
OPDUALAG infuusiokonsentraatti, liuosta varten 240 mg/80 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi ml steriiliä infuusiokonsentraattia liuosta varten sisältää 12 mg nivolumabia ja 4 mg relatlimabia.
Yksi 20 ml:n injektiopullo sisältää 240 mg nivolumabia ja 80 mg relatlimabia.
Nivolumabi ja relatlimabi ovat humaaneja monoklonaalisia immunoglobuliini G4 (IgG4) -vasta-aineita, jotka on tuotettu yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Opdualag on tarkoitettu edenneen melanooman (jota ei voida kirurgisesti poistaa tai joka on metastasoinut) ensilinjan hoidoksi aikuisille ja vähintään 12-vuotiaille nuorille, joilla kasvainsolujen PD‑L1:n ilmentymistaso on < 1 %.
Ehto
Hoitavan lääkärin tulee olla perehtynyt syövän hoitoon.
Annostus ja antotapa
Hoidon aloittavan ja sitä valvovan lääkärin on oltava perehtynyt syövän hoitoon.
Potilaille, joita hoidetaan Opdualag-valmisteella, on annettava potilaskortti ja kerrottava Opdualag-hoidon riskeistä (ks. myös pakkausseloste).
PD‑L1-testi
Opdualag-hoitoa annetaan vain potilaille, joilla kasvaimen PD‑L1-ilmentymistaso on vahvistettu validoidulla testillä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus
Suositeltu annos aikuisille ja vähintään 12‑vuotiaille nuorille on 480 mg nivolumabia ja 160 mg relatlimabia 4 viikon välein infuusiona laskimoon 30 minuutin kuluessa. Tämä annos on varmistettu vähintään 30 kg painaville nuorille potilaille (ks. kohta Farmakokinetiikka).
Opdualag-hoitoa jatketaan niin kauan kuin siitä todetaan olevan kliinistä hyötyä tai kunnes potilas ei enää siedä hoitoa. Annoksen suurentamista tai pienentämistä ei suositella. Annosten siirtäminen myöhemmäksi tai hoidon keskeytys voi olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden vuoksi. Taulukossa 1 on ohjeet hoidon lopettamiseen ja keskeyttämiseen. Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on yksityiskohtaiset ohjeet immuunivälitteisten haittavaikutusten hoitoon.
Taulukko 1: Opdualag-hoidon suositellut muutokset
| Immuunivälitteinen haittavaikutus | Vaikeusaste | Hoidon muutos |
| Immuunivälitteinen keuhkotulehdus | Asteen 2 keuhkotulehdus | Keskeytä hoito, kunnes oireet häviävät, radiologisesti havaittavat poikkeavuudet paranevat ja kortikosteroidihoito on toteutettu kokonaan. |
| Asteen 3 tai 4 keuhkotulehdus | Lopeta hoito pysyvästi. | |
| Immuunivälitteinen koliitti | Asteen 2 tai 3 ripuli tai koliitti | Keskeytä hoito, kunnes oireet häviävät ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
| Asteen 4 ripuli tai koliitti | Lopeta hoito pysyvästi. | |
| Immuunivälitteinen maksatulehdus | Aspartaattiaminotransferaasi (ASAT)- tai alaniiniaminotransferaasi (ALAT) ‑arvojen nousu yli 3 kertaa ja enintään 5 kertaa normaalin ylärajan (ULN) tai Kokonaisbilirubiiniarvon nousu yli 1,5 kertaa ja enintään 3 kertaa ULN | Keskeytä hoito, kunnes laboratorioarvot palaavat ennalleen ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
ASAT- tai ALAT-arvojen nousu yli 5 kertaa ULN riippumatta lähtötilanteesta tai Kokonaisbilirubiiniarvon nousu yli 3 kertaa ULN tai Samanaikaisesti ASAT- tai ALAT-arvojen nousu yli 3 kertaa ULN ja kokonaisbilirubiiniarvon nousu yli 2 kertaa ULN | Lopeta hoito pysyvästi. | |
| Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö | Asteen 2 tai 3 nousu kreatiniiniarvossa. | Keskeytä hoito, kunnes kreatiniiniarvo palaa ennalleen ja kortikosteroidihoito on toteutettu kokonaan. |
| Asteen 4 nousu kreatiniiniarvossa | Lopeta hoito pysyvästi. | |
| Immuunivälitteiset umpierityshäiriöt | Oireiset asteen 2 tai 3 kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, hypofysiitti Asteen 2 lisämunuaisten vajaatoiminta Asteen 3 diabetes | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito (jos sitä tarvitaan akuutin tulehduksen oireisiin) on toteutettu kokonaan. Hoitoa pitää jatkaa yhdessä hormonikorvaushoidona kanssa niin kauan kuin oireita on. |
Asteen 4 kilpirauhasen vajaatoiminta Asteen 4 kilpirauhasen liikatoiminta Asteen 4 hypofysiitti Asteen 3 tai 4 lisämunuaisten vajaatoiminta Asteen 4 diabetes | Lopeta hoito pysyvästi. | |
| Immuunivälitteiset ihohaitat | Asteen 3 ihottuma | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan. |
| Epäilty Stevens–Johnsonin oireyhtymä (SJS) tai toksinen epidermaalinen nekrolyysi (TEN) | Keskeytä hoito. | |
Asteen 4 ihottuma Vahvistettu SJS/TEN | Lopeta hoito pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
| Immuunivälitteinen myokardiitti | Asteen 2 myokardiitti | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaanb |
| Asteen 3 tai 4 myokardiitti | Lopeta hoito pysyvästi. | |
| Muut immuunivälitteiset haittavaikutukset | Asteen 3 (ensimmäistä kertaa) | Keskeytä hoito. |
| Asteen 4 tai uusiutunut asteen 3; jatkuva asteen 2 tai 3 huolimatta hoidon muutoksista; kortikosteroidin vuorokausiannosta ei pystytä vähentämään 10 mg:aan prednisonia tai vastaavaa | Lopeta hoito pysyvästi. | |
| Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäc | Asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaanb |
| Asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Lopeta hoito pysyvästi. |
Huom.: Toksisuus on luokiteltu NCI‑CTCAE-haittavaikutusluokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 5.0 mukaan.
a Suositukset hormonikorvaushoidon käytöstä on annettu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
b Opdualag-hoidon uudelleen aloittamisen turvallisuutta potilaille, joilla on aikaisemmin ollut immuunivälitteinen myokardiitti, ei tunneta.
c Ilmenee näistä joko kahden tai kolmen sairauden limittymisenä. Opdualag-hoitoon suositeltavan muutoksen arvioinnissa on huomioitava yksittäisten tapahtumien vaikein CTCAE-aste.
Erityisryhmät
Pediatriset potilaat
Opdualag-hoidon turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen hoidettaessa iäkkäitä potilaita (≥ 65 vuotta) (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen hoidettaessa lievää tai kohtalaista munuaisten vajaatoimintaa sairastavia (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavista on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä.
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen hoidettaessa lievää tai kohtalaista maksan vajaatoimintaa sairastavia (ks. kohta Farmakokinetiikka). Vaikeaa maksan vajaatoimintaa sairastavista on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä.
Antotapa
Opdualag on tarkoitettu annosteltavaksi vain laskimoon. Se annetaan infuusiona laskimoon 30 minuutin kuluessa.
Opdualag-valmistetta ei saa antaa laskimoon nopeana injektiona eikä boluksena.
Opdualag voidaan antaa laimentamattomana tai laimentaa 9 mg/ml:n (0,9 %) vahvuisella natriumkloridi-injektioliuoksella tai 50 mg/ml:n (5 %) vahvuisella glukoosi-injektioliuoksella (ks. kohta Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen valmistamiseen ja käsittelyyn ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
PD‑L1-statuksen arviointi
Kasvaimen PD‑L1-statuksen arviointiin on ehdottomasti käytettävä hyvin validoitua ja robustia menetelmää.
Immuunivälitteiset haittavaikutukset
Nivolumabi–relatlimabi-yhdistelmähoidossa voi ilmetä immuunivälitteisiä haittavaikutuksia, jotka edellyttävät asianmukaista hoitoa, kuten kortikosteroidihoitoa ja muutosta hoidossa (ks. kohta Annostus ja antotapa).
Useampaan kuin yhteen elinjärjestelmään vaikuttavia immuunijärjestelmään liittyviä haittavaikutuksia voi ilmetä samanaikaisesti.
Potilaita on seurattava jatkuvasti (vähintään viisi kuukautta viimeisen annoksen jälkeen), sillä haittavaikutus voi tulla missä hyvänsä vaiheessa Opdualag-hoitoa tai vasta sen päätyttyä.
Epäiltyjen immuunivälitteisten haittavaikutusten riittävä arviointi on tehtävä syiden vahvistamiseksi tai muiden aiheuttajien poissulkemiseksi. Haittavaikutuksen vakavuuden perusteella Opdualag-hoito on keskeytettävä ja annettava kortikosteroideja. Jos haittavaikutuksen hoitoon käytetään kortikosteroideilla aikaansaatua immunosuppressiota, kortikosteroidiannoksen vähentäminen on aloitettava tilan alkaessa korjaantua, ja siihen on käytettävä vähintään kuukausi. Kortikosteroidihoidon liian nopea vähentäminen voi pahentaa haittavaikutusta tai aiheuttaa haittavaikutuksen uusiutumisen. Muu immunosuppressiivinen lääke on syytä lisätä hoito-ohjelmaan, jos haittavaikutus pahenee tai ei parane kortikosteroidien käytöstä huolimatta.
