
KEVZARA injektioneste, liuos, esitäytetty kynä 150 mg, 200 mg, injektioneste, liuos, esitäytetty ruisku 150 mg, 200 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Kevzara 150 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty kerta-annosruisku sisältää 150 mg sarilumabia 1,14 ml:ssa liuosta (131,6 mg/ml).
Kevzara 150 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kerta-annoskynä sisältää 150 mg sarilumabia 1,14 ml:ssa liuosta (131,6 mg/ml).
Kevzara 200 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty kerta-annosruisku sisältää 200 mg sarilumabia 1,14 ml:ssa liuosta (175 mg/ml).
Kevzara 200 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 200 mg sarilumabia 1,14 ml:ssa liuosta (175 mg/ml).
Sarilumabi on ihmisen monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
Kevzara on tarkoitettu käytettäväksi yhdessä metotreksaatin (MTX) kanssa keskivaikean tai vaikean aktiivisen nivelreuman hoitoon aikuisille, joilla ei ole saatu riittävää vastetta yhdellä tai useammalla taudinkulkua muuttavalla reumalääkkeellä (DMARDs, disease-modifying anti-rheumatic drugs) tai jotka eivät siedä niitä. Kevzara-valmistetta voidaan antaa monoterapiana, jos metotreksaattia ei siedetä tai se ei ole sopiva (ks. kohta Farmakodynamiikka).
Polymyalgia rheumatica
Kevzara on tarkoitettu polymyalgia rheumatican hoitoon aikuisille, joiden vaste kortikosteroidihoitoon on ollut riittämätön tai joilla on ilmennyt relapsi kortikosteroidiannoksen asteittaisen pienentämisen aikana.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Hoidon saa aloittaa vain tämän lääkevalmisteen käyttöaiheena olevan sairauden diagnosointiin ja hoitoon perehtynyt lääkäri, ja hoito on toteutettava tällaisen lääkärin valvonnassa (ks. kohta Käyttöaiheet). Potilaille on annettava potilaskortti.
Annostus
Nivelreuma
Suositeltu sarilumabiannos on 200 mg joka toinen viikko ihonalaisena injektiona.
Polymyalgia rheumatica
Suositeltu sarilumabiannos on 200 mg joka toinen viikko ihonalaisena injektiona yhdistelmänä asteittain vähennettävän systeemisen kortikosteroidihoidon kanssa, minkä jälkeen sarilumabihoitoa voidaan jatkaa monoterapiana.
Tietoja on saatavilla potilaista, joita hoidettiin enintään vuoden ajan. Tämän vuoksi hoidon jatkamisen 52:ta viikkoa pidempään pitää perustua sairauden aktiivisuuteen, lääkärin harkintaan ja potilaan omaan valintaan.
Annosmuutokset
Nivelreuma
Annoksen pienentämistä 200 mg:sta joka toinen viikko 150 mg:aan joka toinen viikko suositellaan, jos on tarpeen hallita neutropeniaa, trombosytopeniaa tai kohonneita maksaentsyymiarvoja.
Jos potilaalle kehittyy vakava infektio, sarilumabihoito on keskeytettävä, kunnes infektio on saatu hallintaan.
Sarilumabihoidon aloittamista ei suositella potilaille, joilla neutrofiilien määrä on vähäinen eli absoluuttinen neutrofiilimäärä (B-Neut) on alle 2 x 109/l.
Sarilumabihoidon aloittamista ei suositella potilaille, joiden verihiutaleiden määrä on alle 150 x 103/µl.
Taulukko 1: Suositellut annosmuutokset nivelreuman hoidossa, jos potilaalla todetaan neutropenia, trombosytopenia tai kohonneita maksaentsyymiarvoja (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset):
| Pieni absoluuttinen neutrofiilimäärä (ks. kohta Farmakodynamiikka) | |
| Laboratorioarvo (soluja x 109/l) | Suositus |
| B-Neut yli 1 | Sarilumabihoitoa jatketaan samalla annoksella. |
| B-Neut 0,5–1 | Sarilumabihoito keskeytetään, kunnes määrä on >1 x 109/l. Sarilumabihoito voidaan aloittaa uudelleen annoksella 150 mg joka toinen viikko ja annos voidaan suurentaa 200 mg:aan joka toinen viikko, jos se on kliinisesti tarkoituksenmukaista. |
| B-Neut alle 0,5 | Sarilumabihoito on lopetettava. |
| Pieni verihiutaleiden määrä | |
| Laboratorioarvo (soluja x 103/µl) | Suositus |
| 50–100 | Sarilumabihoito keskeytetään, kunnes määrä on >100 x 103/µl. Sarilumabihoito voidaan aloittaa uudelleen annoksella 150 mg joka toinen viikko ja annos voidaan suurentaa 200 mg:aan joka toinen viikko, jos se on kliinisesti tarkoituksenmukaista. |
| Alle 50 | Jos tulos on varmistettu toistetulla testillä, sarilumabihoito on lopetettava. |
| Poikkeavat maksaentsyymiarvot | |
| Laboratorioarvo | Suositus |
| ALAT > 1–3 x normaali- arvon yläraja (ULN) | On harkittava samanaikaisesti annetun DMARD-lääkityksen tai immuunivastetta muuntavan hoidon kliinisesti tarkoituksenmukaisia annosmuutoksia. |
| ALAT > 3–5 x ULN | Sarilumabihoito on keskeytettävä, kunnes arvo on < 3 x ULN. Sarilumabihoito voidaan aloittaa uudelleen annoksella 150 mg joka toinen viikko ja annos voidaan suurentaa 200 mg:aan joka toinen viikko, jos se on kliinisesti tarkoituksenmukaista. |
| ALAT > 5 x ULN | Sarilumabihoito on lopetettava. |
Polymyalgia rheumatica
Laboratorioarvojen poikkeavuudet: Sarilumabihoito on lopettava polymyalgia rheumaticaa sairastavilla potilailla, jos heillä ilmenee seuraavia laboratorioarvojen poikkeavuuksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka):
-
neutropenia (B-Neut alle 1 x 109/l annosvälin lopussa)
-
trombosytopenia (verihiutaleiden määrä alle 100 x 103/µl)
-
kohonneet ASAT- tai ALAT-arvot (3‑kertaiset normaaliarvon ylärajaan [ULN] nähden)
Annosmuutoksia ei ole tutkittu polymyalgia rheumaticaa sairastavilla potilailla, joilla ilmenee jokin näistä poikkeavuuksista. Katso hoidon aloittamista koskevat kriteerit kohdasta, jossa on kuvattu valmisteen annostus polymyalgia rheumaticaa sairastavien potilaiden hoidossa.
Unohtunut annos
Jos sarilumabiannos unohtuu ja annoksen unohtamisesta on enintään 3 päivää, seuraava annos otetaan mahdollisimman pian. Sitä seuraava annos pitää ottaa tavanomaisen aikataulun mukaan. Jos annoksen unohtamisesta on kulunut 4 päivää tai yli, seuraava annos otetaan tavanomaisen aikataulun mukaan. Kaksinkertainen annos ei ole sallittu.
Erityisryhmät
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta. Sarilumabia ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Sarilumabin turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla, mukaan lukien potilaat, joilla on saatu serologisissa kokeissa positiivinen tulos hepatiitti B ‑viruksen (HBV) tai hepatiitti C ‑viruksen (HCV) suhteen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät
Yli 65-vuotiaille potilaille ei tarvita annosmuutoksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Esitäytetyssä ruiskussa tai esitäytetyssä kynässä olevan sarilumabin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Ihon alle.
Pistoskohtia (vatsa, reisi, käsivarren yläosa) pitää vaihtaa jokaisella injektiokerralla. Älä pistä sarilumabia aristavaan tai vaurioituneeseen ihoon tai ihoon, jossa on mustelmia tai arpia.
Esitäytetty ruisku ja esitäytetty kynä
Koko esitäytetyn ruiskun / esitäytetyn kynän sisältö (1,14 ml) annetaan ihonalaisena injektiona.
Esitäytettyä ruiskua / esitäytettyä kynää käytettäessä potilas voi pistää sarilumabin itse tai potilaan huoltaja voi antaa sarilumabin, jos hoidosta vastaava terveydenhuollon ammattilainen on katsonut sen tarkoituksenmukaiseksi. Tätä ennen potilaalle ja/tai hänen huoltajalleen on annettava perusteellinen opastus sarilumabin käyttökuntoon saattamisesta ja antamisesta.
Esitäytettyä ruiskua tai kynää ei ole tutkittu pediatrisilla potilailla.
Kattavat ohjeet tämän lääkevalmisteen antoon on esitetty pakkausselosteessa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiiviset, vaikeat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Vakavat infektiot
Potilaita on tarkkailtava huolellisesti infektion merkkien ja oireiden varalta sarilumabihoidon aikana (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Koska iäkkäillä esiintyy yleensä infektioita enemmän kuin nuoremmilla, iäkkäiden potilaiden hoidossa on noudatettava varovaisuutta.
Sarilumabia ei saa antaa potilaille, joilla on aktiivinen infektio. Tämä koskee myös paikallisia infektioita. Hoidon riskejä on harkittava suhteessa hoidon hyötyihin ennen hoidon aloittamista potilaille:
- joilla on krooninen tai toistuva infektio
- joilla on aiemmin ollut vakava tai opportunistinen infektio
- joilla on HIV-infektio
- joilla on jokin muu sairaus, joka saattaa altistaa infektioille
- jotka ovat altistuneet tuberkuloosille tai
- jotka ovat asuneet tai matkustelleet alueilla, joilla esiintyy endeemisesti tuberkuloosia tai mykooseja.
Sarilumabihoito on keskeytettävä, jos potilaalle kehittyy vakava tai opportunistinen infektio. Kun infektio on saatu hallintaan, sarilumabihoito voidaan terveydenhuollon ammattilaisen harkinnan mukaan aloittaa uudelleen.
Potilaalle, jolle kehittyy uusi infektio hoidon aikana, on viipymättä tehtävä immuunipuutteisille potilaille tarkoitetut täydelliset diagnostiset tutkimukset ja aloitettava tarkoituksenmukainen mikrobilääkehoito, ja potilasta on tarkkailtava huolellisesti.
Vakavia ja joskus kuolemaan johtaneita bakteerien, mykobakteerien, invasiivisten sienten, virusten tai muiden opportunististen patogeenien aiheuttamia infektioita on raportoitu immunosuppressiivista lääkehoitoa saaneilla potilailla. Sarilumabihoidon aikana useimmin havaittuja vakavia infektioita nivelreumapotilailla olivat keuhkokuume ja selluliitti (ks. kohta Haittavaikutukset). Kun sarilumabia on käytetty nivelreuman hoitoon, opportunistisina infektioina on raportoitu tuberkuloosia, kandidiaasia ja Pneumocystis-infektioita. Joillakin nivelreumapotilailla, joilla oli samanaikaisesti tuberkuloosi, havaittiin ennemminkin levinneitä kuin paikallisia infektioita. Useimmat näistä potilaista käyttivät samanaikaisesti muita immuunisalpaajia, kuten metotreksaattia tai kortikosteroideja, jotka saattavat suurentaa infektioiden riskiä.
Tuberkuloosi
Potilaiden tuberkuloosin riskitekijät on arvioitava ja heidät on testattava piilevän infektion varalta ennen sarilumabihoidon aloittamista. Potilaat, joilla on piilevä tai aktiivinen tuberkuloosi, on hoidettava tavanomaisilla mykobakteerilääkkeillä ennen hoidon aloittamista. Tuberkuloosilääkitystä on harkittava ennen hoidon aloittamista potilailla, joilla on ollut aiemmin piilevä tai aktiivinen tuberkuloosi ja joiden kohdalla riittävän hoitokuurin toteutumista ei voida varmistaa, sekä potilailla, joiden piilevän tuberkuloosin testi on negatiivinen mutta joilla on tuberkuloosin riskitekijöitä. Terveydenhuollon ammattilaisia muistutetaan tuberkuliini-ihokokeiden ja tuberkuloosin gammainterferoniverikokeiden väärien negatiivisten tulosten riskistä, erityisesti vaikeasti sairaiden tai immuunipuutteisten potilaiden kohdalla. Tuberkuloosilääkitystä harkittaessa saattaa olla tarpeen konsultoida tuberkuloosiin erikoistunutta lääkäriä.
Potilaita on seurattava tarkkaan tuberkuloosin merkkien ja oireiden varalta, myös potilaita, jotka saivat ennen hoidon aloitusta negatiivisen tuloksen piilevän tuberkuloosi-infektion testissä.
Virusten uudelleenaktivoituminen
Virusten uudelleenaktivoitumista on raportoitu immunosuppressiivisten biologisten lääkehoitojen yhteydessä. Sarilumabilla tehdyissä kliinisissä tutkimuksissa on todettu vyöruusutapauksia (ks. kohta Haittavaikutukset). Hepatiitti B ‑viruksen uudelleenaktivoitumista ei ole raportoitu kliinisissä tutkimuksissa, tosin potilaat, joilla oli uudelleenaktivoitumisen riski, suljettiin pois näistä tutkimuksista.
Laboratorioarvot
Neutrofiilien määrä
Sarilumabihoidon yhteydessä havaittiin tavanomaista enemmän absoluuttisen neutrofiilimäärän (B-Neut) pienenemistä (ks. kohta Haittavaikutukset). Absoluuttisen neutrofiilimäärän pienenemiseen ei liittynyt infektioiden lisääntymistä. Tämä koski myös vakavia infektioita.
- Sarilumabihoidon aloittamista ei suositella potilaille, joilla neutrofiilien määrä on vähäinen eli absoluuttinen neutrofiilimäärä (B-Neut) on alle 2 x 109/l. Jos potilaan absoluuttinen neutrofiilimäärä on alle 0,5 x 109/l, sarilumabihoito on suositeltavaa lopettaa (ks. kohta Annostus ja antotapa).
- Neutrofiilien määrä on tarkistettava 4–8 viikon kuluessa hoidon aloittamisen jälkeen ja sen jälkeen kliinisen arvioinnin mukaisesti. Absoluuttisen neutrofiilimäärän tuloksiin perustuvat suositellut annosmuutokset, ks. kohta Annostus ja antotapa.
- Absoluuttisen neutrofiilimäärän muutosten farmakodynamiikan vuoksi annosmuutoksia harkittaessa on käytettävä annosvälin loppuvaiheen tuloksia (ks. kohta Farmakodynamiikka).
Verihiutaleiden määrä
Kliinisissä tutkimuksissa sarilumabihoitoon on liittynyt verihiutaleiden määrän pienenemistä. Verihiutaleiden vähenemiseen ei liittynyt verenvuototapahtumia (ks. kohta Haittavaikutukset).
- Sarilumabihoidon aloittamista ei suositella potilaille, joiden verihiutaleiden määrä on alle 150 x 103/µl. Jos potilaan verihiutaleiden määrä on alle 50 x 103/µl, sarilumabihoito on lopetettava.
- Verihiutaleiden määrää on tarkkailtava 4–8 viikon ajan hoidon aloittamisesta ja sen jälkeen kliinisen arvioinnin mukaisesti. Verihiutaleiden määrään perustuvat suositellut annosmuutokset, ks. kohta Annostus ja antotapa.
Maksaentsyymit
Sarilumabihoidon yhteydessä kohonneiden transaminaasiarvojen ilmaantuvuus suureni. Näiden arvojen kohoaminen oli ohimenevää eikä aiheuttanut kliinisesti ilmeisiä maksavaurioita kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Näiden kohonneiden transaminaasiarvojen esiintyvyyden ja kohoamisen voimakkuuden havaittiin suurentuneen silloin, kun yhdessä sarilumabin kanssa käytettiin mahdollisesti maksatoksisia lääkevalmisteita (kuten metotreksaattia).
Sarilumabihoidon aloittamista ei suositella potilaille, joiden transaminaasi‑, ALAT- tai ASAT-arvot ovat kohonneet yli 1,5-kertaisiksi normaaliarvon ylärajaan (ULN) nähden. Sarilumabihoito on lopetettava potilailla, joiden ALAT on yli 5 x ULN (ks. kohta Annostus ja antotapa).
ALAT- ja ASAT-arvoja on seurattava 4–8 viikon ajan hoidon aloittamisesta ja sen jälkeen 3 kuukauden välein. Kliinisen arvion perusteella voidaan harkita myös muita maksan toimintakokeita, kuten bilirubiinimääritystä. Kohonneisiin transaminaasiarvoihin perustuvat suositellut annosmuutokset, ks. kohta Annostus ja antotapa.
Poikkeavuudet lipidiarvoissa
Kroonista tulehdusta sairastavien potilaiden lipidipitoisuudet saattavat pienentyä. Sarilumabihoitoon liittyi lipidiparametrien, kuten LDL-kolesteroli‑, HDL-kolesteroli- ja/tai triglyseridiarvojen, suurenemista (ks. kohta Haittavaikutukset). Lipidiparametrit on mitattava noin 4–8 viikon kuluttua sarilumabihoidon aloittamisesta ja sen jälkeen noin 6 kuukauden välein.
Potilaita on hoidettava hyperlipidemian hoitosuositusten mukaisesti.
Maha-suolikanavan perforaatio ja divertikuliitti
Sarilumabin käytön yhteydessä on ilmoitettu maha-suolikanavan perforaatioita ja divertikuliittia. Maha-suolikanavan perforaatioita on ilmoitettu potilailla, joilla oli divertikuliitti, ja potilailla, joilla ei ollut divertikuliittia. Jos potilaalle ilmaantuu mahdollisesti divertikuliittiin viittaavia oireita, kuten vatsakipua, maha-suolikanavan verenvuotoa ja/tai selittämättömiä muutoksia suolen toiminnassa ja kuumetta, potilas on tutkittava viipymättä, jotta divertikuliitti ja siihen mahdollisesti liittyvä maha-suolikanavan perforaatio havaitaan mahdollisimman varhain. Sarilumabia on käytettävä varoen potilaille, joilla on aiemmin ollut suolihaavauma tai divertikuliitti (ks. kohta Haittavaikutukset).
Maligniteetit
Hoito immunosuppressiivisilla lääkeaineilla saattaa suurentaa maligniteettien riskiä. Sarilumabihoidon vaikutusta maligniteettien kehittymiseen ei tunneta, mutta maligniteetteja on raportoitu kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset).
Yliherkkyysreaktiot
Sarilumabiin liittyviä yliherkkyysreaktioita on raportoitu (ks. kohta Haittavaikutukset). Pistoskohdan ihottuma, ihottuma ja nokkosrokko olivat kaikkein yleisimmin ilmoitettuja yliherkkyysreaktioita. Potilaita on neuvottava hakeutumaan välittömästi hoitoon, jos heille tulee mitä tahansa yliherkkyysreaktioiden oireita. Jos esiintyy anafylaksiaa tai muita yliherkkyysreaktioita, sarilumabi on lopetettava välittömästi (ks. kohta Vasta-aiheet).
Maksan vajaatoiminta
Sarilumabihoitoa ei suositella potilaille, joilla on aktiivinen maksasairaus tai maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Rokotukset
Eläviä taudinaiheuttajia tai eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden kliinistä turvallisuutta ei ole varmistettu sarilumabihoidon yhteydessä, joten niiden samanaikaista käyttöä on vältettävä. Tietoa ei ole saatavilla infektion sekundaaritartunnasta sarilumabia saaneisiin potilaisiin henkilöistä, jotka ovat saaneet eläviä taudinaiheuttajia sisältäviä rokotteita. Ennen hoidon aloittamista suositellaan, että kaikkien potilaiden rokotussuojat tarkistetaan ajan tasalle voimassa olevien rokotusohjelmien mukaisesti. Eläviä taudinaiheuttajia sisältävien rokotteiden antamisen ja hoidon aloittamisen väliin jäävän ajan on oltava ajantasaisten, immunosuppressiivisia valmisteita koskevien rokotusohjeiden mukainen.
Kardiovaskulaarinen riski
Nivelreumapotilailla on suurentunut sydän- ja verisuonitautiriski, ja riskitekijät (kuten kohonnut verenpaine ja hyperlipidemia) on hoidettava tavanomaisen hoitokäytännön mukaisesti.
Polysorbaatti 20 (E432)
Tämä lääkevalmiste sisältää 2,28 mg polysorbaatti 20:tä per 1,14 ml injektionestettä, liuosta, mikä vastaa pitoisuutta 2 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Metotreksaatin samanaikainen anto ei vaikuttanut sarilumabialtistukseen populaatiofarmakokineettisten analyysien ja tutkimusten ristiinvertailun perusteella. Metotreksaattialtistuksen ei odoteta muuttuvan samanaikaisen sarilumabin annon takia; kerättyä kliinistä tietoa ei kuitenkaan ole. Sarilumabin käyttöä yhdessä Janus-kinaasin (JAK) estäjien tai biologisten DMARD-lääkkeiden, kuten TNF (tumour necrosis factor) -salpaajien, kanssa ei ole tutkittu.
