
JAYPIRCA tabletti, kalvopäällysteinen 100 mg
Huomioitavaa
▼ Tähän lääkkeeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti uutta turvallisuutta koskevaa tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Jaypirca 50 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 50 mg pirtobrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 38 mg laktoosia (monohydraattina).
Jaypirca 100 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 100 mg pirtobrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 77 mg laktoosia (monohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Jaypirca on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on uusiutunut tai huonosti hoitoon reagoiva manttelisolulymfooma (MCL), jota on aiemmin hoidettu Brutonin tyrosiinikinaasin (BTK) estäjällä.
Jaypirca on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on uusiutunut tai huonosti hoitoon reagoiva krooninen lymfaattinen leukemia (KLL), jota on aiemmin hoidettu BTK-estäjällä.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Jaypirca-hoidon aloittaa ja sitä valvoo lääkäri, jolla on kokemusta syöpähoitojen toteuttamisesta.
Annostus
Suositusannos on 200 mg pirtobrutinibia kerran vuorokaudessa.
Jaypirca-hoito on keskeytettävä, kunnes haitat ovat palautuneet asteelle 1 tai lähtötasolle, kun potilaalla ilmenee jokin seuraavista:
- Asteen 3 neutropenia ja kuume ja/tai infektio
- Asteen 4 neutropenia, joka kestää ≥ 7 päivää
- Asteen 3 trombosytopenia ja verenvuoto
- Asteen 4 trombosytopenia
- Asteen 3 tai 4 ei-hematologinen toksisuus
Oireetonta lymfosytoosia ei pidetä haittavaikutuksena. Potilaiden, joilla on tämä tapahtuma, tulee jatkaa Jaypirca-valmisteen käyttöä.
Kliinisissä tutkimuksissa haittatapahtumia hallittiin annosta pienentämällä rajallisella määrällä potilaita (ks. kohta Farmakodynamiikka).
Hoitoa tulee jatkaa, kunnes tauti etenee tai ilmenee toksisuutta, jota ei voida hyväksyä.
Unohtunut annos
Jos yli 12 tuntia on kulunut siitä, kun potilas on unohtanut ottaa annoksen, potilasta neuvotaan ottamaan seuraava annos tavanomaiseen aikaan. Ylimääräistä annosta ei pidä ottaa. Jos potilas oksentaa, hän ei saa ottaa lisäannosta, vaan hänen tulee jatkaa seuraavalla aikataulun mukaisella annoksella.
Erityisryhmät
Vanhukset
Annosta ei tarvitse muuttaa iän perusteella (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta. Dialyysihoidossa olevista potilaista ei ole tietoja (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on lievä, keskivaikea tai vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Jaypirca-valmisteen turvallisuutta ja tehoa alle 18-vuotiaiden lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Jaypirca otetaan suun kautta.
Tabletin kanssa otetaan lasillinen vettä ja tabletti on nieltävä kokonaisena tasaisen tehon varmistamiseksi (potilaat eivät saa pureskella, murskata tai jakaa tabletteja ennen nielemistä). Tabletti voidaan ottaa ruoan kanssa tai ilman ruokaa. Lääkeannos on otettava suunnilleen samaan aikaan joka päivä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Infektiot
Jaypirca-valmisteella hoidetuilla potilailla on esiintynyt vakavia infektioita, myös kuolemaan johtaneita tapauksia. Yleisimmin ilmoitetut asteen 3 tai korkeamman asteen infektiot olivat keuhkokuume, COVID 19 -keuhkokuume, COVID 19 ja sepsis. Ennaltaehkäisevää mikrobilääkehoitoa tulee harkita potilailla, joilla on lisääntynyt opportunististen infektioiden riski. Riippuen infektion asteesta ja siitä, esiintyykö se neutropenian yhteydessä, hoidon keskeyttäminen voi olla tarpeen (ks. kohta Annostus ja antotapa).
Verenvuodot
Jaypirca-valmisteella hoidetuilla potilailla on esiintynyt verenvuototapahtumia, myös kuolemaan johtaneita tapauksia, trombosytopenian kanssa tai ilman. Vakavia asteen 3 tai korkeamman asteen verenvuototapahtumia on havaittu, mukaan lukien maha-suolikanavan verenvuoto ja kallonsisäinen verenvuoto. Potilaita on seurattava verenvuodon oireiden ja löydösten varalta. Potilailla, jotka saavat antikoagulantteja tai verihiutaleiden estäjiä, saattaa olla suurentunut verenvuodon riski. Antikoagulantti- tai verihiutale-estäjähoidon riskit ja hyödyt on otettava huomioon, kun niitä annetaan yhdessä Jaypirca-valmisteen kanssa. Lisäseurantaa verenvuodon löydösten varalta on harkittava. Jaypirca-valmisteen käyttöä varfariinin tai muiden K-vitamiiniantagonistien kanssa ei ole tutkittu.
Hoidon keskeyttäminen voi olla tarpeen asteen 3 tai 4 verenvuototapahtumien vuoksi (ks. kohta Annostus ja antotapa).
Jaypirca-hoidon keskeyttämisen hyötyä ja riskiä 3–5 päivää ennen leikkausta ja sen jälkeen on harkittava huomioiden leikkauksen tyyppi ja verenvuotoriski.
Sytopeniat
Jaypirca-valmisteella hoidetuilla potilailla esiintyi asteen 3 tai 4 sytopeniaa, mukaan lukien neutropenia, anemia ja trombosytopenia. Potilaiden täydellistä verenkuvaa on seurattava hoidon aikana lääketieteellisten perusteiden mukaisesti. Sytopenian asteesta riippuen hoidon keskeyttäminen voi olla tarpeen (ks. kohta Annostus ja antotapa).
Eteisvärinä/-lepatus
Eteisvärinää ja eteislepatusta on havaittu Jaypirca-valmisteella hoidetuilla potilailla, erityisesti potilailla, joilla on ollut anamneesissa eteisvärinää ja/tai useita muita samanaikaisia sydän- ja verisuonisairauksia. Potilaita on seurattava eteisvärinän ja eteislepatuksen oireiden ja löydösten varalta. EKG on otettava lääketieteellisten perusteiden mukaisesti. Eteisvärinän/eteislepatuksen asteesta riippuen hoidon keskeyttäminen voi olla tarpeen (ks. kohta Annostus ja antotapa).
Toinen primaarinen maligniteetti
Toisia primaarisia pahanlaatuisia kasvaimia on yleisesti esiintynyt Jaypirca-valmisteella hoidetuilla potilailla, yleisimmät tyypit ovat ei-melanoottisia ihosyöpiä. Potilaita on seurattava ihosyöpien ilmaantumisen varalta ja neuvottava suojautumaan auringolta.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymää (TLS) on ilmoitettu harvoin Jaypirca-hoidon yhteydessä. Suuri TLS-riski on potilailla, joilla on suuri kasvainkuorma ennen hoitoa. Potilaat on arvioitava mahdollisen TLS-riskin varalta, ja potilaita on seurattava tarkasti kliinisen tarpeen mukaan.
Ehkäisy naisilla, jotka voivat tulla raskaaksi, ja miehillä
Eläimillä tehtyjen löydösten ja pirtobrutinibin genotoksisuuden (ks. kohta Prekliiniset tiedot turvallisuudesta) perusteella pirtobrutinibi voi aiheuttaa sikiövaurioita, kun sitä annetaan raskaana olevalle naiselle. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja 5 viikon ajan viimeisen Jaypirca-annoksen jälkeen. Miehiä neuvotaan käyttämään tehokasta ehkäisymenetelmää ja olemaan siittämättä lasta hoidon aikana ja 3 kuukauden ajan viimeisen Jaypirca-annoksen jälkeen (ks. Kohta Raskaus ja imetys).
Laktoosi
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkevalmistetta.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) 200 mg:n vuorokausiannosta kohden, eli sen voidaan sanoa olevan "natriumiton".
Yhteisvaikutukset
Pirtobrutinibi metaboloituu pääasiassa CYP3A4:n, UGT1A8:n ja UGT1A9:n välityksellä.
Muiden lääkevalmisteiden vaikutus pirtobrutinibin farmakokinetiikkaan
CYP3A:n estäjät
Kliinisessä tutkimuksessa itrakonatsoli, vahva CYP3A4:n estäjä, suurensi pirtobrutinibin AUC-arvoa 48 % eikä muuttanut pirtobrutinibin Cmax-arvoa. Tämä pirtobrutinibialtistuksen suureneminen ei ole kliinisesti merkittävää. Siksi Jaypircan annosta ei tarvitse muuttaa CYP3A-estäjien kanssa.
CYP3A:n induktorit
Kliinisessä tutkimuksessa rifampisiini, vahva CYP3A:n induktori, pienensi pirtobrutinibin AUC-arvoa 71 % ja Cmax-arvoa 42 %. Vaikka tämän pirtobrutinibialtistuksen pienenemisen ei odoteta olevan kliinisesti merkittävää, on voimakkaita CYP3A-induktoreita (esim. rifampisiini, karbamatsepiini, fenytoiini) mahdollisuuksien mukaan vältettävä.
Samanaikainen anto protonipumpun estäjien kanssa
Pirtobrutinibin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja, kun sitä annettiin samanaikaisesti omepratsolin, protonipumpun estäjän, kanssa.
Pirtobrutinibin vaikutukset muiden lääkevalmisteiden farmakokinetiikkaan (pitoisuuden suureneminen plasmassa)
CYP2C8-substraatit
Pirtobrutinibi on kohtalainen CYP2C8:n estäjä. Pirtobrutinibi suurensi repaglinidin (CYP2C8:n substraatti) AUC-arvoa 130 % ja Cmax-arvoa 98 %. Koska pirtobrutinibi voi suurentaa CYP2C8-substraattien pitoisuuksia plasmassa, on noudatettava varovaisuutta annettaessa sitä samanaikaisesti CYP2C8-substraattien (esim. repaglinidi, dasabuviiri, seleksipagi, rosiglitatsoni, pioglitatsoni ja montelukasti) kanssa.
BCRP-substraatit
Pirtobrutinibi on kohtalainen BCRP:n estäjä. Pirtobrutinibi nosti rosuvastatiinin (BCRP-substraatti) AUC-arvoa 140 % ja Cmax-arvoa 146 %. Koska pirtobrutinibi voi suurentaa BCRP-substraattien pitoisuuksia plasmassa, on noudatettava varovaisuutta annettaessa sitä samanaikaisesti BCRP-substraattien (esim. rosuvastatiini) kanssa. Jos samanaikaista antoa kapean terapeuttisen indeksin BCRP-substraattien (esim. suuriannoksinen metotreksaatti, mitoksantroni) kanssa ei voida välttää, on harkittava tarkkaa kliinistä seurantaa.
P-gp-substraatit
Pirtobrutinibi on heikko P-gp:n estäjä. Pirtobrutinibi suurensi digoksiinin (P-gp-substraatti) AUC-arvoa 35 % ja Cmax-arvoa 55 %. Näin ollen pirtobrutinibi voi lisätä P-gp-substraattien pitoisuuksia plasmassa. Jos samanaikaista antoa kapean terapeuttisen indeksin P-gp-substraattien (esim. dabigatraanieteksilaatti ja digoksiini) kanssa ei voida välttää, on harkittava tarkkaa kliinistä seurantaa.
CYP2C19-substraatit
Pirtobrutinibi on heikko CYP2C19:n estäjä. Pirtobrutinibi suurensi omepratsolin (CYP2C19-substraatti) AUC-arvoa 56 % ja Cmax-arvoa 49 %. Näin ollen pirtobrutinibi voi suurentaa CYP2C19-substraattien pitoisuuksia plasmassa. Jos samanaikaista antoa kapean terapeuttisen indeksin CYP2C19-substraattien (esim. fenobarbitaali ja mefenytoiini) kanssa ei voida välttää, on harkittava tarkkaa kliinistä seurantaa.
CYP3A-substraatit
Pirtobrutinibi on heikko CYP3A:n estäjä. Pirtobrutinibi suurensi suun kautta annetun midatsolaamin (herkkä CYP3A-substraatti) AUC-arvoa 70 % ja Cmax-arvoa 58 %. Pirtobrutinibilla ei ollut kliinisesti merkittävää vaikutusta suonensisäisesti annetun midatsolaamin altistukseen. Näin ollen pirtobrutinibi voi suurentaa CYP3A-substraattien pitoisuuksia plasmassa. Jos samanaikaista antoa kapean terapeuttisen indeksin CYP3A-substraattien (esim. alfentaniili, midatsolaami, takrolimuusi) kanssa ei voida välttää, on harkittava tarkkaa kliinistä seurantaa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehillä ja naisilla
Eläimillä tehtyjen löydösten ja pirtobrutinibin genotoksisuuden (ks. kohta Prekliiniset tiedot turvallisuudesta) perusteella pirtobrutinibi voi aiheuttaa sikiövaurioita, kun sitä annetaan raskaana olevalle naiselle. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisymenetelmää hoidon aikana ja 5 viikon ajan viimeisen Jaypirca-annoksen jälkeen. Miehiä neuvotaan käyttämään tehokasta ehkäisymenetelmää ja olemaan siittämättä lasta hoidon aikana ja 3 kuukauden ajan viimeisen Jayprica-annoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Jaypirca-valmisteen käytöstä raskaana oleville naisille ei ole tietoa. Eläinkokeet ovat osoittaneet lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Jaypirca-valmistetta ei tule käyttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö pirtobrutinibi ihmisen rintamaitoon. Imevälle lapselle aiheutuvaa riskiä ei voida sulkea pois. Imetys tulee lopettaa Jaypirca-hoidon ajaksi ja viikon ajaksi viimeisen Jaypirca-annoksen jälkeen.
Hedelmällisyys
Pirtobrutinibin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Jaypirca-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Joillakin potilailla on raportoitu uupumusta, huimausta ja voimattomuutta Jaypirca-hoidon aikana, mikä tulee ottaa huomioon arvioitaessa potilaan kykyä ajaa autoa tai käyttää koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät minkä tahansa asteen haittavaikutukset ovat: neutropenia (27,7 %), uupumus (26,2 %), ripuli (23,8 %), anemia (20,7 %), ihottuma (18,4 %) ja ruhjeet (17,8 %).
Yleisimmät vaikeat (aste ≥ 3) haittavaikutukset ovat: neutropenia (23,9 %), anemia (11,2 %), trombosytopenia (9,7 %) ja keuhkokuume (9,0 %).
Hoidon lopettamisen yleisyys haittavaikutusten vuoksi on 4,2 % ja annoksen pienentämisen yleisyys haittavaikutusten vuoksi 4,8 %.
Yleisimmät haittavaikutukset (ilmoitettu useammalla kuin kahdella potilaalla), jotka johtavat annoksen pienentämiseen, ovat neutropenia (2,5 %), ihottuma (0,6 %), ripuli (0,4 %), uupumus (0,4 %) ja trombosytopenia (0,4 %). Yleisimmät haittavaikutukset (ilmoitettu useammalla kuin kahdella potilaalla), jotka johtavat hoidon lopettamiseen, ovat neutropenia (1,0 %), anemia (1,0 %), keuhkokuume (0,9 %), trombosytopenia (0,7 %) ja ihottuma (0,4 %).
Jaypirca-valmisteeseen liittyviä vakavia haittavaikutuksia on esiintynyt 19,4 %:lla potilaista, ja yleisimmät vakavat haittavaikutukset (esiintyi ≥ 1 %:lla potilaista) olivat keuhkokuume (8,0 %), neutropenia (3,2 %), anemia (2,6 %), eteisvärinä/eteislepatus (1,3 %) ja virtsatieinfektio (1,0 %).
Kuolemaan johtaneita haittavaikutuksia on havaittu 0,4 %:lla potilaista (3 potilasta) keuhkokuumeen vuoksi, 0,3 %:lla potilaista (2 potilasta) verenvuodon vuoksi ja 0,1 %:lla potilaista (1 potilas) virtsatieinfektion vuoksi.
Haittavaikutustaulukko
Taulukossa 1 on lueteltu haittavaikutukset kliinisistä tutkimuksista, joissa Jaypirca-valmistetta käytettiin monoterapiana sekä markkinoille tulon jälkeisen kokemuksen perusteella. Haittavaikutukset kliinisistä tutkimuksista perustuvat 690 potilaan yhdistettyihin tietoihin, kun Jaypirca-valmistetta käytettiin monoterapiana aloitusannoksella 200 mg kerran vuorokaudessa ilman annoksen nostamista vaiheen 1/2 kliinisessä tutkimuksessa ja kun Jaypirca-valmistetta käytettiin monoterapiana 200 mg kerran vuorokaudessa vaiheen 3 kliinisessä tutkimuksessa. Potilaita hoidettiin manttelisolulymfooman, kroonisen lymfaattisen leukemian/pienen lymfosyyttisen lymfooman (KLL/SLL) ja non-Hodgkin-lymfooman (NHL) vuoksi. Potilaiden Jaypirca-hoidon mediaanikesto oli 12 kuukautta. Haittavaikutukset on lueteltu alla MedDRA-elinjärjestelmäluokittain. Yleisyysluokat ovat seuraavat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); erittäin harvinainen (< 1/10 000) ja tuntematon (ei voida arvioida saatavilla olevien tietojen perusteella). Kussakin yleisyysluokassa haittavaikutukset on esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset Jaypircaa-hoitoa saaneilla potilailla
| Elinjärjestelmäluokka (MedDRA) | Haittavaikutus
| Esiintymistiheys (%) (Kaikki asteet) | Aste ≥ 3c (%) |
| Infektiot | Keuhkokuume | Hyvin yleinen (13,8) | 9,0 |
| Ylähengitysteiden infektio | Hyvin yleinen (10,1) | 0,1 | |
| Virtsatieinfektio | Yleinen (9,9) | 1,4 | |
Veri ja imukudos
| Neutropeniab | Hyvin yleinen (27,7) | 23,9 |
| Anemiab | Hyvin yleinen (20,7) | 11,2 | |
| Trombosytopeniab | Hyvin yleinen (16,8) | 9,7 | |
| Lymfosytoosib | Yleinen (6,4) | 3,9 | |
| Hermosto | Päänsärky | Hyvin yleinen (12,6) | 0,7 |
| Sydän | Eteisvärinä/eteislepatus | Yleinen (3,8) | 1,7 |
Verisuonisto
| Verenvuotob | Hyvin yleinen (20,3) | 2,8 |
| Nenäverenvuoto | Yleinen (5,2) | 0 | |
| Hematuria | Yleinen (4,5) | 0,1 | |
| Hematooma | Yleinen (1,7) | 0,1 | |
| Sidekalvon verenvuoto | Yleinen (1,7) | 0,1 | |
| Mustelmatb | Hyvin yleinen (19,7) | 0,3 | |
| Ruhje | Hyvin yleinen (17,8) | 0,1 | |
| Hiussuonipurkauma | Yleinen (5,7) | 0 | |
Ruoansulatuselimistö
| Ripuli | Hyvin yleinen (23,8) | 1,0 |
| Pahoinvointi | Hyvin yleinen (16,7) | 0,4 | |
| Vatsakipu | Hyvin yleinen (10,4) | 1,0 | |
| Maksa ja sappi | Maksan entsyymiarvojen nousu | Tuntematon | Tuntematon |
| Ihon ja ihonalainen kudos | Ihottumab | Hyvin yleinen (18,4) | 1,2 |
| Luusto, lihakset ja sidekudos | Nivelsärky | Hyvin yleinen (14,6) | 1,2 |
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus | Hyvin yleinen (26,2) | 1,9 |
| Perifeerinen turvotus | Hyvin yleinen (11,6) | 0,3 |
a Esiintymistiheydet on johdettu Jaypirca-altistumisesta potilailla, joilla on B-solujen pahanlaatuisia kasvaimia
b Sisältää useita haittavaikutustermejä
cVaikeusasteen määritys perustuu National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) -versioon 5.0
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Vaiheen 1 tutkimuksessa, jossa potilaat saivat toistuvia annoksia enintään 300 mg kerran vuorokaudessa, ei saavutettu suurinta siedettyä annosta. Terveillä vapaaehtoisilla tehdyissä tutkimuksissa ei havaittu annokseen liittyvää toksisuutta, kun annettiin enintään 900 mg:n kerta-annos. Pirtobrutinibin yliannostuksen oireita ja löydöksiä ei ole varmistettu, eikä pirtobrutinibin yliannostukseen ole olemassa erityistä hoitoa.
Yliannostuksen saaneita potilaita on seurattava tarkasti ja annettava asianmukaista tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EL05
Vaikutusmekanismi
Pirtobrutinibi on reversiibeli (palautuva), ei-kovalenttinen Brutonin tyrosiinikinaasin (BTK:n) estäjä. BTK on B-solun antigeenireseptorin ja sytokiinireseptorireittien signalointiproteiini. BTK-signalointi johtaa B-solujen jakautumiseen, kemotaktikseen ja adheesioon tarvittavien reittien aktivoitumiseen sekä elimistössä kulkeutumiseen. Pirtobrutinibi sitoutuu villityypin BTK:hen sekä BTK:hen, jossa on C481-mutaatio, mikä johtaa BTK-aktiivisuuden estoon.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Pirtobrutinibin 900 mg:n kerta-annoksen vaikutusta korjattuun QT-aikaan (QTc) arvioitiin lumelääkettä ja positiivisia kontrolleja sisältäneessä tutkimuksessa 30 terveellä koehenkilöllä. Valittu annos on noin 2 kertaa suurempi kuin pitoisuudet, jotka saavutetaan vakaassa tilassa 200 mg:n suositusannoksella kerran vuorokaudessa. Pirtobrutinibilla ei ollut kliinisesti merkittävää vaikutusta (esim. > 10 ms) sykkeen mukaan Friderician menetelmällä korjatun QT-ajan (QTcF) muutokseen. Pirtobrutinibille altistumisen ja QTc-ajan muutoksen välillä ei ollut yhteyttä.
Kliininen teho ja turvallisuus
Manttelisolulymfooma
Jaypircan tehoa arvioitiin aikuisilla manttelisolulymfoomaa sairastavilla potilailla vaiheen 1/2 avoimessa, yksihaaraisessa kliinisessä monikeskustutkimuksessa: tutkimus 18001 (BRUIN). Tutkimus sisälsi kaksi osaa: vaihe 1 (annoksen nostaminen), jossa tutkittiin pirtobrutinibi-monoterapian annosaluetta 25–300 mg kerran vuorokaudessa ja vaihe 2 (annoksen laajennus). Vaiheessa 1 ensisijaisena tavoitteena oli määrittää pirtobrutinibin vaiheen 2 suositusannos, jonka todettiin olevan 200 mg kerran vuorokaudessa, mutta suurinta siedettyä annosta ei vahvistettu. Vaiheessa 2 ensisijaisena tavoitteena oli arvioida pirtobrutinibin syöpäkasvua estävää aktiivisuutta riippumattoman arviointikomitean arvioiman kokonaisvasteen perusteella. Potilaat saivat Jaypircaa suun kautta päivittäin, kunnes tauti eteni tai ilmeni toksisuutta, joka ei ole hyväksyttävää.
Tutkimukseen 18001 otettiin ja siinä hoidettiin yhteensä 164 potilasta, joilla oli manttelisolulymfoomadiagnoosi. Primaarinen analyysisarja (primary analysis set, PAS) tehon arvioimiseksi perustui ensimmäiseen 90 tutkimukseen otettuun manttelisolulymfoomaa sairastavaan potilaaseen, joilla ei ollut tiedettyä levinneisyyttä keskushermostoon, joita oli hoidettu aiemmalla BTK-estäjällä, jotka olivat saaneet yhden tai useamman annoksen Jaypirca-valmistetta, ja joilla sairaus oli todettavissa radiologisesti vähintään yhdessä kohtaa. Mediaani-ikä oli 70 vuotta (vaihteluväli: 46-87 vuotta), 80 % oli miehiä, 84,4 % oli valkoihoisia, 67,8 %:lla oli lähtötilanteessa ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokka 0 ja 31,1 %:lla ECOG-toimintakykyluokka oli 1. Potilaiden aiempien hoitolinjojen mediaanimäärä oli 3 (vaihteluväli: 1-8). Syynä viimeisimmän aiemman BTK-estäjähoidon lopettamiseen oli sairauden eteneminen 81,1 %:lla potilaista ja intoleranssi 13,3 %:lla potilaista. Potilaista 95,6 % oli saanut aiempaa anti-CD20-hoitoa, 87,8 % kemoterapiaa, 18,9 % autologisen kantasolusiirron, 4,4 % allogeenisen kantasolusiirron, 15,6 % aiempaa BCL2-estäjää ja 4,4 % aiempaa hoitoa kimeerisellä antigeenireseptorilla muunnelluilla T-soluilla (CAR-T). Potilaista 38,9 %:lla oli ekstranodaalinen esiintymä ja 26,7 %:lla oli kasvaimen koko suurempi tai yhtä suuri kuin 5 cm. Yksinkertaistettu MCL International Prognostic Index (sMIPI) -pistemäärä oli matala 22,2 %:lla, keskitasoa 55,6 %:lla ja korkea 22,2 %:lla potilaista.
18001-tutkimukseen otetuista 164 manttelisolulymfoomaa sairastavasta potilaasta 9 potilaalla annosta pienennettiin, mukaan lukien 6 vasteen saanutta potilasta, jotka pystyivät jatkamaan hoitoa ja joilla kestävä vaste säilyi annoksen pienentämisen jälkeen 150 mg:aan kerran vuorokaudessa (3), 100 mg:aan kerran vuorokaudessa (2) ja 50 mg:aan kerran vuorokaudessa (1).
Jaypirca-valmisteen teho perustui vasteeseen, joka arvioitiin malignin lymfooman Lugano 2014 -kriteerien mukaisesti. Taulukossa 2 on yhteenveto tehotuloksista potilailla, jotka olivat saaneet vähintään yhtä aiempaa BTK-estäjää ja jotka sisältyivät primaariseen analyysisarjaan. Primaariseen analyysisarjaan sisältyneistä 90 potilaasta 79 sai vähintään yhden annoksen 200 mg kerran vuorokaudessa. Näistä 79 potilaasta 77 aloitti annoksella 200 mg kerran vuorokaudessa, yhdellä annosta nostettiin pienemmästä annoksesta ja yhdellä annosta pienennettiin suuremmasta annoksesta. Hoitoajan mediaani oli 5,24 kuukautta (vaihteluväli: 0,2-39,6 kuukautta). Vasteen saaneilla 51 potilaalla mediaaniaika vasteen saavuttamiseen oli 1,84 kuukautta (vaihteluväli: 1,0-7,5 kuukautta).
Vaikka alaryhmäanalyyseissä on edustettuna rajoitettu määrä potilaita, kliinisesti merkittäviä tehotuloksia havaittiin kaikissa tärkeissä alaryhmissä, myös potilailla, jotka lopettivat aiemman BTK-estäjähoidon intoleranssin tai taudin etenemisen vuoksi ja riippumatta aiempien hoitojen lukumäärästä ja tyypistä.
Taulukko 2: Yhteenveto tehotiedoista 18001-tutkimuksessa manttelisolulymfoomaa sairastavilla potilailla, jotka olivat saaneet vähintään yhtä aiempaa BTK-estäjää
pirtobrutinibi N = 90 | |
| Objektiivinen vasteosuus (täydellinen vaste + osittainen vaste) | |
| Osuus (%) (95 % Iv) | 56,7 (45,8; 67,1) |
| Täydellinen vaste (%) | 18,9 |
| Osittainen vaste (%) | 37,8 |
| Vasteen kesto | |
| Mediaani (kk) (95 % Iv) | 17,61 (7,29; 27,24) |
Lyhenteet: Iv = luottamusväli
Tiedonkeruun katkaisupäivä: 29.7.2022. Vasteen keston seuranta-ajan mediaani oli 12,68 kuukautta.
Krooninen lymfaattinen leukemia
Jaypircan tehoa arvioitiin kroonista lymfaattista leukemiaa sairastavilla potilailla, joita oli hoidettu BTK-estäjällä, satunnaistetussa, avoimessa, aktiivikontrolloidussa kansainvälisessä monikeskustutkimuksessa (BRUIN CLL-321, tutkimus 20020). Tutkimukseen otettiin 238 kroonista lymfaattista leukemiaa/pienilymfosyyttistä lymfoomaa sairastavaa potilasta, joita oli aiemmin hoidettu BTK-estäjällä. Potilaat satunnaistettiin suhteessa 1:1 saamaan joko Jaypirca-valmistetta 200 mg kerran vuorokaudessa suun kautta, kunnes tauti eteni tai ilmeni toksisuutta, joka ei ole hyväksyttävää, tai tutkijan valintaa:
- Idelalisibi ja rituksimabivalmiste (IR): Idelalisibia 150 mg kaksi kertaa vuorokaudessa suun kautta, kunnes sairaus eteni tai ilmeni toksisuutta, joka ei ole hyväksyttävää, yhdessä 8 rituksimabi-infuusion kanssa (375 mg/m2 laskimoon 1. syklin 1. päivänä, minkä jälkeen 500 mg/m2 2 viikon välein 4 annoksen ajan ja sen jälkeen 4 viikon välein 3 annoksen ajan), 28 vuorokauden mittaisella syklillä.
- Bendamustiini ja rituksimabivalmiste (BR): Bendamustiinia 70 mg/m2 laskimoon (kunkin 28 vuorokauden syklin 1. ja 2. päivänä), yhdessä rituksimabivalmisteen kanssa (375 mg/m2 laskimoon 1. syklin 1. päivänä, sitten 500 mg/m2 seuraavien syklien 1. päivänä) enintään 6 syklin ajan.
Satunnaistaminen ositettiin 17p-deleetiostatuksen (kyllä/ei) ja aiemman venetoklaksihoidon (kyllä/ei) mukaan. Yhteensä 238 potilaasta 119 sai Jaypirca-monoterapiaa, 82 IR-hoitoa ja 37 BR-hoitoa. Kun sairauden eteneminen oli vahvistettu, IR- tai BR-ryhmään satunnaistetuilla potilailla oli mahdollisuus siirtyä Jaypirca-monoterapiaan. Lähtötilanteen ominaisuudet olivat samanlaiset eri hoitoryhmissä. Kaiken kaikkiaan mediaani-ikä oli 67 vuotta (vaihteluväli: 42–90 vuotta), 70 % oli miehiä ja 81 % valkoisia. Lähtötilanteen ECOG-toimintakyky oli 93 %:lla potilaista 0 tai 1 ja 44 %:lla potilaista oli Rai-tautiluokituksen vaiheen III tai IV sairaus. Niistä potilaista, joilla oli keskuslaboratoriotestaus saatavilla, 57 %:lla (101 potilasta 176:sta) oli 17p-deleetio ja/tai TP53-mutaatio, 86 %:lla (164 potilasta 190:stä) oli mutatoitumaton IGHV ja 65 %:lla (97 potilasta 149:stä) oli kompleksinen karyotyyppi.
Potilaiden saamien aiempien hoitojen mediaani oli 3 (vaihteluväli: 1–13). 57 %:lla oli vähintään 3 aiempaa hoitoa ja 51 %:lla oli ollut aiempi BCL2-estäjähoito. Yleisimmät aiemmat BTK-estäjähoidot olivat ibrutinibi (87 %), akalabrutinibi (16 %) ja zanubrutinibi (7 %). 70 % potilaista lopetti viimeisimmän BTK-estäjähoidon huonosti hoitoon reagoivan tai etenevän sairauden vuoksi, 15 % lopetti hoidon toksisuuden vuoksi ja 15 % lopetti hoidon muista syistä.
Teho perustui riippumattoman arviointikomitean arvioimaan etenemättömyysaikaan (PFS) pirtobrutinibi-monoterapialla verrattuna tutkijan valitsemaan hoitohaaraan. Tutkimus saavutti ensisijaisen päätetapahtumansa ennalta määritettynä, riippumattoman arviointitoimikunnan arvioiman etenemättömyysajan lopullisen analyysin ajankohtana (tiedonkeruun katkaisupäivä: 29.8.2023). Päivitetyssä analyysissä (tiedonkeruun katkaisupäivä: 29.8.2024), jossa pirtobrutinibin seuranta-ajan mediaani oli 19,4 kuukautta (vaihteluväli: 0,03–33,3 kuukautta) ja tutkijan valitseman hoitohaaran 17,7 kuukautta (vaihteluväli: 0,03–27,9 kuukautta), pirtobrutinibilla havaittiin parantunut riippumattoman arviointitoimikunnan arvioima etenemättömyysaika verrattuna tutkijan valitsemaan haaraan, mikä on yhdenmukainen primaarianalyysin kanssa. Kliinisesti merkittäviä tehotuloksia havaittiin pirtobrutinibin hyväksi tärkeissä alaryhmissä, mukaan lukien potilaat, jotka keskeyttivät aiemman BTK-estäjähoidon sietämättömyyden tai sairauden etenemisen vuoksi, ja riippumatta aiempien hoitojen määrästä ja tyypistä. Tehoa koskevat tulokset on esitetty taulukossa 3. Etenemättömyysajan Kaplan–Meier-käyrä on esitetty kuvassa 1.
Taulukko 3: Tehotulokset riippumattoman arviointikomitean arvioimana kroonista lymfaattista leukemiaa sairastavilla potilailla, joita on aiemmin hoidettu BTK-estäjällä – ITT-populaatio (tutkimus 20020)
pirtobrutinibi 200 mg kerran vuorokaudessa (N = 119) | Tutkijan valinta: idelalisibi ja rituksimabi tai bendamustiini ja rituksimabi (N = 119) | |
Etenemättömyysaikaa |
|
|
Tapahtumien lukumäärä, n | 74 (62 %) | 79 (66 %) |
Sairauden eteneminen | 60 (50 %) | 66 (55 %) |
Kuolema | 14 (12 %) | 13 (11 %) |
Etenemättömyysajan mediaani (95 % lv), kuukauttab | 14,0 (11,2; 16,6) | 8,7 (8,1; 10,4) |
Riskitiheyssuhde (95 % lv)c | 0,54 (0,39; 0,75) | |
P-arvod | 0,0002 | |
lv = luottamusväli
Tiedonkeruun katkaisupäivä: 29.8.2024
a Tehoa arvioitiin käyttäen vuoden 2018 International Workshop for Chronic Lymphocytic Leukemia (iwCLL) -ohjeita.
b Perustuu Kaplan-Meierin arvioon.
c Perustuu ositettuun Coxin suhteellisten vaarojen malliin.
d 2-puolinen nimellinen p-arvo, joka perustuu ositettuun log-rank-testiin.
Kuva 1: Kaplan-Meier-käyrä riippumattoman arviointikomitean arvioimasta etenemättömyysajasta kroonista lymfaattista leukemiaa sairastavilla potilailla, joita oli aiemmin hoidettu BTK-estäjällä, tutkimus 20020
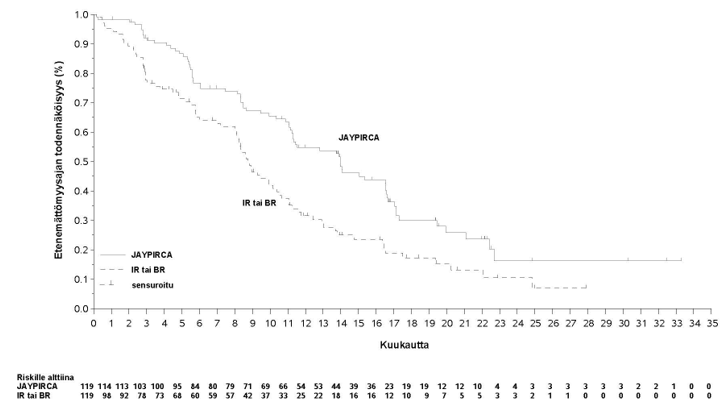
IR = idelalisibi ja rituksimabivalmiste, BR = bendamustiini ja rituksimabivalmiste
Kun kokonaiselossaolon seuranta-ajan mediaani oli pirtobrutinibin osalta 20,4 kuukautta ja tutkijan valitseman haaran osalta 19,2 kuukautta, 38 potilasta (32,0 %) pirtobrutinibihaarassa ja 32 potilasta (27,0 %) tutkijan valitsemassa haarassa kuoli. Kokonaiselossaolon mediaani oli 29,7 kuukautta (95 % luottamusväli: 27,1; ei arvioitavissa) pirtobrutinibihaarassa, eikä sitä saavutettu tutkijan valitsemassa haarassa. Riskitiheyssuhde oli 1,090 (95 % luottamusväli: 0,679; 1,749, p = 0,7202). Kokonaiselossaoloanalyysissä sekoittavana tekijänä voivat olla ne 50 potilasta 119:stä, jotka siirtyivät tutkijan valitsemasta haarasta pirtobrutinibiin.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Jaypirca-valmisteen käytöstä kypsien B-solujen pahanlaatuisten kasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Ehdollinen myyntilupa
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa. Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Pirtobrutinibin farmakokinetiikka karakterisoitiin terveillä koehenkilöillä ja syöpäpotilailla. Annokset vaihtelivat välillä 25–300 mg kerran vuorokaudessa (0,125–1,5 kertaa suositeltu annos 200 mg kerran vuorokaudessa), aina 900 mg:n kerta-annoksiin asti. Plasmapitoisuuden suurentuminen oli suunnilleen suhteessa annokseen. Vakaa tila saavutettiin 5 vuorokauden kuluessa kerran vuorokaudessa otetulla annoksella, ja syöpäpotilailla kumuloitumissuhteen [variaatiokerroin (CV %)] keskiarvo oli 1,63 (26,7 %) AUC:n perusteella 200 mg kerran vuorokaudessa annoksen jälkeen. Pirtobrutinibin farmakokinetiikan muutoksien katsottiin johtuvan kolmesta potilastekijästä: painosta, seerumin albumiinista ja absoluuttisesta eGFR:stä. Painon nousun 70 kilosta 120 kiloon arvioidaan lisäävän pirtobrutinibin puhdistumaa 24 %. Absoluuttisen eGFR:n pienenemisen arvosta 90 ml/min arvoon 30 ml/min arvioidaan vähentävän pirtobrutinibin puhdistumaa 16 %. Seerumin albumiinin pienenemisen 40 g/l:sta 30 g/l:aan arvioidaan lisäävän pirtobrutinibin puhdistumaa 21 %. Nämä tekijät eivät todennäköisesti yksinään aiheuta merkittäviä muutoksia pirtobrutinibin farmakokinetiikkaan, eikä annoksen muuttamista suositella.
Keskiarvo (CV %) vakaan tilan AUC-arvolle oli 92 600 h*ng/ml (39 %) ja Cmax-arvolle 6 500 ng/ml (25 %) syöpäpotilailla suositusannoksella 200 mg kerran vuorokaudessa.
Suositusannoksella pirtobrutinibi saavuttaa farmakokineettisen pitoisuuden, joka voi ylittää BTK IC96:n alimmillakin pitoisuuksilla ja voi siten saada aikaan jatkuvan BTK:n eston kerran päivässä tapahtuvalla annostelulla, riippumatta BTK:n sisäisestä vaihtumisnopeudesta.
Imeytyminen
Pirtobrutinibin absoluuttinen biologinen hyötyosuus suun kautta otetun 200 mg:n kerta-annoksen jälkeen on 85,5 % terveillä koehenkilöillä. Mediaaniaika huippupitoisuuden saavuttamiseen plasmassa (tmax) on noin 2 tuntia sekä syöpäpotilailla että terveillä koehenkilöillä. Imeytymisessä ei ole pH-riippuvuutta.
Ruoan vaikutus
Terveille koehenkilöille annettu runsasrasvainen ja kaloripitoinen ateria pienensi pirtobrutinibin Cmax-arvoa 23 % ja viivästytti tmax-arvoa 1 tunnilla. Pirtobrutinibin AUC-arvoon ei ollut vaikutusta. Pirtobrutinibi voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Pirtobrutinibin näennäisen jakautumistilavuuden keskiarvo on 34,2 l syöpäpotilailla. Sitoutuminen plasman proteiineihin on 96 %:ta, eikä sitoutuminen ollut pitoisuudesta riippuvaista pitoisuusvälillä 0,5-50 µM. Terveiden ja vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden plasmassa proteiineihin sitoutuminen oli 96 %:ta. Veri-plasma-pitoisuussuhteen keskiarvo on 0,79.
Biotransformaatio
Maksametabolia on pirtobrutinibin pääasiallinen puhdistumareitti. Pirtobrutinibi metaboloituu useiksi inaktiivisiksi metaboliiteiksi CYP3A4:n, UGT1A8:n ja UGT1A9:n välityksellä. CYP3A-modulaatiolla ei ollut kliinisesti merkittävää vaikutusta pirtobrutinibialtistuksiin.
Pirtobrutinibi estää CYP2C8:aa, CYP2C9:ää ja CYP3A4:ää in vitro ja estää minimaalisesti CYP1A2:ta, CYP2B6:ta, CYP2C19:ää tai CYP2D6:ta pitoisuudella 60 µM. In vitro pirtobrutinibi indusoi CYP3A4:ää, CYP3A5:ta, CYP2C19:ää ja CYP2B6:ta.
Pirtobrutinibi estää minimaalisesti UGT1A1:tä in vitro, IC50 = 18 µM.
Samanaikainen anto kuljetussubstraattien/estäjien kanssa
In vitro -tutkimukset osoittivat, että pirtobrutinibi on P-gp:n ja BCRP:n substraatti.
Pirtobrutinibi on P-gp:n ja BCRP:n in vitro estäjä. Kliinisissä tutkimuksissa pirtobrutinibi vaikutti digoksiinin (P-gp-substraatti) ja rosuvastatiinin (BCRP-substraatti) farmakokinetiikkaan (ks. kohta Yhteisvaikutukset).
Eliminaatio
Pirtobrutinibin näennäisen puhdistuman keskiarvo on 2,05 l/h ja tehokas puoliintumisaika noin 19,9 tuntia. Kun terveille koehenkilöille annettiin 200 mg:n kerta-annos radioaktiivisesti merkittyä pirtobrutinibia, 37 % annoksesta erittyi ulosteeseen (18 % muuttumatonta) ja 57 % virtsaan (10 % muuttumatonta).
Erityisryhmät
Ikä, sukupuoli, etninen tausta ja paino
Syöpää sairastavien potilaiden populaatiofarmakokineettisen analyysin perusteella iällä (vaihteluväli 22–95 vuotta), etnisellä taustalla, sukupuolella ja painolla (vaihteluväli 35,7–152 kg) ei ollut kliinisesti merkittävää vaikutusta pirtobrutinibialtistukseen.
Munuaisten vajaatoiminta
Syöpäpotilaiden populaatiofarmakokineettisessä analyysissä potilailla, joilla oli lievä (eGFR 60 - < 90 ml/min) tai kohtalainen munuaisten vajaatoiminta (eGFR 30 - < 60 ml/min), pirtobrutinibin puhdistuma oli 16–27 % pienempi kuin potilailla, joilla oli normaali munuaisten toiminta. Tällöin odotettu altistus on AUC = 94 100 ng*h/ml ja Cmax = 6 680 ng/ml potilailla, joilla on lievä munuaisten vajaatoiminta (16–19 % suurempi verrattuna potilaisiin, joilla on normaali munuaisten toiminta), ja AUC = 108 000 ng*h/ml ja Cmax = 7 360 ng/ml potilailla, joilla on kohtalainen munuaisten vajaatoiminta (28–36 % suurempi kuin potilailla, joilla on normaali munuaisten toiminta).
Muutoin terveillä vapaaehtoisilla tehdyssä kliinisen farmakologian tutkimuksessa näennäinen puhdistuma oli 35 % pienempi neljällä vaikeaa munuaisten vajaatoimintaa sairastavalla osallistujalla (eGFR 15 - < 30 ml/min) verrattuna kahdeksaan osallistujaan, joilla oli normaali munuaisten toiminta (eGFR ≥ 90 ml/min), altistuksen ollen AUC0‑inf = 115 000 ng*h/mL (62 % suurempi verrattuna normaaliin munuaisten toimintaan) ja Cmax = 2 980 ng/ml (7 % pienempi verrattuna normaaliin munuaisten toimintaan).
Dialyysihoitoa saavia potilaita, joilla oli loppuvaiheen munuaissairaus, ei ole tutkittu (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Pirtobrutinibin farmakokinetiikassa ei ollut kliinisesti merkittäviä eroja minkään asteisen maksan vajaatoiminnan osalta (Child-Pugh A, B ja C tai mikä tahansa kokonaisbilirubiini ja mikä tahansa ASAT). Erityisessä maksan vajaatoimintatutkimuksessa pirtobrutinibin AUC-arvon keskiarvo ja Cmax-arvon keskiarvo olivat samankaltaisia potilailla, joilla oli lievä maksan vajaatoiminta (Child Pugh A) ja potilailla, joiden maksan toiminta oli normaali. Kohtalaista maksan vajaatoimintaa (Child-Pugh B) sairastavilla potilailla AUC-arvo oli 15 % pienempi verrattuna henkilöihin, joilla oli normaali maksan toiminta ja Cmax-arvo oli samankaltainen. Koehenkilöillä, joilla oli vaikea maksan vajaatoiminta (Child Pugh C), pirtobrutinibin AUC-arvo oli 21 % pienempi ja Cmax-arvon keskiarvo 24 % pienempi kuin henkilöillä, joiden maksan toiminta oli normaali. Sitoutumattoman pirtobrutinibin osuus (fu) koehenkilöillä suureni yleensä maksan vajaatoiminnan vaikeusasteen kasvaessa. Näin ollen, kun pirtobrutinibin farmakokinetiikan altistusparametreja korjattiin sitoutumattoman osuuden osalta, sitoutumattoman pirtobrutinibin farmakokinetiikan altistusparametreissa (AUCu ja Cmax,u) ei havaittu kliinisesti merkittävää eroa sellaisten koehenkilöiden välillä, joilla oli minkä tahansa asteinen maksan vajaatoiminta tai normaali maksan toiminta.
Pediatriset potilaat
Pirtobrutinibilla ei ole tehty farmakokineettisiä tutkimuksia alle 18-vuotiailla potilailla.
Prekliiniset tiedot turvallisuudesta
Toistuvan annoksen tutkimuksissa havaittiin T-soluista riippuvaisen vasta-ainevasteen vähentymistä rotilla (AUC-arvon perusteella 0,69-kertainen altistus verrattuna ihmisen altistukseen 200 mg:n suositusannoksella) ja minimaalisia tai lieviä sarveiskalvovaurioita koirilla (0,42-kertainen altistus verrattuna ihmisen altistukseen). Suurten keuhkoverisuonien lievää tai kohtalaista nekroosia ja verisuoni-/perivaskulaarista tulehdusta havaittiin vain rotilla. Nämä vaikutukset ilmenivät kliinisesti merkityksellisillä altistustasoilla.
Genotoksisuus/karsinogeenisuus
Pirtobrutinibi ei ollut mutageeninen bakteerien mutageenisuustestissä (Ames). Pirtobrutinibi oli aneugeeninen kahdessa in vitro -mikrotumatestissä, joissa käytettiin ihmisen ääreisverenkierron lymfosyyttejä. Pirtobrutinibilla ei ollut vaikutusta in vivo rotan luuytimen mikrotumatestissä annoksilla 2 000 mg/kg asti (kerta-annos), joka on noin 11 kertaa suurempi altistus (ottaen huomioon sitoutumattoman Cmax-arvon naaraseläimillä) kuin ihmisen altistus 200 mg:lla.
Pirtobrutinibilla ei ole tehty karsinogeenisuustutkimuksia.
Alkiotoksisuus/teratogeenisuus
Lisääntymistutkimuksissa eläimillä pirtobrutinibin antaminen tiineille rotille organogeneesin aikana johti sikiön painon laskuun, alkio-sikiökuolleisuuteen ja sikiön epämuodostumiin, kun emon altistus oli AUC-arvon perusteella 3,0-kertainen verrattuna ihmisen altistukseen 200 mg:n suositusannoksella.
Lisääntymistoksisuus
Pirtobrutinibilla ei ole tehty hedelmällisyystutkimuksia. Toistuvan annoksen toksisuustutkimuksissa, jotka kestivät enintään 3 kuukautta, pirtobrutinibilla ei ollut vaikutusta urosrottien lisääntymiselimiin 0,69-kertaisella ja uroskoirien lisääntymiselimiin 0,42-kertaisella altistuksella verrattuna ihmisen altistukseen 200 mg:n suositusannoksella AUC-arvon perusteella. Pirtobrutinibilla ei ollut vaikutusta naarasrottien lisääntymiselimiin 4,0-kertaisella ja naaraskoirien lisääntymiselimiin 0,42-kertaisella altistuksella verrattuna ihmisen altistukseen.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
hypromelloosiasetaattisukkinaatti
selluloosa, mikrokiteinen
laktoosimonohydraatti
kroskarmelloosinatrium
magnesiumstearaatti
piidioksidi, kolloidinen hydratoitu
Kalvopinnoite
hypromelloosi
titaanidioksidi
triasetiini
indigokarmiini (E132)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JAYPIRCA tabletti, kalvopäällysteinen
100 mg (L:kyllä) 56 fol (10963,10 €)
PF-selosteen tieto
Jaypirca 50 mg kalvopäällysteinen tabletti
Polyvinyylikloridi/polyklooritrifluorieteeni läpipainopakkaukset, jotka on suljettu alumiinifoliolla 28, 30 tai 84 kalvopäällysteisen tabletin pakkauksissa.
Jaypirca 100 mg kalvopäällysteinen tabletti
Polyvinyylikloridi/polyklooritrifluorieteeni läpipainopakkaukset, jotka on suljettu alumiinifoliolla 28, 30, 56, 60, 84 tai 168 kalvopäällysteisen tabletin pakkauksissa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Jaypirca 50 mg kalvopäällysteinen tabletti
Sininen, 9 x 9 mm, kaarevan kolmion muotoinen tabletti, jonka toisella puolella on kaiverrus ”Lilly 50” ja toisella puolella ”6902”.
Jaypirca 100 mg kalvopäällysteinen tabletti
Sininen, 10 mm, pyöreä tabletti, jonka toisella puolella on kaiverrus ”Lilly 100” ja toisella puolella ”7026”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JAYPIRCA tabletti, kalvopäällysteinen
100 mg 56 fol
- Ei korvausta.
ATC-koodi
L01EL05
Valmisteyhteenvedon muuttamispäivämäärä
12.01.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240