Havainnoivista tutkimuksista saadut tiedot viittaavat siihen, että immuunivälitteisten haittavaikutusten ilmenemisen riski tarkistuspisteen estäjien saamisen jälkeen saattaa olla suurentunut potilailla, joilla on ennestään jokin autoimmuunisairaus, verrattuna potilaisiin, joilla ei ole ennestään autoimmuunisairautta. Lisäksi taustalla olevan autoimmuunisairauden tilapäinen paheneminen oli yleistä, mutta suurimmassa osassa tapauksista paheneminen oli lievää ja hallittavissa olevaa.
Nimenomaan nivolumabi–relatlimabi-yhdistelmähoitoa koskevia tietoja on kuitenkin saatavilla vain vähän.
Opdualag-hoitoa ei saa aloittaa uudelleen potilaan saadessa immunosuppressiivisia annoksia kortikosteroideja tai muuta immunosuppressiivista hoitoa. Immunosuppressiivista hoitoa saaville potilaille voidaan antaa profylaktista antibioottihoitoa opportunististen infektioiden ehkäisemiseksi.
Opdualag-hoito on lopetettava pysyvästi, jos yksikään vakava immuunivälitteinen haittavaikutus uusiutuu tai jos yksikään immuunivälitteinen haittavaikutus on hengenvaarallinen.
Immuunivälitteinen keuhkotulehdus
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa keuhkotulehdusta tai interstitiaalista keuhkosairautta, myös kuolemaan johtanut tapaus (ks. kohta Haittavaikutukset). Potilaita on seurattava keuhkotulehduksen löydösten ja oireiden varalta. Näitä ovat esimerkiksi radiologiset muutokset (kuten fokaaliset mattalasimuutokset, läiskittäiset infiltraatit), hengenahdistus ja hypoksia. Infektioihin ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 3 tai 4 keuhkotulehdus, Opdualag-hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 2–4 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 (oireinen) keuhkotulehdus, Opdualag-hoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 2–4 mg/kg/vrk metyyliprednisolonia, ja Opdualag-hoito on lopetettava pysyvästi.
Immuunivälitteinen koliitti
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ripulia ja koliittia (ks. kohta Haittavaikutukset). Potilaita on seurattava ripulin ja muiden koliitin oireiden varalta, joita ovat esimerkiksi vatsakipu ja veriset tai limaiset ulosteet. Sytomegalovirus (CMV) ‑infektioita tai niiden uudelleenaktivoitumista on ilmoitettu potilailla, joilla on kortikosteroidihoitoon reagoimaton immuunivälitteinen koliitti. Infektiot ja ripulin muut syyt on suljettava pois, joten asianmukaiset laboratoriotestit ja muut tutkimukset on tehtävä. Jos kortikosteroidihoitoon reagoimattoman immuunivälitteisen koliitin diagnoosi varmistuu, vaihtoehtoisen immunosuppressantin lisäämistä kortikosteroidihoitoon tai kortikosteroidihoidon korvaamista on harkittava.
Jos potilaalla on asteen 4 ripuli tai koliitti, Opdualag-hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 3 ripuli tai koliitti, Opdualag-hoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, Opdualag-hoito on lopetettava pysyvästi.
Jos potilaalla on asteen 2 ripuli tai koliitti, Opdualag-hoito on keskeytettävä. Jos ripuli tai koliitti jatkuu, se on hoidettava antamalla kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja Opdualag-hoito on lopetettava pysyvästi.
Immuunivälitteinen maksatulehdus
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa maksatulehdusta (ks. kohta Haittavaikutukset). Potilaita on seurattava maksatulehduksen löydösten ja oireiden, kuten transaminaasi- ja kokonaisbilirubiiniarvon nousun, varalta. Infektiot ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on ASAT- tai ALAT-arvojen nousu yli 5 kertaa ULN lähtötilanteeseen katsomatta, kokonaisbilirubiiniarvon nousu yli 3 kertaa ULN tai samanaikaisesti ASAT- tai ALAT-arvojen nousu yli 3 kertaa ULN ja kokonaisbilirubiiniarvon nousu yli 2 kertaa ULN, Opdualag-hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on ASAT- tai ALAT-arvojen nousu yli 3 kertaa ULN ja enintään 5 kertaa ULN tai kokonaisbilirubiiniarvon nousu yli 1,5 kertaa ULN ja enintään 3 kertaa ULN, Opdualag-hoito on keskeytettävä. Jos nämä laboratorioarvot ovat jatkuvasti koholla, potilaalle on annettava kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja Opdualag-hoito on lopetettava pysyvästi.
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa munuaistulehdusta ja munuaisten toimintahäiriöitä (ks. kohta Haittavaikutukset). Potilaita on seurattava munuaistulehduksen tai munuaisten toimintahäiriön löydösten ja oireiden varalta. Useimmilla potilailla tila ilmenee seerumin kreatiniiniarvon suurenemisena ilman oireita. Sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 4 seerumin kreatiniiniarvon nousu, Opdualag-hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 tai 3 seerumin kreatiniiniarvon nousu, Opdualag-hoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja Opdualag-hoito on lopetettava pysyvästi.
Immuunivälitteiset umpierityshäiriöt
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeita umpierityshäiriöitä, kuten kilpirauhasen vajaa- ja liikatoimintaa, lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa), hypofysiittiä (mukaan lukien hypopituitarismia) ja diabetes mellitusta. Diabeettista ketoasidoosia on todettu nivolumabi-monoterapian yhteydessä, ja sitä voi mahdollisesti ilmetä nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava umpierityshäiriöiden kliinisten löydösten ja oireiden ja hyperglykemian sekä kilpirauhasen toiminnan muutosten varalta (hoidon alussa, jaksoittain hoidon aikana ja kliinisen arvioinnin pohjalta). Tila saattaa ilmetä potilailla väsymyksenä, päänsärkynä, mielialamuutoksina, vatsakipuina, epätavallisena vatsan toimintana ja matalana verenpaineena tai epäspesifisinä oireina, jotka voivat muistuttaa muita syitä kuten aivojen etäpesäke tai taustasairaus. Jos vaihtoehtoisia syitä ei ole tunnistettu, umpierityshäiriöiden oireet tai löydökset on katsottava immuunivälitteisiksi.
Kilpirauhasen toimintahäiriö
Jos potilaalla on kilpirauhasen vajaatoiminnan oireita, Opdualag-hoito on keskeytettävä ja korvaushoito kilpirauhashormonilla aloitettava tarpeen mukaan. Jos potilaalla on kilpirauhasen liikatoiminnan oireita, Opdualag-hoito on keskeytettävä ja kilpirauhasen toimintaa estävä hoito on aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia kilpirauhastulehdusta. Tilan parannuttua Opdualag-hoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Kilpirauhasen toimintaa on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla. Opdualag-hoito on lopetettava pysyvästi, jos potilaalla on henkeä uhkaava (asteen 4) kilpirauhasen liika- tai vajaatoiminta.
Lisämunuaisten vajaatoiminta
Opdualag-hoito on lopetettava pysyvästi, jos potilaalla on vakava (asteen 3) tai henkeä uhkaava (asteen 4) lisämunuaisten vajaatoiminta. Jos potilaalla on asteen 2 lisämunuaisten vajaatoiminnan oireita, Opdualag-hoito on keskeytettävä ja fysiologinen kortikosteroidikorvaushoito aloitettava tarpeen mukaan. Lisämunuaisten toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että kortikosteroidikorvaushoito on asianmukaisella tasolla.
Hypofysiitti
Opdualag-hoito on lopetettava pysyvästi, jos potilaalla on henkeä uhkaava (asteen 4) hypofysiitti. Jos potilaalla on asteen 2 tai 3 hypofysiitin oireita, Opdualag-hoito on keskeytettävä ja hormonikorvaushoito aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia aivolisäkkeen tulehdusta. Tilan parannuttua Opdualag-hoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Aivolisäkkeen toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla.
Diabetes mellitus
Jos potilaalla on diabeteksen oireita, Opdualag-hoito on keskeytettävä ja insuliinikorvaushoito aloitettava tarpeen mukaan. Verensokeripitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että insuliinikorvaushoito on asianmukaisella tasolla. Opdualag-hoito on lopetettava pysyvästi, jos potilaalla on henkeä uhkaava diabetes.
Immuunivälitteiset ihohaitat
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ihottumaa (ks. kohta Haittavaikutukset). Opdualag-hoito on keskeytettävä asteen 3 ihottumassa ja lopetettava asteen 4 ihottumassa. Vaikeaa ihottumaa on hoidettava suurella kortikosteroidiannoksella, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia.
SJS:ää ja TEN:iä on havaittu harvoin nivolumabi-monoterapian yhteydessä, ja joissain tapauksissa tila on johtanut kuolemaan. Niitä voi mahdollisesti ilmetä nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä. Jos SJS:n tai TEN:n oireita tai löydöksiä epäillään, Opdualag-hoito on keskeytettävä ja potilas on ohjattava niihin erikoistuneeseen yksikköön arviointia ja hoitoa varten. Jos potilaalla varmistetaan SJS tai TEN Opdualag-hoidon aikana, suositellaan hoidon lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Varovaisuutta on noudatettava, kun harkitaan Opdualag-hoitoa potilaalle, jolla on aikaisemmin ollut vaikea tai henkeä uhkaava ihohaitta aiemman toisen immuunijärjestelmää stimuloivan syöpälääkehoidon aikana.
