
ILUMETRI injektioneste, liuos, esitäytetty ruisku 100 mg, 200 mg
Vaikuttavat aineet ja niiden määrät
Ilumetri 100 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 100 mg tildrakitsumabia 1 ml:ssa liuosta.
Ilumetri 200 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 200 mg tildrakitsumabia 2 ml:ssa liuosta.
Tildrakitsumabi on kiinanhamsterin munasarjasoluissa (chinese hamster ovary, CHO) yhdistelmä-DNA-tekniikalla valmistettu humanisoitu monoklonaalinen IgG1/k-vasta-aine.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Apuaine, jonka vaikutus tunnetaan
Yksi Ilumetri 100 mg injektioneste, liuos, esitäytetty ruisku sisältää 0,5 mg polysorbaatti 80:tä (E 433).
Yksi Ilumetri 200 mg injektioneste, liuos, esitäytetty ruisku sisältää 1 mg polysorbaatti 80:tä (E 433).
Lääkemuoto
Injektioneste, liuos (injektio)
Kliiniset tiedot
Käyttöaiheet
Ilumetri on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuisille, joille harkitaan systeemistä hoitoa.
Ehto
Valmiste on tarkoitettu käytettäväksi psoriaasin diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Tämä lääkevalmiste on tarkoitettu käytettäväksi läiskäpsoriaasin diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus
Suositeltu annos on 100 mg injektiona ihon alle viikoilla 0 ja 4 ja sen jälkeen 12 viikon välein.
Jos potilaalla on suuri tautikuorma tai potilaan paino on yli 90 kg, lääkäri saattaa arvioida 200 mg:n annoksen tehokkaammaksi.
Jos potilaalla ei todeta vastetta 28 viikon hoidon jälkeen, hoidon lopettamista on harkittava. Joidenkin potilaiden alussa saama osittainen vaste voi parantua, kun hoitoa jatketaan yli 28 viikkoa.
Väliin jäänyt annos
Jos annos jää väliin, se on annettava mahdollisimman pian. Sen jälkeen annosten antamista jatketaan tavanomaisen aikataulun mukaisesti.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten tai maksan vajaatoiminta
Ilumetria ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida antaa. Lisätietoja tildrakitsumabin eliminaatiosta, ks. kohta Farmakokinetiikka.
Pediatriset potilaat
Ilumetrin turvallisuutta ja tehoa lapsilla ja alle 18-vuotiailla nuorilla ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste annetaan injektiona ihon alle. Injektiokohtaa tulee vaihdella. Ilumetria ei pidä pistää alueelle, jolla on läiskäpsoriaasia tai jonka iho on arka, mustelmainen, punoittava, kova, paksu tai hilseilevä. Ruiskua ei saa ravistaa. Jokainen ruisku on kertakäyttöinen.
Kun potilas on saanut asianmukaisen opastuksen ihon alle annettavien injektioiden antotekniikkaan, hän voi injisoida Ilumetrin itse, jos lääkäri katsoo sen asianmukaiseksi. Lääkärin on kuitenkin varmistettava potilaan asianmukainen seuranta. Potilasta on neuvottava injisoimaan koko tildrakitsumabiannos pakkausselosteen ohjeiden mukaisesti. Pakkausselosteessa on valmisteen antoa koskevat kattavat ohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio, esim. aktiivinen tuberkuloosi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi potilaalle annetun valmisteen kauppanimi ja eränumero on kirjattava selkeästi muistiin.
Infektiot
Tildrakitsumabi saattaa lisätä infektioriskiä (ks. kohta Haittavaikutukset).
Varovaisuutta on noudatettava, kun harkitaan tildrakitsumabin käyttöä potilaille, joilla on krooninen infektio, aiemmin ollut toistuvia infektioita tai hiljattain ollut vakava infektio.
Potilaita on neuvottava hakeutumaan lääkärin hoitoon, jos kliinisesti tärkeän kroonisen tai akuutin infektion oireita tai löydöksiä ilmenee. Jos potilaalle kehittyy vakava infektio, häntä on seurattava huolellisesti, eikä tildrakitsumabia saa antaa ennen kuin infektio on parantunut (ks. kohta Vasta-aiheet).
Tuberkuloosin tutkiminen ennen hoitoa
Potilaalta on tutkittava tuberkuloosi-infektio ennen hoidon aloittamista. Tildrakitsumabihoitoa saavaa potilasta on seurattava tarkasti aktiivisen tuberkuloosin oireiden ja löydösten havaitsemiseksi hoidon aikana ja sen jälkeen. Jos potilaalla on aiemmin ollut piilevä tai aktiivinen tuberkuloosi, jonka riittävää hoitoa ei pystytä varmistamaan, on harkittava tuberkuloosilääkitystä ennen hoidon aloittamista.
Yliherkkyys
Jos potilaalle ilmaantuu vakava yliherkkyysreaktio, tildrakitsumabin antaminen on lopetettava heti ja aloitettava asianmukainen hoito (ks. kohta Vasta-aiheet).
Rokotukset
Kaikkien asianmukaisten rokotusten antamista voimassa olevien rokotusohjeiden mukaisesti on harkittava ennen tildrakitsumabihoidon aloittamista. Jos potilas on saanut eläviä viruksia tai bakteereja sisältävää rokotetta, on suositeltavaa odottaa vähintään neljä viikkoa ennen tildrakitsumabihoidon aloittamista. Tildakitsumabia saaville potilaille ei saa antaa eläviä rokotteita samanaikaisesti eikä vähintään 17 viikkoon hoidon jälkeen (ks. kohta Yhteisvaikutukset).
