
ASPAVELI infuusioneste, liuos 1080 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Yleinen
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 20 ml:n injektiopullo sisältää 1080 mg pegsetakoplaania.
Yksi millilitra sisältää 54 mg pegsetakoplaania.
Apuaine, jonka vaikutus tunnetaan
Yksi millilitra sisältää 41 mg sorbitolia.
Yksi injektiopullo sisältää 820 mg sorbitolia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
ASPAVELI on tarkoitettu monoterapiana sellaisten aikuispotilaiden hoitoon, joilla on kohtauksittainen yöllinen hemoglobiinivirtsaisuus (PNH) ja hemolyyttinen anemia.
ASPAVELI on tarkoitettu sellaisten aikuisten ja 12–17 vuoden ikäisten nuorten potilaiden hoitoon, joilla on C3‑glomerulopatia (C3G) tai primaarinen immunokompleksi membranoproliferatiivinen munuaiskerästulehdus (IC‑MPGN), yhdistelmänä reniini-angiotensiinijärjestelmän (RAS) estäjän kanssa, paitsi jos potilas ei siedä hoitoa RAS:n estäjillä tai se on vasta-aiheista.
Ehto
Hoito on aloitettava hematologisiin sairauksiin perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Hoito on aloitettava hematologisiin sairauksiin tai munuaissairauksiin perehtyneen terveydenhuollon ammattilaisen valvonnassa. Itseannostelua ja koti-infuusiota on harkittava potilaille, jotka ovat sietäneet hoitoa hyvin kokeneissa hoitokeskuksissa. Päätös itseannostelun ja koti-infuusioiden mahdollisuudesta on tehtävä hoitavan lääkärin arvion ja suosituksen jälkeen.
Annostus
Pegsetakoplaanin voi antaa terveydenhuollon ammattilainen tai potilas tai huoltaja voi antaa sen asianmukaisten ohjeiden mukaan.
Kohtauksittainen yöllinen hemoglobiinivirtsaisuus (PNH)
Aikuiset potilaat, joilla on PNH
Pegsetakoplaani annetaan kaksi kertaa viikossa 1 080 mg:n infuusiona ihon alle kaupallisesti saatavilla olevalla infuusiopumpulla tai puettavalla antojärjestelmällä, jolla voidaan antaa enintään 20 ml:n annoksia. Kaksi kertaa viikossa annettava annos on annettava kunkin hoitoviikon päivänä 1 ja päivänä 4.
PNH on krooninen sairaus, ja hoitoa ASPAVELI‑valmisteella suositellaan jatkamaan potilaan eliniän ajan, ellei tämän lääkevalmisteen käytön keskeyttäminen ole kliinisesti aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
ASPAVELI‑hoitoon C5-inhibiittorihoidosta vaihtavat PNH-potilaat
Ensimmäisten neljän viikon ajan pegsetakoplaani annetaan kaksi kertaa viikossa 1 080 mg:n annoksena ihon alle potilaan senhetkisen C5-inhibiittorihoidon lisäksi, jotta saadaan minimoitua äkillisestä hoidon keskeytyksestä aiheutuva hemolyysivaara. Neljän viikon jälkeen potilaan on keskeytettävä C5-inhibiittorihoito ennen ASPAVELI‑monoterapian jatkamista.
Muista komplementin estäjistä kuin ekulitsumabista vaihtamista ei ole tutkittu. Muiden komplementin estäjien käytön lopettaminen ennen pegsetakoplaanin vakaan tilan saavuttamista on tehtävä varoen (ks. kohta Farmakokinetiikka).
PNH-potilaiden annoksen muuttaminen
Annostusohjelma voidaan muuttaa 1 080 mg:aan joka kolmas päivä (esim. päivänä 1, päivänä 4, päivänä 7, päivänä 10, päivänä 13 ja niin edelleen), jos potilaan laktaattidehydrogenaasitaso (LDH-taso) on suurempi kuin 2 x viitevälin yläraja (upper limit of normal, ULN). Mikäli annosta suurennetaan, LDH-tasoa on seurattava kaksi kertaa viikossa ainakin 4 viikon ajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
C3‑glomerulopatia (C3G) ja primaarinen immunokompleksi membranoproliferatiivinen munuaiskerästulehdus (IC‑MPGN)
Pegsetakoplaani annetaan kaksi kertaa viikossa infuusiona ihon alle kaupallisesti saatavilla olevalla infuusiopumpulla tai puettavalla antojärjestelmällä, jolla voidaan antaa enintään 20 ml:n annoksia. Kaksi kertaa viikossa annettava annos on annettava kunkin hoitoviikon päivänä 1 ja päivänä 4.
C3G ja primaarinen IC‑MPGN ovat kroonisia sairauksia. Lääkevalmisteen käytön keskeyttämistä ei suositella, ellei se ole kliinisesti aiheellista.
Aikuiset potilaat, joilla on C3G tai primaarinen IC‑MPGN
Pegsetakoplaani annetaan kaksi kertaa viikossa 1 080 mg:n infuusiona ihon alle.
Nuoret potilaat, joilla on C3G tai primaarinen IC‑MPGN
Nuorten potilaiden annostusohjelma perustuu potilaan painoon seuraavasti:
Paino | Ensimmäinen annos (infuusion tilavuus) | Toinen annos (infuusion tilavuus) | Ylläpitoannos (infuusion tilavuus) |
≥ 50 kg | 1 080 mg kaksi kertaa viikossa (20 ml) | ||
35 – < 50 kg | 648 mg (12 ml) | 810 mg (15 ml) | 810 mg kaksi kertaa viikossa (15 ml) |
30 – < 35 kg | 540 mg (10 ml) | 540 mg (10 ml) | 648 mg kaksi kertaa viikossa (12 ml) |
Annoksen jääminen väliin
Jos PNH:n, C3G:n tai primaarisen IC‑MPGN:n hoitoon annettava pegsetakoplaaniannos jää väliin, se on annettava mahdollisimman pian, ja sen jälkeen jatkettava hoitoa tavallisessa aikataulussa, vaikka korvaavan annoksen ja sitä seuraavan annoksen välinen aika olisi tällöin alle 3 vuorokautta.
Potilaat, joilla on elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN
Elinsiirron jälkeen uusiutuneen C3G:n tai primaarisen IC‑MPGN:n diagnoosi tulee tehdä siirretyn munuaisen biopsian perusteella. C3G:n tai primaarisen IC‑MPGN:n uusiutuminen voidaan havaita rutiininomaisessa elinsiirron jälkeisessä biopsiassa; muutoin biopsia on tehtävä, kun kliiniset oireet viittaavat taudin uusiutumiseen. Pegsetakoplaanihoito voidaan aloittaa ennen kliinisten oireiden ilmaantumista, kuten glomerulusten laskennallisen suodatusnopeuden (eGFR) pienenemistä tai virtsan proteiini-kreatiniinisuhteen (uPCR) suurenemista, kuten tutkimuksessa APL2-C3G-204 tehtiin (ks. kohta Farmakodynamiikka). Kokemus pegsetakoplaanin käytöstä kliinisissä tutkimuksissa potilaille, joilla on elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN, on vähäistä (ks. kohta Farmakodynamiikka).
Erityisryhmät
Iäkkäät
Vaikka kliinisissä tutkimuksissa ei ole havaittu ilmeisiä ikään liittyviä eroja, vähintään 65‑vuotiaiden potilaiden määrä ei riitä määrittämään, reagoivatko he eri tavalla kuin nuoremmat potilaat. Ei ole viitteitä siitä, että tarvittaisiin jonkinlaisia erityisiä varotoimia hoidettaessa iäkkäitä potilaita.
Munuaisten toimintahäiriö
Vaikealla munuaisten toimintahäiriöllä (kreatiniinipuhdistuma < 30 ml/min) ei ollut vaikutusta pegsetakoplaanin farmakokinetiikkaan; siten pegsetakoplaaniannosta ei tarvitse muuttaa potilaille, joilla on munuaisten toimintahäiriö. Pegsetakoplaanin käytöstä potilaille, joilla on loppuvaiheen munuaissairaus (ESRD) ja jotka tarvitsevat dialyysiä, ei ole tietoa (ks. kohta Farmakokinetiikka).
Maksan toimintahäiriö
Pegsetakoplaanin turvallisuutta ja tehoa ei ole tutkittu potilailla, joilla on maksan toimintahäiriö. Annosta ei kuitenkaan suositella muutettavan, koska maksan toimintahäiriön ei odoteta vaikuttavan pegsetakoplaanin puhdistumaan.
Pediatriset potilaat
ASPAVELI‑valmisteen turvallisuutta ja tehoa PNH:ta sairastavien 0–< 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
ASPAVELI‑valmisteen turvallisuutta ja tehoa C3G:tä tai primaarista IC‑MPGN:ää sairastavien alle 12 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Tätä lääkevalmistetta ei pidä käyttää < 12 vuoden ikäisten lasten hoitoon, koska tästä ikäryhmästä ei ole saatavilla prekliinisiä turvallisuustietoja.
Antotapa
ASPAVELI‑valmistetta annetaan vain ihon alle kaupallisesti saatavilla olevalla infuusiopumpulla tai puettavalla antojärjestelmällä.
Potilas voi antaa tämän lääkevalmisteen itse. Kun itseanto aloitetaan, pätevä terveydenhuollon ammattilainen neuvoo potilaalle infuusiotekniikat, infuusiopumpun tai puettavan antojärjestelmän käytön, hoitorekisterin pitämisen, mahdollisten haittavaikutusten tunnistamisen ja toimenpiteet, jos haittavaikutuksia ilmenee.
- Käytettäessä infuusiopumppua ASPAVELI on annettava infuusiona vatsaan, reiteen, lonkkaan tai olkavarteen. Infuusiokohtien tulee olla vähintään 7,5 cm:n päässä toisistaan. Infuusiokohtaa on vaihdettava antokertojen välillä. Infuusion kesto on noin 30 minuuttia (käytettäessä kahta kohtaa) tai noin 60 minuuttia (käytettäessä yhtä kohtaa).
- Käytettäessä puettavaa antojärjestelmää ASPAVELI on annettava infuusiona vatsaan. Infuusiokohtaa on vaihdettava antokertojen välillä laitteen valmistajan ohjeiden mukaan. Infuusion kesto vaihtelee potilaittain ja on tavallisesti 30–60 minuuttia.
Infuusion antamista alueille, joilla iho on arka, punoittava tai kova tai jossa on mustelma, on vältettävä. Infuusion antamista tatuointien, arpien tai venymisjälkien kohdalle on vältettävä. Infuusio on aloitettava viipymättä sen jälkeen, kun tämä lääkevalmiste on otettu ruiskuun. Infuusio on annettava 2 tunnin kuluessa ruiskun valmistelemisesta. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen valmistelusta ja infuusion antamisesta.
Vasta-aiheet
Yliherkkyys pegsetakoplaanille tai kohdassa Apuaineet mainituille apuaineille.