Immuunivälitteinen myokardiitti
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on todettu vaikeaa immuunivälitteistä myokardiittia. Myokardiitin diagnosointi vaatii vahvan epäilyn. Potilaat, joilla on sydän- tai sydän- ja keuhko-oireita, on syytä arvioida mahdollisen myokardiitin varalta. Myokardiittia epäiltäessä on aloitettava ripeästi hoito suurella steroidiannoksella (prednisoni 1–2 mg/kg/vrk tai metyyliprednisoloni 1–2 mg/kg/vrk) ja konsultoitava viipymättä kardiologian asiantuntijaa diagnoosin varmistamista varten nykyisten hoitosuositusten mukaisesti. Myokardiittidiagnoosin varmistuttua Opdualag-hoito on keskeytettävä tai lopetettava pysyvästi alla kuvatun mukaisesti.
Jos potilaalla on asteen 3 tai 4 myokardiitti, Opdualag-hoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 2–4 mg/kg/vrk metyyliprednisolonia (ks. kohta Annostus ja antotapa).
Jos potilaalla on asteen 2 myokardiitti, Opdualag-hoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua Opdualag-hoidon aloittamista uudelleen voi harkita kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannos on suurennettava tasolle, joka vastaa 2–4 mg/kg/vrk metyyliprednisolonia ja Opdualag-hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Muut immuunivälitteiset haittavaikutukset
Seuraavia kliinisesti merkittäviä immuunivälitteisiä haittavaikutuksia on raportoitu nivolumabi–relatlimabi-yhdistelmähoitoa saaneilla potilailla: suonikalvoston tulehdus, haimatulehdus, gastriitti, Guillain–Barrén oireyhtymä, myosiitti/rabdomyolyysi, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä, enkefaliitti, hemolyyttinen anemia, Vogt–Koyanagi–Haradan oireyhtymä (VKH).
Seuraavia muita kliinisesti merkittäviä immuunivälitteisiä haittavaikutuksia on raportoitu nivolumabi-monoterapiaa tai nivolumabia yhdessä muiden myyntiluvallisten lääkkeiden kanssa saaneilla potilailla: demyelinaatio, autoimmuunineuropatia (mukaan lukien kasvo- ja loitontajahermon halvaus), myasteeninen oireyhtymä, aseptinen aivokalvotulehdus, sarkoidoosi, duodeniitti, lisäkilpirauhasten vajaatoiminta ja ei-infektiivinen virtsarakkotulehdus.
Immuunivälitteistä haittavaikutusta epäiltäessä potilaan tila on arvioitava riittävän tarkasti, jotta pystytään vahvistamaan haittavaikutuksen etiologia tai sulkemaan pois muut syyt. Haittavaikutuksen vakavuuden perusteella Opdualag-hoito on keskeytettävä ja annettava kortikosteroideja. Tilan parannuttua Opdualag-hoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Opdualag-hoito on lopetettava pysyvästi, jos yksikään vakava immuunivälitteinen haittavaikutus uusiutuu tai jos yksikään immuunivälitteinen haittavaikutus on hengenvaarallinen.
Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu nivolumabi–relatlimabi-yhdistelmähoidossa. Oireyhtymän varhainen tunnistaminen ja aggressiivinen hoito ovat oleellisia siihen liittyvän sairastuvuuden ja kuolleisuusriskin vuoksi.
Asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymässä Opdualag-hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa). Kortikosteroidien käyttö on aloitettava kliinisen tarpeen mukaan.
Asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymässä Opdualag-hoito on keskeytettävä ja aloitettava kortikosteroidien käyttö kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa). Tilan parannuttua Opdualag-hoidon aloittamista uudelleen voidaan harkita kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on muutettava kliinisen tarpeen mukaan ja Opdualag-hoito on lopetettava pysyvästi.
Muut tärkeät varoitukset ja varotoimet, luokkavaikutukset mukaan lukien
PD‑1:n estäjillä hoidetuilla potilailla valmisteen markkinoille tulon jälkeen on ilmoitettu hyljintäreaktioita kiinteän elimen siirron jälkeen. Nivolumabi–relatlimabi-yhdistelmähoito saattaa suurentaa hyljintäreaktion riskiä kiinteän elimen saaneilla potilailla. Näillä potilailla nivolumabi–relatlimabi-yhdistelmähoidon hyötyjä mahdollisen hyljintäreaktion riskiin nähden on arvioitava huolellisesti.
Hemofagosyyttistä lymfohistiosytoosia (HLH) on havaittu nivolumabi-monoterapian, nivolumabi–relatlimabi-yhdistelmähoidon sekä nivolumabin ja muiden lääkkeiden yhdistelmähoidon yhteydessä. Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä on raportoitu kuolemaan johtanut tapaus. On noudatettava varovaisuutta, kun nivolumabia annetaan yhdistelmähoitona relatlimabin kanssa. Jos potilaalla on vahvistettu HLH, on lopetettava nivolumabi–relatlimabi-yhdistelmähoidon käyttö ja aloitettava HLH:n hoito.
Potilailla, joita hoidettiin nivolumabilla ennen allogeenista hematopoieettista kantasolujen siirtoa (HSCT) tai sen jälkeen, on ilmoitettu nopeasti alkavaa ja vaikea-asteista käänteishyljintää (GVHD), joka on joskus johtanut kuolemaan. Nivolumabi–relatlimabi-yhdistelmähoito saattaa lisätä vaikea-asteisen käänteishyljinnän ja kuoleman riskiä potilailla, jotka ovat saaneet allogeenisen hematopoieettisten kantasolujen siirron; erityisesti potilailla, joilla on aiemminkin ollut käänteishyljintää. Näillä potilailla nivolumabi–relatlimabi-yhdistelmähoidon hyötyjä mahdolliseen riskiin nähden on arvioitava huolellisesti.
Infuusioreaktiot
Kliinisissä tutkimuksissa on raportoitu vaikeita infuusioreaktioita nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä. (ks. kohta Haittavaikutukset). Vaikeissa tai henkeä uhkaavissa infuusioreaktiotapauksissa Opdualag-infuusio täytyy keskeyttää ja antaa asianmukaista lääketieteellistä hoitoa. Potilaat, jotka saavat lievän tai keskivaikean infuusioreaktion, voivat saada Opdualag-hoitoa huolellisessa seurannassa ja käyttämällä infuusioreaktioita estävää hoitoa paikallisten hoitosuositusten mukaisesti.
Edenneen melanooman pivotaalisesta kliinisestä tutkimuksesta poissuljetut potilaat
Potilaat, joilla oli aktiivinen autoimmuunisairaus, keskisuuri- tai suuriannoksista systeemistä kortikosteroidihoitoa tai immunosuppressiivisia lääkevalmisteita vaativa sairaus, uvean melanooma, aktiivisia tai hoitamattomia aivometastaaseja tai leptomeningeaalisia metastaaseja, sekä potilaat, joilla oli anamneesissa myokardiitti, troponiiniarvojen nousu > 2 kertaa ULN tai ECOG-toimintakykyluokka ≥ 2, suljettiin pois nivolumabi–relatlimabi-yhdistelmähoidon pivotaalisesta kliinisestä tutkimuksesta. Tutkimustiedon puuttuessa nivolumabi–relatlimabi-yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Potilaskortti
Lääkettä määräävän lääkärin on keskusteltava Opdualag-hoidon riskeistä potilaan kanssa. Potilaalle annetaan potilaskortti, jota hänen pyydetään pitämään aina mukanaan.
Yhteisvaikutukset
Nivolumabi ja relatlimabi ovat kumpikin humaaneja monoklonaalisia vasta-aineita, eikä yhteisvaikutustutkimuksia ole tehty. Koska monoklonaaliset vasta-aineet eivät metaboloidu sytokromi P450 (CYP) entsyymien eivätkä muiden vaikuttavia aineita metaboloivien entsyymien vaikutuksesta, muiden samaan aikaan käytettyjen lääkevalmisteiden näitä entsyymejä estävien tai indusoivien vaikutusten ei odoteta vaikuttavan relatlimabin tai nivolumabin farmakokinetiikkaan.
Nivolumabin ja relatlimabin ei odoteta vaikuttavan sellaisten vaikuttavien aineiden farmakokinetiikkaan, jotka metaboloituvat CYP-entsyymien vaikutuksesta, koska nivolumabi ja relatlimabi eivät moduloi merkittävästi sytokiineja eikä niillä siten ole vaikutusta sytokromi P450 -entsyymin ilmentymiseen.
Systeeminen immunosuppressio
Systeemisten kortikosteroidien ja muiden immunosuppressiivisten lääkkeiden käyttöä ennen nivolumabi–relatlimabi-yhdistelmähoidon aloitusta on vältettävä, koska ne saattavat vaikuttaa farmakodynaamiseen aktiviteettiin. Systeemisiä kortikosteroideja ja muita immunosuppressiivisia lääkkeitä voi kuitenkin käyttää nivolumabi–relatlimabi-yhdistelmähoidon alettua immuunivälitteisten haittavaikutusten hoitoon.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Opdualag-valmistetta ei suositella käytettäväksi sellaisten naisten hoitoon, jotka saattaisivat tulla raskaaksi mutta eivät käytä tehokasta ehkäisyä, paitsi siinä tapauksessa, että kliininen hyöty on merkittävämpi kuin mahdollinen riski. Tehokasta ehkäisymenetelmää on käytettävä vähintään 5 kuukauden ajan viimeisen Opdualag-annoksen jälkeen.
