
TEPKINLY injektioneste, liuos 4 mg/0,8 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 0,8 ml:n injektiopullo sisältää 4 mg epkoritamabia pitoisuutena 5 mg/ml.
Jokainen injektiopullo on ylitäytetty, jotta siitä voidaan ottaa riittävä määrä lääkettä.
Epkoritamabi on kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla tuotettu humanisoitu immunoglobuliini G1 (IgG1) ‑bispesifinen vasta-aine CD3- ja CD20-antigeenejä vastaan.
Apuaine, jonka vaikutus tunnetaan
Yksi Tepkinly-injektiopullo sisältää 28,8 mg sorbitolia ja 0,42 mg polysorbaatti 80:aa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Tepkinly on tarkoitettu monoterapiana uusiutuneen tai hoitoon reagoimattoman diffuusin suurisoluisen B‑solulymfooman (DLBCL) hoitoon aikuispotilaille vähintään kahden systeemisen hoitolinjan jälkeen.
Tepkinly yhdistelmähoitona lenalidomidin ja rituksimabin kanssa on tarkoitettu uusiutuneen tai hoitoon reagoimattoman follikulaarisen lymfooman (FL) hoitoon aikuispotilaille.
Tepkinly on tarkoitettu monoterapiana uusiutuneen tai hoitoon reagoimattoman follikulaarisen lymfooman (FL) hoitoon aikuispotilaille vähintään kahden systeemisen hoitolinjan jälkeen.
Ehto
Valmistetta saa antaa vain syöpälääkkeiden käyttöön perehtyneen terveydenhuollon ammattilaisen valvonnassa hoitokeskuksessa, jossa on valmius vaikea-asteisten reaktioiden, kuten sytokiinioireyhtymän hoitoon.
Annostus ja antotapa
Tepkinly-valmistetta saa antaa vain syöpälääkkeiden käyttöön perehtyneen terveydenhuollon ammattilaisen valvonnassa. Sytokiinioireyhtymän (CRS) varalta ainakin 1 annos tosilitsumabia on oltava saatavilla ennen epkoritamabin annostusta syklissä 1. Tosilitsumabin lisäannos on myös oltava saatavilla 8 tunnin sisällä edellisestä tosilitsumabi-annoksesta.
Annostus
Suositeltu esilääkitys ja antoaikataulu
Tepkinly monoterapiana
Tepkinly annetaan diffuusia suurisoluista B‑solulymfoomaa sairastaville potilaille taulukossa 1 kuvatun ja follikulaarista lymfoomaa sairastaville potilaille taulukossa 2 kuvatun annoksen suurennusvaiheen sisältävän antoaikataulun mukaisesti 28 vuorokauden sykleinä.
Taulukko 1 Tepkinly-valmisteen 2-vaiheinen suurennusvaiheen sisältävä annosaikataulu diffuusia suurisoluista B‑solulymfoomaa sairastaville potilaille
Antoaikataulu | Hoitosykli | Päivät | Epkoritamabiannos (mg)a |
Viikoittain | Sykli 1 | 1 | 0,16 mg (suurennusvaiheen annos 1) |
8 | 0,8 mg (suurennusvaiheen annos 2) | ||
15 | 48 mg (ensimmäinen täysi annos) | ||
22 | 48 mg | ||
Viikoittain | Syklit 2–3 | 1, 8, 15, 22 | 48 mg |
Kahden viikon välein | Syklit 4–9 | 1, 15 | 48 mg |
Neljän viikon välein | Syklit 10+ | 1 | 48 mg |
a0,16 mg on aloitusannos, 0,8 mg on väliannos ja 48 mg on täysi annos. | |||
Taulukko 2 Tepkinly-valmisteen 3-vaiheinen suurennusvaiheen sisältävä annosaikataulu follikulaarista lymfoomaa sairastaville potilaille
Antoaikataulu | Hoitosykli | Päivät | Epkoritamabiannos (mg)a |
Viikoittain | Sykli 1 | 1 | 0,16 mg (suurennusvaiheen annos 1) |
8 | 0,8 mg (suurennusvaiheen annos 2) | ||
15 | 3 mg (suurennusvaiheen annos 3) | ||
22 | 48 mg (ensimmäinen täysi annos) | ||
Viikoittain | Syklit 2–3 | 1, 8, 15, 22 | 48 mg |
Kahden viikon välein | Syklit 4–9 | 1, 15 | 48 mg |
Neljän viikon välein | Syklit 10+ | 1 | 48 mg |
a0,16 mg on aloitusannos, 0,8 mg on väliannos, 3 mg on toinen väliannos ja 48 mg on täysi annos. | |||
Tepkinly-valmisteen antoa jatketaan, kunnes tauti etenee tai potilaalla ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Tepkinly yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tepkinly annetaan 28 vuorokauden sykleinä 12 syklin ajan tai kunnes tauti etenee tai potilaalla ilmenee ei‑hyväksyttävää toksisuutta, sen mukaan mikä näistä tapahtuu ensin.
Taulukko 3 Tepkinly-valmisteen 3-vaiheinen suurennusvaiheen sisältävä annosaikataulu yhdistelmähoitona lenalidomidin ja rituksimabin kanssa follikulaarista lymfoomaa sairastaville potilaille
Antoaikataulu | Hoitosykli | Päivät | Epkoritamabiannos (mg) |
Viikoittain | Sykli 1 | 1 | 0,16 mg (suurennusvaiheen annos 1) |
8 | 0,8 mg (suurennusvaiheen annos 2) | ||
15 | 3 mg (suurennusvaiheen annos 3) | ||
22 | 48 mg (ensimmäinen täysi annos) | ||
Syklit 2 ja 3 | 1, 8, 15 ja 22 | 48 mg | |
Neljän viikon välein | Syklit 4–12 | 1 | 48 mg |
Tepkinlyn yhdistelmähoitona annetaan 20 mg lenalidomidia suun kautta kerran vuorokaudessa (päivästä 1 päivään 21) syklien 1–12 ajan ja rituksimabia 375 mg/m2 laskimoon viikoittain syklin 1 ajan (päivinä 1, 8, 15 ja 22) ja neljän viikon välein syklien 2–5 ajan (päivänä 1).
Lisätietoja on lenalidomidi- ja rituksimabivalmisteiden valmisteyhteenvedoissa.
Esilääkitys ja estohoito
Taulukossa 4 annetaan yksityiskohtaiset tiedot suositellusta esilääkityksestä sytokiinioireyhtymän (CRS) ehkäisemiseksi.
Taulukko 4 Esilääkitys ennen epkoritamabin antoa
Sykli | Esilääkitystä tarvitsevat potilaat | Esilääkitysa | Anto |
Sykli 1 | Kaikki potilaat | Deksametasoni (15 mg suun kautta tai laskimoon) tai prednisoloni (100 mg suun kautta tai laskimoon) tai vastaava
|
|
|
| ||
Syklistä 2 alkaen | Potilaat, joille kehittyi asteen 2 tai 3b CRS edellisen annoksen yhteydessä | Deksametasoni(15 mg suun kautta tai laskimoon) tai prednisoloni (100 mg suun kautta tai laskimoon) tai vastaava
|
|
aYhdistelmähoidon lääkeaineiden kanssa käytettävä esilääkitys voi toimia epkoritamabin esilääkityksenä hoitavan lääkärin harkinnan mukaan, mikäli annettavat annokset ovat vähintään yhtä suuria. | |||
Lisätietoja valmisteiden esilääkityssuosituksista on lenalidomidin valmisteyhteenvedossa ja rituksimabin valmisteyhteenvedossa.
Pneumocystis jirovecii -keuhkokuumeen (PCP) ja herpesviruksen aiheuttamien infektioiden estohoito on erittäin suositeltavaa epkoritamabihoidon aikana.
Potilaan riittävästä nesteytyksestä on huolehdittava Tepkinly-hoidon aikana.
On erittäin suositeltavaa, että kaikki potilaat noudattavat seuraavia nesteytysohjeita Syklin 1 aikana, ellei se ole lääketieteellisesti vasta-aiheista:
-
2-3 litraa nestettä 24 tunnin aikana ennen jokaista epkoritamabin antokertaa
-
Tauota verenpainetta alentavat lääkkeet 24 tuntia ennen jokaista epkoritamabin antokertaa
-
Anna 500 ml isotonista laskimonsisäistä (iv) nesteytystä epkoritamabin antopäivänä ennen annoksen antamista; JA
-
2-3 litraa nestettä 24 tunnin aikana jokaisen epkoritamabin antokerran jälkeen.
Potilaille, joilla kliinisen tuumorilyysioireyhtymän (CTLS) riski on suurentunut, suositellaan nesteytystä ja estohoitoa virtsahappopitoisuutta pienentävällä lääkkeellä.
Potilaita on seurattava CRS:n ja/tai immuunijärjestelmän efektorisoluihin liittyvän neurotoksisuusoireyhtymän (ICANS) varalta ja hoidettava nykyisten hoitosuositusten mukaisesti epkoritamabin antamisen jälkeen. Potilaille on kerrottava CRS:ään ja ICANS-oireyhtymään liittyvistä merkeistä ja oireista, ja heitä on kehotettava hakeutumaan välittömästi lääkärinhoitoon, jos näitä merkkejä tai oireita ilmenee milloin tahansa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annosmuutokset ja haittavaikutusten hoito
Sytokiinioireyhtymä (CRS)
Epkoritamabihoitoa saaneille potilaille voi kehittyä CRS.
Kuumeen, hypoksian ja hypotension mahdolliset muut syyt on arvioitava ja hoidettava. Jos CRS:ää epäillään, se tulee hoitaa taulukossa 5 annettujen suositusten mukaisesti. Potilaita, joille kehittyy CRS, on seurattava tiiviimmin seuraavan hoitoaikataulun mukaisen epkoritamabin antokerran yhteydessä.
Taulukko 5 CRS:n luokittelu- ja hoito-ohjeet
Astea | Suositeltu hoito | Epkoritamabiannoksen muuttaminenh |
Aste 1
| Anna tukihoitoa, kuten kuumetta alentavia lääkkeitä ja laskimonsisäistä nesteytystä Deksametasoninb antaminen voidaan aloittaa Jos potilas on hyvin iäkäs tai hänellä on suuri kasvaintaakka, kasvainsoluja verenkierrossa tai kuume, joka vastaa huonosti kuumetta alentavaan lääkitykseen
Jos paranemista ei tapahdu 24 tunnissa, tosilitsumabia on harkittavad Jos potilaalla on samanaikaisesti CRS ja ICANS, ks. taulukko 6 | Keskeytä epkoritamabihoito, kunnes CRS-tapahtuma korjaantuu |
Aste 2
ja
ja/tai
| Anna tukihoitoa, kuten kuumetta alentavia lääkkeitä ja laskimonsisäistä nesteytystä Deksametasoninb antamista on syytä harkita Antisytokiinihoitoa tosilitsumabillad suositellaan Jos CRS vastaa huonosti deksametasoni- ja tosilitsumabihoitoon:
Jos potilaalla on samanaikaisesti CRS ja ICANS, ks. taulukko 6 | Keskeytä epkoritamabihoito, kunnes CRS-tapahtuma korjaantuu |
Aste 3
ja
ja/tai
| Anna tukihoitoa, kuten kuumetta alentavia lääkkeitä ja laskimonsisäistä nesteytystä Deksametasoniac tulee antaa Antisytokiinihoitoa tosilitsumabillad suositellaan Jos CRS vastaa huonosti deksametasoni- ja tosilitsumabihoitoon:
Jos potilaalla on samanaikaisesti CRS ja ICANS, ks. taulukko 6 | Keskeytä epkoritamabihoito, kunnes CRS-tapahtuma korjaantuu Lopeta epkoritamabihoito, jos potilaalle kehittyy 3 asteen CRS, joka kestää pidempään kuin 72 tuntia. Lopeta Lopeta epkoritamabihoito, jos potilaalle kehittyy enemmän kuin 2 erillistä 3 asteen CRS tapahtumaa, vaikka kukin korjaantuisi asteen 2 tapahtumaksi 72 tunnin sisällä. |
Aste 4
ja
ja/tai
| Anna tukihoitoa, kuten kuumetta alentavia lääkkeitä ja laskimonsisäistä nesteytystä Deksametasoniac tulee antaa Antisytokiinihoitoa tosilitsumabillad suositellaan Jos CRS vastaa huonosti deksametasoni- ja tosilitsumabihoitoon:
Jos potilaalla on samanaikaisesti CRS ja ICANS, ks. taulukko 6 | Lopeta epkoritamabihoito pysyvästi |
aCRS luokitellaan ASTCT-konsensuskriteerien mukaisesti | ||
Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymä (ICANS)
Potilaita on seurattava ICANS-oireyhtymän merkkien ja oireiden varalta. Muut neurologisten oireiden syyt on suljettava pois. Jos ICANS-oireyhtymää epäillään, se tulee hoitaa taulukossa 6 annettujen suositusten mukaisesti.
