
ZOLGENSMA infuusioneste, liuos 2 x 10exp13 vektorigenomia/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Terveydenhuollon ammattilaisen opas Zolgensma®-hoidon (onasemnogeeniabeparvoveekki) riskien minimointiin
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Onasemnogeeniabeparvoveekki on geenihoitovalmiste, joka ilmentää ihmisen SMN (survival motor neuron) -proteiinia. Se on ei-replikoiva rekombinantti adenoassosioituneen viruksen serotyyppiin 9 (AAV9) perustuva vektori, joka sisältää ihmisen SMN-geenin cDNA:ta, jota kontrolloi sytomegaloviruksen voimistaja/kanan β-aktiini-hybridi-promoottori.
Onasemnogeeniabeparvoveekki on tuotettu ihmisen alkion munuaissoluissa yhdistelmä-DNA-tekniikkaa käyttämällä.
Yksi millilitra sisältää onasemnogeeniabeparvoveekkia, jonka nimellinen pitoisuus on 2 × 1013 vektorigenomia (vg). Injektiopulloista vedettävä määrä on vähintään joko 5,5 ml tai 8,3 ml. Injektiopullojen kokonaislukumäärä ja täyttömäärien yhdistelmä kussakin valmiissa pakkauksessa sovitetaan yksilöllisesti potilaan painon mukaisia annosvaatimuksia noudattaen (ks. kohdat Annostus ja antotapa ja Pakkaukset ja valmisteen kuvaus).
Apuaine, jonka vaikutus tunnetaan
Tämä lääkevalmiste sisältää 0,2 mmol natriumia/ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Zolgensma on tarkoitettu:
- 5q spinaalista lihasatrofiaa (SMA) sairastavien potilaiden hoitoon, joilla on SMN1-geenin bialleelinen mutaatio ja tyypin 1 SMA:n kliininen diagnoosi, tai
- 5q spinaalista lihasatrofiaa (SMA) sairastavien potilaiden hoitoon, joilla on SMN1-geenin bialleelinen mutaatio ja enintään 3 SMN2-geenin kopiota.
Ehto
Hoito on aloitettava ja annettava kliinisissä keskuksissa spinaalista lihasatrofiaa sairastavien potilaiden hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava ja annettava kliinisissä keskuksissa spinaalista lihasatrofiaa sairastavien potilaiden hoitoon perehtyneen lääkärin valvonnassa.
Ennen onasemnogeeniabeparvoveekin antoa on suoritettava ainakin seuraavat lähtötilanteen laboratoriotutkimukset:
- AAV9-vasta-ainetutkimus käyttämällä asianmukaista validoitua määritysmenetelmää
- maksan toimintakoe: alaniiniaminotransferaasi (ALAT), aspartaattiaminotransferaasi (ASAT), kokonaisbilirubiini, albumiini, protrombiiniaika, osittainen tromboplastiiniaika ja INR-arvo,
- kreatiniini,
- täydellinen verenkuva (mukaan lukien hemoglobiini- ja trombosyyttiarvot), ja
- troponiini-I.
Onasemnogeeniabeparvoveekkihoidon ajoitusta vahvistettaessa on otettava huomioon tarvittava maksan toiminnan ja trombosyyttiarvojen huolellinen seuranta lääkkeen annon jälkeen, sekä kortikosteroidihoidon tarve (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vakavaoireisen systeemisen immuunivasteen kohonneen riskin vuoksi on suositeltavaa, että potilaan yleinen terveydentila on kliinisesti vakaa (esim. nesteytyksen ja ravitsemuksen tila, potilaalla ei ole infektiota) ennen onasemnogeeniabeparvoveekki-infuusiota. Akuuttien tai kroonisten hallitsemattomien aktiivisten infektioiden tapauksessa hoitoa on lykättävä siihen saakka, kunnes infektio on parantunut ja potilaan tila on kliinisesti vakaa (ks. alakohdat 4.2 ”Immunomodulatorinen hoito” ja 4.4 ”Systeeminen immuunivaste”).
Annostus
Ainoastaan kertainfuusiona laskimoon.
Potilaat saavat nimellisen annoksen onasemnogeeniabeparvoveekkia 1,1 × 1014 vg painokiloa kohden. Kokonaismäärä määritetään potilaan painon mukaan.
Taulukossa 1 on esitetty suositeltu annostus potilaille, joiden paino on 2,6 kg–21,0 kg.
Taulukko 1 Potilaan painoon perustuva suositeltu annostus
| Potilaan painoalue (kg) | Annos (vg) | Annoksen kokonaismäärä a (ml) |
| 2,6–3,0 | 3,3 × 1014 | 16,5 |
| 3,1–3,5 | 3,9 × 1014 | 19,3 |
| 3,6–4,0 | 4,4 × 1014 | 22,0 |
| 4,1–4,5 | 5,0 × 1014 | 24,8 |
| 4,6–5,0 | 5,5 × 1014 | 27,5 |
| 5,1–5,5 | 6,1 × 1014 | 30,3 |
| 5,6–6,0 | 6,6 × 1014 | 33,0 |
| 6,1–6,5 | 7,2 × 1014 | 35,8 |
| 6,6–7,0 | 7,7 × 1014 | 38,5 |
| 7,1–7,5 | 8,3 × 1014 | 41,3 |
| 7,6–8,0 | 8,8 × 1014 | 44,0 |
| 8,1–8,5 | 9,4 × 1014 | 46,8 |
| 8,6–9,0 | 9,9 × 1014 | 49,5 |
| 9,1–9,5 | 1,05 × 1015 | 52,3 |
| 9,6–10,0 | 1,10 × 1015 | 55,0 |
| 10,1–10,5 | 1,16 × 1015 | 57,8 |
| 10,6–11,0 | 1,21 × 1015 | 60,5 |
| 11,1–11,5 | 1,27 × 1015 | 63,3 |
| 11,6–12,0 | 1,32 × 1015 | 66,0 |
| 12,1–12,5 | 1,38 × 1015 | 68,8 |
| 12,6–13,0 | 1,43 × 1015 | 71,5 |
| 13,1–13,5 | 1,49 × 1015 | 74,3 |
| 13,6–14,0 | 1,54 × 1015 | 77,0 |
| 14,1–14,5 | 1,60 × 1015 | 79,8 |
| 14,6–15,0 | 1,65 × 1015 | 82,5 |
| 15,1–15,5 | 1,71 × 1015 | 85,3 |
| 15,6–16,0 | 1,76 × 1015 | 88,0 |
| 16,1–16,5 | 1,82 × 1015 | 90,8 |
| 16,6–17,0 | 1,87 × 1015 | 93,5 |
| 17,1–17,5 | 1,93 × 1015 | 96,3 |
| 17,6–18,0 | 1,98 × 1015 | 99,0 |
| 18,1–18,5 | 2,04 × 1015 | 101,8 |
| 18,6–19,0 | 2,09 × 1015 | 104,5 |
| 19,1–19,5 | 2,15 × 1015 | 107,3 |
| 19,6–20,0 | 2,20 × 1015 | 110,0 |
| 20,1–20,5 | 2,26 × 1015 | 112,8 |
| 20,6–21,0 | 2,31 × 1015 | 115,5 |
a HUOM: Pakkauksen sisältämien injektiopullojen lukumäärä ja pakkausten lukumäärä määräytyvät painon mukaan. Annosmäärä lasketaan käyttämällä potilaan painoalueen ylärajaa.
Immunomodulatorinen hoito
Immuunivaste AAV9‑kapsidiin syntyy onasemnogeeniabeparvoveekin annon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tämä voi aiheuttaa maksan aminotransferaasien ja troponiini-I:n arvojen kohoamisen tai trombosyyttiarvojen laskun (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Immuunivasteen heikentämiseksi suositellaan immunomodulaatiota kortikosteroidien avulla. Potilaan rokotusohjelmaa on mahdollisuuksien mukaan muutettava, jotta mahdollistetaan samanaikainen kortikosteroidin anto ennen onasemnogeeniabeparvoveekki-infuusiota ja infuusion jälkeen (ks. kohta Yhteisvaikutukset).
Ennen immunomodulatorisen hoidon aloittamista ja ennen onasemnogeeniabeparvoveekin antoa potilas on tutkittava minkä tahansa tyyppisen aktiivisen infektiosairauden oireiden ja löydösten varalta.
Immunomodulatorinen hoito suositellaan aloitettavaksi 24 tuntia ennen onasemnogeeniabeparvoveekin infuusiota noudattamalla alla olevaa hoito-ohjelmaa (ks. taulukko 2). Jos hoito-ohjelman missä tahansa vaiheessa potilailla ei saada riittävää vastetta suun kautta annettavalla vuorokausiannoksella, joka vastaa 1 mg prednisolonia painokiloa kohden, on potilaan kliininen tilanne huomioiden harkittava lasten gastroenterologin tai hepatologin konsultoimista viipymättä ja suositellun immunomodulatorisen hoito-ohjelman muokkaamista mukaan lukien annoksen suurentamista, pidempää hoidon kestoa, tai kortikosteroidiannoksen asteittaiseen pienentämiseen käytettävän aikajakson pidentämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos potilas ei siedä suun kautta otettavaa kortikosteroidihoitoa, voidaan kliinisen tarpeen vaatiessa harkita laskimoon annettavaa kortikosteroidia.
Taulukko 2 Infuusiota edeltävä ja infuusion jälkeinen immunomodulatorinen hoito
| Infuusiota edeltävä hoito | 24 tuntia ennen onasemnogeeniabeparvoveekin antoa | Suun kautta annettava prednisoloni 1 mg/kg/vuorokausi (tai vastaava, jos käytetään jotain muuta kortikosteroidia) |
| Infuusion jälkeinen hoito | 30 vuorokauden ajan (mukaan lukien onasemnogeeniabeparvoveekin antopäivä) | Suun kautta annettava prednisoloni 1 mg/kg/vuorokausi (tai vastaava, jos käytetään jotain muuta kortikosteroidia) |
Minkä jälkeen 28 vuorokauden ajan: Potilaille, joilla löydökset eivät ole merkitseviä (normaali kliininen status, kokonaisbilirubiini ja joiden ALAT- ja ASAT-arvot ovat molemmat alle 2 × normaalin ylärajan [ULN]) 30 vuorokauden ajanjakson lopussa: tai | Systeemisten kortikosteroidien annosta pienennetään asteittain. Prednisolonin (tai vastaavan, jos käytetään jotain muuta kortikosteroidia) asteittainen pienentäminen, esim. 2 viikon ajan 0,5 mg/kg/vuorokausi ja sen jälkeen 2 viikon ajan 0,25 mg/kg/vuorokausi suun kautta annettavaa prednisolonia | |
| Potilaille, joiden maksan toimintakokeiden tulokset ovat poikkeavia 30 vuorokauden ajanjakson lopussa: jatketaan kunnes ASAT- ja ALAT-arvot ovat alle 2 × normaalin ylärajan (ULN) ja kaikki muut analyysiarvot (esim. kokonaisbilirubiini) palautuvat normaalirajoihin, minkä jälkeen kortikosteroidiannosta pienennetään vähitellen 28 vuorokauden ajan tai tarvittaessa pidempään. | Systeemiset kortikosteroidit (vastaten suun kautta annettavaa prednisolonia 1 mg/kg/vuorokausi) Systeemisten kortikosteroidien annosta pienennetään asteittain. |
Maksan toimintaa (ALAT, ASAT, kokonaisbilirubiini) on tarkkailtava säännöllisin väliajoin vähintään 3 kuukauden ajan onasemnogeeniabeparvoveekki-infuusion jälkeen (viikoittain ensimmäisen kuukauden ajan ja koko sen ajan, kun kortikosteroidiannosta pienennetään asteittain, ja sen jälkeen kahden viikon välein vielä yhden kuukauden ajan) ja kliinisen tarpeen mukaan. Jos maksan toimintakokeiden tulokset huononevat ja/tai potilaalla on akuutteja sairauden oireita ja/tai löydöksiä, on potilaan kliininen tila arvioitava viipymättä ja tilaa on seurattava tiiviisti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos lääkäri käyttää jotain muuta kortikosteroidia prednisolonin sijasta, samanlaista harkintaa ja annoksen asteittaista pienentämistapaa 30 vuorokauden jälkeen on noudatettava, kun se on asianmukaista.