Raskaus
On vain vähän tietoja nivolumabi–relatlimabi-yhdistelmähoidon käytöstä raskaana oleville naisille. Vaikutusmekanismin ja eläinkokeista saatujen tietojen perusteella nivolumabi–relatlimabi-yhdistelmähoito voi aiheuttaa sikiötoksisiteettia, kun sitä annetaan raskaana oleville naisille. Eläinkokeissa on todettu alkio- ja sikiötoksisuutta nivolumabia saaneilla eläimillä (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen IgG4:n tiedetään läpäisevän veri-istukkaesteen, ja nivolumabi ja relatlimabi ovat IgG4-luokan vasta-aineita. Siksi nivolumabi ja relatlimabi voivat potentiaalisesti siirtyä äidistä kehittyvään sikiöön. Opdualag-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä tehokasta ehkäisyä, paitsi siinä tapauksessa, että kliininen hyöty on merkittävämpi kuin mahdollinen riski.
Imetys
Ei tiedetä, erittyvätkö nivolumabi ja/tai relatlimabi ihmisen rintamaitoon. Ihmisen immunoglobuliinien tiedetään erittyvän rintamaitoon ensimmäisten synnytyksen jälkeisten päivien aikana, minkä jälkeen pitoisuudet pienenevät pian matalalle tasolle. Imetettävälle lapselle tämän lyhyen ajanjakson aikana koituvaa riskiä ei siis voida sulkea pois. Tämän jälkeen Opdualag-valmistetta voidaan käyttää imetyksen aikana, jos se on kliinisesti tarpeen.
Hedelmällisyys
Nivolumabin ja/tai relatlimabin vaikutusta hedelmällisyyteen ei ole tutkittu. Siksi niiden vaikutusta miehen ja naisen hedelmällisyyteen ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Opdualag-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Koska haittavaikutukset, kuten uupumus ja huimaus, ovat mahdollisia (ks. kohta Haittavaikutukset), potilaita on kehotettava varovaisuuteen ajamisessa ja koneiden käytössä, kunnes he ovat varmoja, ettei Opdualag-valmisteella ole heihin haitallista vaikutusta.
Haittavaikutukset
Tiivistelmä turvallisuusprofiilista
Nivolumabi–relatlimabi-yhdistelmähoitoon liittyy immuunivälitteisiä haittavaikutuksia (ks. alla kohta ”Valikoitujen haittavaikutusten kuvaus”). Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on haittavaikutusten hoito-ohjeet.
Yleisimmät haittavaikutukset ovat uupumus (44 %), muskuloskeletaalinen kipu (34 %), ihottuma (30 %), ripuli (30 %), artralgia (29 %), kutina (28 %), päänsärky (21 %), pahoinvointi (21 %), yskä (18 %), kilpirauhasen vajaatoiminta (18 %), ruokahalun heikkeneminen (16 %), vatsakipu (16 %), vitiligo (14 %), virtsatieinfektio (14 %), kuume (13 %), ummetus (13 %), ylähengitystieinfektio (12 %), hengenahdistus (12 %), oksentelu (12 %), huimaus (10 %) ja turvotus (10 %).
Yleisimmät vakavat haittavaikutukset ovat lisämunuaisten vajaatoiminta (1,4 %), anemia (1,4 %), ripuli (1,4 %), koliitti (1,1 %), myokardiitti (1,1 %) ja virtsatieinfektio (1,1 %). Asteen 3–5 haittavaikutusten ilmaantuvuus edennyttä melanoomaa (jota ei voida kirurgisesti poistaa tai joka on metastasoinut) sairastavilla potilailla oli 47 % nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä ja 38 % nivolumabihoidon yhteydessä.
Tiivistelmä haittavaikutuksista taulukkona
Nivolumabi–relatlimabi-yhdistelmähoidon turvallisuutta arvioitiin 355 potilaalla, jotka sairastivat edennyttä melanoomaa (jota ei voida kirurgisesti poistaa tai joka on metastasoinut) (tutkimus CA224047). Taulukossa 2 esitetään tutkimustiedoista kootut haittavaikutukset potilailla, jotka saivat nivolumabi–relatlimabi-yhdistelmähoitoa (seuranta-ajan mediaani 19,94 kuukautta). Edellä olevat ja taulukossa 2 esitetyt yleisyydet perustuvat mistä tahansa syystä johtuvien haittatapahtumien yleisyyteen. Haittavaikutukset on jaettu elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyydet määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000) ja tuntematon (saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on lueteltu kussakin yleisyysryhmässä vakavimmasta lähtien.
Taulukko 2: Kliinisissä tutkimuksissa todetut haittavaikutukset
| Infektiot | |
| Hyvin yleinen | virtsatieinfektio, ylähengitystieinfektio |
| Yleinen | karvatuppitulehdus |
| Veri ja imukudos | |
| Hyvin yleinen | anemiaa, lymfopeniaa, leukopeniaa, neutropeniaa |
| Yleinen | trombosytopeniaa, eosinofilia |
| Melko harvinainen | hemolyyttinen anemia |
| Umpieritys | |
| Hyvin yleinen | kilpirauhasen vajaatoiminta |
| Yleinen | lisämunuaisten vajaatoiminta, hypofysiitti, kilpirauhasen liikatoiminta, kilpirauhastulehdus |
| Melko harvinainen | hypopituitarismi, hypogonadismi |
| Aineenvaihdunta ja ravitsemus | |
| Hyvin yleinen | ruokahalun heikkeneminen |
| Yleinen | diabetes mellitus, hypoglykemiaa, painon lasku, hyperurikemia, hypoalbuminemia, kuivumistila |
| Psyykkiset häiriöt | |
| Yleinen | sekavuustila |
| Hermosto | |
| Hyvin yleinen | huimaus, päänsärky |
| Yleinen | perifeerinen neuropatia, dysgeusia |
| Melko harvinainen | enkefaliitti, Guillain–Barrén oireyhtymä, näköhermotulehdus, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäc |
| Silmät | |
| Yleinen | uveiitti, näön heikentyminen, kuivat silmät, lisääntynyt kyyneleritys |
| Melko harvinainen | Vogt–Koyanagi–Haradan oireyhtymä, silmän verekkyys |
| Sydän | |
| Yleinen | myokardiitti |
| Melko harvinainen | perikardiaalinen effuusio |
| Verisuonisto | |
| Yleinen | laskimotulehdus |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | hengenahdistus, yskä |
| Yleinen | pneumoniittib, pleuraeffuusio, nenän tukkoisuus |
| Melko harvinainen | astma |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli, oksentelu, pahoinvointi, vatsakipu, ummetus |
| Yleinen | koliitti, haimatulehdus, gastriitti, dysfagia, stomatiitti, suun kuivuminen |
| Melko harvinainen | ruokatorvitulehdus |
| Harvinainen | haiman eksokriininen vajaatoiminta |
| Tuntematon | keliakia |
| Maksa ja sappi | |
| Yleinen | maksatulehdus |
| Melko harvinainen | sappitietulehdus |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | ihottuma, vitiligo, kutina |
| Yleinen | alopesia, likenoidi keratoosi, psoriaasi, nokkosihottuma, valoherkkyysreaktio, kuiva iho |
| Melko harvinainen | pemfigoidi |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | muskuloskeletaalinen kipu, artralgia |
| Yleinen | artriitti, lihaskouristukset, lihasheikkous |
| Melko harvinainen | myosiitti, Sjögrenin oireyhtymä, polymyalgia rheumatica, nivelreuma, systeeminen lupus erythematosus |
| Munuaiset ja virtsatiet | |
| Yleinen | munuaisten vajaatoiminta, proteinuria |
| Melko harvinainen | nefriitti |
| Sukupuolielimet ja rinnat | |
| Melko harvinainen | atsoospermia |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | turvotus, uupumus, kuume |
| Yleinen | influenssan kaltainen sairaus, vilunväristykset |
| Harvinainen | serosiitti |
| Tutkimukset | |
| Hyvin yleinen | lisääntynyt ASATa, lisääntynyt ALATa, hyponatremiaa, lisääntynyt kreatiniinia, lisääntynyt alkalinen fosfataasia, hypokalsemiaa, hypomagnesemiaa, hyperkalemiaa, hyperkalsemiaa, hypokalemiaa, hypernatremiaa, lisääntynyt bilirubiinia |
| Yleinen | hypermagnesemiaa, lisääntynyt troponiini, lisääntynyt glutamyylitransferaasi, lisääntynyt veren laktaattidehydrogenaasi, lisääntynyt lipaasi, lisääntynyt amylaasi, lisääntynyt C‑reaktiivinen proteiini |
| Melko harvinainen | laskon suureneminen |
| Vammat, myrkytykset ja hoitokomplikaatiot | |
| Yleinen | infuusioreaktio |
a Yleisyys laskettiin sen mukaan, kuinka suurella osalla potilaista laboratorioarvo huononi lähtötasolta.
b Kuolemaan johtanut tapaus on ilmoitettu kliinisessä tutkimuksessa.
c Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu nivolumabi–relatlimabi-yhdistelmähoidossa. (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valikoitujen haittavaikutusten kuvaus
Immuunivälitteinen keuhkotulehdus
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä keuhkotulehdusta (interstitiaalinen keuhkosairaus ja keuhkoinfiltraatio mukaan luettuna) ilmeni 5,1 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 0,8 %. Kuolemaan johtaneita tapahtumia ilmeni 0,28 %:ll potilaista. Haittavaikutusten ilmenemisajan mediaani oli 28 viikkoa (vaihteluväli 3,6–94,4). Haittavaikutus korjaantui 88,9 %:lla potilaista. Korjaantumisajan mediaani oli 12,0 viikkoa (vaihteluväli 2,1–133,0+). Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi immuunivälitteisen keuhkotulehduksen takia 1,7 %:lla potilaista, ja 55,6 % potilaista, joilla oli immuunivälitteinen keuhkotulehdus, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Immuunivälitteinen koliitti
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä ripulia, koliittia tai ulostamistiheyden lisääntymistä ilmeni 18,3 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 2,3 %. Haittavaikutusten ilmenemisajan mediaani oli 16 viikkoa (vaihteluväli 0,1–195,4). Haittavaikutus korjaantui 89,1 %:lla potilaista. Korjaantumisajan mediaani oli 7,3 viikkoa (vaihteluväli 0,1–289,1+). Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi immuunivälitteisen koliitin takia 2,5 %:lla potilaista, ja 35,4 % potilaista, joilla oli immuunivälitteinen koliitti, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Immuunivälitteinen maksatulehdus
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä maksan toimintakokeista saatiin poikkeavia tuloksia 14,1 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 4,2 %. Haittavaikutusten ilmenemisajan mediaani oli 11 viikkoa (vaihteluväli 2,0–191,9). Haittavaikutus korjaantui 86,0 %:lla potilaista. Korjaantumisajan mediaani oli 6,0 viikkoa (vaihteluväli 1,0–240,1+). Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi 2,3 %:lla potilaista, ja 40,0 % potilaista tarvitsi suuriannoksista kortikosteroidihoitoa.
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä munuaistulehdusta tai munuaisten toimintahäiriöitä ilmeni 5,6 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 1,4 %. Haittavaikutusten ilmenemisajan mediaani oli 24 viikkoa (vaihteluväli 1,9–210,4). Haittavaikutus korjaantui 85,0 %:lla potilaista. Korjaantumisajan mediaani oli 12,4 viikkoa (vaihteluväli 0,9–235,0+). Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi immuunivälitteisen munuaistulehduksen tai munuaisten toimintahäiriön takia 1,1 %:lla potilaista, ja 20,0 % potilaista, joilla oli immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Immuunivälitteiset umpierityshäiriöt
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä immuunivälitteisiä umpierityshäiriöitä ilmeni 29,0 %:lla potilaista.
Kilpirauhasen häiriöitä, kuten vajaa- ja liikatoimintaa, ilmeni 22,8 %:lla potilaista. Vaikeusasteen 3/4 kilpirauhashäiriöitä ei ilmaantunut. Lisämunuaisten vajaatoimintaa (mukaan lukien akuuttia lisämunuaiskuoren vajaatoimintaa) ilmeni 5,1 %:lla potilaista. Vaikeusasteen 3/4 lisämunuaisten vajaatoiminnan tapahtumia ilmeni 1,4 %:lla. Hypopituitarismia ilmeni 0,8 %:lla potilaista. Vaikeusasteen 3/4 hypopituitarismia ei ilmaantunut. Hypofysiittiä ilmeni 1,1 %:lla potilaista. Vaikeusasteen 3/4 hypofysiitin ilmaantuvuus oli 0,3 %. Diabetes mellitusta (mukaan lukien tyypin 1 diabetes mellitusta) ilmeni 0,3 %:lla potilaista. Vaikeusasteen 3/4 diabetes mellituksen ilmaantuvuus oli 0,3 %.
Näiden umpierityshäiriöiden ilmenemisajan mediaani oli 15 viikkoa (vaihteluväli 1,0–230,4). Haittavaikutus korjaantui 28,2 %:lla potilaista. Korjaantumisajan vaihteluväli oli 0,4–328,0+ viikkoa. Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi immuunivälitteisten umpierityshäiriöiden takia 1,1 %:lla potilaista, ja 6,8 % potilaista, joilla oli immuunivälitteisiä umpierityshäiriöitä, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Immuunivälitteiset ihohaitat
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä ihottumaa, mukaan lukien kutina ja vitiligo, ilmeni 46,8 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 1,4 %. Haittavaikutusten ilmenemisajan mediaani oli 8 viikkoa (vaihteluväli 0,1–142,1). Haittavaikutus korjaantui 47,0 %:lla potilaista. Korjaantumisajan vaihteluväli oli 0,1–318,0+ viikkoa. Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi immuunivälitteisten ihohaittojen takia 0,3 %:lla potilaista, ja 4,2 % potilaista, joilla oli immuunivälitteisiä ihohaittoja, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Immuunivälitteinen myokardiitti
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä myokardiittia ilmeni 1,4 %:lla potilaista. Vaikeusasteen 3/4 tapahtumien ilmaantuvuus oli 0,6 %. Haittavaikutusten ilmenemisajan mediaani oli 4,1 viikkoa (vaihteluväli 2,1–6,3). Haittavaikutus korjaantui 100 %:lla potilaista. Korjaantumisajan mediaani oli 3 viikkoa (vaihteluväli 1,9–14,0). Nivolumabi–relatlimabi-yhdistelmähoito lopetettiin pysyvästi myokardiitin takia 1,4 %:lla potilaista, ja 100 % potilaista, joilla oli immuunivälitteinen myokardiitti, tarvitsi suuriannoksista kortikosteroidihoitoa (≥ 40 mg prednisonia tai vastaavaa vuorokaudessa).
Infuusioon liittyvät reaktiot
Nivolumabi–relatlimabi-yhdistelmähoidon yhteydessä yliherkkyys-/infuusioreaktioita ilmeni 7,3 %:lla potilaista. Vaikeusasteen 3/4 tapahtumia ilmeni 0,3 %:lla potilaista.
Poikkeavat laboratorioarvot
Nivolumabi–relatlimabi-yhdistelmähoitoa saaneista potilaista niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia 3,8 %, lymfopenia 5,3 %, lisääntynyt alkalinen fosfataasi 0,6 %, lisääntynyt ASAT 2,9 %, lisääntynyt ALAT 3,5 %, lisääntynyt kokonaisbilirubiini 0,6 %, lisääntynyt kreatiniini 1,5 %, hyponatremia 1,5 %, hyperkalemia 2,0 %, hypokalemia 0,6 %, hyperkalsemia 0,9 %, hypokalsemia 0,9 %, hypermagnesemia 1,8 % ja hypomagnesemia 1,2 %.
Immunogeenisuus
Tutkimuksessa CA224047 niistä Opdualag-ryhmän potilaista, joilla lääkevasta-aineet pystyttiin arvioimaan, 5,6 %:lle (17/301) kehittyi hoidosta johtuvia relatlimabin vasta-aineita ja 0,3 %:lle (1/301) relatlimabia neutraloivia vasta-aineita. Opdualag-ryhmässä hoidosta johtuvia nivolumabin vasta-aineita kehittyi 4,0 %:lle (12/299) ja nivolumabia neutraloivia vasta-aineita 0,3 %:lle (1/299), mitkä olivat vastaavanlaisia kuin nivolumabiryhmässä todetut luvut 6,7 % (19/283) ja 0,4 % (1/283). Nivolumabin tai relatlimabin vasta-aineiden kehittymisen ei ole havaittu muuttavan farmakokinetiikkaa, tehoa tai turvallisuusprofiilia.
Erityisryhmät
Iäkkäät potilaat
Kaiken kaikkiaan iäkkäiden (≥ 65 vuotta) ja nuorempien potilaiden välillä ei raportoitu olleen eroja lääkkeen turvallisuudessa (ks. kohta Farmakodynamiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen sattuessa potilaita on seurattava tarkasti haittavaikutusten oireiden ja löydösten varalta, ja heille on aloitettava välittömästi tarvittava oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet, ATC-koodi: L01FY02.
Vaikutusmekanismi
Opdualag on nivolumabin, ohjelmoituneen solukuoleman proteiinin 1 estäjän (anti‑PD‑1), ja relatlimabin, lymfosyyttiaktivaation geeni‑3:n estäjän (anti‑LAG‑3), kiinteäannoksinen yhdistelmä.
PD‑1-ligandien (PD‑L1 ja PD‑L2) sitoutuminen T‑solujen PD‑1-reseptoriin estää T‑solujen proliferaation ja sytokiinituotannon. Joissakin kasvaimissa tapahtuu PD‑1-ligandien lisääntymistä; signalointi tämän reitin kautta voi johtaa aktiivisen T‑solu-immuunivalvonnan estymiseen kasvaimissa. Nivolumabi on humaani monoklonaalinen IgG4‑vasta-aine, joka sitoutuu PD‑1-reseptoriin, estää sen interaktion PD‑L1- ja PD‑L2-ligandien kanssa ja vähentää immuunivasteen, mukaan lukien antituumorisen immuunivasteen, PD‑1-reittivälitteistä estymistä. Syngeenisissä kasvainhiirimalleissa PD‑1-aktiivisuuden salpaaminen vähensi kasvainten kasvua.
Relatlimabi on humaani monoklonaalinen IgG4‑vasta-aine, joka sitoutuu LAG‑3-reseptoriin, estää sen interaktion ligandien kanssa, myös MHC II:n, ja vähentää immuunivasteen LAG‑3-reittivälitteistä estymistä. Tämän reitin antagonismi edistää T‑solujen proliferaatiota ja sytokiinieritystä.
Nivolumabin (anti‑PD‑1) ja relatlimabin (anti‑LAG‑3) yhdistelmä lisää T‑solujen aktiivisuutta verrattuna jommankumman vasta-aineen aktiivisuuteen yksinään. Syngeenisissä hiiren kasvainmalleissa LAG‑3:n esto voimistaa PD‑1-toiminnan eston antituumorista aktiivisuutta, mikä estää kasvaimen kasvua ja edistää kasvaimen pienenemistä.
Kliininen teho ja turvallisuus
Satunnaistettu vaiheen 2/3 tutkimus, nivolumabi–relatlimabi-yhdistelmähoito vs. nivolumabi aiemmin hoitamattomassa melanoomassa, joka on metastasoitunut tai jota ei voi kirurgisesti poistaa (CA224047)
Nivolumabi–relatlimabi-yhdistelmähoidon tehoa ja turvallisuutta aiemmin hoitamattoman melanooman (joka on metastasoinut tai jota ei voi kirurgisesti poistaa) hoidossa arvioitiin vaiheen 2/3 satunnaistetussa kaksoissokkotutkimuksessa (CA224047). Tutkimuksessa oli mukana potilaita, joiden ECOG-toimintakykyluokka oli 0 tai 1 ja joilla oli American Joint Committee on Cancer (AJCC) -luokittelun 8. painoksen mukaisesti III asteen (jota ei voi kirurgisesti poistaa) tai IV asteen histologisesti vahvistettu melanooma. Potilaille sallittiin melanooman aikaisempi liitännäishoito tai esiliitännäishoito (anti‑PD‑1, anti‑CTLA‑4 tai BRAF‑MEK-hoito sallittiin, kunhan viimeisen hoitoannoksen ja uusiutumisen välillä oli vähintään kuusi kuukautta; interferonihoito sallittiin, kunhan viimeinen annos oli vähintään kuusi viikkoa ennen satunnaistamista). Potilaita, joilla oli aktiivinen autoimmuunisairaus, anamneesissa myokardiitti, troponiiniarvojen nousu > 2 kertaa ULN tai ECOG-toimintakykyluokka ≥ 2, keskisuuri- tai suuriannoksista systemaattista kortikosteroidihoitoa tai immunosuppressiivisia lääkevalmisteita vaativa sairaus, uvean melanooma tai aktiivisia tai hoitamattomia aivometastaaseja tai leptomeningeaalisia metastaaseja, ei otettu mukaan tutkimukseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yhteensä 714 potilasta satunnaistettiin saamaan joko nivolumabi–relatlimabi-yhdistelmähoitoa (n = 355) tai nivolumabia (n = 359). Yhdistelmähoitohaaran potilaat saivat 480 mg nivolumabia / 160 mg relatlimabia 60 minuutin kuluessa 4 viikon välein. Nivolumabihaarassa potilaille annettiin nivolumabia 480 mg 4 viikon välein. Satunnaistaminen stratifioitiin kasvaimen PD‑L1-statuksen mukaan (≥ 1 % vs. < 1 %) PD‑L1 IHC 28‑8 pharmDx -testausmenetelmällä ja LAG‑3-ilmentymisen mukaan (≥ 1 % vs. < 1) määriteltynä validoidulla LAG‑3 IHC -määrityksellä, BRAF V600 -mutaatiostatuksen mukaan ja AJCC-luokittelun 8. painoksen määrittelemän M-levinneisyysasteen mukaan (M0/M1 mikä tahansa [0] vs. M1 mikä tahansa [1]). Hoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voida hyväksyä. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (Response Evaluation Criteria in Solid Tumours, RECIST) version 1.1 mukaan 12 viikon kuluttua satunnaistamisesta ja sitten enintään 52 viikon ajan joka 8. viikko ja sen jälkeen joka 12. viikko, kunnes tauti eteni tai tutkimushoito lopetettiin, sen mukaan, kumpi näistä tapahtui jälkimmäisenä. Ensisijainen tehokkuusmuuttuja oli etenemisvapaa elinaika (PFS) riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioimana. Toissijaiset tehokkuusmuuttujat olivat kokonaiselinaika (OS) ja BICR:n arvioima kokonaisvaste (overall response rate, ORR). Hierarkkinen tilastollinen testausjärjestys oli PFS, jota seurasivat OS ja sitten ORR. Ensisijaiset ja toissijaiset tehokkuusmuuttujat arvioitiin hoitoaikeen mukaisessa (ITT) populaatiossa. ORR:ää ei testattu muodollisesti, sillä OS:n muodollinen vertailu ei ollut tilastollisesti merkitsevää.
ITT-populaatiossa ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 63 vuotta (vaihteluväli 20–94); 47 % oli iältään ≥ 65 vuotta ja 19 % ≥ 75 vuotta. Suurin osa potilaista oli valkoihoisia (97 %) ja miehiä (58 %). Lähtötason ECOG-toimintakykyluokka oli 0 (67 %) tai 1 (33 %). Useimmilla potilailla oli AJCC-luokittelun mukaisesti levinneisyysasteen IV sairaus (92 %); 38,9 %:lla oli M1c-luokan sairaus, 2,4 %:lla oli M1d-luokan sairaus, 8,7 %:lla oli aikaisempi systeeminen hoito ja 36 %:lla oli tutkimukseen otettaessa lähtötason LDH-arvo suurempi kuin ULN. 39 %:lla potilaista oli BRAF-mutaatiopositiivinen melanooma; 75 %:lla oli LAG‑3 ≥ 1 % ja 41 %:lla oli PD‑L1 ≥ 1 % kasvainsolujen membraaniekspressio. Potilaat, joilla oli määritettävissä oleva kasvaimen PD‑L1:n ilmentyminen, olivat jakaantuneet tasaisesti kahteen hoitoryhmään. Potilaiden taustatiedot ja lähtötason ominaisuudet olivat yleisesti hyvin tasapainossa hoitoryhmien välillä niiden potilaiden osalta, joilla PD‑L1:n ilmentyminen oli < 1 %.
ITT-populaation primaarianalyysissa (seuranta-ajan mediaani 13,21 kuukautta; vaihteluväli 0–33,1 kuukautta) todettiin tilastollisesti merkitsevä paraneminen etenemisvapaassa elinajassa: nivolumabi–relatlimabi-yhdistelmähoitoa saaneiden ryhmässä etenemisvapaan elinajan mediaani oli 10,12 kuukautta, kun taas nivolumabiryhmässä se oli 4,63 kuukautta (riskitiheyssuhde = 0,75, 95 %:n luottamusväli: 0,62, 0,92; p = 0,0055). Kokonaiselinaika ITT-populaatiossa ei ollut tilastollisesti merkitsevä (riskitiheyssuhde = 0,80, 95 %:n luottamusväli: 0,64, 1,01) hetkellä, jolloin ennalta määritelty kokonaiselinajan lopullinen analyysi tehtiin (seurannan mediaani oli 19,3 kuukautta).
Taulukossa 3 ovat eksploratiivisen analyysin keskeiset tehoon liittyvät tulokset niiden potilaiden alaryhmästä, jossa kasvaimen PD‑L1:n ilmentymistaso oli < 1 %.