Pegsetakoplaanihoitoa ei saa aloittaa potilaille
- joilla on kapselillisen bakteerin, kuten Neisseria meningitidis-, Streptococcus pneumoniae- tai Haemophilus influenzae -bakteerin, aiheuttama aktiivinen infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
- joita ei ole parhaillaan rokotettu Neisseria meningitidis-, Streptococcus pneumoniae- ja Haemophilus influenzae -bakteereita vastaan, ellei potilas saa ennaltaehkäisevää hoitoa sopivilla antibiooteilla niin, että antibioottihoito päättyy 2 viikkoa rokotuksen saamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Kapselillisten bakteerien aiheuttamat vakavat infektiot
Pegsetakoplaanihoito voi altistaa potilaat kapselillisen bakteerin, kuten Neisseria meningitidis-, Streptococcus pneumoniae- tai Haemophilus influenzae -bakteerin, aiheuttamille vakaville infektioille. Infektiovaaran pienentämiseksi kaikki potilaat on rokotettava näitä bakteereita vastaan sovellettavien paikallisten ohjeiden mukaisesti vähintään 2 viikkoa ennen pegsetakoplaanin saamista, ellei hoidon viivästymisen aiheuttama riski ole suurempi kuin infektion kehittymisen aiheuttama riski.
Potilaat, joiden rokotushistoria on tiedossa
Jos potilaan rokotushistoria on tiedossa, ennen pegsetakoplaanihoidon aloittamista on varmistettava, että potilas on saanut rokotteet kapselillisia bakteereita, kuten Streptococcus pneumoniae -bakteeria, Neisseria meningitidis ‑seroryhmiä A, C, W, Y ja B sekä Haemophilus influenzae ‑tyyppiä B, vastaan edellisten kahden vuoden sisällä.
Potilaat, joiden rokotushistoria ei ole tiedossa
Jos potilaan rokotushistoria ei ole tiedossa, tarvittavat rokotukset on annettava vähintään 2 viikkoa ennen ensimmäisen pegsetakoplaaniannoksen antamista. Jos tarvitaan välitöntä hoitoa, tarvittavat rokotukset on annettava mahdollisimman pian, ja potilasta on hoidettava sopivilla antibiooteilla niin, että antibioottihoito päättyy 2 viikkoa rokotuksen saamisen jälkeen.
Potilaiden seuraaminen vakavien infektioiden varalta
Rokotus ei ehkä riitä estämään vakavaa infektiota. Viralliset ohjeet bakteerilääkkeiden asianmukaisesta käytöstä on otettava huomioon. Kaikkia potilaita on seurattava kapselillisten bakteerien, kuten Neisseria meningitidis-, Streptococcus pneumoniae- ja Haemophilus influenzae -bakteerien, aiheuttamien varhaisten infektion merkkien varalta ja potilaat on arvioitava välittömästi infektiota epäiltäessä ja hoidettava tarvittaessa asianmukaisilla antibiooteilla. Potilaille on kerrottava näistä merkeistä ja oireista ja kehotettava hakeutumaan välittömästi lääkärin hoitoon. Lääkäreiden on keskusteltava pegsetakoplaanihoidon hyödyistä ja riskeistä potilaiden kanssa.
Yliherkkyys
Yliherkkyysreaktioita on raportoitu. Jos ilmenee vaikea yliherkkyysreaktio (kuten anafylaksia), pegsetakoplaanihoito on lopetettava välittömästi ja potilaalle annettava asianmukaista hoitoa.
Injektiokohdan reaktiot
Injektiokohdan reaktioita on raportoitu ihon alle annetun pegsetakoplaanihoidon yhteydessä (ks. kohta Haittavaikutukset). Potilaille on annettava asianmukaista koulutusta oikeasta injektiotekniikasta.
PNH:n seuranta laboratoriossa
PNH:ta sairastavia, pegsetakoplaanihoitoa saavia potilaita on seurattava säännöllisesti hemolyysin merkkien ja oireiden varalta ja LDH-tasot mitattava. Potilaan annosta on ehkä muutettava suositellun annostusaikataulun sisällä (ks. kohta Annostus ja antotapa).
Vaikutukset laboratoriokokeissa
Koagulaatiopaneelien piioksidireagenssien ja pegsetakoplaanin välillä voi olla yhteisvaikutus, joka pidentää keinotekoisesti aktivoitua osittaista tromboplastiiniaikaa (aPTT); siksi piioksidireagenssien käyttöä koagulaatiopaneeleissa on vältettävä.
PNH:ta sairastavien potilaiden hoidon lopettaminen
Mikäli PNH:ta sairastava potilas lopettaa pegsetakoplaanihoidon, häntä on seurattava tarkasti vakavan intravaskulaarisen hemolyysin merkkien ja oireiden varalta. Vakavan intravaskulaarisen hemolyysin tunnistaa kohonneista LDH-tasoista sekä äkillisestä PNH-kloonin koon pienenemisestä tai hemoglobiinitason (Hb) laskusta tai oireiden, kuten väsymyksen, hemoglobinurian, vatsakivun, hengenahdistuksen, merkittävän haitallisen verisuonitapahtuman (kuten tromboosin), dysfagian tai erektiohäiriön, uusiutumisesta. Mikäli tämän lääkevalmisteen käytön lopettaminen on tarpeen, on harkittava vaihtoehtoista hoitoa. Mikäli vakavaa hemolyysiä ilmenee hoidon lopettamisen jälkeen, on harkittava seuraavia toimenpiteitä/hoitoja: verensiirto (pakatut punasolut), vaihtotransfuusio, antikoagulaatio ja kortikosteroidit. Potilaita on seurattava tarkasti vähintään 8 viikkoa viimeisen annoksen jälkeen, mikä vastaa yli viittä tämän lääkevalmisteen puoliintumisaikaa, jotta lääkevalmiste ehtii poistua elimistöstä (ks. kohta Farmakokinetiikka) ja jotta vakava hemolyysi ja muut reaktiot voidaan havaita. Lisäksi on harkittava hidasta vieroitusta.
Ehkäisy naisilla, jotka voivat tulla raskaaksi
On suositeltavaa, että naiset, jotka voivat tulla raskaaksi, käyttävät tehokasta ehkäisyä raskauden estämiseen pegsetakoplaanihoidon aikana ja vähintään 8 viikon ajan viimeisen pegsetakoplaaniannoksen jälkeen (ks. Kohta Raskaus ja imetys).
Polyetyleeniglykolin (PEG) kertyminen
ASPAVELI on PEGyloitu lääkevalmiste. PEG:n munuaisiin, aivokammion suonipunokseen ja muihin elimiin tapahtuvan kertymisen mahdollisia pitkäaikaisvaikutuksia ei tunneta (ks. Kohta Prekliiniset tiedot turvallisuudesta). On suositeltavaa tutkia munuaisten toiminta säännöllisesti laboratoriokokein.
Koulutusmateriaalit
Kaikkien lääkäreiden, jotka aikovat määrätä ASPAVELI‑valmistetta, on varmistettava, että he ovat saaneet lääkärin koulutusmateriaalin ja tutustuneet siihen. Lääkäreiden on kerrottava potilaalle ASPAVELI-hoidon hyödyistä ja riskeistä, keskusteltava niistä potilaiden kanssa ja annettava potilaille potilaan tietopaketti ja potilaskortti. Potilasta on neuvottava hakeutumaan viipymättä lääkärinhoitoon, jos hänellä ilmenee vakavan infektion tai yliherkkyysreaktion merkkejä tai oireita ASPAVELI‑hoidon aikana, erityisesti jos oireet viittaavat kapselillisten bakteerien aiheuttamaan infektioon.
Apuaine(et), joiden vaikutus tunnetaan
Sisältää sorbitolia
ASPAVELI 1 080 mg sisältää 820 mg sorbitolia per injektiopullo.
Potilaat, joilla on perinnöllinen fruktoosi-intoleranssi (HFI), eivät saa ottaa eikä heille saa antaa tätä lääkevalmistetta.
Sisältää natriumia
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. In vitro ‑tietojen perusteella pegsetakoplaanilla on pieni lääkkeiden välisten kliinisten yhteisvaikutusten potentiaali.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
On suositeltavaa, että naiset, jotka voivat tulla raskaaksi, käyttävät tehokasta ehkäisyä raskauden estämiseen pegsetakoplaanihoidon aikana ja vähintään 8 viikon ajan viimeisen pegsetakoplaaniannoksen jälkeen. Pegsetakoplaanihoitoa voidaan harkita naisille, jotka suunnittelevat raskautta, kun riskit ja hyödyt on arvioitu (ks. Raskaus).
Raskaus
Pegsetakoplaanin käytöstä raskaana oleville naisille on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kappale Prekliiniset tiedot turvallisuudesta).
Pegsetakoplaanin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö pegsetakoplaani ihmisillä äidinmaitoon. Imeväiseen imeytymisen ja imeväiseen kohdistuvan haitan mahdollisuutta ei tunneta. Eläinkokeet viittaavat siihen, että pegsetakoplaania erittyy vähän (alle 1 %, ei farmakologisesti merkittävä määrä) apinan maitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). On epätodennäköistä, että imeväinen saisi kliinisesti merkittävän altistuksen.
On suositeltavaa lopettaa imetys pegsetakoplaanihoidon ajaksi.
Hedelmällisyys
Eläimiä tai ihmisiä koskevia tietoja pegsetakoplaanin vaikutuksesta hedelmällisyyteen ei ole saatavilla. Toksisuustutkimuksissa uros- tai naarasapinoiden lisääntymiselimistöissä ei ollut mikroskooppisia poikkeavuuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
ASPAVELI‑valmisteella ei ole haitallista vaikutusta tai on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
PNH
Yleisimmin pegsetakoplaanihoitoa saaneilla PNH-potilailla raportoitu haittavaikutus oli injektiokohdan reaktio: injektiokohdan eryteema, injektiokohdan kutina, injektiokohdan turvotus, injektiokohdan kipu, injektiokohdan mustelma. Muita kliinisissä tutkimuksissa yli 10 %:lla potilaista raportoituja haittavaikutuksia olivat ylähengitystieinfektio, ripuli, hemolyysi, vatsakipu, päänsärky, väsymys, kuume, yskä, virtsatieinfektio, rokotuskomplikaatio, raajakipu, heitehuimaus, nivelkipu ja selkäkipu. Yleisimmin raportoituja vakavia haittavaikutuksia olivat hemolyysi ja sepsis.
C3G ja primaarinen IC‑MPGN
Yleisimmin pegsetakoplaanihoitoa saaneilla C3G:tä tai primaarista IC‑MPGN:ää sairastavilla potilailla raportoituja haittavaikutuksia olivat infuusiokohdan reaktiot ja ylähengitystieinfektiot. Yleisimmin raportoituja vakavia haittavaikutuksia olivat akuutti munuaisvaurio ja keuhkokuume.
Haittavaikutustaulukko
Taulukossa 1 on lueteltu PNH:ta, C3G:tä ja primaarista IC‑MPGN:ää sairastavien potilaiden pegsetakoplaanihoidon yhteydessä kliinisissä tutkimuksissa ja myyntiintulon jälkeen havaitut haittavaikutukset. Haittavaikutukset on lueteltu MedDRA-elinjärjestelmäluokan ja esiintymistiheyden mukaan käyttämällä seuraavaa luokittelua: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100) tai harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on lueteltu kussakin esiintymistiheysryhmässä vakavuuden mukaan alanevassa järjestyksessä.