Taulukko 6 ICANS-oireyhtymän luokittelu- ja hoito-ohjeet
Astea | Suositeltu hoito | Epkoritamabiannoksen muuttamineng |
Aste 1a | Deksametasonihoitoc Harkitse kouristuskohtauksia estäviä, ei-sedatoivia lääkevalmisteita (esim. levetirasetaamia), kunnes ICANS korjaantuu Ei samanaikaista CRS:ää:
ICANS ja CRS samanaikaisesti:
| Keskeytä epkoritamabihoito, kunnes tapahtuma korjaantuu |
Aste 2a | Deksametasonihoitoe Harkitse kouristuskohtauksia estäviä, ei-sedatoivia lääkevalmisteita (esim. levetirasetaamia), kunnes ICANS korjaantuu Ei samanaikaista CRS:ää:
ICANS ja CRS samanaikaisesti: |
Keskeytä epkoritamabihoito, kunnes tapahtuma korjaantuu |
Aste 3a tai kouristuskohtauksiaa, joko:
tai
kohonnut kallonsisäinen paine: fokaalinen/paikallinen turvotusa, joka näkyy neurokuvantamisessab | Deksametasonihoitof
Harkitse kouristuskohtauksia estäviä, ei-sedatoivia lääkevalmisteita (esim. levetirasetaamia), kunnes ICANS korjaantuu Ei samanaikaista CRS:ää:
ICANS ja CRS samanaikaisesti:
| Lopeta epkoritamabihoito pysyvästi |
Aste 4a tai alentunut tajunnan tasoa, joko:
kouristuksiaa, joko:
tai motorisia löydöksiäa:
kohonnut kallonsisäinen paine / aivoturvotusa, johon liittyy esim. seuraavia oireita/löydöksiä:
tai
| Deksametasonihoitof
Harkitse kouristuskohtauksia estäviä, ei-sedatoivia lääkevalmisteita (esim. levetirasetaamia), kunnes ICANS korjaantuu Ei samanaikaista CRS:ää:
ICANS ja CRS samanaikaisesti:
| Lopeta epkoritamabihoito pysyvästi |
aICANS luokitellaan ASTCT ICANS -konsensusluokituksen mukaisesti. ICANS-aste määräytyy vaikeimman sellaisen tapahtuman mukaan, jolle ei löydy muuta syytä (ICE-pistearvo, tajunnan taso, kouristuskohtaukset, motoriset löydökset, kohonnut kallonsisäinen paine / aivoturvotus) | ||
Taulukko 7 Suositellut annosmuutokset muiden haittavaikutusten varalta
Haittavaikutus1 | Vakavuusaste1 | Toimi |
Infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Asteet 1-4 |
|
Neutropenia tai kuumeinen neutropenia (ks.kohta Haittavaikutukset) | Absoluuttinen neutrofiiliarvo < 0,5 x 109/l |
|
Trombosytopenia (ks. kohta Haittavaikutukset) | Verihiutalemäärä |
|
Muut haittavaikutukset (ks. kohta Haittavaikutukset) | Aste 3 tai korkeampi |
|
1Perustuu National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE)-kriteereihin, Versio 5.0. | ||
Kuumeisen neutropenian ja toistuvan asteen ≥ 3 neutropenian tapauksissa on harkittava granulosyyttiryhmiä stimuloivaa kasvutekijää (G‑CSF). Katso lenalidomidin valmisteyhteenvedosta tarkat ohjeet annosmuutoksista neutropeniatapauksissa. Katso rituksimabin valmisteyhteenvedosta suositukset immunoglobuliinipitoisuuksien määrittämisestä.
Annoksen jääminen väliin tai viivästyminen
Diffuusi suurisoluinen B‑solulymfooma
Uusi aloitussykli (samanlainen kuin sykli 1, tavanomainen CRS-estohoito mukaan lukien) tarvitaan:
-
Jos aloitusannoksen (0,16 mg) ja väliannoksen (0,8 mg) välinen aika on yli 8 vuorokautta.
-
Jos väliannoksen (0,8 mg) ja ensimmäisen täyden annoksen (48 mg) välinen aika on yli 14 vuorokautta.
-
Jos täysien annosten (48 mg) välinen aika on yli 6 viikkoa.
Uuden aloitussyklin jälkeen potilaan hoitoa jatketaan seuraavan suunnitellun hoitosyklin (sen syklin jälkeinen sykli, jolloin annoksen antaminen viivästyi) päivästä 1.
Follikulaarinen lymfooma
Uusi aloitussykli (samanlainen kuin sykli 1, tavanomainen CRS-estohoito mukaan lukien) tarvitaan:
-
Jos aloitusannoksen (0,16 mg) ja väliannoksen (0,8 mg) välinen aika on yli 8 vuorokautta.
-
Jos väliannoksen (0,8 mg) ja toisen väliannoksen (3 mg) välinen aika on yli 8 vuorokautta.
-
Jos toisen väliannoksen (3 mg) ja ensimmäisen täyden annoksen (48 mg) välinen aika on yli 14 vuorokautta.
-
Jos täysien annosten (48 mg) välinen aika on yli 6 viikkoa.
Uuden aloitussyklin jälkeen potilaan hoitoa jatketaan seuraavan suunnitellun hoitosyklin (sen syklin jälkeinen sykli, jolloin annoksen antaminen viivästyi) päivästä 1.
Erityisryhmät
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annoksen muuttamista ei pidetä tarpeellisena. Epkoritamabia ei ole tutkittu vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaistautia sairastavilla potilailla.
Annossuosituksia ei voida antaa vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaistautia sairastaville potilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa sairastavien potilaiden annoksen muuttamista ei pidetä tarpeellisena. Epkoritamabia ei ole tutkittu vaikeaa maksan vajaatoimintaa (määritellään kokonaisbilirubiiniarvoksi > 3 x ULN ja miksi tahansa ASAT-arvoksi) sairastavilla potilailla, ja tietoja keskivaikeaa maksan vajaatoimintaa (määritellään kokonaisbilirubiiniarvoksi > 1,5–3 x ULN ja miksi tahansa ASAT-arvoksi) sairastavista potilaista on vain vähän. Annossuosituksia ei voida antaa keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Iäkkäät
Annoksen muuttaminen ei ole tarpeen ≥ 65-vuotiaille potilaille (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Pediatriset potilaat
Tepkinly-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tepkinly annetaan ihon alle. Sen saa antaa ainoastaan ihonalaisena injektiona mieluiten alavatsan tai reiden alueelle. Pistoskohdan vaihtamista vasemmalta puolelta oikealle tai päinvastoin suositellaan etenkin, kun lääkettä annetaan viikoittain (eli syklien 1–3 aikana).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Sytokiinioireyhtymä (CRS)
Epkoritamabia saaneilla potilailla on esiintynyt CRS:ää, joka voi olla henkeä uhkaava tai johtaa kuolemaan. Yleisimpiä CRS:n merkkejä ja oireita ovat kuume, hypotensio ja hypoksia. Muita CRS:n merkkejä ja oireita, joita on todettu useammalla kuin kahdella potilaalla, ovat vilunväristykset, takykardia, päänsärky ja hengenahdistus.
Useimmat CRS-tapahtumat kehittyivät syklin 1 aikana ja liittyivät ensimmäiseen täyteen epkoritamabiannokseen. Kortikosteroideja tulee antaa estohoitona CRS:n riskin pienentämiseksi (ks. kohta Annostus ja antotapa).
Potilaita on seurattava CRS:n merkkien ja oireiden varalta epkoritamabin annon jälkeen.
CRS:n ensimmäisten merkkien tai oireiden ilmaantuessa on aloitettava tukihoito tosilitsumabilla ja/tai kortikosteroideilla tarpeen mukaan (ks. kohta Annostus ja antotapa, taulukko 5). Potilaille on kerrottava CRS:ään liittyvistä merkeistä ja oireista, ja heitä on kehotettava ottamaan yhteyttä terveydenhuollon ammattilaiseen ja hakeutumaan välittömästi lääkärinhoitoon, jos näitä merkkejä tai oireita ilmenee milloin tahansa.
CRS:n hoito voi edellyttää joko epkoritamabihoidon antamisen viivyttämistä tai lopettamista CRS:n vaikeusasteen mukaan (ks. kohta Annostus ja antotapa).
Hemofagosyyttinen lymfohistiosytoosi (HLH)
Hemofagosyyttistä lymfohistiosytoosia (HLH), myös kuolemaan johtaneita tapauksia, on raportoitu epkoritamabia saaneilla potilailla. HLH on hengenvaarallinen oireyhtymä, jolle on tunnusomaista kuume, ihottuma, lymfadenopatia, maksan ja/tai pernan suurentuma ja sytopeniat. HLH:n mahdollisuutta tulee epäillä, kun CRS:n esiintyminen on epätyypillistä tai pitkittynyttä. Potilaita on seurattava HLH:n kliinisten merkkien ja oireiden varalta. Jos epäillään HLH:ta, epkoritamabin käyttö on keskeytettävä diagnostisen tutkimuksen ja HLH:n hoidon aloittamista varten. Jos HLH on vahvistettu, Tepkinlyn antaminen on lopetettava.
Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymä (ICANS)
Epkoritamabia saaneilla potilailla on esiintynyt ICANS-oireyhtymää, mukaan lukien kuolemaan johtaneita tapauksia. ICANS voi ilmetä afasiana, tajunnan tason muutoksina, kognitiivisten kykyjen heikentymisenä, motorisena heikkoutena, kouristuskohtauksina ja aivoturvotuksena.
Suurin osa ICANS-tapauksista todettiin epkoritamabihoitosyklin 1 aikana, mutta jotkin tapaukset kehittyivät viiveellä.
Potilaita on seurattava ICANS-oireyhtymän merkkien ja oireiden varalta epkoritamabin annon jälkeen.
ICANS-oireyhtymän ensimmäisten merkkien ja oireiden ilmaantuessa on aloitettava hoito kortikosteroideilla ja kouristuskohtauksia estävillä, ei-sedatoivilla lääkkeillä tarpeen mukaan (ks. kohta Annostus ja antotapa, taulukko 6). Potilaille on kerrottava ICANS-oireyhtymän merkeistä ja oireista ja siitä, että tapahtumat voivat kehittyä viiveellä. Potilaita on kehotettava ottamaan yhteyttä terveydenhuollon ammattilaiseen ja hakeutumaan välittömästi lääkärinhoitoon, jos näitä merkkejä tai oireita ilmenee milloin tahansa. Epkoritamabihoidon antamista on viivytettävä tai hoito on lopetettava suositusten mukaisesti (ks. kohta Annostus ja antotapa).
Vakavat infektiot
Epkoritamabihoito voi suurentaa infektioiden riskiä. Kliinisissä tutkimuksissa epkoritamabihoitoa saaneilla potilailla on todettu vakavia tai kuolemaan johtaneita infektioita (ks. kohta Haittavaikutukset).
Epkoritamabin antamista potilaille, joilla on kliinisesti merkittäviä aktiivisia systeemisiä infektioita, on vältettävä.
Potilaille on tarvittaessa annettava estohoitona mikrobilääkkeitä ennen epkoritamabihoidon aloittamista ja hoidon aikana (ks. kohta Annostus ja antotapa). Potilaita on seurattava infektioiden merkkien ja oireiden varalta ennen epkoritamabin antoa ja epkoritamabin annon jälkeen, ja hoitoa on annettava tarvittaessa. Kuumeisen neutropenian kehittyessä potilas on arvioitava infektion varalta ja tila on hoidettava antibiooteilla, nesteytyksellä ja muulla tukihoidolla paikallisten suositusten mukaan.
Epkoritamabia saavilla potilailla on raportoitu myös hypogammaglobulinemiaa (ks. kohta Haittavaikutukset). Immunoglobuliinien (Ig) pitoisuuksia on seurattava ennen hoitoa ja sen aikana. Potilaita on hoidettava paikallisten hoitosuositusten mukaisesti, ottaen huomioon myös infektioihin liittyvät varotoimet sekä mikrobilääkeprofylaksi.
Progressiivista multifokaalista leukoenkefalopatiaa (PML), mukaan lukien kuolemaan johtaneita tapauksia, on raportoitu epkoritamabia saaneilla potilailla, jotka ovat saaneet aiemmin hoitoa myös muilla immunosuppressiivisilla lääkkeillä. Jos epkoritamabihoidon aikana ilmenee PML:ään viittaavia neurologisia oireita, epkoritamabihoito on lopetettava ja asianmukaiset diagnostiset toimenpiteet aloitettava.
Tuumorilyysioireyhtymä (TLS)
TLS:ää on raportoitu epkoritamabia saavilla potilailla (ks. kohta Haittavaikutukset). Potilaille, joiden TLS-riski on suurentunut, suositellaan nesteytystä ja estohoitoa virtsahappopitoisuutta pienentävällä lääkkeellä. Potilaita on seurattava TLS:n merkkien tai oireiden varalta etenkin, jos potilaalla on suuri kasvaintaakka tai nopeasti kasvavia kasvaimia tai jos potilaan munuaisten toiminta on heikentynyt. Potilaiden veriarvoja on seurattava, ja mahdolliset poikkeamat on hoidettava viipymättä.
Tumour flare ‑reaktio
Epkoritamabihoitoa saaneilla potilailla on ilmoitettu tumour flare ‑reaktioita (ks. kohta Haittavaikutukset). Ne voivat ilmetä paikallisena kipuna ja turvotuksena. Tumour flare ‑reaktioiden esiintyminen on linjassa epkoritamabin vaikutusmekanismin kanssa, ja reaktiot johtuvat todennäköisesti T‑solujen kulkeutumisesta kasvaimiin epkoritamabin annon jälkeen.
Tumour flare ‑reaktioille ei ole tunnistettu spesifisiä riskitekijöitä. Tilan heikentymisen ja morbiditeetin riski on kuitenkin tumour flare ‑reaktiosta johtuvan massavaikutuksen vuoksi suurentunut potilailla, joilla on suurimassainen kasvain lähellä hengitysteitä ja/tai vitaalielintä. Epkoritamabihoitoa saavia potilaita on seurattava ja arvioitava anatomiselta sijainniltaan kriittisten tumour flare ‑reaktioiden varalta.
CD20‑negatiivinen tauti
CD20‑negatiivista DLBCL:ää ja CD20-negatiivista FL:ää sairastavia epkoritamabihoitoa saavia potilaita koskevaa tietoa on saatavilla vain vähän. On mahdollista, että CD20‑negatiivista DLBCL:ää ja CD20-negatiivista FL:ää sairastavat potilaat saattavat hyötyä hoidosta vähemmän kuin CD20‑positiivista DLBCL:ää ja CD20-positiivista FL:ää sairastavat. CD20‑negatiivista DLBCL:ää ja FL:ää sairastaville potilaille annettavan epkoritamabihoidon mahdolliset riskit ja hyödyt on punnittava.
Potilaskortti
Lääkärin on kerrottava potilaalle CRS:än ja ICANS-oireyhtymän riskeistä, sekä niiden merkeistä ja oireista. Potilaita on kehotettava hakeutumaan välittömästi lääkärinhoitoon, jos CRS:n ja/tai ICANS-oireyhtymän merkkejä ja oireita ilmenee. Potilaille on annettava potilaskortti ja heitä on kehotettava pitämään potilaskortti aina mukanaan. Tässä kortissa kuvataan CRS:n ja ICANS-oireyhtymän oireita, ja mikäli näitä ilmenee potilaalla, hänen on hakeuduttava välittömästi lääkärinhoitoon.
