tukivat käsitystä, että midostauriinin ja tavanomaisen solunsalpaajahoidon yhdistelmästä on kliinistä hyötyä verrattuna lumelääkkeeseen.
Kokonaiselossaoloajan tulokset kantasolusiirtostatuksen suhteen esitetään kuvassa 2. Tapahtumavapaan elossaoloajan (ottaen huomioon potilaat, jotka saavuttivat täydellisen remission 60 päivän kuluessa tutkimushoidon alusta) HR oli 0,602 [95 % lv: 0,372–0,974] potilailla, joilla kantasolusiirto oli tehty ja 0,827 [95 % lv: 0,689–0,993] potilailla, joilla kantasolusiirtoa ei ollut tehty; midostauriinin hyväksi.
Kuva 2 Kokonaiselossaoloajan Kaplan–Meier-käyrä kantasolusiirtostatuksen mukaisesti AML-potilailla
Alaryhmäanalyysissa ei havaittu ilmeistä elossaoloetua naisilla. Naisilla havaittiin kuitenkin hyötyä kaikkien toissijaisten tehon päätetapahtumien osalta (ks. taulukko 6).
Taulukko 6 OS-, EFS-, CR-, DFS- ja CIR-vasteiden yleiskuva sukupuolen mukaan AML-potilailla
Päätetapahtuma | Yhteensä 95 % lv | Miehet 95 % lv | Naiset 95 % lv |
OS (HR) | 0,774 (0,629–0,953) | 0,533 (0,392–0,725) | 1,007 (0,757–1,338) |
EFS (CR, induktio) (HR) | 0,728 (0,613–0,866) | 0,660 (0,506–0,861) | 0,825 (0,656–1,037) |
CR, induktio (OR) | 0,743* (0,550–1,005) | 0,675* (0,425–1,072) | 0,824* (0,552–1,230) |
DFS (CR, induktio) (HR) | 0,663 (0,516–0,853) | 0,594 (0,408–0,865) | 0,778 (0,554–1,093) |
CIR (CR, induktio) (HR) | 0,676 (0,515–0,888) | 0,662 (0,436–1,006) | 0,742 (0,516–1,069) |
*Vetosuhteen laskutapa: (Ei täydellistä remissiota hoidon aikana/täydellinen remissio hoidon aikana) / (Ei täydellistä remissiota lumehoidon aikana/täydellinen remissio lumehoidon aikana) HR = riskisuhde; OR = vetosuhde | |||
Tehoa ja turvallisuutta > 60–70-vuotiailla potilailla, joilla oli FLT3-ITD-mutatoitunut AML, arvioitiin osana vaiheen II yksiryhmäistä, tutkijalähtöistä tutkimusta, jossa midostauriinia käytettiin intensiivisessä induktiohoidossa ja konsolidaatiohoidossa (sis. allogeeninen kantasolusiirto) yhdistelmänä sekä ylläpitovaiheessa monoterapiana. Loppuanalyysin perusteella tapahtumaton elossaolo 2 v kohdalla (ensisijainen päätetapahtuma) oli 34 % (95 % lv: 27–44), ja kokonaiselossaolon mediaani oli yli 60-vuotiailla potilailla 22,7 kk (128 potilasta 440:stä).
ASM, SM-AHN ja MCL
Midostauriinin tehoa arvioitiin kahdessa avoimessa, yksihaaraisessa monikeskustutkimuksessa potilailla, joilla oli ASM, SM-AHN tai MCL. Näitä tiloja kutsuttiin yhteisnimellä edennyt systeeminen mastosytoosi (SM). Mukana oli yhteensä 142 potilasta.
Avaintutkimus oli yksihaarainen vaiheen II monikeskustutkimus, johon osallistuneilla 116 potilaalla oli edennyt SM (tutkimus CPKC412D2201). Midostauriinia annettiin suun kautta 100 mg kahdesti vuorokaudessa, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta. Mukaan otetuista 116 potilaasta 89:n katsottiin soveltuvan vasteen arviointiin. He muodostivat tutkimuksen ensisijaisen tehopopulaation. Heidän joukossaan oli 73 ASM-potilasta (joista 57 potilaalla oli tautiin liittyvä hematologinen maligniteetti) ja 16 MCL-potilasta (joista 6 potilaalla oli tautiin liittyvä hematologinen maligniteetti). Ensisijaisen tehopopulaation potilaiden mediaani-ikä oli 64 vuotta, ja noin puolet potilaista oli ≥ 65-vuotiaita. Noin kolmannes (36 %) oli saanut aiempaa syöpähoitoa ASM:n, SM-AHN:n tai MCL:n hoitoon. Lähtötilanteessa 65 %:lla ensisijaisen tehopopulaation potilaista oli > 1 mitattavissa oleva C-löydös (trombosytopenia, hypoalbuminemia, anemia, korkea kokonaisbilirubiini, verensiirroilla hoidettava anemia, laihtuminen, neutropenia, korkea ALAT tai korkea ASAT). 82 %:lla potilaista todettiin KIT D816V ‑mutaatio.
Ensisijainen päätetapahtuma oli vasteiden kokonaismäärä. Vasteprosentit arvioitiin muokattujen Valentin ja Chesonin kriteerien perusteella, ja tutkimuksen ohjaustoimikunta vahvisti vasteet. Toissijaisia päätetapahtumia olivat vasteen kesto, vasteen saavuttamiseen kulunut aika ja kokonaiselossaoloaika. Midostauriini-hoidolla saavutetut vasteet esitetään taulukossa 7. Hoidolla todettiin olevan tehoa aiempien hoitojen määrästä ja tautiin liittyvän hematologisen maligniteetin olemassaolosta tai puuttumisesta riippumatta. Vahvistettuja vasteita esiintyi sekä KIT D816V -mutaatiopositiivisilla potilailla (ORR = 63 %) että potilailla, joilla oli villityypin KIT D816V tai joiden mutaatiostatus ei ollut tiedossa (ORR = 43,8 %). KIT D816V positiivisten potilaiden elossaoloajan mediaani oli kuitenkin pidempi, 33,9 kk (95 % lv: 20,7–42), kuin potilailla, joilla oli villityypin KIT D816V tai joiden mutaatiostatus ei ollut tiedossa. Näillä potilailla tämä aika oli 10 kk (95 % lv: 6,9–17,4). 46 %:lla potilaista todettiin yli 50 %:n pieneneminen luuytimen infiltraatiossa ja 58 %:lla todettiin yli 50 %:n pieneneminen seerumin tryptaasipitoisuuksissa. Pernan tilavuuden ≥ 10 % pienenemistä todettiin 68,9 %:lla niistä potilaista, joista oli saatavilla vähintään 1 lähtötilanteen jälkeinen arviointi (26,7 %:lla potilaista pernan tilavuus pieneni ≥ 35 %, mikä vastaa palpoiden arvioidun koon pienenemistä 50 %:lla).
Vasteen saavuttamiseen kulunut mediaaniaika oli 0,3 kk (vaihteluväli 0,1–3,7 kk). Seurannan mediaanikesto oli 43 kk.
Taulukko 7 Midostauriinin teho potilailla, joilla oli ASM, SM-AHN tai MCL: ensisijainen tehopopulaatio
Kaikki | ASM | SM-AHN | MCL | |
N = 89 | N = 16 | N = 57 | N = 16 | |
Ensisijainen päätetapahtuma | ||||
Vasteiden kokonaismäärä, n (%) | 53 (59,6) | 12 (75,0) | 33 (57,9) | 8 (50,0) |
(95 % lv) | (48,6–69,8) | (47,6–92,7) | (44,1–70,9) | (24,7–75,3) |
Merkittävä vaste, n (%) | 40 (44,9) | 10 (62,5) | 23 (40,4) | 7 (43,8) |
Osittainen vaste, n (%) | 13 (14,6) | 2 (12,5) | 10 (17,5) | 1 (6,3) |
Taudin eteneminen pysähtynyt, n (%) | 11 (12,4) | 1 (6,3) | 7 (12,3) | 3 (18,8) |
Etenevä tauti, n (%) | 10 (11,2) | 1 (6,3) | 6 (10,5) | 3 (18,8) |
Toissijaiset päätetapahtumat | ||||
Vasteen mediaanikesto, kk (95 % lv) | 18,6 (9,9–34,7) | 36,8 (5,5–ei arvioitu) | 10,7 (7,4–22,8) | ei saavutettu (3,6–ei arvioitu) |
Kokonaiselossaoloajan mediaani, kk (95 % lv) | 26,8 (17,6–34,7) | 51,1 (28,7–ei arvioitu) | 20,7 (16,3–33,9) | 9,4 (7,5–ei arvioitu) |
Kaplan‑Meier estimaatti 5 vuoden kohdalla (95 % lv) | 26,1 (14,6–39,2) | 34,8 (1,7–76,2) | 19,9 (8,6–34,5) | 33,7 (12,3–56,8) |
Tutkimukseen liittymätöntä syöpähoitoa saaneiden potilaiden taudin katsottiin edenneen uuden hoidon ajankohtana. | ||||
Vaikkakin tutkimus suunniteltiin arvioitavaksi muokattujen Valentin ja Chesonin kriteerien perusteella eksploratiivisena jälkianalyysina, tehoa arvioitiin myös vuoden 2013 IWG-MRT-ECNM-konsensuskriteerien (International Working Group ‑ Myeloproliferative Neoplasms Research and Treatment ‑ European Competence Network on Mastocytosis) perusteella. Vaste Rydapt-hoidolle määritettiin laskennallisella algoritmilla, jota käytettiin ilman arviointia. 116 potilaasta 113:lla oli C-löydös IWG-vastekriteerien perusteella (pois lukien askites C-löydöksenä). Kaikki vasteet huomioitiin, ja 12 viikon vahvistusjakso vaadittiin (ks. taulukko 8).