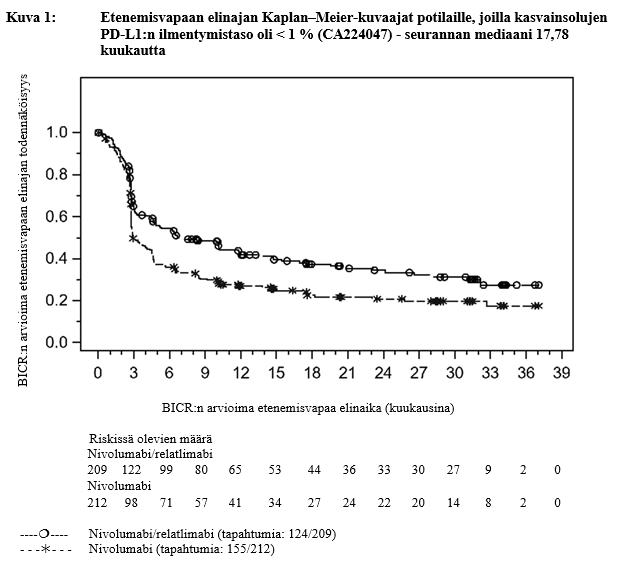
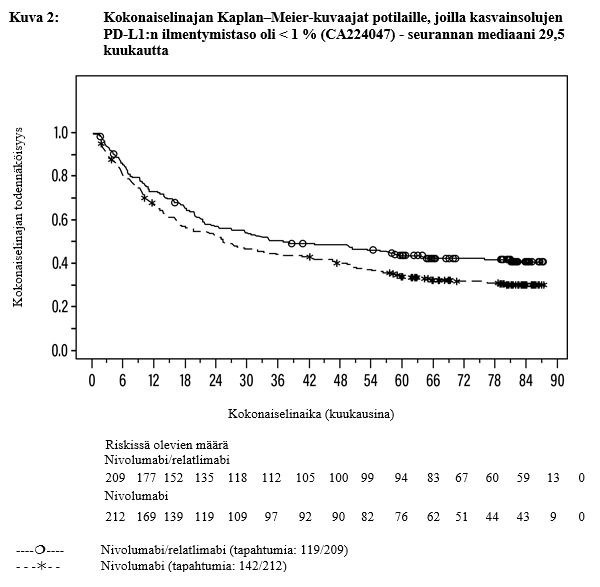
Taulukko 3: Tehoon liittyvät tulokset potilailla, joilla kasvainsolujen PD‑L1-ilmentymistaso oli < 1 % (CA224047)
nivolumabi + relatlimabi (n = 209) | nivolumabi (n = 212) | |
| Etenemisvapaa elinaikaa | ||
| Riskitiheyssuhde (95 %:n luottamusväli) | 0,68 (0,53, 0,86) | |
Mediaani (kuukautta) (95 %:n luottamusväli) | 6,7 (4,7, 12,0) | 3,0 (2,8, 4,5) |
| Kokonaiselinaikab | ||
| Riskitiheyssuhde (95 %:n luottamusväli) | 0,76 (0,60, 0,98) | |
Mediaani (kuukautta) (95 %:n luottamusväli) | 38,3 (24,3, 64,5) | 25,4 (18,7, 42,4) |
| Kokonaisvaste (%)a | 36,4 | 24,1 |
| (95 %:n luottamusväli) | (29,8, 43,3) | (18,5, 30,4) |
a Seurannan mediaani 17,78 kuukautta
b Seurannan mediaani 29,5 kuukautta
Kuvissa 1 ja 2 on esitetty etenemisvapaan elinajan Kaplan–Meier-kuvaajat mediaaniseuranta-ajan ollessa 17,78 kuukautta sekä kokonaiselinajan Kaplan–Meier-kuvaajat mediaaniseuranta-ajan ollessa 29,5 kuukautta PD-L1 ilmentymistasossa < 1 %.
Farmakokinetiikka
Relatlimabin farmakokinetiikkaa nivolumabi–relatlimabi-yhdistelmähoidon annon jälkeen selvitettiin eri syöpiä sairastavilla potilailla, jotka saivat relatlimabia 20–800 mg:n annoksia 2 viikon välein ja 160–1 440 mg:n annoksia 4 viikon välein joko monoterapiana tai yhdistelmähoitona nivolumabin (80 tai 240 mg:n annos 2 viikon välein tai 480 mg:n annos 4 viikon välein) kanssa.
Relatlimabin vakaan tilan pitoisuudet saavutettiin viikkoon 16 mennessä, kun valmistetta annettiin 4 viikon välein. Systeeminen kertymä oli 1,9-kertainen. Relatlimabin keskimääräinen pitoisuus (Cavg) ensimmäisen annoksen jälkeen suureni annosriippuvaisesti annoksilla ≥ 160 mg 4 viikon välein.
Taulukko 4: Nivolumabin ja relatlimabin vakaan tilan altistuksien geometrinen keskiarvo (CV%) sen jälkeen, kun 480 mg nivolumabia ja 160 mg relatlimabia annettiin kiinteäannoksisena yhdistelmänä 4 viikon välein
| Cmax (mikrog/ml) | Cmin (mikrog/ml) | Cavg (mikrog/ml) | |
| Relatlimabi | 62,2 (30,1) | 15,3 (64,3) | 28,8 (44,8) |
| Nivolumabi | 187 (32,9) | 59,7 (58,6) | 94,4 (43,3) |
Populaatiofarmakokineettisten analyysien perusteella nivolumabin ja relatlimabin kiinteäannoksisen yhdistelmän 30 minuuttia ja 60 minuuttia kestävän infuusion ennustettiin tuottavan samankaltaiset nivolumabi- ja relatlimabialtistukset (< 1 %:n ero).
Tutkimuksessa CA224047 nivolumabin vakaan tilan geometrinen keskiarvo Cmin nivolumabi–relatlimabi-yhdistelmähoitoa saaneessa haarassa oli samankaltainen kuin nivolumabihaarassa todettu geometrisen keskiarvon suhde 0,931 (95 %:n luottamusväli: 0,855–1,013).
Jakautuminen
Nivolumabin vakaan tilan jakaantumistilavuuden geometrinen keskiarvo (CV%) on 6,65 l (19,2 %) ja relatlimabin 6,65 l (19,8 %).
Biotransformaatio
Nivolumabi ja relatlimabi ovat lääkkeenä käytettäviä monoklonaalisia IgG4-vasta-aineita, joiden odotetaan hajoavan pieniksi peptideiksi, aminohapoiksi ja pieniksi hiilihydraateiksi lysosomi- tai reseptorivälitteisen endosytoosin kautta.
Eliminaatio
Nivolumabin puhdistuma on vakaassa tilassa 21,1 % pienempi (geometrinen keskiarvo [CV%], 7,57 ml/h [40,1 %]) kuin ensimmäisen annoksen jälkeen (9,59 ml/h [40,3 %]). Sen terminaalinen puoliintumisaika (t1/2) on 26,5 päivää (36,4 %).
Relatlimabin puhdistuma on vakaassa tilassa 9,7 % pienempi (geometrinen keskiarvo [CV%], 5,48 ml/h [41,3 %]) kuin ensimmäisen annoksen jälkeen (6,06 ml/h [38,9 %]). Kun annetaan 160 mg relatlimabia ja 480 mg nivolumabia 4 viikon välein, relatlimabin efektiivisen puoliintumisajan (t1/2) geometrinen keskiarvo (CV%) on 26,2 vuorokautta (37 %).
Erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella seuraavilla tekijöillä ei näyttänyt olevan kliinisesti merkittävää vaikutusta nivolumabin ja relatlimabin puhdistumaan: ikä (vaihteluväli 17–92 vuotta), sukupuoli (miehiä [1 056] ja naisia [657]) tai rotu (valkoihoisia [1 655], afroamerikkalaisia [167] ja aasialaisia [41]). Paino (vaihteluväli 37–170 kg) oli merkittävä muuttuja nivolumabin ja relatlimabin farmakokinetiikassa. Sillä ei kuitenkaan ollut kliinisesti merkityksellistä vaikutusta altistus–vaste-analyysin perusteella.
Pediatriset potilaat
Rajalliset tiedot viittaavat siihen, että nivolumabin puhdistuma oli 36 % pienempi ja jakautumistilavuus 16 % pienempi nuorilla potilailla, joilla oli kiinteitä kasvaimia, kuin vastaavilla aikuisilla verrokkipotilailla. Ei tiedetä, koskeeko tämä myös melanoomapotilaita ja ovatko myös relatlimabin puhdistuma ja jakautumistilavuus nuorilla pienemmät kuin aikuisilla. Populaatiofarmakokineettisten simulaatioiden perusteella turvallisuus ja teho vähintään 30 kg painavilla nuorilla ovat odotettavasti samaa luokkaa kuin saman painoisilla aikuisilla saman suositellun annoksen tuottamilla nivolumabi- ja relatlimabialtistuksilla.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisessä analyysissä arvioitiin munuaisten vajaatoiminnan vaikutusta nivolumabin ja relatlimabin puhdistumaan potilailla, joilla oli lievä tai kohtalainen munuaisten vajaatoiminta, verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Nivolumabin tai relatlimabin puhdistumassa ei todettu kliinisesti merkittäviä eroja munuaisten vajaatoimintaa sairastavien ja niiden potilaiden välillä, joiden munuaiset toimivat normaalisti.
Maksan vajaatoiminta
Populaatiofarmakokineettisessä analyysissä arvioitiin maksan vajaatoiminnan vaikutusta nivolumabin ja relatlimabin puhdistumaan potilailla, joilla oli lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ ULN ja ASAT > ULN tai kokonaisbilirubiini > 1–1,5 kertaa ULN ja mikä tahansa ASAT-arvo) tai kohtalainen maksan vajaatoiminta (kokonaisbilirubiini > 1,5–3 kertaa ULN ja mikä tahansa ASAT-arvo), verrattuna potilaisiin, joiden maksan toiminta oli normaali. Nivolumabin tai relatlimabin puhdistumassa ei todettu kliinisesti merkittäviä eroja maksan vajaatoimintaa sairastavien ja niiden potilaiden välillä, joiden maksan toiminta oli normaali.
Immunogeenisuus
Hoidosta johtuvien nivolumabin vasta-aineiden ja hoidosta johtuvien relatlimabin vasta-aineiden vähäisellä määrällä ei todettu olevan vaikutusta nivolumabin tai relatlimabin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Nivolumabi yhdistelmähoitona relatlimabin kanssa
Eläinkokeita ei ole tehty nivolumabi–relatlimabi-yhdistelmähoidon mahdollisen karsinogeenisuuden, genotoksisuuden tai lisääntymis- ja kehitystoksisuuden arvioimiseksi.
Kuukauden kestäneessä tutkimuksessa, jossa apinoille annettiin nivolumabia ja relatlimabia, todettiin keskushermoston (aivokammion suonipunoksen, verisuoniston, aivokalvojen, selkäytimen) ja lisääntymiselinten (lisäkiveksen, rakkularauhasen ja kivesten) tulehduksia. Vaikka tälle yhdistelmälle ei varmistettu turvallisuusmarginaaleja mainittujen vaikutusten suhteen, näitä ilmeni annoksilla, joista seuraavat altistustasot ovat oletettavasti merkitsevästi suurempia (13‑kertainen nivolumabilla ja 97‑kertainen relatlimabilla) kuin potilaissa saavutettavat altistustasot.
Relatlimabi
Eläinkokeista saatuja tietoja relatlimabin vaikutuksista raskauteen ja lisääntymiseen ei ole. Hiirillä tehdyissä alkion/sikiön toksisuutta koskevissa tutkimuksissa, joissa käytettiin hiiren LAG‑3-vasta-aineita, ei todettu emoon tai kehitykseen kohdistuvia vaikutuksia. Relatlimabin vaikutusta pre‑ ja postnataaliseen kehitykseen ei ole arvioitu; relatlimabin vaikutusmekanismin, LAG‑3:n eston, perusteella sillä voi kuitenkin olla samanlainen negatiivinen vaikutus raskauteen kuin nivolumabilla. Relatlimabin vaikutusta hedelmällisyyteen ei ole tutkittu.
Nivolumabi
PD‑1/PD‑L1-reitin eston on hiirimalleissa todettu huonontavan sikiön sietämistä ja lisäävän sikiökuolemia. Nivolumabin vaikutusta pre‑ ja postnataaliseen kehitykseen arvioitiin apinoilla, joille annettiin nivolumabia 2 kertaa viikossa organogeneesin alusta ensimmäisellä raskauskolmanneksella synnytykseen asti. Altistus oli 8 tai 35 kertaa suurempi kuin on todettu käytettäessä kliinisesti 3 mg/kg nivolumabia (AUC-arvon perusteella). Kolmannesta raskauskolmanneksesta lähtien todettiin annoksen suuruudesta riippuvaa sikiökuolemien lisääntymistä sekä vastasyntyneiden kuolleisuuden lisääntymistä.
Muut nivolumabihoitoa saaneiden naarasapinoiden jälkeläiset säilyivät hengissä suunniteltuun keskeytykseen saakka ilman hoitoon liittyviä kliinisiä merkkejä, normaalin kehityksen muutoksia, vaikutuksia elinten painoon tai makro- tai mikroskooppisia patologisia muutoksia. Tulokset olivat kasvuindeksien sekä teratogeenisten, neurobehavioraalisten, immunologisten ja kliinispatologisten parametrien perusteella koko 6 kuukauden postnataalivaiheen ajan vastaavat kuin verrokkiryhmässä. Vaikutusmekanismiensa perusteella sikiön altistuminen nivolumabille ja samoin relatlimabille voi kuitenkin lisätä immuunivälitteisten sairauksien kehittymisen tai normaalin immuunivasteen muutosten riskiä. PD‑1- ja PD‑1/LAG‑3-poistogeenisillä hiirillä on raportoitu esiintyneen immuunivälitteisiä sairauksia. Nivolumabin vaikutusta hedelmällisyyteen ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Histidiini, histidiinihydrokloridimonohydraatti, sakkaroosi, pentetiinihappo (dietyleenitriamiinipentaetikkahappo), polysorbaatti 80 (E433), injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa. Opdualag-valmistetta ei saa infusoida samaan aikaan saman laskimoletkun kautta kuin muita lääkevalmisteita.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Infuusion valmistamisen jälkeen
Kemiallinen ja fysikaalinen käytönaikainen säilyvyys valmistamisajankohdan jälkeen on osoitettu seuraavasti (aikoihin on sisällytetty valmisteen antoaika):
Infuusion valmistaminen | Kemiallinen ja fysikaalinen käytönaikainen säilyvyys | |
Säilytys 2–8 °C:ssa valolta suojattuna | Säilytys huoneenlämmössä (≤ 25 °C) ja huoneenvalossa | |
Laimentamaton tai laimennettu 9 mg/ml:n vahvuisella (0,9 %) natriumkloridi‑injektioliuoksella | 30 vrk | 24 tuntia (yhteensä 30 vuorokauden säilytyksestä) |
Laimennettu 50 mg/ml:n (5 %) vahvuisella glukoosi-injektioliuoksella | 7 vrk | 24 tuntia (yhteensä 7 vuorokauden säilytyksestä) |
Valmistettu infuusioliuos on mikrobiologiselta kannalta käytettävä välittömästi riippumatta käytetystä laimentimesta. Ellei liuosta käytetä heti, säilytysajat ja -olosuhteet ennen käyttöä ovat käyttäjän vastuulla, eikä niiden normaalisti tulisi ylittää 24:ää tuntia 2–8 °C:ssa, jollei valmistelu ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa (ks. kohta Käyttö- ja käsittelyohjeet).
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Avaamattomat injektiopullot voi säilyttää kontrolloidussa huoneenlämmössä (enintään 25 °C:ssa) enintään 72 tunnin ajan.
Valmistetun infuusion säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OPDUALAG infuusiokonsentraatti, liuosta varten
240 mg/80 mg (L:ei) 1 kpl (20 ml (12 mg/ml / 4 mg/ml)) (7537,08 €)
PF-selosteen tieto
Pakkaus: yksi 25 ml:n injektiopullo (tyypin I lasia), jossa on tulppa (päällystettyä butyylikumia) ja keltainen irti napsautettava (flip-off) alumiinikorkki. Yksi injektiopullo sisältää 21,3 ml liuosta, ylitäyttö 1,3 ml mukaan lukien.
Valmisteen kuvaus:
Kirkas tai opalisoiva, väritön tai kellertävä neste, joka on periaatteessa hiukkaseton.
Liuoksen pH-arvo on noin 5,8 ja osmolaalisuus noin 310 mOsm/kg.
Käyttö- ja käsittelyohjeet
Opdualag toimitetaan yhden annoksen sisältävässä injektiopullossa, eikä se sisällä säilöntäaineita. Valmistelun saa tehdä vain koulutettu henkilökunta hyviä, erityisesti aseptiikkaan liittyviä käytäntöjä noudattaen.
Opdualag-valmisteen voi annostella laskimoon joko
- laimentamatta, kun se on siirretty sopivalla steriilillä ruiskulla infuusiosäiliöön tai
-
laimennettuna seuraavien ohjeiden mukaan:
- lopullisen infuusion pitoisuuden on oltava 3–12 mg/ml nivolumabia ja 1–4 mg/ml relatlimabia
- infuusion kokonaismäärä ei saa ylittää 160 ml:aa. Jos potilas painaa alle 40 kg, infuusion kokonaismäärä ei saa ylittää 4 ml:aa per potilaan yksi painokilo.
Opdualag-konsentraatin laimentamiseen voi käyttää joko
- 9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai
- 50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta.
Infuusion valmistaminen
- Tarkista, onko Opdualag-konsentraatissa hiukkasia tai värjäymiä. Älä ravista injektiopulloa. Opdualag-konsentraatti on kirkasta tai opalisoivaa, väritöntä tai kellertävää liuosta. Hylkää injektiopullo, jos liuos on sameaa, värjäytynyttä, tai sisältää vieraita hiukkasia.
- Ota sopivaan steriiliin ruiskuun tarvittava määrä Opdualag-konsentraattia ja siirrä konsentraatti steriiliin liuospussiin (etyylivinyyliasetaattia [EVA], polyvinyylikloridia [PVC] tai polyolefiinia).
- Laimenna Opdualag-liuos tarvittaessa tarpeellisella määrällä 9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai 50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta. Valmistamisen helpottamiseksi konsentraatin voi myös siirtää suoraan esitäytettyyn pussiin, jossa on oikea määrä 9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai 50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta.
- Sekoita infuusio varovasti käsin pyörittelemällä. Älä ravista.
Annostelu
Opdualag-infuusiota ei saa antaa laskimoon push- eikä bolusinjektiona.
Annostele Opdualag-infuusio laskimoon 30 minuutin kuluessa.
Käytä infuusiolaitetta ja sisäänrakennettua tai lisättävää steriiliä, ei‑pyrogeenistä, vähän proteiineja sitovaa suodatinta (huokoskoko 0,2–1,2 mikrom).
Opdualag-infuusio on yhteensopiva seuraavien kanssa: EVA-, PVC- ja polyolefiinisäiliöt, PVC-infuusiolaitteet ja linjasuodattimet, joiden polyeetterisulfoni-, nailon- tai polypolyvinyylideenifluoridikalvon huokoskoko on 0,2–1,2 mikrom.
Muita lääkevalmisteita ei saa infusoida saman infuusioletkun kautta.
Opdualag-annoksen annostelun jälkeen huuhdo linja 9 mg/ml:n (0,9 %) vahvuisella natriumkloridi-injektioliuoksella tai 50 mg/ml:n (5 %) vahvuisella glukoosi-injektioliuoksella.
Hävittäminen
Älä säilytä käyttämätöntä osaa infuusioliuoksesta uutta käyttöä varten. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OPDUALAG infuusiokonsentraatti, liuosta varten
240 mg/80 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FY02
Valmisteyhteenvedon muuttamispäivämäärä
23.04.2026
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com