Rokotukset
Eläviä ja/tai eläviä heikennettyjä rokotteita ei saa antaa epkoritamabihoidon aikana. Eläviä rokotteita saaneilla potilailla ei ole tehty tutkimuksia.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo eli sen voidaan sanoa olevan "natriumiton".
Tämä lääkevalmiste sisältää 28,8 mg sorbitolia per injektiopullo, joka vastaa 27,33 mg/ml.
Tämä lääkevalmiste sisältää 0,42 mg polysorbaatti 80:aa per injektiopullo, joka vastaa 0,4 mg/ml. Polysorbaatti 80 saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Epkoritamabin aiheuttama tiettyjen proinflammatoristen sytokiinipitoisuuksien ohimenevä nousu saattaa vähentää CYP450-entsyymien aktiivisuutta. Hoidon seurantaa on syytä harkita, jos epkoritamabihoito aloitetaan potilaille, joita hoidetaan kapean terapeuttisen indeksin CYP450-substraateilla.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy naisilla
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisyä epkoritamabihoidon aikana ja vähintään 4 kuukautta viimeisen annoksen jälkeen. Naisille, jotka voivat tulla raskaaksi, on tehtävä raskaustesti ennen epkoritamabihoidon aloittamista.
Raskaus
Vaikutusmekanisminsa perusteella epkoritamabi voi aiheuttaa sikiövaurioita, kuten B‑solujen lymfosytopeniaa ja normaalien immuunivasteiden muutoksia, jos sitä annetaan raskaana oleville naisille. Ei ole olemassa tietoja epkoritamabin käytöstä raskaana oleville naisille. Epkoritamabia ei ole tutkittu eläimille tehdyissä lisääntymistutkimuksissa. IgG1-vasta-aineet, kuten epkoritamabi, voivat läpäistä istukan ja altistaa sikiön. Raskaana oleville naisille on kerrottava sikiöön kohdistuvan riskin mahdollisuudesta.
Epkoritamabin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö epkoritamabi ihmisen rintamaitoon tai vaikuttaako se maidontuotantoon. Rintamaidossa tiedetään olevan IgG:tä, joten vastasyntynyt saattaa altistua epkoritamabille maidon kautta. Rintaruokinta on lopetettava epkoritamabihoidon ajaksi ja vähintään 4 kuukaudeksi viimeisen annoksen jälkeen.
Hedelmällisyys
Epkoritamabilla ei ole tehty hedelmällisyystutkimuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Epkoritamabin vaikutuksia miesten ja naisten hedelmällisyyteen ei tunneta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Epkoritamabilla on merkittävä vaikutus ajokykyyn ja koneidenkäyttökykyyn. ICANS-oireyhtymän mahdollisuuden vuoksi epkoritamabia saavilla potilailla on riski tajunnan tason muuttumiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Potilaita tulee kehottaa noudattamaan varovaisuutta ajaessaan autoa, pyöräillessään tai käyttäessään painavia tai mahdollisesti vaarallisia koneita (tai välttämään näitä toimia, jos heillä esiintyy oireita).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Epkoritamabi monoterapiana
Epkoritamabin turvallisuutta arvioitiin satunnaistamattomassa, yhdellä hoitoryhmällä toteutetussa GCT3013‑01-tutkimuksessa 382 potilaalla, joilla oli uusiutunut tai hoitoon reagoimaton suurisoluinen B-solulymfooma (N = 167), follikulaarinen lymfooma (N = 129) tai follikulaarinen lymfooma (3‑vaiheinen suurennusvaiheen annosaikataulu, N = 86) vähintään kahden systeemisen hoitolinjan jälkeen. Arviointiin sisältyivät kaikki potilaat, jotka otettiin tutkimuksessa 48 mg:n annosta saavaan ryhmään ja jotka saivat vähintään yhden annoksen epkoritamabia.
Seuraavia haittavaikutuksia on raportoitu epkoritamabille kliinisissä tutkimuksissa ja markkinoille tulon jälkeen.
Epkoritamabialtistuksen mediaanikesto oli 4,9 kuukautta (vaihteluväli: < 1 – 30 kuukautta).
Yleisimpiä haittavaikutuksia (≥ 20 %) olivat CRS, pistoskohdan reaktiot, uupumus, virusinfektio, neutropenia, muskuloskeletaalinen kipu, kuume ja ripuli.
Vakavia haittavaikutuksia esiintyi 50 %:lla potilaista. Yleisin vakava haittavaikutus (≥ 10 %) oli sytokiinioireyhtymä (34 %). Neljätoista potilasta (3,7 %) sai kuolemaan johtaneen haittavaikutuksen (keuhkokuume yhdeksällä (2,4 %) potilaalla, virusinfektio neljällä (1,0 %) potilaalla ja ICANS yhdellä (0,3 %) potilaalla).
Hoidon lopettamiseen johtaneita haittavaikutuksia esiintyi 6,8 %:lla potilaista. Neljätoista (3,7 %) potilasta lopetti epkoritamabihoidon keuhkokuumeen takia, kahdeksan (2,1 %) potilasta virusinfektion takia, kaksi (0,5 %) potilasta uupumuksen takia, yksi (0,3 %) potilas CRS:n takia, yksi (0,3 %) potilas ICANS-oireyhtymän takia ja yksi (0,3 %) potilas ripulin takia.
Haittavaikutusten aiheuttamia annosten viivästymisiä esiintyi 42 %:lla potilaista. Annosten viivästymiseen johtaneita haittavaikutuksia (≥ 3 %) olivat virusinfektiot (17 %), CRS (11 %), neutropenia (5,2 %), keuhkokuume (4,7 %), ylähengitystieinfektio (4,2 %) ja kuume (3,7 %).
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Epkoritamabin turvallisuutta yhdistelmähoitona lenalidomidin ja rituksimabin kanssa arvioitiin avoimessa satunnaistetussa M20‑638-monikeskustutkimuksessa, johon osallistui potilaita, joilla oli uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma (FL) yhden aiemman hoitolinjan jälkeen. Potilaat saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa (N = 243) tai pelkästään lenalidomidia ja rituksimabia (N = 238).
CRS:ää ja ICANS-oireyhtymää lukuun ottamatta alla ja taulukossa 9 esitetyt turvallisuustulokset edustavat koko turvallisuustietojoukkoa 243 potilaalta, jotka saivat epkoritamabia 2‑vaiheisen suurennusvaiheen annosaikataulun tai suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. CRS:stä ja ICANS-oireyhtymästä esitetyt tiedot perustuvat 133 potilaaseen, jotka saivat epkoritamabia suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti yhdistelmähoitona lenalidomidin ja rituksimabin kanssa.
Suositeltu 3‑vaiheinen suurennusvaiheen annosaikataulu
Tutkimuksessa M20‑638 minkä tahansa asteen CRS:ää esiintyi 26 %:lla (35/133) potilaista, jotka saivat epkoritamabia suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti yhdistelmähoitona lenalidomidin ja rituksimabin kanssa.
CRS:n aiheuttamia vakavia haittavaikutuksia esiintyi 12 %:lla potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. CRS:n aiheuttamia epkoritamabiannosten viivästymisiä (keskeytyksiä) esiintyi 11 %:lla potilaista. ICANS-oireyhtymää ilmeni 0,8 %:lla potilaista, ja yksi tapahtumista ilmoitettiin asteeksi 1.
Turvallisuustutkimusten koko joukko
Epkoritamabia 2‑vaiheisen suurennusvaiheen annosaikataulun jälkeen tai suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun jälkeen yhdistelmähoitona lenalidomidin ja rituksimabin kanssa saaneessa 243 potilaan joukossa yleisimmät (≥ 20 %) haittavaikutukset olivat neutropenia, ihottuma, ylähengitystieinfektiot, uupumus, ripuli, pistoskohdan reaktiot, anemia, ummetus, trombosytopenia, CRS, hypogammaglobulinemia, COVID‑19, kuume ja keuhkokuume.
Vakavia haittavaikutuksia esiintyi 44 %:lla potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. Vakavia haittavaikutuksia ≥ 5 %:lla potilaista olivat CRS, keuhkokuume, COVID‑19 ja kuumeinen neutropenia.
Epkoritamabin lopettaminen pysyvästi haittavaikutuksen vuoksi tapahtui 6,6 %:lla potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. Haittavaikutuksia, jotka johtivat epkoritamabin lopettamiseen pysyvästi useamman kuin 1 potilaan kohdalla, olivat keuhkokuume, COVID‑19, ylähengitystieinfektiot ja neutropenia.
Epkoritamabiannosten viivästymisiä haittavaikutuksen vuoksi tapahtui 70 %:lla potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. Haittavaikutuksia, jotka johtivat epkoritamabiannosten viivästymisiin ≥ 5 %:lla potilaista, olivat neutropenia, ylähengitystieinfektiot, COVID‑19, keuhkokuume, ihottuma ja trombosytopenia.
Haittavaikutustaulukko
Epkoritamabin käytön yhteydessä ilmenevät haittavaikutukset luetellaan taulukossa 8.
Epkoritamabin yhdistelmähoidossa lenalidomidin ja rituksimabin kanssa ilmenevät haittavaikutukset luetellaan taulukossa 9.
Kliinisissä tutkimuksissa todetut epkoritamabin haittavaikutukset (taulukko 8 ja taulukko 9) on lueteltu MedDRA-elinjärjestelmäluokan ja seuraavien yleisyyksien mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000) ja hyvin harvinainen (< 1/10 000).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 8 Uusiutunutta tai hoitoon reagoimatonta LBCL:ää tai FL:ää sairastavilla, epkoritamabihoitoa saaneilla potilailla raportoidut haittavaikutukset
Elinjärjestelmäluokka / suositeltu termi tai haittavaikutus | Kaikki asteet | Aste 3–4 |
Infektiot | ||
Virusinfektioa | Hyvin yleinen | Yleinen |
Keuhkokuumeb | Hyvin yleinen | Yleinen |
Ylähengitystieinfektioc | Hyvin yleinen | Yleinen |
Sieni-infektiod | Yleinen |
|
Sepsise | Yleinen | Yleinen |
Selluliitti | Yleinen | Yleinen |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | ||
Tumour flare -reaktio | Yleinen |
|
Veri ja imukudos | ||
Neutropeniaf | Hyvin yleinen | Hyvin yleinen |
Anemiag | Hyvin yleinen | Yleinen |
Trombosytopeniah | Hyvin yleinen | Yleinen |
Lymfosytopeniai | Hyvin yleinen | Yleinen |
Kuumeinen neutropenia | Yleinen | Yleinen |
Hemofagosyyttinen lymfohistiosytoosij | Melko harvinainen | Harvinainen |
Immuunijärjestelmä | ||
Sytokiinioireyhtymäj | Hyvin yleinen | Yleinen |
Hypogammaglobulinemia | Hyvin yleinen | Melko harvinainen |
Aineenvaihdunta ja ravitsemus | ||
Ruokahalun väheneminen | Hyvin yleinen | Melko harvinainen |
Hypokalemia | Yleinen | Yleinen |
Hypofosfatemia | Yleinen | Yleinen |
Hypomagnesemia | Yleinen | Melko harvinainen |
Tuumorilyysioireyhtymäk | Yleinen | Melko harvinainen |
Hermosto | ||
Päänsärky | Hyvin yleinen | Melko harvinainen |
Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymäj | Yleinen | Melko harvinainen |
Sydän | ||
Sydämen rytmihäiriötl | Yleinen | Melko harvinainen |
Hengityselimet, rintakehä ja välikarsina | ||
Pleuraeffuusio | Yleinen | Yleinen |
Ruoansulatuselimistö | ||
Ripuli | Hyvin yleinen | Melko harvinainen |
Vatsakipum | Hyvin yleinen | Yleinen |
Pahoinvointi | Hyvin yleinen | Melko harvinainen |
Oksentelu | Yleinen | Melko harvinainen |
Iho ja ihonalainen kudos | ||
Ihottuman | Hyvin yleinen |
|
Kutina | Yleinen |
|
Luusto, lihakset ja sidekudos | ||
Muskuloskeletaalinen kipuo | Hyvin yleinen | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | ||
Pistoskohdan reaktiotp | Hyvin yleinen |
|
Uupumusq | Hyvin yleinen | Yleinen |
Kuumer | Hyvin yleinen | Yleinen |
Edeemas | Hyvin yleinen | Yleinen |
Tutkimukset | ||
Alaniiniaminotransferaasiarvo suurentunut | Yleinen | Yleinen |
Aspartaattiaminotransferaasiarvo suurentunut | Yleinen | Yleinen |
Veren kreatiniiniarvo suurentunut | Yleinen |
|
Alhainen veren natriumarvot | Yleinen | Melko harvinainen |
Alkalinen fosfataasiarvo suurentunut | Yleinen |
|
Haittavaikutukset luokiteltiin NCI CTCAE -kriteerien version 5.0 mukaan | ||
Taulukko 9 Raportoidut haittavaikutukset potilailla, joilla on uusiutunut tai hoitoon reagoimaton FL ja hoitona epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Elinjärjestelmäluokka / suositeltu termi tai haittavaikutus | Kaikki asteet | Aste 3–4 |
Infektiot | ||
Ylähengitystieinfektiota | Hyvin yleinen | Yleinen |
COVID-19b | Hyvin yleinen | Yleinen# |
Keuhkokuumec | Hyvin yleinen | Hyvin yleinen |
Sytomegalovirusinfektiod | Yleinen | Yleinen# |
Herpesinfektioe | Yleinen | Melko harvinainen# |
Sieni-infektiof | Yleinen | Melko harvinainen |
Veri ja imukudos | ||
Neutropeniag | Hyvin yleinen | Hyvin yleinen |
Anemiah | Hyvin yleinen | Yleinen# |
Trombosytopeniai | Hyvin yleinen | Yleinen |
Lymfosytopeniaj | Hyvin yleinen | Hyvin yleinen |
Kuumeinen neutropenia | Yleinen | Yleinen |
Immuunijärjestelmä | ||
Sytokiinioireyhtymä‡ | Hyvin yleinen |
|
Hypogammaglobulinemiak | Hyvin yleinen | Melko harvinainen |
Psyykkiset häiriöt | ||
Unettomuus | Hyvin yleinen |
|
Hermosto | ||
Neurologiset muutoksetl | Hyvin yleinen |
|
Päänsärky | Hyvin yleinen |
|
Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymä‡ | Melko harvinainen |
|
Ruoansulatuselimistö | ||
Ripuli | Hyvin yleinen | Yleinen# |
Ummetus | Hyvin yleinen | Melko harvinainen# |
Pahoinvointi | Hyvin yleinen |
|
Limakalvotulehdusm | Yleinen |
|
Iho ja ihonalainen kudos | ||
Ihottuman | Hyvin yleinen | Hyvin yleinen# |
Yleisoireet ja antopaikassa todettavat haitat | ||
Uupumuso | Hyvin yleinen | Yleinen# |
Pistoskohdan reaktiotp | Hyvin yleinen |
|
Kuume | Hyvin yleinen | Melko harvinainen# |
Tutkimukset | ||
Pienentynyt veren kaliumpitoisuusq | Hyvin yleinen | Yleinen# |
Suurentunut alaniiniaminotransferaasiarvo | Hyvin yleinen | Yleinen# |
Suurentunut aspartaattiaminotransferaasiarvo | Hyvin yleinen | Yleinen# |
§Haittavaikutukset luokiteltiin CTCAE-version 5.0 perusteella. | ||
Valikoitujen haittavaikutusten kuvaus
Sytokiinioireyhtymä
Epkoritamabi monoterapiana
2‑vaiheinen suurennusvaiheen annosaikataulu (suurisoluinen B‑solulymfooma ja follikulaarinen lymfooma)
Tutkimuksessa GCT3013‑01 minkä tahansa asteen CRS:ää esiintyi 58 %:lla (171/296) epkoritamabihoitoa 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti saaneista LBCL- ja FL-potilaista. Asteen 1 CRS:n ilmaantuvuus oli 35 % ja asteen 2 CRS:n ilmaantuvuus 21 %, ja asteen 3 CRS kehittyi 2,4 %:lle potilaista. CRS uusiutui 21 %:lla potilaista. Minkä tahansa asteista CRS:ää esiintyi 9,8 %:lla potilaista aloitusannoksen (syklin 1 päivä 1) jälkeen, 13 %:lla väliannoksen (syklin 1 päivä 8) jälkeen, 51 %:lla ensimmäisen täyden annoksen (syklin 1 päivä 15) jälkeen, 6,5 %:lla toisen täyden annoksen (syklin 1 päivä 22) jälkeen ja 3,7 %:lla kolmannen täyden annoksen (syklin 2 päivä 1) jälkeen tai myöhemmin. Mediaaniaika CRS:n alkamiseen viimeisimmän annetun epkoritamabiannoksen jälkeen oli 2 vuorokautta (vaihteluväli: 1–12 vuorokautta). Mediaaniaika CRS:n alkamiseen ensimmäisen täyden annoksen jälkeen oli 19,3 tuntia (vaihteluväli: < 0,1 – 7 vuorokautta). CRS korjaantui 99 %:lla potilaista, ja CRS-tapahtumien mediaanikesto oli 2 vuorokautta (vaihteluväli: 1–54 vuorokautta).
Yleisimpiä CRS:n merkkejä ja oireita niillä 171 potilaalla, joille kehittyi CRS, olivat kuume 99 %:lla, hypotensio 32 %:lla ja hypoksia 16 %:lla. Muita CRS:n merkkejä ja oireita, joita esiintyi ≥ 3 %:lla potilaista, olivat vilunväristykset (11 %), takykardia (mukaan lukien sinustakykardia (11 %)), päänsärky (8,2 %), pahoinvointi (4,7 %) ja oksentelu (4,1 %). Samanaikaista ohimenevää maksaentsyymiarvojen nousua (ALAT tai ASAT > 3 x ULN) esiintyi 4,1 %:lla CRS:n saaneista potilaista. Seuranta- ja hoito-ohjeet, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
3‑vaiheinen suurennusvaiheen annosaikataulu (follikulaarinen lymfooma)
Tutkimuksessa GCT3013‑01 minkä tahansa asteen CRS:ää esiintyi 49 %:lla (42/86) FL-potilaista, jotka saivat epkoritamabihoitoa suositellun follikulaarista lymfoomaa koskevan 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti. Asteen 1 CRS:n ilmaantuvuus oli 40 % ja asteen 2 CRS:n ilmaantuvuus 9 %. Asteen ≥ 3 CRS-tapahtumia ei raportoitu. CRS uusiutui 23 %:lla potilaista. Suurin osa CRS-tapahtumista esiintyi syklin 1 aikana, jolloin niitä raportoitiin 48 %:lla potilaista. Syklin 1 aikana CRS:ää esiintyi 12 %:lla potilaista aloitusannoksen (syklin 1 päivä 1) jälkeen, 5,9 %:lla väliannoksen (syklin 1 päivä 8) jälkeen, 15 %:lla toisen väliannoksen (syklin 1 päivä 15) jälkeen ja 37 %:lla ensimmäisen täyden annoksen (syklin 1 päivä 22) jälkeen. Mediaaniaika CRS:n alkamiseen viimeisimmän annetun epkoritamabiannoksen jälkeen oli 59 tuntia (vaihteluväli: 1–8 vuorokautta). Mediaaniaika CRS:n alkamiseen ensimmäisen täyden annoksen jälkeen oli 61 tuntia (vaihteluväli: 1–8 vuorokautta). CRS korjaantui 100 %:lla potilaista, ja CRS-tapahtumien mediaanikesto oli 2 vuorokautta (vaihteluväli: 1–14 vuorokautta).
CRS:n aiheuttamia vaikeita haittavaikutuksia esiintyi 28 %:lla epkoritamabia saaneista potilaista. CRS:n aiheuttamia annosten viivästymisiä esiintyi 19 %:lla epkoritamabia saaneista potilaista.
Niillä 42 potilaalla, joilla CRS:ää esiintyi suositellulla annoksella, yleisimpiä (≥ 10 %) CRS:n merkkejä ja oireita olivat kuume (100 %) ja hypotensio (14 %). Kortikosteroidien käytön lisäksi CRS-tapahtumia hoidettiin 12 %:lla potilaista tosilitsumabilla.
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 minkä tahansa asteen CRS:ää esiintyi 26 %:lla (35/133) potilaista, jotka saivat epkoritamabia suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. Asteen 1 CRS:n ilmaantuvuus oli 21 % (28/133) ja asteen 2 ilmaantuvuus 5,3 % (7/133). Analyysihetkellä asteen ≥ 3 CRS-tapahtumia ei ollut raportoitu. CRS uusiutui 10 %:lla (13/133) potilaista. Kaikista CRS-tapahtumista suurin osa (88 %) ilmeni syklin 1 aikana. Syklissä 1 CRS:ää esiintyi 6 %:lla (8/133) potilaista aloitusannoksen (syklin 1 päivä 1) jälkeen, 3,8 %:lla (5/133) potilaista ensimmäisen väliannoksen (syklin 1 päivä 8) jälkeen, 2,3 %:lla (3/132) potilaista toisen väliannoksen (syklin 1 päivä 15) jälkeen ja 19 %:lla (25/132) potilaista ensimmäisen täyden annoksen (syklin 1 päivä 22) jälkeen. Kaikkien annosten mediaaniaika CRS:n alkamiseen viimeisimmän annetun epkoritamabiannoksen jälkeen oli 78 tuntia (vaihteluväli: 0,2–12 vuorokautta). Mediaaniaika CRS:n alkamiseen ensimmäisen täyden 48 mg:n annoksen jälkeen oli 41 tuntia (vaihteluväli: 0,3–12 vuorokautta). CRS korjaantui 100 %:lla potilaista, ja CRS-tapahtumien mediaanikesto oli 2 vuorokautta (vaihteluväli: 0,1–26 vuorokautta).
Immuunijärjestelmän efektorisoluihin liittyvä neurotoksisuusoireyhtymä
Epkoritamabi monoterapiana
Tutkimuksessa GCT3013‑01 ICANS-oireyhtymää esiintyi 4,7 %:lla (18/382) epkoritamabihoitoa saaneista potilaista; 3,1 %:lla esiintyi asteen 1 ja 1,3 %:lla asteen 2 oireyhtymä. Yksi potilas (0,3 %) sai asteen 5 (kuolemaan johtaneen) ICANS-tapahtuman. Mediaaniaika ensimmäisen ICANS-oireyhtymän alkamiseen epkoritamabihoidon aloittamisen (syklin 1 päivä 1) jälkeen oli 18 vuorokautta (vaihteluväli: 8–141 vuorokautta). ICANS korjaantui 94 %:lla (17/18) potilaista tukihoidolla. Mediaaniaika ICANS-oireyhtymän korjaantumiseen oli 2 vuorokautta (vaihteluväli: 1–9 vuorokautta). Niillä 18 potilaalla, jotka saivat ICANS-oireyhtymän, ICANS ilmeni ennen CRS:ää 11 %:lla potilaista, samanaikaisesti CRS:n kanssa 44 %:lla potilaista, CRS:n alkamisen jälkeen 17 %:lla potilaista ja ilman CRS:ää 28 %:lla potilaista.
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 ICANS-oireyhtymää esiintyi 0,8 %:lla (1/133) FL‑potilaista, jotka saivat epkoritamabia suositellun 3‑vaiheisen suurennusvaiheen sisältävän annosaikataulun mukaisesti yhdistelmähoitona lenalidomidin ja rituksimabin kanssa, yhtenä asteeksi 1 raportoituna tapahtumana. Tapahtuma ilmeni 48 mg:n annoksen jälkeen syklin 1 päivänä 22, ja ICANS korjaantui 3 vuorokaudessa.
Vakavat infektiot
Epkoritamabi monoterapiana
Suurisoluinen B‑solulymfooma
Tutkimuksessa GCT3013‑01 minkä tahansa asteisia vakavia infektioita esiintyi 25 %:lla (41/167) epkoritamabihoitoa saaneista LBCL-potilaista. Yleisimpiä vakavia infektioita olivat COVID‑19 (6,6 %), COVID‑19-keuhkokuume (4,2 %), keuhkokuume (3,6 %), sepsis (2,4 %), ylähengitystieinfektio (1,8 %), bakteremia (1,2 %) ja septinen sokki (1,2 %). Mediaaniaika epkoritamabihoidon aloittamisesta (syklin 1 päivä 1) ensimmäisen vakavan infektion kehittymiseen oli 56 vuorokautta (vaihteluväli: 4–631 vuorokautta) ja infektioiden mediaanikesto oli 15 vuorokautta (vaihteluväli: 4–125 vuorokautta). Asteen 5 infektiotapahtumia esiintyi 7 (4,2 %) potilaalla.
Follikulaarinen lymfooma
Tutkimuksessa GCT3013‑01 minkä tahansa asteisia vakavia infektioita esiintyi 32 %:lla (68/215) epkoritamabihoitoa saaneista FL-potilaista. Yleisimpiä vakavia infektioita olivat COVID‑19 (8,8 %), COVID‑19-keuhkokuume (5,6 %), keuhkokuume (3,7 %), virtsatieinfektio (1,9 %) ja Pneumocystis jirovecii -keuhkokuume (1,4 %). Mediaaniaika epkoritamabihoidon aloittamisesta (syklin 1 päivä 1) ensimmäisen vakavan infektion ilmaantumiseen oli 81 vuorokautta (vaihteluväli: 1–636 vuorokautta), ja infektioiden mediaanikesto oli 18 vuorokautta (vaihteluväli: 4–249 vuorokautta). Asteen 5 infektiotapahtumia esiintyi 8 potilaalla (3,7 %), joista 6 (2,8 %) johtui COVID-19:sta tai COVID-19-keuhkokuumeesta.
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 vakavia infektioita, mukaan lukien opportunistisia infektioita, raportoitiin 33 %:lla (81/243) FL‑potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa. Yleisimpiä olivat keuhkokuume (10 %), COVID‑19 (4,5 %) ja COVID‑19-keuhkokuume (3,7 %). Mediaaniaika siitä, kun epkoritamabin yhdistelmähoito lenalidomidin ja rituksimabin kansa aloitettiin (syklin 1 päivä 1), ensimmäisen vakavan infektion ilmaantumiseen oli 91 vuorokautta (vaihteluväli: 2–418 vuorokautta) ja mediaanikesto 13 vuorokautta (vaihteluväli: 1–123 vuorokautta).
Neutropenia
Epkoritamabi monoterapiana
Tutkimuksessa GCT3013‑01 minkä tahansa asteen neutropeniaa esiintyi 28 %:lla (105/382) potilaista, ja 23 % oli asteen 3–4 tapahtumia. Mediaaniaika ensimmäisen neutropeniatapahtuman / neutrofiiliarvon laskun kehittymiseen oli 65 vuorokautta (vaihteluväli: 2–750 vuorokautta) ja tapahtumien mediaanikesto oli 15 vuorokautta (vaihteluväli: 2–415 vuorokautta). Niistä 105 potilaasta, joilla ilmeni neutropeniatapahtuma / neutrofiiliarvojen lasku, 61 % sai G‑CSF-valmistetta tapahtumien hoitoon.
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 minkä tahansa asteen neutropeniaa esiintyi 74 %:lla (180/243) potilaista, ja 27 % (66/243) oli asteen 3 ja 42 % (101/243) asteen 4 tapahtumia. Mediaaniaika ensimmäisen neutropeniatapahtuman / neutrofiiliarvon laskun kehittymiseen oli 57 vuorokautta (vaihteluväli: 2–377 vuorokautta) ja tapahtumien mediaanikesto oli 22 vuorokautta (vaihteluväli: 3–219 vuorokautta). Niistä 167 potilaasta, joilla ilmeni asteen 3–4 neutropeniatapahtuma / neutrofiiliarvojen lasku, 87 % (146/167) sai G‑CSF-valmistetta tapahtumien hoitoon.
Tuumorilyysioireyhtymä
Epkoritamabi monoterapiana
Tutkimuksessa GCT3013‑01 TLS:ää esiintyi 1,0 %:lla (4/382) potilaista. Mediaaniaika ilmaantumiseen oli 18 vuorokautta (vaihteluväli: 8–33 vuorokautta) ja mediaanikesto oli 3 vuorokautta (vaihteluväli: 2–4 vuorokautta).
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 esiintyi TLS‑laboratoriotulos yhdellä (1) potilaalla. Tapahtuma ilmeni syklin 1 päivänä 10 ja korjaantui 6 vuorokaudessa. Kliinistä TLS:ää ei havaittu FL-potilailla, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa.
Tumour flare -reaktio
Epkoritamabi monoterapiana
Tutkimuksessa GCT3013‑01 tumour flare -reaktio esiintyi 1,6 %:lla (6/382) potilaista, joista kaikki oli asteen 2 tapahtumia. Mediaaniaika ensimmäiseen tumour flare -reaktioon oli 19,5 vuorokautta (vaihteluväli 9–34 vuorokautta) ja mediaanikesto oli 9 vuorokautta (vaihteluväli 1–50 vuorokautta).
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Tutkimuksessa M20‑638 tumour flare ‑reaktio esiintyi 1,2 %:lla (3/243) potilaista, ja asteen 2 tapahtumia oli 0,8 % (2/243) ja asteen 3 tapahtumia 0,4 % (1/243). Mediaaniaika ensimmäiseen tumour flare ‑reaktioon oli 8 vuorokautta (vaihteluväli 7–20 vuorokautta) ja mediaanikesto oli 7,5 vuorokautta (vaihteluväli 3–12 vuorokautta).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksessa potilasta on seurattava minkä tahansa haittavaikutusten merkkien ja oireiden varalta, ja asianmukaista tukihoitoa on annettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX27
Vaikutusmekanismi
Epkoritamabi on humanisoitu IgG1‑bispesifinen vasta-aine, joka sitoutuu CD20:n spesifiseen solunulkoiseen epitooppiin B‑solujen pinnalla ja CD3:een T‑solujen pinnalla. Epkoritamabin aktiivisuus riippuu sen CD20:tä ilmentäviin syöpäsoluihin ja CD3:a ilmentäviin endogeenisiin T‑soluihin kohdistuvasta samanaikaisesta vaikutuksesta, joka indusoi spesifisten T‑solujen aktivoitumista ja CD20:tä ilmentävien solujen T‑soluvälitteistä tuhoutumista.
Epkoritamabin Fc-alue on vaimennettu kohteesta riippumattomien immuunijärjestelmän efektorimekanismien estämiseksi. Näitä ovat esimerkiksi vasta-aineesta riippuvainen soluvälitteinen sytotoksisuus (ADCC), komplementista riippuvainen soluvälitteinen sytotoksisuus (CDC) ja vasta-aineesta riippuvainen solujen fagosytoosi (ADCP).
Prekliinisissä tutkimuksissa epkoritamabin ja rituksimabin yhdistelmä ei aiheuttanut toiminnallisia häiriöitä ja tuotti toisiaan täydentävää NK‑soluvälitteistä vasta-aineesta riippuvaista sytotoksisuutta (ADCC) ja T‑soluvälitteistä sytotoksisuutta.
Farmakodynaamiset vaikutukset
Epkoritamabi indusoi verenkierron B‑solujen nopeaa ja pysyvää vähenemistä (määritellään CD19 B‑soluarvoksi ≤ 10 solua/µl) henkilöillä, joilla on havaittavissa olevia B‑soluja hoidon aloitusvaiheessa. DLBCL:ää sairastavista tutkittavista 21 %:lla (n = 33) ja FL:ää sairastavista tutkittavista 50 %:lla (n = 56) oli havaittavissa olevia B‑soluja verenkierrossa hoidon aloitusvaiheessa. Verenkierron T‑solujen ohimenevää vähenemistä todettiin välittömästi jokaisen annoksen jälkeen syklin 1 aikana, ja myöhempien syklien aikana todettiin myös T‑solujen määrän lisääntymistä.
Tutkimuksessa GCT3013‑01 epkoritamabin suositellun 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisen ihonalaisen annon jälkeen LBCL-potilailla esiintyi tiettyjen sytokiinien (IFN‑γ, TNFα, IL‑6, IL‑2 ja IL‑10) pitoisuuksien ohimenevää ja vähäistä nousua pääasiassa ensimmäisen täyden annoksen (48 mg) jälkeen, ja huippupitoisuudet mitattiin 1–4 vuorokautta annoksen antamisen jälkeen. Sytokiinien pitoisuudet palasivat lähtöarvoihin ennen seuraavan täyden annoksen antamista, mutta sytokiinien pitoisuuksien nousua todettiin kuitenkin myös syklin 1 jälkeen.
Tutkimuksessa GCT3013‑01 epkoritamabin suositellun 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisen ihonalaisen annon jälkeen FL-potilaiden CRS:n riskiin yhdistettyjen IL‑6‑pitoisuuksien mediaani pysyi johdonmukaisesti pienenä kunkin syklistä 1 alkaen annetun annoksen jälkeen, etenkin ensimmäisen täyden annoksen jälkeen, verrattuna potilaisiin, jotka saivat epkoritamabia 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti.
Immunogeenisuus
Lääkevasta‑aineita todettiin yleisesti. Hoidon aikana ilmenneiden lääkevasta‑aineiden ilmaantuvuus 2‑vaiheista suurennusvaiheen annosaikataulua (0,16/0,8/48 mg) käytettäessä DLBCL- ja FL-potilaiden yhdistetyssä populaatiossa oli 3,4 % (positiivisia 3,4 %, negatiivisia 93,9 % ja määrittämättömiä 2,7 %; arvioitavissa olleiden potilaiden N = 261) tutkimuksessa GCT3013‑01 ja 3,3 % (positiivisia 3,3 %, negatiivisia 95 % ja määrittämättömiä 1,7 %; arvioitavissa olleiden potilaiden N = 60) tutkimuksessa GCT3013‑04.
Hoidon aikana ilmenneiden lääkevasta‑aineiden ilmaantuvuus 3‑vaiheista suurennusvaiheen annosaikataulua (0,16/0,8/3/48 mg) käytettäessä FL-potilaiden optimointikohortissa oli 7 % (positiivisia 7 %, negatiivisia 91,5 % ja määrittämättömiä 1,4 %; arvioitavissa olevien potilaiden N = 71) tutkimuksessa GCT3013‑01. Määrittämättömiksi määriteltiin ne tutkittavat, joiden positiivisuus lääkevasta-aineille vahvistettiin lähtötilanteessa, mutta joilla ei ollut vahvistettua positiivista tulosta hoidon ajalta tai joilla oli vahvistettu positiivinen tulos hoidon ajalta, mutta titteri oli samansuuruinen tai pienempi kuin lähtötilanteessa.
Epkoritamabivasta-aineita kehittyi 2,1 %:lle (5/238) FL‑potilaista, jotka saivat epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa M20‑638-tutkimuksessa (enintään 12 syklin aikana).
Tutkimuksissa ei saatu näyttöä siitä, että lääkevasta‑aineet vaikuttaisivat valmisteen farmakokinetiikkaan, tehoon tai turvallisuuteen, mutta tietoa on toistaiseksi rajallisesti. Neutraloivia vasta‑aineita ei arvioitu.
Kliininen teho ja turvallisuus
Diffuusi suurisoluinen B‑solulymfooma
Tutkimus GCT3013‑01 oli avoin, useilla kohorteilla ja yhdellä ryhmällä toteutettu monikeskustutkimus, jossa epkoritamabia arvioitiin monoterapiana uusiutunutta tai hoitoon reagoimatonta suurisoluista B‑solulymfoomaa (LBCL) sairastavilla potilailla kahden tai useamman systeemisen hoitolinjan jälkeen. Tutkimuksessa oli mukana myös diffuusia suurisoluista B‑solulymfoomaa (DLBCL) sairastavia potilaita. Tutkimukseen sisältyi annoksen suurentamisvaihe ja hoidon laajennusvaihe. Tutkimuksen laajennusvaiheessa oli mukana aggressiivisen non-Hodgkin-lymfooman (aNHL) kohortti, indolentin NHL:n (iNHL) kohortti ja manttelisolulymfooman (MCL) kohortti. Keskeinen aNHL-kohortti koostui LBCL-potilaista (N = 157), mukaan lukien potilaat, joilla oli DLBCL (N = 139, joista 12 potilaalla oli MYC-, BCL2- ja/tai BCL6-geenien uudelleenjärjestymiä eli DH/TH), korkea-asteinen B‑solulymfooma (HGBCL) (N = 9), asteen 3B follikulaarinen lymfooma (FL) (N = 5) ja potilaat, joilla oli primaarinen mediastinaalinen B‑solulymfooma (PMBCL) (N = 4). DLBCL kohortissa 29 %:lla potilaista (40/139) oli indolentista lymfoomasta muuntunut DLBCL. Tutkimukseen osallistui potilaita, joilla oli oltava dokumentoitu CD20-positiivinen kypsien B‑solujen syöpä WHO:n 2016 tai WHO:n 2008 luokituksen mukaan edustavan patologisen lausunnon perusteella, joiden kohdalla aiempi autologinen hematopoieettinen kantasolusiirto (HSCT) oli epäonnistunut tai jotka eivät voineet saada autologista HSCT-hoitoa, potilaita, joiden lymfosyyttiarvo oli < 5×109/l, ja potilaita, jotka olivat saaneet vähintään yhtä monoklonaalista CD20-vasta-ainetta sisältävää hoitoa.
Tutkimuksesta suljettiin pois potilaat, joiden lymfooma oli levinnyt keskushermostoon, potilaat, joille oli tehty aiemmin allogeeninen HSCT tai kiinteän elimen siirto, ja potilaat, joilla oli jokin krooninen infektiosairaus, tiedossa oleva T‑soluvälitteisen immuniteetin heikkous, kreatiniinipuhdistuma alle 45ml/min, alaniiniaminotransferaasi > 3 kertaa normaaliarvojen ylärajaa suurempi, sydämen ejektiofraktio alle 45 % tai tiedossa oleva kliinisesti merkittävä sydän- ja verisuonitauti. Tehoa arvioitiin 139 potilaalla, joilla oli DLBCL ja jotka olivat saaneet vähintään yhden annoksen epkoritamabia ihon alle 4 viikon eli 28 vuorokauden sykleinä. Epkoritamabia annettiin monoterapiana suositellun 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti seuraavasti:
-
Sykli 1: epkoritamabia 0,16 mg päivänä 1, 0,8 mg päivänä 8, 48 mg päivänä 15 ja päivänä 22
-
Syklit 2–3: epkoritamabia 48 mg päivinä 1, 8, 15 ja 22
-
Syklit 4–9: epkoritamabia 48 mg päivinä 1 ja 15
-
Syklistä 10 alkaen: epkoritamabia 48 mg päivänä 1
Epkoritamabin antoa jatkettiin, kunnes tauti eteni tai potilaalla ilmeni toksisuutta, joka ei ollut hyväksyttävissä.
Demografiset tiedot ja lähtötilanteen ominaisuudet esitetään taulukossa 10.
Taulukko 10 DLBCL-potilaiden demografiset tiedot ja lähtötilanteen ominaisuudet tutkimuksessa GCT3013‑01
Ominaisuudet | (N = 139) |
Ikä | |
Mediaani, vuotta (min., maks.) | 66 (22, 83) |
< 65 vuotta, n (%) | 66 (47) |
65 – < 75 vuotta, n (%) | 44 (32) |
≥ 75 vuotta, n (%) | 29 (21) |
Miehiä, n (%) | 85 (61) |
Rotu, n (%) | |
Valkoihoinen | 84 (60) |
Aasialainen | 27 (19) |
Muu | 5 (4) |
Ei ilmoitettu | 23 (17) |
ECOG-toimintakykyluokka; n (%) | |
0 | 67 (48) |
1 | 67 (48) |
2 | 5 (4) |
Taudin levinneisyysastec diagnoosihetkellä, n (%) | |
III | 16 (12) |
IV | 86 (62) |
Aikaisempien lymfooman hoitolinjojen lukumäärä | |
Mediaani (min., maks.) | 3 (2, 11) |
2, n (%) | 41 (30) |
3, n (%) | 47 (34) |
≥ 4, n (%) | 51 (37) |
DLBCL-tautihistoria, n (%) | |
De novo -DLBCL | 97 (70) |
Indolentista lymfoomasta muuntunut DLBCL | 40 (29) |
Keskuslaboratoriossa tehty FISH-analyysid, N = 88 | |
Double‑hit/triple‑hit -lymfooma, n (%) | 12 (14) |
Aiempi autologinen HSCT | 26 (19) |
Aiempi hoito, n (%) | |
Aiempi CAR‑T | 53 (38) |
Primaarihoitoon reagoimatona | 82 (59) |
Reagoimaton ≥ 2 aiempaan peräkkäiseen lymfooman hoitolinjaanb | 104 (75) |
Reagoimaton viimeiseen systeemiseen antineoplastiseen hoitolinjaanb | 114 (82) |
Reagoimaton aiempaan CD20-vasta-ainehoitoon | 117 (84) |
Reagoimaton CAR‑T-hoitoon | 39 (28) |
a Potilaan taudin katsotaan olevan primaarihoitoon reagoimaton/vastaamaton, jos hän ei saa vastetta ensilinjan lymfoomahoitoon. | |
Ensisijainen tehon päätetapahtuma oli Luganon kriteerien (2014) mukaan määritetty ja riippumattoman arviointilautakunnan (IRC) arvioima kokonaisvasteprosentti (ORR). Seuranta-ajan mediaani oli 15,7 kuukautta (vaihteluväli: 0,3–23,5 kuukautta). Altistuksen mediaanikesto oli 4,1 kuukautta (vaihteluväli: 0–23 kuukautta).
Taulukko 11 Tutkimuksen GCT3013‑01 tehotulokset DLBCL-potilaillaa
Päätetapahtuma | Epkoritamabi |
ORRb, n (%) | 86 (62) |
(95 %:n lv) | (53,3; 70) |
CRb, n (%) | 54 (39) |
(95 %:n lv) | (30,7; 47,5) |
PR, n (%) | 32 (23) |
(95 %:n lv) | (16,3; 30,9) |
DORb |
|
Mediaani (95 %:n lv), kuukautta | 15,5 (9,7; NR) |
DOCRb |
|
Mediaani (95 %:n lv), kuukautta | NR (12,0; NR) |
TTR, mediaani (vaihteluväli), kuukautta | 1,4 (1; 8,4) |
lv = luottamusväli; CR = täydellinen vaste; DOR = vasteen kesto; DOCR = täydellisen vasteen kesto; IRC = riippumaton arviointilautakunta; ORR = kokonaisvasteprosentti; PR = osittainen vaste; TTR = vasteen saavuttamiseen kulunut aika | |
Täydelliseen vasteeseen kuluneen ajan mediaani oli 2,6 kuukautta (vaihteluväli: 1,2–10,2 kuukautta).
Follikulaarinen lymfooma
M20‑638
Tutkimus M20‑638 oli avoin satunnaistettu monikeskustutkimus, jossa arvioitiin epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa potilailla, joilla oli uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma (FL) yhden aiemman hoitolinjan jälkeen. Potilaat satunnaistettiin saamaan epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa tai pelkästään lenalidomidia ja rituksimabia. Tutkimukseen otetuilla potilailla oli histologisesti vahvistettu klassinen FL (aiemmin asteen 1–3a FL), jonka levinneisyysaste oli II, III tai IV ilman näyttöä histologisesta muuntumisesta aggressiiviseksi lymfoomaksi, sekä CD20-positiivinen tauti viimeisimmän edustavan patologiaraportin perusteella WHO:n luokituksen 5. painoksen mukaan, uusiutunut tai hoitoon reagoimaton tauti ainakin yhden sellaisen lymfooman hoitolinjan jälkeen, joka sisälsi hoitoa monoklonaalisilla CD20-vasta-aineilla yhdistettynä kemoterapiaan, Eastern Cooperative Oncology Groupin (ECOG) toimintakykyluokka 0–2, ei dokumentoitua reagoimattomuutta lenalidomidiin, ei lenalidomidialtistusta 12 kuukauteen ennen satunnaistusta ja tarve hoidon aloitukselle tutkijan määrityksen mukaan oireiden ja/tai tautitaakan perusteella (esim. GELF-kriteerit). Tutkimuksesta suljettiin pois potilaat, joilla oli keskushermostoon levinnyt lymfooma, aiempi allografti, tiedossa oleva aktiivinen infektio, tiedossa oleva T-soluvälitteisen immuniteetin heikentyminen, kreatiniinipuhdistuma < 50 ml/min, alaniinitransaminaasi > 3 kertaa normaaliarvojen ylärajaa suurempi tai kliinisesti merkitsevä sydän- ja verisuonisairaus. Potilaat saivat epkoritamabia 28 päivän sykleissä yhteensä 12 syklin ajan tai kunnes tauti eteni tai todettiin ei‑hyväksyttävää toksisuutta, sen mukaan mikä näistä tapahtuikaan ensin.
Epkoritamabin suositeltu 3‑vaiheinen suurennusvaiheen annosaikataulu oli seuraava:
-
Sykli 1: epkoritamabia 0,16 mg päivänä 1, 0,8 mg päivänä 8, 3 mg päivänä 15 ja 48 mg päivänä 22
-
Syklit 2–3: epkoritamabia 48 mg päivinä 1, 8, 15 ja 22
-
Syklit 4–12: epkoritamabia 48 mg päivänä 1
Molemmissa hoitohaaroissa lenalidomidia annettiin suun kautta 20 mg:n annos kerran vuorokaudessa 12 syklin ajan päivinä 1–21 ja rituksimabia annettiin laskimoon annoksella 375 mg/m2 päivinä 1, 8, 15 ja 22 syklin 1 ajan ja sen jälkeen päivänä 1 syklien 2–5 ajan.
Taulukossa 12 esitetyt lähtötilanteen demografiset tiedot ja tautiin liittyvät lähtötilanteen piirteet perustuvat lähtöryhmien mukaiseen (intent-to-treat, ITT) populaatioon.
Taulukko 12 Lähtötilanteen demografiset tiedot ja tautiin liittyvät lähtötilanteen piirteet potilailla, joilla on uusiutunut tai hoitoon reagoimaton FL, M20‑638-tutkimuksessa
Parametri | Epkoritamabi + lenalidomidi ja rituksimabi (N = 243) | Lenalidomidi ja rituksimabi (N = 245) |
Ikä, vuotta |
| |
Mediaani (vaihteluväli) | 60 (30, 84) | 63 (24, 89) |
Ikäjakauma, n (%) |
| |
< 65 vuotta | 155 (64) | 139 (57) |
65 – < 75 vuotta | 68 (28) | 71 (29) |
≥ 75 vuotta | 20 (8) | 35 (14) |
Sukupuoli, n (%) |
| |
Mies | 139 (57) | 138 (56) |
Rotu, n (%) |
| |
Valkoihoinen | 168 (71) | 184 (76) |
Aasialainen | 63 (27) | 54 (22) |
Tummaihoinen tai afroamerikkalainen | 6 (3) | 2 (0.8) |
Amerikan tai Alaskan alkuperäiskansalainen | 0 | 1 (0.4) |
Useita | 1 (0.4) | 1 (0.4) |
Puuttuu | 5 | 3 |
ECOG-toimintakykyluokka, n (%) |
| |
0 | 166 (68) | 170 (69) |
1 | 72 (30) | 68 (28) |
2 | 5 (2) | 7 (3) |
Ann Arbor ‑levinneisyysluokitus, n (%) |
| |
II | 37 (15) | 44 (18) |
III | 74 (31) | 68 (28) |
IV | 132 (54) | 133 (54) |
Suuri kasvainmassaa, n (%) | 76 (32) | 84 (35) |
FLIPI-pisteet lähtötilanteessa, n (%) |
| |
0-1 | 63 (26) | 56 (23) |
2 | 79 (33) | 76 (31) |
3-5 | 100 (41) | 113 (46) |
Aikaisempien hoitolinjojen lukumäärä |
| |
Mediaani (min., maks.) | 1 (1, 7) | 1 (1, 6) |
1, n (%) | 145 (60) | 141 (58) |
2, n (%) | 58 (24) | 61 (25) |
≥ 3, n (%) | 40 (17) | 43 (18) |
Aiempi SCT, n (%) | 23 (10) | 18 (7) |
Reagoimaton viimeisimpään hoitolinjaan, n (%) | 84 (35) | 82 (34) |
Reagoimaton sekä CD20-vasta-ainehoitoon että hoitoon alkyloivalla lääkeaineella, n (%) | 91 (37) | 91 (37) |
POD24, n (%) | 106 (44) | 93 (38) |
ECOG = Eastern Cooperative Oncology Group; FLIPI = follikulaarisen lymfooman kansainvälinen ennusteluokitus (IPI-luokitus); SCT = kantasolusiirre; POD24 = taudin eteneminen 24 kuukauden kuluessa. | ||
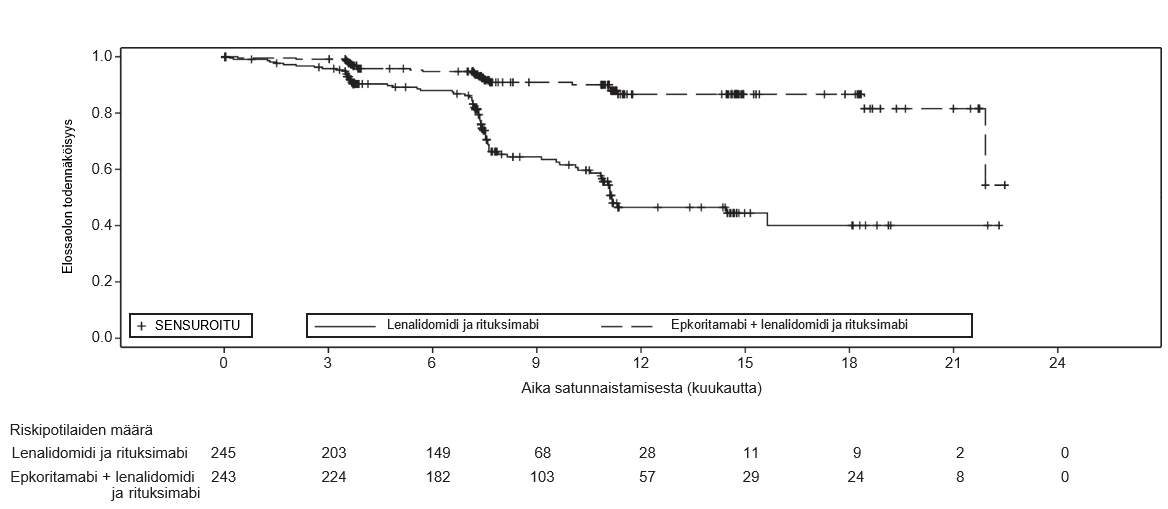
Teho määritettiin kahden ensisijaisen päätetapahtuman perusteella. Nämä olivat elossaoloaika ilman taudin etenemistä (PFS) ja kokonaisvasteprosentti (ORR) Luganon kriteerien (2014) mukaan määritettyinä ja riippumattoman arviointilautakunnan (IRC) arvioimina. Muita tehon mittareita olivat täydellinen vaste (CR) ja kokonaiselossaoloaika (OS). M20‑638-tutkimuksessa IRC:n arvioinnissa havaittiin sekä PFS- että ORR-mittareilla tilastollisesti merkitsevää paranemista epkoritamabilla yhdistelmähoitona lenalidomidin ja rituksimabin kanssa verrattuna pelkkään lenalidomidiin ja rituksimabiin. Lähtöryhmien mukaisessa (ITT) populaatiossa tutkimuksen seurannan mediaanikesto potilailla, jotka satunnaistettiin saamaan epkoritamabia yhdistelmähoitona lenalidomidin ja rituksimabin kanssa, oli 14,8 kuukautta (vaihteluväli: 0–31). Tutkimuksen seurannan mediaanikesto potilailla, jotka satunnaistettiin saamaan pelkästään lenalidomidia ja rituksimabia, oli 14,6 kuukautta (vaihteluväli: 0–30). Yhteenveto tehoa koskevista tuloksista esitetään taulukossa 13.
Taulukko 13: M20‑638-tutkimuksen tehotulokset potilailla, joilla oli uusiutunut tai hoitoon reagoimaton FL
Päätetapahtumaa | Epkoritamabi + lenalidomidi ja rituksimabi | Lenalidomidi ja rituksimabi |
| (N = 243) | (N = 245) |
PFSb |
| |
Tapahtumien lukumäärä, n (%) | 23 (9) | 75 (31) |
Etenevä tauti | 19 (83) | 63 (84) |
Kuolema | 4 (17) | 12 (16) |
Mediaani (95 %:n lv), kuukautta | NR (21.9, NR) | 11.2 (10.5, NR) |
Riskisuhdec (95 %:n lv) | 0.21 (0.13, 0.33) | |
P-arvod | < 0,0001 | |
ORRb,e, n (%) | 111 (96) | 94 (81) |
(95 %:n lv) | (90.2, 98.6) | (72.7, 87.7) |
P-arvof | < 0,0001 | |
CRRb, n (%) | 181 (74) | 106 (43) |
(95 %:n lv) | (68.5, 79.8) | (37.0, 49.7) |
P-arvof | < 0,0001 | |
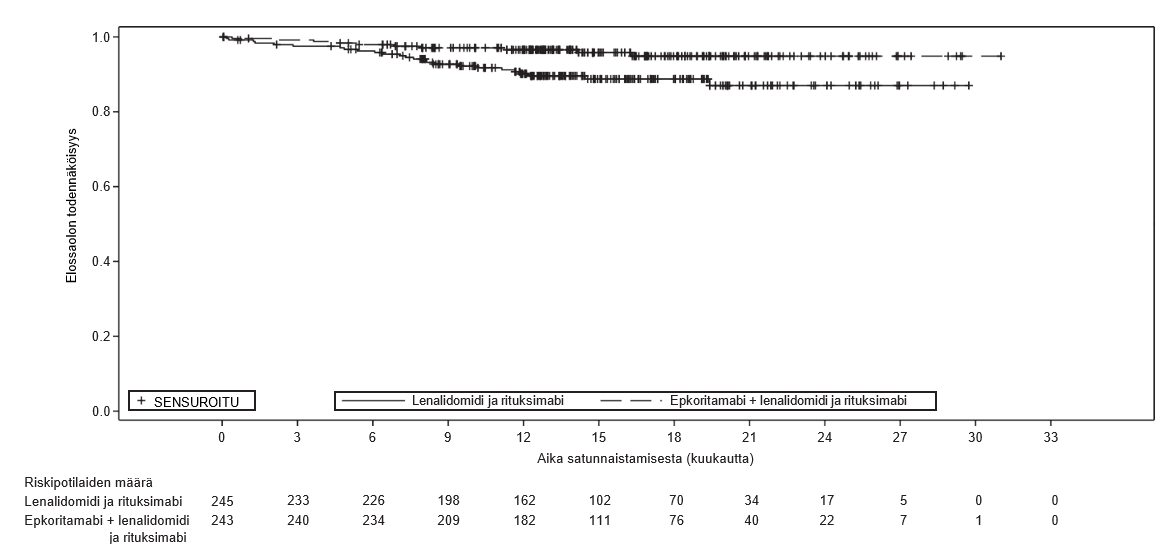
OS |
| |
Tapahtumien lukumäärä, n (%) | 10 (4.1) | 25 (10.2) |
Mediaani (95 %:n lv), kuukautta | NR (NR, NR) | NR (NR, NR) |
HR (95 %:n lv)g | 0.38 (0.18, 0.80) | |
PFS = elossaoloaika ilman taudin etenemistä; lv = luottamusväli; NR = ei saavutettu; CRR = täydellisen vasteen osuus; ORR = kokonaisvasteprosentti; OS = kokonaiselossaoloaika. | ||
Kaplan–Meier-käyrä riippumattoman arviointitoimikunnan arvioimasta Luganon kriteerien (2014) mukaan määritetystä elossaoloajasta ilman taudin etenemistä esitetään kuvassa 1.
Kuva 1: Kaplan–Meier-kaavio elossaoloajasta ilman taudin etenemistä (ITT-populaatio) M20‑638-tutkimuksessa
Kokonaiselossaoloajan Kaplan–Meier-käyrä esitetään kuvassa 2.
Kuva 2: Kaplan–Meier-kaavio kokonaiselossaoloajasta (ITT-populaatio) M20‑638-tutkimuksessa
GCT3013-01
Tutkimus GCT3013‑01 oli avoin, useilla kohorteilla ja yhdellä ryhmällä toteutettu monikeskustutkimus, jossa epkoritamabia arvioitiin monoterapiana annettuna uusiutunutta tai hoitoon reagoimatonta follikulaarista lymfoomaa (FL) sairastavilla potilailla kahden tai useamman systeemisen hoitolinjan jälkeen. Tutkimukseen sisältyi annoksen suurennusvaihe, hoidon laajennusvaihe ja 3‑vaiheinen annoksen optimoimiseksi tehtävä suurennusvaihe. Tutkimuksen laajennusvaiheessa oli mukana aggressiivisen non‑Hodgkin-lymfooman (aNHL) kohortti, indolentin NHL:n (iNHL) kohortti ja manttelisolulymfooman (MCL) kohortti. Keskeiseen iNHL-kohorttiin sisältyi FL-potilaita. Tutkimukseen osallistuvilla potilailla oli oltava dokumentoitu CD20-positiivinen kypsien B‑solujen syöpä WHO:n vuoden 2016 tai 2008 luokituksen mukaan edustavan patologisen lausunnon perusteella sekä alkuperäisen diagnoosin yhteydessä histologisesti vahvistettu FL 1–3A, johon ei liittynyt kliinistä tai patologista näyttöä muuntumisesta. Kaikilla potilailla oli uusiutunut tai viimeisimpään aiempaan hoitolinjaan reagoimaton tauti, johon oli annettu vähintään 2 hoitolinjaa systeemistä antineoplastista hoitoa, mukaan lukien vähintään yhtä monoklonaalista CD20-vasta-ainetta sisältävää hoitoa sekä hoitoa alkyloivalla lääkeaineella tai lenalidomidilla. Tutkimuksesta suljettiin pois potilaat, joiden lymfooma oli levinnyt keskushermostoon, potilaat, joille oli tehty aiemmin allogeeninen HSCT tai kiinteän elimen siirto, potilaat, joilla oli jokin aktiivinen infektiosairaus, ja potilaat, joilla oli tiedossa oleva T‑soluvälitteisen immuniteetin vajaus, kreatiniinipuhdistuma alle 45ml/min, alaniiniaminotransferaasi yli 3 kertaa normaaliarvojen ylärajaa suurempi tai sydämen ejektiofraktio alle 45 %. Tehoa arvioitiin 128 potilaalla, jotka olivat saaneet epkoritamabia ihon alle 4 viikon eli 28 vuorokauden sykleinä. Epkoritamabia annettiin monoterapiana 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti seuraavasti:
-
Sykli 1: epkoritamabia 0,16 mg päivänä 1, 0,8 mg päivänä 8, 48 mg päivänä 15 ja päivänä 22
-
Syklit 2–3: epkoritamabia 48 mg päivinä 1, 8, 15 ja 22
-
Syklit 4–9: epkoritamabia 48 mg päivinä 1 ja 15
-
Syklistä 10 alkaen: epkoritamabia 48 mg päivänä 1.
Epkoritamabin antoa jatkettiin, kunnes tauti eteni tai potilaalla ilmeni toksisuutta, joka ei ollut hyväksyttävissä.
Aloitettujen syklien mediaanilukumäärä oli 8, ja 60 % potilaista sai 6 sykliä hoitoa.
Demografiset tiedot ja lähtötilanteen ominaisuudet on esitetty taulukossa 14.
Taulukko 14 FL-potilaiden demografiset tiedot ja lähtötilanteen ominaisuudet tutkimuksessa GCT3013‑01
Ominaisuudet | (N = 128) |
Ikä |
|
Mediaani, vuotta (min.; maks.) | 65 (39; 84) |
< 65 vuotta, n (%) | 61 (48) |
65 – < 75 vuotta, n (%) | 50 (39) |
≥ 75 vuotta, n (%) | 17 (13) |
Miehiä, (%) | 79 (62) |
Rotu, n (%) |
|
Valkoihoinen | 77 (60) |
Aasialainen | 7 (6) |
Muu | 2 (1,6) |
Ei ilmoitettu | 42 (33) |
ECOG-toimintakykyluokka; n (%) |
|
0 | 70 (55) |
1 | 51 (40) |
2 | 7 (6) |
Aikaisempien hoitolinjojen lukumäärä, n (%) |
|
Mediaani (min.; maks.) | 3 (2; 9) |
2 | 47 (37) |
3 | 41 (32) |
≥ 4 | 40 (31) |
Ann Arbor -levinneisyysluokitus; (%) |
|
Levinneisyysaste III/IV | 109 (85) |
FLIPI-pisteet lähtötilanteessa, n (%) |
|
2 | 31(24) |
3–5 | 78 (61) |
Suuri kasvainmassa, n (%) | 33 (26) |
|
|
Aikaisempi hoito; n (%) |
|
Autologinen kantasolusiirto | 24 (19) |
CAR-T-soluhoito | 6 (5) |
Rituksimabi-lenalidomidihoito | 27 (21) |
PI3K-salpaaja | 29 (23) |
Taudin eteneminen 24 kuukauden kuluessa ensimmäisestä systeemisestä hoidosta | 67 (52) |
Reagoimaton: |
|
≥ 2 aiempaan peräkkäiseen lymfooman hoitolinjaan | 70 (55) |
Viimeiseen systeemiseen antineoplastiseen hoitolinjaan | 88 (69) |
Aiempaan monoklonaaliseen CD20-vasta-ainehoitoon | 101 (79) |
Sekä monoklonaaliseen CD20-vasta-ainehoitoon että hoitoon alkyloivalla lääkeaineella. | 90 (70) |
Tehon arviointiperusteena oli Luganon kriteerien (2014) mukaan määritetty ja riippumattoman arviointilautakunnan (IRC) arvioima kokonaisvasteprosentti (ORR). Vasteen keston (DOR) seuranta-ajan mediaani oli 16,2 kuukautta. Yhteenveto tehoa koskevista tuloksista on esitetty taulukossa 15.
Taulukko 15 Tutkimuksen GCT3013‑01 tehotulokset FL-potilailla
Päätetapahtumaa | Epkoritamabi |
ORRb , n (%) | 106 (83) |
(95 %:n lv) | (75,1; 88,9) |
CRb, n (%) | 81 (63) |
(95 %:n lv) | (54,3; 71,6) |
PRb, n (%) | 25 (20) |
(95 %:n lv) | (13,1; 27,5) |
DORb |
|
Mediaani (95 %:n lv), kuukautta | 21,4 (13,7; NR) |
DOCRb |
|
Mediaani (95 %:n lv), kuukautta | NR (21,4; NR) |
12 kuukauden arvio, % (95 %:n lv) | 78,6 (67,3; 86,4) |
TTR, mediaani (vaihteluväli), kuukautta | 1,4 (1; 3) |
lv = luottamusväli; CR = täydellinen vaste; PR = osittainen vaste; DOR = vasteen kesto; DOCR = täydellisen vasteen kesto; IRC = riippumaton arviointilautakunta; ORR = kokonaisvasteprosentti; PFS = elossaoloaika ilman taudin etenemistä; TTR = vasteen savuttamiseen kulunut aika | |
Täydellisen vasteen saavuttamiseen kuluneen ajan mediaani oli 1,5 kuukautta (vaihteluväli: 1,2–11,1 kuukautta).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset epkoritamabin käytöstä pediatrisen tutkimussuunnitelman päätöksen mukaan myönnetyn käyttöaiheen, kypsien B‑solujen syöpien, hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa. Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Epkoritamabi monoterapiana
Ihon alle annetun epkoritamabin populaatiofarmakokinetiikkaa kuvasi kaksitilamalli, johon sisältyi ensimmäisen vaiheen ihonalainen imeytyminen ja kohdevälitteinen lääkeaineen eliminaatio. Epkoritamabin farmakokineettisen vaihtelun todettiin olevan kohtalaista tai suurta, ja sille oli luonteenomaista yksilöiden välinen vaihtelu (IIV), jossa epkoritamabin farmakokineettisten parametrien variaatiokerroin (CV) oli 25,7–137,5 %.
Tutkimuksen GCT3013‑01 LBCL-potilailla epkoritamabin suositellun 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisen 48 mg:n ihonalaisen annoksen jälkeisten, populaatiofarmakokineettisen, mallinnuksen perusteella arvioitujen yksilöllisten altistusten perusteella epkoritamabin geometrinen Cmax-keskiarvo (% CV) on 10,8 mikrog/ml (41,7 %) ja AUC0‑7d on 68,9 vrk*mikrog/ml (45,1 %) viikoittaisen antoaikataulun lopussa. Ctrough viikolla 12 on 8,4 (53,3 %) mikrog/ml. Epkoritamabin geometrinen Cmax-keskiarvo (% CV) on 7,52 mikrog/ml (41,1 %) ja AUC0‑14d on 82,6 vrk*mikrog/ml (49,3 %) antoaikataulun lopussa, kun lääkettä annetaan kahden viikon välein. Kun lääkettä annetaan kahden viikon välein, Ctrough on 4,1 (73,9 %) mikrog/ml. Epkoritamabin geometrinen Cmax -keskiarvo (% CV) on 4,76 mikrog/ml (51,6 %) ja AUC0‑28d on 74,3 vrk*mikrog/ml (69,5 %) vakaassa tilassa, kun lääkettä annetaan neljän viikon välein. Kun lääkettä annetaan neljän viikon välein, Ctrough on 1,2 (130 %) mikrog/ml.
Epkoritamabialtistuksen parametrit FL-potilailla olivat yhdenmukaisia LBCL-potilailla havaittujen altistusparametrien kanssa. Epkoritamabialtistus oli samankaltainen FL-potilailla, joille käytettiin 3‑vaiheista suurennusvaiheen annosaikataulua, ja FL-potilailla, joille käytettiin 2‑vaiheista suurennusvaiheen annosaikataulua, lukuun ottamatta odotetusti ja ohimenevästi pienempää pienintä epkoritamabipitoisuutta syklin 1 päivänä 15 niillä potilailla, jotka saivat toisen väliannoksen 3‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti, verrattuna potilaisiin, jotka saivat ensimmäisen täyden annoksen (48 mg) 2‑vaiheisen suurennusvaiheen annosaikataulun mukaisesti.
Imeytyminen
Huippupitoisuudet saavutettiin noin 3–4 vuorokaudessa (Tmax) LBCL-potilailla, jotka saivat täyttä 48 mg:n annosta.
Jakautuminen
Populaatiofarmakokineettisen mallinnuksen perusteella sentraalisen jakautumistilavuuden geometrinen keskiarvo (% CV) on 8,27 l (27,5 %) ja näennäinen vakaan tilan jakautumistilavuus on 25,6 l (81,8 %).
Biotransformaatio
Epkoritamabin metaboliareittiä ei ole nimenomaisesti tutkittu. Muiden terapeuttisten proteiinien tavoin epkoritamabin odotetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta.
Eliminaatio
Epkoritamabin puhdistuman odotetaan olevan saturoituvaa ja kohdevälitteistä. Puhdistuman geometrinen keskiarvo (% CV) (l/vrk) on 0,441 (27,8 %). Epkoritamabin puoliintumisaika riippuu pitoisuudesta. Populaatiofarmakokineettisestä mallista saatu täyden epkoritamabiannoksen (48 mg) puoliintumisajan geometrinen keskiarvo oli 22–25 vuorokautta lääkkeen antotiheydestä riippuen.
Epkoritamabi yhdistelmähoitona lenalidomidin ja rituksimabin kanssa
Epkoritamabin farmakokinetiikassa ei havaittu kliinisesti merkitseviä eroja (syklin 1 AUC:n ja syklien 1–3 Cavg geometrisen keskiarvon eron ollessa vastaavasti 2 % ja 23,7 %), kun epkoritamabia annettiin yhdistelmähoitona lenalidomidin ja rituksimabin kanssa verrattuna monoterapiana annettuun epkoritamabiin.
Erityisryhmät
Ikään (20–89 vuotta), sukupuoleen, rotuun / etniseen alkuperään (valkoihoinen, aasialainen ja muu), lievään tai keskivaikeaan munuaisten vajaatoimintaan viittaavaan kreatiniinipuhdistumaan (CLcr ≥ 30 ml/min – CLcr < 90 ml/min) ja lievään maksan vajaatoimintaan (kokonaisbilirubiini ≤ ULN ja ASAT > ULN tai kokonaisbilirubiini 1–1,5 x ULN ja mikä tahansa ASAT-arvo) perustuvia, kliinisesti merkittäviä vaikutuksia epkoritamabin farmakokinetiikkaan ei todettu (syklin 1 AUC noin 36 %), kun potilaiden painoerot otettiin huomioon. Tutkimuksia ei ole tehty vaikeaa tai loppuvaiheen munuaistautia (CLcr < 30 ml/min) tai vaikeaa maksan vajaatoimintaa (kokonaisbilirubiini > 3 x ULN ja mikä tahansa ASAT-arvo) sairastavilla potilailla. Keskivaikeaa maksan vajaatoimintaa sairastavista potilaista (kokonaisbilirubiini > 1,5–3 x ULN ja mikä tahansa ASAT-arvo, N = 1) on vain hyvin vähän tietoa. Siksi epkoritamabin farmakokinetiikkaa näissä ryhmissä ei tunneta.
Muiden terapeuttisten proteiinien tavoin potilaan painolla (39–172 kg) on tilastollisesti merkitsevä vaikutus epkoritamabin farmakokinetiikkaan. Altistus-vasteanalyysin ja kliinisten tietojen perusteella altistuksiin kohdistuva vaikutus ei ole kliinisesti merkittävä, kun otetaan huomioon pienipainoisilla (esim. 46 kg) tai suuripainoisilla (esim. 105 kg) potilailla ja eri painoluokissa (< 65 kg, 65–< 85, ≥ 85) todetut altistukset.
Pediatriset potilaat
Epkoritamabin farmakokinetiikkaa pediatrisilla potilailla ei ole varmistettu.
Prekliiniset tiedot turvallisuudesta
Farmakologia ja/tai toksikologia eläimillä
Epkoritamabia ei ole tutkittu eläimille tehdyissä lisääntymis- tai kehitystoksisuustutkimuksissa.
Jaavanmakakeilla todetut vaikutukset olivat yleisesti ottaen yhdenmukaisia epkoritamabin farmakologisen vaikutusmekanismin kanssa. Näitä löydöksiä olivat esimerkiksi annokseen liittyvät kliiniset haitat (mukaan lukien oksentelu, aktiivisuuden väheneminen ja kuolleisuus suuria annoksia käytettäessä) ja sytokiinien vapautuminen, korjaantuvat hematologiset muutokset, korjaantuva ääreisveren B‑solukato ja korjaantuva imusolujen määrän pieneneminen sekundaarisissa imukudoksissa.
Mutageenisuus
Epkoritamabilla ei ole tehty mutageenisuustutkimuksia.
Karsinogeenisuus
Epkoritamabilla ei ole tehty karsinogeenisuustutkimuksia.
Hedelmällisyyden heikkeneminen
Epkoritamabia ei ole tutkittu eläimille tehdyissä hedelmällisyystutkimuksissa, mutta yleistä toksisuutta selvittäneissä, 5 viikkoa kestäneissä tutkimuksissa epkoritamabi ei aiheuttanut toksikologisia muutoksia jaavanmakakiurosten tai -naaraiden lisääntymiselimissä laskimoon annetuilla annoksilla, jotka olivat enintään 1 mg/kg/viikko. AUC-altistukset (aikakeskiarvo 7 vuorokauden ajalta) jaavanmakakeille annetulla suurella annoksella olivat samaa luokkaa kuin potilailla (AUC0‑7d), jotka saivat suositeltua annosta.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaattitrihydraatti
Etikkahappo
Sorbitoli (E420)
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden ja/tai laimentimien kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
2 vuotta.
Laimennettu tai valmisteltu epkoritamabi
Kemiallinen ja fysikaalinen käytönaikainen säilyvyys on osoitettu 24 tunnin ajalta 2–8 °C:n lämpötilassa, mukaan lukien enintään 12 tunnin ajalta huoneenlämmössä (20–25 °C).
Mikrobiologiselta kannalta valmiste tulee käyttää välittömästi. Jos sitä ei käytetä välittömästi, käytönaikaiset säilytysajat ja -olosuhteet ovat käyttäjän vastuulla, eivätkä ne normaalisti saa olla yli 24 tuntia 2–8 °C:n lämpötilassa, ellei laimennusta ole tehty kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Päivänvalolle altistuminen on minimoitava. Anna epkoritamabiliuoksen lämmetä huoneenlämpöiseksi ennen potilaalle antoa. Hävitä käyttämätön epkoritamabiliuos sallitun säilytysajan päättymisen jälkeen.
Säilytys
Säilytä ja kuljeta kylmässä (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun/avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TEPKINLY injektioneste, liuos
4 mg/0,8 ml (L:ei) 1 kpl (0,8 ml) (734,72 €)
PF-selosteen tieto
Tyypin I lasista valmistettu injektiopullo, jossa on läpäisykohdasta fluoropolymeerillä päällystetty bromobutyylikumitulppa ja alumiinisinetti sekä muovinen vaaleansininen irti napsautettava korkki. Injektiopullo sisältää 4 mg 0,8 ml:ssa injektionestettä, liuosta varten.
Yksi kotelo sisältää yhden injektiopullon.
Valmisteen kuvaus:
Väritön tai kellertävä liuos, jonka pH on 5,5 ja osmolaalisuus noin 211 mOsm/kg.
Käyttö- ja käsittelyohjeet
Epkoritamabin valmistelu
Koko tämä kohta on luettava huolellisesti ennen epkoritamabin valmistelua. Epkoritamabin tietyt annokset (aloitusannos (0,16 mg) ja väliannos (0,8 mg)) on laimennettava ennen antoa. Epkoritamabi voidaan laimentaa kahdella eri menetelmällä: joko injektiopullomenetelmällä tai ruiskumenetelmällä.
Kaikkia alla annettuja ohjeita on noudatettava, sillä epäasianmukainen valmistelu voi johtaa väärään annokseen.
Terveydenhuollon ammattilaisen on valmisteltava epkoritamabi ja annettava se injektiona ihon alle.
Jokainen epkoritamabi-injektiopullo on tarkoitettu vain yhtä käyttökertaa varten.
Jokainen injektiopullo on ylitäytetty, jotta siitä voidaan ottaa riittävä määrä lääkettä.
Epkoritamabia annetaan 28 vuorokauden sykleinä kohdassa Annostus ja antotapa olevan antoaikataulun mukaisesti.
Epkoritamabi on tarkistettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. Injektionesteen tulee olla väritön tai kellertävä liuos. Älä käytä liuosta, jos siinä on värimuutoksia tai hiukkasia tai se on sameaa.
Epkoritamabin valmistelussa on käytettävä aseptista tekniikkaa. Laimennetun liuoksen suodattaminen ei ole tarpeen.
Laimennetun epkoritamabin valmistelu tyhjän ja steriilin injektiopullon menetelmällä
Ohjeet 0,16 mg:n aloitusannoksen valmisteluun – 2 laimennusta tarvitaan – tyhjän ja steriilin injektiopullon menetelmä
Käytä jokaiseen siirtovaiheeseen sopivan kokoista ruiskua, injektiopulloa ja neulaa.
1) Valmistele epkoritamabia sisältävä injektiopullo a) Ota jääkaapista yksi 4 mg/0,8 ml epkoritamabi-injektiopullo, jossa on vaaleansininen korkki. b) Anna injektiopullon lämmetä huoneenlämpöiseksi enintään 1 tunnin ajan. c) Pyörittele epkoritamabia sisältävää injektiopulloa varovasti. Injektiopulloa EI SAA vorteksoida tai ravistaa voimakkaasti. |
2) Suorita ensimmäinen laimennus a) Nimikoi sopivan kokoinen tyhjä injektiopullo ”Laimennus A”. b) Siirrä 0,8 ml epkoritamabia Laimennus A -injektiopulloon. c) Siirrä 4,2 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuosta Laimennus A -injektiopulloon. Kertaalleen laimennettu liuos sisältää 0,8 mg/ml epkoritamabia. d) Pyörittele Laimennus A -injektiopulloa varovasti 30–45 sekunnin ajan. |
3) Suorita toinen laimennus a) Nimikoi sopivan kokoinen tyhjä injektiopullo ”Laimennus B”. b) Siirrä 2 ml liuostaLaimennus A -injektiopullosta Laimennus B -injektiopulloon. Laimennus A -injektiopulloa ei enää tarvita, joten hävitä se. c) Siirrä 8 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuostaLaimennus B -injektiopulloon, jolloin lopulliseksi pitoisuudeksi tulee 0,16 mg/ml. d) Pyörittele Laimennus B -injektiopulloa varovasti 30–45 sekunnin ajan. |
4) Vedä annos ruiskuun |
5) Nimikoi ruisku |
6) Hävitä injektiopullo ja käyttämättä jäänyt epkoritamabi paikallisten vaatimusten mukaisesti. |
Ohjeet 0,8 mg:n väliannoksen valmisteluun – 1 laimennus tarvitaan – tyhjän ja steriilin injektiopullon menetelmä
Käytä jokaiseen siirtovaiheeseen sopivan kokoista ruiskua, injektiopulloa ja neulaa.
1) Valmistele epkoritamabia sisältävä injektiopullo a) Ota jääkaapista yksi 4 mg/0,8 ml epkoritamabi-injektiopullo, jossa on vaaleansininen korkki. b)Anna injektiopullon lämmetä huoneenlämpöiseksi enintään 1 tunnin ajan. c) Pyörittele epkoritamabia sisältävää injektiopulloa varovasti. Injektiopulloa EI SAA vorteksoida tai ravistaa voimakkaasti. |
2) Suorita laimennus a) Nimikoi sopivan kokoinen tyhjä injektiopullo ”Laimennus A”. b) Siirrä 0,8 ml epkoritamabiaLaimennus A -injektiopulloon. c) Siirrä 4,2 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuostaLaimennus A ‑injektiopulloon, jolloin lopulliseksi pitoisuudeksi tulee 0,8 mg/ml. d) Pyörittele Laimennus A -injektiopulloa varovasti 30–45 sekunnin ajan. |
3) Vedä annos ruiskuun |
4) Nimikoi ruisku Merkitse ruiskuun valmisteen nimi, annoksen vahvuus (0,8 mg), päivämäärä ja kellonaika. Laimennetun ja valmistellun epkoritamabin säilytys, ks. kohta Kestoaika. |
5) Hävitä injektiopullo ja käyttämättä jäänyt epkoritamabi paikallisten vaatimusten mukaisesti. |
Laimennetun epkoritamabin valmistelu steriilin ruiskun menetelmällä
Ohjeet 0,16 mg:n aloitusannoksen valmisteluun – 2 laimennusta tarvitaan – steriilin ruiskun menetelmä
Käytä jokaiseen siirtovaiheeseen sopivan kokoista ruiskua ja neulaa.
1) Valmistele epkoritamabia sisältävä injektiopullo a) Ota jääkaapista yksi 4 mg/0,8 ml epkoritamabi-injektiopullo, jossa on vaaleansininen korkki. b) Anna injektiopullon lämmetä huoneenlämpöiseksi enintään 1 tunnin ajan. c) Pyörittele epkoritamabia sisältävää injektiopulloa varovasti. Injektiopulloa EI SAA vorteksoida tai ravistaa voimakkaasti. |
2) Suorita ensimmäinen laimennus a) Nimikoi sopivan kokoinen ruisku ”Laimennus A”. b) Vedä 4,2 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuosta Laimennus A -ruiskuun. Lisää vielä ruiskuun noin 0,2 ml ilmaa. c) Vedä 0,8 ml epkoritamabia uuteen ruiskuun, joka on nimikoitu ”Ruisku 1”. d) Liitä ruiskut toisiinsa, ja ruiskuta 0,8 ml epkoritamabia Laimennus A -ruiskuun. Kertaalleen laimennettu liuos sisältää 0,8 mg/ml epkoritamabia. e) Sekoita varovasti kääntämällä toisiinsa liitetyt ruiskut ylösalaisin 180 astetta 5 kertaa. f) Irrota ruiskut toisistaan, ja hävitä Ruisku 1. |
3) Suorita toinen laimennus a) Nimikoi sopivan kokoinen ruisku ”Laimennus B”. b) Vedä 8 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuostaLaimennus B -ruiskuun. Lisää vielä ruiskuun noin 0,2 ml ilmaa. c) Nimikoi toinen sopivan kokoinen ruisku ”Ruisku 2”. d) Liitä Ruisku 2 Laimennus A -ruiskuun, ja siirrä 2 ml liuosta Ruiskuun 2. Laimennus A -ruiskua ei enää tarvita, joten hävitä se. e) Liitä Ruisku 2 Laimennus B -ruiskuun, ja ruiskuta 2 ml liuostaLaimennus B -ruiskuun, jolloin lopulliseksi pitoisuudeksi tulee 0,16 mg/ml. f) Sekoita varovasti kääntämällä toisiinsa liitetyt ruiskut ylösalaisin 180 astetta 5 kertaa. g) Irrota ruiskut toisistaan, ja hävitä Ruisku 2. |
4) Vedä annos ruiskuun |
5) Nimikoi ruisku |
6) Hävitä injektiopullo ja käyttämättä jäänyt epkoritamabi paikallisten vaatimusten mukaisesti. |
Ohjeet 0,8 mg:n väliannoksen valmisteluun – laimennus tarvitaan – steriilin ruiskun menetelmä
Käytä jokaiseen siirtovaiheeseen sopivan kokoista ruiskua ja neulaa.
1)Valmistele epkoritamabia sisältävä injektiopullo a) Ota jääkaapista yksi 4 mg/0,8 ml epkoritamabi-injektiopullo, jossa on vaaleansininen korkki. b) Anna injektiopullon lämmetä huoneenlämpöiseksi enintään 1 tunnin ajan. c) Pyörittele epkoritamabia sisältävää injektiopulloa varovasti. Injektiopulloa EI SAA vorteksoida tai ravistaa voimakkaasti. |
2) Suorita laimennus a) Nimikoi sopivan kokoinen tyhjä ruisku ”Laimennus A”. b) Vedä 4,2 ml steriiliä 9 mg/ml (0,9 %) natriumkloridiliuostaLaimennus A -ruiskuun. Lisää vielä ruiskuun noin 0,2 ml ilmaa. c) Vedä 0,8 ml epkoritamabia uuteen ruiskuun, joka on nimikoitu ”Ruisku 1”. d) Liitä ruiskut toisiinsa, ja ruiskuta 0,8 ml epkoritamabia Laimennus A -ruiskuun, jolloin lopulliseksi pitoisuudeksi tulee 0,8 mg/ml. e) Sekoita varovasti kääntämällä toisiinsa liitetyt ruiskut ylösalaisin 180 astetta 5 kertaa. f) Irrota ruiskut toisistaan, ja hävitä Ruisku 1. |
3) Vedä annos ruiskuun |
4) Nimikoi ruisku |
5) Hävitä injektiopullo ja käyttämättä jäänyt epkoritamabi paikallisten vaatimusten mukaisesti. |
3 mg:n epkoritamabiannoksen valmistelu
Ohjeet 3 mg:n toisen väliannoksen valmisteluun – laimentaminen ei ole tarpeen
Epkoritamabin 3 mg:n annos on tarpeen ainoastaan FL-potilaille (ks. kohta Annostus ja antotapa).
1) Valmistele epkoritamabia sisältävä injektiopullo a) Ota jääkaapista yksi 4 mg/0,8 ml:n epkoritamabi-injektiopullo, jossa on vaaleansininen korkki. b) Anna injektiopullon lämmetä huoneenlämpöiseksi enintään 1 tunnin ajan. c) Pyörittele epkoritamabia sisältävää injektiopulloa varovasti. Injektiopulloa EI SAA vorteksoida tai ravistaa voimakkaasti. |
2) Vedä annos ruiskuun Vedä 0,6 ml epkoritamabia ruiskuun. |
3) Nimikoi ruisku Merkitse ruiskuun valmisteen nimi, annoksen vahvuus (3 mg), päivämäärä ja kellonaika. Valmistellun epkoritamabin säilytys, ks. kohta Kestoaika. |
4) Hävitä injektiopullo ja käyttämättä jäänyt epkoritamabi paikallisten vaatimusten mukaisesti. |
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TEPKINLY injektioneste, liuos
4 mg/0,8 ml 1 kpl
- Ei korvausta.
ATC-koodi
L01FX27
Valmisteyhteenvedon muuttamispäivämäärä
02.07.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi