
TEPKINLY injektionsvätska, lösning 4 mg/0,8 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje 0,8 ml injektionsflaska innehåller 4 mg epkoritamab med en koncentration på 5 mg/ml.
Varje injektionsflaska innehåller en överfyllning som gör det möjligt att dra upp den angivna mängden.
Epkoritamab är en humaniserad immunglobulin G1 (IgG1) bispecifik antikropp mot CD3- och CD20-antigener som framställs i ovarieceller från kinesisk hamster (CHO) med rekombinant DNA-teknik.
Hjälpämne med känd effekt
Varje injektionsflaska med Tepkinly innehåller 28,8 mg sorbitol och 0,42 mg polysorbat 80. För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Injektionsvätska, lösning (injektion)
Kliniska uppgifter
Terapeutiska indikationer
Tepkinly som monoterapi är avsett för behandling av vuxna patienter med recidiverande eller refraktärt diffust storcelligt B-cellslymfom (DLBCL), vilka tidigare har fått minst två linjer av systemisk behandling.
Tepkinly i kombination med lenalidomid och rituximab är avsett för behandling av vuxna patienter med recidiverande eller refraktärt follikulärt lymfom (FL).
Tepkinly som monoterapi är avsett för behandling av vuxna patienter med recidiverande eller refraktärt follikulärt lymfom (FL), vilka tidigare har fått minst två linjer av systemisk behandling.
Villkor
Valmistetta saa antaa vain syöpälääkkeiden käyttöön perehtyneen terveydenhuollon ammattilaisen valvonnassa hoitokeskuksessa, jossa on valmius vaikea-asteisten reaktioiden, kuten sytokiinioireyhtymän hoitoon.
Dosering och administreringssätt
Tepkinly får endast administreras under överinseende av sjukvårdspersonal som har erfarenhet av diagnos och behandling av cancerpatienter. Det måste finnas tillgängligt minst en dos av tocilizumab innan epkoritamab administreras i cykel 1 för användning i händelse av cytokinfrisättningssyndrom (CRS). Ytterligare en dos tocilizumab måste finnas tillgänglig inom 8 timmar från det att den föregående dosen tocilizumab har använts.
Dosering
Rekommenderad premedicinering och dosschema
Tepkinly som monoterapi
Tepkinly ska administreras i 28-dagarscykler enligt nedanstående upptrappningsschema i tabell 1 för patienter med diffust storcelligt B-cellslymfom och tabell 2 för patienter med follikulärt lymfom.
Tabell 1 Tepkinly doseringsschema med två upptrappningsdoser för patienter med diffust storcelligt B-cellslymfom
Doseringsschema | Behandlingscykel | Dagar | Dos av epkoritamab (mg)a |
Varje vecka | Cykel 1 | 1 | 0,16 mg (upptrappningsdos 1) |
8 | 0,8 mg (upptrappningsdos 2) | ||
15 | 48 mg (första fulla dosen) | ||
22 | 48 mg | ||
Varje vecka | Cykel 2–3 | 1, 8, 15, 22 | 48 mg |
Varannan vecka | Cykel 4–9 | 1, 15 | 48 mg |
Var fjärde vecka | Cykel 10 och därefter | 1 | 48 mg |
a0,16 mg är en förberedande dos, 0,8 mg är en intermediär dos och 48 mg är en full dos. | |||
Tabell 2 Tepkinly doseringsschema med tre upptrappningsdoser för patienter med follikulärt lymfom
Doseringsschema | Behandlingscykel | Dagar | Dos av epkoritamab (mg)a | |
Varje vecka | Cykel 1 | 1 | 0,16 mg (upptrappningsdos 1) | |
8 | 0,8 mg (upptrappningsdos 2) | |||
15 | 3 mg (upptrappningsdos 3) | |||
22 | 48 mg (första fulla dosen) | |||
Varje vecka | Cykel 2–3 | 1, 8, 15, 22 | 48 mg | |
Varannan vecka | Cykel 4–9 | 1, 15 | 48 mg | |
Var fjärde vecka | Cykel 10 och därefter | 1 | 48 mg | |
a0,16 mg är en förberedande dos, 0,8 mg är en intermediär dos, 3 mg är en andra intermediär dos och 48 mg är en full dos. |
| |||
Tepkinly ska administreras till dess att sjukdomsprogress eller oacceptabel toxicitet inträffar.
Tepkinly i kombination med lenalidomid och rituximab
Tepkinly ska administreras i 28-dagarscykler i totalt 12 cykler eller fram till sjukdomsprogress eller oacceptabel toxicitet, beroende på vilket som inträffar först.
Tabell 3 Tepkinly doseringsschema med tre upptrappningsdoser i kombination med lenalidomid och rituximab för patienter med follikulärt lymfom
Doseringsschema | Behandlingscykel | Dagar | Dos av epkoritamab (mg) |
Varje vecka | Cykel 1 | 1 | 0,16 mg (upptrappningsdos 1) |
8 | 0,8 mg (upptrappningsdos 2) | ||
15 | 3 mg (upptrappningsdos 3) | ||
22 | 48 mg (första fulla dosen) | ||
| Cykel 2 och 3 | 1, 8, 15 och 22 | 48 mg |
Var fjärde vecka | Cykel 4 till 12 | 1 | 48 mg |
Tepkinly ska administreras i kombination med lenalidomid 20 mg peroralt en gång dagligen (från dag 1 till dag 21) i cykel 1–12 och rituximab 375 mg/m2 intravenöst varje vecka i cykel 1 (på dag 1, 8, 15 och 22) och var fjärde vecka i cykel 2–5 (på dag 1).
För ytterligare information, se produktresumén för lenalidomid och rituximab.
Premedicinering och profylax
Information om rekommenderad premedicinering mot cytokinfrisättningssyndrom (CRS) visas i tabell 4.
Tabell 4 Premedicinering för epkoritamab
Cykel | Patient som kräver premedicinering | Premedicineringa | Administrering |
Cykel 1 | Alla patienter | Dexametason (15 mg peroralt eller intravenöst) eller prednisolon (100 mg peroralt eller intravenöst) eller motsvarande
|
|
|
| ||
Cykel 2 och därefter | Patienter som uppvisade CRS av grad 2 eller 3b vid tidigare dos | Dexametason (15 mg peroralt eller intravenöst) eller prednisolon (100 mg peroralt eller intravenöst) eller motsvarande
|
|
aPremedicinering som används för kombinationsläkemedel kan användas som premedicinering för epkoritamab enligt den behandlande läkarens bedömning, förutsatt att doserna som administreras är minst likvärdiga. | |||
För ytterligare information om rekommendationer för premedicinering, se produktresumén för lenalidomid respektive rituximab.
Profylax mot Pneumocystis jirovecii-pneumoni (PCP) och herpesvirusinfektioner rekommenderas starkt under behandling med epkoritamab.
Tepkinly ska administreras till adekvat uppvätskade patienter.
Det rekommenderas starkt att alla patienter följer nedanstående riktlinjer för vätskeintag under cykel 1 såvida det inte är medicinskt kontraindicerat:
-
Intag av 2–3 liter vätska under 24 timmar före varje administrering av epkoritamab
-
Inte ta några blodtryckssänkande läkemedel under 24 timmar före varje administrering av epkoritamab
-
Administrera 500 ml isotonisk intravenös (iv) vätska på samma dag som epkoritamab ges; före dosering OCH
-
Intag av 2–3 liter vätska under 24 timmar efter varje administrering av epkoritamab.
För patienter med ökad risk för kliniskt tumörlyssyndrom (CTLS) rekommenderas uppvätskning och profylaktisk behandling med ett urinsyrasänkande medel.
Patienterna ska övervakas för tecken och symtom på CRS och/eller immuneffektorcells-associerat neurotoxicitetssyndrom (ICANS) och hanteras enligt aktuella riktlinjer efter administrering av epkoritamab. Patienterna ska informeras om tecknen och symtomen på CRS och ICANS och att omedelbart söka läkarvård om sådana tecken eller symtom uppstår, oavsett när det händer (se avsnitt Varningar och försiktighet).
Dosändringar och behandling av biverkningar
Cytokinfrisättningssyndrom (CRS)
Patienter som behandlas med epkoritamab kan utveckla CRS.
Utvärdera och behandla andra orsaker till feber, hypoxi och hypotoni. Om CRS misstänks ska behandling ges enligt rekommendationerna i tabell 5. Patienter som uppvisar CRS ska övervakas oftare under nästa planerade administrering av epkoritamab.
Tabell 5 CRS-grader och behandlingsvägledning
Grada | Rekommenderad behandling | Dosändring av epkoritamabh |
Grad 1
| Ge stödjande vård såsom antipyretika och intravenös vätsketillförsel Dexametasonb kan sättas in Vid hög ålder, hög tumörbörda, cirkulerande tumörceller, feber som inte svarar på antipyretika:
Om ingen förbättring ses efter 24 timmar, överväg tocilizumabd För CRS med samtidigt ICANS, se tabell 6 | Pausa epkoritamab tills CRS-händelsen har gått över |
Grad 2
och
och/eller
| Ge stödjande vård såsom antipyretika och intravenös vätsketillförsel Dexametasonb ska övervägas Anti‑cytokinbehandling, tocilizumabd, rekommenderas Om CRS är motståndskraftigt mot dexametason och tocilizumab:
För CRS med samtidigt ICANS, se tabell 6 | Pausa epkoritamab tills CRS-händelsen har gått över |
Grad 3
och
och/eller
| Ge stödjande vård såsom antipyretika och intravenös vätsketillförsel Dexametasonc ska administreras Anti‑cytokinbehandling, tocilizumabd, rekommenderas Om CRS är motståndskraftigt mot dexametason och tocilizumab:
För CRS med samtidigt ICANS, se tabell 6 | Pausa epkoritamab tills CRS-händelsen har gått över I händelse av en grad 3 CRS som varar längre än 72 timmar, ska behandling med epkoritamab avslutas. Om fler än 2 separata händelser av grad 3 CRS inträffar även om varje enskild händelse har behandlats till grad 2 inom 72 timmar, ska behandling med epkoritamab avslutas. |
Grad 4
och
och/eller
| Ge stödjande vård såsom antipyretika och intravenös vätsketillförsel Dexametasonc ska administreras Anti‑cytokinbehandling, tocilizumabd, rekommenderas Om CRS är motståndskraftigt mot dexametason och tocilizumab:
För CRS med samtidigt ICANS, se tabell 6 | Avsluta epkoritamab permanent |
aCRS-grad enligt konsensuskriterier från ASTCT (American Society for Transplantation and Cellular Therapy) | ||
Immuneffektorcells-associerat neurotoxicitetssyndrom (ICANS)
Patienterna ska övervakas beträffande tecken och symtom på ICANS. Andra orsaker till neurologiska symtom ska uteslutas. Om ICANS misstänks ska behandling ges enligt rekommendationerna i tabell 6.