Taulukko 1: Haittavaikutukset, koottu kliinisistä tutkimuksista1 ja myyntiintulon jälkeisistä kokemuksista
MedDRA-elinjärjestelmäluokka Haittavaikutus | Yleisyys PNH-potilailla | Yleisyys C3G:tä tai primaarista IC‑MPGN:ää sairastavilla potilailla |
| Infektiot | ||
| Influenssa | Hyvin yleinen | |
| Ylähengitystieinfektiot2 | Hyvin yleinen | Hyvin yleinen |
| Virtsatieinfektio | Hyvin yleinen | Yleinen |
| Sepsis | Yleinen3 | |
| Opportunistiset infektiot | Yleinen4 | |
| Koronavirustauti (COVID-19), maha-suolikanavan infektio, sieni-infektio, ihoinfektio, suun infektio | Yleinen | |
| Korvainfektio | Yleinen | Yleinen |
| Infektio, hengitystieinfektio5, virusinfektio, bakteeri-infektio, emätininfektio, silmäinfektio | Yleinen | |
| Kohdunkaulan tulehdus, nivusinfektio | Melko harvinainen | |
| Keuhkokuume | Melko harvinainen | Yleinen |
| Nenän absessi, tuberkuloosi, ruokatorven kandidiaasi, koronaviruksen (COVID‑19) aiheuttama keuhkokuume, peräaukon paise | Melko harvinainen | |
| Immuunijärjestelmä | ||
| Yliherkkyysreaktio | Hyvin yleinen6 | |
| Veri ja imukudos | ||
| Hemolyysi | Hyvin yleinen | |
| Trombosytopenia | Yleinen | Yleinen7 |
| Neutropenia | Yleinen | Yleinen |
| Aineenvaihdunta ja ravitsemus | ||
| Hypokalemia | Yleinen | Yleinen |
| Hermosto | ||
| Päänsärky | Hyvin yleinen | Hyvin yleinen |
| Heitehuimaus | Hyvin yleinen | |
| Verisuonisto | ||
| Kohonnut verenpaine | Yleinen | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Yskä | Hyvin yleinen | Yleinen |
| Hengenahdistus, suunielun kipu, nenän tukkoisuus | Yleinen | |
| Nenäverenvuoto | Yleinen | Yleinen |
| Ruoansulatuselimistö | ||
| Vatsakipu | Hyvin yleinen | |
| Ripuli | Hyvin yleinen | Hyvin yleinen |
| Pahoinvointi | Yleinen | Hyvin yleinen |
| Iho ja ihonalainen kudos | ||
| Eryteema, ihottuma, nokkosihottuma | Yleinen | |
| Luusto, lihakset ja sidekudos | ||
| Nivelkipu, selkäkipu | Hyvin yleinen | |
| Raajakipu | Hyvin yleinen | Yleinen |
| Myalgia | Yleinen | Yleinen |
| Lihasspasmit | Yleinen | |
| Munuaiset ja virtsatiet | ||
| Akuutti munuaisvaurio | Yleinen | Hyvin yleinen |
| Kromaturia | Yleinen | |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Kuume | Hyvin yleinen | Hyvin yleinen |
| Väsymys | Hyvin yleinen | Yleinen |
| Infuusiokohdan reaktiot8 | Hyvin yleinen | Hyvin yleinen |
| Tutkimukset | ||
| Suurentunut alaniiniaminotransferaasipitoisuus, suurentunut bilirubiinipitoisuus | Yleinen | |
| Vammat, myrkytykset ja hoitokomplikaatiot | ||
| Rokotuskomplikaatio | Hyvin yleinen | |
1Tutkimukset: APL2-308, APL2-302, APL2-202, APL2-CP-PNH-204 ja PNH-potilaille tehty tutkimus APL‑CP0514 sekä tutkimukset APL2-C3G-310, APL2-C3G-314, APL2-201 ja APL2-C3G-204 C3G:tä ja primaarista IC‑MPGN:ää sairastavilla potilailla.
Lääketieteellisesti samankaltaiset termit on ryhmitelty tarvittaessa lääketieteellisen käsitteen samankaltaisuuden perusteella.
2Sisältää seuraavat: nenänielutulehdus, ylähengitystieinfektio, nielutulehdus, nuha ja sinuiitti.
3Sepsikseen sisältyy yksi septistä sokkia koskeva tapaus ja yksi kapselittoman Neisseria meningitidis -bakteerin aiheuttama tapaus.
4Herpeszoster ‑infektio (mukaan lukien Herpes zoster meningoencephalitis ‑infektio) ja Pneumocystis jirovecii ‑infektio.
5Sisältää seuraavat: hengitystieinfektio ja hengitysteiden virusinfektio.
6Sisältää seuraavat: ihottuma ja ekseema.
7Sisältää seuraavat: pienentynyt verihiutalemäärä.
8Infuusiokohdan reaktioihin sisältyvät suositellut termit (preferred terms): infuusiokohdan eryteema, infuusiokohdan kutina, infuusiokohdan turvotus, infuusiokohdan mustelma, infuusiokohdan kipu, infuusiokohdan kovettuma.
Kuvaus valikoiduista haittavaikutuksista
Infektiot
PNH-tutkimuksen APL2-302 aikana ei raportoitu kapselillisten bakteerien aiheuttamia vakavia infektioita. 48 potilasta sai tutkimuksen aikana jonkin infektion. PNH-tutkimuksen APL2-302 aikana pegsetakoplaanihoitoa saaneilla potilailla yleisimmin raportoituja infektioita olivat ylähengitystieinfektiot (28 tapausta, 35 %). PNH-tutkimuksen APL-302 aikana pegsetakoplaanihoitoa saaneilla potilailla raportoidut infektiot eivät useimmiten olleet vakavia, ja ne olivat vaikeusasteeltaan pääasiassa lieviä. Kymmenelle potilaalle kehittyi vakaviksi raportoituja infektioita, mukaan lukien yhdelle potilaalle kuolemaan johtanut koronavirustauti (COVID-19). Yleisimmät vakavat infektiot olivat sepsis (3 tapausta) (johti pegsetakoplaanihoidon lopettamiseen yhdellä potilaalla) ja gastroenteriitti (3 tapausta); kaikki nämä paranivat.
C3G:tä ja primaarista IC‑MPGN:ää koskevissa tutkimuksissa pegsetakoplaanihoitoa saaneilla potilailla raportoitiin neljä kapselillisten bakteerien aiheuttamaa vakavaa hengitystieinfektiota: kurkunkansitulehdus, pneumokokkipneumonia ja epätyypillinen keuhkokuume, jotka johtivat lääkityksen keskeyttämiseen, ja Haemophilus-keuhkokuume, joka ei vaatinut annoksen muuttamista. Tapahtumat paranivat ja korjaantuivat lukuun ottamatta Haemophilus-keuhkokuumetta ja epätyypillistä keuhkokuumetta, jotka paranivat, mutta joilla oli jälkiseurauksia. Lisäksi raportoitiin yksi vakava Escherichia-virtsatieinfektio. Tämä tapahtuma parani ja korjaantui ilman annoksen muuttamista.
Hemolyysi
PNH-tutkimuksen APL2-302 aikana 19:llä pegsetakoplaanihoitoa saaneella potilaalla raportoitiin hemolyysi. Tapauksista 7 raportoitiin vakavina, 5 tapausta johti pegsetakoplaanihoidon lopettamiseen, ja 10 potilaan pegsetakoplaaniannosta suurennettiin. PNH-tutkimuksessa APL2-308 pegsetakoplaanihoitoa saaneilla potilailla ilmeni kolme hemolyysitapausta. Yksikään raportoiduista tapauksista ei ollut vakava eikä johtanut pegsetakoplaanihoidon lopettamiseen. Kaikkien kolmen potilaan pegsetakoplaaniannosta suurennettiin.
Akuutti munuaisvaurio
C3G:tä ja primaarista IC‑MPGN:ää koskevissa kliinisissä tutkimuksissa raportoitiin 10 akuuttia munuaisvauriota koskevaa vakavaa tapahtumaa 8 potilaalla (5,7 %), jotka saivat pegsetakoplaania. Näistä tapahtumista 5 todettiin neljällä elinsiirteen saaneella potilaalla. Näistä vakavista tapahtumista vain yksi johti lääkityksen lopettamiseen ja yksi hoidon keskeyttämiseen. Yhtä lääkityksen lopettamiseen johtanutta tapahtumaa lukuun ottamatta kaikki tapahtumat paranivat ja korjaantuivat.
Potilaat, joilla on elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN
Tutkimuksiin APL2-C3G-310 ja APL2-C3G-204 osallistuneilla potilailla, joilla oli elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN (N = 22), turvallisuusprofiili vaikutti yhdenmukaiselta kokonaispopulaation kanssa, joskin vaikeita ja vakavia haittatapahtumia esiintyi enemmän, mikä on tämän potilaspopulaation kohdalla odotettavissa.
Pediatriset potilaat
Tutkimukseen APL2‑C3G-310 osallistuneilla nuorilla potilailla, joilla oli C3G tai primaarinen IC‑MPGN (N = 28, ikä 12–17 vuotta) turvallisuusprofiili vaikutti yhdenmukaiselta yleisten tulosten kanssa. Yleisimpiä tässä potilaspopulaatiossa raportoituja haittavaikutuksia olivat infuusiokohdan reaktiot. Pegsetakoplaanin turvallisuutta ei ole tutkittu alle 12 vuoden ikäisillä pediatrisilla potilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
Suomi/Finland
[Finnish]
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
[Swedish]
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Yliannostus
Valmisteen markkinoille tulon jälkeen on raportoitu yliannostustapauksia, mutta uusia turvallisuutta koskevia tapahtumia ei ole todettu. Yliannostustapauksessa on suositeltavaa seurata potilasta haittavaikutusten merkkien tai oireiden varalta ja antaa potilaalle oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, komplementin estäjät, ATC-koodi: L04AJ03
Vaikutusmekanismi
Pegsetakoplaani on symmetrinen molekyyli, joka koostuu kahdesta identtisestä pentadekapeptidistä, jotka ovat kovalenttisesti sitoutuneita lineaarisen 40 kDa:n PEG-molekyylin päihin. Peptidin puolikkaat sitoutuvat komplementteihin C3 ja C3b ja luovat laajan komplementtikaskadin inhibition. 40 kDa:n PEG-puolikkaat mahdollistavat paremman liukoisuuden ja pidemmän pysymisen elimistössä lääkevalmisteen antamisen jälkeen.
Pegsetakoplaani sitoutuu komplementtiproteiiniin C3 ja sen aktivointifragmenttiin C3b suurella affiniteetilla, mikä säätelee C3:n pilkkoutumista ja myöhempien komplementtiaktivaation efektoreiden muodostumista. PNH:ssa ekstravaskulaarista hemolyysiä (EVH) välittää C3b-opsonisaatio, kun taas intravaskulaarista hemolyysiä (IVH) välittää myöhempi membraani-hyökkäyskompleksi (MAC). Pegsetakoplaani säätelee komplementtikaskadia laajasti vaikuttamalla proksimaalisesti sekä C3b:n että MAC:n muodostumiseen, mikä kontrolloi EVH:hon ja IVH:hon johtavia mekanismeja.
C3G:ssä ja primaarisessa IC‑MPGN:ssä esiintyy C3-hajoamistuotteiden liiallista kertymistä munuaisen hiussuonikeräsiin, mikä johtaa munuaisten parenkyymivaurioihin ja munuaistoiminnan heikkenemiseen. Pegsetakoplaanin vaikutus kohdistuu komplementtiaktivaation ylävirran efektoreihin (C3 ja C3b), joten se estää kaikkien komplementtireittien (vaihtoehtoinen, klassinen ja lektiini) kautta käynnistyvää aktivaatiota. Pegsetakoplaani vaikuttaa C3:a estämällä suoraan C3:n epäasianmukaiseen aktivaatioon ja muuntaa perussairautta vähentämällä C3-hajoamistuotteiden liiallista kertymistä munuaisen hiussuonikeräsiin. Pegsetakoplaani kohdentuu myös C3b:hen ja siten se estää myös vaihtoehtoisen reitin C3-konvertaasin aktiivisuutta komplementtikaskadiin liittyvän lisävaikutusmekanismin kautta. Tämä ehkäisee entisestään C3-hajoamistuotteiden kertymistä hiussuonikeräsiin.
Farmakodynaamiset vaikutukset
PNH
Tutkimuksessa APL2‑302 C3-keskipitoisuus seerumissa suureni pegsetakoplaaniryhmässä lähtötilanteen arvosta 0,94 g/l arvoon 3,83 g/l viikkoon 16 mennessä ja pysyi samalla tasolla viikon 48 loppuun asti. Tutkimuksessa APL2-308 C3‑keskipitoisuus seerumissa suureni lähtötilanteen arvosta 0,95 g/l arvoon 3,56 g/l viikkoon 26 mennessä.
Tutkimuksessa APL2-302 PNH-tyypin II + III punasolujen keskimääräinen prosenttiosuus suureni lähtötilanteen arvosta 66,80 % arvoon 93,85 % viikolla 16 ja pysyi samalla tasolla viikon 48 loppuun asti. Tutkimuksessa APL2-308 PNH-tyypin II + III punasolujen keskimääräinen prosenttiosuus suureni lähtötilanteen arvosta 42,4 % arvoon 90,0 % viikolla 26.
Tutkimuksessa APL2-302 niiden PNH-tyypin II + III punasolujen, joihin oli kiinnittynyt C3, keskimääräinen prosenttiosuus pieneni lähtötilanteen arvosta 17,73 % arvoon 0,20 % viikolla 16 ja pysyi samalla tasolla viikon 48 loppuun asti. Tutkimuksessa APL2-308 niiden PNH-tyypin II + III punasolujen, joihin oli kiinnittynyt C3, keskimääräinen prosenttiosuus pieneni lähtötilanteen arvosta 2,85 % arvoon 0,09 % viikolla 26.
C3G ja primaarinen IC‑MPGN
Tutkimuksessa APL2‑C3G-310 C3-keskipitoisuus seerumissa suureni pegsetakoplaaniryhmässä lähtötilanteen arvosta 0,62 g/l arvoon 3,71 g/l viikkoon 26 mennessä ja tämä vaikutus pysyi samalla tasolla viikkoon 52 asti. Lumeryhmässä C3-pitoisuudet pysyivät vakaina viikkoon 26 asti (0,57 g/l lähtötilanteessa; 0,58 g/l viikolla 26) ja pegsetakoplaaniin siirryttäessä suurenivat tasolle 3,59 g/l viikolla 52.
sC5b-9-keskipitoisuus seerumissa pieneni pegsetakoplaaniryhmässä lähtötilanteen arvosta 902,5 ng/ml arvoon 290,2 ng/ml viikkoon 26 mennessä ja vaikutus pysyi samalla tasolla viikkoon 52 asti. Lumeryhmässä sC5b-9-pitoisuudet pysyivät vakaina (768,3 ng/ml lähtötilanteessa; 759,9 ng/ml viikolla 26) ja pegsetakoplaaniin siirryttäessä pienenivät tasolle 272,9 ng/ml viikolla 52.
Hiussuonikerästen C3-kertymien häviämistä 6 kuukauden kohdalla todettiin sen perusteella, että suurempi osuus potilaista pegsetakoplaaniryhmässä (71,4 %) kuin lumeryhmässä (8,8 %) saavutti värjäyksessä pistearvon 0.
Kliininen teho ja turvallisuus
PNH
Pegsetakoplaanin tehoa ja turvallisuutta PNH-potilailla arvioitiin kahdessa avoimessa, satunnaistetussa, kontrolloidussa vaiheen 3 tutkimuksessa: tutkimus APL2-302 potilailla, jotka olivat saaneet aiemmin komplementin estäjiä, ja tutkimus APL2-308 potilailla, jotka eivät olleet saaneet aiemmin komplementin estäjiä. Molemmissa tutkimuksissa pegsetakoplaaniannos oli 1 080 mg kaksi kertaa viikossa. Annos voitiin tarvittaessa muuttaa 1 080 mg:aan joka 3. päivä.
Tutkimus aikuispotilailla, jotka olivat saaneet aiemmin komplementin estäjiä (APL2-302)
Tutkimus APL2-302 oli avoin, satunnaistettu tutkimus, jossa aktiivisella vertailuvalmisteella kontrolloitua 16 viikon jaksoa seurasi 32 viikkoa kestänyt avoin jakso. Tähän tutkimukseen otettiin PNH-potilaita, joita oli hoidettu vakaalla annoksella ekulitsumabia ainakin edelliset 3 kuukautta ja joiden Hb-arvo oli < 10,5 g/dl. Sopivat potilaat siirtyivät neljän viikon aloitusjaksoon, jonka aikana he saivat pegsetakoplaania 1 080 mg ihon alle kaksi kertaa viikossa senhetkisen ekulitsumabiannoksensa lisäksi. Sen jälkeen potilaat satunnaistettiin suhteessa 1:1 saamaan joko 1 080 mg pegsetakoplaania kaksi kertaa viikossa tai senhetkisen ekulitsumabiannoksensa koko 16 viikon satunnaistetun kontrolloidun jakson ajan. Satunnaistus ositettiin päivää ‑28 edeltäneiden 12 kuukauden aikana annetun pakattujen punasolujen siirtomäärän (< 4; > 4) sekä seulontahetken verihiutalemäärän (< 100 000/mm3; ≥ 100 000/mm3) mukaan. Satunnaistetun kontrolloidun jakson loppuun asti mukana olleet potilaat jatkoivat tutkimuksen avoimeen jaksoon, jossa kaikki potilaat saivat pegsetakoplaania enintään 32 viikon ajan (satunnaistetussa kontrolloidussa jaksossa ekulitsumabia saaneet potilaat osallistuivat 4 viikon pituiseen aloitusjaksoon ennen siirtymistä pegsetakoplaanimonoterapiaan).
Ensisijaiset ja toissijaiset tehon päätetapahtumat arvioitiin viikolla 16. Tehon ensisijainen päätetapahtuma oli Hb-tason muutos lähtötilanteesta viikolle 16 (satunnaistetun kontrolloidun jakson aikana). Lähtötilanteeksi määritettiin mittausten keskiarvo ennen ensimmäistä pegsetakoplaaniannosta (aloitusjakson alussa). Tärkeimmät toissijaiset tehon päätetapahtumat olivat verensiirron tarpeettomuus määritettynä niiden potilaiden osuutena, jotka eivät tarvinneet verensiirtoa satunnaistetun kontrolloidun jakson aikana, sekä absoluuttisen retikulosyyttimäärän (ARC), LDH-tason ja kroonisen sairauden hoitoon liittyvän toimintakykyarvioinnin (functional assessment of chronic illness therapy, FACIT) väsymysasteikon pisteiden muutos lähtötilanteesta viikolle 16.
Yhteensä 80 potilasta siirtyi aloitusjaksolle. Aloitusjakson lopussa kaikki 80 satunnaistettiin, 41 saamaan pegsetakoplaania ja 39 ekulitsumabia. Väestötiedot ja lähtötilanteen sairausominaisuudet olivat yleisesti ottaen hyvin tasapainossa hoitoryhmien välillä (ks. taulukko 2). Yhteensä 38 potilasta pegsetakoplaanihoitoa saaneesta ryhmästä ja 39 potilasta ekulitsumabiryhmästä oli mukana 16 viikon satunnaistetun kontrolloidun jakson loppuun ja jatkoi 32 viikon avoimeen jaksoon. Yhteensä 12 potilasta 80:stä (15 %) pegsetakoplaanihoitoa saaneesta lopetti hoidon haittatapahtumien takia. Tutkimussuunnitelman mukaisesti 15 potilaan annos muutettiin 1 080 mg:aan joka 3. päivä. Hyötyä arvioitiin 12 potilaalla, joista 8 potilaan osoitettiin hyötyneen annoksen muuttamisesta.
Taulukko 2: Potilaan lähtötilanteen väestötiedot ja ominaisuudet tutkimuksessa APL2-302
| Parametri | Tilastot | Pegsetakoplaani (N = 41) | Ekulitsumabi (N = 39) |
Ikä (vuotta) 18–64 vuotta ≥ 65 vuotta | Keskiarvo (SD) n (%) n (%) | 50,2 (16,3) 31 (75,6) 10 (24,4) | 47,3 (15,8) 32 (82,1) 7 (17,9) |
Ekulitsumabin annostaso lähtötilanteessa Joka 2. viikko IV 900 mg Joka 11. päivä IV 900 mg Joka 2. viikko IV 1 200 mg Joka 2. viikko IV 1 500 mg | n (%) n (%) n (%) n (%) | 26 (63,4) 1 (2,4) 12 (29,3) 2 (4,9) | 29 (74,4) 1 (2,6) 9 (23,1) 0 |
| Nainen | n (%) | 27 (65,9) | 22 (56,4) |
| Aika PNH-diagnoosista (vuotta) päivään ‑28 | Keskiarvo (SD) | 8,7 (7,4) | 11,4 (9,7) |
| Hemoglobiinitaso (g/dl) | Keskiarvo (SD) | 8,7 (1,1) | 8,7 (0,9) |
| Retikulosyyttimäärä (109/l) | Keskiarvo (SD) | 218 (75,0) | 216 (69,1) |
| LDH-taso (U/l) | Keskiarvo (SD) | 257,5 (97,6) | 308,6 (284,8) |
| Kokonais-FACIT‑väsymys* | Keskiarvo (SD) | 32,2 (11,4) | 31,6 (12,5) |
| Verensiirtojen määrä edellisten 12 kuukauden aikana ennen päivää ‑28 | Keskiarvo (SD) | 6,1 (7,3) | 6,9 (7,7) |
| < 4 | n (%) | 20 (48,8) | 16 (41,0) |
| ≥ 4 | n (%) | 21 (51,2) | 23 (59,0) |
| Verihiutalemäärä seulontahetkellä (109/l) | Keskiarvo (SD) | 167 (98,3) | 147 (68,8) |
| Verihiutalemäärä seulontahetkellä < 100 000/mm3 | n (%) | 12 (29,3) | 9 (23,1) |
| Verihiutalemäärä seulontahetkellä ≥ 100 000/mm3 | n (%) | 29 (70,7) | 30 (76,9) |
| Aiempi aplastinen anemia | n (%) | 11 (26,8) | 9 (23,1) |
| Aiempi myelodysplastinen oireyhtymä | n (%) | 1 (2,4) | 2 (5,1) |
*FACIT‑väsymys mitataan asteikolla 0–52, jossa korkeammat arvot osoittavat vähemmän väsymystä.
Pegsetakoplaani oli ekulitsumabia (P < 0,0001) parempi ensisijaisessa päätepisteessä hemoglobiinin muutos lähtötilanteesta.
Kuva 1. Säädetty keskimääräinen hemoglobiinin muutos (g/dl) tutkimuksen APL2-302 lähtötilanteesta viikolle 16
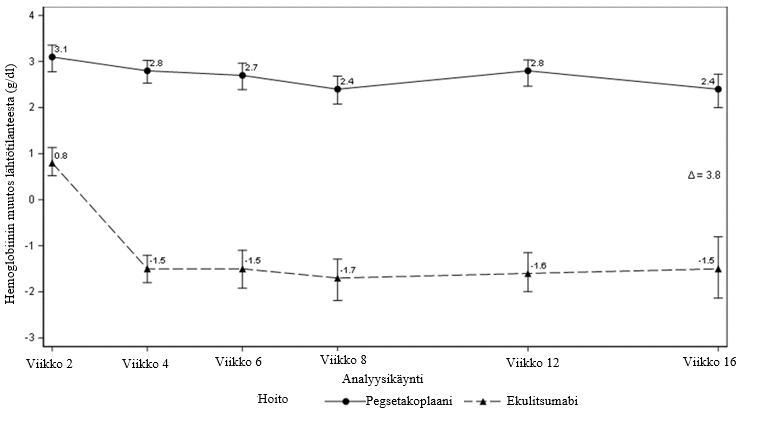
Vertailukelpoisuus (non-inferiority) osoitettiin tärkeimmissä toissijaisissa päätetapahtumissa, verensiirtojen tarpeettomuudessa ja ARC-arvon muutoksessa lähtötilanteesta.
Vertailukelpoisuus (non-inferiority) ei saavutettu LDH-arvon muutoksessa lähtötilanteesta.
Hierarkkisen testauksen vuoksi FACIT‑väsymyspisteiden muutokselle lähtötilanteesta ei tehty muodollista tilastollista testausta.
Tarkempien toissijaisten päätetapahtumien säädetyt keskiarvot, hoitoero, luottamusvälit ja tilastolliset analyysit on esitetty kuvassa 2.
Kuva 2. Tutkimuksen APL2-302 tärkeimpien toissijaisten päätepisteiden analyysi
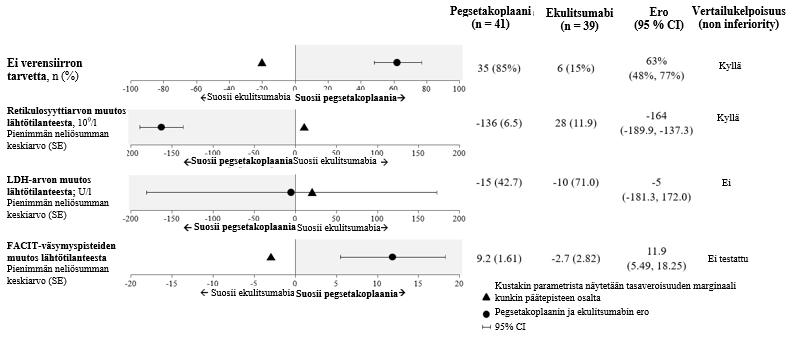
Tulokset olivat yhdenmukaisia kaikissa ensisijaisten ja tärkeimpien toissijaisten vastemuuttujien tukianalyyseissä, mukaan lukien kaikki havaitut tiedot ja verensiirron jälkeiset tiedot.
Hb-tason normalisoituminen saavutettiin 34 %:lla potilaista pegsetakoplaaniryhmässä verrattuna 0 %:iin ekulitsumabiryhmässä viikolla 16. LDH-tason normalisoituminen saavutettiin 71 %:lla potilaista pegsetakoplaaniryhmässä verrattuna 15 %:iin ekulitsumabiryhmässä.
Yhteensä 77 potilasta jatkoi 32 viikon pituiseen avoimeen jaksoon, jossa kaikki potilaat saivat pegsetakoplaania, jolloin kokonaisaltistuksen kesto oli enimmillään 48 viikkoa. Viikolla 48 tulokset olivat yleisesti ottaen yhdenmukaisia viikon 16 tulosten kanssa ja tukivat tehon säilymistä.
Tutkimus aikuispotilailla, jotka eivät olleet aiemmin saaneet komplementin estäjiä (APL2-308)
Tutkimus APL2-308 oli avoin, satunnaistettu, kontrolloitu tutkimus. Siihen otettiin mukaan PNH-potilaita, jotka eivät olleet saaneet komplementin estäjiä tutkimukseen mukaan tuloa edeltäneiden 3 kuukauden aikana ja joiden Hb-arvo oli alle viitevälin alarajan (lower limit of normal, LLN). Sopivat potilaat satunnaistettiin suhteessa 2:1 saamaan joko pegsetakoplaanihoitoa tai tukihoitoa (esim. verensiirrot, kortikosteroidit, ravintolisät, kuten rauta, folaatti ja B12‑vitamiini) 26 viikkoa kestäneen hoitojakson ajan. Tukihoitoa saanutta ryhmää kutsutaan jäljempänä vertailuryhmäksi.
Satunnaistus ositettiin päivää -28 edeltäneiden 12 kuukauden aikana annettujen punasolutiivistesiirtojen lukumäärän (< 4; ≥ 4) mukaan. Jos vertailuryhmään kuuluvan potilaan Hb-arvo laski ≥ 2 g/dl lähtöarvosta tai hänellä ilmeni kohtauksittaiseen yölliseen hemoglobiinivirtsaisuuteen liittyvä tromboembolinen tapahtuma milloin tahansa tutkimuksen aikana, hänet voitiin tutkimussuunnitelman mukaan siirtää tutkimuksen loppuajaksi pegsetakoplaanihoitoon.
Yhteensä 53 potilasta satunnaistettiin: 35 potilasta pegsetakoplaaniryhmään ja 18 potilasta vertailuryhmään. Demografiset tiedot ja sairauden ominaisuudet lähtötilanteessa olivat yleisesti ottaen hyvin tasapainossa hoitoryhmien kesken. Tutkittavien keskimääräinen ikä oli pegsetakoplaaniryhmässä 42,2 vuotta ja vertailuryhmässä 49,1 vuotta. Punasolutiivistesiirtojen määrä seulontaa edeltäneiden 12 kuukauden aikana oli pegsetakoplaaniryhmässä keskimäärin 3,9 ja vertailuryhmässä keskimäärin 5,1. Viidellä potilaalla kummassakin ryhmässä (14,3 % pegsetakoplaaniryhmässä ja 27,8 % vertailuryhmässä) oli ollut aiemmin aplastinen anemia. Muut lähtötilanteen arvot olivat seuraavat: keskimääräinen Hb-taso lähtötilanteessa (pegsetakoplaaniryhmä: 9,4 g/dl vs. vertailuryhmä: 8,7 g/dl), ARC-arvo (pegsetakoplaaniryhmä: 230,2 × 109/l vs. vertailuryhmä: 180,3 × 109/l), LDH-taso (pegsetakoplaaniryhmä: 2 151,0 U/l vs. vertailuryhmä: 1 945,9 U/l) ja verihiutalemäärä (pegsetakoplaaniryhmä: 191,4 × 109/l vs. vertailuryhmä: 125,5 × 109/l). Yksitoista 18:sta vertailuryhmään satunnaistetusta potilaasta siirtyi pegsetakoplaanihoitoon, koska heidän Hb-arvonsa laski ≥ 2 g/dl lähtöarvosta. 52 satunnaistettua potilasta 53:sta (97,8 %) sai ennaltaehkäisevää antibioottihoitoa paikallisten hoito-ohjeistojen mukaisesti.
Ensisijaiset ja toissijaiset tehon päätetapahtumat arvioitiin viikolla 26. Kaksi yhdistettyä ensisijaista tehon päätetapahtumaa olivat Hb-tason vakiintuminen, joka määriteltiin tilanteeksi, jossa Hb-taso ei laskenut > 1 g/dl lähtötilanteesta eikä verensiirtoja annettu, sekä LDH-tason muutos lähtötilanteesta.
Pegsetakoplaanihoitoa saaneessa ryhmässä 30 potilasta 35:stä (85,7 %) ja vertailuryhmässä 0 potilasta saavutti vakaan Hb-tason. Pegsetakoplaaniryhmän ja vertailuryhmän korjattu ero oli 73,1 % (95 %:n luottamusväli 57,2–89,0 %; p < 0,0001).
Pienimmän neliösumman keskiarvon (keskivirhe) muutos lähtötilanteesta viikolla 26 oli LDH-tason osalta -1 870 U/l pegsetakoplaaniryhmässä ja -400 U/l vertailuryhmässä (p < 0,0001). Pegsetakoplaaniryhmän ja vertailuryhmän välinen ero oli -1 470 (95 %:n luottamusväli ‑2 113 – ‑827). Pegsetakoplaaniryhmän ja vertailuryhmän hoitoerot olivat nähtävissä viikolla 2 ja säilyivät viikon 26 loppuun asti (kuva 3). LDH-tasot pysyivät vertailuryhmässä koholla.
Kuva 3. Keskimääräinen (± keskivirhe) LDH-taso (U/l) ajan kuluessa tutkimuksessa APL2‑308 hoitoryhmittäin
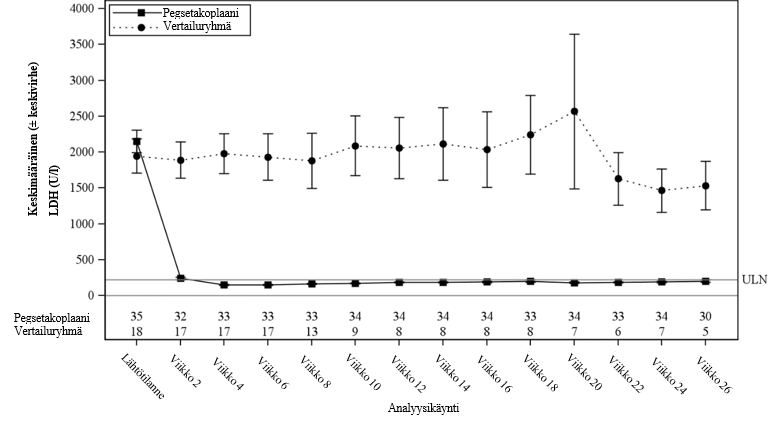
Valikoitujen tärkeimpien toissijaisten tehon päätetapahtumien (hemoglobiinivaste ilman verensiirtoja, hemoglobiinitason muutos ja ARC-arvon muutos) osalta pegsetakoplaanihoitoa saaneessa ryhmässä todettiin merkittävä hoitoero vertailuryhmään nähden (taulukko 3).
Taulukko 3: Tutkimuksen APL2‑308 tärkeimpien toissijaisten päätetapahtumien analyysi
| Parametri | Pegsetakoplaani (N = 35) | Vertailuryhmä (N = 18) | Ero (95 % CI) p-arvo |
Hemoglobiinivaste ilman verensiirtojaa n (%) | 25 (71 %) | 1 (6 %) | 54 % (34 %; 74 %) p < 0,0001 |
Hemoglobiinitason muutos lähtötilanteesta viikkoon 26 (g/dl) Pienimmän neliösumman keskiarvo (SE) | 2,9 (0,38) | 0,3 (0,76) | 2,7 (1,0; 4,4) 9 |
ARC-arvon muutos lähtötilanteesta viikkoon 26 (109/l) Pienimmän neliösumman keskiarvo (SE) | -123 (9,2) | -19 (25,2) | ‑104 (‑159; ‑49) |
a Hemoglobiinivaste määriteltiin hemoglobiiniarvon nousuksi ≥ 1 g/dl lähtötilanteesta viikkoon 26. ARC = absoluuttinen retikulosyyttimäärä, CI = luottamusväli, SE = keskivirhe
C3G ja primaarinen IC‑MPGN
Pegsetakoplaanin tehoa ja turvallisuutta C3G:tä tai primaarista IC‑MPGN:ää sairastavilla potilailla arvioitiin satunnaistetussa, lumekontrolloidussa, kaksoissokkoutetussa vaiheen 3 tutkimuksessa APL2-C3G-310, johon otettiin mukaan aikuisia ja nuoria, joilla oli oman munuaisen C3G tai primaarinen IC‑MPGN tai elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN.
Pegsetakoplaaniannos oli ≥ 50 kg:n painoisille aikuisille tai nuorille 1 080 mg kaksi kertaa viikossa tai < 50 kg:n painoisille nuorille painoon perustuva.
Tutkimus aikuisilla ja nuorilla potilailla, joilla oli C3G tai primaarinen IC‑MPGN (APL2-C3G-310)
Tutkimus APL2-C3G-310 oli satunnaistettu, kaksoissokkoutettu tutkimus, jossa oli 26 viikon pituinen lumekontrolloitu jakso ja sen jälkeen 26 viikon pituinen avoin jakso. Tähän tutkimukseen otettiin mukaan 12–17 vuoden ikäisiä nuoria sekä aikuisia, joilla oli C3G tai primaarinen IC‑MPGN. Tähän tutkimukseen otettiin mukaan potilaita, joilla sairaus oli joko omassa munuaisessa tai se oli uusiutunut elinsiirron jälkeen ja joilla oli proteinuria ≥ 1 g/vrk ja eGFR ≥ 30 ml/min/1,73 m2. Potilaat saivat vakaata ja optimoitua lääkitystä C3G:n / primaarisen IC‑MPGN:n hoitoon (esim. RAS:n estäjiä, natriumin- ja glukoosinkuljettajaproteiini 2:n [SGLT‑2] estäjiä, immunosuppressantteja, systeemisiä kortikosteroideja enintään annoksena, joka vastasi 20 mg:aa/vrk prednisonia) vähintään 12 viikon ajan ennen satunnaistamista.
Sopivat potilaat satunnaistettiin suhteessa 1:1 saamaan joko pegsetakoplaania tai lumelääkettä ihon alle kaksi kertaa viikossa 26 viikon pituisen satunnaistetun kontrolloidun jakson ajan. Satunnaistamiseen sovellettiin kahta ositustekijää: potilaat, joiden sairaus oli uusiutunut elinsiirron jälkeen vs. potilaat, joilla sairaus oli omassa munuaisessa, sekä potilaat, joille oli tehty munuaisbiopsia lähtötilanteessa (joko seulonnassa tai enintään 28 viikkoa ennen satunnaistamista) vs. potilaat, joille ei ollut tehty munuaisbiopsiaa lähtötilanteessa. Satunnaistetun kontrolloidun jakson aikana C3G:n / primaarisen IC‑MPGN:n lähtötilanteen hoito-ohjelmiin tehtiin mahdollisimman vähän muutoksia ja ainoastaan, jos se oli potilaan hyvinvoinnin kannalta välttämätöntä. Satunnaistetun kontrolloidun jakson loppuun saakka mukana olleet potilaat siirtyivät 26 viikon avoimeen jaksoon, jossa kaikki potilaat saivat pegsetakoplaanihoitoa kaksi kertaa viikossa.
Yhteensä 124 potilasta satunnaistettiin, 63 saamaan pegsetakoplaania ja 61 lumelääkettä. Väestötiedot ja lähtötilanteen sairausominaisuudet olivat yleisesti ottaen tasapainossa näiden kahden hoitoryhmän välillä (ks. taulukko 4). Yhteensä 118 potilasta oli mukana 26 viikon satunnaistetun kontrolloidun jakson loppuun, ja näistä 114 potilasta oli mukana avoimen pegsetakoplaanihoitojakson loppuun (N = 59 pegsetakoplaani-pegsetakoplaani; N = 55 lumelääke-pegsetakoplaani).
Taulukko 4: Potilaan lähtötilanteen väestötiedot ja sairausominaisuudet tutkimuksessa APL2‑C3G‑310
| Parametri | Tilastot | Pegsetakoplaani (N = 63) | Lumelääke (N = 61) |
| Ikä (vuotta) | Keskiarvo (SD) | 28,2 (17,1) | 23,6 (14,3) |
| Nuoret (12–17 vuotta) | n (%) | 28 (44,4) | 27 (44,3) |
| Aikuiset ≥ 18 vuotta | n (%) | 35 (55,6) | 34 (55,7) |
Sukupuoli Mies Nainen | n (%) n (%) | 26 (41,3) 37 (58,7) | 28 (45,9) 33 (54,1) |
| Sairauden tyyppi seulonnassa | |||
| C3G | n (%) | 51 (81,0) | 45 (73,8) |
| C3GN | n (%) | 45 (71,4) | 41 (67,2) |
| DDD | n (%) | 4 (6,3) | 4 (6,6) |
| Määrittämätön | n (%) | 2 (3,2) | 0 |
| IC‑MPGN | n (%) | 12 (19,0) | 16 (26,2) |
| Aika C3G-/IC‑MPGN-diagnoosista (vuotta) | Keskiarvo (SD) | 3,64 (3,47) | 3,76 (3,62) |
| Aiempi munuaisensiirto | n (%) | 5 (7,9) | 4 (6,6) |
| Aika viimeisimmästä munuaisensiirrosta (vuotta) | Keskiarvo (SD) | 11,4 (6,7) | 5,8 (6,4) |
| Aika viimeisimmästä elinsiirron jälkeisestä uusiutumisesta (vuotta) | Keskiarvo (SD) | 1,47 (1,49) | 1,38 (1,64) |
| Ensimmäisestä aamuvirtsasta kolme kertaa mitattu uPCR (mg/g) lähtötilanteessa | Keskiarvo (SD) | 3 124 (2 408) | 2 541 (2 015) |
| Lähtötilanteen eGFR (ml/min/1,73 m2) | Keskiarvo (SD) | 78,5 (34,1) | 87,2 (37,2) |
| C3c-värjäys lähtötilanteen biopsiassa | |||
| 3+ | n (%) | 51 (81,0) | 51 (83,6) |
| 2+ | n (%) | 12 (19,0) | 10 (16,4) |
| Seerumin albumiini lähtötilanteessa (g/dl) | Keskiarvo (SD) | 3,31 (0,61) | 3,39 (0,70) |
| Seerumin C3 lähtötilanteessa (mg/dl) | Keskiarvo (SD) | 60,6 (45,7) | 56,3 (35,6) |
| Taudin ilmentymät | |||
| Turvotus | n (%) | 45 (71,4) | 32 (52,5) |
| Väsymys | n (%) | 16 (25,4) | 8 (13,1) |
| Hematuria | n (%) | 37 (58,7) | 39 (63,9) |
| Korkea verenpaine | n (%) | 35 (55,6) | 29 (47,5) |
| Nefroottinen oireyhtymä | n (%) | 32 (50,8) | 27 (44,3) |
| Muiden hoitojen käyttö lähtötilanteessa* | |||
| Reniini-angiotensiinijärjestelmään vaikuttavat aineet | n (%) | 59 (93,7) | 54 (88,5) |
| Immunosuppressantit | n (%) | 49 (77,8) | 45 (73,8) |
| Glukokortikoidit | n (%) | 29 (46,0) | 27 (44,3) |
*Enintään 12 viikkoa ennen tutkimuksessa aloittamista.
C3G = C3-glomerulopatia, C3GN = C3-glomerulonefriitti, DDD = tiiviskertymätauti (dense-deposit disease), IC‑MPGN = immunokompleksi membranoproliferatiivinen munuaiskerästulehdus, uPCR = virtsan proteiini-kreatiniinisuhde, eGFR = glomerulusten laskennallinen suodatusnopeus, SD = keskihajonta (standard deviation)
Ensisijaiset ja tärkeimmät toissijaiset tehon päätetapahtumat arvioitiin viikolla 26. Ensisijainen tehon päätetapahtuma oli ensimmäisen aamuvirtsan uPCR:n log-muunnettu suhde viikolla 26 lähtötilanteeseen verrattuna.
Pegsetakoplaani oli lumelääkettä parempi, sillä uPCR oli pienentynyt (95 %:n luottamusväli: 57,3–76,2 %, p < 0,0001) lähtötilanteesta tilastollisesti merkitsevästi (68,1 %) verrattuna lumelääkkeeseen hoidon jatkuttua 26 viikkoa (pegsetakoplaani ‑67,2 % [95 %:n luottamusväli: -74,9 – -57,2 %] ja lumelääke +2,9 % [95 %:n luottamusväli: -8,6–15,9 %]). Alaryhmissä todettu teho oli samansuuruista iästä (nuoret vs. aikuiset), sairauden tyypistä (C3G vs. primaarinen IC‑MPGN), sairauden statuksesta (sairaus omassa munuaisessa vs. elinsiirron jälkeen uusiutunut tauti) ja immunosuppressanttien/glukokortikoidien samanaikaisesta käytöstä (kyllä vs. ei) riippumatta. Pegsetakoplaanin vaikutus uPCR-arvoon säilyi viikolle 52 (‑67,2 % lähtötilanteesta). Potilailla, jotka siirtyivät lumelääkkeestä pegsetakoplaaniin viikolla 26 (kuva 4), todettiin samankaltainen vähenemä (‑51,3 %) viikolla 52.
Kuva 4. Tutkimuksen APL2-C3G-310 MMRM-mallin mukainen ensimmäisen aamuvirtsan uPCR:n geometristen keskiarvojen suhde (95 %:n luottamusväli) ajan kuluessa verrattuna lähtötilanteeseen hoitoryhmittäin
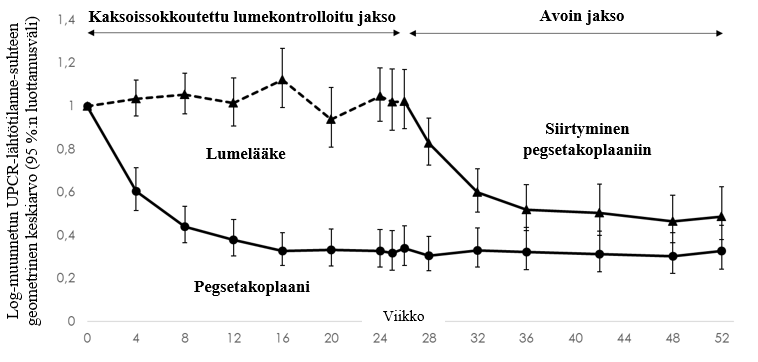
Huomautus: Geometristen keskiarvojen suhde on laskettu potenssiin uudelleen korotetuista LS-keskiarvoista.
LS = pienin neliösumma, uPCR = virtsan proteiini-kreatiniinisuhde, MMRM = toistettujen mittausten sekamalli
26 viikon pegsetakoplaanihoidon osoitettiin parantaneen proteinurian vähenemiseen liittyvää tärkeää toissijaista päätetapahtumaa tilastollisesti merkitsevästi, sillä 60,3 %:lla pegsetakoplaanihoitoa saaneista potilaista uPCR:n todettiin pienentyneen ≥ 50 %, kun vastaava luku lumeryhmässä oli 4,9 %, eli ero oli 52,7 % (95 %:n luottamusväli: 29,2–76,2 %; p < 0,0001).
26 viikon pegsetakoplaanihoidon jälkeen munuaisten C3-värjäytymisvoimakkuudessa todettiin vähintään kahden suuruusluokan vähenemä (asteikolla 0–3) suuremmalla osalla potilaista: 26 (74,3 %) potilaalla pegsetakoplaaniryhmässä vs. 4 (11,8 %) potilaalla lumeryhmässä eli ero oli 64,3 % (95 %:n luottamusväli: 41,4–87,2 %, nimellinen p < 0,0001).
26 viikon pegsetakoplaanihoidon osoitettiin vakauttaneen eGFR-arvoa, sillä muutos lähtötilanteesta oli pegsetakoplaanilla ‑1,497 (2,242) vs. lumelääkkeellä ‑7,808 (1,919) ja hoitojen ero oli 6,312 ml/min/1,73 m2 (95 %:n luottamusväli: 0,501–12,122, nimellinen p = 0,0333). Pegsetakoplaanin vaikutus eGFR:ään säilyi viikon 52 loppuun asti. Potilailla, jotka siirtyivät lumelääkkeestä pegsetakoplaaniin viikolla 26, todettiin samankaltaista vakautumista viikolla 52.
Saman suuruusluokan tehoa proteinurian vähenemisessä ≥ 50 %, C3-värjäyksen puhdistumassa ja eGFR:n vakautumisessa havaittiin viikolla 26 laajalti alaryhmissä iästä (nuoret vs. aikuiset), sairauden tyypistä (C3G vs. primaarinen IC‑MPGN), sairauden statuksesta (sairaus omassa munuaisessa vs. elinsiirron jälkeen uusiutunut tauti) ja immunosuppressanttien/glukokortikoidien samanaikaisesta käytöstä (kyllä vs. ei) riippumatta.
Tutkimus aikuisilla potilailla, joilla oli elinsiirron jälkeen uusiutunut C3G tai primaarinen IC‑MPGN (APL2-C3G-204)
Tutkimus APL2-C3G-204 oli vaiheen 2 avoin, satunnaistettu, 52 viikon pituinen tutkimus 13 aikuispotilaalla, joilla oli elinsiirron jälkeen uusiutunut C3G (N = 10) tai primaarinen IC‑MPGN (N = 3).
Tutkimuksen ensimmäisten 12 viikon aikana 10 potilasta sai pegsetakoplaania tavanomaisen hoidon lisäksi ja 3 potilasta sai pelkkää tavanomaista hoitoa. Kaikki potilaat saivat pegsetakoplaania viikosta 13 viikkoon 52.
Ensisijainen päätetapahtuma oli munuaisbiopsian C3-värjäytymisvoimakkuuden vähenemä viikolla 12, ja se todettiin 50 %:lla pegsetakoplaanihoitoa saaneista potilaista (5 potilasta 10:stä; heistä 4 sai värjäyksessä pistearvon 0) ja 33,3 %:lla verrokkiryhmän potilaista (1 potilas 3:sta; tämä potilas sai värjäyksessä pistearvon 1).
Yleisesti ottaen eGFR:n muutokset ja prosentuaaliset muutokset lähtötilanteesta (toissijainen päätetapahtuma) olivat pieniä. eGFR:n keskiarvo (keskihajonta) muuttui lähtöarvosta 52,3 (12,11) ml/min/1,73 m2 arvoon 57,3 (25,12) ml/min/1,73 m2 viikolla 52, ja eGFR:n mediaani muuttui lähtöarvosta 50,5 ml/min/1,73 m2 arvoon 58,5 ml/min/1,73 m2 viikolla 52. Kaikissa ryhmissä eGFR vakautui tai parani useimmilla potilailla (9 potilasta 13:sta [69,2 %]) viikkoon 52 mennessä.
Immunogeenisuus
Pegsetakoplaanipeptidilääkevasta-aineiden (anti-drug antibody, ADA) toteamiseen käytettiin PNH:ta koskeneissa ja C3G:tä tai primaarista IC‑MPGN:ää koskeneissa kliinisissä tutkimuksissa kahta eri määritystä. C3G:n tai primaarisen IC‑MPGN:n yhteydessä käytetty määritys oli herkempi. Määritysten erojen vuoksi lääkevasta-aineiden esiintyvyyden vertailu jäljempänä kuvatuissa tutkimuksissa ei ole mielekästä.
PNH:tä koskeneissa kliinisissä tutkimuksissa lääkevasta-aineiden esiintyvyys (hoidon aikana ilmaantunut ADA tai tehostettu ADA aiemmasta tasosta) oli vähäistä, ja kun sitä ilmeni, sillä ei ollut huomattavaa vaikutusta pegsetakoplaanin farmakokinetiikkaan/farmakodynamiikkaan, tehoon tai turvallisuusprofiiliin. Tutkimusten APL2‑302 ja APL2-308 kuluessa kolme pegsetakoplaania saanutta potilasta 126:sta varmistui pegsetakoplaanipeptidivasta-ainepositiivisiksi. Kaikilla kolmella potilaalla vasta-aineet olivat neutraloivia (NAb). NAb-vasteella ei ollut ilmeistä vaikutusta farmakokinetiikkaan tai kliiniseen tehoon. Kahdeksalletoista potilaalle 126:sta kehittyi PEG-vasta-aineita; yhdeksän tapausta oli hoidon aikana ilmenneitä ja yhdeksän oli hoidon tehostamia.
C3G:tä ja primaarista IC‑MPGN:ää koskevissa kliinisissä tutkimuksissa lääkevasta-aineiden esiintyvyys (hoidon aikana ilmaantunut ADA tai tehostettu ADA aiemmasta tasosta) tutkimuksessa APL2-C3G-310 oli PEG-vasta-aineiden osalta 23,6 % ja pegsetakoplaanipeptidivasta-aineiden osalta 16,3 %. Populaatiofarmakokineettisen ja -farmakodynaamisen analyysin perusteella lääkevasta-aineilla ei ollut yhdistetyssä analyysipopulaatiossa kliinisesti merkittävää vaikutusta tehoon eikä farmakokinetiikkaan/farmakodynamiikkaan. Viidellä potilaalla todettiin testissä myös neutraloivia vasta-aineita (NAb). NAb-vasteella ei ollut ilmeistä vaikutusta farmakokinetiikkaan tai kliiniseen tehoon. 29 potilaalle 123:sta kehittyi PEG-vasta-aineita: 14:llä ne olivat hoidon aikana ilmenneitä ja 15:llä ne olivat hoidon tehostamia. Tutkimuksessa APL2-C3G-204 potilailla, joilla sairaus oli uusiutunut elinsiirron jälkeen, yhdellekään potilaalle ei kehittynyt positiivista ADA-vastetta (hoidon aikana ilmaantunut ADA tai tehostettu ADA aiemmasta tasosta) pegsetakoplaanipeptidille tai PEG:lle. Tutkimuksessa APL2‑C3G-310 olleen 26 viikon pituisen lumekontrolloidun jakson aikana lääkevasta-aineiden ei havaittu vaikuttavan pegsetakoplaanihoidon turvallisuuteen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset ASPAVELI‑valmisteen käytöstä PNH:n ja C3G:n tai primaarisen IC‑MPGN:n hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Pegsetakoplaani annetaan infuusiona ihon alle, ja se imeytyy asteittain systeemiseen verenkiertoon mediaanin Tmax-ajan ollessa 108–144 tuntia (4,5–6,0 vuorokautta), kun terveille vapaaehtoisille annetaan yksi ihonalainen annos.
Vakaan tilan seerumipitoisuudet annettaessa PNH‑potilaille 1 080 mg:n annos kaksi kertaa viikossa saavutettiin noin 4–6 viikossa ensimmäisen annoksen jälkeen. Potilailla, jotka olivat saaneet aiemmin komplementin estäjiä (tutkimus APL2-302), vakaan tilan seerumipitoisuuden geometrinen keskiarvo (%CV) vaihteli välillä 655–706 mikrog/ml (18,6–15,1 %) 16 viikkoa hoidetuilla potilailla. Vakaan tilan pitoisuus potilailla (n = 22), jotka jatkoivat pegsetakoplaanihoitoa viikkoon 48 saakka, oli 623 mikrog/ml (39,7 %), mikä viittaa pegsetakoplaanin terapeuttisten pitoisuuksien säilymiseen viikkoon 48 saakka. Potilailla, jotka eivät olleet saaneet aiemmin komplementin estäjiä (tutkimus APL2-308), vakaan tilan seerumipitoisuuden geometrinen keskiarvo (%CV) viikolla 26 oli 744 mikrog/ml (25,5 %), kun valmistetta annettiin kaksi kertaa viikossa. Ihonalaisen pegsetakoplaaniannoksen biologisen hyötyosuuden arvioidaan olevan 76 % populaatiofarmakokineettisen analyysin mukaan.
Kun C3G:tä tai primaarista IC‑MPGN:ää sairastaville potilaille annettiin annos 1 080 mg kaksi kertaa viikossa, vakaan tilan pitoisuudet seerumissa saavutettiin noin 4–8 viikkoa ensimmäisen annoksen jälkeen, ja pegsetakoplaanin terapeuttiset pitoisuudet säilyivät viikon 52 loppuun asti. Tutkimuksen APL2-C3G-310 potilailla seerumin vakaan tilan pitoisuuksien keskiarvo (%CV) oli 715,8 mikrog:sta/ml (31,2 %) ja 765,7 mikrog:aan/ml (23,2 %) viikkoon 26 asti ja pysyi välillä 670,1 mikrog/ml (30,1 %) – 726,6 mikrog/ml (30,5 %) viikkoon 52 asti.
Jakautuminen
Pegsetakoplaanin keskimääräinen (%CV) jakautumistilavuus PNH‑potilailla on noin 3,98 l (32 %) populaatiofarmakokineettisen analyysin mukaan.
Pegsetakoplaanin keskimääräinen (%CV) keskustilan jakautumistilavuus aikuisilla potilailla, joilla on C3G tai primaarinen IC‑MPGN, on noin 4,31 l (32,1 %).
Metabolia/eliminaatio
PEGyloidun peptidirakenteensa perusteella pegsetakoplaanin metabolian odotetaan tapahtuvan katabolisia reittejä ja hajoavan pieniksi peptideiksi, aminohapoiksi ja PEGeiksi. Makakiapinoilla tehdyn radioleimaustutkimuksen tulokset viittaavat siihen, että leimatun peptidipuolikkaan ensisijainen eliminaatioreitti on erittyminen virtsaan. Vaikka PEG:n eliminoitumista ei tutkittu, sen tiedetään erittyvän munuaisten kautta.
Pegsetakoplaani ei osoittanut testattujen CYP-entsyymien isoformien inhibitiota tai induktiota in vitro ‑tutkimusten mukaan. Pegsetakoplaani ei ollut ihmisen sisäänotto- tai effluksitransporttereiden substraatti eikä inhibiittori.
Useiden ihonalaisten pegsetakoplaaniannosten jälkeen PNH-potilaiden keskimääräinen (%CV) puhdistuma on 0,015 l/h (30 %) ja mediaani efektiivinen eliminaation puoliintumisaika (t1/2) on 8,6 vuorokautta arvioituna populaatiofarmakokineettisestä analyysistä.
Puhdistuman arvioitu keskiarvo (%CV) aikuisilla potilailla, joilla on C3G tai primaarinen IC‑MPGN, on 0,012 l/h (43 %). Terminaalisen t1/2:n mediaani aikuisilla potilailla, joilla on C3G tai primaarinen IC‑MPGN, on 10,1 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Pegsetakoplaanialtistus suurenee suhteessa annokseen 45 mg:sta 1 440 mg:aan.
Erityisryhmät
Potilailla, joilla on PNH, C3G tai primaarinen IC‑MPGN, ei tunnistettu vaikutusta pegsetakoplaanin farmakokinetiikkaan iän (12–81 vuotta), etnisen taustan tai sukupuolen osalta populaatiofarmakokinetiikka-analyysin tulosten perusteella.
Vakaan tilan keskimääräisen pitoisuuden arvellaan olevan 50 kg painavilla potilailla noin 20 % suurempi 70 kg painavaan viitepotilaaseen verrattuna. 40 kg painavilla PNH-potilailla keskimääräisen pitoisuuden arvellaan olevan 45 % suurempi. Pegsetakoplaanin turvallisuusprofiilista alle 50 kg painavilla PNH-potilailla on hyvin vähän tietoa.
Iäkkäät
Vaikka näissä tutkimuksissa ei ole havaittu ilmeisiä ikään liittyviä eroja, vähintään 65-vuotiaiden potilaiden määrä ei riitä määrittämään, reagoivatko he eri tavalla kuin nuoremmat potilaat. Ks. kohta Annostus ja antotapa.
Pediatriset potilaat
Populaatiofarmakokineettisen analyysin perusteella nuorten potilaiden (12–17-vuotiaiden) paino vaikuttaa puhdistumaan ja jakautumistilavuuteen. C3G:tä tai primaarista IC‑MPGN:ää sairastavien nuorten hoito-ohjelma perustuu potilaan painoon. Ks. kohta Annostus ja antotapa. Mallin perusteella ennustettu altistus C3G:tä tai primaarista IC‑MPGN:ää sairastavilla nuorilla vastaa riittävästi aikuisten viitealtistusta.
Munuaisten toimintahäiriö
Tutkittaessa kahdeksaa vaikeaa munuaisten toimintahäiriötä sairastavaa potilasta, joilla kreatiniinipuhdistuma (CrCl) oli alle 30 ml/l käytettäessä Cockcroft‑Gaultin kaavaa (näistä neljällä potilaalla oli arvo alle 20 ml/min), munuaisten toimintahäiriöllä ei ollut vaikutusta pegsetakoplaanin 270 mg:n kerta-annoksen farmakokinetiikkaan. PNH-potilaista, joilla on munuaisten toimintahäiriö ja jotka ovat saaneet kliinistä annosta 1 080 mg kahdesti viikossa, on hyvin vähän tietoa. Populaatiofarmakokineettisen analyysin perusteella eGFR:llä ei ollut yhdistetyn analyysin populaatiossa kliinisesti merkittävää vaikutusta pegsetakoplaanialtistukseen. Pegsetakoplaanin käytöstä potilaille, joilla on loppuvaiheen ESRD ja jotka tarvitsevat dialyysiä, ei ole saatavilla kliinistä tietoa. Ks. kohta Annostus ja antotapa.
Prekliiniset tiedot turvallisuudesta
In vitro- ja in vivo ‑toksikologiatiedot eivät viittaa erityiseen toksisuuteen ihmisille. Eläimillä havaitut, kliinisiin altistustasoihin verrattavien altistustasojen vaikutukset on kuvattu jäljempänä. Näitä vaikutuksia ei havaittu kliinisissä tutkimuksissa.
Eläinten lisääntyminen
Tiineenä olevien makakiapinoiden pegsetakoplaanihoito ihon alle annettavalla annoksella 28 mg/kg/vrk (2,9 kertaa ihmisen vakaan tilan Cmax) gestaatiojaksosta syntymään asti johti tilastollisesti merkitsevään keskenmenojen ja kuolleena syntyneiden määrän nousuun. Täysiaikaisissa poikasissa ei havaittu emotoksisuus- tai teratogeenisuusvaikutuksia. Poikasilla ei havaittu vaikutuksia kehitykseen 6 kuukauden aikana syntymän jälkeen. Systeemistä altistusta pegsetakoplaanille havaittiin sellaisten apinoiden sikiöissä, joita oli hoidettu annoksella 28 mg/kg/vrk organogeneesistä toiseen kolmannekseen asti, mutta altistus oli minimaalista (alle 1 %, ei farmakologisesti merkitsevää).
Karsinogeneesi
Pegsetakoplaanilla ei ole tehty pitkäkestoisia karsinogeenisuustutkimuksia.
Genotoksisuus
Pegsetakoplaani ei ollut mutageeninen in vitro bakteerin käänteismutaatiomäärityksissä (Ames) eikä genotoksinen in vitro ‑määrityksessä ihmisen TK6-soluissa tai in vivo hiirten mikrotumamäärityksessä.
Eläinten toksikologia
Toistuvien annosten tutkimuksia tehtiin kaniineilla ja makakiapinoilla, ja niissä pegsetakoplaanin ihon alle annettu vuorokausiannos oli enintään 7 kertaa ihmisten annos (1 080 mg kaksi kertaa viikossa). Kummankin lajin histologisissa löydöksissa havaittiin annosriippuvaista epiteelivakuolisaatiota ja vakuolisoitujen makrofagien infiltraatteja useissa kudoksissa. Vastaavia löydöksiä on havaittu muiden myyntiluvallisten PEGyloitujen lääkevalmisteiden yhteydessä, suuriin kumulatiivisiin, pitkäketjuisten PEG:ien annoksiin, mutta niihin ei ole liittynyt kliinisiä löydöksiä, eikä niitä ole siten katsottu haitallisiksi. Löydösten korjaantuvuutta ei tapahtunut pegsetakoplaanilla tehdyissä eläinkokeissa kuukauden kuluessa, eikä sitä arvioitu pidemmällä aikavälillä. Kirjallisuustiedot viittaavat PEG-vakuolien korjaantumiseen.
Munuaisten tubulusdegeneraatiota havaittiin mikroskooppisesti molemmilla lajeilla altistustasoilla, (Cmax ja AUC), joka vastasi korkeintaan ihmisen annostasoa ja se oli minimaalista ja etenemätöntä 4 viikon ja 9 kuukauden välillä, kun pegsetakoplaania annettiin päivittäin. Vaikka eläimillä ei havaittu ilmeisiä merkkejä munuaisten toimintahäiriöstä, näiden löydösten kliinistä merkitystä ja toiminnallisia seuraamuksia ei tunneta.
Farmaseuttiset tiedot
Apuaineet
Sorbitoli (E 420)
Etikkahappo, väkevä
Natriumasetaattitrihydraatti
Natriumhydroksidi (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
30 kuukautta.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ASPAVELI infuusioneste, liuos
1080 mg (L:ei) 1 kpl (20 ml (54 mg/ml)) (3596,95 €), 8 x 1 kpl (20 ml (54 mg/ml)) (27511,40 €)
PF-selosteen tieto
Tyypin I lasista valmistettu injektiopullo, jossa on tulppa (klooributyyliä tai bromibutyyliä) ja sinetti (alumiinia) sekä napsautuskorkki (polypropyleeniä), ja joka sisältää 54 mg/ml steriiliä liuosta.
Yksi pakkaus sisältää yhden injektiopullon.
Kerrannaispakkaus sisältää 8 injektiopulloa (8 yhden injektiopullon pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön tai hieman kellertävä vesipohjainen liuos, jonka pH on 5,0.
Käyttö- ja käsittelyohjeet
ASPAVELI toimitetaan valmiskäyttöisenä liuoksena kertakäyttöisissä injektiopulloissa. Koska liuos ei sisällä säilöntäaineita, tämä lääkevalmiste on infusoitava välittömästi ruiskun valmistelemisen jälkeen.
ASPAVELI on kirkas, väritön tai hieman kellertävä vesipohjainen liuos. Sitä ei saa käyttää, jos neste näyttää samealta, sisältää hiukkasia tai on tummankeltaista.
Injektiopullon on aina annettava tasaantua huoneenlämpöön noin 30 minuutin ajan ennen käyttöä.
Poista suojakorkki injektiopullosta, jotta injektiopullon harmaan kumitulpan keskiosa paljastuu. Puhdista tulppa uudella alkoholipyyhkeellä ja anna tulpan kuivua. Ei saa käyttää, jos suojakorkki puuttuu tai on vahingoittunut.
Ruiskun valmisteleminen:
Vaihtoehto 1: Käytettäessä neulatonta siirtolaitetta (kuten injektiopullon sovitinta) on noudatettava laitteen valmistajan antamia ohjeita.
Vaihtoehto 2: Jos siirto tehdään käyttämällä siirtoneulaa ja ruiskua, noudata seuraavia ohjeita:
- Kiinnitä steriili siirtoneula steriiliin ruiskuun.
- Vedä mäntää taakse, jotta ruisku täyttyy ilmalla, eli noin 20 ml:lla.
- Varmista, että injektiopullo on pystyasennossa. Älä käännä injektiopulloa ylösalaisin.
- Paina ilmalla täytetty ruisku ja kiinnitetty siirtoneula injektiopullon tulpan läpi.
- Siirtoneulan kärjen ei pitäisi olla liuoksessa, jotta ei synny kuplia.
- Paina varovasti ilma ruiskusta injektiopulloon. Tämä injektoi ilman ruiskusta injektiopulloon.
- Käännä injektiopullo ylösalaisin.
- Kun siirtoneulan kärki on liuoksessa, vedä mäntää hitaasti, jotta määrätty ASPAVELI-annos virtaa ruiskuun.
- Poista täytetty ruisku ja siirtoneula injektiopullosta.
- Älä aseta korkkia takaisin siirtoneulaan. Poista neula ja hävitä se terävän jätteen säiliöön.
Antaminen:
ASPAVELI-valmistetta annetaan vain ihon alle infuusiopumpulla tai puettavalla antojärjestelmällä:
- Valmistele infuusiopumppu ja letkut laitteen valmistajan ohjeiden mukaan. Käytettäessä infuusiopumppua infuusion mahdollisia antopaikkoja ovat vatsa, reidet, lantio ja olkavarret. Vaihtele infuusion antopaikkoja infuusiosta toiseen. Jos infuusiopaikkoja on useita, niiden pitää olla vähintään 7,5 cm:n etäisyydellä toisistaan. Infuusion kesto on noin 30 minuuttia (käytettäessä kahta kohtaa) tai noin 60 minuuttia (käytettäessä yhtä kohtaa).
- Valmistele puettava antojärjestelmä valmistajan ohjeiden mukaan. Käytettäessä puettavaa antojärjestelmää ASPAVELI on annettava infuusiona vatsaan. Vaihtele infuusion antopaikkoja infuusiosta toiseen. Infuusion kesto vaihtelee potilaittain ja on tavallisesti 30–60 minuuttia.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ASPAVELI infuusioneste, liuos
1080 mg 1 kpl, 8 x 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Iptakopaani ja pegsetakoplaani: Aikuisten kohtauksittaisen yöllisen hemoglobiinivirtsaisuuden (PNH) hoito erityisin edellytyksin (3078).
ATC-koodi
L04AJ03
Valmisteyhteenvedon muuttamispäivämäärä
15.01.2026
Yhteystiedot
Äyritie 18
01510 Vantaa
0201 558 840
www.sobi.fi
mail.fi@sobi.com