Taulukko 8 Midostauriinin teho ASM:n, SM-AHN:n ja MCL:n hoidossa IWG-MRT-ECNM-konsensuskriteerien perusteella algoritmisella lähestymistavalla
Kaikki arvioidut potilaat | ASM | SM-AHN | MCL | Alatyyppi tuntematon | |
N = 113 | N = 15 | N = 72 | N = 21 | N = 5 | |
Kokonaisvasteprosentti, n (%) | 32 (28,3) | 9 (60,0) | 15 (20,8) | 7 (33,3) | 1 (20,0) |
(95 % lv) | (20,2–37,6) | (32,3–83,7) | (12,2–32,0) | (14,6–57,0) | (0,5–71,6) |
Paras kokonaisvaste, n (%) | |||||
Täydellinen remissio | 1 (0,9) | 0 | 0 | 1 (4,8) | 0 |
Osittainen remissio | 17 (15,0) | 5 (33,3) | 8 (11,1) | 3 (14,3) | 1 (20,0) |
Kliinisen tilan koheneminen | 14 (12,4) | 4 (26,7) | 7 (9,7) | 3 (14,3) | 0 |
Vasteen kesto* | |||||
n/N (%) | 11/32 (34,4) | 4/9 (44,4) | 4/15 (26,7) | 3/7 (42,9) | 0/1 (0,0) |
mediaani (95 % lv) | Ei arv. (27,0–Ei arv.) | 36,8 (10,3–36,8) | Ei arv. (17,3–Ei arv.) | Ei arv. (4,1–Ei arv.) | Ei arv. |
Kokonaiselossaolo | |||||
n/N (%) | 65/113 (57,5) | 4/15 (26,7) | 49/72 (68,1) | 12/21 (57,1) | 0/5 (0,0) |
mediaani (95 % lv) | 29,9 (20,3–42,0) | 51,1 (34,7–Ei arv.) | 22,1 (16,8–32,2) | 22,6 (8,3–Ei arv.) | Ei arv. |
*Vasteen vahvistusjakso: 12 viikkoa Analyysista suljettiin pois askites C-löydöksenä. Tutkimukseen liittymätöntä syöpähoitoa saaneiden potilaiden taudin katsottiin edenneen uuden hoidon ajankohtana. | |||||
Näyttöä tukeva tutkimus oli yksihaarainen, avoin vaiheen II monikeskustutkimus, johon osallistuneilla 26 potilaalla oli ASM, SM-AHN tai MCL (tutkimus CPKC412A2213). Midostauriinia annettiin suun kautta annoksella 100 mg kahdesti vuorokaudessa 28 vuorokauden hoitojaksoissa. Tutkimushoito oli lopetettava, jos potilas ei toisen hoitojakson loppuun mennessä saavuttanut merkittävää vastetta tai osittaista vastetta. Mukana oli 20 ASM-potilasta (76,9 %; heistä 17 potilaalla [85 %] oli AHN) ja 6 MCL-potilasta (23,1 %; heistä 2 potilaalla [33,3 %] oli AHN). Mediaani-ikä oli 64,5 vuotta, ja puolet potilaista oli ≥ 65-vuotiaita. Lähtötilanteessa 88,5 %:lla oli > 1 C-löydös ja 69,2 % oli saanut aiemmin ainakin yhtä syöpähoitoa.
Ensisijainen päätetapahtuma oli vasteiden kokonaismäärä, joka arvioitiin Valentin kriteereillä ensimmäisten kahden hoitojakson aikana. 19 potilasta (73,1 %; 95 % lv: 52,2–88,4) saavutti vasteen ensimmäisten kahden hoitojakson aikana (13 merkittävää ja 6 osittaista vastetta). Seurannan mediaanikesto oli 73 kk, eikä vasteen mediaanikestoa ole saavutettu. Kokonaiselossaoloajan mediaani oli 40,0 kk (potilaiden elossaoloa seurattiin vain yhden vuoden ajan hoidon lopettamisen jälkeen).
Pediatriset potilaat
Vaiheen II tutkimuksessa arvioitiin midostauriinin käyttöä yhdessä solunsalpaajahoidon kanssa pediatrisilla potilailla, joilla oli äskettäin todettu FLT3-mutaatiopositiivinen AML. Tutkimukseen otettiin kolme potilasta, joilla oli FLT3-mutaatiopositiivinen AML. Heistä kahdella potilaalla (joiden iät olivat 10 vuotta ja 14 vuotta) todettiin annosta rajoittavaa toksisuutta heidän saatuaan toisen induktiohoitojakson, johon kuului midostauriinia (30 mg/m2 kahdesti vuorokaudessa) yhdistettynä solunsalpaajahoitoon (johon kuului sytarabiinia, 2 g/m2/vrk päivinä 1–5; fludarabiinia, 30 mg/m2/vrk päivinä 1–5; ja idarubisiinia, 12 mg/m2/vrk päivinä 2, 4 ja 6). Molemmilla potilailla todettiin merkittävästi hidastunut hematologinen toipuminen (eli pitkittynyttä asteen 4 trombosytopeniaa, joka kesti ensimmäisellä potilaalla 44 vrk ja toisella potilaalla 51 vrk, ja asteen 4 neutropeniaa, joka kesti toisella potilaalla 46 vrk). Ensimmäisen induktiohoitojakson aikana molemmat potilaat saivat midostauriinia yhdessä sytarabiinin, etoposidin ja idarubisiinin kanssa.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Rydapt-valmisteen käytöstä malignin mastosytoosin ja syöttösoluleukemian hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Rydapt-valmisteen käytöstä akuutin myelooisen leukemian hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Midostauriini on lääkeaine, joka imeytyy hyvin ja jolla on huono liukoisuus. Kaksi sen metaboliiteista on osoittautunut farmakologisesti aktiivisiksi (CGP52421 ja CGP62221). Usean annoksen annostelun jälkeen midostauriinin ja CGP62221:n farmakokinetiikka oli ajasta riippuvaista. Ensimmäisen viikon aikana havaittiin aluksi pitoisuuden suureneminen, jota seurasi pitoisuuden pieneneminen, kunnes vakaa tila saavutettiin päivänä 28. CGP52421:n pitoisuudet eivät näytä pienenevän yhtä merkittävästi kuin midostauriinin ja CGP62221:n pitoisuudet.
Imeytyminen
Midostauriinin absoluuttinen hyötyosuus suun kautta annostelun jälkeen ei ole tiedossa.
Ihmisellä suun kautta annettu midostauriini imeytyi nopeasti, ja kokonaisradioaktiivisuuden tmax havaittiin 1‑3 tunnin kuluttua annoksesta. Populaatiofarmakokinetiikan analyysin mukaan annoksilla > 50 mg x 2 lääkeaineen imeytyminen oli alle suhteessa annokseen.
Terveillä henkilöillä ruoan kanssa annetun midostauriinin 50 mg kerta-annoksen jälkeen midostauriinin AUC-arvo suureni tasolle 20800 ng*h/ml ja Cmax pieneni tasolle 963 ng/ml (ks. kohta Yhteisvaikutukset). Samaan tapaan CGP52421:n AUC suureni tasolle 19 000 ng*h/ml ja CGP62221:n AUC tasolle 29 200 ng*h/ml. CGP52421:n Cmax pieneni tasolle 172 ng/ml ja CGP62221:n tasolle 455 ng/ml. Samanaikaisesti nautittu runsasrasvainen ateria pidensi myös huippupitoisuuden saavuttamiseen kulunutta aikaa. Kaikkien yhdisteiden Tmax viivästyi: midostauriinin tmax-ajan mediaani oli 3 h, CGP52421:n Tmax oli 6 h ja CGP62221:n 7 h.
Kliinisissä tutkimuksissa, joissa tutkittiin Rydapt-valmisteen tehoa ja turvallisuutta, Rydapt annettiin kevyen aterian kanssa. Kun 100 mg midostauriinikerta-annos annettiin aterioineille ASM-, SM-AHN- ja MCL-potilaille suun kautta, midostauriinin AUCinf oli 49 600 ng*h/ml, Cmax oli 2 940 ng/ml ja Tmax oli 3 h. CGP52421:n AUC0–12h oli 2 770 ng*h/ml ja Cmax 299 ng/ml. CGP62221:n AUC0–12h oli 8 700 ng*h/ml ja Cmax 931 ng/ml. Kun toistuvia 100 mg x 2 midostauriiniannoksia annettiin suun kautta, midostauriinin Cmin,ss plasmassa oli AML-potilailla 919 ng/ml ja ASM-, SM-AHN- ja MCL-potilailla 1 060 ng/ml. CGP62221:n Cmin, ss oli AML-populaatiossa 1 610 ng/ml ja ASM-, SM-AHN- ja MCL-populaatiossa 2 020 ng/ml. CGP52421:n Cmin, ss oli AML-populaatiossa 8 630 ng/ml ja ASM-, SM-AHN- ja MCL-populaatiossa 2 860 ng/ml.
Jakautuminen
Midostauriini jakautuu kudoksiin, jakautumisen geometrinen keskiarvo on 95,2 l (Vz/F-arvo). Midostauriini ja sen metaboliitit jakautuvat lähinnä plasmaan pikemminkin kuin punasoluihin. In vitro -tietojen mukaan yli 98 % midostauriinista sitoutuu plasman proteiineihin kuten albumiiniin, happamaan α1-glykoproteiiniin ja lipoproteiiniin.
Biotransformaatio
Midostauriini metaboloituu CYP3A4-välitteisesti lähinnä hapettumalla. Tärkeimmät plasmassa esiintyvät komponentit ovat midostauriini ja kaksi aktiivista päämetaboliittia, CGP62221 (O-demetylaation välityksellä) ja CGP52421 (hydroksylaation välityksellä), jotka tuottavat 27,7 ± 2,7 % ja 38,0 ± 6,6 % plasman kokonaisaltistuksesta 96 tunnin kohdalla midostauriinin 50 mg kerta-annoksen jälkeen.
Eliminaatio
Midostauriinin ja CGP62221- ja CGP52421-metaboliittien terminaalisten puoliintumisaikojen mediaanit plasmassa ovat noin 20,9 h (midostauriini), 32,3 h (CGP62221) ja 471 h (CGP52421). Näennäisen plasmapuhdistuman (CL/F) keskiarvo terveillä tutkittavilla oli 2,4‑3,1 l/h. Populaatiofarmakokineettinen estimaatti midostauriinin puhdistumalle vakaassa tilassa oli AML-potilaille 5,9 l/h ja ASM:ia, SM-AHN:ia tai MCL:aa sairastaville potilaille 4,4 l/h. Ihmisellä tehdyn massatasapainotutkimuksen tulokset viittaavat siihen, että tärkein eliminaatioreitti on erittyminen ulosteeseen (78 % annoksesta) ja valtaosa lääkkeestä erittyy metaboliitteina (73 % annoksesta). Muuttumattomana midostauriinina erittyy 3 % annoksesta. Vain 4 % annoksesta erittyy virtsaan.
Lineaarisuus/ei-lineaarisuus
Yleisesti ottaen midostauriinin ja sen metaboliittien farmakokinetiikka ei ollut merkittävän epälineaarista suhteessa annokseen 25–100 mg kerta-annosten jälkeen. Toistuvilla 50–225 mg vuorokausiannoksilla altistus suureni kuitenkin vähemmän kuin suhteessa annokseen.
Toistuvien suun kautta otettujen annosten jälkeen midostauriinin farmakokinetiikka oli aikariippuvaista; midostauriinipitoisuudet plasmassa suurenivat aluksi ensimmäisen viikon aikana (Cmin-huippuarvo) ja pienenivät sitten vähitellen vakaan tilan pitoisuuksiin noin 28 päivän jälkeen (2,5-kertainen pieneneminen). Midostauriinipitoisuuksien pienenemisen tarkkaa mekanismia ei tunneta, mutta syynä on todennäköisesti midostauriinin ja sen kahden aktiivisen metaboliitin (CGP52421 ja CGP62221) CYP3A4-entsyymiin kohdistuva autoinduktio. CGP62221-metaboliitin farmakokinetiikassa todettiin samankaltainen trendi. Yhden hoitokuukauden jälkeen CGP52421-pitoisuudet suurenivat midostauriinipitoisuuteen verrattuna jopa 2,5-kertaisiksi potilailla, joilla oli ASM, SM-AHN tai MCL, ja jopa 9-kertaisiksi AML-potilailla.
Lääkeaineinteraktioiden mahdollisuuden arviointi in vitro
In vitro ‑tietojen perusteella midostauriini ja sen aktiiviset metaboliitit CGP52421 ja CGP62221 katsotaan CYP1A2:n ja CYP2E1:n estäjiksi ja CYP2B6:n (induktion välittäjänä CAR) ja CYP1A2:n (induktion välittäjänä AhR) indusoijiksi.
In vitro kokeet osoittivat, että midostauriini, CGP52421 ja CPG62221 voivat estää BCRP:n ja BSEP:n toimintaa. Fysiologiaan perustuviin farmakokineettisiin (PBPK) mallinnuksiin pohjautuvat simulaatiot ennustivat, että kahdesti vuorokaudessa otettu 50 mg tai 100 mg midostauriiniannos ei vakaassa tilassa todennäköisesti kliinisesti merkittävästi estä OATP1B:tä.
Erityisryhmät
Iäkkäät potilaat
Populaatiofarmakokinetiikan analyysien perusteella iällä ei havaittu olevan merkitsevää vaikutusta midostauriinin eikä sen kahden aktiivisen metaboliitin farmakokinetiikkaan 65‑85-vuotiailla potilailla. Midostauriiniannosta ei tarvitse muuttaa iän perusteella, jos kyseessä on aikuinen potilas, jolla on ASM, SM-AHN tai MCL tai AML.
Pediatriset potilaat
Rydapt-valmisteen käyttöä ei suositella lapsille eikä nuorille (ks. kohta Annostus ja antotapa). Midostauriinin farmakokinetiikkaa lapsipotilailla arvioitiin populaatiofarmakokinetiikan menetelmin suurenevilla annoksilla toteutetussa vaiheen I monoterapiatutkimuksessa, johon osallistuneilla 22 potilaalla (12 potilaan ikä 0‑2 vuotta ja 10 potilaan ikä 10‑17 vuotta) oli AML tai MLL-geenin uudelleenjärjestymän omaava ALL. Midostauriinin farmakokinetiikka 30 mg/m2 ja 60 mg/m2 kerta-annosten ja useiden annosten jälkeen oli vähemmän kuin annosriippuvaista. Koska farmakokinetiikasta pediatrisilla potilailla on vain rajoitetusti tietoa, vertailua midostauriinin farmakokinetiikkaan aikuisilla ei voida tehdä.
Sukupuoli
Populaatiofarmakokinetiikan mallien analyysien perusteella sukupuolella ei ollut tilastollisesti merkitsevää vaikutusta midostauriinin eikä sen aktiivisten metaboliittien puhdistumaan, eikä odotettavissa olevien altistusmuutosten (< 20 %) katsottu olevan kliinisesti merkittäviä. Midostauriiniannosta ei tarvitse muuttaa sukupuolen perusteella.
Rotu/etninen tausta
Lääkkeen farmakokineettisessa profiilissa ei todettu eroja kaukaasialaista syntyperää olevien ja mustaihoisten henkilöiden välillä. Terveillä japanilaisilla vapaaehtoisilla tehdyssä vaiheen I tutkimuksessa midostauriinin ja sen metaboliittien (CGP62221 ja CGP52421) farmakokineettiset profiilit vastasivat kaukaasialaista syntyperää olevilla ja mustaihoisilla henkilöillä toteutettujen farmakokinetiikan tutkimusten tuloksia. Midostauriiniannosta ei tarvitse muuttaa etnisen taustan perusteella.
Maksan vajaatoiminta
Maksan vajaatoimintaa koskeneessa spesifisessä tutkimuksessa arvioitiin systeemistä midostauriinialtistusta, kun midostauriinia oli annettu suun kautta 50 mg kahdesti vuorokaudessa 6 vuorokauden ajan ja 50 mg kerta-annos päivänä 7 tutkittaville, joilla oli lähtötilanteessa lievä tai keskivaikea maksan vajaatoiminta (Child–Pugh-luokat A ja B), ja sen jälkeen 50 mg kerta-annos tutkittaville, joilla oli vaikea maksan vajaatoiminta (Child-Pugh-luokka C) verrattuna henkilöihin, joiden maksan toiminta oli normaali. Suurin midostauriinipitoisuus saavutettiin kaikissa ryhmissä 2 ja 3 tunnin välillä kerta-annoksen tai toistuvien annosten jälkeen. Päivänä 1 terveillä tutkittavilla AUC0‑12 oli 8 130 ng*h/ml ja Cmax oli 1 206 ng/ml. AUC0‑12 oli 39 % pienempi tutkittavilla, joilla oli lievä maksan vajaatoiminta ja 36 % pienempi tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta. Päivänä 7 terveillä tutkittavilla AUCCtrough (Ctrough-käyrän alapuolinen altistus päivästä 1 päivään 7) oli 5 410 ng*h/ml. AUCCtrough oli 35 % pienempi tutkittavilla, joilla oli lievä maksan vajaatoiminta ja 20 % pienempi tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta. Päivänä 7 AUCtau oli 28 % pienempi tutkittavilla, joilla oli lievä maksan vajaatoiminta ja 20 % pienempi tutkittavilla, joilla oli keskivaikea maksan vajaatoiminta.
Vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla midostauriinin geometriset Cmax- ja AUCinf -keskiarvot olivat alempia kontrolliryhmään verrattuna (Cmax: 1 360 ng/ml, AUCinf: 30 100 ng.h/ml). Midostauriinin Cmax -arvo laski keskimäärin 78 % ja AUCinf -arvo keskimäärin 59 % vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla.
Lopuksi potilaiden pitkän aikavälin tiedot arvioitiin populaatiofarmakokineettisessä analyysissa. ASM-, SM-AHN-, MCL- ja AML-potilailla, joilla oli lievä tai keskivaikea maksan vajaatoiminta, ei voitu todeta maksan vajaatoiminnan aiheuttamaa vaikutusta.
Kaiken kaikkiaan plasman midostauriinin ja sen metaboliittien (CGP62221 ja CGP52421) altistus (AUC) ei suurentunut, kun lievää, keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavia tutkittavia verrattiin henkilöihin, joiden maksan toiminta oli normaali. Annosta ei tarvitse muuttaa, jos potilaalla on lähtötilanteessa lievä tai keskivaikea maksan vajaatoiminta. Midostauriinin ja sen aktiivisen metaboliitin CGP62221:n altistus on merkittävästi pienempi vaikeaa maksan vajaatoimintaa sairastavilla potilailla kuin potilailla, joiden maksan toiminta on normaalia (ks. kohta Annostus ja antotapa). Tehoon liittyvät tiedot ovat kuitenkin puutteelliset annosmuutosten tarpeen arvioimiseksi vaikeaa maksan vajaatoimintaa sairastavilla potilailla.
Munuaisten vajaatoiminta
Munuaisten kautta tapahtuva eliminoituminen on midostauriinille vähäinen eliminaatioreitti. Midostauriinia ei ole arvioitu munuaisten vajaatoimintaa koskeneessa spesifisessä tutkimuksessa. Populaatiofarmakokinetiikan analyyseissa hyödynnettiin kliinisiä tutkimustietoja potilaista, joilla oli AML (n = 180) ja ASM, SM-AHN ja MCL (n = 141). Näistä 321 potilaasta 177 potilaalla oli entuudestaan lievä (n = 113), keskivaikea (n = 60) tai vaikea (n = 4) munuaisten vajaatoiminta (kreatiniinipuhdistuma ≥ 15 ml/min, mutta < 90 ml/min). 144 potilaan munuaistoiminta oli lähtötilanteessa normaali (kreatiniinipuhdistuma > 90 ml/min). Näiden populaatiofarmakokinetiikan analyysien perusteella munuaisten vajaatoiminta ei vaikuttanut merkitsevästi midostauriinin puhdistumaan. Annosta ei siis tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta.
Prekliiniset tiedot turvallisuudesta
Annosta rajoittavan toksisuuden vuoksi kliinisen hoidon altistustasoja ei voitu saavuttaa eläimillä. Kaikki alla kuvatut eläimillä tehdyt löydökset havaittiin huomattavasti hoitotasoja pienemmällä midostauriinialtistuksella.
Farmakologinen turvallisuus ja kerta-annosten/toistuvan altistuksen aiheuttama toksisuus
Farmakologista turvallisuutta koskevien tutkimusten tulokset viittaavat siihen, että midostauriini ei todennäköisesti häiritse keskushermoston vitaaleja toimintoja. In vitro midostauriini ei estänyt hERG-kanavien toimintaa enintään 12 µM:n pitoisuuksina (liukoisuuden raja). Myös ihmisellä esiintyvät kaksi päämetaboliittia, GGP52421 ja CGP62221, testattiin liukoisuuden rajalla olevilla pitoisuuksilla ja ne estivät hERG-virtausta kohtuullisten turvallisuusrajojen puitteissa. Koirilla tehdyissä toistuvan altistuksen aiheuttamaa toksisuutta arvioivissa tutkimuksissa todettiin yksittäisillä eläimillä syketiheyden laskua, PQ-ajan pitenemistä ja satunnaisesti ilmaantuvia eteis-kammiokatkoksia.
Toistuvan altistuksen aiheuttamaa toksisuutta arvioivissa tutkimuksissa toksisuuden kohde-elimiä olivat ruoansulatuskanava (oksentelu koirilla ja apinoilla, ripuli ja limakalvomuutokset), kivekset (spermatogeneesin väheneminen), luuydin (hyposellulaarisuus) ja lymfaattiset elimet (depleetio/atrofia). Luuytimeen ja lymfaattisiin elimiin kohdistuvan vaikutuksen yhteydessä esiintyi hematologisia muutoksia eli valkosolumäärän ja lymfosyyttimäärän vähenemistä ja erytrosyyttiparametrien laskua. Maksaentsyymiarvojen (ALAT ja ASAT) nousua todettiin rotilla johdonmukaisesti; koirilla ja apinoilla sitä todettiin ≥ 3 kk kestäneissä pitkäaikaistutkimuksissa. Histopatologisia korrelaatteja ei todettu.
Lisääntymistoksisuus
Rotilla tehdyissä hedelmällisyystutkimuksissa midostauriiniin liittyi alentunutta hedelmällisyyttä, kivesten degeneraatiota ja atrofiaa, siittiöiden liikkuvuuden vähentymistä, oligo- ja aspermiaa, alkioiden resorptioiden lisääntymistä, tiineysprosenttien laskua ja implantoituneiden alkioiden ja elävien alkioiden määrän laskua.
Alkion- ja sikiönkehitystutkimuksissa rotalla ja kanilla todettiin alkioiden myöhäisten resorptioiden lisääntymistä, sikiöiden painon pienenemistä ja luuston luutumisen vähenemistä.
Pre- ja postnataalista kehitystä koskeneessa tutkimuksessa emoilla ilmeni dystokiaa ja pentuekoon pienenemistä. Pennuilla ilmeni painon laskua, nopeutunutta silmien täydellistä avautumista ja kuuloärsykkeen tuottaman säpsähdysreaktion kehityksen viivästymistä.
Nuorilla eläimillä tehdyt tutkimukset
Nuorilla rotilla tehdyssä tutkimuksessa midostauriinia annettiin elinpäivinä 7–70 syntymän jälkeen. Rotilla todettiin painon laskua sekä verenvuotoa ja eri solujen infiltraatiota keuhkoissa ja erytrosytoosia/erytrofagosytoosia suoliliepeen imusolmukkeissa. Fyysiseen kehitykseen, aistitoimintoihin tai käytökseen kohdistuvia vaikutuksia ei todettu. Paritteluindeksi, hedelmällisyysindeksi ja hedelmöittymisosuus pienenivät annoksilla 0, 5 ja 15 mg/kg/vrk, mutta eivät annoksella 2 mg/kg/vrk.
Geenitoksisuus
In vitro- ja in vivo ‑geenitoksisuustutkimuksissa, joissa arvioitiin oleellisia geenitoksisuuden päätetapahtumia, ei todettu näyttöä mutageenisuudesta eikä klastogeenisuudesta. Karsinogeenisuustutkimuksia ei ole tehty.
Ympäristöön kohdistuvien riskien arviointi (ERA)
ERA-tutkimusten perusteella midostauriini saattaa säilyä pitkään ympäristössä ja olla biokertyvä ja ympäristölle toksinen.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Makrogoliglyserolihydroksistearaatti
Makrogoli
Vedetön etanoli
Maissiöljyn mono‑di‑triglyseridit
All-rac-alfa-tokoferoli
Kapselin kuori
Liivate
Glyseroli
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Puhdistettu vesi
Painomuste
Karmiini (E120)
Hypromelloosi
Propyleeniglykoli
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RYDAPT kapseli, pehmeä
25 mg (L:ei) 56 fol (2x28) (6634,94 €), 112 fol (4x28) (13089,26 €)
PF-selosteen tieto
PA/alu/PVC/alu-läpipainopakkaukset. Yksi läpipainopakkaus sisältää 4 pehmeää kapselia.
Pakkaukset, joissa 56 (2 kpl 28 kapselin pakkausta) tai 112 (4 kpl 28 kapselin pakkausta) pehmeää kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaalean oranssi, pitkänomainen kapseli, jossa punaisella painettu merkintä ”PKC NVR”. Kapselin koko on noin 25,4 x 9,2 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
RYDAPT kapseli, pehmeä
25 mg 56 fol, 112 fol
- Ylempi erityiskorvaus (100 %). Midostauriini: Akuutin myelooisen leukemian hoito erityisin edellytyksin (1522).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Midostauriini: Akuutin myelooisen leukemian hoito erityisin edellytyksin (3014).
ATC-koodi
L01EX10
Valmisteyhteenvedon muuttamispäivämäärä
11.12.2024
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com