Erityispotilaat
Munuaisten vajaatoiminta
Onasemnogeeniabeparvoveekin turvallisuutta ja tehoa munuaisten vajaatoimintaa sairastavilla potilailla ei ole vahvistettu, ja onasemnogeeniabeparvoveekkihoitoa on harkittava huolellisesti. Annoksen muuttamista ei pidä harkita.
Maksan vajaatoiminta
Onasemnogeeniabeparvoveekkia ei ole tutkittu kliinisissä tutkimuksissa potilailla, joilla ALAT‑, ASAT‑ tai kokonaisbilirubiiniarvo (paitsi jos tämä liittyy vastasyntyneen keltaisuuteen) on > 2 × ULN tai jotka ovat seropositiivisia hepatiitti B‑ tai hepatiitti C‑virukselle. Onasemnogeeniabeparvoveekkihoitoa on harkittava varovaisesti potilaille, jotka sairastavat maksan vajaatoimintaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Annoksen muuttamista ei pidä harkita.
0SMN1/1SMN2-genotyyppi
Annoksen muuttamista ei pidä harkita potilaille, joilla on SMN1-geenin bialleelinen mutaatio ja vain yksi SMN2:n kopio (ks. kohta Farmakodynamiikka).
Anti-AAV9-vasta-aineet
Annoksen muuttamista ei pidä harkita potilaille, joiden lähtötilanteen anti-AAV9-vasta-ainemääritys on yli 1:50 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Onasemnogeeniabeparvoveekin turvallisuutta ja tehoa keskosvauvoille ennen täysiaikaista sikiöikää ei ole varmistettu. Tietoja ei ole saatavilla. Onasemnogeeniabeparvoveekin antoa on harkittava huolellisesti, sillä samanaikainen kortikosteroidihoito saattaa vaikuttaa haitallisesti neurologiseen kehitykseen.
Vähintään 2-vuotiaista tai yli 13,5 kg painavista potilaista on vain niukasti kokemusta. Onasemnogeeniabeparvoveekin turvallisuutta ja tehoa näille potilaille ei ole varmistettu. Tällä hetkellä saatavissa olevat tiedot on annettu kohdassa Farmakodynamiikka. Annoksen muuttamista ei pidä harkita (ks. taulukko 1).
Antotapa
Laskimoon.
Onasemnogeeniabeparvoveekki annetaan kerta-annosinfuusiona. Se on annettava ruiskupumppua käyttämällä kertainfuusiona laskimoon noin 60 minuuttia kestävänä hitaana infuusiona. Sitä ei saa antaa nopeana tai bolusinfuusiona laskimoon.
Toissijaisen (vara-) katetrin laittaminen on suositeltavaa siltä varalta, että ensisijainen katetri tukkiutuu. Infuusion päätyttyä infuusioletku on huuhdeltava 9 mg/ml (0,9 %) NaCl-injektionesteellä.
Ennen lääkevalmisteen käsittelyä tai antoa suoritettavat varotoimet
Tämä lääkevalmiste sisältää geenimuunneltuja organismeja. Tästä syystä terveydenhuoltohenkilöstön on noudatettava asianmukaisia varotoimia (käsineiden, suojalasien, laboratoriotakin ja hihojen käyttö) valmisteen käsittelyssä ja annossa (ks. kohta Käyttö- ja käsittelyohjeet).
Ks. kohdasta Käyttö- ja käsittelyohjeet tarkemmat ohjeet onasemnogeeniabeparvoveekin saattamisesta käyttökuntoon, käsittelystä, vahingossa tapahtuneesta altistuksesta ja hävittämisestä (myös kehon eritteiden asianmukaisesta käsittelystä).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Potilaalla jo entuudestaan oleva immuniteetti AAV9:ta vastaan
Anti-AAV9-vasta-ainetta voi muodostua luonnollisen altistumisen myötä. AAV9:n vasta-aineiden yleisyydestä yleisväestössä on tehty useita tutkimuksia, joissa on osoitettu pediatristen potilaiden vähäistä aikaisemmin tapahtunutta altistumista AAV9:lle. Potilaat on tutkittava AAV9-vasta-aineiden olemassaolon varalta ennen onasemnogeeniabeparvoveekki-infuusion antoa. Tutkimus voidaan tehdä uudelleen, jos ilmoitetut AAV9-vasta-ainemäärät ovat yli 1:50. Ei ole vielä tiedossa, voidaanko ja missä olosuhteissa onasemnogeeniabeparvoveekkia voidaan turvallisesti ja tehokkaasti antaa, kun anti‑AAV9-vasta-aineet ovat yli 1:50 (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Pitkälle edennyt spinaalinen lihasatrofia (SMA)
Koska spinaalinen lihasatrofia aiheuttaa etenevän ja palautumattoman motoristen hermosolujen vaurioitumisen, onasemnogeeniabeparvoveekin hyöty oireenmukaisille potilaille on riippuvainen hoidon aikaisen tautitaakan asteesta. Varhain aloitetusta hoidosta on potentiaalisesti suurempi hyöty.
Siitä huolimatta, että pitkälle edennyttä oireista SMA:ta sairastavat potilaat eivät saavuta samaa liikkeiden kokonaiskehitystä kuin terveet oireettomat verrokit, geenikorvaushoidosta saattaa olla heille kliinistä hyötyä, mikä on riippuvainen taudin etenemisestä hoidon aikana (ks. kohta Farmakodynamiikka).
Hoitavan lääkärin on huomioitava, että hoidosta saatava hyöty on vakavasti vähentynyt potilailla, joilla on merkittävä lihasheikkous ja hengityksen vajaatoiminta, ja potilailla, jotka saavat jatkuvaa ventilaatiota, sekä potilailla, jotka eivät kykene nielemään.
Onasemnogeeniabeparvoveekin hyöty-/riskiprofiilia etenevää spinaalista lihasatrofiaa sairastavilla potilailla, jatkuvan ventilaation avulla elossa pidettävillä potilailla sekä potilailla, joilta puuttuu kyky kasvaa ja kehittyä, ei ole vahvistettu.
Infuusioon liittyvät reaktiot ja anafylaktiset reaktiot
Infuusioon liittyviä reaktioita, myös anafylaktisia reaktioita, on esiintynyt onasemnogeeniabeparvoveekki-infuusion aikana ja/tai pian sen jälkeen (ks. kohta Haittavaikutukset). Potilaita on seurattava huolellisesti infuusioon liittyvien reaktioiden kliinisten oireiden ja löydösten varalta. Jos reaktio ilmenee, infuusio on keskeytettävä ja hoitoa annettava tarpeen mukaan. Kliinisen arvion perusteella ja tavanomaisia hoitokäytäntöjä noudattaen infuusiota voidaan jatkaa varovaisesti.
Immunogeenisuus
Onasemnogeeniabeparvoveekki-infuusion jälkeen AAV9‑kapsidia kohtaan syntyy immuunivaste, mukaan lukien vasta-aineen muodostuminen AAV9‑kapsidia kohtaan ja T‑soluvälitteinen immuunivaste, kohdassa Annostus ja antotapa suositellusta immunomodulatorisesta hoito-ohjelmasta huolimatta (ks. myös alakohta ”Systeeminen immuunivaste” jäljempänä).
Maksatoksisuus
Immuunivälitteinen maksatoksisuus ilmenee yleensä ALAT‑ ja/tai ASAT‑arvon nousuna. Onasemnogeeniabeparvoveekkihoidon yhteydessä on ilmoitettu akuutteja vakavia maksavaurioita ja akuuttia maksan vajaatoimintaa, myös kuolemaan johtaneita tapauksia. Tapaukset ovat tyypillisesti ilmaantuneet 2 kuukauden kuluessa infuusiosta siitä huolimatta, että potilas on saanut kortikosteroidihoitoa ennen infuusiota ja sen jälkeen. Immuunivälitteinen maksatoksisuus saattaa edellyttää immunomodulatorisen hoidon muokkaamista mukaan lukien pidempää hoidon kestoa, annoksen suurentamista tai kortikosteroidiannoksen asteittaiseen pienentämiseen käytettävän aikajakson pidentämistä (ks. kohta Haittavaikutukset).
- Onasemnogeeniabeparvoveekkihoidon riskit ja hyödyt on huolellisesti punnittava sellaisten potilaiden kohdalla, joilla on entuudestaan maksan vajaatoiminta.
- Potilailla, joilla on entuudestaan maksan vajaatoiminta tai akuutti maksan virusinfektio, voi olla kohonnut akuutin vakavan maksavaurion kehittymisen riski (ks. kohta Annostus ja antotapa).
- Pienestä tutkimuksesta, johon osallistui noin 1,5–9-vuotiaita ≥ 8,5 – ≤ 21 kg painavia lapsia, saadut tiedot viittaavat siihen, että ASAT- tai ALAT-arvojen nousu on näillä potilailla yleisempää (23 potilaalla 24:stä) verrattuna < 8,5 kg painavilla potilailla tehtyihin tutkimuksiin (31 potilaalla 99:stä) (ks. kohta Haittavaikutukset).
- AAV-vektorin antaminen aiheuttaa usein aminotransferaasiarvojen kohoamista.
- Akuuttia vakavaa maksavauriota ja akuuttia maksan vajaatoimintaa on esiintynyt onasemnogeeniabeparvoveekkihoidon yhteydessä. Kuolemaan johtaneita akuutteja maksan vajaatoimintatapauksia on ilmoitettu (ks. kohta Haittavaikutukset).
- Maksan toiminta on tarkistettava kaikilta potilailta lääkärintutkimuksen ja laboratoriokokeiden avulla ennen infuusiota (ks. kohta Annostus ja antotapa).
- Kaikille potilaille on annettava systeemistä kortikosteroidia ennen onasemnogeeniabeparvoveekki-infuusiota ja infuusion jälkeen, jotta mahdollinen aminotransferaasiarvojen nousu jäisi mahdollisimman pieneksi (ks. kohta Annostus ja antotapa).
- Maksan toimintaa on seurattava tasaisin väliajoin vähintään 3 kuukauden ajan infuusion jälkeen, ja kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa).
- Jos maksan toimintakokeiden tulokset huononevat ja/tai potilaalla on akuutteja sairauden oireita tai löydöksiä, on potilaan kliininen tila arvioitava viipymättä ja tilaa on seurattava tiiviisti.
- Maksavauriota epäiltäessä suositellaan viipymättä konsultoimaan lasten gastroenterologia tai hepatologia, suositellun immunomodulatorisen hoito-ohjelman muokkaamista ja lisäkokeiden tekemistä (esim. albumiini, protrombiiniaika, osittainen tromboplastiiniaika ja INR‑arvo).
ASAT/ALAT/kokonaisbilirubiiniarvo on määritettävä viikoittain ensimmäisen kuukauden ajan onasemnogeeniabeparvoveekki-infuusion jälkeen ja koko kortikosteroidiannoksen asteittaiseen pienentämiseen käytettävän aikajakson ajan. Kortikosteroidiannoksen asteittaista pienentämistä ei pidä harkita ennen kuin ASAT/ALAT‑arvot ovat alle 2 × normaalin ylärajan (ULN) ja kaikki muut testitulokset (esim. kokonaisbilirubiini) ovat palanneet normaaleiksi (ks. kohta Annostus ja antotapa). Jos potilaan tila on kliinisesti vakaa eikä merkittäviä löydöksiä ole kortikosteroidiannoksen asteittaiseen pienentämiseen käytetyn aikajakson lopussa, on maksan toiminnan seuraamista jatkettava kahden viikon välein vielä yhden kuukauden ajan.
Trombosytopenia
Ohimenevää trombosyyttien määrän laskua, joista muutamat tapaukset täyttivät trombosytopenian kriteerit, havaittiin onasemnogeeniabeparvoveekkiin liittyvissä kliinisissä tutkimuksissa. Useimmissa tapauksissa alhaisin trombosyyttiarvo esiintyi ensimmäisellä viikolla onasemnogeeniabeparvoveekki-infuusion jälkeen.
Valmisteen markkinoille tulon jälkeen on ilmoitettu tapauksia, joissa on ilmennyt trombosyyttiarvoja < 25 x 109/l kolmen viikon kuluessa valmisteen antamisen jälkeen.
Trombosyyttien määrä on määritettävä ennen onasemnogeeniabeparvoveekki-infuusion antoa ja määrää on seurattava tiiviisti infuusion jälkeisten kolmen viikon aikana, ja tämän jälkeen säännöllisesti, vähintään viikoittain ensimmäisen kuukauden ajan ja joka toinen viikko vielä toisen ja kolmannen kuukauden aikana, kunnes trombosyyttien määrä palautuu lähtötasoon.
Pienestä tutkimuksesta, johon osallistui noin 1,5–9-vuotiaita ≥ 8,5 – ≤ 21 kg painavia lapsia, saadut tiedot viittaavat siihen, että trombosytopenia on näillä potilailla yleisempää (20 potilaalla 24:stä) verrattuna < 8,5 kg painavilla potilailla tehtyihin tutkimuksiin (22 potilaalla 99:stä) (ks. kohta Haittavaikutukset).
Kohonnut troponiini-I-pitoisuus
Sydäntroponiini-I:n pitoisuuden nousua on havaittu onasemnogeeniabeparvoveekki-infuusion jälkeen (ks. kohta Haittavaikutukset). Troponiini-I:n pitoisuuden nousu joillakin potilailla saattaa olla merkki mahdollisesta sydänlihasvauriosta. Näiden löydösten ja hiirillä havaitun sydäntoksisuuden vuoksi troponiini-I:n pitoisuus on määritettävä ennen onasemnogeeniabeparvoveekki-infuusion antoa ja pitoisuutta on seurattava kliinisen tarpeen mukaan. Kardiologian erikoislääkärin konsultointia on harkittava tarpeen mukaan.
Tromboottinen mikroangiopatia
Onasemnogeeniabeparvoveekki-infuusion yhteydessä on ilmoitettu useampia tromboottinen mikroangiopatia (TMA) -tapauksia (ks. kohta Haittavaikutukset). Yleensä tapaukset ilmaantuivat kahden ensimmäisen viikon kuluessa onasemnogeeniabeparvoveekki-infuusiosta. Tromboottinen mikroangiopatia on akuutti ja henkeä uhkaava tila, johon liittyvät trombosytopenia ja mikroangiopaattinen hemolyyttinen anemia. Kuolemaan johtaneita tapauksia on ilmoitettu. Myös akuutteja munuaisvaurioita on havaittu. Joissakin tapauksissa on ilmoitettu samanaikaisesta immuunijärjestelmän aktivaatiosta (esim. infektiot, rokotukset) (ks. tiedot rokotteiden antamisesta kohdista Annostus ja antotapa ja Yhteisvaikutukset).
Trombosytopenia on olennainen tekijä tromboottisessa mikroangiopatiassa, minkä vuoksi trombosyyttiarvoja on seurattava tiiviisti infuusion jälkeisten kolmen viikon aikana ja tämän jälkeen säännöllisesti (katso kohta ”Trombosytopenia”). Jos potilaalle ilmaantuu trombosytopeniaa, on tilannetta viipymättä arvioitava laajemmin ja potilas tutkittava hemolyyttisen anemian ja munuaistoiminnan häiriöiden havaitsemiseksi. Jos potilaalla on tromboottiseen mikroangiopatiaan sopivia kliinisiä löydöksiä, oireita tai laboratoriolöydöksiä, on välittömästi konsultoitava erikoislääkäriä tromboottisen mikroangiopatian hoitamiseksi kliinisen tarpeen mukaan. Potilaan huoltajille on kerrottava tromboottisen mikroangiopatian merkeistä ja oireista. Oireiden ilmaantuessa on kehotettava hakeutumaan kiireellisesti lääkärin hoitoon.
Systeeminen immuunivaste
Vakavaoireisen systeemisen immuunivasteen kohonneen riskin vuoksi on suositeltavaa, että potilaan yleinen terveydentila on kliinisesti vakaa (esim. nesteytyksen ja ravitsemuksen tila, potilaalla ei ole infektiota) ennen onasemnogeeniabeparvoveekki-infuusiota. Hoitoa ei pidä aloittaa silloin, jos potilaalla on aktiivinen joko akuutti (kuten akuutti hengitystieinfektio tai akuutti hepatiitti) tai hallitsematon krooninen (kuten krooninen aktiivinen B-hepatiitti) infektio. On odotettava infektion paranemista ja potilaan kliinisen tilan vakaantumista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Immunomodulatorinen hoito (ks. kohta Annostus ja antotapa) saattaa myös vaikuttaa immuunivasteeseen infektioita (esim. hengitystieinfektioita) kohtaan, mikä voi mahdollisesti aiheuttaa infektion vaikeampia taudinkuvia. Onasemnogeeniabeparvoveekilla tehtyihin kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli infektio. Tehostettua valppautta infektioiden ennalta ehkäisemisessä, seurannassa ja hoidossa suositellaan ennen onasemnogeeniabeparvoveekki-infuusiota ja sen jälkeen. Ennalta ehkäisevää RSV (respiratory syncytial virus) -kausirokotetta suositellaan ja se on pidettävä ajan tasalla. Potilaan rokotusohjelmaa on muutettava mahdollisuuksien mukaan, jotta kortikosteroidi voidaan antaa samanaikaisesti onasemnogeeniabeparvoveekki-infuusiota ennen ja infuusion jälkeen (ks. kohta Yhteisvaikutukset).
Jos kortikosteroidihoito on pitkäkestoinen tai annosta suurennetaan, hoitavan lääkärin on oltava tietoinen lisämunuaisten vajaatoiminnan riskistä.
Vektori-integraatiosta johtuva tuumorigeenisuusriski
On olemassa teoreettinen riski vektori-DNA:n integroitumisesta genomiin, ja tämän seurauksena tuumorigeenisuudesta.
Onasemnogeeniabeparvoveekki koostuu ei-replikoivasta AAV9-vektorista, jonka DNA säilyy suurelta osin episomaalisessa muodossa. Harvinaisissa tapauksissa on mahdollista, että vektori integroituu satunnaisesti ihmisen DNA:han, kun käytetään rekombinantti-AAV-vektoria. Yksittäisten integraatiotapahtumien kliinistä merkitystä ei tunneta, mutta tiedetään, että ne voivat mahdollisesti lisätä tuumorigeenisuuden riskiä.
Tähän mennessä ei ole raportoitu onasemnogeeniabeparvoveekkihoitoon liittyviä maligniteetteja. Jos potilaalle ilmaantuu kasvain, tulee ottaa yhteyttä myyntiluvan haltijaan, joka antaa ohjeet potilasnäytteiden keräämisestä tutkittavaksi.
Erittyminen
Onasemnogeeniabeparvoveekin ohimenevää erittymistä saattaa esiintyä, pääasiassa elimistön eritteiden mukana. Huoltajia ja potilaiden perheitä on neuvottava potilaan elimistön nesteiden ja eritteiden asianmukaisesta käsittelystä:
- hyvää käsihygieniaa on noudatettava vähintään 1 kuukauden ajan onasemnogeeniabeparvoveekkihoidon jälkeen, kun ollaan kontaktissa potilaan kehon eritteisiin.
- kertakäyttövaipat voidaan laittaa sinetöitäviin kaksinkertaisiin muovipusseihin ja hävittää talousjätteen mukana (ks. kohta Farmakokinetiikka).
Veren, elinten, kudosten ja solujen luovutus
Zolgensma‑hoitoa saaneet potilaat eivät saa luovuttaa verta, elimiä, kudoksia tai soluja siirteiksi.
Natriumpitoisuus
Tämä lääkevalmiste sisältää 4,6 mg natriumia per ml, joka vastaa 0,23 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Yksi 5,5 ml:n injektiopullo sisältää 25,3 mg natriumia, ja yksi 8,3 ml:n injektiopullo sisältää 38,2 mg natriumia.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Kokemusta onasemnogeeniabeparvoveekin käytöstä potilaille, jotka saavat maksatoksisia lääkevalmisteita tai käyttävät maksatoksisia aineita, on niukasti. Onasemnogeeniabeparvoveekin turvallisuutta näille potilaille ei ole varmistettu.
Kokemusta samanaikaisesti käytettävien, 5q SMA:aan kohdistuvien lääkkeiden käytöstä on niukasti.
Rokotukset
Mikäli mahdollista, potilaan rokotusohjelmaa on säädettävä, jotta voidaan samanaikaisesti antaa kortikosteroidia ennen onasemnogeeniabeparvoveekki-infuusiota ja infuusion jälkeen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Ennalta ehkäisevää RSV (respiratory syncytial virus) -kausirokotetta suositellaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Eläviä rokotteita kuten MPR ja vesirokkorokote ei pidä antaa potilaille, jotka saavat immunosuppressiivisia steroidiannoksia (so. prednisolonia tai vastaavaa ≥ 2 viikon ajan kerran vuorokaudessa annoksella 20 mg tai 2 mg/painokiloa kohden).
Raskaus ja imetys
Tietoa käytöstä raskauden tai imetyksen aikana ihmisellä ei ole, eikä eläinkokeita ole tehty hedelmällisyyden tai lisääntymisen selvittämiseksi.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Onasemnogeeniabeparvoveekilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Onasemnogeeniabeparvoveekin turvallisuutta arvioitiin 5 avoimessa kliinisessä tutkimuksessa 99 potilaalla, jotka saivat onasemnogeeniabeparvoveekkia suositusannoksena (1,1 x 1014 vg/kg). Yleisimmin ilmoitettuja annon jälkeisiä haittavaikutuksia olivat maksaentsyymiarvojen kohoaminen (24,2 %), maksatoksisuus (9,1 %), oksentelu (8,1 %), trombosytopenia (6,1 %), troponiiniarvon kohoaminen (5,1 %) ja kuume (5,1 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutusten taulukkomuotoinen luettelo
Onasemnogeeniabeparvoveekkiin liittyneet haittavaikutukset kaikilla potilailla, joiden saamalla laskimoinfuusiolla (suositusannos) oli syysuhde hoitoon nähden, on esitetty taulukossa 3. Haittavaikutukset on lueteltu MedDRA:n elinjärjestelmäluokituksen ja yleisyyden mukaan. Yleisyysluokat ovat hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3 Onasemnogeeniabeparvoveekin käyttöön liittyneet haittavaikutukset
| Haittavaikutukset MedDRA:n elinjärjestelmäluokkien, suositeltujen termien ja esiintyvyystiheyden mukaisesti | |
| Veri ja imukudos | |
| Yleinen | Trombosytopenia1) |
| Melko harvinainen | Tromboottinen mikroangiopatia2)3) |
| Ruoansulatuselimistö | |
| Yleinen | Oksentelu |
| Immuunijärjestelmä | |
| Harvinainen | Anafylaktiset reaktiot |
| Maksa ja sappi | |
| Yleinen | Maksatoksisuus4) |
| Melko harvinainen | Akuutti maksan vajaatoiminta2)3) |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Yleinen | Kuume |
| Melko harvinainen | Infuusioon liittyvät reaktiot |
| Tutkimukset | |
| Hyvin yleinen | Maksaentsyymiarvon kohoaminen5) |
| Yleinen | Troponiiniarvon kohoaminen6) |
1)Trombosytopenia kattaa trombosytopenian ja trombosyyttiarvon laskun. 2)Hoitoon liittyviä haittavaikutuksia, joita on ilmoitettu ennen markkinoille tuloa tehtyjen kliinisten tutkimusten ulkopuolella, mukaan lukien markkinoille tulon jälkeisessä käytössä. 3)Sisältää kuolemaan johtaneita tapauksia. 4)Maksatoksisuus kattaa rasvamaksan ja suuret transaminaasiarvot. 5)Maksaentsyymiarvon kohoaminen kattaa ALAT‑arvon kohoamisen, ammoniakkiarvon kohoamisen, ASAT‑arvon kohoamisen, GGT‑arvon kohoamisen, maksaentsyymiarvon kohoamisen, maksan toimintakoearvon kohoamisen ja transaminaasiarvojen kohoamisen. 6)Troponiiniarvon kohoaminen kattaa troponiiniarvon kohoamisen, troponiini-T-arvon kohoamisen ja troponiini‑I‑arvon kohoamisen (ilmoitettu kliinisten tutkimusten ulkopuolella, mukaan lukien markkinoille tulon jälkeisessä käytössä). | |
Valittujen haittavaikutusten kuvaus
Maksa ja sappi
Kliinisessä kehitysohjelmassa (ks. kohta Farmakodynamiikka) transaminaasiarvojen kohoamista > 2 × normaalin ylärajan (ULN) (ja joissakin tapauksissa > 20 × ULN) havaittiin 31 %:lla suositeltua annosta saaneista potilaista. Potilaat olivat kliinisesti oireettomia eikä heillä esiintynyt kliinisesti merkitsevää bilirubiinin nousua. Seerumin transaminaasipitoisuuksien nousu korjaantui yleensä prednisolonihoidolla, ja potilaat toipuivat ilman kliinisiä seuraamuksia (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Markkinoille tulon jälkeisessä käytössä, on ilmoitettu tapauksia, joissa lapsille ilmaantui akuutin maksan vajaatoiminnan merkkejä ja oireita (kuten keltaisuutta, koagulopatiaa, enkefalopatiaa) tyypillisesti 2 kuukauden kuluessa onasemnogeeniabeparvoveekkihoidosta siitä huolimatta, että he olivat saaneet kortikosteroidihoitoa ennen ja jälkeen infuusion. Kuolemaan johtaneita akuutteja maksan vajaatoimintatapauksia on ilmoitettu.
Tutkimuksessa (COAV101A12306), johon osallistui 24 lasta (paino ≥ 8,5 – ≤ 21 kg, ikä noin 1,5–9 vuotta; 21 lopetti aiemman SMA-hoidon), havaittiin transaminaasiarvojen nousua 23 potilaalla 24:stä. Potilaat olivat oireettomia, eivätkä bilirubiiniarvot kohonneet. ASAT- ja ALAT-arvojen nousua hoidettiin kortikosteroideilla, tyypillisesti pitkäkestoisesti (17 potilasta jatkoi prednisolonihoitoa viikolla 26 ja 6 potilasta sai prednisolonihoitoa edelleen viikolla 52) ja/tai suuremmalla annoksella.
Ohimenevä trombosytopenia
Kliinisessä kehitysohjelmassa (ks. kohta Farmakodynamiikka) havaittiin ohimenevää trombosytopeniaa useana annoksen jälkeisenä ajankohtana. Arvot palautuivat yleensä kahden viikon kuluessa. Trombosyyttiarvojen laskut olivat selvempiä hoidon ensimmäisellä viikolla. Valmisteen markkinoille tulon jälkeen on ilmoitettu tapauksista, joissa trombosyyttitasot ovat laskeneet kolmen viikon sisällä valmisteen antamisen jälkeen ohimenevästi tasolle < 25 x 109/l (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tutkimuksessa (COAV101A12306), johon osallistui 24 lasta (paino ≥ 8,5 – ≤ 21 kg, ikä noin 1,5–9 vuotta), trombosytopeniaa havaittiin 20 potilaalla 24:stä.
Troponiini-I:n tasojen nousu
Sydäntroponiini-I:n tasojen nousua enintään 0,2 µg/l:aan havaittiin onasemnogeeniabeparvoveekki-infuusion jälkeen. Kliinisessä tutkimusohjelmassa ei havaittu mitään kliinisesti ilmeisiä sydämeen liittyviä löydöksiä onasemnogeeniabeparvoveekin annon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Geenihoitoa edeltävät ja hoitoa seuraavat anti-AAV9-vasta-ainemääritykset tehtiin kliinisissä tutkimuksissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kaikilla onasemnogeeniabeparvoveekkia saaneilla potilailla anti-AAV9:n vasta-ainetitteri ennen hoitoa oli enintään 1:50. AAV9:n vasta-ainetitterin keskimääräistä nousua lähtötilanteesta havaittiin kaikilla potilailla kaikkina paitsi yhtenä ajankohtana AAV9 peptidin vasta-ainetitteritasoilla, mikä kuvastaa normaalia vastetta elimistölle vieraalle virusantigeenille. Muutamilla potilailla esiintyi AAV9:n vasta-ainetitterin suurentumista yli määritystason, mutta useimmilla näistä potilaista ei esiintynyt potentiaalisesti kliinisesti merkitseviä haittavaikutuksia. Sen vuoksi korkean anti-AAV9:n vasta-ainetitterin ja potentiaalisten haittavaikutusten tai tehon parametrien välistä suhdetta ei ole voitu vahvistaa.
Kliinisessä AVXS-101-CL-101-tutkimuksessa 16 potilasta seulottiin anti-AAV9:n vasta-ainetitterin suhteen: 13 potilaan vasta-ainetitterit olivat alle 1:50 ja nämä otettiin mukaan tutkimukseen; vasta-ainetitterit olivat yli 1:50 kolmella potilaalla, joista kaksi arvioitiin uudelleen imetyksen lopettamisen jälkeen, jolloin näiden mitatut vasta-ainetitterit olivat alle 1:50, ja kumpikin otettiin mukaan tutkimukseen. Ei ole tietoa siitä, pitäisikö imettämistä rajoittaa äideillä, jotka saattavat olla seropositiivisia anti-AAV9-vasta-aineille. Kaikkien potilaiden AAV9:n vasta-ainetitteri oli alle tai tasan 1:50 ennen onasemnogeeniabeparvoveekkihoitoa ja osoittivat vastaavasti nousua anti-AAV9:n vasta-ainetitteritasoissa vähintään 1:102 400:een ja enintään > 1:819 200:een.
Vasta-ainemuodostuksen toteaminen on suuresti riippuvainen tutkimuksen herkkyydestä ja spesifisyydestä. Lisäksi vasta-ainepositiivisuuden (ml. neutraloivat vasta-aineet) havaittuun ilmaantuvuuteen tutkimuksessa saattavat vaikuttaa useat tekijät, mukaan lukien tutkimuksen metodiikka, näytteiden käsittely, näytteiden keräämisen ajoitus, samanaikaisesti käytetyt lääkevalmisteet ja taustalla olevat sairaudet.
Kukaan onasemnogeeniabeparvoveekkihoitoa saaneesta potilaasta ei osoittanut immuunivastetta transgeenille.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Onasemnogeeniabeparvoveekin yliannostuksesta ei ole kliinisiä tutkimuksia saatavissa. Prednisolonin annoksen säätämistä, huolellista kliinistä seurantaa ja laboratoriotulosten seurantaa (ml. kliininen kemia ja hematologia) suositellaan systeemisen immuunivasteen varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut tuki- ja liikuntaelinten sairauksien lääkkeet, ATC-koodi: M09AX09
Vaikutusmekanismi
Onasemnogeeniabeparvoveekki on geenihoito, jonka tarkoituksena on luoda ’survival motor neuron’ (SMN1) -geenin toimiva kopio transdusoiduissa soluissa sairauden monogeenisen perussyyn hoitamiseksi. Tarjoamalla SMN-proteiinin ilmentymän vaihtoehtoisen lähteen liikehermosoluissa, niiden eloonjäännin ja transdusoitujen liikehermosolujen toiminnan paraneminen on odotettavissa.
Onasemnogeeniabeparvoveekki on ei-replikoiva rekombinantti AAV-vektori, joka käyttää AAV9-kapsidia vakaan, täysin toimivan ihmisen SMN-transgeenin aikaansaamiseksi. AAV9-kapsidin kyky läpäistä veri-aivoeste ja transdusoida liikehermosoluja on osoitettu. Onasemnogeeniabeparvoveekissa oleva SMN1-geenin on tarkoitus sijaita DNA-episomina transdusoitujen solujen ytimessä ja sen odotetaan ekspressoituvan vakaasti pidemmän aikaa post-mitoottisissa soluissa. AAV9-viruksen ei tiedetä aiheuttavan sairautta ihmiselle. Transgeeni viedään kohdesoluihin itselleen komplementaarisena kaksijuosteisena molekyylinä. Transgeenin ilmentymistä aktivoi peruspromoottori (sytomegaloviruksen vahvistama kanan β‑aktiinin hybridi), mikä aikaansaa jatkuvan ja pysyvän SMN-proteiinin ilmentymisen. Näyttöä vaikutusmekanismista tukevat ei-kliiniset tutkimukset ja tiedot biodistribuutiosta ihmisellä.
Kliininen teho ja turvallisuus
AVXS-101-CL-303 vaiheen 3 tutkimus potilaille, joilla on tyypin 1 SMA
AVXS-101-CL-303 (tutkimus CL-303) on vaiheen 3, avoin, yksihaarainen kerta-annostutkimus laskimoon annetun onasemnogeeniabeparvoveekin hoitoannoksella (1,1 × 1014 vg/kg). Tutkimukseen otettiin mukaan kaksikymmentäkaksi potilasta, joilla oli tyypin 1 SMA ja SMN2:n 2 kopiota. Yksikään 22 potilaasta ei tarvinnut noninvasiivista ventilaatiotukea ja kaikki potilaat pystyivät syömään suun kautta (eivät tarvinneet muuta kuin peroraalista ravitsemusta) ennen onasemnogeeniabeparvoveekkihoitoa. CHOP‑INTEND‑pistemäärän (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) keskiarvo oli lähtötilanteessa 32,0 (vaihteluväli 18–52). Kyseisten 22 potilaan ikäkeskiarvo hoitoajankohtana oli 3,7 kuukautta (0,5–5,9 kuukautta).
Näistä 22:sta mukaan otetusta potilaasta 21 saavutti ilman jatkuvaa ventilaatiota (tapahtumavapaa elossaolo, event-free survival) ≥ 10,5 kuukauden iän, 20 potilasta saavutti ≥ 14 kuukauden iän (rinnakkainen ensisijainen tehon päätetapahtuma) ja 20 potilasta saavutti 18 kuukauden iän ilman tapahtumia.
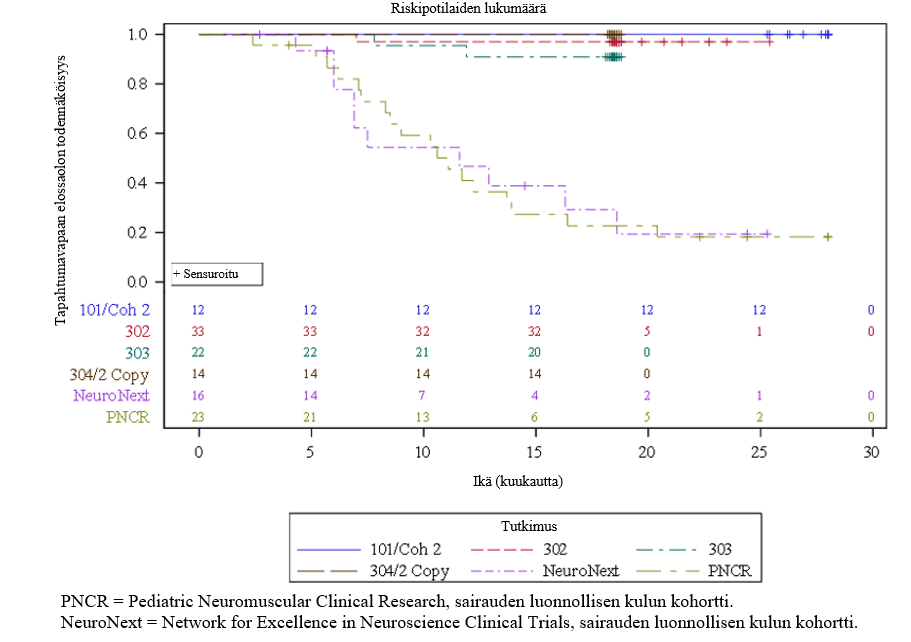
Kolme potilasta ei ollut tutkimuksessa mukana loppuun asti. Näistä 2:lla oli tapahtuma (kuolema tai jatkuva ventilaatio), mikä johti tapahtumavapaan elossaolon todennäköisyyteen 90,9 % (95 %:n luottamusväli: 79,7 %; 100,0 %) (elossa ilman jatkuvaa ventilaatiota) 14 kuukauden iässä, ks. kuva 1.
Kuva 1 Aika (kuukausina) ennen potilaan kuolemaa tai jatkuvaa ventilaatiota, yhdistetyt tiedot onasemnogeeniabeparvoveekki IV -tutkimuksista (CL-101, CL-302, CL‑303, CL-304:n 2 kopion kohortti)
Mediaani-ikä niillä CL-303-tutkimuksen 14 potilaalla, jotka saavuttivat ensimmäisinä kyvyn istua itsenäisesti ilman tukea vähintään 30 sekunnin ajan millä tahansa käynnillä tutkimuksen aikana, oli 12,6 kuukautta (aikaväli: 9,2 ja 18,6 kuukauden välillä). Kolmetoista potilasta (59,1 %) saavutti kyvyn istua itsenäisesti vähintään 30 sekunnin ajan, mikä todettiin 18 kuukauden iässä tehdyllä käynnillä (yhdistetty päätetapahtuma, p<0,0001). Yksi potilas saavutti kyvyn istua itsenäisesti 30 sekunnin ajan 16 kuukauden iässä, mutta tätä saavutusta ei ole vahvistettu 18 kuukauden iässä tehdyllä käynnillä. Yhteenveto videotallenteelta vahvistetuista saavutuksista kehityksessä CL-303-tutkimukseen osallistuneilla potilailla on esitetty taulukossa 4. Kolme potilasta ei saavuttanut mitään motorisista kehitystasoista (13,6 %) ja toiset 3 potilasta (13,6 %) saavutti pään hallintakyvyn, mikä oli heidän parhain motorinen kehitystaso, ennen viimeistä tutkimuskäyntiä 18 kuukauden ikäisenä.
Taulukko 4 Kulunut mediaaniaika videotallenteelle dokumentoituihin motorisiin kehitystasoihin, tutkimus CL‑303
| Videotallenteelle dokumentoitu kehitystaso | Kehitystason saavuttaneiden potilaiden lukumäärä n/N (%) | Mediaani-ikä kehitystason saavuttamiseen (kuukautta) | 95 %:n luottamusväli |
| Pään hallintakyky | 17/20* (85,0) | 6,8 | (4,77; 7,57) |
| Selinmakuulta kääntyminen kyljelleen | 13/22 (59,1) | 11,5 | (7,77; 14,53) |
| Kyky istua ilman tukea 30 sekunnin ajan (Beyley) | 14/22 (63,6) | 12,5 | (10,17; 15,20) |
| Kyky istua ilman tukea vähintään 10 sekunnin ajan (WHO) | 14/22 (63,6) | 13,9 | (11,00; 16,17) |
* 2 potilaalla ilmoitettiin olevan pään hallintakyky lähtötilanteessa tehdyn kliinisen arvion mukaan.
Yksi potilas (4,5 %) kykeni myös kävelemään tuen avulla 12,9 kuukauden iässä. Sairauden luonnollisen kulun mukaan potilaiden, jotka täyttivät tutkimukseen mukaanoton kriteerit, ei odoteta saavuttavan kykyä istua ilman tukea. Lisäksi 18 potilasta 22:sta ei ollut riippuvaisia ventilaatiotuesta 18 kuukauden iässä.
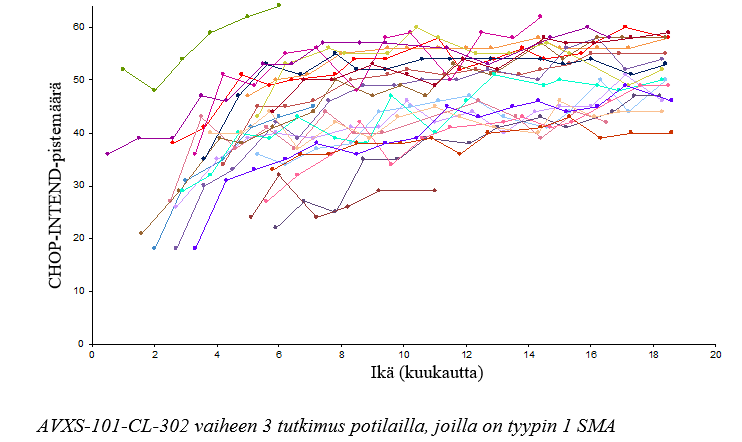
Liiketoiminnoissa havaittiin myös tapahtuneen parannuksia mitattuna CHOP-INTEND-pistemäärillä, ks. kuva 2. Kaksikymmentäyksi potilasta (95,5 %) saavutti CHOP-INTEND-pistemäärän ≥ 40, neljätoista potilasta (63,6 %) saavutti CHOP-INTEND-pistemäärän ≥ 50, ja yhdeksän potilasta (40,9 %) saavutti CHOP-INTEND-pistemäärän ≥ 58. Potilaat, joilla on hoitamaton tyypin 1 SMA eivät lähes koskaan saavuta CHOP-INTEND-pistemäärää ≥ 40. Motorisen kehitystason saavuttaminen havaittiin joillakin potilailla siitä huolimatta, että CHOP-INTEND-pistemäärät saavuttivat tasannevaiheen. Selvää yhteyttä CHOP-INTEND-pistemäärien ja motoristen kehitystasojen saavuttamisen välillä ei havaittu.
Kuva 2 Motorisen toimintakyvyn CHOP-INTEND-pistemäärät, tutkimus CL-303 (N=22)
AVXS-101-CL-302 (tutkimus CL-302) on vaiheen 3 avoin yksihaarainen kerta-annostutkimus laskimoon annetun onasemnogeeniabeparvoveekin hoitoannoksella (1,1 × 1014 vg/kg). Tutkimukseen otettiin 33 potilasta, joilla on tyypin 1 SMA ja kaksi SMN2-geenin kopiota. Yhdeksän potilaan (27,3 %) ilmoitettiin tarvinneen ventilaatiohoitoa ja yhdeksän potilaan (27,3 %) tukea ruokailuun ennen onasemnogeeniabeparvoveekkihoitoa. Lähtötilanteessa 33 potilaan keskimääräinen CHOP-INTEND-pistemäärä oli 27,9 (vaihteluväli 14–55). 33 potilaan keskimääräinen ikä hoidon antoajankohtana oli 4,1 kuukautta (vaihteluväli 1,8–6,0 kuukautta).
Tutkimukseen otetuista 33 potilaasta (Efficacy Completers -populaatio) yhden (3 %) ikä oli annoksen antohetkellä tutkimussuunnitelman mukaisen ikävälin ulkopuolella, joten potilasta ei otettu mukaan hoitoaikomuspopulaatioon (intent-to-treat [ITT] -populaatio). ITT-populaation 32 potilaasta yksi kuoli tutkimuksen aikana taudin etenemisen vuoksi.
ITT-populaation 32 potilaasta 14 (43,8 %) kykeni istumaan ilman tukea vähintään 10 sekunnin ajan millä tahansa tutkimuskäynnillä, mukaan lukien 18 kuukauden käynti (ensisijainen tehon päätetapahtuma). Tämä kehitystaso saavutettiin ensimmäisen kerran 15,9 kuukauden mediaani-iässä (vaihteluväli 7,7–18,6 kuukautta). 31 ITT-populaation potilasta (96,9 %) pärjäsi ilman jatkuvaa ventilaatiohoitoa (tapahtumavapaa elossaolo) 14 kuukauden ikään saakka tai kauemmin (toissijainen tehon päätetapahtuma).
Muut videotallenteelta vahvistetut saavutetut kehitystasot tutkimuksen CL-302 Efficacy Completers-populaation potilailla millä tahansa tutkimuskäynnillä, mukaan lukien 18 kuukauden käynti kuvataan taulukossa 5.
Taulukko 5 Kulunut mediaaniaika videotallenteelle dokumentoituihin motorisiin kehitystasoihin, tutkimus CL-302 (Efficacy Completers -populaatio)
| Videotallenteelle dokumentoitu kehitystaso | Kehitystason saavuttaneiden potilaiden lukumäärä n/N (%) | Mediaani-ikä kehitystason saavuttamiseen (kuukautta) | 95 %:n luottamusväli |
| Pään hallintakyky | 23/30* (76,7) | 8,0 | (5,8; 9,2) |
| Selinmakuulta kääntyminen kyljelleen | 19/33 (57,6) | 15,3 | (12,5; 17,4) |
| Kyky istua ilman tukea 30 sekunnin ajan | 16/33 (48,5) | 14,3 | (8,3; 18,3) |
* 3 potilaalla ilmoitettiin olevan pään hallintakyky lähtötilanteessa tehdyn kliinisen arvion mukaan.
Yksi potilas (3 %) saavutti ryömimisen, avustajan avulla seisomisen, omin avuin seisomisen, tuettuna kävelemisen ja omin avuin kävelemisen motoriset kehitystasot 18 kuukauden ikään mennessä.
33 mukaan otetusta potilaasta 24 (72,7 %) saavutti CHOP-INTEND-pistemäärän ≥ 40, 14 potilasta (42,4 %) CHOP-INTEND-pistemäärän ≥ 50 ja 3 potilasta (9,1 %) CHOP-INTEND-pistemäärän ≥ 58 (ks. kuva 3). Potilaat, joilla on hoitamaton tyypin 1 SMA eivät lähes koskaan saavuta CHOP-INTEND-pistemäärää ≥ 40.
Kuva 3 Motorisen toimintakyvyn CHOP-INTEND-pistemäärät, tutkimus CL-302 (Efficacy Completers -populaatio, N=33)*
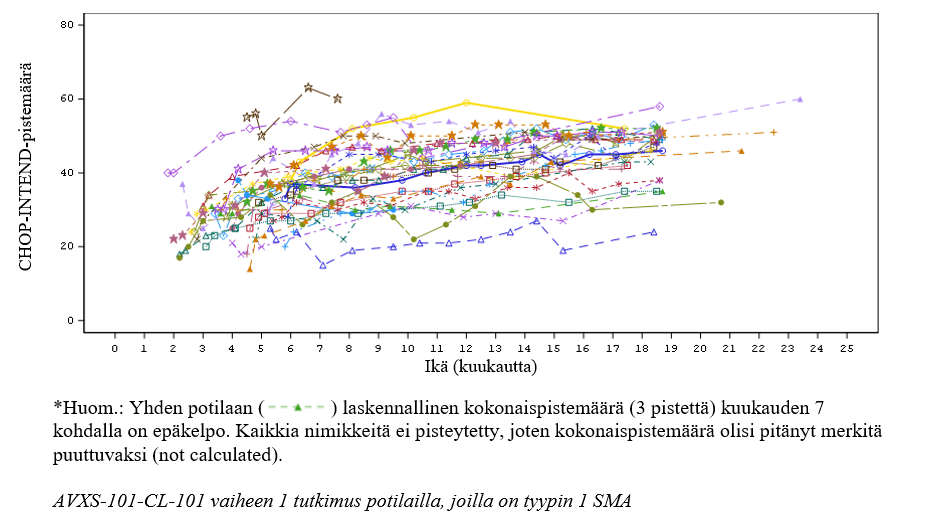
Tutkimuksessa CL‑303 saatuja tuloksia tukee AVXS-101-CL-101-tutkimus (tutkimus CL-101; vaiheen 1 tutkimus tyypin 1 SMA ‑potilailla), jossa onasemnogeeniabeparvoveekki annettiin kertainfuusiona laskimoon 12 potilaalle, joiden paino oli 3,6 kg–8,4 kg (ikä 0,9–7,9 kk). Saavuttaessaan 14 kuukauden iän kaikki hoitoa saaneet potilaat olivat tapahtumavapaita eli olivat elossa ilman jatkuvaa ventilaatiota verrattuna 25 prosenttiin sairauden luonnollisen kulun kohortissa. Tutkimuksen päätyttyä (24 kuukauden kuluttua annoksesta) kaikki hoitoa saaneet potilaat olivat tapahtumavapaita verrattuna alle 8 prosenttiin sairauden luonnollisen kulun kohortissa, ks. kuva 1.
Annoksen jälkeisessä seurannassa 24. kuukauteen mennessä 10 potilasta 12:sta kykeni istumaan ilman tukea ≥ 10 sekunnin ajan, 9 potilasta kykeni istumaan ilman tukea ≥ 30 sekunnin ajan ja 2 potilasta kykeni seisomaan ja kävelemään ilman avustajaa. Yksi potilas 12:sta ei saavuttanut pään hallintakykyä parhaimpana motorisena kehitystasona ennen 24 kuukauden ikää. Kymmenen CL-101-tutkimuksen 12 potilaasta jatkavat seurannassa pitkäaikaisessa tutkimuksessa (enintään 6,6 vuoden ajan annostelusta) ja kaikki kymmenen potilasta olivat elossa ilman jatkuvaa ventilaatiota tarkasteluajankohtana 23.5.2021. Kaikki potilaat ovat joko säilyttäneet aikaisemmat kehitystasot tai yltäneet uusiin kehitystasoihin, esimerkiksi istuminen tuen avulla, seisominen avustajan avulla ja kävely omin avuin. Viisi näistä 10 potilaasta sai samanaikaista nusinerseeni- tai risdiplaamihoitoa tämän pitkäaikaisen tutkimuksen jossakin vaiheessa. Tehon ylläpidon ja kehitystasojen saavuttamisen ei siksi voida sanoa olevan yksin onasemnogeeniabeparvoveekin ansiota kaikilla potilailla. Seisominen avustajan avulla oli uusi saavutettu kehitystaso 2 potilaalla, jotka eivät olleet saaneet nusinerseeniä tai risdiplaamia missään vaiheessa ennen tämän kehitystason saavuttamisajankohtaa.
AVXS-101-CL-304 vaiheen 3 tutkimus potilailla, joilla on presymptomaattinen SMA
CL-304-tutkimus on globaali, vaiheen 3, avoin, yksihaarainen, yhden annoksen tutkimus, jossa annetaan onasemnogeeniabeparvoveekkia laskimoon presymptomaattisille vastasyntyneille potilaille, jotka ovat iältään korkeintaan 6 viikkoa ja joilla on kaksi (kohortti 1, n=14) tai kolme (kohortti 2, n=15) SMN2-kopiota.
Kohortti 1
Neljäätoista hoidettua potilasta, joilla oli kaksi SMN2-kopiota, seurattiin 18 kuukauden ikään saakka. Kaikki potilaat pysyivät tapahtumavapaina ≥ 14 kuukauden ikään saakka eivätkä tarvinneet jatkuvaa ventilaatiota.
Kaikki neljätoista potilasta saavuttivat kyvyn istua omin avuin vähintään 30 sekunnin ajan 5,7–11,8 kuukauden iässä, kun huomioitiin kaikki käynnit 18 kuukauden ikäkäyntiin asti (tehon ensisijainen päätetapahtuma). Näistä 14 potilaasta 11 saavutti kyvyn istua omin avuin ennen 279 päivän ikää tai kyseisessä iässä, mikä oli 99 persentiiliä tämän kehitystason saavuttamisesta. Yhdeksän potilasta saavutti kyvyn kävellä omin avuin (64,3 %). Kaikki neljätoista potilasta saavuttivat CHOP-INTEND-pistemäärän ≥ 58, kun huomioitiin kaikki käynnit 18 kuukauden ikäkäyntiin asti. Yksikään potilaista ei tarvinnut tutkimuksen aikana ventilaatiotukea eikä ravitsemustukea.
Kohortti 2
Viittätoista hoidettua potilasta, joilla oli kolme SMN2-kopiota, seurattiin 24 kuukauden ikään saakka. Kaikki potilaat pysyivät tapahtumavapaina 24 kuukauden ikään saakka eivätkä tarvinneet jatkuvaa ventilaatiota.
Kaikki 15 potilasta kykenivät seisomaan omin avuin ilman tukea vähintään 3 sekunnin ajan (tehon ensisijainen päätetapahtuma) 9,5–18,3 kuukauden iässä. Näistä 15 potilaasta 14 saavutti kyvyn seistä ilman tukea ennen 514 päivän ikää tai kyseisessä iässä, mikä oli 99 persentiiliä tämän kehitystason saavuttamisesta. Neljätoista potilasta (93,3 %) kykeni ottamaan vähintään viisi askelta omin avuin. Kaikki 15 potilasta saavuttivat skaalatun Bayley-III-pistemäärän ≥ 4 karkea- ja hienomotoristen taitojen alatesteissä (2 keskihajonnan sisällä ikäkeskiarvosta), kun huomioitiin kaikki lähtötilanteen jälkeiset käynnit 24 kuukauden ikään saakka. Yksikään potilaista ei tarvinnut tutkimuksen aikana ventilaatiotukea eikä ravitsemustukea.
COAV101A12306 vaiheen 3 tutkimus SMA-potilailla, joiden paino oli ≥ 8,5 – ≤ 21 kg
Tutkimus COAV101A12306 on loppuun saatettu, vaiheen 3 avoin, yksihaarainen, yhden annoksen monikeskustutkimus. Tutkimuksessa onasemnogeeniabeparvoveekki-hoitoannos (1,1 × 1014 vg/kg) annettiin laskimoon 24 pediatriselle SMA-potilaalle, joiden paino oli ≥ 8,5 – ≤ 21 kg (mediaanipaino: 15,8 kg). Potilaiden ikä antohetkellä vaihteli noin 1,5 vuodesta 9 vuoteen. Potilailla oli 2–4 SMN2-kopiota (kaksi [n = 5], kolme [n = 18], neljä [n = 1] kopiota). Ennen onasemnogeeniabeparvoveekki-hoitoa 19 potilasta 24:stä oli aiemmin saanut nusinerseenia keskimäärin 2,1 vuoden ajan (vaihteluväli 0,17–4,81 vuotta) ja kaksi potilasta 24:stä oli aiemmin saanut risdiplamia keskimäärin 0,48 vuoden ajan (vaihteluväli 0,11–0,85 vuotta). Lähtötilanteessa potilaiden Hammersmith Functional Motor Scale - Expanded (HFMSE) -pisteiden keskiarvo oli 28,3 ja Revised Upper Limb Module (RULM) -pisteiden keskiarvo 22,0. Lisäksi kaikki potilaat olivat saavuttaneet pään hallintakyvyn ja tuen avulla istumisen kehitystasot, 21 pystyi istumaan ilman tukea ja kuusi potilasta oli saavuttanut korkeimmat mahdolliset saavutettavissa olevat, omin avuin seisomisen ja omin avuin kävelemisen kehitystasot.
Viikolla 52 HFMSE-kokonaispistemäärän keskimääräinen muutos lähtötilanteesta oli 3,7 (18 potilasta 24:stä). RULM-kokonaispistemäärän keskimääräinen nousu oli 2,0 (17 potilasta 24:stä) viikolla 52. Neljä potilasta saavutti uusia kehitystasoja. Lähtötilanteen käynnillä havaitut kehitystasot säilyivät suurimmalla osalla potilaista viikkoon 52. Kahdella potilaalla, jotka eivät osoittaneet aiemmin saavuttamiaan kehitystasoja, HFMSE-pisteet paranivat lähtötilanteesta viikkoon 52.
Onasemnogeeniabeparvoveekkia ei ole tutkittu potilailla, joilla on SMN1-geenin bialleelinen mutaatio ja vain yksi SMN2-kopio kliinisissä tutkimuksissa.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset onasemnogeeniabeparvoveekin käytöstä spinaalisen lihasatrofian hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Onasemnogeeniabeparvoveekin vektorin erittymistutkimuksissa on arvioitu elimistöstä syljen, virtsan, ulosteen ja nenän eritteiden mukana poistuvien vektorien määrää.
Onasemnogeeniabeparvoveekkivektori-DNA:ta oli havaittavissa infuusion jälkeisissä eritenäytteissä. Onasemnogeeniabeparvoveekkia erittyi ensisijaisesti ulosteen kautta. Useimmilla potilailla suurin erittymisen määrä infuusion jälkeen havaittiin ulosteissa 7 vuorokauden kuluessa, ja syljessä, virtsassa ja nenän eritteissä 2 vuorokauden kuluessa. Suurin osa vektorista poistui 30 vuorokauden kuluessa annoksen antamisesta.
Biodistribuutiota arvioitiin kahdella potilaalla, jotka kuolivat 5,7 kuukauden ja 1,7 kuukauden iässä, kun onasemnogeeniabeparvoveekkia oli infusoitu annoksella 1,1 x 1014 vg/kg. Molemmat tapaukset osoittivat suurimpien vektori-DNA-pitoisuuksien löytyvän maksasta. Vektori-DNA:ta todettiin myös pernasta, sydämestä, haimasta, nivuksien imurauhasista, luustolihaksista, perifeerisistä hermoista, munuaisista, keuhkoista, suolistosta, sukupuolirauhasista, selkäytimestä, aivoista ja kateenkorvasta. SMN-proteiinin värjäys osoitti yleistynyttä SMN-ekspressiota selkäytimen liikehermosoluissa, aivojen hermo- ja gliasoluissa sekä sydämessä, maksassa, luustolihaksissa ja muissa analysoiduissa kudoksissa.
Prekliiniset tiedot turvallisuudesta
Vastasyntyneelle hiirelle laskimoon annon jälkeen vektorin jakaantuminen oli laajaa, ja eniten vektori-DNA:ta havaittiin yleensä sydämessä, maksassa, keuhkoissa ja luustolihaksissa. Transgeeninen mRNA ilmentyi vastaavalla tavalla. Nuorille kädellisille laskimoon annon jälkeen vektorin jakaantuminen oli laajaa ja sitä seurasi transgeenisen mRNA:n ilmentyminen. Vektori-DNA:n ja transgeenisen mRNA:n pitoisuudet olivat yleensä suurimmat maksassa, lihaksissa ja sydämessä. Vektori-DNA:ta ja transgeenistä mRNA:ta havaittiin molempien lajien selkäytimessä, aivoissa ja sukurauhasissa.
Keskeisissä, 3 kuukauden pituisissa toksikologisissa tutkimuksissa hiirellä toksisuuden pääasialliset kohde-elimet olivat sydän ja maksa. Onasemnogeeniabeparvoveekkiin liittyvät löydökset sydänkammioissa olivat annosriippuvainen tulehdus, edeema ja fibroosi. Sydämen eteisissä niitä olivat tulehdus, tromboosi, sydänlihaksen rappeuma/nekroosi ja fibroplasia. Taso, joka ei aiheuta havaittavaa haittavaikutusta (NoAEL), ei ollut tutkimuksissa tunnistettavissa onasemnogeeniabeparvoveekin käytössä hiirellä, sillä pienintä annosta käyttämällä (1,5 × 1014 vg/kg) havaitut vaikutukset olivat kammion sydänlihaksen tulehdus/edeema/fibroosi ja sydämen eteisen tulehdus. Tätä annosta pidetään suurimpana siedettynä annoksena ja se on noin 1,4-kertainen suositeltuun kliiniseen annokseen verrattuna. Onasemnogeeniabeparvoveekin aiheuttamaan kuolleisuuteen suurimmalla osalla hiiristä liittyi sydämen eteisten tukos, ja sitä esiintyi annoksella 2,4 × 1014 vg/kg. Kuolleisuuden syy muilla eläimillä ei ole selvä, mutta näiden eläinten sydämen mikroskooppista degeneraatiota/regeneraatiota on todettu.
Hiiren maksassa havaittiin maksasolujen hypertrofiaa, Kupfferin solujen aktivoitumista ja hajanaista maksasolujen nekroosia. Pitkäaikaistoksisuustutkimuksissa, joissa onasemnogeeniabeparvoveekkia annettiin nuorille kädellisille laskimoon ja (käyttöaiheen vastaisesti) selkäydinnesteeseen, maksan löydökset, mm. yksittäisten maksasolujen nekroosi ja soikeiden solujen liikakasvu, olivat osittain (anto laskimoon) tai kokonaan (anto selkäydinnesteeseen) palautuvia.
Kuusi kuukautta kestäneessä toksikologisessa tutkimuksessa nuorille kädellisille laskimoon annettu, kliinisesti suositellun suuruinen kerta-annos onasemnogeeniabeparvoveekkia, yhdessä kortikosteroidihoidon kanssa tai ilman, aiheutti akuutin, minimaalisen tai lievän mononukleaarisoluvälitteisen tulehdusreaktion sekä hermosolujen degeneraatiota selkäydinhermosolmussa ja puolikuuhermosolmussa sekä selkäytimen aksonien degeneraatiota ja/tai glioosia. Kuuden kuukauden kohdalla nämä ei-progressiiviset löydökset olivat täysin hävinneet puolikuuhermosolmun osalta ja osittain hävinneet (alhaisempi esiintyvyys ja/tai vaikeusaste) selkäydinhermosolmun ja selkäytimen osalta. Onasemnogeeniabeparvoveekin intratekaalisen annon jälkeen (käyttöaiheen vastaisesti) näitä akuutteja, ei-progressiivisia löydöksiä havaittiin nuorilla kädellisillä. Löydösten vaikeusaste vaihteli minimaalisesta keskivaikeaan ja löydökset olivat osittain tai täysin hävinneet 12 kuukauden kohdalla. Nämä kädellisillä havaitut löydökset eivät korreloineet kliinisten havaintojen kanssa, ja siksi niiden kliinistä merkitsevyyttä ihmiselle ei tunneta.
Onasemnogeeniabeparvoveekin genotoksisuutta, karsinogeenisuutta ja lisääntymistoksisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Trometamiini
Magnesiumkloridi
Natriumkloridi
Poloksameeri 188
Suolahappo (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta
Sulatuksen jälkeen
Kun lääkevalmiste on sulanut, sitä ei saa pakastaa uudelleen, vaan se on säilytettävä jääkaapissa 2 °C‑8 °C:n lämpötilassa alkuperäisessä pakkauksessa 14 vuorokauden ajan.
Kun annosmäärä on vedetty ruiskuun, se on infusoitava 8 tunnin kuluessa. Hävitä vektoria sisältävä ruisku, jos sitä ei ole infusoitu 8 tunnin sisällä.
Säilytys
Säilytä ja kuljeta pakastettuna (≤ -60 °C).
Säilytä jääkaapissa (2 °C‑8 °C) välittömästi vastaanoton jälkeen.
Säilytä alkuperäispakkauksessa.
Sulatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Vastaanottopäivämäärä on merkittävä alkuperäiseen pakkaukseen ennen kuin valmiste laitetaan jääkaappiin.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZOLGENSMA infuusioneste, liuos
2 x 10exp13 vektorigenomia/ml (L:ei) 2x5,5 ml+10x8,3 ml (-), 5,5 ml+10x8,3 ml (-), 2x5,5 ml+11x8,3 ml (-), 5,5 ml+11x8,3 ml (-), 2x5,5 ml+12x8,3 ml (-), 5,5 ml+12x8,3 ml (-), 5,5 ml+13x8,3 ml (-), 2x5,5 ml+2x8,3 ml (-), 5,5 ml+2x8,3 ml (-), 2x5,5 ml+3x8,3 ml (-), 5,5 ml+3x8,3 ml (-), 2x5,5 ml+4x8,3 ml (-), 5,5 ml+4x8,3 ml (-), 2x5,5 ml+5x8,3 ml (-), 5,5 ml+5x8,3 ml (-), 2x5,5 ml+6x8,3 ml (-), 5,5 ml+6x8,3 ml (-), 2x5,5 ml+7x8,3 ml (-), 5,5 ml+7x8,3 ml (-), 2x5,5 ml+8,3 ml (-), 2x5,5 ml+8x8,3 ml (-), 5,5 ml+8x8,3 ml (-), 2x5,5 ml+9x8,3 ml (-), 5,5 ml+9x8,3 ml (-), 2 x 8,3 ml (-), 3 x 8,3 ml (-), 4 x 8,3 ml (-), 5 x 8,3 ml (-), 6 x 8,3 ml (-), 7 x 8,3 ml (-), 8 x 8,3 ml (-), 9 x 8,3 ml (-), 10 x 8,3 ml (-), 11 x 8,3 ml (-), 12 x 8,3 ml (-), 13 x 8,3 ml (-), 14 x 8,3 ml (-)
PF-selosteen tieto
Onasemnogeeniabeparvoveekki toimitetaan injektiopullossa (10 ml, polymeeri Crystal Zenith), joka on varustettu tulpalla (20 mm:n paksuinen klorobutyylikumi) ja sinetillä (alumiininen, repäistävä), jossa on värillinen (muovi-) korkki, kahdessa eri volyymikoossa, sisältäen joko 5,5 ml tai 8,3 ml.
Onasemnogeeniabeparvoveekin annos ja injektiopullojen tarkka tarvittava lukumäärä kullekin potilaalle lasketaan potilaan painon mukaan (ks. kohta Annostus ja antotapa ja taulukko 6 alla).
Taulukko 6 Pakkausten sisällön määrä
| Potilaan paino (kg) | 5,5 ml:n injektiopulloa | 8,3 ml:n injektiopullob | Pakkauksen sisältämien injektiopullojen kokonaislukumäärä |
| 2,6–3,0 | 0 | 2 | 2 |
| 3,1–3,5 | 2 | 1 | 3 |
| 3,6–4,0 | 1 | 2 | 3 |
| 4,1–4,5 | 0 | 3 | 3 |
| 4,6–5,0 | 2 | 2 | 4 |
| 5,1–5,5 | 1 | 3 | 4 |
| 5,6–6,0 | 0 | 4 | 4 |
| 6,1–6,5 | 2 | 3 | 5 |
| 6,6–7,0 | 1 | 4 | 5 |
| 7,1–7,5 | 0 | 5 | 5 |
| 7,6–8,0 | 2 | 4 | 6 |
| 8,1–8,5 | 1 | 5 | 6 |
| 8,6–9,0 | 0 | 6 | 6 |
| 9,1–9,5 | 2 | 5 | 7 |
| 9,6–10,0 | 1 | 6 | 7 |
| 10,1–10,5 | 0 | 7 | 7 |
| 10,6–11,0 | 2 | 6 | 8 |
| 11,1–11,5 | 1 | 7 | 8 |
| 11,6–12,0 | 0 | 8 | 8 |
| 12,1–12,5 | 2 | 7 | 9 |
| 12,6–13,0 | 1 | 8 | 9 |
| 13,1–13,5 | 0 | 9 | 9 |
| 13,6–14,0 | 2 | 8 | 10 |
| 14,1–14,5 | 1 | 9 | 10 |
| 14,6–15,0 | 0 | 10 | 10 |
| 15,1–15,5 | 2 | 9 | 11 |
| 15,6–16,0 | 1 | 10 | 11 |
| 16,1–16,5 | 0 | 11 | 11 |
| 16,6–17,0 | 2 | 10 | 12 |
| 17,1–17,5 | 1 | 11 | 12 |
| 17,6–18,0 | 0 | 12 | 12 |
| 18,1–18,5 | 2 | 11 | 13 |
| 18,6–19,0 | 1 | 12 | 13 |
| 19,1–19,5 | 0 | 13 | 13 |
| 19,6–20,0 | 2 | 12 | 14 |
| 20,1–20,5 | 1 | 13 | 14 |
| 20,6–21,0 | 0 | 14 | 14 |
a Injektiopullon sisältämä nimellinen pitoisuus on 2 × 1013 vg/ml ja sen sisältämä ruiskuun vedettävä määrä on vähintään 5,5 ml.
b Injektiopullon sisältämä nimellinen pitoisuus on 2 × 1013 vg/ml ja sen sisältämä ruiskuun vedettävä määrä on vähintään 8,3 ml.
Valmisteen kuvaus:
Kirkas tai lähes opaakki, väritön tai himmeän valkoinen liuos.
Käyttö- ja käsittelyohjeet
Injektiopullojen vastaanottaminen ja sulattaminen
- Injektiopullot kuljetetaan pakastettuina (≤‑60 ºC:n lämpötilassa). Vastaanottamisen jälkeen injektiopullot on välittömästi laitettava jääkaappiin 2 °C‑8 °C:n lämpötilaan alkuperäisessä pakkauksessa. Onasemnogeeniabeparvoveekkihoito on aloitettava 14 vuorokauden kuluessa injektiopullojen vastaanottamisesta.
- Injektiopullot on sulatettava ennen käyttöä. Älä käytä onasemnogeeniabeparvoveekkia, ellei se ole sulanut.
- Enintään 9 injektiopulloa sisältävissä pakkauksissa oleva valmiste on sulanut noin 12 tunnin kuluttua jääkaapissa. Enintään 14 injektiopulloa sisältävissä pakkauksissa oleva valmiste on sulanut noin 16 tunnin kuluttua jääkaapissa. Vaihtoehtoisesti ja välitöntä käyttöä varten valmisteen voi antaa sulaa huoneenlämpötilassa.
- Enintään 9 injektiopulloa sisältävissä pakkauksissa oleva valmiste on sulanut pakasteesta noin 4 tunnin kuluttua huoneen lämpötilassa (20 °C‑25 °C). Enintään 14 injektiopulloa sisältävissä pakkauksissa oleva valmiste on sulanut noin 6 tunnin kuluttua huoneenlämpötilassa (20 °C‑25 °C)
- Ennen annosmäärän vetämistä ruiskuun, pyöritä sulanutta valmistetta kevyesti. ÄLÄ ravistele.
- Älä käytä tätä lääkettä, jos huomaat siinä partikkeleita tai värjääntymistä sulatuksen jälkeen ja ennen antamista.
- Kun lääkevalmiste on sulatettu, sitä ei pidä pakastaa uudelleen.
- Onasemnogeeniabeparvoveekki on sulatuksen jälkeen annettava potilaalle mahdollisimman pian. Kun annosmäärä on vedetty ruiskuun, se on annettava 8 tunnin kuluessa. Hävitä vektoria sisältävä ruisku, ellei sitä ole infusoitu 8 tunnin kuluessa.
Onasemnogeeniabeparvoveekin antaminen potilaalle
- Vedä koko vedettävä annosmäärä injektiopullosta ruiskuun onasemnogeeniabeparvoveekin antamiseksi. Poista ilma ruiskusta ennen laskimoinfuusion antoa potilaalle käyttämällä laskimokatetria.
Varotoimenpiteet, joita on noudatettava lääkevalmisteen käsittelyssä, hävittämisessä ja vahingossa tapahtuneessa altistumisessa
Tämä lääkevalmiste sisältää geenimuunneltuja organismeja. Asianmukaisia varotoimenpiteitä liittyen onasemnogeeniabeparvoveekin käsittelyyn, hävittämiseen ja vahingossa tapahtuneeseen altistumiseen on noudatettava:
- Onasemnogeeniabeparvoveekkia sisältävää ruiskua on käsiteltävä aseptisesti steriileissä olosuhteissa.
- Henkilökohtaisia suojavarusteita (ml. käsineet, suojalasit, laboratoriotakki ja hihat) on käytettävä onasemnogeeniabeparvoveekkia käsiteltäessä ja annettaessa potilaalle. Hoitohenkilökunnan jäsenen, jonka ihossa on haavoja tai naarmuja, ei pidä käsitellä onasemnogeeniabeparvoveekkia.
- Kaikki onasemnogeeniabeparvoveekin roiskeet on pyyhittävä imukykyisellä harsotaitoksella ja roiskealue on desinfioitava käyttämällä natriumhypokloriittiliuosta ja sen jälkeen alkoholilla kostutettuja puhdistuspyyhkeitä. Kaikki puhdistusmateriaalit on laitettava kaksinkertaiseen pussiin ja hävitettävä noudattaen biologisten jätteiden käsittelystä annettuja paikallisia ohjeita.
- Käyttämätön lääkevalmiste tai jäte on hävitettävä biologisten jätteiden käsittelystä annettujen paikallisten ohjeiden mukaisesti.
- Kaikki materiaalit, jotka ovat saattaneet tulla kosketukseen onasemnogeeniabeparvoveekin kanssa (esim. injektiopullo, kaikki injektioon käytetyt materiaalit, mukaan luettuna steriilit liinat ja neulat), on hävitettävä biologisten jätteiden käsittelystä annettujen paikallisten ohjeiden mukaisesti.
- Vahingossa tapahtuvaa altistumista onasemnogeeniabeparvoveekille on vältettävä. Ihoaltistumisen tapauksessa altistunut alue on puhdistettava perusteellisesti vedellä ja saippualla vähintään 15 minuutin ajan. Jos silmät ovat altistuneet, silmät on huuhdeltava huolellisesti vedellä vähintään 15 minuutin ajan.
Erittyminen
Onasemnogeeniabeparvoveekin ohimenevää erittymistä saattaa esiintyä, pääasiassa elimistön eritteiden mukana. Huoltajia ja potilaiden perheitä on kehotettava noudattamaan seuraavia ohjeita potilaan elimistön nesteiden ja eritteiden asianmukaisesta käsittelystä.
- Hyvä käsihygienia on välttämätöntä (suojakäsineiden käyttö ja käsien huolellinen pesu juoksevalla lämpimällä vedellä ja saippualla käsittelyn jälkeen tai käyttämällä alkoholipohjaista käsien desinfiointiainetta), kun joudutaan suoraan kontaktiin potilaan elimistön nesteiden ja eritteiden kanssa vähintään 1 kuukauden ajan onasemnogeeniabeparvoveekkihoidon jälkeen.
- Kertakäyttövaipat on laitettava sinetöitäviin kaksinkertaisiin muovipusseihin, jotka voidaan hävittää talousjätteen mukana.
Korvattavuus
ZOLGENSMA infuusioneste, liuos
2 x 10exp13 vektorigenomia/ml 2x5,5 ml+10x8,3 ml, 5,5 ml+10x8,3 ml, 2x5,5 ml+11x8,3 ml, 5,5 ml+11x8,3 ml, 2x5,5 ml+12x8,3 ml, 5,5 ml+12x8,3 ml, 5,5 ml+13x8,3 ml, 2x5,5 ml+2x8,3 ml, 5,5 ml+2x8,3 ml, 2x5,5 ml+3x8,3 ml, 5,5 ml+3x8,3 ml, 2x5,5 ml+4x8,3 ml, 5,5 ml+4x8,3 ml, 2x5,5 ml+5x8,3 ml, 5,5 ml+5x8,3 ml, 2x5,5 ml+6x8,3 ml, 5,5 ml+6x8,3 ml, 2x5,5 ml+7x8,3 ml, 5,5 ml+7x8,3 ml, 2x5,5 ml+8,3 ml, 2x5,5 ml+8x8,3 ml, 5,5 ml+8x8,3 ml, 2x5,5 ml+9x8,3 ml, 5,5 ml+9x8,3 ml, 2 x 8,3 ml, 3 x 8,3 ml, 4 x 8,3 ml, 5 x 8,3 ml, 6 x 8,3 ml, 7 x 8,3 ml, 8 x 8,3 ml, 9 x 8,3 ml, 10 x 8,3 ml, 11 x 8,3 ml, 12 x 8,3 ml, 13 x 8,3 ml, 14 x 8,3 ml
- Ei korvausta.
ATC-koodi
M09AX09
Valmisteyhteenvedon muuttamispäivämäärä
12.06.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com