Tabell 6 ICANS-grader och behandlingsvägledning
Grada | Rekommenderad behandling | Dosändring av epkoritamabg |
Grad 1a ICE-poängb 7–9a eller sänkt medvetandea: vaknar spontant | Behandling med dexametasonc Överväg icke‑sederande läkemedel mot anfall (t.ex. levetiracetam) tills ICANS har gått över Inget samtidigt CRS:
För ICANS med samtidigt CRS:
| Pausa epkoritamab tills händelsen har gått över |
Grad 2a ICE-poängb 3–6 eller sänkt medvetandea: vaknar av röst | Behandling med dexametasone Överväg icke‑sederande läkemedel mot anfall (t.ex. levetiracetam) tills ICANS har gått över Inget samtidigt CRS:
För ICANS med samtidigt CRS:
| Pausa epkoritamab tills händelsen har gått över |
Grad 3a ICE-poängb 0–2 eller sänkt medvetandea: vaknar endast vid taktil stimulering, eller anfalla, antingen:
eller
| Behandling med dexametasonf
Överväg icke‑sederande läkemedel mot anfall (t.ex. levetiracetam) tills ICANS har gått över Inget samtidigt CRS:
För ICANS med samtidigt CRS:
| Avsluta epkoritamab permanent |
Grad 4a ICE-poänga, b 0 eller sänkt medvetandea, antingen:
anfalla, antingen:
eller motoriska fynda:
eller
| Behandling med dexametasonf
Överväg icke‑sederande läkemedel mot anfall (t.ex. levetiracetam) tills ICANS har gått över Inget samtidigt CRS:
För ICANS med samtidigt CRS:
| Avsluta epkoritamab permanent |
aICANS-grad enligt konsensusgradering för ICANS från ASTCT. ICANS-graden fastställs av den allvarligaste händelsen (ICE-poäng, medvetandenivå, anfall, motoriska fynd, förhöjt intrakraniellt tryck/cerebralt ödem) som inte kan tillskrivas någon annan orsak | ||
Tabell 7 Rekommenderade dosändringar för andra biverkningar
Biverkning1 | Allvarlighetsgrad1 | Åtgärd |
Infektioner (se avsnitt Varningar och försiktighet) | Grad 1–4 |
|
Neutropeni eller febril neutropeni (se avsnitt Biverkningar) | Absolut antal neutrofila granulocyter mindre än 0,5 x 109/liter |
|
Trombocytopeni (se avsnitt Biverkningar) | Trombocyttal mindre än 50 x 109/liter |
|
Andra biverkningar (se avsnitt Biverkningar) | Grad 3 eller högre |
|
1Baserat på National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), Version 5.0. | ||
Vid febril neutropeni och återkommande neutropeni av grad ≥ 3 ska granulocytkolonistimulerande faktor (G-CSF) övervägas. Specifika anvisningar om dosändringar vid neutropeni finns i produktresumén för lenalidomid. Rekommendationer beträffande fastställande av immunglobulinnivåer finns i produktresumén för rituximab.
Missad eller försenad dos
Diffust storcelligt B-cellslymfom
En ny upptrappningscykel (identisk med cykel 1 med standardprofylax mot CRS) krävs:
-
Om det är mer än 8 dagar mellan den förberedande dosen (0,16 mg) och den intermediära dosen (0,8 mg), eller
-
Om det är mer än 14 dagar mellan den intermediära dosen (0,8 mg) och den första fulla dosen (48 mg), eller
-
Om det är mer än 6 veckor mellan fulla doser (48 mg)
Efter den nya upptrappningscykeln ska patienten återuppta behandlingen med dag 1 i nästa planerade behandlingscykel (som kommer efter cykeln under vilken dosen försenades).
Follikulärt lymfom
En ny upptrappningscykel (identisk med cykel 1 med standardprofylax mot CRS) krävs:
-
Om det är mer än 8 dagar mellan den förberedande dosen (0,16 mg) och den intermediära dosen (0,8 mg), eller
-
Om det är mer än 8 dagar mellan den intermediära dosen (0,8 mg) och den andra intermediära dosen (3 mg), eller
-
Om det är mer än 14 dagar mellan den andra intermediära dosen (3 mg) och den första fulla dosen (48 mg), eller
-
Om det är mer än 6 veckor mellan två fulla doser (48 mg)
Efter den nya upptrappningscykeln ska patienten återuppta behandlingen med dag 1 i nästa planerade behandlingscykel (som kommer efter cykeln under vilken dosen försenades).
Särskilda populationer
Nedsatt njurfunktion
Dosjusteringar anses inte vara nödvändiga hos patienter med lindrigt till måttligt nedsatt njurfunktion. Epkoritamab har inte studerats hos patienter med svårt nedsatt njurfunktion eller terminal njursjukdom.
Inga dosrekommendationer kan göras för patienter med svårt nedsatt njurfunktion eller terminal njursvikt (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Dosjusteringar anses inte vara nödvändiga hos patienter med lindrigt nedsatt leverfunktion. Epkoritamab har inte studerats hos patienter med svårt nedsatt leverfunktion (definierat som totalt bilirubin > 3 gånger ULN oavsett ASAT) och data är begränsade för patienter med måttligt nedsatt leverfunktion (definierat som totalt bilirubin > 1,5 till 3 gånger ULN oavsett ASAT). Inga dosrekommendationer kan göras för patienter med måttligt till svårt nedsatt leverfunktion (se avsnitt Farmakokinetiska egenskaper).
Äldre
Ingen dosjustering krävs hos patienter i åldern ≥ 65 år (se avsnitt Farmakodynamiska egenskaper och Farmakokinetiska egenskaper).
Pediatrisk population
Säkerhet och effekt för Tepkinly hos barn som är yngre än 18 år har ännu inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Tepkinly är avsett för subkutan användning. Det ska endast administreras som en subkutan injektion, helst i nedre delen av buken eller låret. Det rekommenderas att byta injektionsställe från höger till vänster sida eller vice versa, särskilt vid veckovis administrering (dvs. cykel 1–3).
Anvisningar om spädning av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Cytokinfrisättningssyndrom (CRS)
CRS, som kan vara livshotande eller leda till dödsfall, förekom hos patienter som fick epkoritamab. De vanligaste tecknen och symtomen på CRS innefattar pyrexi, hypotoni och hypoxi. Andra tecken och symtom på CRS som förekom hos fler än två patienter innefattar frossa, takykardi, huvudvärk och andnöd.
De flesta CRS-händelserna förekom i cykel 1 och förknippades med den första fulla dosen epkoritamab. Administrera profylaktiska kortikosteroider för att minska risken för CRS (se avsnitt Dosering och administreringssätt).
Patienterna ska övervakas beträffande tecken och symtom på CRS efter administrering av epkoritamab.
Vid de första tecknen eller symtomen på CRS ska behandling med stödjande vård med tocilizumab och/eller kortikosteroider inledas, i tillämpliga fall (se avsnitt Dosering och administreringssätt, tabell 5). Patienterna ska informeras om tecknen och symtomen på CRS och patienterna ska instrueras att kontakta läkare och omedelbart söka läkarvård om sådana tecken eller symtom uppstår, oavsett när det händer. Behandlingen av CRS kan kräva att behandlingen med epkoritamab antingen tillfälligt försenas eller avbryts, beroende på allvarlighetsgraden av CRS (se avsnitt Dosering och administreringssätt).
Hemofagocyterande lymfohistiocytos (HLH)
Hemofagocyterande lymfohistiocytos (HLH), inklusive fatala fall, har rapporterats hos patienter som behandlats med epkoritamab. HLH är ett livshotande tillstånd som kännetecknas av feber, hudutslag, lymfadenopati, hepato- och/eller splenomegali och cytopenier. HLH bör övervägas vid atypisk eller förlängd CRS. Patienter ska övervakas beträffande kliniska tecken och symtom på HLH. Vid misstänkt HLH måste behandlingen med epkoritamab avbrytas för diagnostisk undersökning och behandling mot HLH påbörjas. Vid bekräftad HLH ska administreringen av Tepkinly avslutas.
Immuneffektorcells‑associerat neurotoxicitetssyndrom (ICANS)
ICANS, inklusive dödsfall, har förekommit hos patienter som har fått epkoritamab. ICANS kan manifesteras som afasi, förändrad medvetandenivå, nedsatt kognitiv förmåga, motorisk svaghet, anfall, och cerebralt ödem.
Majoriteten av ICANS-fallen uppträdde inom cykel 1 i behandlingen med epkoritamab, vissa hade dock en fördröjd debut.
Patienter ska övervakas beträffande tecken och symtom på ICANS efter administrering av epkoritamab. Vid de första tecknen eller symtomen på ICANS ska behandling med kortikosteroider och icke‑sederande läkemedel mot anfall inledas i tillämpliga fall (se avsnitt Dosering och administreringssätt, tabell 6). Patienterna ska informeras om tecknen och symtomen på ICANS och att debuten av symtomen kan vara fördröjd. Patienterna ska instrueras att kontakta läkare och omedelbart söka läkarvård om sådana tecken eller symtom uppstår, oavsett när det händer. Behandlingen med epkoritamab ska senareläggas eller avbrytas enligt rekommendationer (se avsnitt Dosering och administreringssätt).
Allvarliga infektioner
Behandling med epkoritamab kan leda till en ökad risk för infektioner. Allvarliga eller fatala infektioner har observerats hos patienter som behandlats med epkoritamab i kliniska studier (se avsnitt Biverkningar).
Administrering av epkoritamab ska undvikas hos patienter med kliniskt signifikanta aktiva systemiska infektioner.
Vid behov ska profylaktiska antimikrobiella medel administreras före och under behandling med epkoritamab (se avsnitt Dosering och administreringssätt). Patienter ska övervakas beträffande tecken och symtom på infektioner, före och efter administrering av epkoritamab, och behandlas på lämpligt sätt. Vid febril neutropeni ska patienter utvärderas beträffande infektion och behandlas med antibiotika, vätska och annan stödjande vård, enligt lokala riktlinjer.
Hypogammaglobulinemi har också rapporterats hos patienter som får epkoritamab (se avsnitt Biverkningar). Nivåerna av immunglobulin (Ig) ska övervakas före och under behandlingen. Patienterna ska behandlas i enlighet med lokala riktlinjer, inklusive försiktighetsåtgärder med avseende på infektioner och antimikrobiell profylax.
Fall av progressiv multifokal leukoencefalopati (PML), inklusive fatala fall, har rapporterats hos patienter som behandlats med epkoritamab och som tidigare behandlats med andra immunsuppressiva läkemedel. Om patienten får neurologiska symtom på PML under behandling med epkoritamab ska behandlingen med epkoritamab avslutas och lämpliga diagnostiska åtgärder initieras.
Tumörlyssyndrom (TLS)
TLS har rapporterats hos patienter som fått epkoritamab (se avsnitt Biverkningar). För patienter med ökad risk för TLS rekommenderas hydrering och profylaktisk behandling med ett urinsyrasänkande medel. Patienterna ska övervakas avseende tecken eller symtom på TLS, i synnerhet patienter med hög tumörbörda eller tumörer med snabb proliferation samt patienter med nedsatt njurfunktion. Patienternas blodstatus ska övervakas och avvikelser ska hanteras omgående.
Tumörexacerbationer (tumour flare)
Tumörexacerbationer har rapporterats hos patienter som behandlats med epkoritamab (se avsnitt Biverkningar). Manifestationer kan innefatta lokal smärta och svullnad. I överensstämmelse med epkoritamabs verkningsmekanism, beror tumörexacerbationer sannolikt på inflödet av T-celler i tumörområdet efter administrering av epkoritamab.
Inga specifika riskfaktorer för tumörexacerbationer har identifierats, men risken för försämrad hälsa och ökad morbiditet är förhöjd på grund av de sekundära effekterna av tumörexacerbationer hos patienter som har bulkiga tumörer i närheten av luftvägar och/eller vitala organ. Patienter som behandlas med epkoritamab ska övervakas och utvärderas avseende tumörexacerbationer nära kritiska anatomiska områden.
CD20-negativ sjukdom
Det finns begränsad data tillgänglig om patienter med CD20-negativ DLBCL och patienter med CD20-negativ FL som behandlas med epkoritamab och det är möjligt att patienter med CD20-negativ DLBCL och patienter med CD20-negativ FL kan ha mindre nytta jämfört med patienter med CD20-positiv DLBCL respektive patienter med CD20-positiv FL. De potentiella riskerna och fördelarna med behandling av patienter med CD20-negativ DLBCL och FL med epkoritamab bör övervägas.
Patientkort
Läkaren måste informera patienten om risken för CRS och ICANS och eventuella tecken eller symtom på CRS och ICANS. Patienter måste instrueras att omedelbart söka läkarvård om de upplever tecken eller symtom på CRS och/eller ICANS. Patienterna ska förses med ett patientkort och instrueras att alltid bära kortet. Detta kort beskriver symtom på CRS och ICANS och, om de upplevs, uppmanar patienten att omedelbart söka läkarvård.
Immunisering
Levande och/eller levande försvagade vaccin ska inte ges under behandling med epkoritamab. Inga studier har utförts med patienter som fått levande vaccin.
Hjälpämnen med känd effekt
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per injektionsflaska, d.v.s. är näst intill ”natriumfritt”.
Detta läkemedel innehåller 28,8 mg sorbitol per injektionsflaska motsvarande 27,33 mg/ml.
Detta läkemedel innehåller 0,42 mg polysorbat 80 per injektionsflaska motsvarande 0,4 mg/ml. Polysorbat 80 kan orsaka allergiska reaktioner.
Interaktioner
Inga interaktionsstudier har utförts.
Övergående förhöjning av vissa proinflammatoriska cytokiner orsakad av epkoritamab kan hämma CYP450-enzymaktivitet. När behandling med epkoritamab inleds hos patienter som behandlas med CYP450-substrat med ett smalt terapeutiskt index ska terapeutisk övervakning övervägas.
Fertilitet, graviditet och amning
Fertila kvinnor/preventivmedel hos kvinnor
Fertila kvinnor ska rådas att använda effektiv preventivmetod under behandling med epkoritamab och i minst 4 månader efter den sista dosen. Verifiera graviditetsstatus hos fertila kvinnor innan behandling med epkoritamab inleds.
Graviditet
På grund av dess verkningsmekanism kan epkoritamab orsaka fosterskador, inklusive B-cellslymfocytopeni och förändrat normalt immunsvar, vid administrering till gravida kvinnor. Det finns inga data från användningen av epkoritamab hos gravida kvinnor. Fortplantningsstudier på djur har inte utförts med epkoritamab. IgG1-antikroppar som epkoritamab kan passera placentan vilket leder till att fostret exponeras. Informera gravida kvinnor om den potentiella risken för fostret.
Epkoritamab rekommenderas inte under graviditet och till fertila kvinnor som inte använder preventivmedel.
Amning
Det är okänt om epkoritamab utsöndras i bröstmjölk eller dess effekt på mjölkproduktion. Eftersom det är känt att IgG-antikroppar finns i mjölk kan neonatal exponering för epkoritamab förekomma via bröstmjölk. Amning ska avbrytas under behandling med epkoritamab och i minst 4 månader efter den sista dosen.
Fertilitet
Inga fertilitetsstudier har utförts med epkoritamab (se avsnitt Prekliniska säkerhetsuppgifter). Effekten av epkoritamab på manlig och kvinnlig fertilitet är okänd.
Effekter på förmågan att framföra fordon och använda maskiner
Epkoritamab har stor effekt på förmågan att framföra fordon och använda maskiner. På grund av risken för ICANS finns det en risk att medvetandenivån påverkas hos patienter som behandlas med epkoritamab (se avsnitt Varningar och försiktighet). Patienter ska tillrådas att vara försiktiga när de kör, cyklar eller använder tunga eller potentiellt farliga maskiner (eller undvika det helt om de upplever symtom).
Biverkningar
Sammanfattning av säkerhetsprofilen
Epkoritamab som monoterapi
Säkerheten för epkoritamab utvärderades i den icke-randomiserade, enarmade studien GCT3013-01 med 382 patienter med recidiverat eller refraktärt storcelligt B-cellslymfom (N=167), follikulärt lymfom (N=129) och follikulärt lymfom (doseringsschema med tre upptrappningsdoser, N=86) efter två eller fler linjer av systemisk behandling och inkluderade alla patienter som tilldelades 48 mg-dosen och fick minst en dos epkoritamab.Följande biverkningar har rapporterats för epkoritamab under kliniska studier och efter påbörjad marknadsföring.
Medianvärdet för exponeringstiden för epkoritamab var 4,9 månader (intervall <1 till 30 månader).
De vanligaste biverkningarna (≥ 20 %) var CRS, reaktioner vid injektionsstället, trötthet, virala infektioner, neutropeni, muskuloskeletal smärta, pyrexi, och lös avföring.
Allvarliga biverkningar förekom hos 50 % av patienterna. Den vanligaste allvarliga biverkningen (≥ 10 %) var cytokinfrisättningssyndrom (34 %). 14 patienter (3,7 %) drabbades av en fatal biverkning (pneumoni hos 9 (2,4 %) patienter, virala infektioner hos 4 (1,0 %) patienter, och ICANS hos 1 (0,3 %) patient).
Biverkningar som ledde till att behandlingen avbröts förekom hos 6,8 % av patienterna. Avbruten behandling med epkoritamab på grund av pneumoni förekom hos 14 (3,7 %) patienter, virala infektioner hos 8 (2,1 %) patienter, trötthet hos 2 (0,5 %) patienter och CRS, ICANS eller lös avföring förekom hos 1 (0,3 %) patient vardera.
Uppskjutna doser på grund av biverkningar förekom hos 42 % av patienterna. Biverkningar som ledde till uppskjutna doser (≥ 3 %) var virala infektioner (17 %), CRS (11 %), neutropeni (5,2 %), pneumoni (4,7 %), övre luftvägsinfektion (4,2 %) och pyrexi (3,7 %).
Epkoritamab i kombination med lenalidomid och rituximab
Säkerheten för epkoritamab i kombination med lenalidomid och rituximab utvärderades i M20-638, en öppen, randomiserad multicenterstudie som inkluderade patienter med recidiverande eller refraktärt follikulärt lymfom (FL) efter en tidigare behandlingslinje. Patienter fick epkoritamab i kombination med lenalidomid och rituximab (N=243) eller endast lenalidomid och rituximab (N=238).
Med undantag för CRS och ICANS avser säkerhetsresultaten som presenteras nedan och i tabell 9 säkerhetsdata från sammanlagt 243 patienter som fick epkoritamab enligt doseringsschemat med två upptrappningsdoser eller det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab. Data som presenteras för CRS och ICANS representerar de 133 patienter som fick epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab.
Rekommenderat doseringsschema med tre upptrappningsdoser
I studie M20-638 förekom CRS av någon grad hos 26 % (35/133) av patienter som behandlades med epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab.
Allvarliga biverkningar på grund av CRS förekom hos 12 % av patienterna som fick epkoritamab i kombination med lenalidomid och rituximab. Uppskjutna doser (avbrott) av epkoritamab på grund av CRS förekom hos 11 % av patienterna. ICANS förekom hos 0,8 % av patienterna, med en enstaka händelse rapporterad som grad 1.
Total säkerhetsdata
Hos de 243 patienter som fick epkoritamab enligt doseringsschemat med två upptrappningsdoser eller det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab var de vanligaste (≥ 20 %) biverkningarna neutropeni, hudutslag, övre luftvägsinfektioner, trötthet, lös avföring, reaktioner vid injektionsstället, anemi, förstoppning, trombocytopeni, CRS, hypogammaglobulinemi, covid‑19, pyrexi och pneumoni.
Allvarliga biverkningar förekom hos 44 % av patienterna som fick epkoritamab i kombination med lenalidomid och rituximab. Allvarliga biverkningar hos ≥ 5 % av patienterna inkluderade CRS, pneumoni, covid‑19 och febril neutropeni.
Permanent utsättning av epkoritamab på grund av en biverkning förekom hos 6,6 % av patienterna som fick epkoritamab i kombination med lenalidomid och rituximab. Biverkningar som ledde till permanent utsättning av epkoritamab hos mer än 1 patient inkluderade pneumoni, covid‑19, övre luftvägsinfektioner och neutropeni.
Uppskjutna doser av epkoritamab på grund av en biverkning förekom hos 70 % av patienterna som fick epkoritamab i kombination med lenalidomid och rituximab. Biverkningar som ledde till uppskjutna doser av epkoritamab hos ≥ 5 % av patienterna inkluderade neutropeni, övre luftvägsinfektioner, covid‑19, pneumoni, hudutslag och trombocytopeni.
Lista över biverkningar i tabellform
Biverkningar som rapporterats med epkoritamab anges i tabell 8.
Biverkningar som rapporterats med epkoritamab i kombination med lenalidomid och rituximab anges i tabell 9.
Biverkningar för epkoritamab från kliniska studier (tabell 8 och tabell 9) anges efter MedDRA-organsystem och baseras på följande konvention: mycket vanliga (≥1/10), vanliga (≥1/100, <1/10), mindre vanliga (≥1/1 000, <1/100), sällsynta (≥1/10 000, <1/1 000) och mycket sällsynta (<1/10 000).
Inom varje frekvensgrupp anges biverkningarna efter fallande allvarlighetsgrad.
Tabell 8 Biverkningar som rapporterats hos patienter med recidiverande eller refraktärt LBCL eller FL som behandlades med epkoritamab
Klassificering av organsystem/föredragen term eller biverkning | Alla grader | Grad 3–4 | ||
Infektioner och infestationer | ||||
Virala infektionera | Mycket vanliga | Vanliga | ||
Pneumonib | Mycket vanliga | Vanliga | ||
Övre luftvägsinfektionc | Mycket vanliga | Vanliga | ||
Svampinfektiond | Vanliga |
| ||
Sepsise | Vanliga | Vanliga | ||
Cellulit | Vanliga | Vanliga | ||
Neoplasier; benigna, maligna och ospecificerade tumörer (inkl. cystor och polyper) | ||||
Tumörexacerbationer (tumour flare) | Vanliga |
| ||
Blodet och lymfsystemet | ||||
Neutropenif | Mycket vanliga | Mycket vanliga | ||
Anemig | Mycket vanliga | Vanliga | ||
Trombocytopenih | Mycket vanliga | Vanliga | ||
Lymfopenii | Mycket vanliga | Vanliga | ||
Febril neutropeni | Vanliga | Vanliga | ||
Hemofagocyterande lymfohistiocytosj | Mindre vanliga | Sällsynta | ||
Immunsystemet | ||||
Cytokinfrisättningssyndromj | Mycket vanliga | Vanliga | ||
Hypogammaglobulinemi | Mycket vanliga | Mindre vanliga | ||
Metabolism och nutrition | ||||
Minskad aptit | Mycket vanliga | Mindre vanliga | ||
Hypokalemi | Vanliga | Vanliga | ||
Hypofosfatemi | Vanliga | Vanliga | ||
Hypomagnesemi | Vanliga | Mindre vanliga | ||
Tumörlyssyndromk | Vanliga | Mindre vanliga | ||
Centrala och perifera nervsystemet | ||||
Huvudvärk | Mycket vanliga | Mindre vanliga | ||
Immuneffektorcells‑associerat neurotoxicitetssyndromj | Vanliga | Mindre vanliga | ||
Hjärtat | ||||
Hjärtrytmrubbningarl | Vanliga | Mindre vanliga | ||
Andningsvägar, bröstkorg och mediastinum | ||||
Pleural effusion | Vanliga | Vanliga | ||
Magtarmkanalen | ||||
Lös avföring | Mycket vanliga | Mindre vanliga | ||
Buksmärtam | Mycket vanliga | Vanliga | ||
Illamående | Mycket vanliga | Mindre vanliga | ||
Kräkningar | Vanliga | Mindre vanliga | ||
Hud och subkutan vävnad | ||||
Hudutslagn | Mycket vanliga |
| ||
Pruritus | Vanliga |
| ||
Muskuloskeletala systemet och bindväv | ||||
Muskuloskeletal smärtao | Mycket vanliga | Vanliga | ||
Allmänna symtom och/eller symtom vid administreringsstället | ||||
Reaktioner vid injektionsställetp | Mycket vanliga |
| ||
Trötthetq | Mycket vanliga | Vanliga | ||
Pyrexir | Mycket vanliga | Vanliga | ||
Ödems | Mycket vanliga | Vanliga | ||
Undersökningar och provtagningar | ||||
Förhöjt alaninaminotransferas | Vanliga | Vanliga | ||
Förhöjt aspartataminotransferas | Vanliga | Vanliga | ||
Ökning av blodkreatinin | Vanliga |
| ||
Minskat natrium i blodett | Vanliga | Mindre vanliga | ||
Förhöjt alkaliskt fosfatas | Vanliga |
| ||
Biverkningarna graderades med NCI CTCAE version 5.0 | ||||
Tabell 9 Biverkningar som rapporterats hos patienter med recidiverande eller refraktärt FL som behandlades med epkoritamab i kombination med lenalidomid och rituximab
Klassificering av organsystem/föredragen term eller biverkning | Alla grader | Grad 3–4 |
Infektioner och infestationer | ||
Övre luftvägsinfektionera | Mycket vanliga | Vanliga |
Covid‑19b | Mycket vanliga | Vanliga# |
Pneumonic | Mycket vanliga | Mycket vanliga |
Cytomegalovirusinfektiond | Vanliga | Vanliga# |
Herpesvirusinfektione | Vanliga | Mindre vanliga# |
Svampinfektionf | Vanliga | Mindre vanliga |
Blodet och lymfsystemet | ||
Neutropenig | Mycket vanliga | Mycket vanliga |
Anemih | Mycket vanliga | Vanliga# |
Trombocytopenii | Mycket vanliga | Vanliga |
Lymfopenij | Mycket vanliga | Mycket vanliga |
Febril neutropeni | Vanliga | Vanliga |
Immunsystemet | ||
Cytokinfrisättningssyndrom‡ | Mycket vanliga |
|
Hypogammaglobulinemik | Mycket vanliga | Mindre vanliga |
Psykiatriska tillstånd | ||
Insomni | Mycket vanliga |
|
Centrala och perifera nervsystemet | ||
Neurologiska förändringarl | Mycket vanliga |
|
Huvudvärk | Mycket vanliga |
|
Immuneffektorcells-associerat neurotoxicitetssyndrom‡ | Mindre vanliga |
|
Magtarmkanalen | ||
Lös avföring | Mycket vanliga | Vanliga# |
Förstoppning | Mycket vanliga | Mindre vanliga# |
Illamående | Mycket vanliga |
|
Mukositm | Vanliga |
|
Hud och subkutan vävnad | ||
Hudutslagn | Mycket vanliga | Mycket vanliga# |
Allmänna symtom och/eller symtom vid administreringsstället | ||
Tröttheto | Mycket vanliga | Vanliga# |
Reaktioner vid injektionsställetp | Mycket vanliga |
|
Pyrexi | Mycket vanliga | Mindre vanliga# |
Undersökningar och provtagningar | ||
Minskat kalium i blodq | Mycket vanliga | Vanliga# |
Förhöjt alaninaminotransferas | Mycket vanliga | Vanliga# |
Förhöjt aspartataminotransferas | Mycket vanliga | Vanliga# |
§Biverkningarna graderades med CTCAE version 5.0. | ||
Beskrivning av utvalda biverkningar
Cytokinfrisättningssyndrom
Epkoritamab som monoterapi
Doseringsschema med två upptrappningsdoser (storcelligt B-cellslymfom och follikulärt lymfom)
I studie GCT3013-01 förekom CRS av någon grad hos 58 % (171/296) av patienterna med storcelligt B-cellslymfom och follikulärt lymfom som behandlades med epkoritamab med doseringsschemat med två upptrappningsdoser. Incidensen av grad 1 var 35 %, grad 2 var 21 % och grad 3 förekom hos 2,4 % av patienterna. Återkommande CRS förekom hos 21 % av patienterna. CRS av någon grad förekom hos 9,8 % av patienterna efter den förberedande dosen (cykel 1, dag 1), 13 % efter den intermediära dosen (cykel 1, dag 8), 51 % efter den första fulla dosen (cykel 1, dag 15), 6,5 % efter den andra fulla dosen (cykel 1, dag 22) och 3,7 % efter den tredje fulla dosen (cykel 2, dag 1) eller därefter. Mediantiden till debuten av CRS från den senaste administrerade epkoritamabdosen var 2 dagar (intervall: 1 till 12 dagar). Mediantiden till debuten efter den första fulla dosen var 19,3 timmar (intervall: < 0,1 till 7 dagar). CRS gick över hos 99 % av patienterna och medianvaraktigheten av CRS-händelserna var 2 dagar (intervall 1 till 54 dagar).
Hos de 171 patienter som uppvisade CRS inkluderade de vanligaste tecknen och symtomen på CRS pyrexi (99 %), hypotoni (32 %) och hypoxi (16 %). Andra tecken och symtom på CRS som förekom hos ≥ 3% patienter inkluderade frossa (11 %), takykardi (inklusive sinustakykardi (11 %)), huvudvärk (8,2 %), illamående (4,7 %) och kräkningar (4,1 %). Övergående förhöjda leverenzymer (ALAT eller ASAT > 3xULN) förekom samtidigt med CRS hos 4,1 % av patienterna med CRS. Se avsnitt Dosering och administreringssätt och Varningar och försiktighet för vägledning om övervakning och behandling.
Doseringsschema med tre upptrappningsdoser vid follikulärt lymfom
I studie GCT3013-01 förekom CRS av någon grad hos 49 % (42/86) av patienterna som behandlades med epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser. Incidensen av grad 1 var 40 % och av grad 2 9 %. Inga CRS-händelser av grad ≥ 3 rapporterades. Återkommande CRS förekom hos 23 % av patienterna. Flertalet CRS-händelser inträffade under cykel 1, då 48 % av patienterna erfor en sådan händelse. I cykel 1 förekom CRS hos 12 % av patienterna efter den förberedande dosen (cykel 1, dag 1), 5,9 % av patienterna efter den intermediära dosen (cykel 1, dag 8), 15 % av patienterna efter den andra intermediära dosen (cykel 1, dag 15) och 37 % av patienterna efter den första fulla dosen (cykel 1, dag 22). Mediantiden till debut av CRS efter den senast administrerade epkoritamabdosen var 59 timmar (intervall: 1 till 8 dagar). Mediantiden till debut efter den första fulla dosen var 61 timmar (intervall: 1 till 8 dagar). CRS gick över hos 100 % av patienterna och medianvaraktigheten på CRS-händelserna var 2 dagar (intervall: 1 till 14 dagar).
Allvarliga biverkningar på grund av CRS förekom hos 28 % av patienterna som fick epkoritamab.
Uppskjutna doser på grund av CRS förekom hos 19 % av patienterna som fick epkoritamab.
Hos de 42 patienter som uppvisade CRS vid den rekommenderade dosen inkluderade de vanligaste (≥ 10 %) tecknen och symtomen på CRS pyrexi (100 %) och hypotoni (14 %). Utöver kortikosteroider användes tocilizumab för att hantera CRS-händelsen hos 12 % av patienterna.
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 förekom CRS av någon grad hos 26 % (35/133) av patienterna som behandlades med epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab. Incidensen av CRS av grad 1 var 21 % (28/133) och 5,3 % för grad 2 (7/133). Vid tidpunkten för analysen hade inga CRS-händelser av grad ≥ 3 rapporterats. Återkommande CRS förekom hos 10 % (13/133) av patienterna. Av alla CRS-händelser inträffade flertalet (88 %) under cykel 1. I cykel 1 förekom CRS hos 6 % (8/133) av patienterna efter den förberedande dosen (cykel 1, dag 1), 3,8 % (5/133) av patienterna efter den första intermediära dosen (cykel 1, dag 8), 2,3 % (3/132) av patienterna efter den andra intermediära dosen (cykel 1, dag 15) och 19 % (25/132) av patienterna efter den första fulla dosen (cykel 1, dag 22). Mediantiden till debut av CRS efter den senast administrerade epkoritamabdosen för alla doser var 78 timmar (intervall: 0,2 till 12 dagar). Mediantiden till debut efter den första fulla 48 mg-dosen var 41 timmar (intervall: 0,3 till 12 dagar). CRS gick över hos 100 % av patienterna och medianvaraktigheten på CRS-händelserna var 2 dagar (intervall: 0,1 till 26 dagar).
Immuneffektorcells-associerat neurotoxicitetssyndrom
Epkoritamab som monoterapi
I studie GCT3013-01 förekom ICANS hos 4,7 % (18/382) av patienterna som behandlades med epkoritamab, 3,1 % uppvisade grad 1 och 1,3 % uppvisade grad 2. En patient (0,3 %) uppvisade en ICANS-händelse av grad 5 (fatal). Mediantiden till första ICANS-debuten från starten av behandlingen med epkoritamab (cykel 1, dag 1) var 18 dagar (intervall: 8 till 141 dagar). ICANS gick över hos 94 % (17/18) av patienterna med stödjande vård. Mediantiden till när ICANS gått över var 2 dagar (intervall: 1 till 9 dagar). Hos de 18 patienterna med ICANS ägde debuten av ICANS rum före CRS hos 11 % av patienterna, samtidigt med CRS hos 44 %, efter debuten av CRS hos 17 % och i frånvaro av CRS hos 28 %.
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 förekom ICANS hos 0,8 % (1/133) av patienterna med FL som behandlades med epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser i kombination med lenalidomid och rituximab, med en enstaka händelse rapporterad som grad 1. Händelsen inträffade efter 48 mg-dosen på dag 22 i cykel 1 och ICANS gick över inom 3 dagar.
Allvarliga infektioner
Epkoritamab som monoterapi
Storcelligt B-cellslymfom
I studie GCT3013-01 förekom allvarliga infektioner av någon grad hos 25 % (41/167) av patienter med storcelligt B-cellslymfom som behandlades med epkoritamab. De mest frekventa allvarliga infektionerna inkluderade covid-19 (6,6 %), covid-19-pneumoni (4,2 %), pneumoni (3,6 %), sepsis (2,4 %), övre luftvägsinfektion (1,8 %), bakteriemi (1,2 %) och septisk chock (1,2 %). Mediantiden till debut av första allvarliga infektion från behandlingsstart med epkoritamab (cykel 1, dag 1) var 56 dagar (intervall: 4 till 631 dagar), med medianvaraktighet på 15 dagar (intervall: 4 till 125 dagar). Infektion av grad 5 förekom hos 7 (4,2 %) av patienterna.
Follikulärt lymfom
I studien GCT3013-01 förekom allvarliga infektioner av någon grad hos 32 % (68/215) av patienterna med follikulärt lymfom som behandlades med epkoritamab. De vanligaste allvarliga infektionerna inkluderade covid-19 (8,8 %), covid-19-pneumoni (5,6 %), pneumoni (3,7 %), urinvägsinfektion (1,9 %), och pneumocystis jirovecii-pneumoni (1,4 %). Mediantiden till debut av första allvarliga infektion från behandlingsstart med epkoritamab (cykel 1, dag 1) var 81 dagar (intervall: 1 till 636 dagar), med en medianduration på 18 dagar (intervall: 4 till 249 dagar). Infektion av grad 5 förekom hos 8 (3,7 %) av patienterna, varav 6 (2,8 %) ansågs ha orsakats av covid-19 eller covid-19-pneumoni.
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 rapporterades allvarliga infektioner, inklusive opportunistiska infektioner, hos 33 % (81/243) av patienterna med FL som behandlades med epkoritamab i kombination med lenalidomid och rituximab. De vanligaste var pneumoni (10 %), covid-19 (4,5 %) och covid-19-pneumoni (3,7 %). Mediantiden till debut av första allvarliga infektion från behandlingsstart med epkoritamab i kombination med lenalidomid och rituximab (cykel 1, dag 1) var 91 dagar (intervall: 2 till 418 dagar), med en medianduration på 13 dagar (intervall: 1 till 123 dagar).
Neutropeni
Epkoritamab som monoterapi
I studie GCT3013-01 förekom neutropeni av någon grad hos 28 % (105/382) av patienterna, inklusive 23 % grad 3–4-händelser. Mediantiden till debuten av den första händelsen med neutropeni/minskat antal neutrofila granulocyter var 65 dagar (intervall: 2 till 750 dagar) och händelsen varade i median 15 dagar (intervall: 2 till 415 dagar). Av de 105 patienterna som uppvisade neutropeni/minskat antal neutrofila granulocyter fick 61 % G‑CSF för att behandla händelserna.
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 förekom neutropeni av någon grad hos 74 % (180/243) av patienterna, inklusive 27 % (66/243) grad 3-händelser och 42 % (101/243) grad 4-händelser. Mediantiden till debuten av den första händelsen med neutropeni/minskat antal neutrofila granulocyter var 57 dagar (intervall: 2 till 377 dagar) och händelsen varade i median 22 dagar (intervall: 3 till 219 dagar). Av de 167 patienterna som uppvisade neutropeni av grad 3–4/minskat antal neutrofila granulocyter fick 87 % (146/167) G‑CSF för att behandla händelserna.
Tumörlyssyndrom
Epkoritamab som monoterapi
I studie GCT3013-01 förekom TLS förekom hos 1,0 % (4/382) av patienterna. Mediantiden till debut var 18 dagar (intervall: 8 till 33 dagar) och mediandurationen var 3 dagar (intervall: 2 till 4 dagar).
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 förekom laboratoriemässigt TLS hos en (1) patient. Händelsen inträffade på dag 10 i cykel 1 och gick över inom 6 dagar. Kliniskt TLS sågs inte hos patienter med FL som behandlades med epkoritamab i kombination med lenalidomid och rituximab.
Tumörexacerbationer (Tumour Flare)
Epkoritamab som monoterapi
I studie GCT3013-01 förekom tumörexacerbationer hos 1,6 % (6/382) av patienterna, alla av dem var av typen grad 2. Mediantiden till debut var 19,5 dagar (intervall: 9 till 34 dagar) och händelsen varade i median 9 dagar (intervall: 1 till 50 dagar).
Epkoritamab i kombination med lenalidomid och rituximab
I studie M20-638 förekom tumörexacerbationer hos 1,2 % (3/243) av patienterna, 0,8 % (2/243) upplevde grad 2 och 0,4 % (1/243) grad 3. Mediantiden till debut var 8 dagar (intervall: 7 till 20 dagar) och medianvaraktigheten var 7,5 dagar (intervall: 3 till 12 dagar).
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
I händelse av överdos ska patienten övervakas beträffande tecken eller symtom på biverkningar och ges lämplig understödjande behandling.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel, övriga monoklonala antikroppar och antikroppsläkemedelskonjugat, ATC-kod: L01FX27
Verkningsmekanism
Epkoritamab är en humaniserad IgG1-bispecifik antikropp som binder till en specifik extracellulär epitop hos CD20 på B-celler och till CD3 på T-celler. Epkoritamab verkar genom att samtidigt binda till CD20-uttryckande cancerceller och CD3-uttryckande endogena T-celler, vilket inducerar specifik T-cellsaktivering och T-cellsmedierat dödande av CD20-uttryckande celler.
Fc (kristalliserbart fragment)-regionen för epkoritamab är tystad för att förhindra måloberoende immuneffektormekanismer, till exempel antikroppsberoende cellulär cytotoxicitet (ADCC), komplementberoende cellulär cytotoxicitet (CDC) och antikroppsberoende cellulär fagocytos (ADCP).
I prekliniska studier ledde kombinationen av epkoritamab och rituximab inte till funktionella störningar och resulterade i kompletterande NK-cellsmedierad antikroppsberoende cellulär cytotoxicitet (ADCC) och T-cellsmedierad cytotoxicitet.
Farmakodynamisk effekt
Epkoritamab inducerar snabb och varaktig utarmning av cirkulerande B-celler (definierat som CD19 B-cellsantal ≤ 10 celler/µl) hos patienter med detekterbara B-celler vid behandlingsstart. 21 % av patienterna (n=33) med DLBCL och 50 % av patienterna (n=56) med FL hade detekterbara cirkulerande B-celler vid behandlingsstart. En övergående minskning av cirkulerande T-celler observerades omedelbart efter varje dos i cykel 1 och följdes av T-cellsexpansion i efterföljande cykler.
I studie GCT3013-01 förekom, efter subkutan administrering av epkoritamab enligt det rekommenderade doseringsschemat med två upptrappningsdoser till patienter med LBCL, övergående och något förhöjda nivåer av cirkulerande utvalda cytokiner (IFN‑γ, TNFα, IL‑6, IL‑2 och IL‑10) huvudsakligen efter den första fulla dosen (48 mg), med toppnivåer mellan 1 till 4 dagar efter dosen. Cytokinnivåerna återgick till baslinjen före nästa fulla dos. Dock kunde även förhöjda nivåer av cytokiner observeras efter cykel 1.
I studie GCT3013-01 förblev, efter subkutan administrering av epkoritamab enligt det rekommenderade doseringsschemat med tre upptrappningsdoser till patienter med FL, mediannivåerna av IL-6 som associeras med risk för CRS stadigt låga efter varje dos i cykel 1 och därefter, i synnerhet efter den första fulla dosen, jämfört med patienter vars dos trappades upp i två steg.
Immunogenicitet
Anti-läkemedelsantikroppar (ADA) detekterades ofta. Incidensen av behandlingsrelaterad ADA vid doseringsschemat med två upptrappningsdoser (0,16/0,8/48 mg) i den kombinerade populationen med DLBCL och FL var 3,4 % (3,4 % positiva, 93,9 % negativa och 2,7 % obestämda, N = 261 utvärderbara patienter) och 3,3 % (3,3 % positiva, 95 % negativa och 1,7 % obestämda, N = 60 utvärderbara patienter), i studie GCT3013-01 respektive GCT3013-04.
Incidensen av behandlingsrelaterad ADA vid doseringsschemat med tre upptrappningsdoser (0,16/0,8/3/48 mg) i FL-optimeringskohorten var 7 % (7 % positiva, 91,5 % negativa och 1,4 % obestämda, N = 71 utvärderbara patienter) i studie GCT3013-01. En patient klassificeras som obestämd om patienten är bekräftat ADA-positiv vid baslinjen men det inte finns något bekräftat positivt resultat registrerat i journalen under behandling eller om titrarna under behandling hos bekräftat ADA-positiva patienter registrerats som lika med eller lägre än vid baslinjen.
Anti-epkoritamab-antikroppar utvecklades hos 2,1 % av patienterna (5 av 238) med FL som behandlades med epkoritamab i kombination med lenalidomid och rituximab i studien M20-638 (upp till 12 cykler).
Inga bevis för att ADAs påverkade farmakokinetik, effekt eller säkerhet observerades, dock är data fortfarande begränsad. Neutraliserande antikroppar utvärderades inte.
Klinisk effekt och säkerhet
Diffust storcelligt B-cellslymfom
Studie GCT3013‑01 var en öppen, enarmad multicenterstudie med flera kohorter som utvärderade epkoritamab som monoterapi hos patienter med recidiverat eller refraktärt storcelligt B-cellslymfom (LBCL) efter två eller fler linjer av systemisk behandling, inklusive diffust storcelligt B-cellslymfom (DLBCL). Studien innefattade en dosupptrappningsdel och en expansionsdel. Studiens expansionsdel innefattade en kohort med aggressiva non-Hodgkins lymfom (aNHL), en kohort med indolenta non-Hodgkins lymfom (iNHL) och en kohort med mantelcellslymfom (MCL). Den pivotala aNHL-kohorten bestod av patienter med LBCL (N = 157), inklusive patienter med DLBCL (N = 139, 12 av dessa patienter hade rearrangemang av MYC, BCL2 och/eller BCL6, dvs. dubbelpositivt/trippelpositivt), med höggradigt B-cellslymfom (HGBCL) (N = 9), med follikulärt lymfom (FL) grad 3B (N = 5) och patienter med primärt mediastinalt B-cellslymfom (PMBCL) (N = 4). I DLBCL-kohorten hade 29 % (40/139) av patienterna transformerat DLBCL till följd av indolent lymfom. Enligt WHO-klassificering 2016 eller 2008 var kriterierna, för patienterna som inkluderades, att de hade dokumenterad CD20+ mogen B-cellsneoplasi baserat på en representativ patologirapport, att tidigare autolog hematopoetisk stamcellstransplantation (HSCT) inte hade fungerat eller att de inte var lämpliga för autolog HSCT, att de hade lymfocytantal < 5 × 109/l och att de hade fått minst en tidigare behandling med monoklonala anti‑CD20-antikroppar.
Studien uteslöt patienter med lymfom i centrala nervsystemet (CNS), tidigare behandling med allogen HSCT eller transplantation av solida organ, pågående kroniska infektionssjukdomar, patienter med känd nedsatt T-cellsimmunitet, kreatininclearance på mindre än 45ml/min, alaninaminotransferas > 3 gånger övre normalgränsen, kardiell ejektionsfraktion mindre än 45 % och känd kliniskt signifikant hjärt- och kärlsjukdom. Effektiviteten utvärderades i 139 patienter med DLBCL som fått minst en dos epkoritamab s.c. i 4-veckorscykler, dvs. 28 dagar. Epkoritamab som monoterapi administrerades enligt det rekommenderade doseringsschemat med två upptrappningsdoser på följande sätt:
-
Cykel 1: epkoritamab 0,16 mg på dag 1, 0,8 mg på dag 8, 48 mg på dag 15 och dag 22
-
Cykel 2–3: epkoritamab 48 mg på dag 1, 8, 15 och 22
-
Cykel 4–9: epkoritamab 48 mg på dag 1 och 15
-
Cykel 10 och därefter: epkoritamab 48 mg på dag 1
Patienterna fortsatte få epkoritamab till dess att sjukdomsprogress eller oacceptabel toxicitet inträffade.
Demografiska data och baslinjedata visas i tabell 10.
Tabell 10 Demografiska data och baslinjedata hos patienter med DLBCL i studie GCT3013‑01
Egenskaper | (N = 139) |
Ålder | |
Median, år (min, max) | 66 (22, 83) |
< 65 år, n (%) | 66 (47) |
65 till < 75 år, n (%) | 44 (32) |
≥ 75 år, n (%) | 29 (21) |
Män, n (%) | 85 (61) |
Etnicitet n (%) | |
Vit | 84 (60) |
Asiatisk | 27 (19) |
Annan | 5 (4) |
Ej rapporterat | 23 (17) |
ECOG-skattningskala, n (%) | |
0 | 67 (48) |
1 | 67 (48) |
2 | 5 (4) |
Sjukdomsstadiumc vid initial diagnos, n (%) | |
III | 16 (12) |
IV | 86 (62) |
Antal tidigare linjer av lymfombehandling | |
Median (min, max) | 3 (2, 11) |
2, n (%) | 41 (30) |
3, n (%) | 47 (34) |
≥ 4, n (%) | 51 (37) |
Sjukdomshistoria DLBCL, n (%) | |
De novo DLBCL | 97 (70) |
DLBCL transformerat från indolent lymfom, n (%) | 40 (29) |
FISH-analys av centralt labbd, N = 88 | |
Dubbelpositivt/trippelpositivt lymfom, n (%) | 12 (14) |
Tidigare autolog HSCT | 26 (19) |
Tidigare behandling, n (%) | |
Tidigare CAR‑T | 53 (38) |
Primär refraktär sjukdoma | 82 (59) |
Refraktär mot ≥ 2 på varandra följande linjer av tidigare lymfombehandlingb | 104 (75) |
Refraktär mot sista linjen av systemisk antineoplastisk behandlingb | 114 (82) |
Refraktär mot tidigare anti-CD20-behandling | 117 (84) |
Refraktär mot CAR‑T | 39 (28) |
aEn patient anses vara primärt refraktär om patienten är refraktär mot första linjens behandling av lymfom. | |
Det primära effektmåttet var total responsfrekvens (ORR) enligt Lugano-kriterierna (2014), utifrån bedömning av den oberoende granskningskommittén (Independent Review Committee, IRC). Medianvärdet för uppföljningstiden var 15,7 månader (intervall 0,3 till 23,5 månader). Exponeringen varade i median 4,1 månader (intervall 0 till 23 månader).
Tabell 11 Effektresultat i studie GCT3013‑01 hos patienter med DLBCLa
Effektmått | Epkoritamab |
ORRb, n (%) | 86 (62) |
(95 % KI) | (53,3; 70) |
CRb, n (%) | 54 (39) |
(95 % KI) | (30,7; 47,5) |
PR, n (%) | 32 (23) |
(95 % KI) | (16,3; 30,9) |
DORb |
|
Median (95 % KI), månader | 15,5 (9,7; NR) |
DOCRb |
|
Median (95 % KI), månader | NR (12,0; NR) |
TTR, median (intervall), månader | 1,4 (1; 8,4) |
KI = konfidensintervall, CR = komplett respons, DOR = responsduration, DOCR = duration av komplett respons, IRC = oberoende granskningskommitté, ORR = total responsfrekvens, PR = partiell respons, TTR = tid till respons | |
Mediantiden till CR var 2,6 månader (intervall 1,2 till 10,2 månader).
Follikulärt lymfom
M20-638
Studie M20-638 var en öppen, randomiserad multicenterstudie som utvärderade epkoritamab i kombination med lenalidomid och rituximab hos patienter med recidiverande eller refraktärt follikulärt lymfom (FL) efter en tidigare behandlingslinje. Patienter randomiserades att få epkoritamab i kombination med lenalidomid och rituximab eller endast lenalidomid och rituximab. Studien inkluderade patienter med histologiskt bekräftat klassiskt FL (tidigare grad 1 till 3a FL) i stadium II, III eller IV utan evidens för histologisk transformation till ett aggressivt lymfom och CD20+ sjukdom baserat på den senaste representativa patologirapporten, enligt WHO-klassificering av 5:e upplagan, R/R sjukdom efter behandling med minst en tidigare regim för lymfom som innehöll en monoklonal anti-CD20-antikropp i kombination med kemoterapi, Eastern Cooperative Oncology Group-skattningsskala (ECOG) 0 till 2, ingen dokumenterad refraktäritet mot lenalidomid, ingen exponering för lenalidomid inom 12 månader före randomisering samt behov av behandlingsstart enligt prövarens bedömning baserat på symtom och/eller sjukdomsbörda (t.ex. GELF-kriterier). Studien uteslöt patienter med känt CNS-engagemang av lymfom, tidigare allograft, känd aktiv infektion, känd nedsatt T‑cellsimmunitet, kreatininclearance < 50 ml/min, alaninaminotransferas > 3 gånger övre normalgränsen och kliniskt signifikant hjärt- och kärlsjukdom. Patienter fick epkoritamab i 28-dagarscykler i totalt 12 cykler eller tills sjukdomsprogress eller oacceptabel toxicitet, beroende på vilket som inträffade först.
Det rekommenderade doseringsschemat med tre upptrappningsdoser av epkoritamab var
-
cykel 1: epkoritamab 0,16 mg på dag 1, 0,8 mg på dag 8, 3 mg på dag 15 och 48 mg på dag 22
-
cykel 2–3: epkoritamab 48 mg på dag 1, 8, 15 och 22
-
cykel 4–12: epkoritamab 48 mg på dag 1
I båda behandlingsarmarna gavs lenalidomid peroralt i dosen 20 mg en gång dagligen från dag 1 till 21 i 12 cykler medan rituximab administrerades intravenöst i dosen 375 mg/m2 på dag 1, 8, 15 och 22 i cykel 1, följt av administrering på dag 1 i cykel 2 till 5.
Demografi och sjukdomsrelaterade karakteristika vid baslinjen som visas i tabell 12 är baserade på intent-to-treat-populationen (ITT).
Tabell 12 Demografi och sjukdomsrelaterade karakteristika vid baslinjen för patienter med recidiverande eller refraktärt FL i studie M20-638
Parameter | epkoritamab + lenalidomid och rituximab (N=243) | lenalidomid och rituximab (N=245) |
Ålder, år |
| |
Median (intervall) | 60 (30, 84) | 63 (24, 89) |
Åldersfördelning, n (%) |
| |
< 65 år | 155 (64) | 139 (57) |
65 till < 75 år | 68 (28) | 71 (29) |
≥ 75 år | 20 (8) | 35 (14) |
Kön, n (%) |
| |
Man | 139 (57) | 138 (56) |
Etnicitet, n (%) |
| |
Vit | 168 (71) | 184 (76) |
Asiatisk | 63 (27) | 54 (22) |
Svart eller afroamerikansk | 6 (3) | 2 (0,8) |
Amerikansk indian eller Alaskas ursprungsbefolkning | 0 | 1 (0,4) |
Flera | 1 (0,4) | 1 (0,4) |
Saknas | 5 | 3 |
ECOG-skattningsskala, n (%) |
| |
0 | 166 (68) | 170 (69) |
1 | 72 (30) | 68 (28) |
2 | 5 (2) | 7 (3) |
Ann Arbor-stadium, n (%) |
| |
II | 37 (15) | 44 (18) |
III | 74 (31) | 68 (28) |
IV | 132 (54) | 133 (54) |
Bulkig sjukdoma, n (%) | 76 (32) | 84 (35) |
FLIPI-poäng vid baslinjen, n (%) |
| |
0-1 | 63 (26) | 56 (23) |
2 | 79 (33) | 76 (31) |
3-5 | 100 (41) | 113 (46) |
Antal tidigare behandlingslinjer |
| |
Median (min, max) | 1 (1, 7) | 1 (1, 6) |
1, n (%) | 145 (60) | 141 (58) |
2, n (%) | 58 (24) | 61 (25) |
≥ 3, n (%) | 40 (17) | 43 (18) |
Tidigare SCT, n (%) | 23 (10) | 18 (7) |
Refraktär mot senaste behandlingslinje, n (%) | 84 (35) | 82 (34) |
Refraktär mot både anti-CD20-behandling och alkylerande medel, n (%) | 91 (37) | 91 (37) |
POD24, n (%) | 106 (44) | 93 (38) |
ECOG = Eastern Cooperative Oncology Group; FLIPI = Follicular Lymphoma International Prognostic Index; SCT = stamcellstransplantation; POD24 = sjukdomsprogress inom 24 månader. | ||
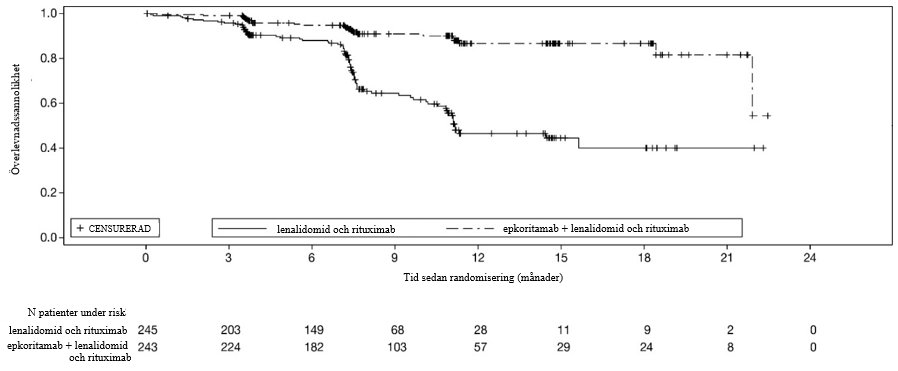
Effekten fastställdes baserat på dubbla primära effektmått; progressionsfri överlevnad (PFS) och total responsfrekvens (ORR) enligt Lugano-kriterierna från 2014, utifrån bedömning av den oberoende granskningskommittén (IRC). Ytterligare effektmått inkluderade komplett respons (CR) och total överlevnad (OS). Studie M20-638 visade en statistiskt signifikant förbättring av både PFS och ORR genom IRC-bedömning för epkoritamab i kombination med lenalidomid och rituximab jämfört med endast lenalidomid och rituximab. I ITT-populationen var medianvärdet för uppföljningstiden i studien för patienter som randomiserades att få epkoritamab i kombination med lenalidomid och rituximab 14,8 månader (intervall: 0 till 31). Medianvärdet för uppföljningstiden i studien för patienter som randomiserades att få enbart lenalidomid och rituximab var 14,6 månader (intervall: 0 till 30). Effektresultaten sammanfattas i tabell 13.
Tabell 13: Effektresultat i M20-638 hos patienter med recidiverande eller refraktärt FL
Effektmåtta | epkoritamab + lenalidomid och rituximab | lenalidomid och rituximab |
| (N=243) | (N=245) |
PFSb |
| |
Antal händelser, n (%) | 23 (9) | 75 (31) |
Progressiv sjukdom | 19 (83) | 63 (84) |
Dödsfall | 4 (17) | 12 (16) |
Median (95 % KI), månader | NR (21,9; NR) | 11,2 (10,5; NR ) |
Riskkvotc (95 % KI) | 0,21 (0,13; 0,33) | |
P-värded | < 0,0001 | |
ORRb,e, n (%) | 111 (96) | 94 (81) |
(95 % KI) | (90,2; 98,6 ) | (72,7; 87,7) |
P-värdef | < 0,0001 | |
CRRb, n (%) | 181(74) | 106 (43) |
(95 % KI) | (68,5; 79,8 ) | (37,0; 49,7 ) |
P-värdef | < 0,0001 | |
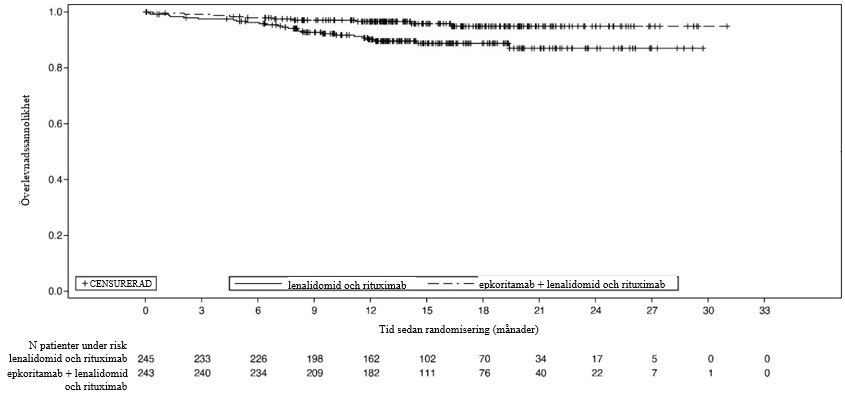
OS |
| |
Antal händelser, n (%) | 10 (4,1) | 25 (10,2) |
Median (95 % KI), månader | NR (NR; NR) | NR (NR; NR) |
HR (95 % KI)g | 0,38 (0,18; 0,80) | |
PFS = progressionsfri överlevnad; KI = konfidensintervall; NR = nåddes inte; CRR = komplett responsfrekvens; ORR = total responsfrekvens; OS = total överlevnad; HR = riskkvot | ||
Kaplan-Meier-kurvan över PFS som fastställts enligt Lugano-kriterierna (2014) utifrån IRC-bedömning visas i figur 1.
Figur 1: Kaplan-Meier-diagram över PFS (ITT-population) i studie M20-638
Kaplan-Meier-kurvan över OS visas i figur 2.
Figur 2: Kaplan-Meier-diagram över OS (ITT-population) i studie M20-638
GCT3013-01
Studie GCT3013-01 var en öppen, enarmad multicenterstudie med flera kohorter som utvärderade epkoritamab som monoterapi hos patienter med recidiverande eller refraktärt follikulärt lymfom (FL) efter två eller fler linjer av systemisk behandling. Studien innefattade en dosupptrappningsdel, en expansionsdel och en dosoptimeringsdel med tre upptrappningsdoser. Studiens expansionsdel innefattade en kohort med aggressiva non-Hodgkins lymfom (aNHL), en kohort med indolenta non-Hodgkins lymfom (iNHL) och en kohort med mantelcellslymfom (MCL). Den pivotala iNHL-kohorten inkluderade patienter med FL. Enligt WHO-klassificering 2016 eller 2008 var kriterierna, för patienterna som inkluderades, att de hade dokumenterad CD20+ mogen B-cellsneoplasi baserat på en representativ patologirapport med histologiskt bekräftat FL 1–3A vid den initiala diagnosen utan klinisk eller patologisk evidens för transformation. Alla patienter hade recidiverande eller refraktär sjukdom efter den senast föregående behandlingslinjen och hade tidigare behandlats med minst två linjer av systemisk antineoplastisk behandling, inklusive minst en behandling med monoklonala anti‑CD20-antikroppar och ett alkylerande medel eller lenalidomid. Studien uteslöt patienter med lymfom i centrala nervsystemet (CNS), allogen HSCT eller transplantation av solida organ, pågående aktiva infektionssjukdomar, patienter med känd nedsatt T-cellsimmunitet, kreatininclearance på mindre än 45ml/min, alaninaminotransferas > 3 gånger övre normalgränsen och kardiell ejektionsfraktion på mindre än 45 %. Effekten utvärderades hos 128 patienter som fått epkoritamab s.c. i 4-veckorscykler, dvs. 28 dagar. Epkoritamab som monoterapi administrerades enligt det rekommenderade doseringsschemat med två upptrappningsdoser på följande sätt:
-
Cykel 1: epkoritamab 0,16 mg på dag 1, 0,8 mg på dag 8, 48 mg på dag 15 och 48 mg på dag 22
-
Cykel 2–3: epkoritamab 48 mg på dag 1, 8, 15 och 22
-
Cykel 4–9: epkoritamab 48 mg på dag 1 och 15
-
Cykel 10 och därefter: epkoritamab 48 mg på dag 1
Patienterna fortsatte få epkoritamab till dess att sjukdomsprogress eller oacceptabel toxicitet inträffade.
Medianantalet cykler som initierades var 8 och 60 % genomgick 6 cykler.
Demografiska data och baslinjedata visas i tabell 14.
Tabell 14 Demografiska data och baslinjedata hos patienter med FL i studie GCT3013-01
Egenskaper | (N = 128) |
Ålder |
|
Median, år (min, max) | 65 (39, 84) |
< 65 år, n (%) | 61 (48) |
65 till < 75 år, n (%) | 50 (39) |
≥ 75 år, n (%) | 17 (13) |
Män, (%) | 79 (62) |
Etnicitet, n (%) |
|
Vit | 77 (60) |
Asiatisk | 7 (6) |
Annan | 2 (1,6) |
Ej rapporterat | 42 (33) |
ECOG-skattningsskala; n (%) |
|
0 | 70 (55) |
1 | 51 (40) |
2 | 7 (6) |
Antal tidigare behandlingslinjer, n (%) |
|
Median (min, max) | 3 (2, 9) |
2 | 47 (37) |
3 | 41 (32) |
≥4 | 40 (31) |
Ann Arbor-stadieindelning; (%) |
|
Stadium III/IV | 109 (85) |
FLIPI vid baslinjen, n (%) |
|
2 | 31 (24) |
3–5 | 78 (61) |
Bulkig sjukdom, n (%) | 33 (26) |
|
|
Tidigare behandling; n (%) |
|
Autolog stamcellstransplantation | 24 (19) |
T-cellsbehandling med chimär antigenreceptor (CAR-T) | 6 (5) |
Behandling med rituximab plus lenalidomid | 27 (21) |
PI3K-hämmare | 29 (23) |
Sjukdomsprogression inom 24 månader efter första systemiska behandling | 67 (52) |
Refraktär mot: |
|
≥ 2 på varandra följande linjer av tidigare lymfombehandling | 70 (55) |
Sista linjen av systemisk antineoplastisk behandling | 88 (69) |
Tidigare behandling med monoklonala anti-CD20-antikroppar | 101 (79) |
Behandling med både monoklonala anti-CD20-antikroppar och alkylerande medel | 90 (70) |
Effekten fastställdes baserat på total responsfrekvens (ORR) enligt Lugano-kriterierna (2014), utifrån bedömning av den oberoende granskningskommittén (Independent Review Committee, IRC). Medianvärdet för uppföljningstiden var 16,2 månader. Effektresultaten har sammanställts i tabell 15.
Tabell 15 Effektresultat i studie GCT3013-01 hos patienter med FL
Effektmått a | Epkoritamab |
ORRb , n (%) | 106 (83) |
(95 % KI) | (75,1; 88,9) |
CRb, n (%) | 81 (63) |
(95 % KI) | (54,3; 71,6) |
PRb, n (%) | 25 (20) |
(95 % KI) | (13,1; 27,5) |
DORb |
|
Median (95 % KI), månader | 21,4 (13,7; NR) |
DOCRb |
|
Median (95 % KI), månader | NR (21,4; NR) |
Uppskattning vid 12 månader, % (95 % KI) | 78,6 (67,3; 86,4) |
TTR, median (intervall), månader | 1,4 (1; 3) |
KI = konfidensintervall; CR = komplett respons; DOR = responsduration; DOCR = duration av komplett respons; IRC = oberoende granskningskommitté; ORR = total responsfrekvens; PFS = progressionsfri överlevnad; TTR = tid till respons | |
Mediantiden till CR var 1,5 månader (intervall 1,2 till 11,1 månader).
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för epkoritamab för en eller flera grupper av den pediatriska populationen för behandling av mogna B‑cellsmaligniteter, som definieras i gällande Paediatric Investigation Plan (PIP), för godkänd indikation (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Villkorat godkännande för försäljning
Detta läkemedel har godkänts enligt reglerna om ”villkorat godkännande för försäljning”. Detta innebär att det ska inkomma ytterligare evidens för detta läkemedel. Europeiska läkemedelsmyndigheten går igenom ny information om detta läkemedel minst varje år och uppdaterar denna produktresumé när så behövs.
Farmakokinetiska egenskaper
Epkoritamab som monoterapi
Populationsfarmakokinetiken efter subkutan administrering av epkoritamab utvärderades med en två-kompartementmodell med första ordningens subkutan absorption och målmedierad läkemedelseliminering. Den måttliga till höga farmakokinetiska variabiliteten för epkoritamab observerades och kännetecknades av interindividuell variabilitet (IIV) med variationskoefficienter från 25,7 % till 137,5 % för farmakokinetikparametrar för epkoritamab.
Hos patienter med LBCL i studie GCT3013-01, baserat på individuellt uppskattade exponeringar med populationsfarmakokinetisk modellering och enligt det rekommenderade doseringsschemat med två subkutana upptrappningsdoser av epkoritamab 48 mg, är det geometriska medelvärdet (% CV) för Cmax för epkoritamab 10,8 mikrogram/ml (41,7 %) och AUC0‑7d är 68,9 dag*mikrogram/ml (45,1 %) i slutet av det veckovisa doseringsschemat. Ctrough vid vecka 12 är 8,4 (53,3 %) mikrogram/ml. Det geometriska medelvärdet (% CV) för Cmax för epkoritamab är 7,52 mikrogram/ml (41,1 %) och AUC0‑14d är 82,6 dag*mikrogram/ml (49,3 %) i slutet av q2w-schemat (varannan vecka). Ctrough för q2w-schemat är 4,1 (73,9 %) mikrogram/ml. Det geometriska medelvärdet (% CV) för Cmax för epkoritamab är 4,76 mikrogram/ml (51,6 %) och AUC0‑28d är 74,3 dag*mikrogram/ml (69,5 %) vid steady-state under q4w-schemat (var fjärde vecka). Ctrough för q4w-schemat är 1,2 (130 %) mikrogram/ml.
Exponeringsparametrarna för epkoritamab hos patienter med FL överensstämde med de exponeringsparametrar som sågs hos patienter med LBCL. Exponeringen för epkoritamab är likartad för FL-patienter som följde doseringsschemat med tre upptrappningsdoser och doseringsschemat med två upptrappningsdoser, med undantag för övergående lägre dalkoncentrationer, som förväntat, på dag 15 i cykel 1 efter den andra intermediära dosen (3 mg) i doseringsschemat med tre upptrappningsdoser jämfört med den första fulla 48 mg-dosen i doseringsschemat med två upptrappningsdoser.
Absorption
Maximala koncentrationer förekom omkring 3–4 dagar (Tmax) hos patienter med LBCL som fick den fulla dosen på 48 mg.
Distribution
Det geometriska medelvärdet (% CV) för den centrala distributionsvolymen är 8,27 liter (27,5 %) och den skenbara distributionsvolymen vid steady-state är 25,6 liter (81,8 %) baserat på populationsfarmakokinetisk modellering.
Metabolism
Metabolismen för epkoritamab har inte studerats direkt. Liksom andra terapeutiska proteiner förväntas epkoritamab brytas ned till små peptider och aminosyror via katabolism.
Eliminering
Epkoritamab förväntas genomgå mättnadsbar målmedierad clearance. Det geometriska medelvärdet (% CV) för clearance (liter/dag) är 0,441 (27,8 %). Halveringstiden för epkoritamab är koncentrationsberoende. Det geometriska medelvärdet för halveringstiden, som härletts från den populationsfarmakokinetiska modellen, för full dos av epkoritamab (48 mg) varierade från 22 till 25 dagar baserat på doseringsfrekvensen.
Epkoritamab i kombination med lenalidomid och rituximab
Det fanns inga kliniskt signifikanta skillnader i farmakokinetiken för epkoritamab (cykel 1 AUC och cykel 1–3 Cavg med en skillnad i geometriskt medelvärde inom 2 % respektive 23,7 %) när epkoritamab administrerades i kombination med lenalidomid och rituximab jämfört med epkoritamab administrerat som monoterapi.
Speciella populationer
Inga kliniskt viktiga effekter på farmakokinetiken för epkoritamab (cykel 1 AUC inom cirka 36 %) observerades baserat på ålder (20 till 89 år), kön eller etnicitet (vit, asiatisk eller annan), kreatininclearance vid mild till måttligt nedsattnjurfunktion (CLcr ≥ 30 ml/min till CLcr < 90 ml/min) och milt nedsatt leverfunktion (total bilirubin≤ ULN och ASAT > ULN eller total bilirubin 1 till 1,5 gånger ULN oavsett ASAT) efter att ha tagit hänsyn till skillnader i kroppsvikt. Inga patienter med svår eller terminal njursjukdom (CLcr < 30 ml/min) eller svår nedsatt leverfunktion (total bilirubin > 3 gånger ULN oavsett ASAT) har studerats. Det finns mycket begränsade data för måttligt nedsatt leverfunktion (total bilirubin > 1,5 till 3 gånger ULN oavsett ASAT, N = 1). Därför är farmakokinetiken för epkoritamab okänd i dessa populationer.
Liksom för andra terapeutiska proteiner har kroppsvikten (39 till 172 kg) en statistiskt signifikant effekt på farmakokinetiken för epkoritamab. Baserat på exponering-responsanalys och kliniska data, med beaktande av exponeringar hos patienter med antingen låg kroppsvikt (t.ex. 46 kg) eller hög kroppsvikt (t.ex. 105 kg) och för alla kroppsviktskategorier (< 65 kg, 65–< 85, ≥ 85), är effekten på exponeringar inte kliniskt relevant.
Pediatrisk population
Farmakokinetiken för epkoritamab hos pediatriska patienter har inte fastställts.
Prekliniska säkerhetsuppgifter
Farmakologi och/eller toxikologi hos djur
Inga studier om reproduktions- eller utvecklingstoxicitet hos djur har utförts med epkoritamab.
Effekter som generellt överensstämmer med den farmakologiska verkningsmekanismen för epkoritamab har observerats hos cynomolgusapor. Dessa fynd inkluderar dosrelaterade ogynnsamma kliniska tecken (inklusive kräkningar, minskad aktivitet och mortalitet vid höga doser) och cytokinfrisättning, reversibla hematologiska förändringar, reversibel B-cellsutarmning i perifert blod och reversibel minskad lymfoid cellularitet i sekundära lymfoida vävnader.
Mutagenicitet
Mutagenicitetsstudier har inte utförts med epkoritamab.
Karcinogenicitet
Karcinogenicitetsstudier har inte utförts med epkoritamab.
Nedsatt fertilitet
Fertilitetsstudier med djur har inte utförts med epkoritamab. Epkoritamab orsakade dock inte toxikologiska förändringar i hanars och honors fortplantningsorgan hos cynomolgusapor i doser upp till 1 mg/kg/vecka i en intravenös allmän toxicitetsstudie som varade i 5 veckor. AUC-exponeringar (tidsgenomsnitt för 7 dagar) vid den höga dosen hos cynomolgusapor var ungefär samma som i patienter (AUC0‑7d) som fick den rekommenderade dosen.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Natriumacetattrihydrat
Ättiksyra
Sorbitol (E420)
Polysorbat 80
Vatten för injektioner
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel och/eller spädningsmedel förutom de som nämns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Hållbarhet
Oöppnad injektionsflaska
2 år.
Utspädd eller beredd epkoritamab
Kemisk och fysisk stabilitet vid användning har påvisats i 24 timmar vid 2 °C till 8 °C inklusive upp till 12 timmar i rumstemperatur (20–25 °C).
Ur mikrobiologisk synvinkel ska produkten användas omedelbart. Om den inte används omedelbart är förvaringstider och -förhållanden vid användning användarens ansvar och ska normalt inte överskrida 24 timmar vid 2 °C till 8 °C, såvida inte spädningen har ägt rum under kontrollerade och validerade aseptiska förhållanden.
Minimera exponering för dagsljus. Låt lösningen med epkoritamab nå rumstemperatur före administrering. Kassera oanvänd lösning med epkoritamab när den tillåtna förvaringstiden har passerat.
Särskilda förvaringsanvisningar
Förvaras och transporteras kallt (2 °C till 8 °C).
Får ej frysas.
Förvara injektionsflaskan i ytterkartongen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter spädning/första öppnande finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
TEPKINLY injektioneste, liuos
4 mg/0,8 ml (L:ei) 1 kpl (0,8 ml) (734,72 €)
PF-selosteen tieto
Injektionsflaska av glas typ I med en propp av bromobutylgummi belagd med fluoropolymer vid kontaktstället och aluminiumförsegling med ett ljusblått flip-off-lock av plast, som innehåller 4 mg per 0,8 ml injektionsvätska, lösning.
Varje kartong innehåller en injektionsflaska.
Läkemedlets utseende:
Färglös till något gul lösning, pH 5,5 och osmolalitet på cirka 211 mOsm/kg.
Särskilda anvisningar för destruktion och övrig hantering
Beredning av epkoritamab
Hela detta avsnitt måste läsas noga före beredning av epkoritamab. Vissa doser (den förberedande dosen (0,16 mg) och den intermediära dosen (0,8 mg)) av epkoritamab måste spädas före administrering. Epkoritamab kan spädas med hjälp av två olika metoder; antingen med injektionsflaskor eller med sprutor.
Alla anvisningar nedan måste följas eftersom felaktig beredning kan leda till felaktig dos.
Epkoritamab måste beredas och administreras av hälso- och sjukvårdspersonal som en subkutan injektion.
Varje injektionsflaska med epkoritamab är endast avsedd för engångsbruk.
Varje injektionsflaska innehåller en överfyllning som gör det möjligt att dra upp den angivna mängden.
Administreringen av epkoritamab sker under 28‑dagarscykler, enligt doseringsschemat i avsnitt Dosering och administreringssätt
Epkoritamab ska inspekteras visuellt avseende partiklar och missfärgning före administrering. Injektionsvätskan ska vara en färglös till något gul lösning. Lösningen får inte användas om den är missfärgad eller grumlig eller om den innehåller främmande partiklar.
Epkoritamab måste beredas med aseptisk teknik. Den utspädda lösningen behöver inte filtreras.
Beredning av spädd epkoritamab med hjälp av metoden med tomma sterila injektionsflaskor
Beredningsanvisningar för 0,16 mg förberedande dos – 2 spädningar krävs– metod med tomma sterila injektionsflaskor
Använd spruta, injektionsflaska och nål av lämplig storlek för varje överföringssteg.
1) Förbered injektionsflaskan med epkoritamab |
2) Utför första spädningen |
3) Utför andra spädningen |
4) Dra upp dos |
5) Märk sprutan |
6) Kassera injektionsflaskan och eventuellt oanvänt epkoritamab enligt gällande anvisningar. |
Beredningsanvisningar för 0,8 mg intermediär dos – 1 spädning krävs – metod med tom steril injektionsflaska
Använd spruta, injektionsflaska och nål av lämplig storlek för varje överföringssteg.
1) Förbered injektionsflaskan med epkoritamab |
2) Utför spädning |
3) Dra upp dos |
4) Märk sprutan |
5) Kassera injektionsflaskan och eventuellt oanvänt epkoritamab enligt gällande anvisningar. |
Beredning av spädd epkoritamab med hjälp av metoden med sterila sprutor
Beredningsanvisningar för 0,16 mg förberedande dos – 2 spädningar krävs – metod med sterila sprutor
Använd spruta och nål av lämplig storlek för varje överföringssteg.
1)Förbered injektionsflaskan med epkoritamab |
2) Utför första spädningen |
3) Utför andra spädningen |
4) Dra upp dos |
5) Märk sprutan |
6) Kassera injektionsflaskan och eventuellt oanvänt epkoritamab enligt gällande anvisningar. |
Beredningsanvisningar för 0,8 mg intermediär dos – 1 spädning krävs – metod med steril spruta
Använd spruta och nål av lämplig storlek för varje överföringssteg.
1)Förbered injektionsflaskan med epkoritamab |
2) Utför spädning |
3) Dra upp dos |
4) Märk sprutan |
5) Kassera injektionsflaskan och eventuellt oanvänt epkoritamab enligt gällande anvisningar. |
Beredning av dosen 3 mg epkoritamab
Beredningsanvisningar för 3 mg andra intermediär dos – ingen spädning krävs
Epkoritamabdosen på 3 mg krävs endast för FL-patienter (se avsnitt Dosering och administreringssätt).
1) Förbered injektionsflaskan med epkoritamab |
2) Dra upp dos |
3) Märk sprutan |
4) Kassera injektionsflaskan och eventuellt oanvänt epkoritamab enligt gällande anvisningar. |
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
TEPKINLY injektioneste, liuos
4 mg/0,8 ml 1 kpl
- Ei korvausta.
Atc-kod
L01FX27
Datum för översyn av produktresumén
02.07.2026
Yhteystiedot
 ABBVIE OY
ABBVIE OY Veturitie 11 T 132
00520 Helsinki
010 2411 200
www.abbvie.fi