
TEZSPIRE injektioneste, liuos, esitäytetty kynä 210 mg, injektioneste, liuos, esitäytetty ruisku 210 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 210 mg tetsepelumabia 1,91 ml:ssa liuosta (110 mg/ml).
Esitäytetty kynä
Yksi esitäytetty kynä sisältää 210 mg tetsepelumabia 1,91 ml:ssa liuosta (110 mg/ml).
Tetsepelumabi on humaani monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 48 mg L‑proliinia ja 0,19 mg polysorbaatti 80:tä per 210 mg:n annos (1,91 ml).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Astma
Tezspire on tarkoitettu lisälääkkeeksi vaikean astman ylläpitohoitoon aikuisille ja vähintään 12‑vuotiaille nuorille, joilla ei ole saavutettu riittävää hoitotasapainoa suurella annoksella annetun inhaloitavan kortikosteroidin ja jonkin muun ylläpitohoitoon tarkoitetun lääkevalmisteen yhdistelmän käytöstä huolimatta.
Krooninen rinosinuiitti, johon liittyy nenäpolyyppeja (krooninen polypoottinen rinosinuiitti)
Tezspire on tarkoitettu intranasaalisten kortikosteroidien lisälääkkeeksi vaikean kroonisen polypoottisen rinosinuiitin hoitoon aikuisille, joiden tautia ei saada riittävän hyvin hallintaan systeemisillä kortikosteroideilla ja/tai leikkaushoidolla.
Ehto
Hoidon saa aloittaa vain vaikean astman diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon saa aloittaa vain lääkäri, jolla on kokemusta niiden sairauksien diagnosoinnista ja hoidosta, joihin Tezspire on tarkoitettu (ks. kohta Käyttöaiheet).
Annostus
Tezspire on tarkoitettu pitkäaikaishoitoon. Päätös hoidon jatkamisesta on tehtävä vähintään kerran vuodessa, ja päätöksen on perustuttava potilaan sairauden hoitotasapainon arviointiin.
Astma
Aikuiset ja nuoret (vähintään 12‑vuotiaat)
Suositeltu annos on 210 mg tetsepelumabia injektiona ihon alle 4 viikon välein.
Krooninen polypoottinen rinosinuiitti
Suositeltu annos aikuisille potilaille on 210 mg tetsepelumabia injektiona ihon alle 4 viikon välein.
Annoksen jääminen väliin
Jos annos jää väliin, se on annettava mahdollisimman pian. Sen jälkeen potilas voi jatkaa hoitoa ottamalla seuraavan annoksen aikataulun mukaisena päivänä. Jos on jo seuraavan annoksen aika, se annetaan suunnitelman mukaan. Kaksinkertaista annosta ei saa antaa.
Erityiset potilasryhmät
Iäkkäät (≥ 65‑vuotiaat)
Annoksen muuttaminen ei ole tarpeen iäkkäillä potilailla (ks. kohta Farmakokinetiikka).
Munuaisten tai maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen munuaisten tai maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Tezspire-valmisteen turvallisuutta ja tehoa alle 12‑vuotiaiden lasten astman hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Tezspire-valmisteen turvallisuutta ja tehoa alle 18‑vuotiaiden lasten kroonisen polypoottisen rinosinuiitin hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tezspire annetaan injektiona ihon alle.
Potilas itse tai potilaan huoltaja voi antaa tämän lääkevalmisteen, kun hänelle on neuvottu, miten ihonalaiset injektiot annetaan. Ennen kuin Tezspire annetaan käyttöohjeiden mukaan, potilaalle ja/tai hänen huoltajalleen on annettava asianmukainen opastus Tezspire-valmisteen saattamisesta käyttökuntoon ja antamisesta.
Tezspire annetaan injektiona ihon alle reiteen tai vatsaan, mutta vähintään 5 cm:n etäisyydelle navasta. Jos terveydenhuollon ammattilainen tai huoltaja antaa injektion, voidaan käyttää myös olkavartta. Potilas ei saa itse pistää olkavarteen. Valmistetta ei saa pistää aristaville, punoittaville tai kovettuneille ihoalueille eikä alueille, joilla on mustelmia. Pistoskohdan vaihtamista suositellaan jokaisella pistoskerralla.
Käyttöohjeissa on tarkat ohjeet valmisteen antamisesta esitäytetyllä ruiskulla tai esitäytetyllä kynällä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Astman akuutit pahenemisvaiheet
Tezspire-valmistetta ei pidä käyttää astman akuuttien pahenemisvaiheiden hoitoon.
Hoidon aikana saattaa ilmetä astmaan liittyviä oireita tai pahenemisvaiheita. Potilaita on neuvottava ottamaan yhteys lääkäriin, jos astma ei ole hoitotasapainossa tai pahenee hoidon aloittamisen jälkeen.
Kortikosteroidit
Kortikosteroidien käytön äkillinen lopettaminen Tezspire-hoidon aloittamisen jälkeen ei ole suositeltavaa. Jos kortikosteroidiannoksen pienentäminen on tarpeen, se on tehtävä asteittain ja lääkärin valvonnassa.
Yliherkkyysreaktiot
Tetsepelumabin antamisen jälkeen saattaa ilmetä yliherkkyysreaktioita (esim. anafylaksia tai ihottumaa) (ks. kohta Haittavaikutukset). Tällaiset reaktiot saattavat ilmetä tuntien kuluessa antamisesta, mutta joissakin tapauksissa ne ilmaantuvat viiveellä (vuorokausien kuluessa).
Aiempi tetsepelumabiin liittymätön anafylaksi saattaa olla Tezspire-valmisteen antamisen jälkeisen anafylaksin riskitekijä. Kliinisen käytännön mukaisesti potilaiden tilaa on seurattava riittävän pitkään Tezspire-valmisteen antamisen jälkeen.
Jos potilaalla ilmenee vakava yliherkkyysreaktio (esim. anafylaksi), tetsepelumabihoito on keskeytettävä välittömästi ja on aloitettava asianmukainen hoito kliinisten oireiden mukaan.
Vakavat infektiot
Kateenkorvan strooman lymfopoietiinin (TSLP) estäminen voi teoriassa suurentaa vakavien infektioiden riskiä. Lumekontrolloiduissa tutkimuksissa ei todettu vakavien infektioiden lisääntymistä tetsepelumabihoidon yhteydessä.
Potilaan mahdollinen vakava infektio on hoidettava ennen tetsepelumabihoidon aloittamista. Jos potilas saa vakavan infektion tetsepelumabihoidon aikana, tetsepelumabihoito on keskeytettävä, kunnes vakava infektio on hävinnyt.
Vakavat sydäntapahtumat
Pitkän aikavälin kliinisessä tutkimuksessa tetsepelumabihoitoa saaneilla potilailla todettiin numeerisesti enemmän vakavia sydämeen liittyviä haittatapahtumia kuin lumelääkettä saaneilla. Syy-yhteyttä tetsepelumabin käytön ja näiden tapahtumien välillä ei ole varmistettu, eikä ole tunnistettu potilasryhmää, jossa näiden tapahtumien riski olisi suurentunut.
Potilaille on kerrottava sydäntapahtumaan viittaavista merkeistä ja oireista (esimerkiksi rintakipu, hengenahdistus, yleinen sairauden tunne, huimaus tai pyörrytys), ja heitä on neuvottava hakeutumaan välittömästi lääkärin hoitoon, jos tällaisia oireita ilmenee. Jos potilaalla ilmenee vakava sydäntapahtuma tetsepelumabihoidon aikana, tetsepelumabihoito on keskeytettävä, kunnes akuutti tapahtuma on saatu hallintaan.
Tällä hetkellä ei ole tietoa uusintahoidosta potilailla, joilla ilmenee vakava sydäntapahtuma tai vakava infektio.
Loisinfektio (matoinfektio)
TSLP saattaa olla osallisena immunologisessa vasteessa joillekin loismatoinfektioille. Potilaat, joilla oli todettu loismatoinfektio, suljettiin pois kliinisistä tutkimuksista. Ei tiedetä, voiko tetsepelumabi vaikuttaa potilaan vasteeseen loismatoinfektioita kohtaan.
Potilaan mahdollinen loismatoinfektio on hoidettava ennen tetsepelumabihoidon aloittamista. Jos potilas saa infektion tetsepelumabihoidon aikana eikä loismatolääkkeillä saada hoitovastetta, tetsepelumabihoito on keskeytettävä, kunnes infektio on hävinnyt.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per 210 mg:n annos eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää 48 mg L‑proliinia per 210 mg:n annos (1,91 ml). L‑proliini voi olla haitallista potilaille, joilla on hyperprolinemia.
Tämä lääkevalmiste sisältää 0,19 mg polysorbaatti 80:tä per 210 mg:n annos (1,91 ml). Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Eläviä heikennettyjä taudinaiheuttajia sisältävien rokotteiden käyttöä on vältettävä tetsepelumabia saavilla potilailla.
Satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa tutkimuksessa 70 potilaalla, joiden ikä oli 12–21 vuotta ja joilla oli keskivaikea tai vaikea astma, tetsepelumabihoito ei näyttänyt vaikuttavan nelivalenttisella kausi-influenssarokotuksella aikaansaatuihin humoraalisiin vasta-ainevasteisiin.
Tetsepelumabilla ei odoteta olevan kliinisesti merkittäviä vaikutuksia samanaikaisesti annettujen muiden astman hoitoon käytettävien lääkevalmisteiden farmakokinetiikkaan. Populaatiofarmakokineettisen analyysin perusteella yleisesti samanaikaisesti annetut astman hoitoon käytettävät lääkevalmisteet (kuten leukotrieeninsalpaajat, teofylliini/teofyllamiini ja suun kautta annettavat kortikosteroidit) eivät vaikuttaneet tetsepelumabin puhdistumaan.
Raskaus ja imetys
Raskaus
Tetsepelumabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta). Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Tetsepelumabin kaltaiset humaanit IgG‑vasta-aineet läpäisevät istukan, joten Tezspire saattaa siirtyä äidistä sikiöön.
Varmuuden vuoksi Tezspire-valmisteen käyttöä on suositeltavaa välttää raskauden aikana, elleivät hoidosta odotettavissa olevat hyödyt raskaana olevalle äidille ole suurempia kuin mahdolliset sikiöön kohdistuvat riskit.
Imetys
Ei tiedetä, erittyykö tetsepelumabi ihmisillä äidinmaitoon. Ihmisen IgG‑vasta-aineiden tiedetään erittyvän äidinmaitoon muutamien vuorokausien ajan synnytyksen jälkeen, minkä jälkeen niiden pitoisuus pienenee nopeasti. Siksi imetettävään lapseen tällä lyhyellä ajanjaksolla kohdistuvia riskejä ei voida sulkea pois.
Tätä tiettyä ajanjaksoa varten on päätettävä, pidättäydytäänkö tetsepelumabihoidosta, ottaen huomioon imetyksen hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Myöhemmin tetsepelumabia voidaan käyttää imetyksen aikana, jos se on kliinisesti tarpeen.
Ks. kohdasta Prekliiniset tiedot turvallisuudesta tietoa tetsepelumabin erittymisestä maitoon eläimillä (jaavanmakakeilla).
Hedelmällisyys
Ihmisten hedelmällisyyttä koskevia tietoja ei ole. Eläinkokeissa tetsepelumabihoidolla ei ole todettu olevan haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tezspire-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimpiä hoidon aikana ilmoitettuja haittavaikutuksia ovat astman hoidossa nivelkipu (3,8 %) ja nielutulehdus (4,1 %) ja kroonisen polypoottisen rinosinuiitin hoidossa nielutulehdus (5,4 %).
Haittavaikutustaulukko
Taulukossa 1 on esitetty haittavaikutukset, jotka todettiin vaikeaa astmaa tai kroonista polypoottista rinosinuiittia sairastavilla potilailla tehdyissä kliinisissä tutkimuksissa, joissa potilaat saivat ainakin yhden annoksen Tezspire-valmistetta 52 viikon pituisissa tutkimuksissa, sekä myyntiluvan myöntämisen jälkeen ilmoitetut haittavaikutukset.
Haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 1Luettelo haittavaikutuksista | ||
| Elinjärjestelmä | Haittavaikutus | Esiintymistiheys |
| Infektiot | Nielutulehdusa | Yleinen |
| Immuunijärjestelmä | Yliherkkyys (mukaan lukien anafylaktinen reaktio) | Tuntematon |
| Iho ja ihonalainen kudos | Ihottumab | Yleinen |
| Luusto, lihakset ja sidekudos | Nivelkipu | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktioc | Yleinen |
a Nielutulehdus määriteltiin seuraavilla suositelluilla termeillä: nielutulehdus, bakteerin aiheuttama nielutulehdus, streptokokin aiheuttama nielutulehdus ja viruksen aiheuttama nielutulehdus.
b Ihottuma määriteltiin seuraavilla suositelluilla termeillä: ihottuma, kutiava ihottuma, punoittava ihottuma, makulopapulaarinen ihottuma ja makulaarinen ihottuma.
c Ks. ”Valikoitujen haittavaikutusten kuvaus”.
Valikoitujen haittavaikutusten kuvaus
Pistoskohdan reaktiot
PATHWAY- ja NAVIGATOR-tutkimusten yhdistetyissä turvallisuustiedoissa ilmeni pistoskohdan reaktioita (esim. pistoskohdan punoitusta, pistoskohdan turvotusta tai pistoskohdan kipua) 3,8 %:lla potilaista, jotka olivat saaneet tetsepelumabia 210 mg ihon alle 4 viikon välein.
Pediatriset potilaat
52 viikon pituiseen vaiheen 3 NAVIGATOR-tutkimukseen osallistui yhteensä 82 nuorta, 12–17-vuotiasta tutkittavaa, joilla oli vaikea, huonossa hoitotasapainossa oleva astma (ks. kohta Farmakodynamiikka). Nuorilla todettu turvallisuusprofiili oli yleisesti ottaen samankaltainen kuin koko tutkimuspopulaatiolla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa astmaa sairastaville potilaille annettiin enintään 280 mg:n annoksia ihon alle 2 viikon välein ja enintään 700 mg:n annoksia laskimoon 4 viikon välein. Näissä tutkimuksissa ei todettu näyttöä annosriippuvaisesta toksisuudesta.
Tetsepelumabin yliannostukseen ei ole spesifistä hoitoa. Yliannostustapauksessa potilaalle on annettava asianmukaista tukihoitoa ja hänen tilaansa on seurattava tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Ahtauttavien hengitystiesairauksien lääkkeet, muut systeemisesti käytettävät ahtauttavien hengitystiesairauksien lääkkeet, ATC-koodi: R03DX11
Vaikutusmekanismi
Tetsepelumabi on monoklonaalinen vasta-aine (IgG2λ), joka on suunnattu kateenkorvan strooman lymfopoietiinia (TSLP) vastaan ja estää sen vuorovaikutusta heterodimeerisen TSLP-reseptorin kanssa. Astmassa ja kroonisessa polypoottisessa rinosinuiitissa TSLP:n tuotantoa indusoivat sekä allergiaan liittyvät että allergiaan liittymättömät laukaisevat tekijät. TSLP:n estäminen tetsepelumabilla vähentää astmaan ja krooniseen polypoottiseen rinosinuiittiin liittyvään hengitystie- ja limakalvotulehdukseen liittyvien lukuisten eri biomerkkiaineiden ja sytokiinien (esim. veren eosinofiilien, hengitysteiden limakalvonalaisten eosinofiilien, IgE:n, FeNO:n, IL‑5:n ja IL‑13:n) määrää. Tetsepelumabin vaikutusmekanismia astmassa ja kroonisessa polypoottisessa rinosinuiitissa ei ole kuitenkaan lopullisesti selvitetty.
Farmakodynaamiset vaikutukset
Vaikutukset veren eosinofiileihin, tulehdusmerkkiaineisiin ja sytokiineihin
Kun tetsepelumabia annettiin astmaa koskeneissa kliinisissä tutkimuksissa 210 mg ihon alle 4 viikon välein, veren eosinofiilimäärät ja FeNO‑, IL‑5- ja IL‑13-pitoisuudet sekä seerumin IgE‑pitoisuus pienenivät lähtötilanteesta lumelääkkeeseen verrattuna. Näiden merkkiaineiden pitoisuudet olivat pienentyneet lähes maksimaalisesti kahden viikon hoidon jälkeen lukuun ottamatta IgE:tä, jonka pitoisuus pieneni hitaammin. Nämä vaikutukset säilyivät koko hoidon ajan.
Kun tetsepelumabia annettiin kroonista polypoottista rinosinuiittia koskeneessa kliinisessä tutkimuksessa 210 mg ihon alle 4 viikon välein, tulehduksen biomerkkiaineet (veren eosinofiilimäärät, FeNO-pitoisuus [osallistujilla, joilla oli samanaikaisesti astma] ja seerumin IgE-pitoisuus) pienenivät.
Vaikutukset hengitysteiden limakalvonalaisiin eosinofiileihin
Kun tetsepelumabia annettiin kliinisissä tutkimuksissa 210 mg ihon alle 4 viikon välein, limakalvonalaiset eosinofiilimäärät pienenivät 89 %. Lumelääkkeen käytön yhteydessä havaittu pieneneminen oli vastaavasti 25 %. Pieneneminen oli johdonmukaista lähtötilanteen tulehdusmerkkiaineista riippumatta.
Immunogeenisuus
Astmapotilailla tehdyssä tutkimuksessa (NAVIGATOR) todettiin lääkevasta-aineita jossakin vaiheessa 52 viikon pituisen tutkimusjakson aikana 26 potilaalla (4,9 %:lla) 527 potilaasta, jotka saivat tetsepelumabia suositellulla annostusohjelmalla. Näistä 26 potilaasta 10 potilaalla (1,9 %:lla tetsepelumabia saaneista potilaista) kehittyi hoidon aikana lääkevasta-aineita ja yhdellä potilaalla (0,2 %:lla tetsepelumabia saaneista potilaista) kehittyi neutraloivia vasta-aineita. Lääkevasta-aineiden titterit olivat yleensä pieniä ja niiden ilmeneminen usein ohimenevää. Lääkevasta-aineiden vaikutuksista farmakokinetiikkaan, farmakodynamiikkaan, tehoon tai turvallisuuteen ei ole havaittu näyttöä.
Kroonista polypoottista rinosinuiittia sairastavilla potilailla tehdyssä tutkimuksessa (WAYPOINT) 6 potilaalla (4 %:lla) 164 potilaasta, jotka saivat tetsepelumabia 210 mg ihon alle 4 viikon välein, todettiin hoidon aikana kehittynyt lääkevasta-ainevaste 52 viikon pituisen tutkimusjakson aikana. Yhdellä lääkevasta-ainepositiivisista potilaista todettiin neutraloivaa vasta-aineaktiivisuutta. Lääkevasta-aineiden vaikutuksista farmakokinetiikkaan, farmakodynamiikkaan, tehoon tai turvallisuuteen ei havaittu selvää näyttöä. Hoidon aikana kehittyneitä lääkevasta-aineita todettiin kuitenkin niin pienellä määrällä potilaita, että asiaa ei voitu muodollisesti arvioida kroonista polypoottista rinosinuiittia sairastavilla.
Kliininen teho
Astma
Tetsepelumabin tehoa arvioitiin kahdessa 52 viikon pituisessa satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa lumekontrolloidussa kliinisessä tutkimuksessa (PATHWAY ja NAVIGATOR), joihin osallistui yhteensä 1 609 vähintään 12‑vuotiasta potilasta, joilla oli vaikea astma. Kumpaankaan tutkimukseen osallistuvilta potilailta ei edellytetty veren eosinofiilipitoisuuden tai muiden tulehdusmerkkiaineiden (kuten FeNO tai IgE) lähtötilanteen vähimmäisarvoja.
PATHWAY oli 52 viikon pituinen pahenemisvaiheita koskeva tutkimus, johon osallistui 550 potilasta (vähintään 18‑vuotiaita), joilla oli vaikea, huonossa hoitotasapainossa oleva astma. Potilaat saivat tetsepelumabia 70 mg ihon alle 4 viikon välein, tetsepelumabia 210 mg ihon alle 4 viikon välein, tetsepelumabia 280 mg ihon alle 2 viikon välein tai lumelääkettä. Potilailla edellytettiin olleen viimeksi kuluneiden 12 kuukauden aikana vähintään kaksi astman pahenemisvaihetta, jotka olivat vaatineet suun kautta otettavien tai systeemisten kortikosteroidien käyttöä, tai yksi astman pahenemisvaihe, joka oli johtanut sairaalahoitoon.
NAVIGATOR oli 52 viikon pituinen pahenemisvaiheita koskeva tutkimus, johon osallistui yhteensä 1 061 potilasta (aikuisia ja vähintään 12‑vuotiaita nuoria), joilla oli vaikea, huonossa hoitotasapainossa oleva astma. Potilaat saivat tetsepelumabia 210 mg ihon alle 4 viikon välein tai lumelääkettä. Potilailla edellytettiin olleen viimeksi kuluneiden 12 kuukauden aikana vähintään kaksi astman pahenemisvaihetta, jotka olivat vaatineet suun kautta otettavien tai systeemisten kortikosteroidien käyttöä tai johtaneet sairaalahoitoon.
Sekä PATHWAY- että NAVIGATOR-tutkimuksessa edellytettiin, että potilaiden pistemäärä astman hallintaa arvioivalla ACQ‑6-mittarilla (Asthma Control Questionnaire 6) oli vähintään 1,5 seulontavaiheessa ja keuhkofunktio heikentynyt lähtötilanteessa (ennen bronkodilataatiota mitatut FEV1-arvot alle 80 % ennustearvosta aikuisilla ja alle 90 % ennustearvosta nuorilla). Potilaiden edellytettiin saaneen säännöllistä hoitoa keskisuurella tai suurella annoksella inhaloitavaa kortikosteroidia ja ainakin yhtä astman hallintaan annettua lisähoitoa suun kautta annettavien kortikosteroidien kanssa tai ilman niitä. Suureksi inhaloitavan kortikosteroidin annokseksi määriteltiin > 500 mikrogrammaa flutikasonipropionaattia tai vastaava lääkitys vuorokaudessa. Keskisuureksi inhaloitavan kortikosteroidin annokseksi määriteltiin > 250 – 500 mikrogrammaa flutikasonipropionaattia tai vastaava lääkitys vuorokaudessa PATHWAY-tutkimuksessa ja 500 mikrogrammaa flutikasonipropionaattia tai vastaava lääkitys vuorokaudessa NAVIGATOR-tutkimuksessa. Potilaat jatkoivat astman vuoksi annettua taustahoitoa koko tutkimusten ajan.
Näiden kahden tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa on esitetty alla olevassa taulukossa 2.
| Taulukko 2Astmatutkimusten demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa | ||
PATHWAY N = 550 | NAVIGATOR N = 1 059 | |
| Ikä, keskiarvo (vuotta) (keskihajonta) | 52 (12) | 50 (16) |
| Naisia (%) | 66 | 64 |
| Valkoihoisia (%) | 92 | 62 |
| Mustaihoisia tai afrikkalaisamerikkalaisia (%) | 3 | 6 |
| Aasialaisia (%) | 3 | 28 |
| Taustaltaan espanjankielisiä tai latinalaisamerikkalaisia (%) | 1 | 15 |
| Keskimääräinen astman kesto (vuotta) (keskihajonta) | 17 (12) | 22 (16) |
| Ei koskaan tupakoineita (%) | 81 | 80 |
| Inhaloitavan kortikosteroidin käyttö suurella annoksella (%) | 49 | 75 |
| Suun kautta annettavan kortikosteroidin käyttö (%) | 9 | 9 |
| Pahenemisvaiheiden määrä edellisenä vuonna, keskiarvo (keskihajonta) | 2,4 (1,2) | 2,8 (1,4) |
| Keskimääräinen FEV1 ennustearvosta lähtötilanteessa, % (keskihajonta) | 60 (13) | 63 (18) |
| Keskimääräinen FEV1 ennen bronkodilataatiota (l) (keskihajonta) | 1,9 (0,6) | 1,8 (0,7) |
| FEV1-arvon keskimääräinen palautuvuus bronkodilataation jälkeen (%) (keskihajonta) | 23 (20) | 15 (15) |
| Veren EOS-määrä lähtötilanteessa, keskiarvo (solua/mikrol) (keskihajonta) | 371 (353) | 340 (403) |
| Veren EOS-määrä ≥ 150 solua/mikrol (%) | 76 | 74 |
| Positiivinen allergiastatus (%)a | 46 | 64 |
| FeNO, keskiarvo (ppb) (keskihajonta) | 35 (39) | 44 (41) |
| FeNO ≥ 25 ppb (%) | 44 | 59 |
| ACQ‑6-pistemäärän keskiarvo (keskihajonta) | 2,7 (0,8) | 2,8 (0,8) |
| Veren EOS-määrä ≥ 150 solua/mikrol ja FeNO ≥ 25 ppb (%) | 38 | 47 |
a Positiivinen allergiastatus määriteltiin mille tahansa ympärivuotiselle aeroallergeenille spesifisen seerumin IgE:n positiiviseksi tulokseksi FEIA-paneelissa.
ACQ‑6 = astman hallintaa arvioiva mittari (Asthma Control Questionnaire 6); EOS = eosinofiilit; FEIA = fluoroentsyymi-immuunimääritys (Fluorescent enzyme immunoassay); FeNO = uloshengitysilman typpioksidi (Fractional exhaled nitric oxide); FEV1 = uloshengityksen sekuntikapasiteetti (Forced expiratory volume in one second); IgE = immunoglobuliini E; ppb = miljardisosia.
Jäljempänä esitetty yhteenveto on tehty tuloksista, jotka on saatu tetsepelumabin suositellulla annostusohjelmalla 210 mg ihon alle 4 viikon välein.
Pahenemisvaiheet
Ensisijainen päätemuuttuja PATHWAY- ja NAVIGATOR-tutkimuksissa oli vaikeiden astman pahenemisvaiheiden ilmaantuvuus 52 viikon aikana. Vaikeiksi astman pahenemisvaiheiksi määriteltiin vaikeutunut astma, joka edellytti vähintään 3 vuorokauden mittaista hoitoa suun kautta annettavilla tai systeemisillä kortikosteroideilla tai niiden annosten suurentamista tai kerta-injektiota kortikosteroidia sisältävällä depotvalmisteella ja/tai päivystyskäyntiä, jolla on tarvittu suun kautta annettavia tai systeemisiä kortikosteroideja ja/tai sairaalahoitoa.
Sekä PATHWAY- että NAVIGATOR-tutkimuksissa vaikeiden astman pahenemisvaiheiden vuotuinen ilmaantuvuus oli tetsepelumabia saaneilla potilailla merkittävästi pienempi kuin lumelääkettä saaneilla (taulukko 3 ja taulukko 4). Tetsepelumabia saaneilla potilailla oli myös vähemmän päivystyskäyntejä ja/tai sairaalahoitoa vaatineita pahenemisvaiheita kuin lumelääkettä saaneilla. Päivystyskäyntejä ja/tai sairaalahoitoa vaatineet vaikeat astman pahenemisvaihteet vähenivät PATHWAY-tutkimuksessa 85 % ja NAVIGATOR-tutkimuksessa 79 % potilailla, jotka saivat tetsepelumabia 210 mg ihon alle 4 viikon välein.
Taulukko 3Vaikeiden pahenemisvaiheiden ilmaantuvuus viikon 52 kohdalla NAVIGATOR-tutkimuksessaa
| Tetsepelumabi (N = 528) | Lumelääke (N = 531) | |
| Vaikeiden astman pahenemisvaiheiden vuotuinen ilmaantuvuus | ||
| Ilmaantuvuus | 0,93 | 2,10 |
| Ilmaantuvuuksien suhde (95 %:n luottamusväli) | 0,44 (0,37, 0,53) | |
| p‑arvo | < 0,001 | |
a ”Riskiaika” määriteltiin kokonaisajaksi, jolloin uusi pahenemisvaihe voi ilmetä (eli kokonaisseuranta-ajaksi, josta on vähennetty pahenemisvaiheeseen kulunut aika ja sen jälkeiset 7 vuorokautta).
Taulukko 4Vaikeiden pahenemisvaiheiden ilmaantuvuus viikon 52 kohdalla PATHWAY-tutkimuksessaa
| Tetsepelumabi (N = 137) | Lumelääke (N = 138) | |
| Vaikeiden astman pahenemisvaiheiden vuotuinen ilmaantuvuus | ||
| Ilmaantuvuus | 0,20 | 0,72 |
| Ilmaantuvuuksien suhde (95 %:n luottamusväli) | 0,29 (0,16, 0,51) | |
| p‑arvo | < 0,001 | |
a ”Riskiaika” määriteltiin kokonaisseuranta-ajaksi.
Alaryhmäanalyysi
NAVIGATOR-tutkimuksessa tetsepelumabilla todettiin vaikeiden astman pahenemisvaiheiden vähentyneen riippumatta lähtötilanteen veren eosinofiilimääristä, FeNO-arvoista ja allergiastatuksesta (ympärivuotisille aeroallergeeneille spesifisen IgE:n avulla määritettynä). PATHWAY-tutkimuksen tulokset olivat samankaltaiset. Ks. kuva 1.
NAVIGATOR-tutkimuksessa vaikeiden astman pahenemisvaiheiden ilmaantuvuus pieneni sitä enemmän, mitä suuremmat veren eosinofiilimäärät ja FeNO-arvot olivat lähtötilanteessa (ilmaantuvuuksien suhde = 0,79 [95 %:n luottamusväli: 0,48, 1,28] potilailla, joilla veren eosinofiilimäärä oli < 150 solua/µl ja FeNO < 25 ppb lähtötilanteessa; ilmaantuvuuksien suhde = 0,30 [95 %:n luottamusväli: 0,23, 0,40] potilailla, joilla veren eosinofiilimäärä oli ≥ 150 solua/µl ja FeNO ≥ 25 ppb lähtötilanteessa).
Kuva 1 Vaikeiden astman pahenemisvaiheiden vuotuisten ilmaantuvuuksien suhde 52 viikon aikana lähtötilanteen biomerkkiaineiden mukaan täydellisessä analyysijoukossa (NAVIGATOR- ja PATHWAY-tutkimusten yhdistetyt tiedot)a
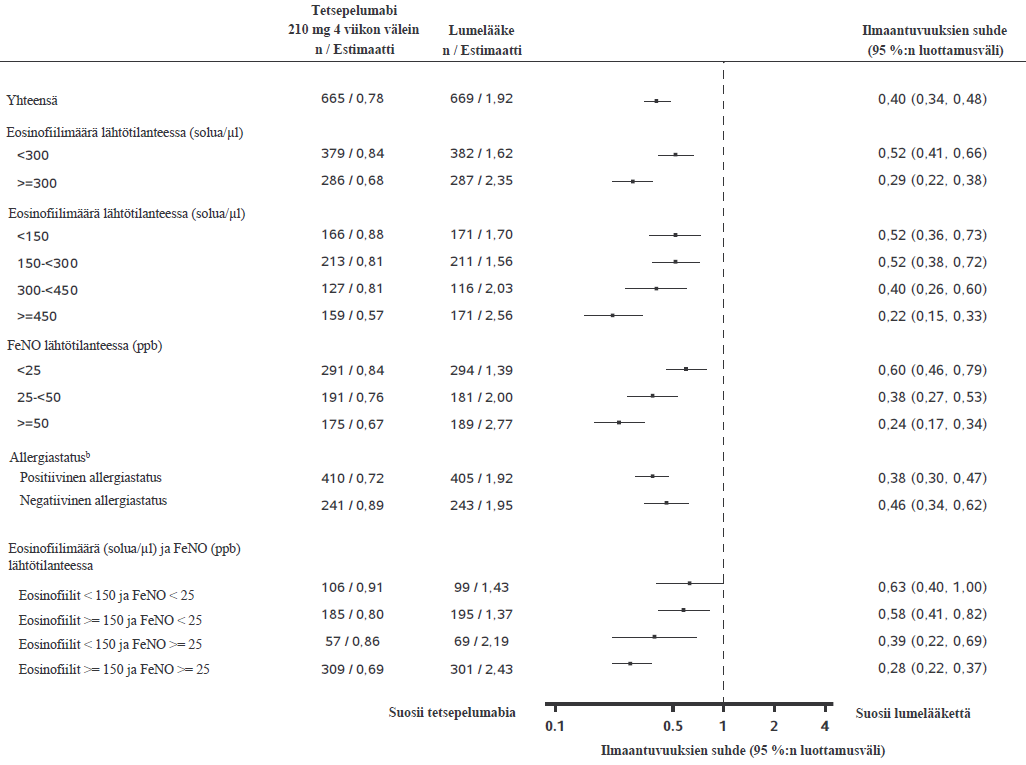
a "Riskiaika” määriteltiin kokonaisajaksi, jolloin uusi pahenemisvaihe voi ilmetä (eli kokonaisseuranta-ajaksi, josta on vähennetty pahenemisvaiheeseen kulunut aika ja sen jälkeiset 7 vuorokautta).
b Allergiastatus määriteltiin mille tahansa ympärivuotiselle aeroallergeenille spesifisen seerumin IgE:n tuloksen perusteella FEIA-paneelissa.
Keuhkofunktio
NAVIGATOR-tutkimuksessa arvioitiin toissijaisena päätemuuttujana FEV1-arvon keskimääräistä muutosta lähtötilanteesta. Lumelääkkeeseen verrattuna tetsepelumabi paransi kliinisesti merkittävästi keskimääräisiä FEV1-arvoja lähtötilanteeseen nähden (taulukko 5).
Potilaiden ilmoittamat hoitotulokset
NAVIGATOR-tutkimuksessa arvioitiin toissijaisina päätemuuttujina ACQ‑6-mittarilla ja AQLQ(S)+12-mittarilla (vähintään 12‑vuotiaille astmapotilaille standardoitu elämänlaatumittari, Standardised Asthma Quality of Life Questionnaire) saatujen pistemäärien sekä astmaoirepäiväkirjan (Asthma Symptom Diary, ASD) keskimääräisten viikoittaisten pistemäärien muutoksia lähtötilanteesta. Hengityksen vinkumisen, hengenahdistuksen, yskän ja rinnassa ilmenevän puristavan tunteen vaikeusasteet arvioitiin kaksi kertaa vuorokaudessa (aamulla ja illalla). Yölliset heräämiset ja oireiden vaikutus päiväaikaisiin toimintoihin arvioitiin päivittäin. ASD-kokonaispistemäärä laskettiin 10 kohdan keskiarvona (taulukko 5).
ACQ‑6-pistemäärien todettiin parantuneen jo kahden viikon kuluttua ja AQLQ(S)+12-pistemäärien jo 4 viikon kuluttua tetsepelumabin antamisesta, ja nämä vaikutukset säilyivät viikkoon 52 asti molemmissa tutkimuksissa.
Taulukko 5Keskeisten toissijaisten päätemuuttujien tulokset viikon 52 kohdalla NAVIGATOR-tutkimuksessaa
| Tetsepelumabi | Lumelääke | |
| FEV1 ennen bronkodilataatiota | ||
| N | 527 | 531 |
| Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta (l) | 0,23 | 0,10 |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (l) (95 %:n luottamusväli) | 0,13 (0,08, 0,18) | |
| p‑arvo | < 0,001 | |
| AQLQ(S)+12-kokonaispistemäärä | ||
| N | 525 | 526 |
| Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta | 1,48 | 1,14 |
| Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | 0,33 (0,20, 0,47) | |
| p‑arvo | < 0,001 | |
| ACQ‑6-pistemäärä | ||
| N | 527 | 531 |
| Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta | ‑1,53 | ‑1,20 |
| Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | ‑0,33 (‑0,46, ‑0,20) | |
| p-arvo | < 0,001 | |
| ASD | ||
| N | 525 | 531 |
| Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta | ‑0,70 | ‑0,59 |
| Ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | ‑0,11 (‑0,19, ‑0,04) | |
| p-arvo | 0,004 | |
a Estimaatit on johdettu toistuville mittauksille tarkoitetusta sekamallista (MMRM) käyttämällä kaikkia saatavilla olevia tietoja potilaista, joilla oli todettu ainakin yksi muutos lähtötilanteen arvosta, keskeyttämisen jälkeen saadut tiedot mukaan lukien.
ACQ‑6 = astman hallintaa arvioiva mittari (Asthma Control Questionnaire 6); AQLQ(S)+12 = vähintään 12‑vuotiaille astmapotilaille standardoitu elämänlaatumittari, Standardised Asthma Quality of Life Questionnaire; ASD = astmaoirepäiväkirja (Asthma Symptom Diary); FEV1 = uloshengityksen sekuntikapasiteetti (Forced expiratory volume in one second); N = lopullisessa analyysissä mukana olleiden sellaisten potilaiden määrä, joilla oli todettu ainakin yksi muutos lähtötilanteen arvosta
Iäkkäät (≥ 65‑vuotiaat) potilaat
Tetsepelumabia 210 mg ihon alle 4 viikon välein PATHWAY- ja NAVIGATOR-tutkimuksissa saaneista 665 astmapotilaasta yhteensä 119 potilasta oli vähintään 65‑vuotiaita, ja heistä 32 potilasta oli vähintään 75‑vuotiaita. Näillä ikäryhmillä todetut turvallisuustiedot olivat samankaltaiset kuin koko tutkimuspopulaatiolla. Näillä ikäryhmillä todettu teho oli samankaltainen kuin NAVIGATOR-tutkimuksen koko tutkimuspopulaatiolla. PATHWAY-tutkimuksessa ei ollut riittävästi vähintään 65‑vuotiaita potilaita tehon määrittämiseksi tässä ikäryhmässä.
Krooninen rinosinuiitti, johon liittyy nenäpolyyppeja (krooninen polypoottinen rinosinuiitti)
Tetsepelumabin tehoa arvioitiin 52 viikon pituisessa satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmillä toteutetussa lumekontrolloidussa kliinisessä monikeskustutkimuksessa (WAYPOINT), johon osallistui 408 vähintään 18-vuotiasta potilasta, jotka saivat tavanomaista hoitoa krooniseen polypoottiseen rinosinuiittiin. Tutkimukseen otettiin oireista kroonista polypoottista rinosinuiittia sairastavia potilaita, joille oli annettu systeemistä kortikosteroidihoitoa viimeksi kuluneiden 12 kuukauden aikana ja/tai tehty jokin nenä- tai sivuonteloleikkaus tai joilla jompikumpi näistä kroonisen polypoottisen rinosinuiitin hoidoista oli vasta-aiheinen tai huonosti siedetty.
Potilaat saivat 210 mg tetsepelumabia tai lumelääkettä ihon alle 4 viikon välein 52 viikon ajan ja lisäksi intranasaalista kortikosteroidihoitoa (esim. mometasonifuroaattinenäsumutetta) kroonisen polypoottisen rinosinuiitin hoitoon.
WAYPOINT-tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa on esitetty taulukossa 6.
Taulukko 6 WAYPOINT-tutkimuksen demografiset tiedot ja potilaiden ominaisuudet lähtötilanteessa
WAYPOINT N = 408a | |
| Ikä, keskiarvo (vuotta) (keskihajonta) | 50 (14) |
| Miehiä (%) | 65 |
| Keskimääräinen kroonisen polypoottisen rinosinuiitin kesto (vuotta) (keskihajonta) | 13 (10) |
| Potilaat, joilla ≥1 aiempi leikkaus (%) | 71 |
| Systeemistä kortikosteroidia kroonisen polypoottisen rinosinuiitin hoitoon edellisen vuoden aikana saaneet potilaat (%) | 58 |
| Keskimääräinen NPS-kokonaispistemääräb (keskihajonta), vaihteluväli 0–8 | 6,1 (1,2) |
| Keskimääräinen kahden viikon ajalta arvioitu NCS-pistemääräb, c (keskihajonta), vaihteluväli 0–3 | 2,6 (0,5) |
| Sinusten TT-kuvista arvioitu keskimääräinen LMK-kokonaispistemääräb (keskihajonta), vaihteluväli 0–24 | 19 (4) |
| Keskimääräinen kahden viikon ajalta arvioitu hajuaistin heikkeneminenb, d (keskihajonta), vaihteluväli 0–3 | 2,9 (0,4) |
| Keskimääräinen kokonaispistemäärä SNOT-22-mittarillab (keskihajonta), vaihteluväli 0–110 | 69 (18) |
| Veren keskimääräinen eosinofillipitoisuus (solua/µl) (keskihajonta) | 360 (235) |
| Keskimääräinen kokonais‑IgE, IU/ml (keskihajonta) | 176 (285) |
| Astma/NSAID-ERD/AERDe (%) | 61 |
| NSAID-ERD/AERD (%) | 17 |
| Allerginen nuha (%) | 14 |
a Potilaiden määrä (N) = 407 keskimääräisen NPS-kokonaispistemäärän osalta; N = 406 kahden viikon ajalta arvioidun keskimääräisen NCS-pistemäärän ja kahden viikon ajalta arvioidun keskimääräisen hajuaistin heikkenemisen osalta; N = 404 sinusten TT-kuvista arvioidun keskimääräisen LMK-kokonaispistemäärän ja veren keskimääräisen eosinofiilipitoisuuden osalta; N = 389 keskimääräisen kokonais-IgE:n osalta.
b Suuremmat pistemäärät viittaavat vaikeampaan tautiin tai vaikeampiin oireisiin.
c Arvioitu nenäpolypoosin oireita koskevan päiväkirjan (NPSD, Nasal Polyposis Symptom Diary) osana.
d Arvioitu NPSD-päiväkirjan hajuaistivaikeuksia koskevan pistemäärän perusteella.
e Tässä alaryhmässä kaikilla potilailla, joilla oli AERD tai NSAID‑ERD, oli ilmoitettu olevan myös astmadiagnoosi, kolmea potilasta lukuun ottamatta.
AERD, asetyylisalisyylihapon pahentama hengitystiesairaus (aspirin exacerbated respiratory disease); IgE = immunoglobuliini E; IU = kansainvälinen yksikkö; LMK = Lund–Mackay; NCS = nenän tukkoisuutta kuvaava pistemäärä (nasal congestion score); NPS = nenäpolyyppeja kuvaava pistemäärä (nasal polyp score); NSAID-ERD = tulehduskipulääkkeiden pahentama hengitystiesairaus (nonsteroidal anti-inflammatory drug exacerbated respiratory disease); SNOT-22 = 22-kohtainen nenään ja sivuonteloihin liittyviä tuloksia arvioiva testi (22-item Sino-Nasal Outcome Test); TT = tietokonekerroskuvaus.
Rinnakkaiset ensisijaiset tehoa koskevat päätemuuttujat olivat nenäpolyyppeja kuvaavan pistemäärän (NPS) muutos lähtötilanteesta, kun riippumattomat sokkoutetut arvioijat määrittivät pistemäärän nenän endoskopian perusteella viikon 52 kohdalla, ja kahden viikon ajalta arvioidun nenän tukkoisuutta kuvaavan pistemäärän (NCS) muutos lähtötilanteesta, kun pistemäärä arvioitiin nenäpolypoosin oireita koskevan päiväkirjan (NPSD, Nasal Polyposis Symptom Diary) osana viikon 52 kohdalla. NPS-kokonaispistemäärä arvioitiin kategorisella asteikolla (0–8). Potilaat arvioivat nenän tukkoisuuden päivittäin käyttäen vaikeusastetta kuvaavaa kategorista 0–3 pisteen asteikkoa. WAYPOINT-tutkimuksesta esitettyjä p‑arvoja ei ole korjattu.
Potilailla, jotka saivat tetsepelumabia, todettiin tilastollisesti merkitsevää paranemista NPS-kokonaispistemäärissä ja kahden viikon ajalta arvioiduissa keskimääräisissä NCS-pistemäärissä viikon 52 kohdalla verrattuna lumelääkkeeseen (ks. taulukko 7).
Rinnakkaisten ensisijaisten päätemuuttujien ja keskeisten toissijaisten päätemuuttujien tulokset WAYPOINT-tutkimuksessa on esitetty taulukossa 7.
Taulukko 7 Rinnakkaisten ensisijaisten päätemuuttujien ja keskeisten toissijaisten päätemuuttujien tulokset WAYPOINT-tutkimuksessa
Tetsepelumabi (N = 203) | Lumelääke (N = 205) | p-arvoa | |
| Rinnakkaiset ensisijaiset päätemuuttujat | |||
| NPS-pistemäärä viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 6,1 | 6,1 | |
| Pienimmän neliösumman keskimääräinen muutos | -2,46 | -0,38 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -2,08 (-2,40, -1,76) | < 0,0001 | |
| NCS-pistemäärä viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 2,59 | 2,55 | |
| Pienimmän neliösumman keskimääräinen muutos | -1,74 | -0,70 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -1,04 (-1,21, -0,87) | < 0,0001 | |
| Keskeiset toissijaiset päätetapahtumat | |||
| Hajuaistin heikkeneminenb viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 2,9 | 2,8 | |
| Pienimmän neliösumman keskimääräinen muutos | -1,26 | -0,26 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -1,01 (-1,18, -0,83) | < 0,0001 | |
| SNOT-22-pistemäärä viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 68,2 | 69,2 | |
| Pienimmän neliösumman keskimääräinen muutos | -45,02 | -17,58 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -27,44 (-32,51, -22,37) | < 0,0001 | |
| Lund–Mackay-pistemäärä (LMK) viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 18,9 | 18,5 | |
| Pienimmän neliösumman keskimääräinen muutos | -6,27 | -0,57 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -5,70 (-6,37, -5,03) | < 0,0001 | |
| Aika ensimmäiseen kroonisesta polypoottisesta rinosinuiitista johtuvaan nenä- tai sivuonteloleikkausta koskevaan päätökseen ja/tai systeemiseen kortikosteroidihoitoon viikkoon 52 mennessä | |||
| Osuus potilaista (%)c | 5,7 | 31,4 | |
| Pieneneminen verrattuna lumelääkkeeseen, % [riskitiheyksien suhde (95 %:n luottamusväli)] | 92 % [0,08 (0,03, 0,16)] | < 0,0001 | |
| Aika ensimmäiseen nenä- tai sivuonteloleikkausta koskevaan päätökseen viikkoon 52 mennessä | |||
| Osuus potilaista (%)c | 0,5 | 22,0 | |
| Pieneneminen verrattuna lumelääkkeeseen, % [riskitiheyksien suhde (95 %:n luottamusväli)] | 98 % [0,02 (0,00, 0,09)] | < 0,0001 | |
| Aika ensimmäiseen systeemiseen kortikosteroidihoitoon kroonisen polypoottisen rinosinuiitin vuoksi viikkoon 52 mennessä | |||
| Osuus potilaista (%)c | 5,2 | 19,3 | |
| Pieneneminen verrattuna lumelääkkeeseen, % [riskitiheyksien suhde (95 %:n luottamusväli)] | 89 % [0,11 (0,04, 0,25)] | < 0,0001 | |
| Oireita koskeva kokonaispistemäärä (TSS, total symptom score) viikon 52 kohdalla | |||
| Lähtötilanteen keskiarvo | 16,3 | 16,4 | |
| Pienimmän neliösumman keskimääräinen muutos | -10,39 | -3,43 | |
| Pienimmän neliösumman keskimääräinen ero lumelääkkeeseen verrattuna (95 %:n luottamusväli) | -6,96 (-8,09, -5,83) | < 0,0001 | |
a Esitetyt p-arvot ovat korjaamattomia. Tilastollisesti merkitsevä monivertailukorjauksen jälkeen.
b Hajuaistin heikkenemisen muutos lähtötilanteesta, kun arviointiperusteena on kahden viikon ajalta arvioitu keskimääräinen NPSD-päiväkirjan hajuaistivaikeuksia koskevan kohdan pistemäärä.
c Kaplan–Meier-estimaatit niiden potilaiden osuudesta, joilla todettiin tapahtuma.
Pienimmän neliösumman keskimääräinen muutos, pienimmän neliösumman menetelmällä laskettu keskimääräinen muutos lähtötilanteesta; pistemäärän pieneneminen viittaa tilanteen paranemiseen; NCS, nenän tukkoisuutta koskeva pistemäärä (nasal congestion score); NPS, nenän polyyppeja koskeva pistemäärä (nasal polyp score); SNOT-22, 22-kohtainen nenään ja sivuonteloihin liittyviä tuloksia arvioiva testi (22-item Sino-Nasal Outcome Test).
Kuva 2 Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta nenän polyyppeja koskevan NPS-kokonaispistemäärän ja kahden viikon ajalta arvioidun keskimääräisen nenän tukkoisuutta koskevan NCS-pistemäärän kohdalla viikkoon 52 mennessä
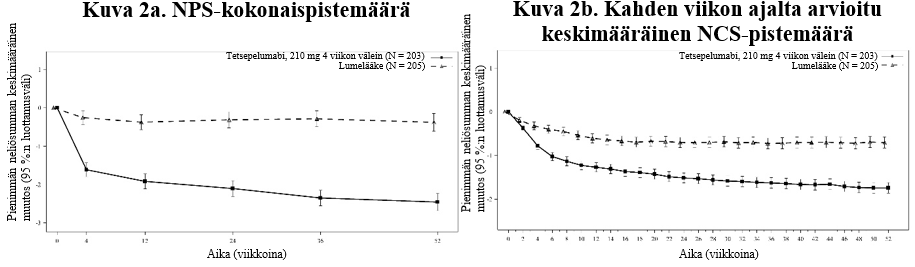
Hajuaistin heikkenemisen todettiin lievittyneen tetsepelumabia saaneilla potilailla jo ensimmäisen arvioinnin eli 2 viikon kohdalla verrattuna lumelääkettä saaneisiin.
Pediatriset potilaat
Astma
NAVIGATOR-tutkimukseen osallistui yhteensä 82 nuorta, 12–17-vuotiasta tutkittavaa, joilla oli vaikea, huonossa hoitotasapainossa oleva astma ja jotka saivat tetsepelumabia (n = 41) tai lumelääkettä (n = 41). Tetsepelumabihoitoa saaneista 41 nuoresta 15 sai lähtötilanteessa inhaloitavaa kortikosteroidia suurella annoksella. Tetsepelumabia saaneilla nuorilla todettu astman pahenemisvaiheiden vuotuinen ilmaantuvuus oli 0,68 ja lumelääkettä saaneilla 0,97 (ilmaantuvuuksien suhde 0,70; 95 %:n luottamusväli 0,34, 1,46). FEV1-arvojen pienimmän neliösumman keskimääräinen muutos lähtötilanteesta oli tetsepelumabia saaneilla nuorilla 0,44 l ja lumelääkettä saaneilla 0,27 l (pienimmän neliösumman keskimääräinen ero 0,17 l; 95 %:n luottamusväli ‑0,01, 0,35). Nuorilla tutkittavilla todetut farmakodynaamiset vasteet olivat yleisesti ottaen samankaltaiset kuin koko tutkimuspopulaatiolla.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Tezspire-valmisteen käytöstä astman hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Krooninen polypoottinen rinosinuiitti
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Tezspire-valmisteen käytöstä kroonisen polypoottisen rinosinuiitin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Tetsepelumabin farmakokinetiikka on samankaltaista astmapotilailla ja potilailla, joilla on krooninen polypoottinen rinosinuiitti.
Tetsepelumabin farmakokinetiikka oli annoksesta riippuvaista ihon alle annetuilla 2,1–420 mg:n annoksilla.
Imeytyminen
Kun annettiin kerta-annos ihon alle, huippupitoisuus seerumissa saavutettiin noin 3–10 päivässä. Populaatiofarmakokineettisen analyysin perusteella arvioitu absoluuttinen hyötyosuus oli noin 77 %. Hyötyosuudessa ei todettu kliinisesti merkittäviä eroja, kun valmiste annettiin eri pistoskohtiin (vatsan alueelle, reiteen tai olkavarteen).
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella 70 kg painavalla henkilöllä tetsepelumabin sentraalinen jakautumistilavuus oli 3,9 l ja perifeerinen jakautumistilavuus 2,2 l.
Metabolia
Tetsepelumabi on humaani monoklonaalinen vasta-aine (IgG2λ), jota hajottavat laajalti elimistössä esiintyvät proteolyyttiset entsyymit. Tetsepelumabi ei metaboloidu maksaentsyymien välityksellä.
Eliminaatio
Koska tetsepelumabi on humaani monoklonaalinen vasta-aine, se eliminoituu intrasellulaarisella katabolialla, eikä kohdevälitteisestä puhdistumasta ole näyttöä. Populaatiofarmakokineettisen analyysin mukaan tetsepelumabin arvioitu puhdistuma 70 kg painavalla henkilöllä oli 0,17 l/vrk. Eliminaation puoliintumisaika oli noin 26 vuorokautta.
Erityiset potilasryhmät
Ikä, sukupuoli ja etninen tausta
Populaatiofarmakokineettisen analyysin perusteella iällä, sukupuolella tai etnisellä taustalla ei ole kliinisesti merkittäviä vaikutuksia tetsepelumabin farmakokinetiikkaan.
Paino
Populaatiofarmakokineettisen analyysin perusteella suurempaan painoon liittyi pienempi altistus. Painon vaikutus altistukseen ei kuitenkaan merkittävästi vaikuttanut tehoon tai turvallisuuteen, eikä annoksen muuttaminen painon mukaan ole tarpeen.
Pediatriset potilaat
Populaatiofarmakokineettisen analyysin perusteella tetsepelumabin farmakokinetiikassa ei ole kliinisesti merkittäviä ikään liittyviä eroja aikuisten ja 12–17-vuotiaiden nuorten astmapotilaiden välillä. Tetsepelumabia ei ole tutkittu alle 12‑vuotiailla lapsilla astman hoidossa eikä alle 18-vuotiailla lapsilla kroonisen polypoottisen rinosinuiitin hoidossa (ks. kohta Annostus ja antotapa).
Iäkkäät (≥ 65‑vuotiaat) potilaat
Populaatiofarmakokineettisen analyysin perusteella tetsepelumabin farmakokinetiikassa ei ole kliinisesti merkittäviä eroja vähintään 65‑vuotiaiden ja sitä nuorempien potilaiden välillä.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutuksia tetsepelumabiin ei ole arvioitu erityisissä kliinisissä tutkimuksissa. Populaatiofarmakokineettisen analyysin perusteella tetsepelumabin puhdistuma oli samankaltainen riippumatta siitä, oliko potilailla lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma 60 – < 90 ml/min) tai kohtalainen munuaisten vajaatoiminta (kreatiniinipuhdistuma 30 – < 60 ml/min) vai toimivatko heidän munuaisensa normaalisti (kreatiniinipuhdistuma ≥ 90 ml/min). Tetsepelumabia ei ole tutkittu vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavilla potilailla, mutta tetsepelumabi ei poistu munuaisten kautta.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutuksia tetsepelumabiin ei ole arvioitu erityisissä kliinisissä tutkimuksissa. Monoklonaaliset IgG‑vasta-aineet eivät ensisijaisesti poistu maksan kautta, joten maksan toiminnan muutosten ei odoteta vaikuttavan tetsepelumabin puhdistumaan. Populaatiofarmakokineettisen analyysin perusteella maksan toimintaa kuvaavien biomerkkiaineiden (ALAT, ASAT ja bilirubiini) pitoisuuksilla lähtötilanteessa ei ollut vaikutuksia tetsepelumabin puhdistumaan.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten (mukaan lukien farmakologista turvallisuutta ja hedelmällisyyttä koskevat arvioinnit) sekä jaavanmakakeilla tehdyn lisääntymistoksisuutta koskevan ePPND-tutkimuksen (enhanced Pre- and Post-Natal Development) ei-kliiniset tulokset eivät viitanneet erityiseen vaaraan ihmisille. ePPND-tutkimuksessa jaavanmakakien saamat annokset olivat enintään 300 mg/kg/viikko (saavat aikaan altistuksia, jotka ovat yli 100‑kertaisia verrattuna kliiniseen altistukseen käytettäessä ihmiselle suositeltua enimmäisannosta).
Tetsepelumabi erittyy apinoilla maitoon, mutta pieninä pitoisuuksina (< 1 %).
Tetsepelumabi on monoklonaalinen vasta-aine, joten genotoksisuutta tai karsinogeenisuutta koskevia tutkimuksia ei ole tehty.
Farmaseuttiset tiedot
Apuaineet
Etikkahappo (E 260)
L-proliini
Polysorbaatti 80 (E 433)
Natriumhydroksidi (pH:n säätelyyn)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Tezspire-valmistetta voidaan säilyttää huoneenlämmössä (20–25 °C) enintään 30 vuorokauden ajan. Kun Tezspire on otettu jääkaapista, se täytyy käyttää 30 vuorokauden kuluessa tai hävittää.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C). Jääkaapista otetun valmisteen säilytys, ks. kohta Kestoaika.
Säilytä esitäytetty ruisku tai esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Ei saa jäätyä. Älä ravista. Ei saa altistaa kuumuudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TEZSPIRE injektioneste, liuos, esitäytetty kynä
210 mg (L:ei) 1 kpl (1,91 ml (110 mg/ml)) (1161,01 €)
TEZSPIRE injektioneste, liuos, esitäytetty ruisku
210 mg (L:ei) 1 kpl (1,91 ml (110 mg/ml)) (1161,01 €)
PF-selosteen tieto
Esitäytetty ruisku
1,91 ml liuosta silikonoidussa tyypin I lasista valmistetussa esitäytetyssä ruiskussa, jossa on lisäksi ruostumattomasta teräksestä valmistettu erityisen ohutseinäinen 27 gaugen, 12,7 mm:n neula, jossa on jäykkä neulansuojus, ja bromibutyylistä valmistettu männän pysäytin. Esitäytettyyn ruiskuun on asennettu neulan suojamekanismi, ja ruiskussa on pidennetty sormituki.
Pakkauskoot:
Pakkaus, joka sisältää 1 esitäytetyn ruiskun.
Monipakkaus, joka sisältää 3 (3 x 1) esitäytettyä ruiskua.
Esitäytetty kynä
1,91 ml liuosta silikonoidussa tyypin I lasista valmistetussa esitäytetyssä ruiskussa, jossa on lisäksi ruostumattomasta teräksestä valmistettu erityisen ohutseinäinen 27 gaugen, 12,7 mm:n neula, jossa on neulansuojus ja männän pysäytin. Esitäytettyyn kynään on asennettu esitäytetty ruisku ja käsikäyttöinen mekaaninen (jouseen perustuva) injektiolaite.
Pakkauskoot:
Pakkaus sisältää 1 esitäytetyn kynän.
Monipakkaus, joka sisältää 3 (3 x 1) esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai vaaleankeltainen liuos.
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste on tarkoitettu vain yhtä käyttökertaa varten.
Ennen Tezspire-valmisteen antamista ota pakkaus jääkaapista ja anna valmisteen lämmetä huoneenlämpöön. Tämä kestää yleensä 60 minuuttia.
Tarkista ennen Tezspire-injektion antamista, näkyykö valmisteessa hiukkasia tai värimuutoksia. Tezspire on kirkasta tai opaalinhohtoista ja väritöntä tai vaaleankeltaista. Älä käytä tätä lääkevalmistetta, jos neste on sameaa, siinä näkyy värimuutoksia tai se sisältää suuria hiukkasia tai vierasaineita.
Pakkausselosteessa ja käyttöohjeissa on lisätietoa Tezspire-valmisteesta ja ohjeet injektion valmisteluun ja antamiseen esitäytetyllä ruiskulla tai esitäytetyllä kynällä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TEZSPIRE injektioneste, liuos, esitäytetty kynä
210 mg 1 kpl
TEZSPIRE injektioneste, liuos, esitäytetty ruisku
210 mg 1 kpl
- Alempi erityiskorvaus (65 %). Tetsepelumabi: Vaikean astman hoito aikuisille ja 12 vuotta täyttäneille nuorille erityisin edellytyksin (259).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Tetsepelumabi: Vaikean astman hoito aikuisille ja 12 vuotta täyttäneille nuorille erityisin edellytyksin (3089).
ATC-koodi
R03DX11
Valmisteyhteenvedon muuttamispäivämäärä
20.10.2025
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi