
ELOCTA injektiokuiva-aine ja liuotin, liuosta varten 250 IU, 500 IU, 750 IU, 1000 IU, 1500 IU, 2000 IU, 3000 IU, 4000 IU
Vaikuttavat aineet ja niiden määrät
ELOCTA 250 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 250 IU efmoroktokogi alfaa (efmoroctocogum alfa).
Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 83 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 500 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 167 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 750 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 750 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 250 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 1000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 1 000 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 333 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 1500 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 1 500 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 500 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 2000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 2 000 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 667 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 3000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 3 000 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 1 000 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
ELOCTA 4000 IU injektiokuiva-aine ja liuotin, liuosta varten
Kukin injektiopullo sisältää nimellisesti 4 000 IU efmoroktokogi alfaa (efmoroctocogum alfa). Käyttökuntoon saattamisen jälkeen ELOCTA sisältää noin 1 333 IU/ml rekombinanttia ihmisen hyytymistekijä VIII:tä, efmoroktokogi alfaa.
Teho (kansainvälinen yksikkö (International Units, IU)) on määritetty käyttämällä Euroopan farmakopean kromogeenista määritystä. ELOCTA-valmisteen spesifinen aktiivisuus on 4 000‑10 200 IU/mg proteiinia.
Efmoroktokogi alfa (rekombinantti hyytymistekijä VIII Fc-fuusioproteiini (rFVIIIFc)) sisältää 1 890 aminohappoa. Sitä valmistetaan yhdistelmä-DNA-tekniikalla ihmisalkion munuaisten (HEK) solulinjassa siten, että solunviljelyprosessissa, puhdistamisessa tai lopullisessa valmisteessa ei ole käytetty lainkaan eksogeenista, ihmisestä tai eläimestä peräisin olevaa proteiinia.
Apuaine, jonka vaikutus tunnetaan
0,6 mmol (tai 14 mg) natriumia / injektiopullo.
0,4 mg polysorbaatti a 20 / injektiopullo.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja liuotin, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Hemofilia A:ta (synnynnäinen hyytymistekijä VIII:n puute) sairastavien potilaiden verenvuodon hoito ja ennaltaehkäisy.
ELOCTA-valmistetta voidaan käyttää kaikille ikäryhmille.
Ehto
Hoito tulee aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää aloittaa hemofilian hoitoon perehtyneen lääkärin valvonnassa.
Hoidon valvonta
Hyytymistekijä VIII:n pitoisuudet suositellaan määrittämään hoidon aikana tarkoituksenmukaisin väliajoin (yksivaiheisilla hyytymismäärityksillä tai kromogeenisilla määrityksillä), jotta annettava annos ja toistettujen injektioiden antotiheys voidaan määrittää. Potilaiden FVIII-vasteet saattavat vaihdella yksilöllisesti siten, että puoliintumisajat ja saanto ovat erilaisia eri potilailla. Potilaan painoon perustuvaa annosta on ehkä muutettava ali- ja ylipainoisilla potilailla. Korvaushoidon tarkka seuranta hyytymistekijämääritysten (plasman FVIII -aktiivisuus) avulla on välttämätöntä erityisesti suurten leikkausten yhteydessä.
Käytettäessä in vitro tromboplastiiniaikaan (aPTT) perustuvaa yksivaiheista hyytymismääritystä tekijän VIII aktiivisuuden määrittämiseksi potilaiden verikokeista, plasman tekijä VIII:n aktiivisuuden tuloksiin saattavat vaikuttaa huomattavasti sekä aPTT-reagenssin tyyppi että määrityksessä käytettävä viitestandardi. Määritystulosten välillä voi olla huomattavia eroja myös riippuen siitä, onko ne saatu aPTT:hen perustuvalla yksivaiheisella hyytymismäärityksellä vai Ph. Eur. -mukaisella kromogeenisella määrityksellä. Tällä on merkitystä etenkin vaihdettaessa laboratoriota ja/tai määrityksessä käytettävää reagenssia.
Annostus
Korvaushoidon annostus ja kesto riippuvat hyytymistekijä VIII:n puutoksen vaikeusasteesta, verenvuodon laajuudesta ja vuotokohdasta sekä potilaan kliinisestä tilasta.
Annettavien rekombinanttitekijä VIII Fc -yksiköiden määrä ilmaistaan IU‑yksikköinä, mikä perustuu FVIII -valmisteita koskevaan voimassaolevaan WHO-standardiin. FVIII -aktiivisuus plasmassa ilmaistaan joko prosentteina (suhteessa normaaliin ihmisen plasmaan) tai IU-yksikköinä (plasman FVIII -pitoisuuden kansainvälisen standardin mukaan).
Yksi kansainvälinen yksikkö (IU) rekombinanttitekijä VIII Fc -aktiivisuutta vastaa FVIII -määrää yhdessä millilitrassa normaalia ihmisen plasmaa.
Tarvittaessa toteutettava hoito
Tarvittavan rekombinanttitekijä VIII Fc -annoksen laskeminen perustuu empiiriseen havaintoon, että 1 IU‑yksikkö FVIII -valmistetta painokiloa kohden suurentaa plasman FVIII -aktiivisuutta 2 IU:lla desilitraa kohden. Tarvittava annos lasketaan seuraavalla laskukaavalla:
Tarvittava yksikkömäärä = potilaan paino (kg) × haluttu tekijä VIII:n pitoisuuden lisäys (%) (IU/dl) × 0,5 (IU/kg per IU/dl)
Annettavalla annoksella ja antotiheydellä on aina pyrittävä yksilölliseen kliiniseen tehokkuuteen.
Jos potilaalla on jokin seuraavista verenvuototapahtumista, FVIII -aktiivisuus ei saa laskea taulukossa mainitun pitoisuuden alapuolelle (% normaalista tai IU/dl) vastaavan hoitojakson aikana. Taulukkoa 1 voidaan käyttää annostusohjeena verenvuotojen ja kirurgisten toimenpiteiden yhteydessä:
Taulukko 1: ELOCTA-annostusohje verenvuototapausten hoitoa ja leikkauksia varten
Verenvuodon määrä / kirurgisen toimenpiteen tyyppi | Tarvittava FVIII-pitoisuus (%) (IU/dl) | Antotiheys (tuntia) / hoidon kesto (vrk) |
Verenvuoto | ||
Alkava hemartroosi, lihasverenvuoto tai suun limakalvovuoto | 20‑40 | Injektio toistetaan 12‑24 tunnin välein vähintään 1 päivän ajan, kunnes kipuna ilmenevä vuotoepisodi on mennyt ohi tai paraneminen on tapahtunut. 1 |
Laajempi hemartroosi, lihasverenvuoto tai hematooma | 30‑60 | Injektio toistetaan 12‑24 tunnin välein 3-4 tai useamman päivän ajan, kunnes kipu lakkaa ja akuutti toimintakyvyn vajaus korjautuu. 1 |
Hengenvaaralliset verenvuodot | 60‑100 | Injektio toistetaan 8‑24 tunnin välein, kunnes vaara on ohi. |
Leikkaus | ||
Pieni leikkaus kuten hampaanpoisto | 30‑60 | Injektio toistetaan 24 tunnin välein, vähintään 1 päivän ajan, kunnes paraneminen on tapahtunut. |
Suuri leikkaus | 80‑100 (pre- ja postoperatiivisesti) | Injektio toistetaan 8‑24 tunnin välein tarpeen mukaan, kunnes haava on parantunut riittävästi. Hoitoa jatketaan sen jälkeen vielä vähintään 7 vuorokauden ajan, jotta tekijä VIII -aktiivisuus pysyy 30‑60 %:n (IU/dl) tasolla. |
1 Joillakin potilailla ja joissakin tilanteissa annosväliä voidaan pidentää korkeintaan 36 tuntiin. Ks. farmakokineettiset tiedot kohdasta Farmakokinetiikka.
Estohoito eli profylaksi
Pitkäkestoisessa estohoidossa suositeltu annos on 50 IU FVIII:tä painokiloa kohden 3‑5 vuorokauden välein. Annosta voidaan muuttaa potilaan vasteen mukaan välillä 25‑65 IU/kg (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Joissakin tapauksissa, etenkin nuorille potilaille, voi olla tarpeen käyttää lyhyempiä antovälejä tai suurempia annoksia.
Iäkkäät
Valmisteen käytöstä ≥ 65-vuotiaille on vain vähän kokemusta.
Pediatriset potilaat
Alle 12‑vuotiaille lapsille tiheämmät antovälit tai suuremmat annokset voivat olla tarpeen (ks. kohta Farmakodynamiikka). 12 vuotta täyttäneille nuorille annossuositukset ovat samat kuin aikuisille.
Antotapa
ELOCTA annetaan laskimoon.
ELOCTA injisoidaan laskimoon usean minuutin aikana. Antonopeus on määritettävä potilaan sietämän kiputason mukaan ja se saa olla enintään 10 ml/min.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yliherkkyys
Allergistyyppiset yliherkkyysreaktiot ovat mahdollisia ELOCTA-valmistetta käytettäessä. Potilaita on neuvottava lopettamaan lääkevalmisteen käyttö välittömästi ja ottamaan yhteyttä lääkäriin, jos yliherkkyysoireita ilmaantuu.
Potilaille on kerrottava yliherkkyysreaktioiden oireista, joita ovat esim. paukamat, yleistynyt nokkosihottuma, puristuksen tunne rinnassa, hengityksen vinkuminen, verenpaineen lasku ja anafylaksia.
Sokin saanutta potilasta on hoidettava yleisten sokinhoito-ohjeiden mukaisesti.
Vasta-aineet eli inhibiittorit
Hyytymistekijää VIII neutraloivien vasta-aineiden (inhibiittorien) kehittyminen on A-hemofilian hoidon tunnettu komplikaatio. Nämä vasta-aineet ovat yleensä tekijä VIII:n prokoagulanttiaktiivisuutta vastaan vaikuttavia IgG-immunoglobuliineja, ja niiden määrä ilmaistaan Bethesda-yksikköinä (BU) millilitrassa plasmaa. Määrityksessä käytetään modifioitua menetelmää. Inhibiittoreiden muodostumisen riski riippuu taudin vaikeusasteesta ja altistumisesta tekijä VIII:lle. Riski on suurin 50 ensimmäisen altistuspäivän aikana mutta koko elämän ajan, vaikkakin riski on melko harvinainen.
Inhibiittorien muodostumisen kliininen merkitys riippuu inhibiittori titteristä. Riittämättömän kliinisen vasteen riski on pienempi alhaisen titterin kuin korkean titterin inhibiittoreilla.
Kaikkia hyytymistekijä VIII -hoitoa saavia potilaita on seurattava huolellisesti vasta-aineiden kehittymisen varalta sekä kliinisten arviointien että laboratoriokokeiden avulla. Jos odotettua tekijä VIII:n aktiivisuutta plasmassa ei saavuteta tai jos verenvuotoa ei saada hallintaan asianmukaisella annoksella, hyytymistekijä VIII:n vasta-aineet on testattava. Potilailla, joilla vasta-ainepitoisuus on korkea, hyytymistekijä VIII -hoito ei välttämättä tehoa ja on harkittava muita hoitovaihtoehtoja. Tällöin potilas on ohjattava sellaisen lääkärin hoitoon, jolla on kokemusta hyytymistekijä VIII:lle inhibiittoreita kehittäneiden hemofiliapotilaiden hoitamisesta.
Kardiovaskulaariset tapahtumat
Potilailla, joilla on ennestään kardiovaskulaarisia riskitekijöitä, korvaushoito hyytymistekijä VIII:lla voi suurentaa kardiovaskulaarista riskiä.
Katetreihin liittyvät komplikaatiot
Jos toimenpide edellyttää keskuslaskimokatetria, on huomioitava keskuslaskimokatetriin liittyvät komplikaatiot, mukaan lukien paikalliset infektiot, bakteremia ja katetrointikohdan tromboosi.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Pediatriset potilaat
Luetellut varoitukset ja varotoimet koskevat sekä aikuisia, lapsia että nuoria.
Apuaineeseen liittyvät huomioitavat seikat
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per injektiopullo eli sen voidaan sanoa olevan ”natriumiton”.
Riippuen potilaan painosta ja annostuksesta tätä valmistetta voidaan kuitenkin antaa enemmän kuin yksi injektiopullo (ks. kohdasta Vaikuttavat aineet ja niiden määrät tiedot sisällön määrästä / injektiopullo). Tämä on otettava huomioon potilailla, joilla on ruokavaliossa natriumrajoitus.
Polysorbaatti
Tämä lääkevalmiste sisältää 0,4 mg polysorbaattia 20 per injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutuksia ihmisen hyytymistekijä VIII:n (rDNA) ja muiden lääkevalmisteiden välillä ei ole raportoitu. Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Tekijä VIII:llä ei ole tehty lisääntymistä koskevia eläinkokeita. Hiirillä on tehty koe, jossa tutkittiin ELOCTA‑valmisteen siirtymistä istukan läpi (ks. kohta Prekliiniset tiedot turvallisuudesta). Koska hemofilia A on naisilla harvinainen, raskauden ja imetyksen aikaisesta tekijä VIII -hoidosta ei ole kokemuksia. Tämän vuoksi tekijä VIII -valmistetta saa käyttää raskauden ja imetyksen aikana vain, jos se on selvästi välttämätöntä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ELOCTA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Harvinaisissa tapauksissa on havaittu yliherkkyyttä tai allergisia reaktioita (näitä saattavat olla mm. angioedeema, pistoskohdan polte ja kirvely, vilunväristykset, punoitus, yleistynyt nokkosihottuma, päänsärky, paukamat, alhainen verenpaine, letargia, pahoinvointi, levottomuus, takykardia, puristuksen tunne rinnassa, kihelmöinti, oksentelu, vinkuva hengitys), jotka voivat joissakin tapauksissa kehittyä vaikeaksi anafylaksiaksi (mukaan lukien sokki).
Neutraloivia vasta-aineita (inhibiittoreita) voi kehittyä hemofilia A -potilaille, jotka saavat tekijä VIII ‑hoitoa, kuten ELOCTA-valmistetta. Jos näitä vasta-aineita muodostuu, potilaan kliininen hoitovaste on riittämätön. Tällaisissa tapauksissa suositellaan yhteydenottoa hemofiliaan erikoistuneeseen hoitoyksikköön.
Taulukoitu yhteenveto haittavaikutuksista
Alla oleva taulukko 2 on laadittu MedDRA-elinjärjestelmäluokituksen mukaisesti. Haittavaikutusten esiintymistiheydet perustuvat kliinisiin tutkimuksiin, joihin osallistui yhteensä 379 vaikeaa hemofilia A:ta sairastavaa potilasta, joista 276 oli aiemmin hoidettuja potilaita (PTP) ja 103 oli aiemmin hoitamattomia potilaita (PUP). Kohdassa Farmakodynamiikka on lisätietoja näistä kliinisistä tutkimuksista.
Esiintymistiheydet on arvioitu seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2: ELOCTA-valmisteen kliinisissä tutkimuksissa ilmoitetut haittavaikutukset1
MedDRA-järjestelmän elinluokka | Haittavaikutukset | Esiintymistiheys1 |
Veri ja imukudos | Tekijä VIII:n inhibitio | Melko harvinainen (PTP)2 Hyvin yleinen (PUP)2 |
Hermosto | Päänsärky | Melko harvinainen |
Huimaus | Melko harvinainen | |
Makuhäiriöt | Melko harvinainen | |
Sydän | Bradykardia | Melko harvinainen |
Verisuonisto | Korkea verenpaine | Melko harvinainen |
Kuumat aallot | Melko harvinainen | |
Angiopatia4 | Melko harvinainen | |
Hengityselimet, rintakehä ja välikarsina | Yskä | Melko harvinainen |
Ruoansulatuselimistö | Vatsakipu, alavatsa | Melko harvinainen |
Iho ja ihonalainen kudos | Papulaarinen ihottuma | Yleinen (PUP)3 |
Ihottuma | Melko harvinainen | |
Luusto, lihakset ja sidekudos | Nivelkipu | Melko harvinainen |
Lihaskipu | Melko harvinainen | |
Selkäkipu | Melko harvinainen | |
Nivelturvotus | Melko harvinainen | |
Yleisoireet ja antopaikassa todettavat haitat | Laitteeseen liittyvä tromboosi | Yleinen (PUP)3 |
Huonovointisuus | Melko harvinainen | |
Rintakipu | Melko harvinainen | |
Vilun tunne | Melko harvinainen | |
Kuumuuden tunne | Melko harvinainen | |
Vammat, myrkytykset ja hoitokomplikaatiot | Toimenpiteeseen liittyvä alhainen verenpaine | Melko harvinainen |
PTP = aiemmin hoidetut potilaat, PUP = aiemmin hoitamattomat potilaat.
1 Lääkkeen haittavaikutukset ja yleisyys perustuvat esiintyvyyteen vain aiemmin hoidetuilla potilailla (PTP), ellei muuta ilmoiteta.
2 Yleisyys perustuu kaikilla tekijä VIII -valmisteilla tehtyihin tutkimuksiin, joihin osallistui vaikeaa hemofilia A:ta sairastavia potilaita.
3 Lääkkeen haittavaikutukset ja yleisyys perustuvat esiintyvyyteen vain aiemmin hoitamattomilla potilailla (PUP).
4 Tutkijan käyttämä termi: verisuonissa tuntuva kipu ELOCTA-valmisteen injisoinnin jälkeen.
Pediatriset potilaat
Haittavaikutuksissa ei havaittu ikään liittyviä eroja pediatristen potilaiden ja aikuispotilaiden välillä.
Haittavaikutusten esiintymistiheyksien, tyyppien ja vaikeusasteiden oletetaan olevan lapsilla samoja kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta:
www-sivusto:
Suomi/Finland
[Finnish]
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
[Swedish]
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Yliannostus
Yliannostusoireita ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antihemorraginen veren hyytymistekijä VIII, ATC-koodi: B02BD02
Vaikutusmekanismi
Tekijä VIII:n / von Willebrand -tekijän kompleksi koostuu kahdesta molekyylistä (tekijä VIII ja von Willebrandin -tekijä), joiden fysiologiset tehtävät ovat erilaiset. Hemofiliapotilaaseen infusoituna tekijä VIII sitoutuu potilaan verenkierrossa von Willebrand -tekijään. Aktivoitunut tekijä VIII toimii aktivoituneen tekijä IX:n kofaktorina kiihdyttäen tekijä X:n muuttumista aktivoituneeksi tekijä X:ksi. Aktivoitunut tekijä X muuttaa protrombiinin trombiiniksi. Trombiini muuttaa tämän jälkeen fibrinogeenin fibriiniksi, jolloin hyytymä voi muodostua.
Hemofilia A on sukupuolisidonnainen perinnöllinen veren hyytymissairaus, joka johtuu tekijä VIII:C:n pienentyneistä pitoisuuksista ja johtaa verenvuotoihin nivelissä, lihaksissa tai sisäelimissä joko spontaanisti tai tapaturman tai leikkausvamman seurauksena. Korvaushoidon ansiosta tekijä VIII:n pitoisuudet suurenevat, jolloin hyytymistekijän puutos ja vuototaipumus hetkellisesti korjaantuvat.
On huomattava, että vuositasolla lasketut vuotomäärät eivät ole vertailukelpoisia eri vahvuuksien ja eri kliinisten tutkimusten välillä.
ELOCTA (efmoroktokogi alfa) on täysin rekombinantti fuusioproteiini, jonka puoliintumisaika on pidentynyt. ELOCTA koostuu ihmisen hyytymistekijä VIII:sta, jonka B-domeenit on poistettu, ja joka on kovalenttisesti kiinnittynyt ihmisen immunoglobuliini G1:n Fc-domeeniin. Ihmisen immunoglobuliini G1:n Fc-osa sitoutuu vastasyntyneen Fc-reseptoriin. Tämä reseptori ilmentyy koko elämän ajan ja on osa luonnollisesti esiintyvää reittiä, joka suojaa immunoglobuliineja lysosomaaliselta hajoamiselta kuljettamalla näitä proteiineja takaisin verenkiertoon, mikä johtaa niiden pitempään puoliintumisaikaan plasmassa. Efmoroktokogi alfa sitoutuu vastasyntyneen Fc-reseptoriin käyttäen siinä samaa luonnollisesti esiintyvää reittiä; tämä viivästyttää lysosomaalista hajoamista ja mahdollistaa pitemmän puoliintumisajan plasmassa kuin endogeeninen tekijä VIII.
Kliininen teho ja turvallisuus
ELOCTA-valmisteen turvallisuutta, tehoa ja farmakokinetiikkaa aiemmin hoidetuilla potilailla (PTP) on arvioitu kahdessa monikansallisessa avoimessa faasin 3 avaintutkimuksessa (tutkimus I ja tutkimus II) (ks. Pediatriset potilaat) ja korkeintaan neljä vuotta kestäneessä jatkotutkimuksessa (tutkimus III). Yhteensä 276:ta aiemmin hoidettua potilasta seurattiin yhteensä 80 848 altistuspäivän ajan mediaanin ollessa 294 (vaihteluväli 1–735) altistuspäivää potilasta kohti. Lisäksi suoritettiin faasin 3 tutkimus (tutkimus IV) ELOCTA-valmisteen turvallisuuden ja tehon arvioimiseksi aiemmin hoitamattomilla potilailla (PUP) (ks. Pediatriset potilaat).
Tutkimukseen I otettiin 165 aiemmin hoitoa saanutta miespotilasta (12‑65-vuotiaita), joilla oli vaikea hemofilia A. Ennen tutkimukseen tuloa estohoitoa käyttäneet tutkimuspotilaat valittiin tässä tutkimuksessa yksilöllisen estohoidon haaraan. Ennen tutkimukseen tuloa tarvittaessa toteutettavaa hoitoa saaneet henkilöt valittiin joko yksilöllisen estohoidon haaraan tai heidät satunnaistettiin joko viikoittaisen estohoidon tai tarvittaessa toteutettavan hoidon haaraan.
Estohoidot:
Yksilöllinen estohoito: 25–65 IU/kg 3–5 päivän välein.
Viikoittainen estohoito: 65 IU/kg
153:sta tutkimuksen I loppuun suorittaneesta tutkittavasta 150 otettiin tutkimukseen III (jatkotutkimus). Tutkimuksiin I+III kokonaisosallistumisajan mediaani oli 4,2 vuotta ja altistuspäivien mediaani oli 309.
Yksilöllinen estohoito: Tekijän vuosikulutuksen mediaani oli 4 212 IU/kg (vaihteluväli 2 877–7 943) tutkimuksessa I ja 4 233 IU/kg (vaihteluväli 2 668–8 317) tutkimuksessa III. Vuositasolla lasketut mediaanivuotomäärät olivat vastaavasti 1,60 (vaihteluväli 0–18,2) ja 0,74 (vaihteluväli 0–15,6).
Viikoittainen estohoito: Tekijän vuosikulutuksen mediaani oli 3 805 IU/kg (vaihteluväli 3 353–6 196) tutkimuksessa I ja 3 510 IU/kg (vaihteluväli 2 758–3 984) tutkimuksessa III. Vuositasolla lasketut mediaanivuotomäärät olivat vastaavasti 3,59 (vaihteluväli 0–58) ja 2,24 (vaihteluväli 0–17,2).
Tarvittaessa toteutettava hoito: Tekijän vuosikulutuksen mediaani oli 1 039 IU/kg (vaihteluväli 280–3 571) 23 potilaalla, jotka oli satunnaistettu tarvittavan hoidon haaraan tutkimuksessa 1, ja 671 IU/kg (vaihteluväli 286–913) niillä 6 potilaalla, jotka pysyivät tarvittaessa toteutettavan hoidon haarassa vähintään yhden vuoden tutkimuksessa III.
Tarvittaessa toteutettavasta hoidosta viikoittaiseen estohoitoon siirtyneiden potilaiden vuositasolla laskettu mediaanivuotomäärä tutkimuksessa III oli 1,67.
Verenvuodon hoito: Tutkimusten I ja III aikana 2 490 verenvuototapahtumaa hoidettiin kunkin vuodon hallitsemiseksi mediaaniannoksella 43,8 IU/kg (vaihteluväli 13,0–172,8). Potilaat arvioivat 79,2 % ensimmäisistä injektioista erinomaisiksi tai hyviksi.
Perioperatiivinen hoito (leikkaukseen liittyvä estohoito): Tutkimuksissa I ja III yhteensä 34 tutkittavalle suoritettiin 48 suurta leikkausta, jotka kaikki myös arvioitiin. Lääkärit arvioivat hemostaasivasteen erinomaiseksi 41:ssä ja hyväksi 3:ssa yhteensä 44:sta suuresta leikkauksesta. Hemostaasin ylläpitämiseksi annettu mediaaniannos leikkauksen aikana oli 60,6 IU/kg (vaihteluväli 38–158).
Pediatriset potilaat
Tutkimukseen II otettiin yhteensä 71 aiemmin hoitoa saanutta miespuolista pediatrista potilasta, jotka olivat < 12‑vuotiaita ja joilla oli vaikea hemofilia A. Näistä 71 tutkimuksen osallistuneesta henkilöstä 69 sai ainakin yhden (1) annoksen ELOCTA-valmistetta, jolloin valmisteen tehoa ko. henkilöillä voitiin arvioida (35 henkilöä oli < 6-vuotiaita ja 34 henkilöä oli 6 - < 12-vuotiaita). Aloitusvaiheessa estohoito käsitti 25 IU/kg ensimmäisenä päivänä, jonka jälkeen annettiin 50 IU/kg neljäntenä päivänä. Sallittu ja käytetty annos oli korkeimmillaan 80 IU/kg ja sallittu ja käytetty annosteluväli lyhimmillään 2 päivää rajoitetulla määrällä potilaita. Tutkimuksen II loppuun asti suorittaneista 67 tutkittavasta yhteensä 61 tutkittavaa otettiin tutkimukseen III (jatkotutkimus). Tutkimuksiin II+III osallistumisen mediaaniaika oli 3,4 vuotta ja altistuspäivien mediaani oli 332.
Estohoito, ikä < 6 vuotta: Tutkimuksissa II ja III mediaani annosväli oli 3,50 päivää. Tekijän vuosikulutuksen mediaani oli 5 146 IU/kg (vaihteluväli 3 695–8 474) tutkimuksessa II ja 5 418 IU/kg (vaihteluväli 3 435–9 564) tutkimuksessa III. Vuositasolla lasketut mediaanivuotomäärät olivat vastaavasti 0,00 (vaihteluväli 0–10,5) ja 1,18 (vaihteluväli 0–9,2).
Estohoito, ikä 6–12 vuotta: Tutkimuksessa II mediaani annosväli oli 3,49 päivää ja tutkimuksessa III se oli 3,50 päivää. Tekijän vuosikulutuksen mediaani oli 4 700 IU/kg (vaihteluväli 3 819–8 230) tutkimuksessa II ja 4 990 IU/kg (vaihteluväli 3 856–9 527) tutkimuksessa III. Vuositasolla lasketut mediaanivuotomäärät olivat vastaavasti 2,01 (vaihteluväli 0–27,2) ja 1,59 (vaihteluväli 0–8,0).
12 iältään 12–18‑vuotiasta nuorta otettiin mukaan estohoidon aikuisten tutkittavien populaatioon. Tekijän vuosikulutuksen mediaani oli 5 572 IU/kg (vaihteluväli 3 849–7 035) tutkimuksessa I ja 4 456 IU/kg (vaihteluväli 3 563–8 011) tutkimuksessa III. Vuositasolla lasketut mediaanivuotomäärät olivat vastaavasti 1,92 (vaihteluväli 0–7,1) ja 1,25 (vaihteluväli 0–9,5).
Verenvuodon hoito: Tutkimusten II ja III aikana 447 verenvuototapahtumaa hoidettiin kunkin vuodon hallitsemiseksi mediaaniannoksella 63 IU/kg (vaihteluväli 8–186). Potilaat ja heidän hoitajansa arvioivat 90,2 % ensimmäisistä injektioista erinomaisiksi tai hyviksi.
Tutkimuksessa IV arvioitiin 103:a aiemmin hoitamatonta miespuolista potilasta (PUP), jotka olivat < 6‑vuotiaita ja joilla oli vaikea hemofilia A. Potilaita seurattiin yhteensä 11 255 altistuspäivän ajan mediaanin ollessa 100 (vaihteluväli 0-649) altistuspäivää potilasta kohti. Useimmilla tutkimushenkilöillä hoito alkoi episodihoidoilla (N=81), josta myöhemmin siirryttiin estohoitoon (N=69). 89 aiemmin hoitamatonta potilasta (PUP) sai estohoitoa milloin tahansa tutkimuksen aikana. Suositeltu estohoidon aloitusannos oli 25–80 IU/kg 3–5 päivän välein. Estohoitoa saaneilla tutkittavilla keskimääräisen viikkoannoksen mediaani oli 101,4 IU/kg (vaihteluväli: 28,5–776,3 IU/kg) ja annostusvälin mediaani oli 3,87 päivää (vaihteluväli 1,1–7 päivää). Tekijän vuosikulutuksen mediaani oli 3971,4 IU/kg. Vuositasolla laskettu vuotomäärä oli 1,49 (vaihteluväli 0,0–18,7).
Farmakokinetiikka
Kaikki ELOCTA-valmisteella tehdyt farmakokineettiset tutkimukset suoritettiin aiemmin hoitoa saaneilla potilailla, joilla oli vaikea hemofilia A. Tässä kohdassa esitettävät tiedot on saatu kromogeenisen määrityksen ja yksivaiheisen hyytymismäärityksen avulla. Kromogeenisen määrityksen datasta saadut farmakokineettiset muuttujat olivat samanlaisia verrattuna yksivaiheisen hyytymismäärityksen datasta saatuihin muuttujiin.
Farmakokineettisiä ominaisuuksia arvioitiin ELOCTA-valmistetta (rFVIIIFc) saavilla 28 tutkimushenkilöllä (≥ 15-vuotiaat). Vähintään 96 tuntia (4 päivää) kestäneen lääkkeettömän jakson jälkeen tutkimushenkilöille annettiin yksi 50 IU/kg ELOCTA-annos. Farmakokineettiset näytteet kerättiin ennen annosta ja 7 eri ajankohtana enintään 120 tunnin (5 päivän) kuluttua annoksen jälkeen. 50 IU/kg ELOCTA-annoksen jälkeiset farmakokineettiset muuttujat esitetään taulukoissa 3 ja 4.
Taulukko 3: ELOCTA-valmisteen farmakokineettiset muuttujat yksivaiheisen hyytymismäärityksen avulla määritettyinä
Farmakokineettiset muuttujat1 | ELOCTA (95 %:n CI) |
N=28 | |
Inkrementaalinen saanto (IU/dl per IU/kg) | 2,24 (2,11‑2,38) |
AUC/annos (IU*h/dl per IU/kg) | 51,2 (45,0‑58,4) |
Cmax (IU/dl) | 108 (101‑115) |
CL (ml/h/kg) | 1,95 (1,71‑2,22) |
t½ (h) | 19,0 (17,0‑21,1) |
MRT (h) | 25,2 (22,7‑27,9) |
Vss (ml/kg) | 49,1 (46,6‑51,7) |
1 Farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI)
Lyhenteet: CI = luottamusväli; Cmax= maksimiaktiivisuus; AUC = FVIII:n aktiivisuuden aikakäyrän alle jäävä alue; t½= terminaalinen puoliintumisaika; CL = puhdistuma; Vss = jakaantumistilavuus vakaan tason vaiheessa; MRT = keskimääräinen elimistössä oloaika.
Taulukko 4: ELOCTA-valmisteen farmakokineettiset muuttujat kromogeenisen määrityksen avulla määritettyinä
Farmakokineettiset muuttujat1
| ELOCTA (95 %:n CI) |
N=27 | |
Inkrementaalinen saanto (IU/dl per IU/kg) | 2,49 (2,28‑2,73) |
AUC/annos (IU*h/dl per IU/kg) | 47,5 (41,6‑54,2) |
Cmax (IU/dl) | 131 (104‑165) |
CL (ml/h/kg) | 2,11 (1,85‑2,41) |
t½ (h) | 20,9 (18,2‑23,9) |
MRT (h) | 25,0 (22,4‑27,8) |
Vss (ml/kg) | 52,6 (47,4‑58,3) |
1 Farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI)
Lyhenteet: CI = luottamusväli; Cmax= maksimiaktiivisuus; AUC = FVIII:n aktiivisuuden aikakäyrän alle jäävä alue; t½= terminaalinen puoliintumisaika; CL = puhdistuma; Vss = jakautumistilavuus vakaan tason vaiheessa; MRT = keskimääräinen elimistössä oloaika.
Farmakokineettiset tiedot osoittavat, että ELOCTA-valmisteella on pidentynyt puoliintumisaika verenkierrossa.
Pediatriset potilaat
ELOCTA-valmisteen farmakokineettiset muuttujat määritettiin nuorilla tutkimuksessa I (farmakokineettisiä näytteitä otettiin ennen annoksen antamista, minkä jälkeen arvioita tehtiin useina ajankohtina kunnes annoksen antamisesta oli kulunut 120 tuntia (5 päivää)) ja lapsilla tutkimuksessa II (farmakokineettisiä näytteitä otettiin ennen annoksen antamista, minkä jälkeen arvioita tehtiin useina ajankohtina kunnes annoksen antamisesta oli kulunut 72 tuntia (3 päivää)). Taulukoissa 5 ja 6 esitetään alle 18-vuotiaiden tutkimushenkilöiden pediatrisesta datasta lasketut farmakokineettiset muuttujat.
Taulukko 5: ELOCTA-valmisteen farmakokineettiset muuttujat pediatrisilla potilailla yksivaiheisen hyytymismäärityksen avulla määritettyinä
Farmakokineettiset muuttujat1 | Tutkimus II | Tutkimus I* | ||
< 6-vuotiaat | 6 - < 12-vuotiaat | 12 - < 18-vuotiaat | ||
N = 23 | N = 31 | N = 11 | ||
Inkrementaalinen saanto (IU/dl per IU/kg) | 1,90 (1,79‑2,02) | 2,30 (2,04‑2,59) | 1,81 (1,56‑2,09) | |
AUC/annos (IU*h/dl per IU/kg) | 28,9 (25,6‑32,7) | 38,4 (33,2‑44,4) | 38,2 (34,0‑42,9) | |
t½ (h) | 12,3 (11,0‑13,7) | 13,5 (11,4‑15,8) | 16,0 (13,9‑18,5) | |
MRT (h) | 16,8 (15,1‑18,6) | 19,0 (16,2‑22,3) | 22,7 (19,7‑26,1) | |
CL (ml/h/kg) | 3,46 (3,06‑3,91) | 2,61 (2,26‑3,01) | 2,62 (2,33‑2,95) | |
Vss (ml/kg) | 57,9 (54,1‑62,0) | 49,5 (44,1‑55,6) | 59,4 (52,7‑67,0) | |
1 Farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI) Lyhenteet: CI = luottamusväli; AUC = FVIII:n aktiivisuuden alle jäävä aikakäyrä; t½ = terminaalinen puoliintumisaika; CL = puhdistuma; MRT = keskimääräinen elimistössä oloaika; Vss = jakautumistilavuus vakaan tason vaiheessa *12 - < 18-vuotiaita koskevissa farmakokineettisissä muuttujissa oli mukana tutkimushenkilöitä tutkimuksen I kaikista haaroista ja näytteenoton aikataulut olivat erilaisia | ||||
Taulukko 6: ELOCTA-valmisteen farmakokineettiset muuttujat pediatrisilla potilailla kromogeenisen määrityksen avulla määritettyinä
Farmakokineettiset muuttujat1 | Tutkimus II | Tutkimus I* | ||
< 6-vuotiaat | 6 - < 12-vuotiaat | 12 - < 18-vuotiaat | ||
N = 24 | N = 27 | N = 11 | ||
Inkrementaalinen saanto (IU/dl per IU/kg) | 1,88 (1,73‑2,05) | 2,08 (1,91‑2,25) | 1,91 (1,61‑2,27) | |
AUC/annos (IU*h/dl per IU/kg) | 25,9 (23,4‑28,7) | 32,8 (28,2‑38,2) | 40,8 (29,3‑56,7) | |
t½ (h) | 14,3 (12,6‑16,2) | 15,9 (13,8‑18,2) | 17,5 (12,7‑24,0) | |
MRT (h) | 17,2 (15,4‑19,3) | 20,7 (18,0‑23,8) | 23,5 (17,0‑32,4) | |
CL (ml/h/kg) | 3,86 (3,48‑4,28) | 3,05 (2,62‑3,55) | 2,45 (1,76‑3,41) | |
Vss (ml/kg) | 66,5 (59,8‑73,9) | 63,1 (56,3‑70,9) | 57,6 (50,2‑65,9) | |
1 Farmakokineettiset parametrit esitetään geometrisena keskiarvona (95 %:n CI) Lyhenteet: CI = luottamusväli; AUC = FVIII:n aktiivisuuden alle jäävä aikakäyrä; t½ = terminaalinen puoliintumisaika; CL = puhdistuma; MRT = keskimääräinen elimistössä oloaika; Vss = jakautumistilavuus vakaan tason vaiheessa *12 - < 18-vuotiaita koskevissa farmakokineettisissä muuttujissa oli mukana tutkimushenkilöitä tutkimuksen I kaikista haaroista ja näytteenoton aikataulut olivat erilaisia | ||||
Alle 12-vuotiailla lapsilla puhdistuma voi olla suurempi ja puoliintumisaika lyhyempi kuin teini-ikäisillä ja aikuisilla, mikä on johdonmukaista muita hyytymistekijöitä koskevien havaintojen kanssa. Nämä erot pitää ottaa annostuksessa huomioon.
Prekliiniset tiedot turvallisuudesta
Akuutin ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten (joihin sisältyivät myös paikallisen toksisuuden ja farmakologisen turvallisuuden arvioinnit) tulokset eivät viittaa erityiseen vaaraan ihmisille. Genotoksisuutta, karsinogeenisuutta, lisääntymistoksisuutta tai alkion-/sikiönkehitystä koskevia tutkimuksia ei ole tehty. Hiirillä tehdyssä kokeessa, jossa tutkittiin valmisteen siirtymistä istukan läpi, ELOCTA-valmisteen on osoitettu läpäisevän istukan pieninä määrinä.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine
Sakkaroosi
Natriumkloridi
Histidiini
Kalsiumklorididihydraatti
Polysorbaatti 20
Natriumhydroksidi (pH:n säätämiseen)
Suolahappo (pH:n säätämiseen)
Liuotin
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Vain mukana toimitettua infuusiovälineistöä saa käyttää, koska hyytymistekijä VIII:n tarttuminen joidenkin injektiovälineiden sisäpintoihin voi aiheuttaa hoidon epäonnistumisen.
Kestoaika
Avaamaton injektiopullo
5 vuotta
Kestoajan puitteissa valmistetta voidaan säilyttää huoneenlämmössä (enintään 30 °C:ssa) yhtäjaksoisesti enintään 6 kuukautta. Päivämäärä, jolloin valmiste otetaan pois jääkaapista, on syytä merkitä pakkauksen päälle. Valmistetta ei saa siirtää enää takaisin jääkaappiin huoneenlämmössä säilyttämisen jälkeen. Ei saa käyttää injektiopullon päälle painetun viimeisen käyttöpäivämäärän jälkeen tai 6 kuukauden kuluttua siitä, kun pakkaus on otettu jääkaapista, riippuen siitä, kumpi päivä on aikaisempi.
Käyttökuntoon saattamisen jälkeen
Käyttökuntoon saattamisen jälkeen kemiallisen ja fysikaalisen säilyvyyden on osoitettu olevan 6 tuntia, kun valmistetta säilytetään huoneenlämmössä (enintään 30 °C:ssa) Suojaa valmiste suoralta auringonvalolta. Jos valmistetta ei käyttökuntoon saattamisen jälkeen käytetä 6 tunnin kuluessa, se on hävitettävä. Mikrobiologiselta kannalta valmiste on käytettävä heti käyttökuntoon saattamisen jälkeen. Ellei sitä käytetä heti, käytönaikainen varastointi ja käyttöä edeltävät olosuhteet ovat käyttäjään vastuulla.
Säilytys
Säilytä jääkaapissa (2°C ‑ 8°C). Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ELOCTA injektiokuiva-aine ja liuotin, liuosta varten
250 IU (L:ei) 1 pakkaus (83 IU/ml) (199,84 €)
500 IU (L:ei) 1 pakkaus (167 IU/ml) (383,65 €)
750 IU (L:ei) 1 pakkaus (250 IU/ml) (567,45 €)
1000 IU (L:ei) 1 pakkaus (333 IU/ml) (747,11 €)
1500 IU (L:ei) 1 pakkaus (500 IU/ml) (1098,48 €)
2000 IU (L:ei) 1 pakkaus (667 IU/ml) (1449,83 €)
3000 IU (L:ei) 1 pakkaus (1000 IU/ml) (2132,59 €)
4000 IU (L:ei) 1 pakkaus (1333 IU/ml) (2783,25 €)
PF-selosteen tieto
Jokainen pakkaus sisältää:
- kuiva-ainetta lasisessa (tyyppi 1) injektiopullossa, jossa on klorobutyylikumitulppa
- 3 ml liuotinta lasisessa (tyyppi 1) esitäytetyssä ruiskussa, jossa on bromobutyylikumista valmistettu mäntätulppa
- männän varsi
- steriili injektiopullon adapteri käyttökuntoon saattamista varten
- steriili infuusiovälineistö
- kaksi alkoholipyyhettä
- kaksi laastaria
- yksi sideharsotaitos.
Pakkauskoko 1.
Valmisteen kuvaus:
Kuiva-aine: kylmäkuivattu, valkoinen tai luonnonvalkoinen jauhe tai kakku.
Liuotin: injektionesteisiin käytettävä vesi, kirkas, väritön liuos.
Käyttö- ja käsittelyohjeet
Injektiopullossa oleva kylmäkuivattu injektiokuiva-aine liuosta varten valmistetaan käyttökuntoon lisäämällä siihen pakkauksessa olevan esitäytetyn ruiskun sisältämä liuotin (injektionesteisiin käytettävä vesi) käyttäen tähän tarkoitettua injektiopullon steriiliä adapteria.
Injektiopulloa on pyöriteltävä varovasti, kunnes injektiokuiva-aine on kokonaan liuennut.
Käyttökuntoon saatettu lääkevalmiste on tarkastettava silmämääräisesti ennen antoa mahdollisten hiukkasten ja värimuutosten varalta. Liuoksen on oltava kirkasta tai hiukan opaalinhohtoista sekä väritöntä. Älä käytä liuosta, jos se on sameaa tai sisältää sakkaa.
Lisätietoja käyttökuntoon saattamisesta ja antamisesta
ELOCTA annetaan injektiona laskimoon (i.v.) sen jälkeen, kun se on tehty käyttökuntoon liuottamalla injektiokuiva-aine pakkauksessa olevasta esitäytetystä ruiskusta saatavaan liuottimeen. ELOCTA-pakkaus sisältää:
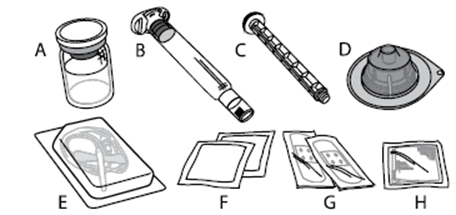
A) 1 injektiokuiva-ainepullo B) 3 ml liuotinta esitäytetyssä ruiskussa C) 1 männän varsi D) 1 injektiopullon adapteri E) 1 infuusiovälineistö F) 2 alkoholipyyhettä G) 2 laastaria H) 1 sideharsotaitos
ELOCTA-valmistetta ei saa sekoittaa muiden injektio- tai infuusionesteiden kanssa.
Pese kädet ennen pakkauksen avaamista.
Valmistus:
| 1. Tarkista pakkauksesta valmisteen nimi ja vahvuus varmistaaksesi, että lääke on oikea. Tarkista viimeinen käyttöpäivämäärä ELOCTA-pakkauksesta. Älä käytä, jos lääke on vanhentunut. | |
| 2. Jos ELOCTA-valmistetta on säilytetty jääkaapissa, odota jonkin aikaa, jotta ELOCTA-valmistetta sisältävä injektiopullo (A) ja liuotinta sisältävä ruisku (B) lämpiävät huoneenlämpöisiksi ennen käyttöä. Älä käytä ulkoisia lämmönlähteitä. | |
| 3. Aseta injektiopullo puhtaalle, tasaiselle alustalle. Poista muovinen irti napsautettava korkki ELOCTA-injektiopullosta. | 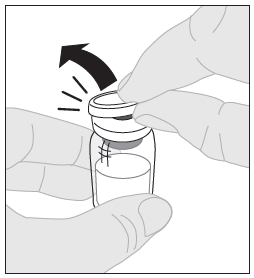 |
| 4. Pyyhi injektiopullon yläosa pakkauksessa olevalla alkoholipyyhkeellä (F) ja anna sen kuivua. Älä koske injektiopullon yläosaan äläkä anna sen koskettaa mihinkään pyyhkimisen jälkeen. | 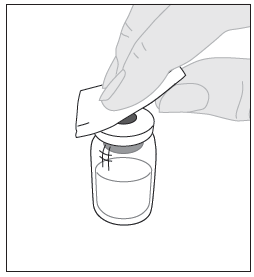 |
| 5. Irrota paperinen suojus injektiopullon kirkkaasta, muovisesta adapterista (D). Älä irrota adapteria sen suojakorkista. Älä koske injektiopullon adapteripakkauksen sisäpuolta. | |
| 6. Aseta injektiopullo tasaiselle alustalle. Pitele injektiopullon adapteria suojakorkista ja aseta se suoraan injektiopullon yläosan päälle. Paina tiukasti alaspäin, kunnes adapteri napsahtaa paikalleen injektiopullon yläosaan siten, että adapterin piikki läpäisee injektiopullon tulpan. | 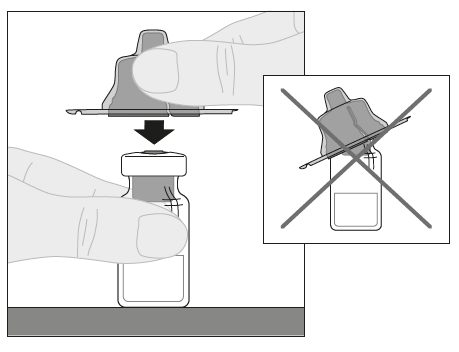 |
| 7. Kiinnitä männän varsi (C) liuotinruiskuun työntämällä männän varren kärki ruiskun männässä olevaan aukkoon. Käännä männän vartta tiukasti myötäpäivään, kunnes se on varmasti kiinni ruiskun männässä. | 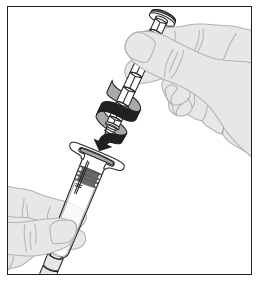 |
| 8. Katkaise valkoinen, avaamattomuuden osoittava muovinen korkki liuotinruiskusta taivuttamalla sitä lävistyksen kohdalta, kunnes se irtoaa naksahtaen. Siirrä korkki syrjään asettamalla se nurinpäin tasaiselle alustalle. Älä koske korkin sisäpuolta tai ruiskun kärkeä. | 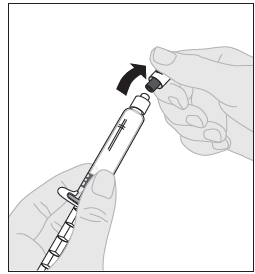 |
| 9. Ota suojakorkki pois adapterista ja hävitä se. | 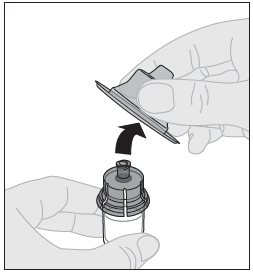 |
| 10. Liitä liuotinruisku injektiopullon adapteriin työntämällä ruiskun kärki adapterin aukkoon. Paina ja käännä ruiskua tiukasti myötäpäivään, kunnes se on varmasti kiinni. | 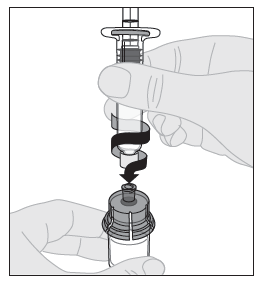 |
| 11. Paina männän vartta hitaasti alaspäin kunnes kaikki liuotin on ELOCTA-injektiopullossa. | 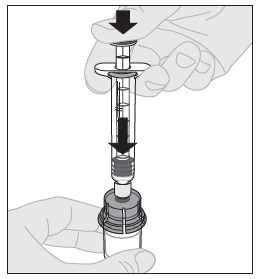 |
| 12. Ruiskun ollessa yhä kiinni adapterissa ja männän varren ollessa alhaalla pyörittele injektiopulloa varovasti, kunnes kuiva-aine on kokonaan liuennut. Ei saa ravistaa. | 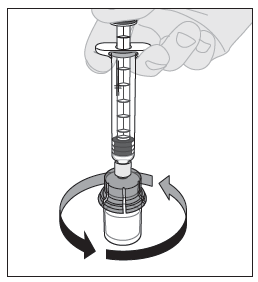 |
| 13. Lopullinen liuos pitää tarkastaa silmämääräisesti ennen antoa. Liuoksen on oltava kirkasta tai hieman opaalinhohtoista sekä väritöntä. Älä käytä liuosta, jos se on sameaa tai jos siinä näkyy hiukkasia. | |
| 14. Varmista, että ruiskun männän varsi on edelleen painettuna kokonaan alas ja käännä injektiopullo ylösalaisin. Vedä männän vartta hitaasti niin, että kaikki liuos virtaa injektiopullon adapterin läpi ruiskuun. | 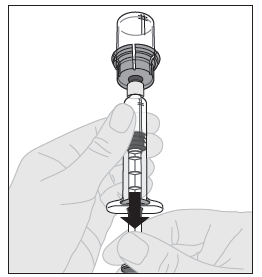 |
| 15. Irrota ruisku injektiopullon adapterista vetämällä sitä varovasti ja kääntämällä ruiskua vastapäivään. | 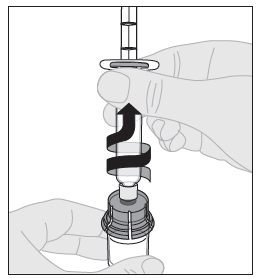 |
| Huom! Jos käytät yhdellä injektiokerralla enemmän kuin yhden injektiopullon ELOCTA-valmistetta, kukin injektiopullo on saatettava käyttökuntoon erikseen edellä esitettyjen ohjeiden mukaisesti (vaiheet 1‑13) ja liuotinruisku on vedettävä pois, mutta injektiopullon adapteri jätettävä paikalleen. Käyttövalmiit liuokset voidaan vetää kustakin injektiopullosta käyttämällä yhtä isoa luer-lukollista ruiskua. | |
16. Hävitä injektiopullo ja adapteri. Huom! Jos liuosta ei oteta heti käyttöön, ruiskun korkki pitää laittaa varovasti takaisin ruiskun kärkeen. Älä koske ruiskun kärkeä tai korkin sisäpuolta. Käyttövalmista ELOCTA-valmistetta voidaan säilyttää huoneenlämmössä enintään 6 tunnin ajan ennen antoa. Tämän ajan kuluttua käyttövalmis ELOCTA on hävitettävä. Suojaa valmiste suoralta auringonvalolta. | |
Antotapa (injektio laskimoon):
ELOCTA annetaan käyttäen pakkauksessa olevaa infuusiovälineistöä (E).
| 1. Avaa infuusiovälineistön pakkaus ja poista letkun päässä oleva korkki. Kiinnitä käyttövalmista ELOCTA-liuosta sisältävä ruisku infuusiovälineistön letkun päähän kääntämällä ruiskua myötäpäivään. | 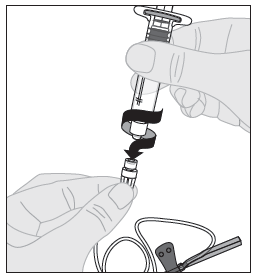 |
2. Käytä tarvittaessa kiristyssidettä ja valmistele injektiokohta pyyhkimällä iho hyvin pakkauksen toisella alkoholipyyhkeellä.
| |
| 3. Poista kaikki ilma infuusioletkustosta painamalla männän vartta hitaasti alaspäin kunnes neste on infuusiovälineistön neulan kohdalla. Älä työnnä liuosta neulan läpi. Poista neulasta sen kirkas, muovinen suojus. | |
| 4. Työnnä infuusiovälineistön neula laskimoon lääkärin tai sairaanhoitajan opettamalla tavalla ja poista kiristysside. Halutessasi voit kiinnittää toisella pakkauksen laastareista (G) neulan muovisiivekkeet paikalleen injektiokohtaan. Käyttövalmis valmiste on injisoitava laskimoon usean minuutin aikana. Lääkäri saattaa muuttaa suositeltua injektionopeutta, jotta olosi tuntuisi mukavammalta. | |
| 5. Injektion lopettamisen ja neulan poistamisen jälkeen neulan suojus on käännettävä takaisin ja napsautettava neulan päälle. | 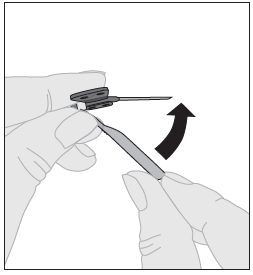 |
| 6. Hävitä käytetty neula, käyttämättä jäänyt liuos, ruisku ja tyhjä injektiopullo turvallisesti asianmukaiseen sairaalajätteen säiliöön, koska nämä materiaalit voivat vahingoittaa toisia, ellei niitä hävitetä oikeaoppisesti. Tarvikkeita ei saa käyttää uudelleen. | |
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ELOCTA injektiokuiva-aine ja liuotin, liuosta varten
250 IU 1 pakkaus
500 IU 1 pakkaus
750 IU 1 pakkaus
1000 IU 1 pakkaus
1500 IU 1 pakkaus
2000 IU 1 pakkaus
3000 IU 1 pakkaus
4000 IU 1 pakkaus
- Ylempi erityiskorvaus (100 %). Krooniset hyytymishäiriöt (126).
- Peruskorvaus (40 %).
ATC-koodi
B02BD02
Valmisteyhteenvedon muuttamispäivämäärä
25.04.2025
Yhteystiedot
Äyritie 18
01510 Vantaa
0201 558 840
www.sobi.fi
mail.fi@sobi.com