Apuaineet
Tämä lääkevalmiste sisältää 0,5 mg polysorbaatti 80:tä (E 433) per 100 mg:n esitäytetty ruisku ja 1 mg polysorbaatti 80:tä (E 433) per 200 mg:n esitäytetty ruisku, joka vastaa 0,5 mg/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Rokotteet
Vasteesta eläville tai inaktivoituja taudinaiheuttajia sisältäville rokotteille ei ole tietoja. Eläviä rokotteita ei saa antaa tildrakitsumabihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Interaktiot sytokromi P450:n kanssa
Samanaikaisesti käytettävät lääkevalmisteet eivät oletettavasti vaikuta tildrakitsumabin farmakokinetiikkaan, koska lääkeaine poistuu kehosta yleisten proteiinikataboliaprosessien kautta. Sytokromi P450 (CYP450) ‑entsyymit eivät osallistu näihin prosesseihin, eikä lääkeaine poistu munuaisten tai maksan kautta. Tildrakitsumabi ei myöskään vaikuta sellaisten samanaikaisesti käytettyjen lääkevalmisteiden farmakokinetiikkaan, joita CYP450-entsyymit metaboloivat joko suorilla tai epäsuorilla mekanismeilla (ks. kohta Farmakokinetiikka).
Yhteisvaikutukset muiden immunosuppressiivisten aineiden tai valohoidon kanssa
Tildrakitsumabin turvallisuutta ja tehoa yhdessä muiden immunosuppressiivisten aineiden, mukaan lukien biologiset lääkkeet, tai valohoidon kanssa ei ole tutkittu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 17 viikon ajan hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta) tildrakitsumabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varotoimena suositellaan, ettei Ilumetria käytetä raskauden aikana.
Imetys
Ei tiedetä, erittyykö tildrakitsumabi ihmisen rintamaitoon. Saatavissa olevat toksikologiset tiedot cynomolgus-apinoista ovat osoittaneet vähäpätöisiä Ilumetri-pitoisuuksia maidossa 28. syntymänjälkeisenä päivänä (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisillä vasta-aineita voi siirtyä vastasyntyneeseen rintamaidon kautta ensimmäisinä syntymänjälkeisinä päivinä. Tämän lyhyen jakson aikana vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Ilumetri-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ilumetrin vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ilumetrilla ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset ovat ylähengitysteiden infektiot (12,6 %), päänsärky (4,0 %), ripuli (1,6 %), gastroenteriitti (1,5 %), selkäkipu (1,5 %), pahoinvointi (1,3 %) ja injektiokohdan kipu (1,3 %).
Haittavaikutustaulukko
Haittavaikutukset kliinisistä lääketutkimuksista (taulukko 1) on luokiteltu MedDRA-elinjärjestelmän ja esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutusluettelo
MedDRA-elinjärjestelmä | Suositeltu termi | Esiintymistiheys |
Infektiot | Ylähengitysteiden infektiota | Hyvin yleinen |
Hermosto | Päänsärky | Yleinen |
Ruoansulatuselimistö | Gastroenteriitti | Yleinen |
Ripuli | Yleinen | |
Pahoinvointi | Yleinen | |
Yleisoireet ja antopaikassa todettavat haitat | Selkäkipu | Yleinen |
Injektiokohdan kipu | Yleinen |
aMukaan lukien nasofaryngiitti.
Pitkäaikainen turvallisuus
reSURFACE 1- ja reSURFACE 2 -tutkimusten pitkäkestoisten jatkojaksojen aikana havaittu tildrakitsumabin turvallisuusprofiili oli yhdenmukainen kaksoissokkoutettujen jaksojen turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskekus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa laskimoon on annettu turvallisesti enintään 10 mg/kg annoksia.
Yliannostustapauksessa on suositeltavaa seurata potilaan vointia haittavaikutusten oireiden tai löydösten varalta ja aloittaa viipymättä asianmukainen oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC17
Vaikutusmekanismi
Tildrakitsumabi on humanisoitu monoklonaalinen IgG1/k:n vasta-aine, joka sitoutuu spesifisesti interleukiini-23 (IL-23) -sytokiinin p19-proteiinialayksikköön sitoutumatta IL-12:een ja estää sen vuorovaikutuksen IL-23:n reseptorin kanssa.
IL-23 on luonnollisesti esiintyvä sytokiini, joka on osallisena tulehduksellisissa ja immuunivasteissa. Tildrakitsumabi estää tulehdusta edistävien sytokiinien ja kemokiinien vapautumista.
Kliininen teho ja turvallisuus
Satunnaistettuihin, kaksoissokkoutettuihin, lumekontrolloituihin monikeskustutkimuksiin reSURFACE 1 ja reSURFACE 2 otettiin mukaan yhteensä 1 862 potilasta, jotka olivat iältään vähintään 18-vuotiaita ja joilla oli läiskäpsoriaasi. Näillä potilailla sairaan ihon alue oli vähintään 10 %, lääkärin tekemässä yleisarviossa (Physician Global Assessment, PGA) kokonaisarvio (läiskän paksuus, punoitus ja hilseily) oli ≥ 3, kun psoriaasin vaikeusasteen arviointiasteikko oli 0–5, PASI-vastepisteiden (Psoriasis Area and Severity Index) oli ≥ 12, ja potilaille suunniteltiin valohoitoa tai systeemistä hoitoa.
Potilaat satunnaistettiin näissä tutkimuksissa joko lumelääke- tai tildrakitsumabiryhmään (mukaan lukien 200 mg ja 100 mg viikoilla 0 ja 4, ja tämän jälkeen 12 viikon välein) enintään 52 tai 64 viikon ajaksi. Tutkimuksessa, jossa oli käytössä aktiivinen vertailuvalmiste (reSURFACE 2), potilaat satunnaistettiin myös saamaan etanerseptiä 50 mg kahdesti viikossa 12 viikon ajan sekä tämän jälkeen viikoittain viikkoon 28 asti. Jos potilas ei saanut vastetta etanerseptihoitoon (PASI-vastepisteiden pieneneminen < 75 % lähtötilanteesta), hoidoksi vaihdettiin 200 mg tildrakitsumabia 12 viikon välein enintään 52 viikon ajan, kun taas etanerseptihoitoon vasteen saaneiden potilaiden tutkimukseen osallistuminen keskeytettiin.
Tutkimuksiin soveltuneet potilaat, jotka olivat tutkimuksissa reSURFACE 1 ja reSURFACE 2 mukana niiden kaksoissokkoutettujen jaksojen loppuun saakka ja joiden PASI-vastepisteet paranivat ≥ 50 % lähtötilanteesta, saivat osallistua näiden tutkimusten avoimiin jatkojaksoihin, jotta jatkuvan tildrakitsumabihoidon pitkäaikaista turvallisuutta ja tehon säilymistä arvioitiin. reSURFACE 1- ja reSURFACE 2 -tutkimusten jatkojaksoihin mukaan tulleet potilaat jatkoivat hoitoa samalla tildrakitsumabiannoksella (joko 100 mg tai 200 mg), jota he saivat viikolla 64 (reSURFACE 1) tai viikolla 52 (reSURFACE 2). Tietoja on saatavilla enimmillään 6 vuoden pituisesta seurannasta.
Potilaiden demografiset ja lähtötilanteen ominaisuudet olivat tutkimuksissa reSURFACE1 ja reSURFACE2 yleisesti yhdenmukaiset kaikissa yksittäisissä kokeissa. Potilaat olivat 18–82-vuotiaita, ja heidän keskimääräinen ikänsä oli 45,9 vuotta. Lähtötilanteen PASI-vastepisteiden mediaani kaikissa tutkimusryhmissä oli 17,7–18,4. Potilaista 33,4 %:lla oli lähtötilanteessa merkittävän tai vaikea-asteisen sairauden PGA-pisteet. 35,8 % potilaista oli saanut aiemmin läiskäpsoriaasin hoitoon valohoitoa, 41,1 % oli saanut aiemmin tavanomaista systeemistä hoitoa, ja 16,7 % oli saanut aiemmin biologista hoitoa. Yhteensä 15,4 %:lla tutkimuspotilaista oli ollut nivelpsoriaasi. Ihosairautta koskevan elämänlaatuindeksin (Dermatology Life Quality Index, DLQI) pisteet olivat lähtötilanteessa keskimäärin 13,0–14,8.
Tutkimuksissa reSURFACE 1 ja reSURFACE 2 arvioitiin kahden ensisijaisen päätetapahtuman muutosta lähtötilanteesta viikkoon 12: 1) PASI 75 -vastepisteet ja 2) PGA-pisteet 0 (parantunut) tai 1 (vähäinen), ja vähintään 2 pisteen parannus lähtötilanteesta. Muita arvioituja hoitotuloksia olivat niiden potilaiden osuus, jotka saavuttivat PASI 90 ‑vasteen tai PASI 100 ‑vasteen, niiden potilaiden osuus, joiden DLQI-pisteet olivat 0 tai 1, sekä tehon säilyminen viikkoon 52 tai 64 saakka.
Tulokset viikoilla 12, 28 ja sen jälkeen (reSURFACE 1 -tutkimuksessa viikkoon 64 saakka ja reSURFACE 2 ‑tutkimuksessa viikkoon 52 saakka) on esitetty taulukossa 2 ja taulukossa 3.
Taulukko 2. Yhteenveto vasteluvuista tutkimuksissa reSURFACE 1 ja reSURFACE 2
| Viikko 12 (2 annosta)* | Viikko 28 (3 annosta)* | |||||
200 mg | 100 mg | Lumelääke | Etanersepti | 200 mg | 100 mg | Etanersepti | |
reSURFACE1 | |||||||
Potilaiden lukumäärä | 308 | 309 | 154 | - | 298 | 299 | - |
PASI 75a (%) | 62,3†b | 63,8†b | 5,8b | - | 81,9c | 80,4c | - |
Lääkärin yleisarvion perusteella tauti ”parantunut” tai ”vähäinen”, ja ≥ 2 asteen parannus lähtötilanteestaa (%) | 59,1†b | 57,9†b | 7,1b | - | 69,1c | 66,0c | - |
PASI 90 (%) | 35,4†b | 34,6†b | 2,6b | - | 59,0c | 51,6c | - |
PASI 100 (%) | 14,0†b | 13,9†b | 1,3b | - | 31,5c | 23,5c | - |
DLQI-pisteet 0 tai 1 (%) | 44,2† | 41,5 † | 5,3 | - | 56,7c | 52,4c | - |
reSURFACE2 | |||||||
Potilaiden lukumäärä | 314 | 307 | 156 | 313 | 299 | 294 | 289 |
PASI 75a (%) | 65,6†‡b | 61,2†‡b | 5,8b | 48,2b | 72,6‡b | 73,5‡b | 53,6b |
Lääkärin yleisarvion perusteella tauti ”parantunut” tai ”vähäinen”, ja ≥ 2 asteen parannus lähtötilanteestaa (%) | 59,2†¥b | 54,7†b | 4,5b | 47,6b | 69,2‡b | 64,6‡b | 45,3b |
PASI 90 (%) | 36,6†‡b | 38,8†‡b | 1,3b | 21,4b | 57,7‡c | 55,5‡c | 29,4 c |
PASI 100 (%) | 11,8†‡b | 12,4†‡b | 0 | 4,8b | 27,0‡c | 22,8‡c | 10,7c |
DLQI-pisteet 0 tai 1 (%) | 47,4†¥ | 40,2† | 8,0 | 35,5 | 65,0‡c | 54,1‡c | 39,4c |
a Toinen ensisijainen tehon päätetapahtuma viikolla 12.
b Hoitoon vastaamattomien potilaiden puuttuvien tietojen korvaaminen.
c Puuttuvia tietoja ei korvattu.
* Annettujen annosten määrä viittaa vain tildrakitsumabiryhmiin.
n = tietojen korvaamisen jälkeen, jos sovellettavissa, niiden potilaiden lukumäärä koko analyysiaineistossa, joista tietoja oli saatavilla.
p-arvot laskettu Cochran-Mantel-Haenszelin (CMH) testillä painon mukaan (≤ 90 kg, > 90 kg) ositettuna ja ennen altistusta psoriaasin biologiselle hoidolle (kyllä/ei).
† p ≤ 0,001 verrattaessa lumelääkkeeseen; ‡ p ≤ 0,001 verrattaessa etanerseptiin; ¥ p ≤ 0,05 verrattaessa etanerseptiin.
Vasteen säilyminen
Vasteen säilyminen tutkimuksissa reSURFACE 1 ja reSURFACE 2 esitetään taulukossa 3. PASI 90 ‑vasteen säilyminen ja kestoaika esitetään kuvassa 1.
Taulukko 3. Vasteen säilyminen tutkimuksissa reSURFACE 1 ja reSURFACE 2
| Pitkäaikaisvastea,b | |||
200 mg | 100 mg | |||
reSURFACE 1 | Viikko 28 | Viikko 64 | Viikko 28 | Viikko 64 |
Potilaiden lukumäärä | 116 | 114 | 115 | 112 |
Lääkärin yleisarvion perusteella tauti ”parantunut” tai ”vähäinen”, ja ≥ 2 asteen parannus lähtötilanteesta (%) | 80,2 | 76,3 | 80,9 | 61,6 |
PASI 90 (%) | 70,7 | 74,6 | 65,2 | 58,0 |
PASI 100 (%) | 38,8 | 40,4 | 25,2 | 32,1 |
reSURFACE 2 | Viikko 28 | Viikko 52 | Viikko 28 | Viikko 52 |
Potilaiden lukumäärä | 108 | 105 | 213 | 204 |
Lääkärin yleisarvion perusteella tauti ”parantunut” tai ”vähäinen”, ja ≥ 2 asteen parannus lähtötilanteesta (%) | 88,0 | 84,8 | 84,0 | 79,4 |
PASI 90 (%) | 75,0 | 81,9 | 74,2 | 78,4 |
PASI 100 (%) | 34,3 | 46,7 | 30,2 | 35,3 |
a Pitkäaikaisvaste potilailla, jotka olivat saaneet vasteen (olivat saaneet vähintään PASI 75 ‑vasteen) tildrakitsumabille viikolla 28.
b Puuttuvia tietoja ei korvattu.
Kuva 1. PASI 90 ‑vasteen säilyminen ja kestoaika. Niiden potilaiden osuus, joilla PASI 90 -vaste säilyi viikkoon 64 saakka (täydellinen analyysiaineisto osa 3*)
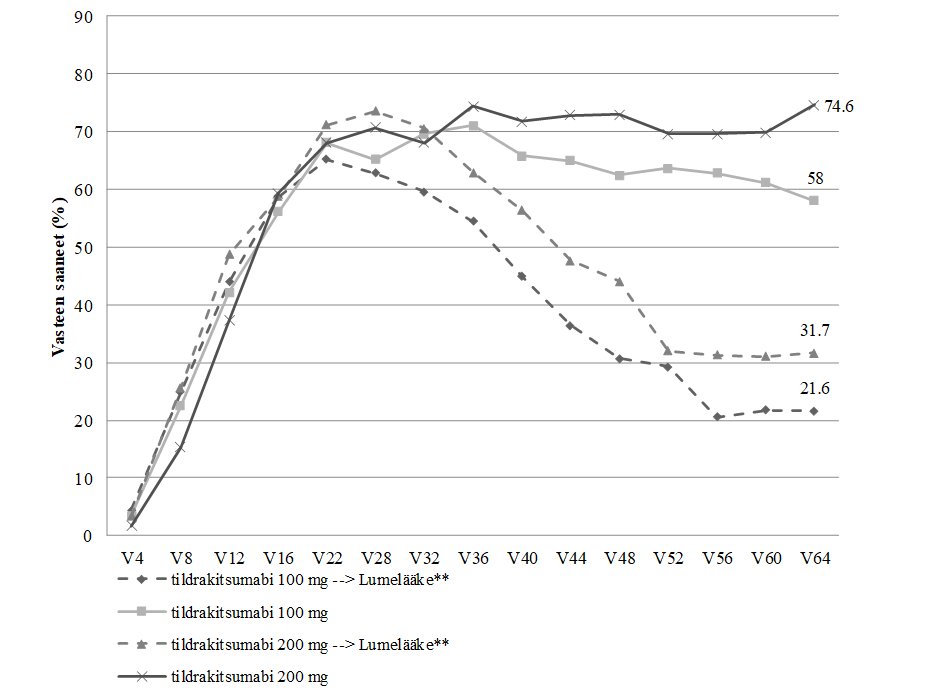
Tutkimuksen osassa 1 tildrakitsumabiannoksia 100 mg tai 200 mg saamaan satunnaistetut potilaat, jotka olivat saaneet PASI 75 ‑vasteen viikolla 28 (reSURFACE 1).
* Puuttuvia tietoja ei korvattu.
** Nämä potilaat siirtyivät saamaan lumelääkettä viikolla 28.
Kaksoissokkoutetussa jaksossa sen loppuun saakka mukana olleista potilaista 506 (79 %) potilasta reSURFACE 1 ‑tutkimuksessa ja 730 (97 %) potilasta reSURFACE 2 ‑tutkimuksessa tuli mukaan jatkojaksoon. Näissä tutkimuksissa potilaista, joilla oli PASI 90 ‑vaste kaksoissokkoutetun jakson lopussa, vähintään 76 %:lla PASI 90 ‑vaste säilyi jatkojakson aikana, kun 100 mg:n tai 200 mg:n tildrakitsumabihoitoa jatkettiin 192 viikon ajan (kuvat 2 ja 3).
Kuva 2. Niiden potilaiden prosenttiosuus käynneittäin, joilla PASI 90 -vaste säilyi reSURFACE 1 ‑tutkimuksen avoimen jatkojakson aikana (täydellinen analyysiaineisto, jatkojakso*)
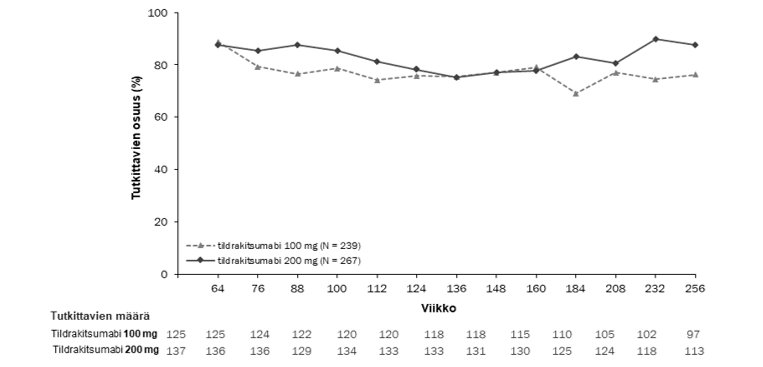
*PASI 90 -vasteen saaneilla kaksoissokkoutetun tutkimusjakson päättyessä. Puuttuvia tietoja ei korvattu.
Huomautus: Käyntiviikko on nimellinen, koska tutkittavilla oli viikosta 64 alkaen noin 12 viikon aikaikkuna jatkojakson aloittamiseen.
Kuva 3. Niiden potilaiden prosenttiosuus käynneittäin, joilla PASI 90 -vaste säilyi reSURFACE 2 ‑tutkimuksen avoimen jatkojakson aikana (täydellinen analyysiaineisto, jatkojakso*)
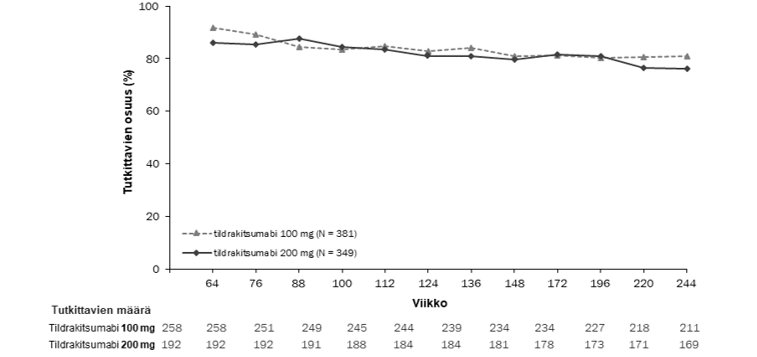
*PASI 90 -vasteen saaneilla kaksoissokkoutetun tutkimusjakson päättyessä. Puuttuvia tietoja ei korvattu.
Elämänlaatu / potilaiden raportoimat hoitotulokset
Tildrakitsumabihoitoon liittyi viikolla 12 ja kaikissa tutkimuksissa terveyteen liittyvän elämänlaadun tilastollisesti merkitsevää paranemista DLQI-indeksillä (taulukko 2) arvioituna. Elämänlaadun paraneminen säilyi viikkoon 52 siten, että 63,7 %:lla (100 mg) ja 73,3 %:lla (200 mg) reSURFACE 1-potilaista, ja 68,8 %:lla (100 mg) ja 72,4 %:lla (200 mg) reSURFACE 2-potilaista, jotka olivat saaneet PASI 75 -vasteen viikolla 28, DLQI oli 0 tai 1.
Päänahan läiskäpsoriaasi
Satunnaistetussa, lumekontrolloidussa tutkimuksessa arvioitiin tildrakitsumabin tehoa ja turvallisuutta 231 potilaalla, joilla oli keskivaikea tai vaikea päänahan läiskäpsoriaasi, jonka määritelmänä oli päänahkaa koskeva tutkijan yleisarvion (Investigator’s Global Assessment, IGA) muokattu (mod 2011) pistemäärä ≥ 3, Psoriasis Scalp Severity Index (PSSI) ‑vaikeusasteindeksin pistemäärä ≥ 12 ja ≥ 30 % päänahan pinta-alasta psoriaasin peitossa lähtötilanteessa. Viikon 16 aikapisteessä tildrakitsumabihoitoon liittyi lumelääkkeeseen verrattuna tilastollisesti merkittävää parannusta kummankin päätepisteen osalta, jotka olivat pelkän päänahan vastetta koskeva IGA mod 2011 ‑pistemäärä 0 tai 1 (49 % tildrakitsumabia saaneilla ja 7 % lumelääkettä saaneilla) ja PSSI 90 ‑vaste (56 % tildrakitsumabia saaneilla ja 4 % lumelääkettä saaneilla). Nämä vaikutukset säilyivät tildrakitsumabia saaneilla potilailla, jotka jatkoivat hoitoa viikolle 52 saakka.
Kynsipsoriaasi
Satunnaistetussa, lumekontrolloidussa tutkimuksessa arvioitiin tildrakitsumabin tehoa ja turvallisuutta 99 aikuisella, joilla oli keskivaikea tai vaikea kynsipsoriaasi, jonka määritelmänä oli modifioidun kynsipsoriaasin vaikeusasteindeksin (modified Nail Psoriasis Severity Index, mNAPSI) pistemäärä ≥ 20 ja vähintään 50 % kynsistä tuhoutunut. Tutkittavat satunnaistettiin saamaan 100 mg tildrakitsumabia viikolla 0, viikolla 4 ja sen jälkeen 12 viikon välein tai lumelääkettä enintään 28 viikon ajan. Tildrakitsumabilla osoitettiin olevan tilastollisesti merkitsevää hyötyä keskivaikeaa tai vaikeaa kynsipsoriaasia sairastavilla potilailla (taulukko 4).
Taulukko 4. Tehotulokset viikolla 28
Päätetapahtuma | Tildrakitsumabi 100 mg 12 viikon välein | Lumelääke | Vasteen ero (95 %:n luottamusväli) |
N = 51 | N = 48 | ||
mNAPSI 75, n (%) | 13 (25,5)a | 2 (4,2)a | 22,0 % (7,97; 36,06)b |
Keskimääräinen muutos sormenkynsiä koskevassa mNAPSI-kokonaispistemäärässä | -21,8 | -7,7 | -14,1 (-19,86; -8,42)c |
aHoitoon vastaamattomien potilaiden puuttuvien tietojen korvaaminen.
bVasteen ero ja luottamusväli laskettiin käyttäen Miettinen-Nurminen-menetelmää painon ja aiemman TNF-estäjälle altistumisen perusteella ositettuna.
cMMRM-malliin sisältyivät kiinteinä vaikutuksina hoito, käynti, hoidon vaikutus käynneittäin, TNF-alfa-estäjien aiempi käyttö, painoluokka ja aiempi altistuminen TNF-estäjille.
Immunogeenisuus
Yhdistetyissä vaiheen 2b ja vaiheen 3 analyyseissä 7,3 %:lle tildrakitsumabihoitoa saaneista potilaista kehittyi vasta-aineita tildrakitsumabille viikolle 64 mennessä. Potilaista, joille kehittyi vasta-aineita tildrakitsumabille, 38 %:lla (22/57 potilasta) oli neutraloivia vasta-aineita. Tämä vastaa 2,8 % kaikista tildrakitsumabia saaneista tutkittavista.
Yhdistetyissä vaiheen 3 analyyseissä 8,3 %:lle tildrakitsumabihoitoa saaneista potilaista kehittyi vasta-aineita tildrakitsumabille 420 hoitoviikkoon mennessä. Tildrakitsumabihoitoa saaneista potilaista, joille kehittyi vasta-aineita tildrakitsumabille, 35 %:lla (36/102 potilasta) oli neutraloiviksi luokiteltuja vasta-aineita. Tämä vastaa 2,9 % kaikista tildrakitsumabihoitoa saaneista potilaista.
Neutraloivien vasta-aineiden kehittymiseen tildrakitsumabia vastaan liittyi pienempi tildrakitsumabin pitoisuus seerumissa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Ilumetrin käytöstä yhden tai useamman pediatrisen potilasryhmän läiskäpsoriaasin hoidossa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Tildrakitsumabin ihon alle annettava lääkemuoto osoitti absoluuttisen biologisen hyötyosuuden olevan arvojen 73 % (90 % LV: 46–115 %, 200 mg ihon alle vs. 3 mg/kg laskimoon) ja 80 % (90 % LV: 62–103 %, 50 mg ihon alle vs. 0,5 mg/kg laskimoon) välillä terveillä tutkittavilla tutkimusten välisessä yhden annoksen vertailussa. Maksimipitoisuus saavutettiin 6,2 vuorokautta injektion jälkeen. Populaatiofarmakokineettinen analyysi osoitti, että biologinen hyötyosuus oli 31 % suurempi terveillä tutkittavilla potilaisiin verrattuna.
100 mg:n tildrakitsumabiannosten jälkeen keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavien tutkittavien vakaan tilan AUC0-τ-arvon geometrinen keskiarvo (variaatiokerroin, %) ja Cmax-arvon geometrinen keskiarvo (variaatiokerroin, %) oli 305 μg·vrk/ml (41 %) ja 8,1 μg/ml (34 %), tässä järjestyksessä, kun se oli 612 μg·vrk/ml (40 %) ja 16,3 μg/ml (33 %) 200 mg:n annoksen antamisen jälkeen.
Jakautuminen
Tildrakitsumabi jakautuu niukasti solunulkoiseen tilaan, ja jakautumistilavuus (Vd) on 76,9–106 ml/kg.
Biotransformaatio
Tildrakitsumabi kataboloituu aminohappokomponenteiksi yleisten proteiinin hajoamisprosessien välityksellä. Pienimolekyyliset metaboliset reitit (esim. CYP450-entsyymit, glukuronyylitransferaasit) eivät osallistu sen puhdistumaan.
Eliminaatio
Läiskäpsoriaasia sairastavien tutkittavien puhdistuma-arvojen vaihteluväli oli 2,04–2,52 ml/vrk/kg ja puoliintumisaika oli 23,4 vrk (23 % variaatiokerroin).
Lineaarisuus/ei-lineaarisuus
Läiskäpsoriaasia sairastavilla tutkittavilla tildrakitsumabin farmakokinetiikka oli annoksilla 50–400 mg ihon alle suhteessa annokseen; puhdistuma oli annoksesta riippumaton.
Vakaa tila saavutettiin viikkoon 16 mennessä, kun annoksia annettiin viikoilla 0 ja 4 sekä tämän jälkeen 12 viikon välein; elimistöön kertyminen oli 1,1-kertaista viikon 1 ja viikon 12 välisessä altistuksessa annoksesta riippumatta.
Farmakokinetiikka erityisryhmissä
Iäkkäät potilaat
Populaatiofarmakokineettinen analyysi osoitti, ettei iällä ollut kliinisesti merkittävää vaikutusta tildrakitsumabin puhdistumaan aikuisilla tutkittavilla, jotka sairastivat läiskäpsoriaasia. Kun vähintään 65-vuotiaat tutkittavat saivat 100 mg (n = 81) tai 200 mg (n = 81) tildrakitsumabia, tildrakitsumabin puhdistuma oli heillä samankaltainen kuin alle 65-vuotiailla tutkittavilla (n = 884).
Munuaisten ja maksan vajaatoiminta
Maksan tai munuaisten vajaatoiminnan vaikutuksesta tildrakitsumabin farmakokinetiikkaan ei ole tehty varsinaisia tutkimuksia. Tildrakitsumabi kataboloituu aminohappokomponentiksi yleisten proteiinien hajoamisprosessien kautta, eikä se eliminoidu munuais- tai maksareittien kautta.
Kehon paino
Populaatiofarmakokineettinen mallinnus osoitti, että altistus heikkeni kehon painon kasvaessa. Geometrisen keskimääräisen altistuksen (vakaan tilan AUC0-τ-arvo) > 90 kg:n painoisilla aikuispotilailla ihon alle annetun 100 mg tai 200 mg annoksen jälkeen ennustettiin olevan noin 30 % alhaisempi kuin < 90 kg:n painoisella aikuispotilaalla (ks. kohta Annostus ja antotapa).
Lääkkeiden yhteisvaikutukset
Läiskäpsoriaasia sairastavilla tutkittavilla tehtyjen yhteisvaikutustutkimusten tulokset viittaavat siihen, että tildrakitsumabilla ei ollut kliinisesti oleellista vaikutusta CYP1A2-, CYP2C9-, CYP2C19-, CYP2D6- ja CYP3A4-entsyymeihin. Tildrakitsumabi ei siten vaikuta samanaikaisesti käytettävien CYP-entsyymin välityksellä metaboloituvien lääkevalmisteiden farmakokinetiikkaan (ks. kohta Yhteisvaikutukset).
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Tildrakitsumabilla ei ole tehty mutageenisuutta ja karsinogeenisuutta koskevia eläinkokeita. Tutkimukset hiiren tuumorimalleilla osoittivat, ettei IL-23p19:n selektiivinen estyminen lisää syöpäriskiä.
Cynomolgus-apinoilla lääkeaineen erittyminen maitoon oli merkityksetöntä. Kuukausi syntymän jälkeen maito-seerumisuhde oli ≤ 0,002. Tildrakitsumabin osoitettiin läpäisevän istukkaesteen. Kun tiineenä olleet cynomolgus-apinat saivat lääkeainetta toistuvasti, pitoisuudet sikiön seerumissa olivat määritettävissä, mutta lisääntymistoksisuustutkimuksissa ei havaittu haitallisia vaikutuksia.
Cynomolgus-apinauroksilla ja -naarailla ei havaittu vaikutuksia hedelmällisyyttä koskeviin parametreihin, kuten lisääntymiselimiin, kiimakierron pituuteen ja/tai hormoneihin, kun apinat saivat tildrakitsumabia > 100-kertaisina annoksina verrattuna kliinisestä annoksesta AUC-arvon perusteella ihmiselle aiheutuvaan altistukseen nähden.
Pre- ja postnataalista kehitystä koskevassa toksisuustutkimuksessa apinoilla ei havaittu keskenmenojen lisääntymistä altistuksella, joka oli enintään 85-kertainen ihmiselle suositellusta annoksesta aiheutuvaan altistukseen nähden. Vastasyntyneillä poikasilla ei havaittu haitallisia vaikutuksia, kun emon altistus oli enimmillään 9-kertainen ihmiselle suositellusta annoksesta aiheutuvaan altistukseen nähden. Kaksi vastasyntynyttä apinanpoikasta kuoli, kun emoille oli annettu tildrakitsumabia annoksina, joista aiheutuva altistus oli 85-kertainen ihmiselle suositellusta annoksesta aiheutuvaan altistukseen nähden. Kuolemat liittyivät mahdolliseen virusinfektioon, ja niiden yhteyden hoitoon katsottiin olevan epävarma. Näiden löydösten kliinistä merkitystä ei tiedetä.
Farmaseuttiset tiedot
Apuaineet
L-histidiini
L-histidiinihydrokloridimonohydraatti
Polysorbaatti 80 (E 433)
Sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Avaamaton ruisku voidaan ottaa jääkaapista ja säilyttää enintään 25 °C:ssa yhden enintään 30 päivän jakson ajan. Kun Ilumetri on otettu jääkaapista ja säilytetty näissä olosuhteissa, hävitä se 30 päivän kuluttua tai pakkauksessa mainittuun viimeiseen käyttöpäivämäärään mennessä, kumpi tapahtuu ensin. Pakkauksessa on tila, johon jääkaapista poistamispäivä voidaan kirjoittaa ylös.
Pidä ulkopakkauksessa. Herkkä valolle.
Ei saa ravistaa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ILUMETRI injektioneste, liuos, esitäytetty ruisku
100 mg (L:ei) 1 kpl (1 ml (100 mg/ml)) (2861,32 €)
200 mg (L:ei) 1 kpl (2 ml (100 mg/ml)) (2861,32 €)
PF-selosteen tieto
Ilumetri 100 mg injektioneste, liuos, esitäytetty ruisku
1 ml liuosta tyypin I lasista valmistetussa esitäytetyssä ruiskussa, jossa on ruostumatonta terästä oleva 29G x ½ tuuman neula, joka on suojattu neulansuojuksella, sekä fluropolymeerillä laminoitu jäykkä polypropeenisuojus; männän pysäytin, joka on asennettu passiiviseen turvalaitteeseen.
Pakkaus, jossa 1 esitäytetty ruisku tai 2 esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Ilumetri 200 mg injektioneste, liuos, esitäytetty ruisku
2 ml liuosta tyypin I lasista valmistetussa esitäytetyssä ruiskussa, jossa on ruostumatonta terästä oleva 27G x ½ tuuman neula, joka on suojattu neulansuojuksella, sekä fluropolymeerillä laminoitu jäykkä polypropeenisuojus; männän pysäytin, joka on asennettu passiiviseen turvalaitteeseen.
Pakkaus, jossa 1 esitäytetty ruisku.
Valmisteen kuvaus:
Liuos on kirkasta tai hieman opaalinhohtoista ja väritöntä tai kellertävää. Liuoksen pH on 5,7–6,3 ja sen osmolaliteetti on 258–311 mOsm/kg.
Käyttö- ja käsittelyohjeet
Ilumetri on steriili injektioneste, liuos, esitäytetyssä ruiskussa. Ruiskut ovat kertakäyttöisiä.
Ei saa ravistaa. Ei saa jäätyä. 100 mg:n tai 200 mg:n ruisku tulee ottaa jääkaapista 30 minuuttia ennen injektiota, jotta se ehtii lämmetä huoneenlämpöiseksi (enintään 25 °C). Ruisku suositellaan tarkistamaan silmämääräisesti ennen käyttöä. Pieniä ilmakuplia saattaa olla nähtävissä: tämä on normaalia. Älä käytä liuosta, jos se sisältää silmin havaittavia hiukkasia tai se on sameaa tai selvästi ruskeaa.
Pakkausselosteessa olevaa käyttöohjetta on noudatettava tarkoin.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ILUMETRI injektioneste, liuos, esitäytetty ruisku
100 mg 1 kpl
200 mg 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AC17
Valmisteyhteenvedon muuttamispäivämäärä
19.03.2026
Yhteystiedot
 ORION OYJ ORION PHARMA
ORION OYJ ORION PHARMA Orionintie 1, PL 65
02101 Espoo
010 4261
www.orion.fi
etunimi.sukunimi@orionpharma.com