Erilaiset in vitro- ja ihmisillä tehdyt rajalliset in vivo ‑tutkimukset ovat osoittaneet, että sytokiinit ja sytokiinimodulaattorit voivat vaikuttaa spesifisten sytokromi P450 (CYP) ‑entsyymien (CYP1A2, CYP2C9, CYP2C19 ja CYP3A4) toimintaan ja siten mahdollisesti muuttaa sellaisten samanaikaisesti annettujen lääkevalmisteiden farmakokinetiikkaa, jotka ovat näiden entsyymien substraatteja. Suurentuneet interleukiini 6 (IL‑6) ‑pitoisuudet saattavat vaimennussäädellä CYP-aktiivisuutta esimerkiksi nivelreumaa tai polymyalgia rheumaticaa sairastavilla potilailla ja siten suurentaa lääkkeen pitoisuuksia verrattuna tutkittaviin, joilla ei ole nivelreumaa tai polymyalgia rheumaticaa. Kun IL-6Rα-antagonistit, kuten sarilumabi, salpaavat IL-6-signaalivälityksen, IL-6:n estovaikutus saattaa kumoutua ja CYP-aktiivisuus palautua, mikä johtaa muuttuneisiin lääkevalmisteiden pitoisuuksiin.
Sarilumabin kyvyllä muuntaa IL-6:n vaikutusta CYP-entsyymeihin saattaa olla kliinistä merkitystä käytettäessä CYP-substraatteja, joiden terapeuttinen indeksi on kapea ja annos säädetään potilaskohtaisesti. Jos potilas saa lääkevalmistetta, joka on CYP-substraatti, sarilumabia aloitettaessa tai lopetettaessa on tarpeen seurata hoidon vaikutusta (esim. varfariinin kohdalla) tai lääkepitoisuutta (esim. teofylliinin kohdalla) ja tarvittaessa muuttaa lääkevalmisteen annosta potilaskohtaisesti.
Varovaisuutta on noudatettava, jos potilas aloittaa sarilumabihoidon käyttäessään CYP3A4‑substraatteja (kuten ehkäisytabletteja tai statiineja), sillä sarilumabi voi kumota IL-6:n estovaikutuksen ja palauttaa CYP3A4-aktiivisuuden, joka pienentää altistusta CYP3A4-substraatille ja CYP3A4‑substraatin aktiivisuutta (ks. kohta Farmakokinetiikka). Sarilumabin yhteisvaikutuksia muiden CYP-entsyymien (CYP2C9, CYP2C19, CYP2D6) substraattien kanssa ei ole tutkittu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukauden ajan hoidon päättymisen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaus
Sarilumabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja.
Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria eikä epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Sarilumabia ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa sarilumabilla.
Imetys
Ei tiedetä, erittyykö sarilumabi ihmisillä äidinmaitoon tai imeytyykö se systeemisesti maha-suolikanavasta. Sarilumabin erittymistä maitoon ei ole tutkittu koe-eläimillä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Koska IgG1 erittyy ihmisillä äidinmaitoon, on päätettävä, lopetetaanko imetys vai lopetetaanko sarilumabihoito, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ei ole olemassa tietoja sarilumabin vaikutuksesta ihmisen hedelmällisyyteen. Eläintutkimuksissa ei ole osoitettu urosten tai naaraiden hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kevzara-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimpiä haittavaikutuksia potilailla, joilla on nivelreuma (n = 661) tai polymyalgia rheumatica (n = 59), ovat neutropenia (14,3 %), ylähengitystieinfektiot (6,8 %), ALAT-arvon nousu (6,3 %), virtsatieinfektiot (5,3 %) ja pistoskohdan punoitus (5,0 %). Yleisimpiä vakavia haittavaikutuksia ovat infektiot (3,1 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Taulukossa luetellut haittavaikutukset on raportoitu kontrolloiduissa kliinisissä tutkimuksissa. Tässä lueteltujen haittavaikutusten esiintymistiheydet on määritelty seuraavasti:
hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: Haittavaikutukset nivelreumaa tai polymyalgia rheumaticaa sairastavilla potilailla
| MedDRA-elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | Yleinen | Ylähengitystieinfektio |
| Virtsatieinfektio | ||
| Huuliherpes | ||
| Selluliitti | ||
| Keuhkokuume | ||
| Melko harvinainen | Nasofaryngiitti | |
| Divertikuliitti | ||
| Veri ja imukudos | Hyvin yleinen | Neutropenia* |
| Yleinen | Leukopenia* | |
| Trombosytopenia | ||
| Aineenvaihdunta ja ravitsemus | Yleinen | Hypertriglyseridemia |
| Hyperkolesterolemia | ||
| Ruoansulatuselimistö | Harvinainen | Maha-suolikanavan perforaatio |
| Maksa ja sappi | Yleinen | Suurentuneet transaminaasiarvot |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Pistoskohdan punoitus |
| Pistoskohdan kutina* |
* SAPHYR-tutkimuksessa polymyalgia rheumaticaa sairastavilla potilailla ilmoitettuja haittavaikutuksia olivat neutropenia, leukopenia ja pistoskohdan kutina.
Valikoitujen haittavaikutusten kuvaus
Nivelreuma
Infektiot
Lumekontrolloidussa tutkimuspopulaatiossa todettiin 100:aa potilasvuotta kohti 84,5 infektiota sarilumabia 200 mg:n annoksella + DMARD-lääkkeitä saaneiden ryhmässä, 81,0 infektiota sarilumabia 150 mg:n annoksella + DMARD-lääkkeitä saaneiden ryhmässä ja 75,1 infektiota lumelääkettä + DMARD-lääkkeitä saaneiden ryhmässä. Yleisimmin raportoituja infektioita (5–7 %:lla potilaista) olivat ylähengitystieinfektiot, virtsatieinfektiot ja nasofaryngiitti. Vakavia infektioita todettiin 100:aa potilasvuotta kohti 4,3 tapausta sarilumabia 200 mg:n annoksella + DMARD-lääkkeitä saaneiden ryhmässä, 3,0 tapausta sarilumabia 150 mg:n annoksella + DMARD-lääkkeitä saaneiden ryhmässä ja 3,1 tapausta lumelääkettä + DMARD-lääkkeitä saaneiden ryhmässä.
Tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, infektioiden määrä oli 57,3 tapahtumaa 100:aa potilasvuotta kohti ja vakavien infektioiden määrä 3,4 tapahtumaa 100:aa potilasvuotta kohti.
Useimmin havaittuja vakavia infektioita olivat keuhkokuume ja selluliitti. Opportunistisia infektioita on raportoitu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektioiden ja vakavien infektioiden kokonaismäärät olivat sarilumabimonoterapiaa saaneessa populaatiossa yhdenmukaiset sarilumabin + DMARD-lääkkeiden yhdistelmää saaneessa populaatiossa todettujen määrien kanssa.
Maha-suolikanavan perforaatio
Maha-suolikanavan perforaatiota raportoitiin potilailla, joilla oli divertikuliitti, ja potilailla, joilla ei ollut divertikuliittia. Useimmat potilaat, joille kehittyi maha-suolikanavan perforaatio, käyttivät samanaikaisesti tulehduskipulääkkeitä (NSAIDs, nonsteroidal anti-inflammatory medications), kortikosteroideja tai metotreksaattia. Näiden samanaikaisesti annettujen lääkkeiden osuutta maha-suolikanavan perforaatioiden kehittymisessä sarilumabihoidon aikana ei tunneta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yliherkkyysreaktiot
Lumekontrolloidussa tutkimuspopulaatiossa yliherkkyysreaktioiden vuoksi hoidon lopettaneiden potilaiden osuus oli suurempi sarilumabia saaneiden ryhmässä (0,9 % 200 mg:n annosta saaneilla ja 0,5 % 150 mg:n annosta saaneilla) lumeryhmään verrattuna (0,2 %). Yliherkkyyden vuoksi keskeyttäneiden osuudet tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, ja tutkimuspopulaatiossa, jossa annettiin sarilumabia monoterapiana, olivat yhdenmukaiset lumekontrolloidussa populaatiossa todetun osuuden kanssa. Lumekontrolloidussa tutkimuspopulaatiossa 0,2 %:lla potilaista, jotka saivat sarilumabia 200 mg joka toinen viikko + DMARD-lääkettä, ilmoitettiin vakava yliherkkyysreaktioon liittyvä haittatapahtuma, kun taas ryhmässä, jossa potilaat saivat sarilumabia 150 mg joka toinen viikko + DMARD-lääkettä, niitä ei ilmoitettu yhtään.
Pistoskohdan reaktiot
Pistoskohdan reaktioita ilmoitettiin lumekontrolloidussa tutkimuspopulaatiossa 9,5 %:lla 200 mg:n sarilumabiannoksia saaneista, 8 %:lla 150 mg:n sarilumabiannoksia saaneista ja 1,4 %:lla lumelääkettä saaneista potilaista. Nämä pistoskohdan reaktiot (kuten punoitus ja kutina) olivat suurimmalla osalla potilaista vaikeusasteeltaan lieviä tai keskivaikeita (99,5 %:lla 200 mg:n sarilumabiannoksia saaneista, 100 %:lla 150 mg:n sarilumabiannoksia saaneista ja 100 %:lla lumelääkettä saaneista). Kaksi sarilumabia saanutta potilasta (0,2 %) lopetti hoidon pistoskohdan reaktioiden vuoksi.
Laboratorioarvojen poikkeavuudet
Laboratorioarvojen poikkeavuuksien esiintyvyyden suorassa vertailussa lumehoidon ja aktiivisen hoidon välillä käytettiin viikoilla 0–12 saatuja tietoja, jolloin lumelääkkeen käytöstä sarilumabihoitoon siirtyminen ei vielä ollut sallittua.
Neutrofiilien määrä
Neutrofiilien määrän pienenemistä alle arvoon 1 x 109/l ilmeni 6,4 %:lla 200 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista potilaista ja 3,6 %:lla 150 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista, mutta ei yhdelläkään potilaalla lumelääkettä + DMARD-lääkkeitä saaneiden ryhmässä. Neutrofiilien määrän pienenemistä alle arvoon 0,5 x 109/l ilmeni 0,8 %:lla 200 mg:n sarilumabiannoksia + DMARD-lääkkeitä ja 0,6 %:lla 150 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneiden potilaiden ryhmässä. Potilailla, joilla absoluuttinen neutrofiilimäärä (B-Neut) pieneni, hoito-ohjelman muuttaminen, kuten sarilumabin tilapäinen keskeyttäminen tai annoksen pienentäminen, suurensi absoluuttista neutrofiilimäärää tai palautti sen normaaliksi (ks. kohta Annostus ja antotapa). Absoluuttisen neutrofiilimäärän pienenemiseen ei liittynyt infektioiden lisääntymistä. Tämä koski myös vakavia infektioita.
Tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, ja tutkimuspopulaatiossa, jossa sarilumabia annettiin monoterapiana, havaitut neutrofiilimäärät olivat yhdenmukaiset lumekontrolloidussa populaatiossa havaittujen neutrofiilimäärien kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verihiutaleiden määrä
Verihiutaleiden määrän pienenemistä alle arvoon 100 x 103/µl ilmeni 1,2 %:lla 200 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista ja 0,6 %:lla 150 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista potilasta, mutta ei yhdelläkään lumelääkettä + DMARD lääkkeitä saaneella potilaalla.
Tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, ja tutkimuspopulaatiossa, jossa sarilumabia annettiin monoterapiana, havaitut verihiutaleiden määrät olivat yhdenmukaiset lumekontrolloidussa populaatiossa havaittujen määrien kanssa.
Verihiutaleiden määrän pienenemiseen ei liittynyt verenvuototapahtumia.
Maksaentsyymit
Maksaentsyymien poikkeavuudet on lueteltu taulukossa 3. Potilailla, joilla todettiin kohonneita maksaentsyymiarvoja, hoito-ohjelman muuttaminen, kuten hoidon tilapäinen keskeyttäminen tai annoksen pienentäminen, laski maksaentsyymiarvoja tai palautti ne normaaleiksi (ks. kohta Annostus ja antotapa). Tähän maksaentsyymiarvojen kohoamiseen ei liittynyt kliinisesti merkityksellistä konjugoituneen bilirubiinin lisääntymistä eikä kliinistä näyttöä hepatiitista tai maksan vajaatoiminnasta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 3: Poikkeavien maksaentsyymiarvojen ilmaantuvuus kontrolloiduissa kliinisissä tutkimuksissa
| Lume + DMARD N = 661 | Sarilumabi 150 mg + DMARD N = 660 | Sarilumabi 200 mg + DMARD N = 661 | Sarilumabi-monoterapia millä tahansa annoksella N = 467 | |
| ASAT | ||||
>3 x ULN – 5 x ULN | 0 % | 1,2 % | 1,1 % | 1,1 % |
| > 5 x ULN | 0 % | 0,6 % | 0,2 % | 0 % |
| ALAT | ||||
>3 x ULN – 5 x ULN | 0,6 % | 3,2 % | 2,4 % | 1,9 % |
| > 5 x ULN | 0 % | 1,1 % | 0,8 % | 0,2 % |
Lipidit
Lipidiparametrit (LDL, HDL ja triglyseridit) tutkittiin ensimmäisen kerran 4 viikon kuluttua sarilumabin ja DMARD-lääkkeiden yhdistelmähoidon aloittamisesta lumekontrolloidussa tutkimuspopulaatiossa. Viikolla 4 keskimääräinen LDL-pitoisuus oli suurentunut 14 mg/dl, keskimääräinen triglyseridipitoisuus 23 mg/dl ja keskimääräinen HDL-pitoisuus 3 mg/dl. Viikon 4 jälkeen ei enää havaittu pitoisuuksien suurenemista. Annosten välillä ei ollut merkityksellisiä eroja.
Tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, ja tutkimuspopulaatiossa, jossa sarilumabia annettiin monoterapiana, havaitut lipidiparametrien arvot olivat yhdenmukaiset lumekontrolloidussa populaatiossa havaittujen arvojen kanssa.
Maligniteetit
Lumekontrolloidussa tutkimuspopulaatiossa maligniteetteja ilmeni sarilumabia yhdistelmänä DMARD-lääkkeiden kanssa saaneilla potilailla yhtä paljon kuin lumelääkettä yhdistelmänä DMARD-lääkkeiden kanssa saaneilla potilailla (1,0 tapahtumaa 100:aa potilasvuotta kohti).
Tutkimuspopulaatiossa, jossa arvioitiin sarilumabin ja DMARD-lääkkeiden yhdistelmän pitkän aikavälin turvallisuutta, ja tutkimuspopulaatiossa, jossa sarilumabia annettiin monoterapiana, havaitut maligniteettien määrät olivat yhdenmukaiset lumekontrolloidussa populaatiossa havaittujen määrien kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Kuten kaikkiin terapeuttisiin proteiineihin, myös sarilumabiin liittyy immunogeenisuuden mahdollisuus.
Lumekontrolloidussa tutkimuspopulaatiossa 4,0 %:lla 200 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista, 5,6 %:lla 150 mg:n sarilumabiannoksia + DMARD-lääkkeitä saaneista ja 2,0 %:lla lumelääkettä + DMARD-lääkkeitä saaneista potilaista todettiin positiivinen vaste lääkevasta-aine- (ADA‑) määrityksessä. Neutraloivan vasta-aineen (NAb) analyysissä todettiin positiiviset vasteet 1,0 %:lla 200 mg:n sarilumabiannoksia saaneista, 1,6 %:lla 150 mg:n sarilumabiannoksia saaneista ja 0,2 %:lla lumelääkettä saaneista potilaista.
Sarilumabi monoterapiana saaneiden tutkimuspopulaatiossa tehdyt havainnot olivat yhdenmukaisia sarilumabia yhdessä DMARD-lääkkeiden kanssa saaneiden potilaiden tutkimuspopulaatiossa tehtyjen havaintojen kanssa.
Lääkevasta-aineiden (ADA, anti drug antibody) muodostuminen saattaa vaikuttaa sarilumabin farmakokinetiikkaan. Lääkevasta-aineiden kehittymisen ja lääkkeen tehon heikkenemisen tai haittatapahtumien välillä ei havaittu korrelaatiota.
Polymyalgia Rheumatica
Sarilumabin turvallisuutta tutkittiin vaiheen 3 tutkimuksessa (SAPHYR) 117:llä polymyalgia rheumaticaa sairastavalla potilaalla, joista 59 sai sarilumabia 200 mg:n annoksella ihon alle (ks. kohta Farmakodynamiikka). Kyseessä oli 12 kuukauden pituinen kaksoissokkoutettu lumekontrolloitu tutkimus. Tutkimuspopulaatiossa, jossa polymyalgia rheumaticaa sairastavat potilaat saivat sarilumabia, potilasvuosia oli yhteensä 47,37. Turvallisuustietoja on saatavilla enintään 1 vuoden ajalta.
Infektiot
SAPHYR-tutkimuksessa niiden potilaiden osuus, joilla ilmeni infektioita, oli pienempi ryhmässä, jossa potilaat saivat sarilumabia 200 mg:n annoksella sekä prednisonihoitoa asteittain pienenevällä annoksella 14 viikon ajan (37,3 %), kuin ryhmässä, jossa potilaat saivat lumelääkettä sekä prednisonihoitoa asteittain pienenevällä annoksella 52 viikon ajan (50,0 %). Vakavia infektioita ilmoitettiin 3 potilaalla (5,1 %:lla), jotka olivat saaneet sarilumabia 200 mg:n annoksella sekä prednisonihoitoa asteittain pienenevällä annoksella 14 viikon ajan (kaikki tapaukset olivat bakteeri-infektioita), ja 3 potilaalla (5,2 %:lla), jotka olivat saaneet lumelääkettä sekä prednisonihoitoa asteittain pienenevällä annoksella 52 viikon ajan (kaikki tapaukset olivat COVID‑19-infektioita).
Laboratorioarvojen poikkeavuudet
Neutrofiilien määrä
SAPHYR-tutkimuksessa neutrofiilien määrän pienenemistä alle arvoon 1 x 109/l ilmeni sarilumabiryhmässä 7 potilaalla (12 %:lla), joista kahdella (3,4 %:lla) tapahtuma oli vakava (neutrofiilimäärä pieneni alle arvoon 0,5 x 109/l).
Maksaentsyymit
SAPHYR-tutkimuksessa yhdelläkään sarilumabia saaneista potilaista ALAT- tai ASAT-arvot eivät olleet yli 3‑kertaisia normaaliarvon ylärajaan (ULN) nähden. Lumeryhmässä kahdella potilaalla ALAT-arvot kohosivat yli 3‑kertaisiksi normaaliarvon ylärajaan nähden.
Immunogeenisuus
Kuten kaikkiin terapeuttisiin proteiineihin, myös sarilumabiin liittyy immunogeenisuuden mahdollisuus.
Polymyalgia rheumaticaa sairastavien potilaiden populaatiossa yhdellä potilaalla (1,8 %:lla), joka oli saanut sarilumabia 200 mg:n annoksella, todettiin pitkäkestoinen lääkevasta-aine- (ADA‑) vaste. Yhdelläkään lumeryhmän potilaalla ei todettu lääkevasta-ainevastetta. Neutraloivan vasta-aineen analyysissä todettiin positiivinen vaste sarilumabia 200 mg:n annoksella saaneella polymyalgia rheumaticaa sairastavalla potilaalla, jolla oli todettu lääkevasta-ainevaste. Lääkevasta-aineiden pienen ilmaantuvuuden vuoksi näiden vasta-aineiden vaikutuksia sarilumabin turvallisuuteen ja/tai tehoon ei tunneta.
Pediatriset potilaat
Esitäytetyssä ruiskussa tai esitäytetyssä kynässä olevan sarilumabin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistetttu. Tietoja ei ole saatavilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kevzara-valmisteen yliannostukseen ei ole erityistä hoitoa. Yliannostustapauksessa potilasta on seurattava tarkasti ja hoidettava oireenmukaisesti, ja tarvittaessa on aloitettava peruselintoimintoja tukeva hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC14
Vaikutusmekanismi
Sarilumabi on ihmisen monoklonaalinen vasta-aine (IgG1-alatyyppi), joka sitoutuu spesifisesti sekä liukoisiin että membraaniin sitoutuneihin IL‑6-reseptoreihin (IL-6Rα) ja estää IL‑6-välitteistä signaalivälitystä, johon osallistuvat kaikkialla elimistössä esiintyvät signaaleja transdusoiva glykoproteiini 130 (gp130) ja STAT-3-proteiini (Signal Transducer and Activator of Transcription-3).
Funktionaalisissa ihmisen soluihin perustuvissa määrityksissä sarilumabi pystyi salpaamaan IL‑6-signaalivälitysreitin, mikä mitattiin STAT-3-inhibitiona, ainoastaan IL‑6:n läsnä ollessa.
IL‑6 on pleiotrooppinen sytokiini, joka stimuloi erilaisia soluvasteita, kuten solujen lisääntymistä, erilaistumista, elossapysymistä ja apoptoosia, ja voi aktivoida maksasolut vapauttamaan akuutin vaiheen proteiineja, kuten C-reaktiivista proteiinia (CRP) ja seerumin amyloidi A:ta. IL‑6:n suurentuneita pitoisuuksia todetaan nivelreumaa tai idiopaattista juveniilia polyartriittia sairastavien potilaiden nivelnesteessä, ja niillä on tärkeä tehtävä sekä patologisessa tulehduksessa että niveltuhossa, jotka ovat nivelreuman ja idiopaattisen juveniilin polyartriitin tunnusmerkkejä. IL‑6 osallistuu erilaisiin fysiologisiin prosesseihin, kuten T‑solujen, B‑solujen, monosyyttien ja osteoklastien migraatioon ja aktivaatioon, mikä johtaa nivelreumaa tai idiopaattista juveniilia polyartriittia sairastavilla potilailla systeemiseen tulehdukseen, synoviaaliseen tulehdukseen ja luusyöpymään.
Sarilumabin tulehdusta vähentävään vaikutukseen liittyy muutoksia laboratoriokokeissa mitatuissa arvoissa, kuten absoluuttisen neutrofiilimäärän pienenemistä ja lipidiarvojen kohoamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakodynaamiset vaikutukset
Sarilumabin 200 mg:n tai 150 mg:n kerta-annoksen ihon alle antamisen jälkeen nivelreumapotilailla havaittiin nopea CRP-arvojen pieneneminen. Arvot laskivat normaalille tasolle jo 4 päivän kuluttua hoidon aloittamisesta. Sarilumabin kerta-annoksen antamisen jälkeen nivelreumapotilaiden absoluuttinen neutrofiilimäärä oli alimmillaan 3–4 päivän kuluttua, minkä jälkeen se alkoi palautua kohti lähtötasoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sarilumabihoito pienensi fibrinogeenin ja seerumin amyloidi A:n määriä ja lisäsi hemoglobiinia ja seerumin albumiinia. Sarilumabihoidon (200 mg joka toinen viikko) vaikutukset farmakodynaamisten merkkiaineiden profiileihin (CRP ja B-Neut) ajan kuluessa olivat polymyalgia rheumaticaa sairastavilla potilailla samankaltaiset kuin nivelreumapotilailla.
Kliininen teho
Nivelreuma
Sarilumabin tehoa ja turvallisuutta arvioitiin kolmessa satunnaistetussa, kaksoissokkoutetussa, kontrolloidussa monikeskustutkimuksessa (MOBILITY ja TARGET olivat lumekontrolloituja tutkimuksia ja MONARCH oli aktiiviverrokkikontrolloitu tutkimus), joihin osallistui yli 18-vuotiaita potilaita, joilla oli ACR-kriteeristön (American College of Rheumatology) mukaisesti diagnosoitu keskivaikea tai vaikea aktiivinen nivelreuma. Potilailla oli lähtötilanteessa vähintään 8 aristavaa ja 6 turvonnutta niveltä.
Lumekontrolloidut tutkimukset
MOBILITY-tutkimukseen osallistui 1 197 nivelreumapotilasta, jotka eivät olleet saaneet riittävää hoitovastetta metotreksaatilla. Potilaat saivat samanaikaisesti metotreksaatin kanssa joka toinen viikko joko 200 mg tai 150 mg sarilumabia tai lumelääkettä. Ensisijaiset päätemuuttujat olivat viikolla 24 ACR20-vasteen saavuttaneiden potilaiden osuus, viikolla 16 HAQ-DI-mittarilla (Health Assessment Questionnaire – Disability Index) arvioidut muutokset lähtötilanteeseen verrattuna ja viikolla 52 mTSS-mittarilla (van der Heijde-modified Total Sharp Score) arvioitu muutos lähtötilanteeseen verrattuna.
TARGET-tutkimukseen osallistui 546 nivelreumapotilasta, jotka eivät olleet saaneet riittävää hoitovastetta yhdellä tai useammalla TNFα-salpaajalla tai jotka eivät sietäneet yhtä tai useampaa TNFα-salpaajaa. Potilaat saivat samanaikaisesti perinteisten DMARD-lääkkeiden kanssa joka toinen viikko joko 200 mg tai 150 mg sarilumabia tai lumelääkettä. Ensisijaiset päätemuuttujat olivat viikolla 24 ACR20-vasteen saavuttaneiden potilaiden osuus ja viikolla 12 HAQ-DI-mittarilla arvioidut muutokset lähtötilanteeseen verrattuna.
Kliininen vaste
Sarilumabin ja DMARD-lääkkeiden yhdistelmää saaneiden ACR20‑, ACR50- tai ACR70-vasteen saavuttaneiden potilaiden prosentuaaliset osuudet MOBILITY- ja TARGET-tutkimuksissa on esitetty taulukossa 4. Molemmissa tutkimuksissa potilailla, jotka saivat yhdistelmänä joko 200 mg tai 150 mg sarilumabia ja DMARD-lääkkeitä, ACR20‑, ACR50- ja ACR70-vasteiden määrät olivat viikolla 24 suurempia kuin lumelääkettä saaneilla potilailla. Nämä vasteet säilyivät avoimessa jatkotutkimuksessa koko kolme vuotta jatkuneen hoidon ajan.
MOBILITY-tutkimuksessa suurempi osuus potilaista, jotka saivat joka toinen viikko 200 mg tai 150 mg sarilumabia ja metotreksaattia, oli saavuttanut remission viikolla 52 verrattuna lumelääkkeen ja metotreksaatin yhdistelmään. Remissio määriteltiin DAS28-CRP-mittarin (Disease Activity Score 28-C-Reactive Protein) pistearvona, joka oli < 2,6. TARGET-tutkimuksen tulokset viikon 24 kohdalla olivat samanlaisia kuin MOBILITY-tutkimuksen tulokset viikon 52 kohdalla (ks. taulukko 4).
Taulukko 4:Kliininen vaste viikoilla 12, 24 ja 52 lumekontrolloiduissa MOBILITY‑ ja TARGET‑tutkimuksissa
| Potilaiden osuus (%) | ||||||
| MOBILITY Riittämätön vaste metotreksaattihoitoon | TARGET Riittämätön vaste hoitoon TNF-salpaajalla | |||||
| Lume + MTX N = 398 | Sarilumabi 150 mg + MTX N = 400 | Sarilumabi 200 mg + MTX N = 399 | Lume + DMARD* N = 181 | Sarilumabi 150 mg + DMARD* N = 181 | Sarilumabi 200 mg + DMARD* N = 184 | |
| Viikko 12 | ||||||
| DAS28-CRP remissio (< 2,6) | 4,8 % | 18,0 %††† | 23,1 %††† | 3,9 % | 17,1 %††† | 17,9 %††† |
ACR20 ACR50 ACR70 | 34,7 % 12,3 % 4,0 % | 54,0 %††† 26,5 %††† 11,0 %†† | 64,9 %††† 36,3 %††† 17,5 %††† | 37,6 % 13,3 % 2,2 % | 54,1 %† 30,4 %††† 13,8 %††† | 62,5 %††† 33,2 %††† 14,7 %††† |
| Viikko 24 | ||||||
| DAS28-CRP remissio (< 2,6) | 10,1 % | 27,8 %††† | 34,1 %††† | 7,2 % | 24,9 %††† | 28,8 %††† |
ACR20‡ ACR50 ACR70 | 33,4 % 16,6 % 7,3 % | 58,0 %††† 37,0 %††† 19,8 %††† | 66,4 %††† 45,6 %††† 24,8 %††† | 33,7 % 18,2 % 7,2 % | 55,8 %††† 37,0 %††† 19,9 %†† | 60,9 %††† 40,8 %††† 16,3 %† |
| Viikko 52 | ||||||
| DAS28-CRP remissio (< 2,6) | 8,5 % | 31,0 %††† | 34,1 %††† | Ei oleellinen§ | Ei oleellinen§ | Ei oleellinen§ |
ACR20 ACR50 ACR70 | 31,7 % 18,1 % 9,0 % | 53,5 %††† 40,0 %††† 24,8 % | 58,6 %††† 42,9 %††† 26,8 % | |||
| Merkittävä kliininen vaste¶ | 3,0 % | 12,8 %††† | 14,8 %††† | |||
| * TARGET-tutkimuksessa käytetyt DMARD-lääkkeet olivat metotreksaatti, sulfasalatsiini, leflunomidi ja hydroksiklorokiini † p-arvo <0,01, ero lumelääkkeeseen verrattuna †† p-arvo <0,001, ero lumelääkkeeseen verrattuna ††† p-arvo <0,0001, ero lumelääkkeeseen verrattuna ‡ Ensisijainen päätemuuttuja § Ei oleellinen, koska TARGET oli 24 viikon pituinen tutkimus ¶ Merkittävä kliininen vaste = ACR70-vaste vähintään 24 peräkkäisellä viikolla 52 viikon pituisen jakson aikana | ||||||
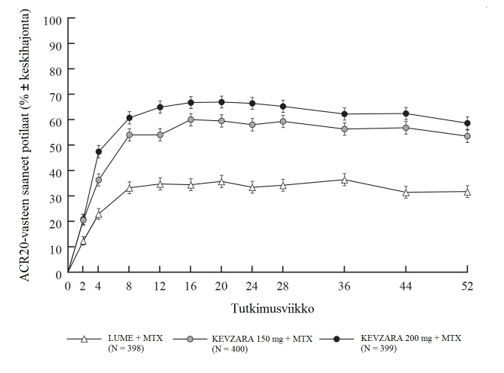
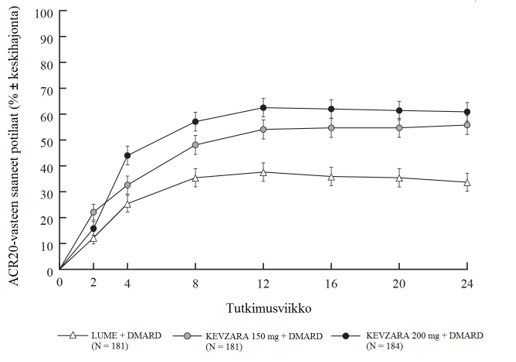
Sekä MOBILITY- että TARGET-tutkimuksissa kahden viikon kuluessa todettu ACR20-vasteen saaneiden määrä oli suurempi lumelääkkeeseen verrattuna ja säilyi tutkimusten loppuun saakka (ks. kuvat 1 ja 2).
Kuva 1: ACR20-vasteen saaneiden potilaiden prosentuaalinen osuus tutkimuskäynneillä MOBILITY-tutkimuksessa
Kuva 2: ACR20-vasteen saaneiden potilaiden prosentuaalinen osuus tutkimuskäynneillä TARGET-tutkimuksessa
Viikolla 24 MOBILITY-ja TARGET-tutkimuksissa saadut ACR-kriteeristön mukaiset vasteet osa-alueittain on esitetty taulukossa 5. MOBILITY-tutkimuksen tulokset olivat viikolla 52 samanlaisia kuin TARGET-tutkimuksessa viikon 24 kohdalla.
Taulukko 5:ACR-osa-alueiden pistemäärien keskimääräinen pieneneminen lähtötilanteesta viikolle 24
| MOBILITY | TARGET | |||||
| Osa-alue (vaihteluväli) | Lume + MTX (N = 398) | Sarilumabi 150 mg joka 2. viikko + MTX (N = 400) | Sarilumabi 200 mg joka 2. viikko + MTX (N = 399) | Lume + DMARD (N = 181) | Sarilumabi 150 mg joka 2. viikko + DMARD (N = 181) | Sarilumabi 200 mg joka 2. viikko + DMARD (N = 184) |
| Aristavat nivelet (0–68) | ‑14,38 | ‑19,25††† | ‑19,00††† | ‑17,18 | ‑17,30† | ‑20,58††† |
| Turvonneet nivelet (0–66) | ‑8,70 | ‑11,84††† | ‑12,43††† | ‑12,12 | ‑13,04†† | ‑14,03††† |
| Kipu VAS- mittarilla‡ (0–100 mm) | ‑19,43 | ‑30,75††† | ‑34,35††† | ‑27,65 | ‑36,28†† | ‑39,60††† |
| Lääkärin yleisarvio VAS-mittarilla‡ (0–100 mm) | ‑32,04 | ‑40,69††† | ‑42,65††† | ‑39,44 | ‑45,09††† | ‑48,08††† |
| Potilaan yleisarvio VAS-mittarilla‡ (0–100 mm) | ‑19,55 | ‑30,41††† | ‑35,07††† | ‑28,06 | ‑33,88†† | ‑37,36††† |
| HAQ‑DI (0–3) | ‑0,43 | ‑0,62††† | ‑0,64††† | ‑0,52 | ‑0,60† | ‑0,69†† |
| CRP | ‑0,14 | ‑13,63††† | ‑18,04††† | ‑5,21 | ‑13,11††† | ‑29,06††† |
| ‡ VAS = kivun voimakkuuden arvioinnissa käytettävä asteikko (Visual analogue scale) † p-arvo < 0,01, ero lumelääkkeeseen verrattu †† p-arvo < 0,001, ero lumelääkkeeseen verrattuna ††† p-arvo < 0,0001, ero lumelääkkeeseen verrattuna | ||||||
Röntgentutkimuksilla todettu vaste
MOBILITY-tutkimuksessa arvioitiin röntgentutkimuksilla nivelrakenteen vaurioitumista, joka ilmaistiin mTSS-mittarilla (van der Heijde-modified Total Sharp Score) ja sen osa-alueilla todettuina muutoksina sekä eroosion laajuutta ja nivelraon kaventumista kuvaavina pistemäärinä viikolla 52. Käsistä ja jalkateristä otettiin röntgenkuvat lähtötilanteessa, viikolla 24 ja viikolla 52. Kuvat pisteytti toisistaan riippumatta vähintään kaksi hyvin koulutettua arvioitsijaa, jotka oli sokkoutettu hoitoryhmän ja tutkimuskäynnin numeron suhteen.
Molemmat yhdessä metotreksaatin kanssa annetut sarilumabiannokset olivat parempia lumelääkkeen ja metotreksaatin yhdistelmään verrattuna arvioitaessa muutosta lähtötilanteesta mTSS-mittarilla viikolla 24 ja viikolla 52 (ks. taulukko 6). Sarilumabiryhmissä raportoitiin viikoilla 24 ja 52 lumelääkeryhmään verrattuna vähemmän eroosion ja nivelraon kaventumisen etenemistä niitä kuvaavien pistemäärien perusteella.
Sarilumabin ja metotreksaatin yhdistelmähoitoon liittyi merkittävästi vähemmän röntgentutkimuksella todettua rakenteellisten vaurioiden etenemistä lumelääkkeeseen verrattuna. Viikolla 52 sarilumabia 200 mg:n annoksella saaneista potilaista 55,6 %:lla ja sarilumabia 150 mg:n annoksella saaneista potilaista 47,8 %:lla ei todettu rakenteellisten vaurioiden etenemistä (määriteltiin TSS-mittarin arvoksi nolla tai vähemmän). Lumelääkettä saaneilla potilailla vastaava luku oli 38,7 %.
Viikolla 52 sarilumabi + metotreksaatti -hoito oli estänyt rakenteellisten vaurioiden etenemistä 91 % sarilumabiannoksella 200 mg ja 68 % sarilumabiannoksella 150 mg verrattuna lumelääkkeen ja metotreksaatin yhdistelmään.
MOBILITY-tutkimuksessa viikolla 52 todettu sarilumabin ja samanaikaisesti käytettyjen DMARD-lääkkeiden teho röntgentutkimuksella todettavan etenemisen estämisessä, jota arvioitiin osana ensisijaista päätemuuttujaa, säilyi jopa kolme vuotta hoidon aloittamisesta.
Taulukko 6:MOBILITY-tutkimuksessa viikoilla 24 ja 52 keskimääräinen röntgentutkimuksella todettu muutos lähtötilanteeseen verrattuna
| MOBILITY Riittämätön vaste metotreksaattihoitoon | |||
| Lume + MTX (N = 398) | Sarilumabi 150 mg joka 2. viikko + MTX (N = 400) | Sarilumabi 200 mg joka 2. viikko + MTX (N = 399) | |
Keskimääräinen muutos viikolla 24
|
1,22 0,68 0,54 |
0,54† 0,26† 0,28 |
0,13†† 0,02†† 0,12† |
Keskimääräinen muutos viikolla 52
| 2,78 1,46 1,32 | 0,90†† 0,42†† 0,47† | 0,25†† 0,05†† 0,20†† |
| † p-arvo <0,001 †† p-arvo <0,0001 ‡ Ensisijainen päätemuuttuja | |||
Fyysisessä toimintakyvyssä saavutettu vaste
MOBILITY- ja TARGET-tutkimuksissa arvioitiin potilaiden fyysistä toimintakykyä ja toimintakyvyn heikkenemistä HAQ-DI-mittarilla (Health Assessment Questionnaire Disability Index). Potilailla, jotka saivat yhdistelmänä sarilumabia 200 mg:n tai 150 mg:n annoksella ja DMARD-lääkkeitä joka toinen viikko, todettiin fyysisen toimintakyvyn parantuneen lähtötilanteesta enemmän kuin lumelääkettä saaneilla potilailla viikolla 16 MOBILITY-tutkimuksessa ja viikolla 12 TARGET-tutkimuksessa.
MOBILITY-tutkimuksessa HAQ-DI-mittarilla arvioidun fyysisen toimintakyvyn osoitettiin parantuneen merkittävästi lumelääkkeeseen verrattuna viikolla 16 (sarilumabi 200 mg ja metotreksaatti: ‑0,58; sarilumabi 150 mg ja metotreksaatti: ‑0,54; lumelääke ja metotreksaatti: ‑0,30; annostus joka toinen viikko). TARGET-tutkimuksessa HAQ-DI-mittarin pistemäärien osoitettiin parantuneen merkittävästi lumelääkkeeseen verrattuna viikolla 12 (sarilumabi 200 mg ja DMARD: ‑0,49; sarilumabi 150 mg ja DMARD: ‑0,50; lumelääke ja DMARD: ‑0,29; annostus joka toinen viikko).
MOBILITY-tutkimuksessa HAQ-DI-mittarilla arvioidun parantuneen fyysisen toimintakyvyn osoitettiin säilyneen viikolle 52 (sarilumabi 200 mg ja metotreksaatti: ‑0,75; sarilumabi 150 mg ja metotreksaatti: ‑0,71; lumelääke ja metotreksaatti: ‑0,46).
Sarilumabin ja metotreksaatin yhdistelmää saaneilla potilailla (47,6 %:lla 200 mg:n annosta saaneiden ryhmässä ja 47,0 %:lla 150 mg:n annosta saaneiden ryhmässä) saavutettiin kliinisesti merkittävä paraneminen HAQ-DI-pisteissä (≥ 0,3 yksikön muutos lähtötilanteeseen nähden) viikolla 52. Lumelääkkeen ja metotreksaatin yhdistelmää saaneiden potilaiden ryhmässä vastaava luku oli 26,1 %.
Potilaiden ilmoittamat hoitotulokset
Potilaiden yleistä terveydentilaa arvioitiin SF-36-lomakkeella (Short Form health survey). MOBILITY- ja TARGET-tutkimuksissa 200 mg:n sarilumabiannosta yhdessä DMARD-lääkkeiden kanssa joka toinen viikko tai 150 mg:n sarilumabiannosta yhdessä DMARD-lääkkeiden kanssa joka toinen viikko saaneilla potilailla havaittiin viikolla 24 enemmän paranemista fyysistä ulottuvuutta kuvaavassa summapistemäärässä lähtötasoon nähden lumelääkkeen ja DMARD-lääkkeiden yhdistelmään verrattuna. Psyykkistä ulottuvuutta kuvaavassa summapistemäärässä ei havaittu huononemista. Sarilumabia 200 mg:n annoksella yhdessä DMARD-lääkkeiden kanssa saaneilla potilailla raportoitiin lumelääkkeeseen verrattuna enemmän paranemista seuraavilla osa-alueilla: fyysinen toimintakyky, roolitoiminta/fyysinen, kivuttomuus, koettu terveys, tarmokkuus, sosiaalinen toimintakyky ja psyykkinen hyvinvointi.
Väsymystä arvioitiin FACIT-Fatigue ‑mittarilla (Functional Assessment of Chronic Illness Therapy-Fatigue). MOBILITY- ja TARGET-tutkimuksissa joka toinen viikko sarilumabia 200 mg:n annoksella yhdessä DMARD-lääkkeiden kanssa tai joka toinen viikko sarilumabia 150 mg:n annoksella yhdessä DMARD-lääkkeiden kanssa saaneilla potilailla todettiin enemmän paranemista lähtötilanteeseen verrattuna kuin lumelääkkeen ja DMARD-lääkkeiden yhdistelmällä.
Aktiiviverrokkikontrolloitu tutkimus
MONARCH oli 24 viikkoa kestänyt satunnaistettu, kaksoissokkoutettu, kaksoislumetekniikalla toteutettu tutkimus, jossa verrattiin sarilumabi 200 mg -monoterapiaa adalimumabi 40 mg -monoterapiaan ihon alle annettuna joka toinen viikko. Tutkimuksessa oli 369 potilasta, joilla oli keskivaikea tai vaikea aktiivinen nivelreuma ja jotka eivät soveltuneet hoitoon metotreksaatilla, mukaan lukien potilaat, jotka eivät sietäneet metotreksaattia tai eivät saaneet metotreksaatilla riittävää hoitovastetta.
Sarilumabihoito 200 mg:n annoksella oli parempi kuin adalimumabihoito 40 mg:n annoksella sairauden aktiivisuuden vähentämisessä ja fyysisen toimintakyvyn parantamisessa, ja suurempi osa potilaista saavutti kliinisen remission 24 viikon kuluessa (ks. taulukko 7).
Taulukko 7: MONARCH-tutkimuksen tehoa koskevat tulokset
| Adalimumabi 40 mg joka 2. viikko* (N = 185) | Sarilumabi 200 mg joka 2. viikko* (N = 184) | |
DAS28-ESR
| ‑2,20 (0,106) | ‑3,28 (0,105) < 0,0001 |
DAS28-ESR-remissio (< 2,6), n (%)
| 13 (7,0 %) | 49 (26,6 %) < 0,0001 |
ACR20-vaste, n (%)
| 108 (58,4 %) | 132 (71,7 %) 0,0074 |
ACR50-vaste, n (%)
| 55 (29,7 %) | 84 (45,7 %) 0,0017 |
ACR70-vaste, n (%)
| 22 (11,9 %) | 43 (23,4 %) 0,0036 |
HAQ-DI
| ‑0,43 (0,045) | ‑0,61 (0,045) 0,0037 |
*Sisältää potilaat, joilla adalimumabin 40 mg:n annoksen antotiheys suurennettiin yhteen kertaan viikossa riittämättömän vasteen vuoksi
Polymyalgia rheumatica
Sarilumabin tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (SAPHYR), johon osallistui vähintään 50‑vuotiaita polymyalgia rheumaticaa sairastavia potilaita, joiden tauti oli diagnosoitu ACR/EULAR (American College of Rheumatology/European Union League against Rheumatism) ‑luokituskriteerien mukaisesti. Potilailla oli ilmennyt ainakin yksi yksiselitteinen polymyalgia rheumatican pahenemisvaihe sinä aikana, kun kortikosteroidihoitoa oli yritetty asteittain vähentää.
SAPHYR-tutkimuksessa aktiivista polymyalgia rheumaticaa sairastavat potilaat satunnaistettiin saamaan sarilumabia 200 mg joka toinen viikko niin, että prednisonihoitoa vähennettiin ennalta määritellyllä tavalla asteittain 14 viikon aikana (n = 60), tai lumelääkettä joka toinen viikko niin, että prednisonihoitoa vähennettiin ennalta määritellyllä tavalla asteittain 52 viikon aikana (n = 58). Yksi potilas satunnaistettiin sarilumabi 200 mg ‑haaraan, mutta hän ei saanut hoitoa. Potilaita, jotka jatkoivat tutkimushoitojakson loppuun asti, oli sarilumabiryhmässä 42 (70 %) ja lumeryhmässä 36 (62,1 %). Potilaat, joilla ilmeni taudin pahenemisvaihe tai jotka eivät pystyneet sitoutumaan määrättyyn aikatauluun, jossa prednisonihoitoa asteittain vähennettiin, saattoivat saada kortikosteroideja oirelääkityksenä.
Tutkimusasetelman mukaisesti prednisonihoidon asteittainen vähentäminen tehtiin hoitohaaroissa eri aikatauluissa. Todellinen kumulatiivinen prednisonia vastaava kortikosteroidien kokonaisannos oli sarilumabihaarassa pienempi (mediaani 777 mg) kuin lumehaarassa (mediaani 2 044 mg).
Ensisijainen päätemuuttuja oli niiden potilaiden osuus, joilla todettiin pitkäkestoinen remissio viikolla 52. Pitkäkestoinen remissio määriteltiin niin, että taudin remissio saavutettiin viimeistään viikolla 12, viikon 12 ja viikon 52 välillä ei ilmennyt taudin pahenemisvaiheita, CRP-arvot pienenivät pitkäkestoisesti (pitoisuudeksi < 10 mg/l) viikosta 12 viikkoon 52 ja sitoutuminen prednisonihoidon asteittaiseen vähentämiseen viikosta 12 viikkoon 52 onnistui. Muita päätemuuttujia olivat kortikosteroidien kumulatiivinen kokonaisannos 52 viikon ajalta, aika ensimmäiseen polymyalgia rheumatican pahenemisvaiheeseen ja potilaiden ilmoittamat hoitotulokset.
Kliininen vaste
Niiden potilaiden osuus, jotka olivat saavuttaneet pitkäkestoisen remission viikkoon 52 mennessä, oli suurempi sarilumabihaarassa kuin lumehaarassa (p = 0,0193). 52 viikon kohdalla niiden potilaiden osuus, joilla oli saavutettu pitkäkestoisen remission päätemuuttujan jokainen osatekijä, oli sarilumabihaarassa suurempi kuin lumehaarassa. Kumulatiivinen kortikosteroidiannos 52 viikon pituisen hoitojakson aikana oli sarilumabihaarassa pienempi kuin lumehaarassa (ks. taulukko 8).
Taulukko 8:Kliininen vaste aktiivista polymyalgia rheumaticaa sairastavilla aikuisilla (SAPHYR-tutkimus)
Lumelääke (N = 58) | Sarilumabi (N = 60) | p‑arvo vs. lumelääke | |||
| Pitkäkestoinen remissio viikon 52 kohdalla | |||||
| Potilaita, joilla todettiin pitkäkestoinen remissio | n (%) | 6 (10,3) | 17 (28,3) | ||
| Osuuden ero (95 %:n luottamusväli) vs. lumelääke | 18,0 (4,15; 31,82) | 0,0193 | |||
| Pitkäkestoisen remission osatekijät viikolla 52 | |||||
| Ei merkkejä ja oireita ja CRP < 10 mg/l (taudin remissio*) viimeistään viikolla 12 | n (%) | 22 (37,9) | 28 (46,7) | Ei laskettu | |
| Ei taudin pahenemisvaihetta‡ viikkojen 12 ja 52 välillä | n (%) | 19 (32,8) | 33 (55,0) | Ei laskettu | |
| CRP-arvojen pitkäkestoinen pieneneminen (< 10 mg/l) viikosta 12 viikkoon 52 | n (%) | 26 (44,8) | 40 (66,7) | Ei laskettu | |
| Onnistunut sitoutuminen prednisonihoidon asteittaiseen vähentämiseen viikosta 12 viikkoon 52 | n (%) | 14 (24,1) | 30 (50,0) | Ei laskettu | |
* Taudin remission määritelmänä oli polymyalgia rheumatican merkkien ja oireiden häviäminen ja CRP-arvojen normalisoituminen (< 10 mg/l).
‡ Pahenemisvaiheen määritelmänä oli kortikosteroidiannoksen suurentamista edellyttävä aktiivisen polymyalgia rheumatican aiheuttamien merkkien ja oireiden uusiutuminen tai aktiivisen polymyalgia rheumatican aiheuttama laskon kohoaminen yhdistettynä kortikosteroidiannoksen suurentamiseen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Kevzara-valmisteen (sarilumabin) käytöstä polymyalgia rheumatican hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Kevzara-valmisteen (sarilumabin) käytöstä kroonisen idiopaattisen reuman (mukaan lukien nivelreuma, spondylartriitti, psoriaasiartriitti ja juveniili nivelreuma) hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Nivelreuma
Sarilumabin farmakokinetiikkaa arvioitiin 2 186 aikuisella potilaalla, joilla oli nivelreuma ja jotka saivat sarilumabia. Näistä potilaista 751 sai 150 mg:n ja 891 sai 200 mg:n hoitoannoksia ihon alle kahden viikon välein 52 viikon ajan.
Imeytyminen
Sarilumabin absoluuttisen biologisen hyötyosuuden arvioitiin olevan 80 % ihonalaisen injektion jälkeen farmakokinetiikan analyysissä. tmax-arvon mediaani ihon alle injisoidun kerta-annoksen antamisen jälkeen todettiin 2–4 vuorokauden kuluessa. Kun annettiin kahden viikon välein toistuvia 150 mg:n ja 200 mg:n annoksia, vakaa tila saavutettiin 12–16 viikossa ja lääkeainekertymä oli 2–3-kertainen verrattuna kerta-annoksen tuottamaan altistukseen.
Kun hoito toteutettiin kahden viikon välein annetulla 150 mg:n annoksella, sarilumabin arvioitu keskimääräinen (± keskihajonta) vakaan tilan AUC (area under the curve, pinta-ala käyrän alla) oli 210 ± 115 mg⋅vrk/l, Cmin 6,95 ± 7,60 mg/l ja Cmax 20,4 ± 8,27 mg/l.
Kun hoito toteutettiin kahden viikon välein annetulla 200 mg:n annoksella, sarilumabin arvioitu keskimääräinen (± keskihajonta) vakaan tilan AUC oli 396 ± 194 mg⋅vrk/l, Cmin 16,7 ± 13,5 mg/l ja Cmax 35,4 ± 13,9 mg/l.
Käytettävyystutkimuksessa sarilumabialtistus annoksella 200 mg joka 2. viikko oli hieman suurempi (Cmax +24 – +34 %, AUC(0–2w) +7 – +21 %) esitäytyn kynän käytön jälkeen esitäytettyyn ruiskuun verrattuna.
Jakautuminen
Nivelreumaa sairastavilla potilailla näennäinen jakautumistilavuus vakaassa tilassa oli 8,3 litraa.
Biotransformaatio
Sarilumabin metaboliareittia ei ole määritelty. Monoklonaalisena vasta-aineena sarilumabin oletetaan hajoavan pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta samalla tavalla kuin endogeenisen IgG:n.
Eliminaatio
Sarilumabi eliminoituu sekä lineaarista että ei-lineaarista reittiä. Suurilla pitoisuuksilla eliminoituminen tapahtuu lähinnä lineaarisen, ei-saturoituvan proteolyyttisen reitin kautta, kun taas pienillä pitoisuuksilla pääasiallisena reittinä on ei-lineaarinen saturoituva kohdeproteiinin välittämä eliminaatio. Nämä rinnakkaiset eliminaatioreitit johtavat siihen, että alkuvaiheen puoliintumisaika on 8–10 vuorokautta ja efektiivinen puoliintumisaika vakaassa tilassa arviolta 21 vuorokautta.
Mediaani ajalle, jolloin pitoisuus ei ollut enää mitattavissa, oli sarilumabin viimeisen vakaan tilan 150 mg:n annoksen antamisen jälkeen 30 vuorokautta ja 200 mg:n annoksen antamisen jälkeen 49 vuorokautta.
Monoklonaaliset vasta-aineet eivät eliminoidu munuaisten tai maksan kautta.
Lineaarisuus/ei-lineaarisuus
Nivelreumapotilailla farmakokineettisen altistuksen havaittiin suurenevan suhteellisesti annosta enemmän. Vakaassa tilassa AUC-arvona mitattu altistus annosvälillä suureni suunnilleen kaksinkertaiseksi, kun annos suurennettiin 1,33-kertaiseksi kahden viikon välein annettavasta 150 mg:sta 200 mg:aan.
Yhteisvaikutus CYP450-substraattien kanssa
Simvastatiini on CYP3A4- ja OATP1B1-substraatti. Yhden viikon kuluttua 200 mg:n sarilumabikerta-annoksen ihon alle pistämisestä 17 nivelreumapotilaan altistus simvastatiinille oli pienentynyt 45 % ja simvastatiinihapolle 36 % (ks. kohta Yhteisvaikutukset).
Polymyalgia rheumatica
Ihon alle annetun sarilumabin farmakokineettiset ominaisuudet polymyalgia rheumaticaa sairastavilla potilailla määritettiin populaatiofarmakokineettisellä analyysillä. Analyysissä oli mukana niukasti Ctrough-havaintoja, jotka oli saatu 58 potilaalta, joilla oli polymyalgia rheumatica ja jotka olivat saaneet toistuvasti ihon alle sarilumabia 200 mg joka toinen viikko. Tällä annostusohjelmalla sarilumabin arvioitu keskimääräinen (± keskihajonta) vakaan tilan AUC oli 551 ± 321 mg⋅vrk/l, Cmin 27,0 ± 21,5 mg/l ja Cmax 46,5 ± 23,0 mg/l. Farmakokineettisten tietojen analyysit viittaavat siihen, että mediaaniaika vakaan tilan saavuttamiseen on polymyalgia rheumaticaa sairastavilla potilailla noin 24 viikkoa. Sarilumabin kertymistä tapahtui ihon alle antamisen jälkeen niin, että kertymäsuhde oli 5–6‑kertainen keskimääräisistä minimipitoisuuksista laskettuna.
Erityisryhmät
Ikä, sukupuoli, etninen ryhmä ja kehon paino
Aikuisilla nivelreumapotilailla (joiden ikä vaihteli 18 vuodesta 88 vuoteen ja joista 14 % oli yli 65‑vuotiaita) tehdyt populaatiofarmakokineettiset analyysit osoittivat, että ikä, sukupuoli ja rotu eivät merkittävästi vaikuttaneet sarilumabin farmakokinetiikkaan.
Kehon paino vaikutti sarilumabin farmakokinetiikkaan aikuisilla potilailla. Sekä 150 mg:n että 200 mg:n annokset osoittautuivat tehokkaiksi potilaille, joilla oli suurempi kehon paino (> 100 kg), mutta yli 100 kg painavat potilaat saivat paremman terapeuttisen hyödyn 200 mg:n annoksella.
Munuaisten vajaatoiminta
Virallisia tutkimuksia munuaisten vajaatoiminnan vaikutuksesta sarilumabin farmakokinetiikkaan ei ole tehty. Lievä tai kohtalainen munuaisten vajaatoiminta ei vaikuttanut sarilumabin farmakokinetiikkaan. Annosta ei tarvitse muuttaa lievää tai kohtalaista munuaisten vajaatoimintaa sairastavilla potilailla. Vaikeaa munuaisten vajaatoimintaa sairastavia potilaita ei ole tutkittu.
Maksan vajaatoiminta
Virallisia tutkimuksia maksan vajaatoiminnan vaikutuksesta sarilumabin farmakokinetiikkaan ei ole tehty (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta, karsinogeenisuusriskin arviointia sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Sarilumabin karsinogeenisuuden arvioimiseksi ei ole tehty pitkäaikaistutkimuksia eläimillä. IL-6Rα-inhibitiota koskevan näytön vahvuus viittaa pääasiassa antituumorivaikutuksiin, joita välittävät useat mekanismit, joihin yleensä sisältyy STAT-3-inhibitio. Sarilumabilla tehdyt in vitro- ja in vivo ‑tutkimukset, joissa käytettiin ihmisen kasvainsolulinjoja, osoittivat STAT-3-aktivaation inhibition ja kasvaimen kasvun estymisen ihmisestä siirrettyjen kasvainsiirteiden eläinmalleissa.
Uros- ja naarashiirillä tehdyissä hedelmällisyystutkimuksissa, joissa käytettiin hiiren surrogaattivasta-ainetta hiiren IL-6Rα:aa vastaan, ei todettu hedelmällisyyden heikkenemistä.
Tehostetussa pre-/postnataalisessa kehitystoksisuustutkimuksessa tiineille Cynomolgus-apinoille annettiin sarilumabia kerran viikossa laskimoon tiineyden varhaisvaiheesta luonnolliseen synnytykseen asti (noin 21 viikon ajan). Emojen altistuksilla, jotka olivat AUC-arvojen perusteella enintään noin 83-kertaiset verrattuna ihmisen altistukseen kahden viikon välein ihon alle annettujen 200 mg:n annosten jälkeen, ei ollut mitään vaikutuksia emoihin tai alkioihin/sikiöihin. Sarilumabi ei vaikuttanut tiineyden jatkumiseen eikä vastasyntyneisiin, jotka tutkittiin yhden kuukauden ikään asti punnitsemalla, arvioimalla toiminnallisen ja morfologisen kehityksen muuttujat, kuten luusto, tekemällä perifeerisen veren lymfosyyteille immunofenotyypitys ja suorittamalla mikroskooppitutkimus. Sarilumabia todettiin vastasyntyneiden seerumissa enintään yhden kuukauden ikään asti. Sarilumabin erittymistä maitoon Cynomolgus-apinoilla ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Arginiini
Polysorbaatti 20 (E432)
Sakkaroosi
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Kevzara 150 mg injektioneste, liuos, esitäytetty ruisku; Kevzara 150 mg injektioneste, liuos, esitäytetty kynä; Kevzara 200 mg injektioneste, liuos, esitäytetty ruisku; ja Kevzara 200 mg injektioneste, liuos, esitäytetty kynä
3 vuotta.
Kun Kevzara on otettu jääkaapista, se on käytettävä 14 vrk:n kuluessa ja säilytettävä alle 25 °C:n lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 ºC – 8 ºC). Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KEVZARA injektioneste, liuos, esitäytetty kynä
150 mg (L:ei) 2 x 1 kpl (1,14 ml) (836,51 €)
200 mg (L:ei) 2 x 1 kpl (1,14 ml) (836,51 €)
KEVZARA injektioneste, liuos, esitäytetty ruisku
200 mg (L:ei) 1 kpl (1,14 ml) (430,40 €), 2 x 1 kpl (1,14 ml) (836,51 €)
PF-selosteen tieto
Esitäytetyissä kynissä ja esitäytetyissä ruiskussa on 1,14 ml liuosta ruiskussa (tyypin 1 lasia), jossa on ruostumattomasta teräksestä valmistettu kiinteä neula ja elastomeeristä valmistettu männän pysäytin.
Kevzara 150 mg injektioneste, liuos, esitäytetty ruisku
Kertakäyttöisen esitäytetyn ruiskun neulansuojus on styreeni-butadieenielastomeeriä, valkoinen männänvarsi polystyreeniä ja vaaleanoranssi sormituki polypropeenia.
Kevzara 200 mg injektioneste, liuos, esitäytetty ruisku
Kertakäyttöisen esitäytetyn ruiskun neulansuojus on styreeni-butadieenielastomeeriä, valkoinen männänvarsi polystyreeniä ja tummanoranssi sormituki polypropeenia.
Kevzara 150 mg injektioneste, liuos, esitäytetty kynä
Ruiskun osat on koottu kertakäyttöiseksi esitäytetyksi kynäksi, jossa on keltainen neulansuojus ja vaaleanoranssi korkki.
Kevzara 200 mg injektioneste, liuos, esitäytetty kynä
Ruiskun osat on koottu kertakäyttöiseksi esitäytetyksi kynäksi, jossa on keltainen neulansuojus ja tummanoranssi korkki.
Pakkauskoot:
- 1 esitäytetty ruisku
- 2 esitäytettyä ruiskua
- monipakkaus, joka sisältää 6 esitäytettyä ruiskua (3 kpl 2 ruiskun pakkauksia)
- 1 esitäytetty kynä
- 2 esitäytettyä kynää
- monipakkaus, joka sisältää 6 esitäytettyä kynää (3 kpl 2 kynän pakkauksia)
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön tai vaaleankeltainen steriili liuos, jonka pH-arvo on noin 6,0.
Kevzara 150 mg injektioneste, liuos
298–346 mmol/kg
Kevzara 200 mg injektioneste, liuos
306–371 mmol/kg
Käyttö- ja käsittelyohjeet
Liuos on tarkastettava ennen käyttöä. Liuosta ei pidä käyttää, jos se on sameaa tai värjäytynyttä tai sisältää hiukkasia tai mikä tahansa laitteen osa näyttää vaurioituneelta.
Jääkaapista pois ottamisen jälkeen esitäytetyn ruiskun/esitäytetyn kynän annetaan lämmetä huoneenlämpöiseksi (< 25 °C). Esitäytetyn ruiskun annetaan lämmetä 30 minuuttia ja esitäytetyn kynän annetaan lämmetä 60 minuuttia ennen Kevzara-valmisteen pistämistä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. Laita esitäytetty ruisku / esitäytetty kynä käytön jälkeen terävälle jätteelle tarkoitettuun astiaan ja hävitä se paikallisten määräysten mukaisesti.
Korvattavuus
KEVZARA injektioneste, liuos, esitäytetty kynä
150 mg 2 x 1 kpl
200 mg 2 x 1 kpl
KEVZARA injektioneste, liuos, esitäytetty ruisku
200 mg 1 kpl
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313).
KEVZARA injektioneste, liuos, esitäytetty ruisku
150 mg 1 kpl
200 mg 2 x 1 kpl
- Ei korvausta.
ATC-koodi
L04AC14
Valmisteyhteenvedon muuttamispäivämäärä
19.02.2026
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi