
SUNOSI tabletti, kalvopäällysteinen 75 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Sunosi 75 mg tabletti, kalvopäällysteinen
Yksi tabletti sisältää solriamfetolihydrokloridia määrän, joka vastaa 75 mg:aa solriamfetolia.
Sunosi 150 mg tabletti, kalvopäällysteinen
Yksi tabletti sisältää solriamfetolihydrokloridia määrän, joka vastaa 150 mg:aa solriamfetolia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Sunosi on tarkoitettu narkolepsiaa (johon voi liittyä katapleksiaa) sairastavien aikuispotilaiden valvetilan edistämiseen ja liiallisen päiväaikaisen uneliaisuuden vähentämiseen.
Sunosi on tarkoitettu valvetilan edistämiseen ja liiallisen päiväaikaisen uneliaisuuden vähentämiseen sellaisille obstruktiivista uniapneaa sairastaville aikuispotilaille, joiden liiallinen päiväaikainen uneliaisuus ei ole parantunut riittävästi obstruktiivisen uniapnean ensisijaisella hoidolla, kuten jatkuvalla positiivisella hengitystiepaineella (CPAP).
Ehto
Hoidon saa aloittaa vain narkolepsian tai obstruktiivisen uniapnean hoitoon perehtynyt terveydenhuollon ammattilainen.
Annostus ja antotapa
Hoidon saa aloittaa vain narkolepsian tai obstruktiivisen uniapnean hoitoon perehtynyt terveydenhuollon ammattilainen.
Sunosi ei ole hoito obstruktiivista uniapneaa sairastavien potilaiden taustalla olevaan hengitystieahtaumaan. Obstruktiivisen uniapnean ensisijaista hoitoa tulee jatkaa näille potilaille.
Verenpaine ja syke tulee mitata ennen solriamfetolihoidon aloittamista ja niitä tulee seurata säännöllisesti hoidon aikana, etenkin annoksen suurentamisen jälkeen. Jo olemassa oleva hypertensio tulee saada hallintaan ennen solriamfetolihoidon aloittamista, ja varovaisuutta on noudatettava hoidettaessa potilaita, joilla on tavallista suurempi vakavien sydänperäisten haittatapahtumien (MACE) riski. Tällaisia ovat etenkin potilaat, joilla on entuudestaan hypertensio, potilaat, joilla on diagnosoitu sydän- ja verisuonitauti tai aivoverisuonten sairaus, ja iäkkäät potilaat.
Solriamfetolihoidon jatkamisen tarve tulee arvioida säännöllisesti. Jos potilaalla esiintyy verenpaineen tai sykkeen kohoamista, jota ei saada hallintaan solriamfetoliannosta pienentämällä tai muulla asianmukaisella lääketieteellisellä interventiolla, on harkittava solriamfetolihoidon lopettamista. Muita verenpainetta ja sykettä kohottavia lääkevalmisteita tulee käyttää varoen (ks. kohta Yhteisvaikutukset).
Annostus
Narkolepsia
Suositeltu aloitusannos on 75 mg kerran päivässä, heräämisen yhteydessä. Potilaille, jotka kärsivät vaikeammasta uneliaisuudesta, voidaan harkita 150 mg:n aloitusannosta, jos se on kliinisesti aiheellista. Annosta voidaan suurentaa asteittain kliinisen vasteen mukaan kaksinkertaistamalla annos 3 päivän välein tai tätä harvemmin. Suurin suositeltu vuorokausiannos on 150 mg kerran päivässä.
Obstruktiivinen uniapnea
Suositeltu aloitusannos on 37,5 mg kerran päivässä, heräämisen yhteydessä. Annosta voidaan suurentaa asteittain kliinisen vasteen mukaan kaksinkertaistamalla annos 3 päivän välein tai tätä harvemmin. Suurin suositeltu vuorokausiannos on 150 mg kerran päivässä.
Sunosi-valmisteen ottamista alle 9 tuntia ennen nukkumaanmenoaikaa on vältettävä, koska se saattaa vaikuttaa yöuneen.
Pitkäaikainen käyttö
Pitkäaikaisessa solriamfetolihoidossa hoidon jatkamisen tarve ja annoksen sopivuus on arvioitava säännöllisesti.
Erityisryhmät
Iäkkäät potilaat (> 65 vuotta)
Iäkkäitä potilaita koskevia tietoja on niukasti. Pienempien annosten käyttöä ja tiivistä seurantaa on harkittava tässä potilasryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koska solriamfetoli eliminoituu pääasiassa munuaisten kautta ja koska heikentynyt munuaisten toiminta on todennäköisempi iäkkäillä potilailla, annosta saattaa olla tarpeen muuttaa kreatiniinin puhdistuman perusteella.
Munuaisten vajaatoiminta
Lievä munuaisten vajaatoiminta (kreatiniinin puhdistuma 60–89 ml/min): Annosta ei tarvitse muuttaa.
Kohtalainen munuaisten vajaatoiminta (kreatiniinin puhdistuma 30–59 ml/min): Suositeltu aloitusannos on 37,5 mg kerran päivässä. Annosta voidaan viiden vuorokauden jälkeen suurentaa enintään 75 mg:aan kerran päivässä.
Vaikea munuaisten vajaatoiminta (kreatiniinin puhdistuma 15–29 ml/min): Suositeltu annos on 37,5 mg kerran päivässä.
Loppuvaiheen munuaissairaus (kreatiniinin puhdistuma < 15 ml/min): Solriamfetolin käyttöä ei suositella potilaille, joilla on loppuvaiheen munuaissairaus.
Pediatriset potilaat
Sunosi-valmisteen turvallisuutta ja tehoa lasten ja nuorten (< 18 vuotta) hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Sunosi on tarkoitettu suun kautta annettavaksi.
Sunosi voidaan ottaa ruoan kanssa tai ilman.
37,5 mg:n annos saadaan puolittamalla 75 mg:n tabletti siinä olevan jakouurteen avulla.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Sydäninfarkti viimeisen vuoden aikana, epävakaa angina pectoris, huonossa hoitotasapainossa oleva hypertensio, vakavat sydämen rytmihäiriöt ja muut vakavat sydänongelmat.
- Samanaikainen käyttö monoamiinioksidaasin estäjien (MAO:n estäjien) kanssa tai käyttö MAO:n estäjähoidon lopettamisen jälkeisten 14 vuorokauden aikana (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Psykiatriset oireet
Solriamfetolia ei ole arvioitu potilailla, joilla on tai on ollut psykoosi tai kaksisuuntainen mielialahäiriö. Varovaisuutta on noudatettava tällaisilla potilailla, sillä psykiatriset haittavaikutukset saattavat pahentaa olemassa olevien psykiatristen häiriöiden oireita (esim. maanisia jaksoja).
Solriamfetolihoitoa saavia potilaita on seurattava huolellisesti haittavaikutusten kuten ahdistuneisuuden, unettomuuden ja ärtyneisyyden varalta. Näitä haittavaikutuksia havaittiin yleisesti hoidon aloitusvaiheessa, mutta ne hävisivät yleensä hoidon jatkuessa. Jos tällaiset oireet pitkittyvät tai pahenevat, on harkittava annoksen pienentämistä tai hoidon lopettamista.
Verenpaine ja syke
Kliinisten tutkimusten tulokset osoittivat solriamfetolihoidon johtavan annosriippuvaiseen systolisen ja diastolisen verenpaineen sekä sykkeen kohoamiseen.
Epidemiologiset tiedot osoittavat, että kroonisesti koholla oleva verenpaine lisää vakavien sydän- ja verisuonihaittatapahtumien (MACE), mukaan lukien aivohalvauksen, sydänkohtauksen ja sydän- ja verisuoniperäisen kuoleman, riskiä. Absoluuttisen riskin lisääntymisen suuruus riippuu verenpaineen kohoamisen suuruudesta ja taustalla olevasta vakavien sydän- ja verisuonihaittatapahtumien riskistä hoidettavassa väestössä. Monilla narkolepsiaa tai obstruktiivista uniapneaa sairastavilla potilailla on useita vakavien sydän- ja verisuonihaittatapahtumien riskitekijöitä, kuten hypertensio, diabetes, hyperlipidemia ja korkea painoindeksi (BMI).
Käyttö on vasta-aiheista potilaille, joilla on epävakaa sydän- ja verisuonitauti, vakava sydämen rytmihäiriö tai muita vakavia sydänongelmia (ks. kohta Vasta-aiheet).
Kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla saattaa olla tavallista suurempi verenpaineen ja sykkeen kohoamisen riski solriamfetolin pidentyneen puoliintumisajan vuoksi.
Väärinkäyttö
Sunosi-valmisteen väärinkäytön mahdollisuutta ihmisellä arvioiva tutkimus osoitti väärinkäyttöriskin olevan pieni. Lääkkeestä pitämistä mittaavat Drug Liking ‑pisteet tässä kliinisessä tutkimuksessa olivat solriamfetolilla suuremmat kuin lumelääkkeellä, mutta yleisesti ottaen samankaltaiset tai pienemmät kuin fentermiinillä (heikolla piristeellä). Varovaisuutta on noudatettava hoidettaessa potilaita, joiden anamneesissa on ollut piristeiden (esim. metyylifenidaatin, amfetamiinin) tai alkoholin väärinkäyttöä, ja tällaisia potilaita on seurattava solriamfetolin virheellisen käytön tai väärinkäytön merkkien varalta.
Ahdaskulmaglaukooma
Solriamfetolia ottavilla potilailla saattaa esiintyä mustuaisten laajentumista. Varovaisuutta suositellaan hoidettaessa potilaita, joilla on kohonnut silmänpaine tai jotka ovat vaarassa sairastua ahdaskulmaglaukoomaan.
Naiset, jotka voivat tulla raskaaksi, tai heidän kumppaninsa
Naisten, jotka voivat tulla raskaaksi, tai heidän mieskumppaniensa on käytettävä tehokasta ehkäisyä solriamfetolihoidon aikana (ks. kohta Raskaus ja imetys).
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty (ks. kohta Farmakokinetiikka).
Solriamfetolia ei saa antaa samanaikaisesti MAO:n estäjien kanssa eikä MAO:n estäjähoidon lopettamisen jälkeisten 14 vuorokauden aikana, sillä se voi suurentaa hypertensiivisen reaktion riskiä (ks. kohta Vasta-aiheet).
Samanaikaisesti käytettäviä verenpainetta ja sykettä kohottavia lääkevalmisteita tulee käyttää varoen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääkevalmisteilla, jotka suurentavat dopamiinipitoisuutta tai sitoutuvat suoraan dopamiinireseptoreihin, saattaa olla farmakodynaamisia yhteisvaikutuksia solriamfetolin kanssa. Tällaisten lääkevalmisteiden samanaikaisessa käytössä tulee noudattaa varovaisuutta.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja solriamfetolin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Sunosi-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Solriamfetolia erittyy rintamaitoon noin 4 % äidin painoon suhteutetusta annoksesta (ks. kohta Farmakokinetiikka). Solriamfetolin vaikutusta vastasyntyneisiin/imeväisiin tai sen vaikutusta maidontuotantoon ei tunneta. Riskiä imetettävälle lapselle ei voida sulkea pois.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Sunosi-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Solriamfetolin vaikutusta ihmisen hedelmällisyyteen ei tunneta. Eläinkokeissa ei ole havaittu suoria tai epäsuoria vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Valmisteella odotetaan olevan vähäinen vaikutus ajokykyyn potilailla, jotka saavat solriamfetolia vakaalla annostuksella. Solriamfetolin annon jälkeen saattaa esiintyä huimausta ja tarkkaavuuden häiriintymistä (ks. kohta Haittavaikutukset).
Solriamfetolia ottaville potilaille, joilla esiintyy poikkeavan voimakasta uneliaisuutta, tulee kertoa, ettei heidän valvetilansa välttämättä palaudu normaaliksi. Liiallisesta päiväaikaisesta uneliaisuudesta kärsivien potilaiden –myös solriamfetolia ottavien potilaiden – uneliaisuuden aste tulee arvioida tiheästi, ja potilaita on tarvittaessa neuvottava välttämään ajamista tai muita mahdollisesti vaarallisia toimia, varsinkin hoidon alussa tai kun annosta muutetaan.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoidut haittavaikutukset olivat päänsärky (11,1 %), pahoinvointi (6,6 %) ja ruokahalun heikentyminen (6,8 %).
Haittavaikutusten taulukkomuotoinen luettelo
Haittavaikutusten yleisyydet on määritetty seuraavien MedDRA-yleisyysluokkien mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Elinjärjestelmä | Haittavaikutukset | Yleisyys |
Aineenvaihdunta ja ravitsemus | Ruokahalun heikentyminen | Yleinen |
Psyykkiset häiriöt | Ahdistuneisuus | Yleinen |
Unettomuus | Yleinen | |
Ärtyneisyys | Yleinen | |
Bruksismi | Yleinen | |
Kiihtyneisyys | Melko harvinainen | |
Levottomuus | Melko harvinainen | |
Hermosto | Päänsärky | Hyvin yleinen |
Huimaus | Yleinen | |
Tarkkaavuuden häiriintyminen | Melko harvinainen | |
Vapina | Melko harvinainen | |
Sydän | Palpitaatio | Yleinen |
Takykardia | Melko harvinainen | |
Verisuonisto | Hypertensio | Melko harvinainen |
Hengityselimet, rintakehä ja välikarsina | Yskä | Yleinen |
Hengenahdistus | Melko harvinainen | |
Ruoansulatuselimistö | Pahoinvointi | Yleinen |
Ripuli | Yleinen | |
Suun kuivuminen | Yleinen | |
Vatsakipu | Yleinen | |
Ummetus | Yleinen | |
Oksentelu | Yleinen | |
Iho ja ihonalainen kudos | Liikahikoilu | Yleinen |
Yleisoireet ja antopaikassa todettavat haitat | Hermostuneisuus | Yleinen |
Epämiellyttävät tuntemukset rinnassa | Yleinen | |
Rintakipu | Melko harvinainen | |
Jano | Melko harvinainen | |
Tutkimukset | Sykkeen kohoaminen | Melko harvinainen |
Verenpaineen kohoaminen | Yleinen | |
Painonlasku | Melko harvinainen |
Tiettyjen haittavaikutusten kuvaus
Hoidon aloitusvaihe
Suurin osa yleisimmin raportoiduista haittavaikutuksista ilmeni ensimmäisten 2 viikon kuluessa hoidon aloittamisesta ja suurimmalla osalla potilaista ne hävisivät alle 2 viikossa (mediaanikesto).
Yliherkkyysreaktiot
Valmisteen markkinoille tulon jälkeen on raportoitu yliherkkyysreaktioita, joiden yhteydessä on esiintynyt vähintään yhtä seuraavista: erytematoottinen ihottuma, ihottuma, nokkosihottuma (ks. kohta Vasta-aiheet).
Annosriippuvaiset haittavaikutukset
12 viikon pituisissa kliinisissä tutkimuksissa, joissa 37,5 mg:n, 75 mg:n ja 150 mg:n vuorokausiannoksia solriamfetolia verrattiin lumelääkkeeseen, annosriippuvaisia haittavaikutuksia olivat päänsärky, pahoinvointi, ruokahalun heikentyminen, ahdistuneisuus, ripuli ja suun kuivuminen. Annosriippuvuudet olivat yleisesti ottaen samankaltaisia obstruktiivista uniapneaa ja narkolepsiaa sairastavien potilaiden välillä. Tiettyjä tapahtumia, kuten ahdistuneisuutta, unettomuutta, ärtyneisyyttä ja kiihtyneisyyttä, havaittiin yleisesti hoidon aloitusvaiheessa, mutta ne hävisivät yleensä hoidon jatkuessa. Jos tällaiset oireet pitkittyvät tai pahenevat, on harkittava annoksen pienentämistä tai hoidon lopettamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hoidon lopettaminen
12 viikon pituisissa lumekontrolloiduissa kliinisissä tutkimuksissa 11 potilasta solriamfetolihoitoa saavasta 396 potilaasta (3 %) lopetti hoidon haittavaikutusten takia. Vastaava määrä lumelääkettä saavilla potilailla oli 1 potilas 226:sta (< 1 %). Hoidon lopettamiseen johtaneet haittavaikutukset, joita esiintyi useammalla kuin yhdellä solriamfetolihoitoa saavalla potilaalla ja joiden esiintyvyys oli suurempi kuin lumelääkettä saavilla potilailla, olivat ahdistuneisuus, palpitaatio ja levottomuus. Kaikkien näiden haittavaikutusten esiintyvyys oli alle 1 %.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kautta:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Solriamfetolin yliannostuksia ei ole raportoitu kliinisissä tutkimuksissa.
Terveillä tutkittavilla esiintyi haittavaikutuksina yksi lievä tardiivin dyskinesian tapaus ja yksi kohtalaisen akatisian tapaus, jotka ilmenivät terapeuttisia annoksia suuremmalla, 900 mg:n annoksella. Oireet hävisivät hoidon lopettamisen jälkeen.
Erityistä vastalääkettä ei ole saatavilla. Tahattomassa yliannostustapauksessa tulee antaa oireenmukaista ja elintoimintoja tukevaa hoitoa, ja potilasta on seurattava huolellisesti tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Masennuslääkkeet ja keskushermostoa stimuloivat lääkeaineet, keskushermostoon vaikuttavat sympatomimeetit, ATC-koodi: N06BA14
Vaikutusmekanismi
Mekanismia/mekanismeja, jo(i)lla solriamfetoli parantaa narkolepsiaan tai obstruktiiviseen uniapneaan liittyvästä liiallisesta päiväaikaisesta uneliaisuudesta kärsivien potilaiden valvetilaa, ei ole täysin selvitetty. Teho saattaa kuitenkin olla seurausta solriamfetolin aktiivisuudesta noradrenaliinin ja dopamiinin takaisinoton estäjänä (NDRI).
Farmakodynaamiset vaikutukset
In vitro -tiedot
Radioligandin sitoutumista arvioivissa kokeissa, joissa käytettiin kloonattuja ihmisen reseptoreita/kuljettajaproteiineja ilmentäviä soluja, solriamfetoli osoitti affiniteettia dopamiinin kuljettajaan (replikaatin Ki = 6,3 ja 14,2 µM) ja noradrenaliinin kuljettajaan (replikaatin Ki = 3,7 ja > 10 µM), mutta ei merkittävää affiniteettia serotoniinin kuljettajaan. Solriamfetoli esti näiden solujen dopamiinin takaisinottoa (replikaatin IC50 = 2,9 ja 6,4 µM) ja noradrenaliinin takaisinottoa (IC50 = 4,4 µM), mutta ei serotoniinin takaisinottoa.
In vivo -tiedot eläinkokeista
Autoradiografiakokeessa parenteraalisilla annoksilla, joilla oli selvä rottien valvetilaa edistävä vaikutus, solriamfetoli suurensi yksilöllisiä aivojuovion dopamiinipitoisuuksia ja prefrontaalisen aivokuoren noradrenaliinipitoisuuksia eikä sitoutunut merkittävästi rottien dopamiinin ja noradrenaliinin kuljettajiin.
Kliininen teho ja turvallisuus
Narkolepsia
Tutkimus 1 oli 12 viikon pituinen satunnaistettu, kaksoissokkoutettu, lumekontrolloitu rinnakkaisryhmätutkimus, jossa arvioitiin solriamfetolin tehoa narkolepsiaa (johon saattoi liittyä katapleksiaa) sairastavilla aikuispotilailla.
Tutkimuksen mukaanottokriteereinä olivat, että potilaat kärsivät liiallisesta päiväaikaisesta uneliaisuudesta (Epworth Sleepiness Scale ‑uneliaisuusasteikon [ESS] pisteet vähintään 10) ja että valvetilan ylläpitäminen tuotti potilaille vaikeuksia (ensimmäisten neljän lähtötilanteessa tehdyn 40 minuutin hereilläpysymistestin [Maintenance of Wakefulness Test, MWT] nukahtamisviiveiden keskiarvo alle 25 minuuttia).
Tehon mittareina olivat muutokset lähtötilanteesta viikkoon 12 seuraavissa: kyky pysyä hereillä MWT-testien keskimääräisellä nukahtamisviiveellä mitattuna, liiallinen päiväaikainen uneliaisuus ESS-pisteillä mitattuna ja yleisen kliinisen tilan parannus Patient Global Impression of Change (PGIc) -asteikolla mitattuna. ESS on kahdeksankohtainen potilaskysely, joka mittaa nukahtamisen todennäköisyyttä tavanomaisten päivittäisten toimien yhteydessä. PGIc on seitsemän vaihtoehtoa käsittävä asteikkokysely, jonka avulla arvioidaan potilaan raportoimaa muutosta kliinisessä tilassaan. Asteikon kaksi ääripäätä ovat ”erittäin paljon parempi” ja ”erittäin paljon huonompi”.
Narkolepsiaa sairastavien potilaiden heikentynyt valvetila ja liiallinen päiväaikainen uneliaisuus arvioitiin lähtötilanteessa MWT-testin nukahtamisviiveen keskiarvon ja ESS-pisteiden avulla (taulukko 1). Useimmat potilaat olivat aiemmin käyttäneet keskushermostostimulantteja. Katapleksiaa esiintyi kaiken kaikkiaan noin puolella potilaista. Demografiset tekijät ja lähtötilanteen arvot olivat samankaltaisia potilailla, joilla esiintyi katapleksiaa, ja potilailla, joilla ei esiintynyt katapleksiaa.
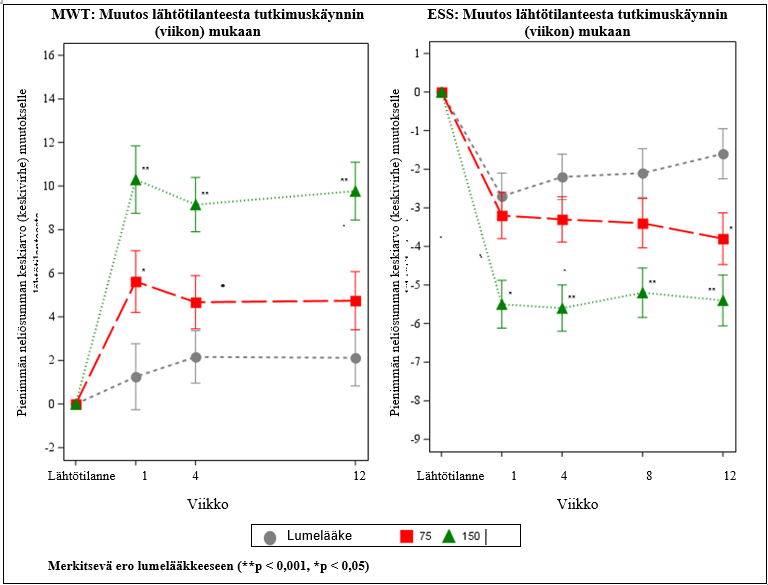
Narkolepsiaa sairastavat potilaat satunnaistettiin tässä tutkimuksessa saamaan solriamfetolia 75 mg, 150 mg tai 300 mg (kaksi kertaa suurin suositeltu vuorokausiannos) tai lumelääkettä kerran päivässä. Viikolla 12 potilailla, jotka oli satunnaistettu 150 mg:n annosryhmään, todettiin tilastollisesti merkitseviä parannuksia MWT-testien ja ESS-kyselyn tuloksissa (rinnakkaiset ensisijaiset päätetapahtumat) sekä PGIc-kyselyn tuloksissa (tärkein toissijainen päätetapahtuma) lumelääkeryhmään nähden. Potilailla, jotka oli satunnaistettu 75 mg:n annosryhmään, todettiin tilastollisesti merkitsevä parannus ESS-kyselyn, mutta ei MWT-testien ja PGIc-kyselyn, tuloksissa (taulukko 1). Nämä vaikutukset olivat annosriippuvaisia, ne havaittiin viikolla 1 ja ne säilyivät tutkimuksen loppuun asti (kuva 1). Vaikutuksen voimakkuuden havaittiin yleisesti ottaen olevan pienempi niillä potilailla, joiden uneliaisuus oli lähtötilanteessa vaikeampi, verrattuna saman annosryhmän potilaisiin, joiden uneliaisuus oli lähtötilanteessa lievempi. Viikolla 12 valvetila niillä potilailla, jotka oli satunnaistettu solriamfetolin 150 mg:n annosryhmään, oli tilastollisesti merkitsevästi parempi kuin lumelääkeryhmässä koko päivän ajan kaikissa viidessä MWT-testissä. Testit toteutettiin noin 9 tunnin ajanjaksolla, joka alkoi lääkkeen annosta. Kyky suorittaa päivittäisiä toimia parani annosriippuvaisesti Functional Outcomes of Sleep Questionnaire Short Version (FOSQ‑10) -kyselyllä mitattuna. Yli 150 mg:n vuorokausiannosten teho ei ole riittävän paljon suurempi annosriippuvaisiin haittavaikutuksiin nähden.
Solriamfetolin käyttö ei vaikuttanut yöuneen unipolygrafialla mitattuna.
Taulukko 1.Yhteenveto tehoa koskevista tuloksista viikolla 12 narkolepsiaa sairastavilla potilailla tutkimuksessa 1
| Hoitoryhmä (N) | Pisteiden keskiarvo lähtö-tilanteessa (SD) | Keski-määräinen muutos lähtö-tilanteesta | Ero lumelääkkeeseen (95 %:n luottamusväli) | P-arvo | |
| MWT (min) | Tutkimus 1 Lumelääke (58) Sunosi 75 mg (59) Sunosi 150 mg (55) | 6,15 (5,68) 7,50 (5,39) 7,85 (5,74) | LS-keskiarvo (SE) 2,12 (1,29) 4,74 (1,34) 9,77 (1,33) | 2,62 (-1,04, 6,28) 7,65 (3,99, 11,31) | 0,1595 < 0,0001 |
| ESS | Tutkimus 1 Lumelääke (58) Sunosi 75 mg (59) Sunosi 150 mg (55) | 17,3 (2,86) 17,3 (3,53) 17,0 (3,55) | LS-keskiarvo (SE) -1,6 (0,65) -3,8 (0,67) -5,4 (0,66) | -2,2 (-4,0, -0,3) -3,8 (-5,6, -2,0) | 0,0211 < 0,0001 |
| Niiden potilaiden osuus, joiden kliininen tila parantui* | Prosenttiero lumelääkkeeseen (95 %:n luottamusväli) | P-arvo | |||
| PGIc | Tutkimus 1 Lumelääke (58) Sunosi 75 mg (59) Sunosi 150 mg (55) | 39,7 % 67,8 % 78,2 % | 28,1 (10,8, 45,5) 38,5 (21,9, 55,2) | 0,0023† < 0,0001 | |
SD = keskihajonta; SE = keskivirhe; LS-keskiarvo = pienimmän neliösumman keskiarvo; Ero lumelääkkeeseen = pienimmän neliösumman keskiarvo vaikuttavan lääkkeen ja lumelääkkeen väliselle erolle muutoksessa lähtötilanteesta. MWT-tulokset perustuvat ensimmäisiin neljään MWT-testiin, ja positiivinen muutos lähtötilanteesta tarkoittaa parannusta nukahtamisviiveessä. ESS-pisteiden negatiivinen muutos lähtötilanteesta tarkoittaa parannusta liiallisessa päiväaikaisessa uneliaisuudessa. *Niiden potilaiden prosenttiosuus, joiden kliininen tila parantui, käsittää ne potilaat, jotka raportoivat PGIc-kyselyssä tilanteensa parantuneen erittäin paljon, paljon tai hieman;
†Nimellinen p-arvo.
Kuva 1: Rinnakkaiset ensisijaiset tehon päätetapahtumat narkolepsiaa sairastavilla potilailla tutkimuksessa 1
Obstruktiivinen uniapnea
Tutkimus 2 oli 12 viikon pituinen satunnaistettu, kaksoissokkoutettu, lumekontrolloitu rinnakkaisryhmätutkimus, jossa arvioitiin solriamfetolin tehoa obstruktiivista uniapneaa sairastavilla aikuispotilailla. Tutkimuksen rinnakkaiset ensisijaiset päätetapahtumat ja tärkeimmät toissijaiset päätetapahtumat olivat samat kuin tutkimuksessa 1. Tutkimus 3 oli 6 viikon pituinen, kaksoissokkoutettu, lumekontrolloitu, satunnaistetun tutkimuslääkkeettömän vaiheen sisältävä tutkimus, jossa arvioitiin solriamfetolin tehoa obstruktiivista uniapneaa sairastavilla aikuispotilailla. Tehon mittareina satunnaistetussa tutkimuslääkkeettömässä vaiheessa olivat muutokset MWT-testien ja ESS-kyselyn tuloksissa ja huonontuminen kliinistä yleistilaa arvioivan PGIc-kyselyn tuloksissa satunnaistetun tutkimuslääkkeettömän vaiheen alusta vaiheen loppuun.
Kummankin tutkimuksen mukaanottokriteereinä olivat, että potilaat kärsivät liiallisesta päiväaikaisesta uneliaisuudesta (ESS-pisteet ≥ 10) ja että valvetilan ylläpitäminen tuotti potilaille vaikeuksia (ensimmäisten neljän lähtötilanteessa tehdyn MWT-testin nukahtamisviiveiden keskiarvo < 30 minuuttia). Lisäksi potilaiden piti: 1) parhaillaan käyttää jotakin obstruktiivisen uniapnean ensisijaista hoitoa (riippumatta hoitoon sitoutumisen asteesta); 2) olla aiemmin käyttänyt jotakin ensisijaista hoitoa vähintään yhden kuukauden ajan siten, että hoitoon oli tehty vähintään yksi dokumentoitu muutos; tai 3) olla saanut leikkaushoitoa taustalla olevaan ahtaumaansa. Potilaita kehotettiin jatkamaan parhaillaan käyttämäänsä obstruktiivisen uniapnean ensisijaista hoitoa samalla tavalla koko tutkimuksen ajan. Potilaat suljettiin pois tutkimuksesta ensisijaisen hoidon käyttönsä perusteella vain siinä tapauksessa, että he eivät olleet suostuneet kokeilemaan jotakin ensisijaista hoitoa, kuten CPAP-hoitoa, suuhun asetettavaa laitetta tai leikkaushoitoa taustalla olevan ahtaumansa hoitoon.
Tutkimuksessa 2 obstruktiivista uniapneaa sairastavien potilaiden heikentynyt valvetila ja liiallinen päiväaikainen uneliaisuus arvioitiin lähtötilanteessa MWT-testin nukahtamisviiveiden keskiarvon ja ESS-pisteiden avulla (taulukko 2). Noin 71 % potilaista noudatti jotakin obstruktiivisen uniapnean ensisijaista hoitoa (esim. ≥ 4 tuntina yössä ≥ 70 %:na öistä). Potilaiden demografiset tekijät ja lähtötilanteen arvot olivat samankaltaisia riippumatta siitä, noudattivatko he jotakin obstruktiivisen uniapnean ensisijaista hoitoa vai eivät. Lähtötilanteessa obstruktiivisen uniapnean ensisijaista hoitoa käytti noin 73 % potilaista. Näistä potilaista 92 % käytti ylipainehoitoa (PAP).
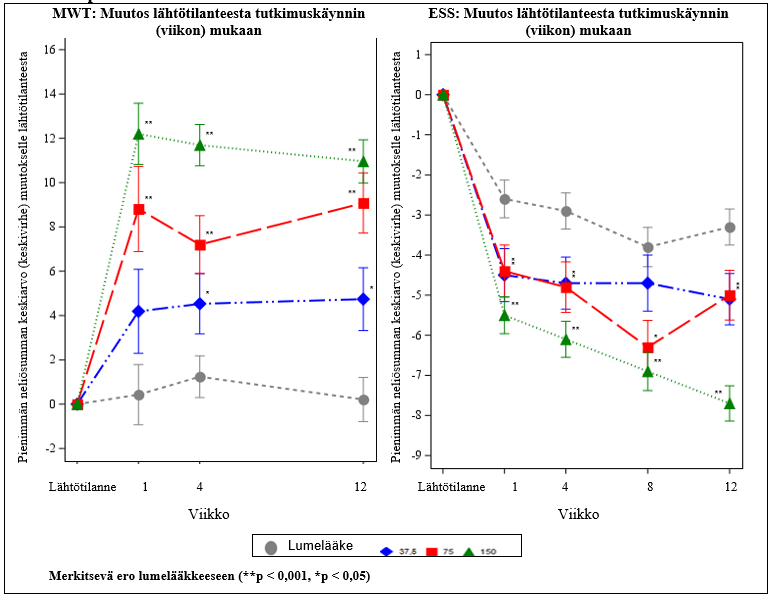
Potilaat satunnaistettiin saamaan solriamfetolia 37,5 mg, 75 mg, 150 mg tai 300 mg (kaksi kertaa suurin suositeltu vuorokausiannos) tai lumelääkettä kerran päivässä. Viikolla 12 potilailla, jotka oli satunnaistettu 75 mg:n ja 150 mg:n annosryhmiin, todettiin tilastollisesti merkitseviä parannuksia MWT-testien ja ESS-kyselyn tuloksissa (rinnakkaiset ensisijaiset päätetapahtumat) sekä PGIc-kyselyn tuloksissa (tärkein toissijainen päätetapahtuma) lumelääkeryhmään nähden (taulukko 2). Potilailla, jotka oli satunnaistettu 37,5 mg:n solriamfetoliryhmään, todettiin tilastollisesti merkitseviä parannuksia MWT-testien ja ESS-kyselyn tuloksissa. Nämä vaikutukset havaittiin viikolla 1, ne säilyivät tutkimuksen loppuun asti ja ne olivat annosriippuvaisia (kuva 2). Viikolla 12 valvetila niillä potilailla, jotka oli satunnaistettu 75 mg:n ja 150 mg:n Sunosi-annosryhmiin, oli tilastollisesti merkitsevästi parempi koko päivän ajan kuin lumelääkeryhmässä kaikissa viidessä MWT-testissä. Testit toteutettiin noin 9 tunnin ajanjaksolla, joka alkoi lääkkeen annosta. Kyky suorittaa päivittäisiä toimia parani annosriippuvaisesti FOSQ‑10-kyselyllä mitattuna. Yli 150 mg:n vuorokausiannosten teho ei ole riittävän paljon suurempi annosriippuvaisiin haittavaikutuksiin nähden.
Solriamfetolin käyttö tutkimuksessa 2 ei vaikuttanut yöuneen unipolygrafialla mitattuna. Kliinisesti merkittäviä muutoksia potilaiden obstruktiivisen uniapnean ensisijaisen hoidon käytössä ei havaittu 12-viikkoisen tutkimuksen aikana missään hoitoryhmässä. Sen, noudattivatko potilaat obstruktiivisen uniapnean ensisijaista hoitoa vai ei, ei havaittu vaikuttavan tehoon.
Tutkimuksessa 3 potilaiden lähtötilanteen demografiset ja sairauteen liittyvät ominaisuudet olivat samankaltaisia kuin tutkimuksen 2 potilailla. Hoito aloitettiin 75 mg:n annoksella kerran päivässä, minkä jälkeen annosta voitiin suurentaa tehon ja siedettävyyden mukaan yhdellä annostasolla 3 vuorokauden välein tai tätä harvemmin 150 mg:aan tai 300 mg:aan. Potilaan annosta voitiin myös asteittain pienentää 75 mg:aan tai 150 mg:aan. Neljä viikkoa kestäneen avoimen hoitojakson jälkeisessä satunnaistetussa tutkimuslääkkeettömässä vaiheessa solriamfetolihoitoa saavien potilaiden parantunut tila säilyi, kun taas lumelääkettä saavien potilaiden tila huonontui (hoitoeron pienimmän neliösumman keskiarvo oli MWT-testien osalta 11,2 minuuttia ja ESS-kyselyn osalta ‑4,6; p < 0,0001 kummallekin). Harvempi solriamfetolia saava potilas raportoi PGIc-kyselyssä kliinisen tilansa huonontuneen verrattuna lumelääkettä saaviin potilaisiin (prosentuaalinen ero oli 30 %; p = 0,0005).
Taulukko 2. Yhteenveto tehoa koskevista tuloksista viikolla 12 obstruktiivista uniapneaa sairastavilla potilailla tutkimuksessa 2
| Hoitoryhmä (N) | Pisteiden keskiarvo lähtö-tilanteessa (SD) | Keski-määräinen muutos lähtö-tilanteesta | Ero lumelääkkeeseen (95 %:n luottamusväli) | P-arvo | |
MWT (min) | Lumelääke (114) Sunosi 37,5 mg (56) Sunosi 75 mg (58) Sunosi 150 mg (116) | 12,58 (7,14) 13,6 (8,15) 12,44 (6,91) 12,54 (7,18) | LS-keskiarvo (SE) 0,21 (1,0) 4,74 (1,42) 9,08 (1,36) 10,96 (0,97) | - 4,53 (1,16, 7,90) 8,87 (5,59, 12,14) 10,74 (8,05, 13,44) | - 0,0086 < 0,0001 < 0,0001 |
| ESS | Lumelääke (114) Sunosi 37,5 mg (56) Sunosi 75 mg (58) Sunosi 150 mg (116) | 15,6 (3,32) 15,1 (3,53) 15,0 (3,51) 15,1 (3,37) | LS-keskiarvo (SE) -3,3 (0,45) -5,1 (0,64) -5,0 (0,62) -7,7 (0,44) | - -1,9 (-3,4, -0,3) -1,7 (-3,2, -0,2) -4,5 (-5,7, -3,2) | - 0,0161 0,0233 < 0,0001 |
| Niiden potilaiden osuus, joiden kliininen tila parantui* | Prosenttiero lumelääkkeeseen (95 %:n luottamusväli) | P-arvo | |||
| PGIc | Lumelääke (114) Sunosi 37,5 mg (56) Sunosi 75 mg (58) Sunosi 150 mg (116) | 49,1 % 55,4 % 72,4 % 89,7 % | - 6,2 (-9,69, 22,16) 23,3 (8,58, 38,01) 40,5 (29,81, 51,25) | - 0,4447 0,0035 < 0,0001 | |
SD = keskihajonta; SE = keskivirhe; LS-keskiarvo = pienimmän neliösumman keskiarvo; Ero lumelääkkeeseen = pienimmän neliösumman keskiarvo vaikuttavan lääkkeen ja lumelääkkeen väliselle erolle muutoksessa lähtötilanteesta. MWT-tulokset perustuvat ensimmäisiin neljään MWT-testiin, ja positiivinen muutos lähtötilanteesta tarkoittaa parannusta nukahtamisviiveessä. ESS-pisteiden negatiivinen muutos tarkoittaa parannusta liiallisessa päiväaikaisessa uneliaisuudessa. *Niiden potilaiden prosenttiosuus, joiden kliininen tila parantui, käsittää ne potilaat, jotka raportoivat PGIc-kyselyssä tilanteensa parantuneen erittäin paljon, paljon tai hieman.
Kuva 2: Rinnakkaiset ensisijaiset tehon päätetapahtumat obstruktiivista uniapneaa sairastavilla potilailla tutkimuksessa 2
Pitkän aikavälin teho narkolepsiassa ja obstruktiivisessa uniapneassa
Tutkimus 4 oli pitkäaikainen turvallisuutta ja tehon säilymistä arvioiva tutkimus narkolepsiaa tai obstruktiivista uniapneaa sairastavilla aikuispotilailla, jotka olivat suorittaneet aiemman tutkimuksen kokonaan. Tutkimuksessa annettiin solriamfetolihoitoa enintään vuoden ajan, ja siihen sisältyi 2 viikon pituinen satunnaistettu tutkimuslääkkeetön, lumelääkekontrolloitu vaihe, joka aloitettiin, kun solriamfetolihoitoa oli annettu vähintään 6 kuukauden ajan.
Tehon mittareina satunnaistetussa tutkimuslääkkeettömässä vaiheessa olivat muutos ESS-kyselyn tuloksissa ja huonontuminen kliinistä yleistilaa arvioivan PGIc-kyselyn tuloksissa satunnaistetun tutkimuslääkkeettömän vaiheen alusta vaiheen loppuun. Aloitusannos ja annoksen asteittaisen muuttamisen periaate olivat samat kuin tutkimuksessa 3.
Vähintään 6 kuukautta kestäneen avoimen hoitojakson jälkeisessä satunnaistetussa tutkimuslääkkeettömässä vaiheessa solriamfetolihoitoa saaneiden potilaiden parantunut tila säilyi, kun taas lumelääkettä saaneiden potilaiden tila huonontui (hoitoeron pienimmän neliösumman keskiarvo ESS-kyselyn osalta oli ‑3,7; p < 0,0001). Harvempi solriamfetolia saanut potilas raportoi PGIc-kyselyssä kliinisen tilansa huonontuneen verrattuna lumelääkettä saaneisiin potilaisiin (prosentuaalinen ero oli ‑36,2 %; p < 0,0001). Nämä tulokset osoittavat solriamfetolin tehon säilyvän pitkällä aikavälillä, kun hoitoa jatketaan, ja hoidosta saatavan hyödyn häviävän, kun hoito lopetetaan.
Obstruktiivisen uniapnean ensisijaisen hoidon käyttö niillä potilailla, jotka käyttivät tällaista hoitoa tutkimuksen alussa, ei muuttunut pitkäaikaisen tutkimuksen kuluessa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Sunosi-valmisteen käytöstä narkolepsiasta johtuvan liiallisen päiväaikaisen uneliaisuuden oireenmukaisessa hoidossa kaikissa 6 – < 18-vuotiaiden pediatristen potilaiden potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Paastotilassa suun kautta otetun solriamfetolin biologinen hyötyosuus on noin 95 % ja huippupitoisuus plasmassa saavutetaan noin 2 tunnissa (Tmax-arvon mediaani; vaihteluväli 1,25–3 tuntia).
Solriamfetolin ottamisella runsasrasvaisen aterian kanssa oli minimaalinen vaikutus Cmax- ja AUC-arvoihin, mutta Tmax-ajassa todettiin noin 1 tunnin viive. Nämä tulokset osoittavat, että solriamfetoli voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Solriamfetolin näennäinen jakautumistilavuus on noin 198,7 l, mikä osoittaa lääkeaineen jakautuvan laajasti kudoksiin verenkierrosta. Plasman proteiineihin sitoutuminen oli 13,3–19,4-prosenttista solriamfetolin pitoisuusvälillä 0,059–10,1 µg/ml ihmisen plasmassa. Veren ja plasman solriamfetolipitoisuuksien suhde vaihteli välillä 1,16–1,29, mikä viittaa vähäiseen verisoluihin sitoutumiseen.
Biotransformaatio
Solriamfetolin metaboloituminen on minimaalista ihmisellä.
Yhteisvaikutukset
Lukuun ottamatta lievää CYP2D6:n estoa (IC50: 360 µM) solriamfetoli ei ole kliinisesti merkittävillä pitoisuuksilla minkään tärkeän CYP-entsyymin substraatti eikä estäjä eikä CYP1A2-, CYP2B6-, CYP3A4- eikä UGT1A1 -entsyymien indusoija. Solriamfetoli ei vaikuta olevan seuraavien kuljettajaproteiinien substraatti eikä estäjä: P‑gp, BCRP, OATP1B1, OATP1B3, OAT1 ja OAT3. Solriamfetoli erittyy pääasiassa muuttumattomana virtsaan, ja se on useiden munuaisten kationisia vaikuttavia aineita kuljettavien proteiinien substraatti heikolla affiniteetilla, ilman voimakasta affiniteettia mihinkään yksittäiseen testattuun kuljettajaproteiiniin (OCT2, MATE1, OCTN1 ja OCTN2). Solriamfetoli ei estä munuaisten kuljettajaproteiineja OCT1, MATE2-K, OCTN1 ja OCTN2, mutta sillä on heikko estävä vaikutus kuljettajaproteiineihin OCT2 (IC50: 146 µM) ja MATE1 (IC50: 211 µM). Kaiken kaikkiaan nämä tulokset osoittavat kliinisesti merkittävien farmakokineettisten lääkeyhteisvaikutusten olevan epätodennäköisiä solriamfetolia ottavilla potilailla.
Eliminaatio
Solriamfetolin näennäisen eliminaation puoliintumisajan keskiarvo on 7,1 tuntia ja näennäinen kokonaispuhdistuma noin 19,5 l/h. Munuaispuhdistuma on noin 18,2 l/h.
Ihmisillä tehdyssä massatasapainotutkimuksessa noin 95 % annoksesta erittyi muuttumattomana solriamfetolina virtsaan ja 1 % tai vähemmän poistui vähäisenä, inaktiivisena N‑asetyylisolriamfetoli-metaboliittina. Munuaispuhdistuma muodosti suurimman osan näennäisestä kokonaispuhdistumasta ja oli noin kolme kertaa kreatiniinin puhdistumaa suurempi, mikä viittaa siihen, että kanta-aineen aktiivinen tubulaarinen eritys on todennäköisesti tärkein eliminaatioreitti.
Lineaarisuus/ei-lineaarisuus
Solriamfetolin farmakokinetiikka on lineaarista kliinisellä annosalueella. Vakaa tila saavutetaan 3 vuorokaudessa, ja kerran päivässä annettujen 150 mg:n annosten aiheuttaman solriamfetolin kumuloitumisen odotetaan olevan minimaalista (1,06-kertainen altistus kerta-annokseen nähden).
Erityisryhmät
Munuaisten vajaatoiminta
Lievää munaisten vajaatoimintaa (eGFR 60–89 ml/min/1,73 m2), kohtalaista munuaisten vajatoimintaa (eGFR 30–59 ml/min/1,73 m2) ja vaikeaa munuaisten vajatoimintaa (eGFR < 30 ml/min/1,73 m2) sairastavien potilaiden solriamfetolin AUC-arvo oli noin 1,5-, 2,3- ja 4,4-kertainen ja t1/2 noin 1,2-, 1,9- ja 3,9-kertainen verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (eGFR ≥ 90 ml/min/1,73 m2). Munuaisten vajaatoiminta ei yleisesti ottaen vaikuttanut keskimääräiseen Cmax-arvoon eikä Tmax-arvon mediaaniin.
Hemodialyysihoitoa saamattomien ja hemodialyysihoitoa saavien loppuvaiheen munuaissairautta sairastavien potilaiden solriamfetolin AUC-arvo oli noin 6,2- ja 4,6-kertainen ja t1/2 vähintään 13‑kertainen verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (eGFR ≥ 90 ml/min/1,73 m2). Potilailla, joilla oli loppuvaiheen munuaissairaus, keskimäärin 21 % solriamfetolista poistui hemodialyysissa.
Imetys ja rintaruokinta
Yhden annoksen maito- ja plasmatutkimus suoritettiin kuudella terveellä imettävällä aikuisella naisella 15–37 viikkoa synnytyksen jälkeen. Heille annettiin yksi suun kautta otettu 150 mg:n annos Sunosi-valmistetta. Rintamaitoon erittynyt kumulatiivinen keskimääräinen määrä oli 0,59 mg 24 tunnin aikana, mikä vastaa noin 4 % äidin painoon suhteutetusta annoksesta. Koko 72 tunnin aikana rintamaitoon erittyneestä solriamfetolimäärästä noin 78 % erittyi 8 tunnin kuluessa ja 98 % 24 tunnin kuluessa. Näennäinen keskimääräinen eliminaation puoliintumisaika rintamaidossa oli noin 5 tuntia.
Ikä, sukupuoli ja rotu
Populaatiofarmakokineettinen analyysi osoitti, etteivät ikä, sukupuoli ja rotu vaikuta kliinisesti merkittävällä tavalla solriamfetolin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Genotoksisuutta ja urosten ja naaraiden hedelmällisyyttä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Toistuvan altistuksen aiheuttamaa toksisuutta koskevia tutkimuksia, joissa solriamfetolia annettiin päivittäin suun kautta, tehtiin hiirillä (kesto 3 kuukautta, NOAEL-arvo 17 mg/kg/vrk), rotilla (kesto 6 kuukautta sekä 3 kuukauden lääkkeetön jakso, NOAEL-arvoa ei määritetty, LOAEL-arvo 29 mg/kg/vrk) ja koirilla (kesto 12 kuukautta sekä 3 kuukauden lääkkeetön jakso, NOAEL-arvoa ei määritetty, LOAEL-arvo 8 mg/kg/vrk). Näistä tutkimuksista saadut AUC-arvoon perustuvat solriamfetolin turvallisuuskertoimet (verrattaessa kliiniseen AUC-arvoon suurimmalla ihmisille suositellulla, 150 mg:n vuorokausiannoksella) olivat hiirillä < 1 (NOAEL-arvon perusteella) ja rotilla ja koirilla < 2 (LOAEL-arvon perusteella), pääasiassa solriamfetolin keskushermoston toimintaan kohdistuvien liiallisten farmakologisten vaikutusten vuoksi.
Pitkäaikaisia karsinogeenisuustutkimuksia on tehty hiirillä, joille solriamfetolia annettiin suun kautta 20 mg/kg:n, 65 mg/kg:n ja 200 mg/kg:n vuorokausiannoksina enintään 104 viikon ajan, ja rotilla, joille solriamfetolia annettiin suun kautta 35 mg/kg:n, 80 mg/kg:n ja 200 mg/kg:n vuorokausiannoksina enintään 101 viikon ajan. Solriamfetoli ei lisännyt neoplastisten löydösten ilmaantuvuutta näissä eläinten eliniän kattavissa karsinogeenisuutta koskevissa analyyseissa. Suurimpien annosten ja suurimman ihmisille suositellun annoksen (150 mg/vrk) välinen AUC-arvoihin perustuva turvallisuusmarginaali oli hiirillä noin 7,8 ja rotilla noin 20,7. Ottaen huomioon genotoksisuustulosten negatiivisuus ja se, ettei kasvainten ilmaantuvuus lisääntynyt kummassakaan karsinogeenisuustutkimuksessa, solriamfetolin ei katsota muodostavan karsinogeenista riskiä ihmisille. Elossaolo-osuus verrokkeihin nähden pieneni solriamfetolia saaneilla (uros)hiirillä, eniten 65 mg/kg:n vuorokausiannoksia käytettäessä (AUC-arvoihin perustuva turvallisuusmarginaali noin 2,9 suurimpaan ihmisille suositeltuun annokseen nähden), mutta ei solriamfetolia saaneilla rotilla.
Alkion ja sikiön kehitys
Mahdollisia vaikutuksia alkion ja sikiön kehitykseen tutkittiin tiineillä rotilla ja kaniineilla. Toksisuutta alkiolle ja sikiölle (lisääntyneet implantaation jälkeiset alkio- ja sikiömenetykset rotilla; seuraavien luustomuutosten ilmaantuvuuden lisääntyminen: rintalastan luiden virheasento rotilla ja kaniineilla ja takaraajojen rotaatio ja luiden käyristymä rotilla; sikiöiden painonlasku kummallakin lajilla) sekä rotilla situs inversusta havaittiin vasta emoon kohdistuvan toksisuuden ilmetessä (painonlasku). Ei tiedetä, oliko alkiotoksisuus seurausta toksisuudesta emolle vai suora solriamfetolin vaikutus. Jakautumista koskevassa tutkimuksessa tiineillä rotilla 14C-solriamfetolia havaittiin sikiökalvossa (noin kaksi kertaa suurempana pitoisuutena kuin veressä) sekä istukassa ja koko sikiössä (melkein samana pitoisuutena kuin veressä), minkä vuoksi sikiöön kohdistuvaa suoraa toksista vaikutusta ei voida poissulkea. Rotilla emon ja alkion/sikiön kehityksen NOAEL-arvojen mukaiset altistusmarginaalit ovat pienemmät kuin ihmisen altistus suurimmalla ihmisille suositellulla annoksella (0,6–0,7 AUC-arvojen perusteella). Kaniineilla emon ja alkion/sikiön kehityksen NOAEL-arvojen mukaiset altistusmarginaalit ovat < 6 (milligrammoina kehon pinta-alan neliömetriä kohden laskettuna).
Prenataalinen ja postnataalinen kehitys
Rotilla tiineyden ja imetyksen aikainen altistus (AUC), joka ylitti 0,6–0,7-kertaisen ihmisen altistuksen (AUC) suurimmalla ihmisille suositellulla annoksella, aiheutti toksisuutta emolle sekä poikasten kasvuun ja kehitykseen kohdistuvia haittavaikutuksia. Altistustasoilla (AUC), jotka olivat 8–12 kertaa suurempia kuin ihmisen altistus (AUC) suurimmalla ihmisille suositellulla annoksella, ei havaittu pitkäaikaisia vaikutuksia oppimiseen ja muistiin, mutta jälkeläisten hedelmällisyyttä ja lisääntymiskykyä mittaavat indeksit pienenivät.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Hydroksipropyyliselluloosa
Magnesiumstearaatti
Kalvopäällyste
Poly(vinyylialkoholi)
Makrogoli
Talkki
Titaanidioksidi (E 171)
Keltainen rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
5 vuotta
Pullot ensimmäisen avaamisen jälkeen: 120 vuorokautta
Säilytys
Läpipainopakkaukset: Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pullot: Käytettävä 4 kuukauden kuluessa avaamisesta. Pidä pakkaus tiiviisti suljettuna. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SUNOSI tabletti, kalvopäällysteinen
75 mg (J) (L:ei) 28 fol (287,64 €)
150 mg (L:ei) 28 fol (395,00 €)
PF-selosteen tieto
7 x 1 kalvopäällysteistä tablettia yksittäispakatuissa PVC/PCTFE/alumiini-läpipainopakkauksissa.
PVC/PCTFE/alumiini-läpipainopakkaukset.
Pakkauskoot: 7, 28 tai 56 kalvopäällysteistä tablettia.
Suurtiheyspolyeteenistä (HDPE) valmistettu pullo, jossa on polypropeenista (PP) valmistettu turvakorkki. Korkkiin on integroitu piidioksidigeeliä kuivatusaineeksi. Yksi pullo sisältää 30 tai 100 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Sunosi 75 mg tabletti, kalvopäällysteinen
Keltainen tai tummankeltainen, pitkulainen, 7,6 mm pitkä ja 4,4 mm leveä tabletti, jonka toiselle puolelle on kaiverrettu ”75” ja jonka vastakkaisella puolella on jakouurre.
Tabletin voi jakaa yhtä suuriin annoksiin.
Sunosi 150 mg tabletti, kalvopäällysteinen
Keltainen, pitkulainen, 9,5 mm pitkä ja 5,6 mm leveä tabletti, jonka toiselle puolelle on kaiverrettu ”150”.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Korvattavuus
SUNOSI tabletti, kalvopäällysteinen
75 mg 28 fol
150 mg 28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Pitolisantti ja solriamfetoli: Aikuisten narkolepsian hoito erityisin edellytyksin (3035).
ATC-koodi
N06BA14
Valmisteyhteenvedon muuttamispäivämäärä
10.07.2025
Yhteystiedot
Copenhagen Towers, Oerestads Boulevard 108
DK-2300 Copenhagen S
Denmark
+45 33 33 76 33
info@pharmanovia.com