
ONUREG tabletti, kalvopäällysteinen 200 mg, 300 mg
Vaikuttavat aineet ja niiden määrät
Onureg 200 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 200 mg atsasitidiinia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 3,61 mg laktoosia (monohydraattina).
Onureg 300 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 300 mg atsasitidiinia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 5,42 mg laktoosia (monohydraattina). Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Onureg on tarkoitettu ylläpitohoidoksi aikuispotilaille, joilla on akuutti myelooinen leukemia (AML) ja jotka ovat saavuttaneet täydellisen remission (CR) tai täydellisen remission ilman verisolujen määrän täydellistä palautumista (CRi) induktiohoidon jälkeen joko ilman konsolidaatiohoitoa tai sen kanssa ja joille ei voida tehdä tai jotka eivät halua hematopoieettisten kantasolujen siirtoa (haematopoietic stem cell transplantation, HSCT).
Ehto
Valmistetta saa antaa vain syövän kemoterapian antoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Onureg-hoito tulee aloittaa ja sitä tulee seurata kemoterapeuttisten aineiden käyttöön perehtyneen lääkärin valvonnassa.
Potilaille on annettava pahoinvointilääkettä 30 minuuttia ennen jokaista Onureg-annosta ensimmäisen kahden hoitosyklin ajan. Pahoinvointia ehkäisevä lääkitys voidaan jättää pois kahden hoitosyklin jälkeen, jos pahoinvointia ja oksentelua ei ole ilmennyt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Suositeltu annos on 300 mg atsasitidiinia suun kautta kerran päivässä. Jokaiseen toistuvaan hoitosykliin kuuluu 14 päivän hoitojakso, jota seuraa 14 päivän jakso, jonka aikana valmistetta ei käytetä (28 päivän hoitosykli).
Onureg-hoitoa on jatkettava, kunnes perifeerisessä veressä tai luuytimessä havaitaan korkeintaan 15 % blasteja tai kunnes ilmenee ei-hyväksyttävää toksisuutta (ks. ohjeet annostuksen muuttamisesta sairauden uusiutuessa).
Onureg-valmistetta ei saa käyttää vaihdellen injektoitavan atsasitidiinin kanssa, koska lääkemuotojen altistus, annostus ja hoidon aikataulu eroavat toisistaan. Terveydenhuollon ammattilaisia kehotetaan varmistamaan lääkevalmisteen nimi, annos ja antotapa.
Laboratoriotutkimukset
Ennen hoidon aloittamista on otettava täydellinen verenkuva. Lisäksi verenkuvan seurantaa suositellaan joka toinen viikko ensimmäisen kahden syklin (56 päivän) ajan, joka toinen viikko seuraavan kahden syklin ajan aina annoksen muuttamisen jälkeen ja kuukausittain tämän jälkeen ennen seuraavien hoitosyklien aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostuksen muuttaminen akuutin myelooisen leukemian uusiutuessa
Jos sairaus uusiutuu ja perifeerisessä veressä tai luuytimessä on 5–15 % blasteja, kliinisen arvion yhteydessä on harkittava hoitojakson pidentämistä 14 päivästä 21 päivään toistuvissa 28 päivän hoitosykleissä. Annostus ei saa minkään 28 päivän syklin aikana ylittää 21:tä päivää. Onureg-hoito on keskeytettävä, jos perifeerisessä veressä tai luuytimessä havaitaan yli 15 % blasteja tai lääkärin harkinnan mukaan.
Annoksen muuttaminen haittavaikutusten takia
Hematologisista ja ei-hematologisista haittavaikutuksista johtuvia annosmuutoksia suositellaan kliinisten löydösten ja laboratoriolöydösten perusteella (ks. taulukko 1).
Taulukko 1: Annosmuutokset hematologisten ja ei-hematologisten haittavaikutusten perusteella
Kriteerit* | Suositeltu toimenpide |
Asteen 4 neutropenia tai kuumeinen asteen 3 neutropenia | Ensimmäinen kerta
Esiintyminen kahdessa peräkkäisessä syklissä
|
Asteen 4 trombosytopenia tai asteen 3 trombosytopenia, johon liittyy verenvuotoa | Ensimmäinen kerta
Esiintyminen kahdessa peräkkäisessä syklissä
|
| Vähintään asteen 3 tason pahoinvointi, oksentelu tai ripuli |
|
Muut asteen 3 tai tätä vakavammat ei-hematologiset tapahtumat |
|
* Aste 1 on lievä, aste 2 keskivaikea, aste 3 vaikea ja aste 4 henkeä uhkaava. Haittavaikutusten asteet on esitetty National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events -haittatapahtumaluokituksen version 4.3 (NCI-CTCAE v4.3) mukaisesti.
Unohtunut tai viivästynyt annos
Jos potilas unohtaa ottaa Onureg-annoksen tai ei ota annosta oikeaan aikaan, hänen on otettava annos mahdollisimman pian saman päivän aikana. Seuraava aikataulun mukainen annos on otettava normaaliin aikaan seuraavana päivänä. Samana päivänä ei pidä ottaa kahta annosta.
Jos potilas oksentaa annoksen, samana päivänä ei pidä ottaa toista annosta, vaan seuraavana päivänä annos on otettava normaalin aikaan annosaikataulun mukaisesti.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttamista ei suositella yli 65-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Onureg-valmistetta voidaan antaa aloitusannosta muuttamatta potilaille, joilla on lievä, keskivaikea tai vaikea munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella potilaille, joilla on lievä maksan vajaatoiminta (kokonaisbilirubiini (BIL) ≤ normaalin yläraja (ULN) ja aspartaattiaminotransferaasi (ASAT) > ULN, tai BIL-arvo 1–1,5 × ULN ja mikä tahansa ASAT-arvo) (ks. kohta Farmakokinetiikka).
Potilaita, joilla on keskivaikea (BIL-arvo > 1,5–3 × ULN) tai vaikea maksan vajaatoiminta (BIL-arvo > 3 × ULN), on seurattava useammin haittavaikutusten varalta, ja annosta on muutettava tarpeen mukaan (ks. taulukko 1).
Pediatriset potilaat
Onureg-valmisteen turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Onureg-valmiste otetaan suun kautta.
Onureg-valmiste voidaan ottaa ruokailun yhteydessä tai erikseen. Tabletit on nieltävä kokonaisina veden (lasillinen vettä) kanssa aina suunnilleen samaan aikaan päivästä. Tabletteja ei saa jakaa, murskata, liuottaa tai pureskella (ks. kohta Käyttö- ja käsittelyohjeet).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Hematologiset haittavaikutukset
Onureg-hoitoon on liittynyt neutropeniaa, trombosytopeniaa ja kuumeista neutropeniaa (ks. esiintymistiheydet kohdasta Haittavaikutukset). Onureg-annoksen pienentäminen tai Onureg-hoidon keskeyttäminen tai lopettaminen saattaa olla tarpeen hematologisten haittavaikutusten hoitamiseksi. Potilaita on kehotettava ilmoittamaan kuumejaksoista välittömästi. Potilaita, joilla on matalat verihiutalearvot, on lisäksi kehotettava ilmoittamaan verenvuodon varhaisista merkeistä ja oireista. Potilaalle on annettava potilaan yksilöllisten ominaisuuksien, hoitovasteen ja hoitosuositusten mukaista tukihoitoa, kuten antibiootteja ja/tai kuumelääkkeitä tulehdusten/kuumeen hoitoon ja granulosyyttikasvutekijää neutropenian hoitoon (ks. kohta Annostus ja antotapa, taulukko 1).
Ennen hoidon aloittamista on otettava täydellinen verenkuva. Lisäksi verenkuvan seurantaa suositellaan joka toinen viikko ensimmäisen kahden syklin (56 päivän) ajan, joka toinen viikko seuraavan kahden syklin ajan aina annoksen muuttamisen jälkeen ja kuukausittain tämän jälkeen ennen seuraavien hoitosyklien aloittamista.
Potilaita, joilla on keskivaikea (BIL-arvo > 1,5–3 × ULN) tai vaikea maksan vajaatoiminta (BIL-arvo > 3 × ULN), on seurattava useammin haittavaikutusten varalta, ja annosta on muutettava tarpeen mukaan (ks. kohta Annostus ja antotapa, taulukko 1).
Erilaistumisoireyhtymä
Atsasitidiinia suun kautta saaneilla potilailla on raportoitu erilaistumisoireyhtymää (tunnetaan myös retinoiinihappo-oireyhtymänä). Erilaistumisoireyhtymä voi johtaa kuolemaan, ja sen oireita ja kliinisiä löydöksiä ovat muun muassa hengitysvaikeudet, keuhkoinfiltraatit, kuume, ihottuma, keuhkoedeema, perifeerinen edeema, nopea painonnousu, keuhkopussin nestekertymät, sydänpussin nestekertymät, matala verenpaine ja munuaisten toimintahäiriö (ks. kohta Haittavaikutukset). Hoitoa suonensisäisesti annetuilla suuriannoksisilla kortikosteroideilla sekä hemodynamiikan monitorointia tulisi harkita heti erilaistumisoireyhtymään viittaavien oireiden tai merkkien ilmaannuttua. Atsasitidiinin antaminen suun kautta on ehkä keskeytettävä tilapäisesti oireiden häviämiseen saakka, ja hoidon jatkamista tulee harkita huolella.
Ruuansulatuselimistöön liittyvät haittavaikutukset
Ruuansulatuselimistöön liittyvät vaikutukset olivat yleisimmin ilmoitettuja haittavaikutuksia Onureg- hoitoa saaneilla potilailla (ks. kohta Haittavaikutukset). Potilaille on annettava pahoinvointia ehkäisevää hoitoa Onureg-hoidon kahden ensimmäisen syklin ajan (ks. kohta Annostus ja antotapa). Ripulia on hoidettava pikaisesti oireiden alkaessa. Onureg-annoksen pienentäminen tai Onureg-hoidon keskeyttäminen tai lopettaminen saattaa olla tarpeen ruuansulatuselimistöön liittyvien vaikutusten hoitamiseksi (ks. kohta Annostus ja antotapa).
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 6 kuukautta hoidon päättymisen jälkeen. Miesten on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen (ks. kohta Raskaus ja imetys).
Laktoosi-intoleranssi
Onureg-tabletit sisältävät laktoosimonohydraattia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi- intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Atsasitidiinilla ei ole tehty kliinisiä lääkkeiden yhteisvaikutustutkimuksia.
Jos lääkettä annetaan samanaikaisesti muiden antineoplastisten aineiden kanssa, on suositeltavaa noudattaa varovaisuutta ja seurata tilannetta, koska antagonistisia, additiivisia tai synergistisiä farmakodynaamisia vaikutuksia ei voida poissulkea. Vaikutukset voivat olla riippuvaisia annoksesta, valmisteiden antamisen järjestyksestä ja antamisen aikatauluista.
Samanaikainen anto protonipumpun estäjän (omepratsolin) kanssa vaikutti hyvin vähän Onureg- valmisteen altistukseen. Tästä syystä annosta ei tarvitse muuttaa annettaessa Onureg-valmistetta samanaikaisesti protonipumpun estäjien tai muiden pH-arvoon vaikuttavien aineiden kanssa.
Atsasitidiinilla tehty in vitro -tutkimus ihmisen maksan fraktioissa osoitti, että atsasitidiini ei metaboloidu sytokromi P450:n isoformien (CYP) välityksellä. Tästä syystä yhteisvaikutuksia CYP:n induktorien tai estäjien kanssa pidetään epätodennäköisinä (ks. kohta Farmakokinetiikka).
Atsasitidiinin kliinisesti merkittävät inhibitio- tai induktiovaikutukset sytokromi P450:n substraattien metaboliaan ovat epätodennäköisiä (ks. kohta Farmakokinetiikka). Kliinisesti merkittäviä lääkkeiden välisiä yhteisvaikutuksia ei ole odotettavissa annettaessa Onureg-valmistetta samanaikaisesti P-glykoproteiinin (P-gp), rintasyövän resistenssiproteiinin (BCRP), orgaanisten anionien kuljettajaproteiiniien (OAT) OAT1:n ja OAT3:n, orgaanisten anioneja kuljettavien polypeptidien (OATP) OATP1B1:n and OATP1B3:n tai orgaanisten kationien kuljettajan (OCT) OCT2:n substraattien kanssa.
Atsasitidiini ei ole P-gp:n substraatti, joten sillä ei oleteta olevan yhteisvaikutuksia P-gp:n indusoijien tai estäjien kanssa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehille ja naisille
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 6 kuukautta hoidon päättymisen jälkeen. Miehiä on neuvottava olemaan siittämättä lasta hoidon aikana, ja miesten on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta).
Raskaus
Onureg-valmisteen käytöstä raskaana olevilla naisilla ei ole olemassa riittävästi tietoa Hiirillä ja rotilla tehdyissä eläinkokeissa on havaittu lisääntymistoksisuutta ja kehitystoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Mahdollista vaaraa ihmisille ei tunneta. Eläinkokeiden tulosten perusteella ja vaikutusmekanisminsa vuoksi Onureg-valmisteen käyttöä ei suositella raskauden aikana (etenkään ensimmäisen raskauskolmanneksen aikana, ellei se ole selvästi välttämätöntä) eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä. Hoidon hyötyjä tulee punnita sikiölle mahdollisesti aiheutuvaan riskiin nähden jokaisessa yksittäistapauksessa. Jos potilas tai potilaan kumppani tulee raskaaksi Onureg-hoidon aikana, potilaalle on kerrottava hoidon mahdollisista riskeistä sikiölle.
Imetys
Ei tiedetä, erittyvätkö atsasitidiini tai sen metaboliitit ihmisen rintamaitoon. Imetettävälle lapselle mahdollisesti aiheutuvien vakavien haittavaikutusten vuoksi imetys on vasta-aiheista Onureg-hoidon aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Atsasitidiinin vaikutuksesta ihmisten hedelmällisyyteen ei ole tietoja. Eläimillä on dokumentoitu atsasitidiinin käytöstä aiheutuneita haittavaikutuksia urosten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Potilaita, jotka haluavat tulla raskaaksi tai siittää lapsen, on neuvottava hakeutumaan lisääntymistä koskevaan neuvontaan ja siittiöiden tai munasolujen talteenottoon ennen Onureg-hoidon aloittamista.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Onureg-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Onureg-valmisteen käytön yhteydessä on raportoitu väsymystä. Sen vuoksi suositellaan varovaisuutta ajettaessa tai käytettäessä koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät haittavaikutukset ovat pahoinvointi (64,8 %), oksentelu (59,7 %), ripuli (50,4 %), neutropenia (44,5 %), väsymys/voimattomuus (44,1 %)5, ummetus (38,6 %), trombosytopenia (33,5 %), vatsakipu (21,6 %)4, hengitystieinfektio (17 %)2, nivelkivut (13,6 %), heikentynyt ruokahalu (12,7 %), kuumeinen neutropenia (11,9 %), selkäkipu (11,9 %), leukopenia (10,6 %), raajojen särky (10,6 %) ja keuhkokuume (10,2 %)1.
Vakavia haittavaikutuksia ilmeni 16,1 %:lla Onureg-hoitoa saavista potilaista. Yleisimmät vakavat haittavaikutukset ovat kuumeinen neutropenia (6,8 %) ja keuhkokuume (5,1 %)1.
Onureg-hoito jouduttiin lopettamaan pysyvästi haittavaikutuksen takia 6,8 %:lla potilaista. Yleisimmät hoidon pysyvää lopettamista vaativat haittavaikutukset ovat pahoinvointi (2,1 %), ripuli (1,7 %) ja oksentelu (1,3 %).
Hoito jouduttiin keskeyttämään haittavaikutuksen vuoksi 36,4 %:lla Onureg-valmistetta saaneista potilaista. Hoidon keskeyttämistä vaativia haittavaikutuksia ovat mm. neutropenia (19,9 %), trombosytopenia (8,5 %), pahoinvointi (5,5 %), ripuli (4,2 %), oksentelu (3,8 %), keuhkokuume (3,4 %)1, leukopenia (2,5 %), kuumeinen neutropenia (2,1 %) ja vatsakipu (2,1 %)4.
Annosta pienennettiin haittavaikutuksen vuoksi 14 %:lla Onureg-valmistetta saaneista potilaista. Annoksen pienentämistä vaativia haittavaikutuksia olivat mm. neutropenia (5,5 %), ripuli (3,4 %), trombosytopenia (1,7 %) ja pahoinvointi (1,7 %).
Haittavaikutustaulukko
Taulukossa 2 esitetään kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä käytössä raportoitujen Onureg-valmisteen haittavaikutusten yleisyysluokat. Yhtensä 236 potilasta sai Onureg-valmistetta keskeisessä vaiheen 3 tutkimuksessa. Hoidon mediaanikesto oli 11,6 kuukautta (vaihteluväli: 0,5–74,3 kuukautta) Onureg-haarassa.
Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Haittavaikutukset on esitetty seuraavassa taulukossa suurimman havaitun esiintymistiheyden mukaan.
Taulukko 2: Haittavaikutukset Onureg-ylläpitohoitoa saavilla akuuttia myelooista leukemiaa (AML) sairastavilla potilailla
| Elinjärjestelmäluokka | Esiintymistiheys, kaikki asteeta |
| Infektiot | Hyvin yleinen Keuhkokuume1, 6 Hengitystieinfektio2 Yleinen Sepsis Influenssa Virtsatietulehdus3 Keuhkoputkitulehdus Nuha Melko harvinainen Neutropeeninen sepsis |
| Hyvänlaatuiset, pahanlaatuiset ja määrittelemättömät kasvaimet (ml. kystat ja polyypit) | Tuntematon Erilaistumisoireyhtymä |
| Veri ja imukudos | Hyvin yleinen Neutropenia Trombosytopenia6 Kuumeinen neutropenia6 Leukopenia |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen Ruokahalun väheneminen |
| Elinjärjestelmäluokka | Esiintymistiheys, kaikki asteeta |
| Psyykkiset häiriöt | Yleinen Ahdistuneisuus |
| Ruoansulatuselimistö | Hyvin yleinen Pahoinvointi Oksentelu Ripuli Ummetus Vatsakipu4 |
| Luusto, lihakset ja sidekudos | Hyvin yleinen Nivelkipu Selkäkipu Raajojen kipu |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen Väsymys/voimattomuus5 |
| Tutkimukset | Yleinen Painon lasku |
a Kaikki haittatapahtumat, joita ilmeni vähintään 5,0 %:lla Onureg-haaran potilaista ja vähintään 2,0 % useammin kuin lumelääkehaarassa.
1 Käsite kattaa keuhkokuumeen, bronkopulmonaalisen aspergilloosin, keuhkotulehduksen, Pneumocystis jiroveci - keuhkokuumeen, atyyppisen keuhkokuumeen, bakteeripneumonian ja sienipneumonian.
2 Käsite kattaa ylähengitystieinfektion, hengitystieinfektion ja viruksen aiheuttaman hengitystieinfektion.
3 Käsite kattaa virtsatietulehduksen, bakteerin aiheuttaman virtsatietulehduksen, Escherichia-bakteerin aiheuttaman virtsatietulehduksen ja kystiitin.
4 Käsite kattaa vatsakivun, ylävatsakivun, epämiellyttävän tunteen vatsassa ja ruuansulatuskanavan kivun.
5 Käsite kattaa väsymyksen ja voimattomuuden.
6 Haittavaikutukset, joista ainakin yksi oli henkeä uhkaava (jos haittavaikutuksen lopputulos oli kuolema, se on mukana kuolemantapauksissa).
Valikoitujen haittavaikutusten kuvaus
Hematologiset haittavaikutukset
Onureg-valmisteella hoidetuilla potilailla raportoitiin yleisenä haittavaikutuksena uutta tai pahenevaa asteen 3 tai tätä vakavampaa neutropeniaa (41,1 %), trombosytopeniaa (22,5 %) tai kuumeista neutropeniaa (11,4 %). Asteen 3 tai 4 neutropenia ilmaantui ensimmäisen kerran ensimmäisen kahden syklin aikana 19,9 %:lla Onureg-hoitoa saaneista potilaista, asteen 3 tai 4 trombosytopenia ilmaantui ensimmäisen kerran ensimmäisen kahden syklin aikana 10,6 %:lla Onureg-hoitoa saaneista potilaista ja asteen 3 tai 4 kuumeinen neutropenia ilmaantui ensimmäisen kerran ensimmäisen kahden syklin aikana 1,7 %:lla Onureg-hoitoa saaneista potilaista (ks. kohta Annostus ja antotapa).
Ruuansulatuselimistöön liittyvät haittavaikutukset
Yleisimmät Onureg-hoidon yhteydessä raportoidut haittavaikutukset olivat ruuansulatuselimistöön liittyvät vaikutukset. Onureg-valmisteella hoidetuilla potilailla raportoitiin pahoinvointia (64,8 %), oksentelua (59,7 %) ja ripulia (50,4 %). Asteen 3 tai tätä vakavampaa ripulia ilmeni 5,1 %:lla potilaista, asteen 3 tai tätä vakavampaa oksentelua 3,0 %:lla Onureg-hoitoa saaneista potilaista ja asteen 3 tai tätä vakavampaa pahoinvointia 2,5 %:lla Onureg-hoitoa saaneista potilaista. Asteen 3 tai 4 pahoinvointi ilmaantui ensimmäisen kerran kahden ensimmäisen syklin aikana 1,7 %:lla Onureg- hoitoa saaneista potilaista, asteen 3 tai 4 oksentelu ilmaantui ensimmäisen kerran kahden ensimmäisen syklin aikana 3,0 %:lla Onureg-hoitoa saaneista ja asteen 3 tai 4 ripuli ilmaantui ensimmäisen kerran kahden ensimmäisen syklin aikana 1,3 %:lla Onureg-hoitoa saaneista potilaista (ks. kohta Annostus ja antotapa).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen sattuessa potilaan verenkuvaa on tarkkailtava asianmukaisesti, ja hänelle on annettava tukihoitoa tarpeen mukaan paikallisten suositusten mukaisesti. Onureg-valmisteen yliannostukselle ei tunneta spesifistä vastalääkettä.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, antimetaboliitit, pyrimidiinianalogit; ATC- koodi: L01BC07
Vaikutusmekanismi
Atsasitidiini on DNA:n metyylitransferaasin estäjä ja epigenetiikkaa muuntava lääkeaine. Atsasitidiini liittyy DNA:han ja RNA:han soluunoton seurauksena muunnuttuaan nukleotiditrifosfaateiksi entsymaattisen biotransformaation kautta. Atsasitidiinin liittyminen AML-solujen DNA:han muunsi epigeneettisiä reittejä estämällä DNA:n metyylitransferaaseja ja vähentämällä DNA:n metylaatiota. Tämä johti muutoksiin geenien ilmentymisessä, muun muassa kasvainten kasvun estämistä, immuunireittejä, solukiertoa ja solujen erilaistumista säätelevien geenien uudelleenilmentymisessä. Atsasitidiinin liittyminen AML-solujen RNA:han esti RNA:n metyylitransferaasia ja vähensi RNA:n metylaatiota, RNA:n stabiliteettia ja proteiinisynteesiä.
Kliininen teho ja turvallisuus
Onureg-valmisteen tehoa ja turvallisuutta tutkittiin lumekontrolloidussa, kaksoissokkoutetussa, satunnaistetussa vaiheen 3 monikeskus- ja rinnakkaisryhmätutkimuksessa QUAZAR AML-001 (CC-486-AML-001), jossa arvioitiin Onureg-valmistetta ja lumelääkettä ylläpitohoitona akuuttia myelooista leukemiaa sairastavilla potilailla. Tutkimukseen otettiin mukaan potilaita, joilla oli de novo akuutti myelooinen leukemia, aiemman diagnosoidun myelodysplastisen oireyhtymän jälkeinen akuutti myelooinen leukemia tai krooninen myelomonosyyttileukemia. Potilaat olivat iältään ≥ 55-vuotiaita ja saavuttaneet ensimmäisen täydellisen remission (CR) tai täydellisen remission ilman verisolujen määrän täydellistä palautumista (CRi) neljän kuukauden kuluessa (+/- 7 päivää) intensiivisen induktiokemoterapian jälkeen joko ilman konsolidaatiohoitoa tai sen kanssa. Potilaat eivät satunnaistamisen aikaan soveltuneet kantasolusiirtoon, mukaan lukien potilaat, joille ei ollut siirteen luovuttajaa ja potilaat, jotka eivät halunneet kantasolusiirtoa.
Molempien hoitohaarojen potilaat saivat tutkijan arvioiman tarpeen mukaista, parasta mahdollista tukihoitoa. Parhaaseen tukihoitoon kuului muun muassa punasolusiirtoja, trombosyyttisiirtoja, erytropoieesia stimuloivien lääkeaineiden käyttöä, antibioottihoitoa, viruslääkitystä ja/tai sienilääkitystä, granulosyyttikasvutekijää, pahoinvointia ehkäisevää lääkitystä ja ravitsemustukea.
Niille potilaille, jotka saavuttivat täydellisen remission tai täydellisen remission ilman verisolujen määrän täydellistä palautumista intensiivisen induktiokemoterapian päättymisen jälkeen joko ilman konsolidaatiohoitoa tai sen kanssa, annettiin 300 mg Onureg-valmistetta (N = 236) tai lumelääkettä (N = 233) kerran päivässä päivinä 1–14 jokaisessa 28-päivän syklissä. Sairauden uusiutuessa (5–15 % blasteja perifeerisessä veressä tai luuytimessä) hoitojaksoa pidennettiin 21 päivään toistuvissa 28 päivän hoitosykleissä lääketieteellisen harkinnan mukaisesti. Hoitoa jatkettiin, kunnes tauti eteni (perifeerisessä veressä tai luuytimessä havaittiin yli 15 % blasteja) tai kunnes kehittyi sietämätöntä toksisuutta.
Yhteensä 472 potilasta satunnaistettiin suhteessa 1:1 saamaan joko Onureg-valmistetta tai lumelääkettä. Lähtötilanteen demografiset tekijät ja sairauden ominaisuudet olivat samankaltaiset molemmissa akuuttia myelooista leukemiaa sairastavien potilaiden hoitohaaroissa, kuten taulukossa 3 esitetään. Hoidon mediaanikesto oli 11,6 kuukautta (vaihteluväli: 0,5–74,3 kuukautta) Onureg- haarassa ja 5,7 kuukautta (vaihteluväli: 0,7–68,5 kuukautta) lumelääkehaarassa. Yhteensä 51:llä (21 %) Onureg-valmistetta saaneella potilaalla ja 40:llä (17 %) lumelääkettä saaneella potilaalla hoitojaksoa (annostus 300 mg päivässä) pidennettiin 21 päivään akuutin myelooisen leukemian uusiutumisen takia.
Vaiheen 3 tutkimuksessa hoitoa saaneesta 469:stä potilaasta 61 % (285/469) oli 65-vuotiaita tai tätä vanhempia ja 11 % (51/469) oli 75-vuotiaita tai tätä vanhempia. Onureg-valmisteen tehossa ja turvallisuudessa ei havaittu eroja näiden potilaiden ja nuorempien potilaiden välillä.
Taulukko 3: Lähtötilanteen demografiset tekijät ja sairauteen liittyvät ominaisuudet tutkimuksessa CC-486-AML-001
Muuttuja | Onureg (N = 238) | Lumelääke (N = 234) |
Ikä (vuosina) | ||
Mediaani (minimi, maksimi) | 68,0 (55, 86) | 68,0 (55, 82) |
Ikäryhmä, n (%) | ||
< 65 vuotta | 66 (27,7) | 68 (29,1) |
≥ 65 – < 75 vuotta | 144 (60,5) | 142 (60,7) |
≥ 75 vuotta | 28 (11,8) | 24 (10,3) |
Sukupuoli, n (%) | ||
Mies | 118 (49,6) | 127 (54,3) |
Nainen | 120 (50,4) | 107 (45,7) |
Etninen tausta, n (%) | ||
Valkoihoinen | 216 (90,8) | 197 (84,2) |
Mustaihoinen tai afroamerikkalainen | 2 (0,8) | 6 (2,6) |
Aasialainen | 6 (2,5) | 20 (8,5) |
Muu | 12 (5,0) | 11 (4,7) |
Ei kerätty tai raportoitu | 2 (0,8) | 0 (0) |
ECOG-toimintakykyluokka, n (%) | ||
0 | 116 (48,7) | 111 (47,4) |
1 | 101 (42,4) | 106 (45,3) |
2 | 21 (8,8) | 15 (6,4) |
3 | 0 (0) | 2 (0,9) |
Sytogeneettisen riskin status diagnoosin yhteydessä, n (%) | ||
Keskisuuri riski1 | 203 (85,3) | 203 (86,6) |
Suuri riski2 | 35 (14,7) | 31 (13,2) |
Akuutin myelooisen leukemian (AML) luokitus alussa, n (%) | ||
AML, johon liittyy toistuvia geneettisiä poikkeavuuksia | 39 (16,4) | 46 (19,7) |
AML, johon liittyy myelodysplastisia muutoksia | 49 (20,6) | 42 (17,9) |
Aikaisempiin hoitoihin (MPN) liittyvä AML | 2 (0,8) | 0 (0) |
Muutoin spesifioimaton AML | 148 (62,2) | 145 (62,0) |
Puuttuu | 0 (0) | 1 (0,4) |
Akuutin myelooisen leukemian (AML) tyyppi, n (%) | ||
Primaarinen (de novo) | 213 (89,5) | 216 (92,3) |
Sekundaarinen | 25 (10,5) | 18 (7,7) |
MRD:n tila satunnaistamisen aikana3, n (%) | ||
Negatiivinen | 133 (55,9) | 111 (47,4) |
Positiivinen | 103 (43,3) | 116 (49,6) |
Puuttuu | 2 (0,8) | 7 (3,0) |
AML = akuutti myelooinen leukemia, MDS = myelodysplastinen oireyhtymä, CMML = krooninen myelomonosyyttileukemia, ECOG = Eastern cooperative oncology group, CR = täydellinen morfologinen remissio, CRi = täydellinen morfologinen remissio ilman verisolujen määrän täydellistä palautumista
1 Keskisuureksi riskiksi määriteltiin normaali sytogenetiikka +8, t(9;11) tai muu määrittelemätön.
2 Suureksi riskiksi määriteltiin kompleksinen karyotyyppi (≥ 3 poikkeavuutta): -5; 5q-; -7; 7q-; 11q23 - non t(9;11); inv(3); t(3;3); t(6;9); tai t(9;22). Keskisuuren riskin ja suuren riskin lähde: National comprehensive cancer network clinical practice guidelines in oncology for AML.
3 MRD:n (minimaalinen jäännöstauti) tila luuytimessä mitattiin seulontavaiheessa virtaussytometrisellä analyysillä 0,1 %:n herkkyystasolla.
Useimmat potilaat saivat induktiohoidon jälkeen konsolidaatiohoitoa sekä Onureg-haarassa (78 %) että lumelääkehaarassa (82 %), ja yli 90 % näistä potilaista kummassakin haarassa sai 1 tai 2 sykliä konsolidaatiohoitoa induktiohoidon jälkeen (taulukko 4).
Taulukko 4: Konsolidaatiohoito tutkimuksessa CC-486-AML-001
Muuttuja | Onureg (N = 238) | Lumelääke (N = 234) |
Induktiohoidon jälkeen saatu konsolidaatiohoitoa | ||
Kyllä, n (%) | 186 (78,2) | 192 (82,1) |
1 sykli, n (%) | 110 (46,2) | 102 (43,6) |
2 sykliä, n (%) | 70 (29,4) | 77 (32,9) |
3 sykliä, n (%) | 6 (2,5) | 13 (5,6) |
Ei, n (%) | 52 (21,8) | 42 (17,9) |
CR:n/CRi:n status satunnaistamisen aikana | ||
CR, n (%) | 183 (76,9) | 177 (75,6) |
CRi, n (%) | 50 (21,0) | 44 (18,8) |
Ei CR:ää/CRi:tä a, n (%) | 5 (2,1) | 11 (4,7) |
Puuttuu, n (%) | 0 (0) | 2 (0,9) |
CR = täydellinen remissio, CRi = täydellinen morfologinen remissio ilman verisolujen määrän täydellistä palautumista
a Lähtötilanteessa näillä potilailla oli alle 5 % blasteja luuytimessä, absoluuttinen neutrofiilien määrä (ANC) < 1 x 109 ja verihiutaleiden määrä < 100 x 109.
Onureg-valmisteen teho akuuttia myelooista leukemiaa sairastavilla aikuispotilailla määritettiin kokonaiselossaolon (OS) ja relapsivapaan elossaolon (RFS) perusteella.
Tulokset tehosta on koottu taulukkoon 5.
Taulukko 5: Tutkimuksen CC-486-AML-001 tulokset tehosta (hoitoaikeen mukainen [ITT] populaatio)
Päätetapahtumat | Onureg (N = 238) | Lumelääke (N = 234) |
Kokonaiselossaolo | ||
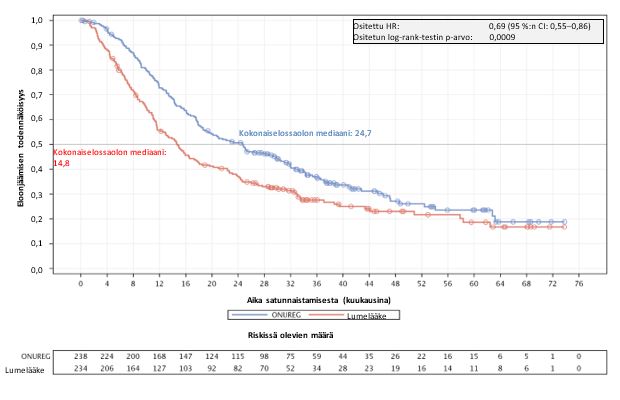
Kokonaiselossaolon tapahtumat, n (%) | 158 (66,4) | 171 (73,1) |
Kokonaiselossaolon mediaani kuukausina (95 %:n CI) | 24,7 (18,7; 30,5) | 14,8 (11,7; 17,6) |
Riskisuhde (95 %:n CI) p-arvo | 0,69 (0,55; 0,86) 0,0009 | |
Relapsivapaa elossaolo | ||
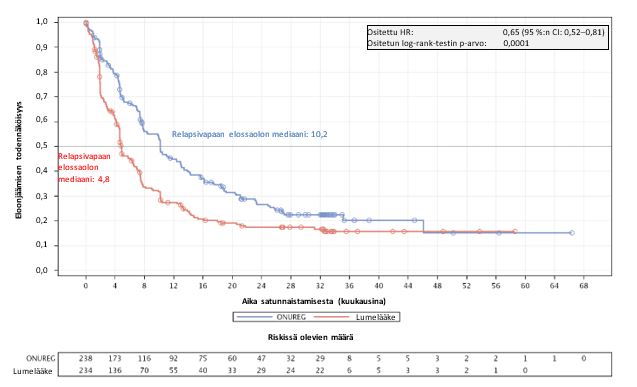
Tapahtumat, n (%) | 164 (68,9) | 181 (77,4) |
Relapsivapaan elossaolon mediaani kuukausina (95 %:n CI) | 10,2 (7,9; 12,9) | 4,8 (4,6; 6,4) |
Riskisuhde (95 %:n CI) p-arvo | 0,65 (0,52; 0,81) 0,0001 | |
Taudin uusiutumiseen kulunut aika | ||
Tauti uusiutunut, n (%) | 154 (64,7) | 179 (76,5) |
Taudin uusiutumiseen kuluneen ajan mediaani kuukausina (95 %:n CI) | 10,2 (8,3; 13,4) | 4,9 (4,6; 6,4) |
| Hoidon keskeyttämiseen kulunut aika | ||
| Hoito keskeytetty, n (%) | 193 (81,1) | 208 (88,9) |
| Hoidon keskeyttämiseen kuluneen ajan mediaani, kuukausina (95 %:n CI) | 11,4 (9,8; 13,6) | 6,1 (5,1; 7,4) |
Hoito keskeytetty – sairaus uusiutunut, n (%) | 143 (60,1) | 180 (76,9) |
CI = luottamusväli (confidence interval)
Kokonaiselossaolon ja relapsivapaan elossaolon analyysit ennalta määritetyissä alaryhmissä osoittivat Onureg-valmisteen yhtäläisen hoitotehon kaikissa demografisissa ja sairauteen liittyvissä alaryhmissä, mukaan lukien lähtötason sytogeneettinen riski, aiempien konsolidaatiosyklien lukumäärä ja CR:n/CRi:n status.
Kaplan–Meier-kuvaajat kuvaavat kokonaiselossaolon (ks. kuva 1) ja relapsivapaan elossaolon (ks. kuva 2) tulokset.
Kuva 1: Kokonaiselossaolon Kaplan–Meier-kuvaaja: Onureg ja lumelääke (hoitoaikeen mukainen [ITT] populaatio)
Kuva 2: Relapsivapaan elossaolon Kaplan–Meier-kuvaaja: Onureg ja lumelääke (hoitoaikeen mukainen [ITT] populaatio)
Potilailla, joiden annostusta suurennettiin relapsin takia 300 mg:aan 21 päivän ajaksi, kokonaiselossaolon mediaani (22,8 kuukautta Onureg-haarassa ja 14,6 kuukautta lumelääkehaarassa) ja relapsivapaan elossaolon mediaani (7,4 kuukautta Onureg-haarassa ja 4,6 kuukautta lumelääkehaarassa) olivat verrannollisia koko tutkimuksen tulosten kanssa.
Onureg-valmisteella oli lumelääkkeeseen verrattuna suotuisa hoidollinen vaikutus kokonaiselossaoloon sekä MRD-positiivisilla että MRD-negatiivisilla potilailla (MRD, minimaalinen jäännöstauti). Kokonaiselossaoloon vaikuttava hoitoteho oli voimakkaampi MRD-positiivisilla potilailla (HR = 0,69; 95 %:n CI: 0,51; 0,93) kuin MRD-negatiivisilla potilailla (HR = 0,81; 95 %:n CI: 0,59; 1,12).
Terveyteen liittyvä elämänlaatu (Health related quality of life, HRQoL)
Terveyteen liittyvää elämänlaatua arvioitiin FACIT-F-asteikolla (Functional assessment of chronic illness therapy-fatigue), EQ-5D-3L-mittarilla (Five dimensions three levels health utility index) ja visuaalis-analogisella asteikolla (VAS). Lähtötasolla potilaiden väsymys oli vähäistä ja terveyteen liittyvän elämänlaadun taso hyvä. Lähtötaso oli yleisesti ottaen verrannollinen saman ikäisen muun väestön kanssa. Tämä terveyteen liittyvän elämänlaadun taso säilyi Onureg-valmistetta saaneilla niin lähtötasoon kuin lumelääkettä saaneisiinkin verrattuna. Selvään heikkenemiseen kulunut aika ja niiden potilaiden osuus, joilla oli kliinisesti merkittävää heikkenemistä, olivat samankaltaisia Onureg- valmistetta ja lumelääkettä saaneilla potilailla. Yleisesti ottaen löydökset osoittavat, että terveyteen liittyvä elämänlaatu oli samankaltainen Onureg-haarassa ja lumelääkehaarassa eikä siinä ajan myötä tapahtunut kliinisesti merkittävää heikkenemistä.
Farmakokinetiikka
Imeytyminen
Altistus oli yleisesti ottaen lineaarista ja systeeminen altistus suureni suhteessa annokseen; tutkittavien välillä havaittiin kuitenkin suuria eroja. Geometrinen keskiarvo (variaatiokerroin [% CV]) Cmax oli 145,1 ng/ml (63,7) ja AUC-arvo 241,6 ng h/ml (64,5) suun kautta annetun 300 mg:n kerta-annoksen jälkeen. Toistuva anto suositellulla annoksella ei johtanut lääkeaineen kertymiseen. Atsasitidiinin imeytyminen oli nopeaa, ja Tmax-arvojen mediaani saavutettiin 1 tunnin kuluttua annoksesta. Keskimääräinen biologinen hyötyosuus suun kautta annon jälkeen suhteessa ihon alle antoon oli noin 11 %.
Ruuan vaikutus
Ruualla oli hyvin vähäinen vaikutus Onureg-valmisteen altistukseen. Tästä syystä Onureg-valmisteen voi ottaa ruokailun yhteydessä tai erikseen.
Jakautuminen
Suun kautta tapahtuneen annostelun jälkeen näennäisen jakautumistilavuuden geometrinen keskiarvo oli 70 kg painavalla henkilöllä 12,6 l/kg. Atsasitidiinin sitoutuminen plasman proteiineihin oli 6–12 %.
Biotransformaatio
In vitro -tietojen perusteella atsasitidiinin metabolia ei vaikuta välittyvän sytokromi P450:n isoentsyymien (CYP) kautta. Atsasitidiini läpikäy spontaanin hydrolyysin ja sytidiinideaminaasin välittämän deaminaation.
Eliminaatio
Näennäisen puhdistuman geometrinen keskiarvo oli 1 242 l/tunnissa ja geometrinen keskimääräinen puoliintumisaika oli noin 0,5 tuntia. Kun viidelle syöpäpotilaalle annettiin 14C-atsasitidiinia laskimoon, kumulatiivinen erittyminen virtsaan oli 85 % radioaktiivisesta annoksesta. Annetusta radioaktiivisuudesta < 1% erittyi ulosteeseen kolmen päivän aikana. Keskimääräinen radioaktiivisuuden erittyminen virtsaan 14C-atsasitidiinin ihon alle annon jälkeen oli 50 %. Virtsasta mitatun muuttumattoman atsasitidiinin määrä suhteessa annokseen oli ihon alle tai suun kautta annostelun jälkeen < 2 %. Erittymistä ulosteeseen suun kautta annostelun jälkeen ei ole mitattu.
Farmakodynaamiset vaikutukset
Atsasitidiinin epigeneettistä säätelevää vaikutusta DNA:n globaalin metylaation vähenemiseen veressä pidettiin yllä pitkittyneellä altistuksella, jossa 300 mg annettiin päivittäin 14 tai 21 päivän ajan 28-päivän syklissä myelooisia syöpiä, mukaan lukien AML:ää, sairastaville potilaille vaiheen 1/2 tutkimuksessa. Atsasitidiinin plasma-altistuksen ja veren globaalin DNA:n metylaation vähentymisen farmakodynaamisen vaikutuksen välillä havaittiin positiivinen korrelaatio.
Erityisryhmät
Iäkkäät
286 AML-potilaalla tehdyn populaatiofarmakokineettisen analyysin mukaan iällä (46–93 vuotta) ei ollut kliinisesti merkitsevää vaikutusta Onureg-valmisteen farmakokinetiikkaan. Tästä syystä Onureg-valmisteen annosta ei ole tarpeen muuttaa potilaan iän mukaan.
Maksan vajaatoiminta
Muodollisia tutkimuksia potilailla, joilla on maksan vajaatoiminta, ei ole tehty. Maksan vajaatoiminnalla ei todennäköisesti ole kliinisesti merkittävää vaikutusta farmakokinetiikkaan, koska atsasitidiini läpikäy spontaanin hydrolyysin ja sytidiinideaminaasin välittämän deaminaation. Populaatiofarmakokineettisen analyysin mukaan ASAT-arvolla (8–155 U/L), ALAT-arvolla (5–185 U/L) ja lievällä maksan vajaatoiminnalla (BIL ≤ ULN ja ASAT > ULN tai BIL 1–1,5 × ULN ja mikä tahansa ASAT-arvo) ei ollut kliinisesti merkittävää vaikutusta atsasitidiinin farmakokinetiikkaan. Kohtalaisen tai vaikean maksan vajaatoiminnan vaikutusta (BIL > 1,5 × ULN ja mikä tahansa ASAT- arvo) atsasitidiinin farmakokinetiikkaan ei tunneta.
Munuaisten vajaatoiminta
Atsasitidiinin farmakokinetiikkaa verrattiin päivittäisen ihon alle annetun (päivät 1–5) 75 mg/m2:n päiväannoksen jälkeen kuudella syöpäpotilaalla, joilla oli normaali munuaisten toiminta (CLcr >80 ml/min), ja kuudella syöpäpotilaalla, joilla oli vaikea munuaisten vajaatoiminta (CLcr <30 ml/min). Vaikea munuaisten vajaatoiminta lisäsi atsasitidiinin altistusta noin 70 %:lla kerta-annoksen jälkeen ja 41 %:lla toistuvien ihon alle annettujen annosten jälkeen. Altistuksen suurenemisella ei ollut korrelaatiota haittatapahtumien lisääntymisen kanssa.
Populaatiofarmakokineettinen analyysi osoitti, että 300 mg:n Onureg-annoksen annon jälkeen atsasitidiinin AUC-arvo plasmassa nousi lievää munuaisten vajaatoimintaa (CLcr: ≥ 60–< 90 ml/min) sairastavilla potilailla 19 %, kohtalaista munuaisten vajaatoimintaa (CLcr: ≥ 30–< 60 ml/min) sairastavilla 25 % ja vaikeaa munuaisten vajaatoimintaa (CLcr: < 30 ml/min) sairastavilla 38 %. Vaikean munaisten vajaatoiminnan vaikutus Onureg-valmisteeseen oli samankaltainen kuin edellä kuvatussa injektoitavalla atsasitidiinilla tehdyssä kliinisessä, munuaisten vajaatoimintaa koskevassa tutkimuksessa (∼ 40 %:n lisäys AUC-arvossa). Atsasitidiinin altistus (AUC) on noin 75 % pienempi suun kautta annon jälkeen kuin ihon alle annon jälkeen, joten noin 40 %:n altistuksen lisäys suun kautta annon jälkeen katsotaan edelleen turvalliseksi ja siedettäväksi. Tästä syystä Onureg-annosta ei tarvitse muuttaa lievää, kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastaville potilaille.
Rotu ja etninen tausta
Rodun tai etnisen taustan vaikutuksia Onureg-valmisteen farmakokinetiikkaan ei tunneta.
Prekliiniset tiedot turvallisuudesta
Koirilla tehdyssä 14 päivän toksisuustutkimuksessa, jossa atsasitidiinia annettiin koirille suun kautta, kuolleisuutta ilmeni annoksilla 8 ja 16 mg/m2 päivässä. Suurin siedetty annos oli 4 mg/m2/päivä. Yhden tai kaikkien annosten annon jälkeen pansytopenia korreloi luuytimen hypoplasian, imukudoskadon, rauhasen/luumenin laajentumisen ja paksu- ja ohutsuolen limakalvon suolirauhasten solukuoleman kanssa ja/tai havaittiin sentrilobulaarista maksasolujen vakuolisaatiota. Suurimmalla siedetyllä annoksella nämä löydökset paranivat kokonaan tai osittain kolmen viikon kuluttua. Kun atsasitidiinia annettiin parenteraalisesti vertailukelpoisilla annoksilla jyrsijöille, koirille ja apinoille, niillä havaittiin kuolleisuutta ja vastaavaa kohde-elintoksisuutta. Atsasitidiinin toistuvan altistuksen aiheuttamaa toksisuutta koskevien tutkimusten ei-kliiniset tiedot eivät viittaa erityiseen vaaraan ihmisille.
Atsasitidiini indusoi sekä geenimutaatioita että kromosomipoikkeavuuksia bakteeri- ja nisäkäslajien solujärjestelmissä in vitro. Atsasitidiinin mahdollista karsinogeenisuutta arvioitiin hiirillä ja rotilla. Atsasitidiini indusoi hematopoieettisen järjestelmän kasvaimia naarashiirissä, kun sitä annettiin vatsakalvonsisäisesti 3 kertaa viikossa 52 viikon ajan. 50 viikkoa vatsakalvonsisäisesti atsasitidiinia saaneilla hiirillä todettiin lymforetikulaarisen järjestelmän, keuhkojen, rintarauhasen ja ihon kasvainten esiintyvyyden lisääntyneen. Rotilla suoritetussa tuumorigeenisuutta koskevassa kokeessa havaittiin kiveskasvainten esiintyvyyden lisääntyneen.
Hiirillä suoritetuissa varhaisen vaiheen sikiötoksisuutta koskevissa kokeissa todettu kohtukuolemien (lisääntynyt imeytyminen) esiintyvyys oli 44 % organogeneesin aikana annetun yksittäisen vatsakalvonsisäisen atsasitidiinipistoksen jälkeen. Aivojen kehitysvaurioita on todettu hiirillä, joille annettiin atsasitidiinia kovan suulaen sulkeutumisen aikana tai ennen sitä. Rotilla atsasitidiini ei aiheuttanut haittavaikutuksia annettaessa ennen implantaatiota, mutta se oli selvästi embryotoksinen annettaessa organogeneesin aikana. Organogeneesin aikaisia sikiövaurioita rotilla olivat: keskushermoston anomaliat (eksenkefalia/enkefaloseele), raajojen anomaliat (mikromelia, kampurajalka, syndaktylia, oligodaktylia) ja muut (mikroftalmia, mikrognatia, vatsahalkio, ödeema ja kylkiluiden epämuodostumat).
Atsasitidiinin anto uroshiirille ennen parittelua naarashiirien kanssa, jotka eivät saaneet atsasitidiinia, johti heikentyneeseen hedelmällisyyteen ja keskenmenoon tai jälkeläisten menetykseen hedelmöitystä seuraavassa embryonaalisessa ja syntymän jälkeisessä kehityksessä. Atsasitidiinin antamisesta urosrotille seurasi kivesten ja lisäkivesten painon pieneneminen, siittiöiden määrän väheneminen, raskauksien väheneminen, epämuodostuneiden alkioiden lisääntyminen ja alkiomenetysten lisääntyminen paritelluilla naarailla (ks. kohta Raskaus ja imetys).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kroskarmelloosinatrium (E468), magnesiumstearaatti (E572), mannitoli (E421), silisifioitu mikrokiteinen selluloosa (E460, E551)
Onureg 200 mg:n tabletin päällyste
Opadry II (vaaleanpunainen), jonka aineet ovat:
Hypromelloosi (E464), titaanidioksidi (E171), laktoosimonohydraatti, polyeteeniglykoli/makrogolit (E1521), triasetiini (E1518), punainen rautaoksidi (E172)
Onureg 300 mg:n tabletin päällyste
Opadry II (ruskea), jonka aineet ovat:
Hypromelloosi (E464), titaanidioksidi (E171), laktoosimonohydraatti, polyeteeniglykoli/makrogolit (E1521), triasetiini (E1518), punainen rautaoksidi (E172), keltainen rautaoksidi (E172), musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ONUREG tabletti, kalvopäällysteinen
200 mg (L:kyllä) 7 fol (6766,59 €)
300 mg (L:kyllä) 7 fol (7498,37 €)
PF-selosteen tieto
Kalvopäällysteiset tabletit on pakattu OPA/PVC/alumiini-läpipainopakkauksiin.
Pakkauksessa on 7 tai 14 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Onureg 200 mg kalvopäällysteiset tabletit
Vaaleanpunainen, soikea kalvopäällysteinen tabletti, jonka koko on 17,0 x 7,6 mm ja jonka toisella puolella on merkintä ”200” ja toisella puolella ”ONU”.
Onureg 300 mg kalvopäällysteiset tabletit
Ruskea, soikea kalvopäällysteinen tabletti, jonka koko on 19,0 x 9,0 mm ja jonka toisella puolella on merkintä ”300” ja toisella puolella ”ONU”.
Käyttö- ja käsittelyohjeet
Onureg on sytotoksinen lääkevalmiste. Jos kalvopäällysteisten tablettien sisältämää jauhetta joutuu iholle, iho on pestävä välittömästi ja huolellisesti vedellä ja saippualla. Jos jauhetta joutuu limakalvoille, alue on huuhdeltava huolellisesti vedellä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ONUREG tabletti, kalvopäällysteinen
200 mg 7 fol
300 mg 7 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Atsasitidiini-tabletti: Aikuisten akuutin myelooisen leukemian ylläpitohoito erityisin edellytyksin (3076).
ATC-koodi
L01BC07
Valmisteyhteenvedon muuttamispäivämäärä
08.01.2026
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com