
ZOLGENSMA infusionsvätska, lösning 2 x 10exp13 vektorigenomia/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Terveydenhuollon ammattilaisen opas Zolgensma®-hoidon (onasemnogeeniabeparvoveekki) riskien minimointiin
Observera
▼ Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Onasemnogen-abeparvovek är ett genterapiläkemedel som uttrycker det humana proteinet överlevnadsmotorneuron (SMN). Det är en icke‑replikerande rekombinant adenoassocierad virusbaserad vektor av serotyp 9 (AAV9) som innehåller cDNA av den humana SMN‑genen under kontroll av cytomegalovirusförstärkaren/β‑aktin‑hybridpromotorn från kyckling.
Onasemnogen-abeparvovek produceras i humana embryonala njurceller med rekombinant DNA‑teknik.
Varje ml innehåller onasemnogen-abeparvovek med en nominell koncentration på 2 × 1013 vektorgenom (vg). Injektionsflaskor kommer att innehålla en extraherbar volym som antingen är minst 5,5 ml eller 8,3 ml. Det totala antalet injektionsflaskor och kombinationen av fyllnadsvolymer i varje färdig förpackning kommer att anpassas för att uppfylla doseringskraven hos enskilda patienter baserat på deras vikt (se avsnitt Dosering och administreringssätt och Förpackningstyp och innehåll).
Hjälpämne med känd effekt
Detta läkemedel innehåller 0,2 mmol natrium per ml.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Infusionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
Zolgensma är avsett för behandling av:
- patienter med 5q spinal muskelatrofi (SMA) med en biallelisk mutation i SMN1‑genen och en klinisk diagnos på SMA typ 1 eller
- patienter med 5q SMA med en biallelisk mutation i SMN1‑genen och upp till 3 kopior av SMN2‑genen.
Villkor
Hoito on aloitettava ja annettava kliinisissä keskuksissa spinaalista lihasatrofiaa sairastavien potilaiden hoitoon perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandling ska initieras och administreras på kliniska centra och övervakas av en läkare med erfarenhet av behandling av patienter med SMA.
Före administrering av onasemnogen-abeparvovek krävs laboratorietester vid baslinjen som inkluderar, men inte är begränsade till:
- test av AAV9‑antikroppar med en lämplig validerad analys
- leverfunktion: alaninaminotransferas (ALAT), aspartataminotransferas (ASAT), totalt bilirubin, albumin, protrombintid, partiell tromboplastintid (PTT) och internationellt normaliserat förhållande (INR)
- kreatinin
- fullständig blodstatus (inklusive hemoglobin och trombocytantal) och
- troponin I
Behovet av noggrann övervakning av leverfunktion och trombocytantal efter administrering och behovet av kortikosteroidbehandling ska beaktas när tidpunkten för behandling med onasemnogen-abeparvovek fastställs (se avsnitt Varningar och försiktighet).
På grund av den ökade risken för ett allvarligt systemiskt immunsvar, rekommenderas det att patienter är kliniskt stabila i sitt allmänna hälsotillstånd (t.ex. vätskebalans och näringstillstånd, frånvaro av infektion) före infusion av onasemnogen-abeparvovek. Vid akuta eller kroniska okontrollerade aktiva infektioner ska behandlingen skjutas upp tills infektionen har försvunnit och patienten är kliniskt stabil (se underavsnitt 4.2 ”Immunmodulerande regim” och 4.4 ”Systemiskt immunsvar”).
Dosering
Endast för infusion av en intravenös engångsdos.
Patienter kommer att få en nominell dos på 1,1 × 1014 vg/kg onasemnogen-abeparvovek. Den totala volymen bestäms av patientens kroppsvikt.
I tabell 1 anges den rekommenderade dosen för patienter som väger 2,6 kg till 21,0 kg.
Tabell 1 Rekommenderad dos baserad på patientens kroppsvikt
| Intervall för patientvikt (kg) | Dos (vg) | Total dosvolyma (ml) |
| 2,6‑3,0 | 3,3 × 1014 | 16,5 |
| 3,1‑3,5 | 3,9 × 1014 | 19,3 |
| 3,6‑4,0 | 4,4 × 1014 | 22,0 |
| 4,1‑4,5 | 5,0 × 1014 | 24,8 |
| 4,6‑5,0 | 5,5 × 1014 | 27,5 |
| 5,1‑5,5 | 6,1 × 1014 | 30,3 |
| 5,6‑6,0 | 6,6 × 1014 | 33,0 |
| 6,1‑6,5 | 7,2 × 1014 | 35,8 |
| 6,6‑7,0 | 7,7 × 1014 | 38,5 |
| 7,1‑7,5 | 8,3 × 1014 | 41,3 |
| 7,6‑8,0 | 8,8 × 1014 | 44,0 |
| 8,1‑8,5 | 9,4 × 1014 | 46,8 |
| 8,6‑9,0 | 9,9 × 1014 | 49,5 |
| 9,1‑9,5 | 1,05 × 1015 | 52,3 |
| 9,6‑10,0 | 1,10 × 1015 | 55,0 |
| 10,1‑10,5 | 1,16 × 1015 | 57,8 |
| 10,6‑11,0 | 1,21 × 1015 | 60,5 |
| 11,1‑11,5 | 1,27 × 1015 | 63,3 |
| 11,6‑12,0 | 1,32 × 1015 | 66,0 |
| 12,1‑12,5 | 1,38 × 1015 | 68,8 |
| 12,6‑13,0 | 1,43 × 1015 | 71,5 |
| 13,1‑13,5 | 1,49 × 1015 | 74,3 |
| 13,6–14,0 | 1,54 × 1015 | 77,0 |
| 14,1–14,5 | 1,60 × 1015 | 79,8 |
| 14,6–15,0 | 1,65 × 1015 | 82,5 |
| 15,1–15,5 | 1,71 × 1015 | 85,3 |
| 15,6–16,0 | 1,76 × 1015 | 88,0 |
| 16,1–16,5 | 1,82 × 1015 | 90,8 |
| 16,6–17,0 | 1,87 × 1015 | 93,5 |
| 17,1–17,5 | 1,93 × 1015 | 96,3 |
| 17,6–18,0 | 1,98 × 1015 | 99,0 |
| 18,1–18,5 | 2,04 × 1015 | 101,8 |
| 18,6–19,0 | 2,09 × 1015 | 104,5 |
| 19,1–19,5 | 2,15 × 1015 | 107,3 |
| 19,6–20,0 | 2,20 × 1015 | 110,0 |
| 20,1–20,5 | 2,26 × 1015 | 112,8 |
| 20,6–21,0 | 2,31 × 1015 | 115,5 |
a ANM: Antal injektionsflaskor per kit och det antal kit som krävs är viktberoende. Dosvolym beräknas med hjälp av den övre gränsen av intervallet för patientvikt.
Immunmodulerande regim
Ett immunsvar mot AAV9-kapsiden kommer att uppkomma efter administrering av onasemnogen-abeparvovek (se avsnitt Varningar och försiktighet). Detta kan leda till ökningar av leveraminotransferaser, ökningar av troponin I eller minskat trombocytantal (se avsnitt Varningar och försiktighet och Biverkningar). För att dämpa immunsvaret rekommenderas immunmodulering med kortikosteroider. Om möjligt ska patientens vaccinationsschema anpassas för att ge utrymme för samtidig administrering av kortikosteroid före och efter infusion av onasemnogen-abeparvovek (se avsnitt Interaktioner).
Innan den immunmodulerande regimen initieras och före administrering av onasemnogen-abeparvovek måste patienten kontrolleras för tecken och symtom på någon typ av aktiv infektionssjukdom.
Med början 24 timmar före infusion av onasemnogen-abeparvovek rekommenderas initiering av en immunmodulerande regim enligt schemat nedan (se tabell 2). Om patienterna vid något tillfälle inte svarar adekvat på oralt prednisolon 1 mg/kg/dag (eller motsvarande), ska omedelbar konsultation med en pediatrisk gastroenterolog eller hepatolog övervägas, baserat på patientens kliniska förlopp. Justering av den rekommenderade immunmodulerande regimen, inklusive ökad dos, längre varaktighet eller förlängning av kortikosteroidnedtrappningen ska också övervägas (se avsnitt Varningar och försiktighet). Om oral kortikosteroidbehandling inte tolereras kan kortikosteroid intravenöst anses vara kliniskt indicerat.
Tabell 2 Immunmodulerande regim före och efter infusion
| Före infusion | 24 timmar före onasemnogen-abeparvovek | Prednisolon oralt 1 mg/kg/dag (eller motsvarande om annan kortikosteroid används) |
| Efter infusion | 30 dagar (inklusive den dag onasemnogen-abeparvovek administreras) | Prednisolon oralt 1 mg/kg/dag (eller motsvarande om annan kortikosteroid används) |
Följt av 28 dagar: För patienter med icke anmärkningsvärda resultat (normal klinisk undersökning, normalt totalt bilirubin och hos vilka både ALAT- och ASAT‑värden är under 2 × övre normalgränsen (ULN)) efter avslutad 30‑dagarsperiod: eller | Systemiska kortikosteroider ska trappas ned gradvis. Nedtrappning av prednisolon (eller motsvarande om annan kortikosteroid används), t.ex. 2 veckor med 0,5 mg/kg/dag och därefter 2 veckor med 0,25 mg/kg/dag oralt prednisolon | |
| För patienter med avvikande leverfunktionsvärden efter avslutad 30‑dagarsperiod: fortsätt tills ASAT- och ALAT‑värden är under 2 × ULN och alla andra värden (t.ex. totalt bilirubin) återgår till normalintervallet, följt av nedtrappning under 28 dagar eller längre om det behövs. | Systemiska kortikosteroider (motsvarande oralt prednisolon 1 mg/kg/dag) Systemiska kortikosteroider ska trappas ned gradvis. |
Leverfunktionen (ASAT, ALAT, totalt bilirubin) ska kontrolleras med regelbundna intervaller under minst 3 månader efter infusion av onasemnogen-abeparvovek (varje vecka under den första månaden och under hela nedtrappningsperioden av kortikosteroiden och därefter varannan vecka i ytterligare en månad), och vid andra tidpunkter om det anses vara kliniskt indicerat. Patienter med försämrade resultat vid leverfunktionstest och/eller tecken eller symtom på akut sjukdom ska omedelbart utvärderas kliniskt och övervakas noggrant (se avsnitt Varningar och försiktighet).
Om en annan kortikosteroid än prednisolon används ska liknande överväganden och metoder användas för att trappa ned dosen efter 30 dagar.
Särskilda populationer
Nedsatt njurfunktion
Säkerhet och effekt för onasemnogen-abeparvovek har inte fastställts för patienter med nedsatt njurfunktion och behandling med onasemnogen-abeparvovek ska övervägas noggrant. En dosjustering ska inte övervägas.
Nedsatt leverfunktion
Patienter med ALAT-, ASAT-, totala bilirubinnivåer >2 × ULN (utom på grund av neonatal gulsot) eller positiv serologi för hepatit B eller hepatit C har inte studerats i kliniska studier med onasemnogen-abeparvovek. Behandling med onasemnogen-abeparvovek ska övervägas noggrant hos patienter med nedsatt leverfunktion (se avsnitt Varningar och försiktighet och Biverkningar). En dosjustering ska inte övervägas.
0SMN1/1SMN2‑genotyp
Ingen dosjustering ska övervägas hos patienter med en biallelisk mutation av SMN1‑genen och bara en kopia av SMN2 (se avsnitt Farmakodynamiska egenskaper).
Anti‑AAV9‑antikroppar
Ingen dosjustering ska övervägas hos patienter med anti‑AAV9‑antikroppstitrar vid baslinjen över 1:50 (se avsnitt Varningar och försiktighet).
Pediatrisk population
Säkerhet och effekt för onasemnogen-abeparvovek hos förtidigt födda barn innan de nått normal gestationsålder har inte fastställts. Inga data finns tillgängliga. Administrering av onasemnogen-abeparvovek ska övervägas noggrant eftersom samtidig behandling med kortikosteroider kan ha en negativ effekt på neurologisk utveckling.
Det finns begränsad erfarenhet hos patienter 2 år och äldre eller med en kroppsvikt över 13,5 kg. Säkerhet och effekt för onasemnogen-abeparvovek för dessa patienter har inte fastställts. Tillgänglig information finns i avsnitt Farmakodynamiska egenskaper. En dosjustering ska inte övervägas (se tabell 1).
Administreringssätt
För intravenös användning.
Onasemnogen-abeparvovek administreras som en intravenös engångsinfusion. Det ska administreras med en sprutpump som en intravenös engångsinfusion i form av en långsam infusion under cirka 60 minuter. Det får inte administreras som en intravenös stötdos eller bolusdos.
Insättning av en andra kateter (back‑up‑kateter) rekommenderas vid en blockering i den primära katetern. Efter avslutad infusion ska slangen spolas med natriumklorid 9 mg/ml (0,9 %) injektionsvätska, lösning.
Försiktighetsåtgärder före hantering eller administrering av läkemedlet
Detta läkemedel innehåller en genetiskt modifierad organism. Hälso- och sjukvårdspersonal ska därför vidta lämpliga försiktighetsåtgärder (användning av handskar, säkerhetsglasögon, laboratorierock och ärmskydd) vid hantering eller administrering av produkten (se avsnitt Särskilda anvisningar för destruktion och övrig hantering).
Detaljerade anvisningar om beredning, hantering, oavsiktlig exponering och kassering (inklusive korrekt hantering av kroppsavfall) av onasemnogen-abeparvovek finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Befintlig immunitet mot AAV9
Bildande av anti‑AAV9‑antikropp kan ske efter naturlig exponering. Det finns flera studier av prevalensen av AAV9‑antikroppar hos den allmänna populationen som visar låga frekvenser före exponering för AAV9 hos den pediatriska populationen. Patienter ska testas för förekomst av AAV9‑antikroppar före infusion av onasemnogen-abeparvovek. Nytt test ska utföras om AAV9‑antikroppstitrar rapporteras vara över 1:50. Det är ännu okänt om eller under vilka förhållnaden onasemnogen-abeparvovek kan administreras på ett säkert och effektivt sätt vid förekomst av anti‑AAV9‑antikroppar över 1:50 (se avsnitt Dosering och administreringssätt och Farmakodynamiska egenskaper).
Avancerad SMA
Eftersom SMA leder till progressiv och icke‑reversibel skada på motorneuroner beror nyttan av onasemnogen-abeparvovek hos symtomatiska patienter på sjukdomsbördans omfattning vid tidpunkten för behandling. Tidig behandling kan eventuellt leda till större nytta. Patienter med avancerad symtomatisk SMA uppnår inte en lika omfattande motorisk utveckling som oberörda friska jämlikar. De kan ha klinisk nytta av genersättningsterapi, beroende på hur avancerad sjukdomen är vid tidpunkten för behandling (se avsnitt Farmakodynamiska egenskaper).
Behandlande läkare ska beakta att nyttan är kraftigt nedsatt hos patienter med uttalad muskelsvaghet och andningssvikt, patienter som står på permanent ventilation och patienter som inte kan svälja.
Nytta/riskprofilen för onasemnogen-abeparvovek hos patienter med avancerad SMA, som hålls vid liv genom permanent ventilation och utan förmåga att utvecklas, är inte fastställd.
Infusionsrelaterade reaktioner och anafylaktiska reaktioner
Infusionsrelaterade reaktioner, inklusive anafylaktiska reaktioner, har inträffat under och/eller strax efter infusion av onasemnogen-abeparvovek (se avsnitt Biverkningar). Patienter ska övervakas noggrant med avseende på kliniska tecken och symtom på infusionsrelaterade reaktioner. Om en reaktion inträffar ska infusionen avbrytas och behandling ges efter behov. Baserat på klinisk utvärdering och standardpraxis kan administreringen återupptas försiktigt.
Immunogenicitet
Ett immunsvar mot AAV9-kapsiden kommer att uppkomma efter infusion av onasemnogen-abeparvovek, inklusive antikroppsbildning mot AAV9‑kapsiden och T‑cellsmedierat immunsvar, trots den immunmodulerande regim som rekommenderas i avsnitt Dosering och administreringssätt (se även underavsnitt ”Systemiskt immunsvar” nedan).
Levertoxicitet
Immunmedierad levertoxicitet manifesteras i allmänhet som förhöjda ALAT- och/eller ASAT-nivåer. Akut allvarlig leverskada och akut leversvikt, inklusive dödsfall, har rapporterats med användning av onasemnogen-abeparvovek, vanligtvis inom 2 månader efter infusion trots behandling med kortikosteroider före och efter infusionen. Immunmedierad levertoxicitet kan kräva justering av den immunmodulerande regimen inklusive längre varaktighet, ökad dos eller förlängning av kortikosteroidnedtrappningen (se avsnitt Biverkningar).
- Riskerna och fördelarna med behandling med onasemnogen-abeparvovek ska övervägas noggrant hos patienter med befintlig nedsatt leverfunktion.
- Patienter med befintlig nedsatt leverfunktion eller akut virusinfektion i levern kan löpa större risk för akut allvarlig leverskada (se avsnitt Dosering och administreringssätt).
- Data från en liten studie på barn som väger ≥8,5 kg till ≤21 kg (i åldern cirka 1,5 till 9 år) tyder på en högre frekvens av ASAT- eller ALAT-ökningar (hos 23 av 24 patienter) jämfört med frekvenser av ASAT/ALAT-ökningar som observerats i andra studier på patienter som väger <8,5 kg (hos 31 av 99 patienter) (se avsnitt Biverkningar).
- Administrering av AAV‑vektor leder ofta till aminotransferasökning.
- Akut allvarlig leverskada och akut leversvikt har uppkommit med onasemnogen-abeparvovek. Fall av akut leversvikt med dödlig utgång har rapporterats (se avsnitt Biverkningar).
- Före infusion ska leverfunktion hos alla patienter bedömas genom klinisk undersökning och laboratorietester (se avsnitt Dosering och administreringssätt).
- För att mildra eventuella aminotransferasökningar ska en systemisk kortikosteroid administreras till alla patienter före och efter infusion av onasemnogen-abeparvovek (se avsnitt Dosering och administreringssätt).
- Leverfunktion ska kontrolleras med regelbundna intervaller under minst 3 månader efter infusion och vid andra tidpunkter om det anses vara kliniskt indicerat (se avsnitt Dosering och administreringssätt).
- Patienter med försämrade resultat vid leverfunktionstest och/eller tecken eller symtom på akut sjukdom ska omedelbart utvärderas kliniskt och övervakas noggrant.
- Om leverskada misstänks rekommenderas omedelbar konsultation med en pediatrisk gastroenterolog eller hepatolog. Justering av den rekommenderade immunmodulerande regimen och ytterligare tester (t.ex. albumin, protrombintid, PTT och INR) rekommenderas också.
ASAT/ALAT/totalt bilirubin ska bedömas varje vecka under den första månaden efter infusion av onasemnogen-abeparvovek och under hela nedtrappningsperioden av kortikosteroiden. Nedtrappning av prednisolon ska inte övervägas förrän ASAT/ALAT-nivåerna är under 2 × ULN och alla andra värden (t.ex. totalt bilirubin) återgår till normalintervallet (se avsnitt Dosering och administreringssätt). Om patienten är kliniskt stabil med icke anmärkningsvärda resultat i slutet av perioden för nedtrappning av kortikosteroiden ska leverfunktionen fortsätta att kontrolleras varannan vecka i ytterligare en månad.
Trombocytopeni
Övergående minskning av trombocytantalet, en del som uppfyllde kriterierna för trombocytopeni, observerades i kliniska studier med onasemnogen-abeparvovek. I de flesta fall uppkom det lägsta trombocytvärdet första veckan efter infusion av onasemnogen-abeparvovek.
Efter godkännande för försäljning har fall med trombocytantal <25 x 109/l rapporterats inträffa inom tre veckor efter administrering.
Trombocytantalet ska mätas före infusion med onasemnogen-abeparvovek och kontrolleras noggrant inom loppet av de tre första veckorna efter infusionen och regelbundet därefter, åtminstone varje vecka den första månaden och varannan vecka under den andra och tredje månaden tills trombocytantalet återgår till baslinjen.
Data från en liten studie på barn som väger ≥8,5 kg till ≤21 kg (i åldern cirka 1,5 till 9 år) tyder på en högre frekvens av trombocytopeni (hos 20 av 24 patienter) jämfört med frekvenser av trombocytopeni som observerats i andra studier på patienter som väger <8,5 kg (hos 22 av 99 patienter) (se avsnitt Biverkningar).
Ökning av troponin-I
Ökningar av kardiellt troponin-I‑nivåer efter infusion av onasemnogen-abeparvovek observerades (se avsnitt Biverkningar). De ökade nivåerna av troponin I som sågs hos vissa patienter kan eventuellt tyda på skadad hjärtvävnad. Baserat på dessa resultat och den observerade kardiotoxiciteten hos möss ska troponin I‑nivåer bestämmas före infusion av onasemnogen-abeparvovek och kontrolleras om det anses vara kliniskt indicerat. Överväg att konsultera en hjärtexpert vid behov.
Trombotisk mikroangiopati
Flera fall av trombotisk mikroangiopati (TMA) har rapporterats inträffa med onasemnogen-abeparvovek (se avsnitt Biverkningar). Fallen inträffade vanligtvis inom de två första veckorna efter infusion med onasemnogen-abeparvovek. TMA är ett akut och livshotande tillstånd som kännetecknas av trombocytopeni och mikroangiopatisk hemolytisk anemi. Dödsfall har rapporterats. Akut njurskada har också observerats. I vissa fall har samtidig immunsystemaktivering rapporterats (t.ex. infektioner, vaccinationer) (se avsnitt Dosering och administreringssätt och Interaktioner för information om administrering av vaccinationer).
Trombocytopeni är ett centralt inslag i TMA, därför ska trombocytantalet övervakas noggrant inom loppet av de tre första veckorna efter infusionen och regelbundet efteråt (se underavsnittet ”Trombocytopeni”). Vid trombocytopeni ska ytterligare utvärdering inklusive diagnostisk testning för hemolytisk anemi och nedsatt njurfunktion göras omedelbart. Om patienter visar kliniska tecken, symtom eller laboratoriefynd som överensstämmer med TMA, ska en specialist omedelbart konsulteras för att hantera TMA som kliniskt indicerat. Vårdgivare ska informeras om tecken och symtom på TMA och ska uppmanas att söka akut medicinsk vård om sådana symtom uppstår.
Systemiskt immunsvar
På grund av den ökade risken för ett allvarligt systemiskt immunsvar, rekommenderas det att patienter är kliniskt stabila i sitt allmänna hälsotillstånd (t.ex. vätskebalans och näringstillstånd, frånvaro av infektion) före infusion av onasemnogen-abeparvovek. Behandling ska inte initieras samtidigt med aktiva infektioner, varken vid akuta (såsom akuta respiratoriska infektioner eller akut hepatit) eller kroniska okontrollerade (såsom kroniskt aktiv hepatit B), förrän infektionen har försvunnit och patienten är kliniskt stabil (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Den immunmodulerande regimen (se avsnitt Dosering och administreringssätt) kan också påverka immunsvaret på infektioner (t.ex. respiratoriska), vilket potentiellt kan resultera i allvarligare kliniskt förlopp för infektionen. Patienter med infektion uteslöts från deltagande i kliniska prövningar med onasemnogen-abeparvovek. Ökad vaksamhet vid förebyggande, övervakning och behandling av infektion rekommenderas före och efter infusion av onasemnogen-abeparvovek. Profylaktiska säsongsbehandlingar, som förhindrar infektioner med respiratoriska syncytialvirus (RSV), rekommenderas och ska vara aktuella. Om möjligt ska patientens vaccinationsschema anpassas för att ge utrymme för samtidig administrering av kortikosteroid före och efter infusion av onasemnogen-abeparvovek (se avsnitt Interaktioner).
Om behandlingstiden med kortikosteroider förlängs eller dosen ökas, ska behandlande läkare känna till risken för binjurebarksvikt.
Risk för tumörutveckling till följd av vektorintegration
Det finns en teoretisk risk för tumörutveckling på grund av integration av AAV-vektor-DNA i genomet.
Onasemnogen-abeparvovek består av en icke-replikerande AAV9-vektor vars DNA till stor del kvarstår i episomal form. Sällsynta fall av slumpmässig vektorintegration i humant DNA är möjliga med rekombinant AAV. Den kliniska relevansen av individuella integrationshändelser är okänd, men det är vedertaget att individuella integrationshändelser potentiellt kan bidra till en risk för tumörutveckling.
Hittills har inga fall av maligniteter associerade med behandling med onasemnogen-abeparvovek rapporterats. I händelse av tumör ska innehavaren av godkännandet för försäljning kontaktas för vägledning om insamling av patientprover för testning.
Utsöndring
Tillfällig utsöndring av onasemnogen-abeparvovek uppkommer, primärt via kroppsavfall. Vårdgivare och patientens familj ska få råd om att använda följande anvisningar för korrekt hantering av patientens kroppsvätskor och avföring:
- god handhygien krävs vid direktkontakt med patientens kroppsvätskor och avföring under minst 1 månad efter behandling med onasemnogen-abeparvovek.
- engångsblöjor ska förseglas i dubbla plastpåsar och kastas i hushållssoporna (se avsnitt Farmakokinetiska egenskaper).
Blod, organ, vävnad och celldonation
Patienter som behandlas med Zolgensma ska inte donera blod, organ, vävnad eller celler för transplantation.
Natriuminnehåll
Detta läkemedel innehåller 4,6 mg natrium per ml motsvarande 0,23 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna). Varje injektionsflaska à 5,5 ml innehåller 25,3 mg natrium och varje injektionsflaska à 8,3 ml innehåller 38,2 mg natrium.
Interaktioner
Inga interaktionsstudier har utförts.
Erfarenhet av användning av onasemnogen-abeparvovek hos patienter som får levertoxiska läkemedel eller som använder levertoxiska substanser är begränsad. Säkerhet för onasemnogen-abeparvovek hos dessa patienter har inte fastställts.
Erfarenhet av samtidig användning av preparat inriktade mot 5q SMA är begränsad.
Vaccinationer
Om möjligt ska patientens vaccinationsschema anpassas för att ge utrymme för samtidig administrering av kortikosteroid före och efter infusion av onasemnogen-abeparvovek (se avsnitt Dosering och administreringssätt och Varningar och försiktighet). Säsongsprofylax mot RSV rekommenderas (se avsnitt Varningar och försiktighet). Levande vacciner, såsom MMR och vattkoppor, ska inte administreras till patienter som står på en immunsuppressiv steroiddos (dvs. ≥ 2 veckors daglig behandling med 20 mg eller 2 mg/kg/kroppsvikt prednisolon eller motsvarande).
Fertilitet, graviditet och amning
Det finns inga data om användning hos kvinnor under graviditet eller amning.
Fertilitets- eller reproduktionsstudier på djur har inte utförts.
Effekter på förmågan att framföra fordon och använda maskiner
Onasemnogen-abeparvovek har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
Onasemnogen-abeparvoveks säkerhet utvärderades för 99 patienter som fick onasemnogen-abeparvovek med den rekommenderade dosen (1,1 x 1014 vg/kg) i 5 öppna kliniska studier. De vanligaste rapporterade biverkningarna efter administrering var ökning av leverenzymer (24,2 %), levertoxicitet (9,1 %), kräkningar (8,1 %), trombocytopeni (6,1 %), ökning av troponin (5,1 %) och feber (5,1 %) (se avsnitt Varningar och försiktighet).
Tabell med lista över biverkningar
De biverkningar som identifierats hos alla patienter som behandlats med en intravenös infusion av onasemnogen-abeparvovek med den rekommenderade dosen och som har ett orsakssamband med behandling visas i Tabell 3. Biverkningar listas enligt MedDRA:s klassificering av organsystem och frekvens. Frekvenskategorier är baserade på följande konventioner: mycket vanliga (≥1/10); vanliga (≥1/100, <1/10); mindre vanliga (≥1/1 000, <1/100); sällsynta (≥1/10 000, <1/1 000); mycket sällsynta (<1/10 000); ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp visas biverkningarna i fallande allvarlighetsgrad.
Tabell 3 Lista över biverkningar mot onasemnogen-abeparvovek
| Biverkningar enligt MedDRA SOC/PT och frekvens | |
| Blodet och lymfsystemet | |
| Vanliga | Trombocytopeni1) |
| Mindre vanliga | Trombotisk mikroangiopati2)3) |
| Immunsystemet | |
| Sällsynta | Anafylaktiska reaktioner |
| Magtarmkanalen | |
| Vanliga | Kräkningar |
| Lever och gallvägar | |
| Vanliga | Levertoxicitet4) |
| Mindre vanliga | Akut leversvikt2)3) |
| Allmänna symtom och/eller symtom vid administreringsstället | |
| Vanliga | Feber |
| Mindre vanliga | Infusionsrelaterade reaktioner |
| Undersökningar och provtagningar | |
| Mycket vanliga | Ökning av leverenzymer5) |
| Vanliga | Ökning av troponin6) |
1)Trombocytopeni inkluderar trombocytopeni och minskat antal trombocyter. 2)Behandlingsrelaterade biverkningar rapporterade utanför kliniska studier före godkännande för försäljning, inklusive tiden efter godkännande för försäljning. 3)Inkluderar dödsfall. 4)Levertoxicitet inkluderar leversteatos och hypertransaminasemi. 5)Ökning av leverenzymer inkluderar: ökning av alaninaminotransferas, ökning av ammoniak, ökning av aspartataminotransferas, ökning av gamma-glutamyltransferas, ökning av leverenzymer, ökning av leverfunktionstest och ökning av transaminaser. 6)Ökning av troponin inkluderar ökning av troponin, ökning av troponin T och troponin I (rapporterat utanför kliniska studier inklusive efter godkännande för försäljning). | |
Beskrivning av utvalda biverkningar
Lever och gallvägar
I det kliniska utvecklingsprogrammet (se avsnitt Farmakodynamiska egenskaper) observerades ökning av transaminaser > 2 × ULN (och i vissa fall > 20 × ULN) hos 31 % av patienterna behandlade med den rekommenderade dosen. Dessa patienter var kliniskt asymtomatiska och ingen av dem hade kliniskt signifikanta ökningar av bilirubin. Ökning av serumtransaminaser försvann vanligtvis vid behandling med prednisolon, och patienterna återhämtade sig utan kliniska följdtillstånd (se avsnitt Dosering och administreringssätt och Varningar och försiktighet).
Efter godkännande för försäljning har det förekommit rapporter om barn som utvecklat tecken och symtom på akut leversvikt (t.ex. gulsot, koagulopati, encefalopati) vanligtvis inom 2 månader av behandling med onasemnogen-abeparvovek, trots behandling med kortikosteroider före och efter infusionen. Fall av akut leversvikt med dödlig utgång har rapporterats.
I en studie (COAV101A12306) med 24 barn som vägde ≥8,5 kg till ≤21 kg (i åldern cirka 1,5 till 9 år; 21 hade avbrutit tidigare SMA-behandling) observerades ökning av transaminaser hos 23 av 24 patienter. Patienterna var asymtomatiska och inga ökningar av bilirubin observerades. ASAT- och ALAT-ökningarna hanterades med användning av kortikosteroider, vanligtvis med förlängd varaktighet (vid vecka 26 fortsatte 17 patienter med prednisolon, vid vecka 52 fick 6 patienter fortfarande prednisolon) och/eller en högre dos.
Övergående trombocytopeni
I det kliniska utvecklingsprogrammet (se avsnitt Farmakodynamiska egenskaper) observerades övergående trombocytopeni vid flera tidpunkter efter dosering och försvann normalt inom två veckor. Minskning av trombocytantal var mer uttalad under den första behandlingsveckan. Efter godkännande för försäljning har fall med övergående minskning av antal trombocyter till nivåer <25 x 109/l rapporterats inträffa inom tre veckor efter administrering (se avsnitt Varningar och försiktighet).
I en studie (COAV101A12306) med 24 barn som vägde ≥8,5 kg till ≤21 kg (i åldern cirka 1,5 till 9 år) observerades trombocytopeni hos 20 av 24 patienter.
Ökningar av troponin I‑nivåer
Ökningar av kardiellt troponin I‑nivåer upp till 0,2 mikrogram/l efter infusion av onasemnogen-abeparvovek observerades. I det kliniska studieprogrammet observerades inga tydliga kliniska kardiella resultat efter administrering av onasemnogen-abeparvovek (se avsnitt Varningar och försiktighet).
Immunogenicitet
Anti‑AAV9‑antikroppstitrar mättes före och efter genterapi i de kliniska studierna (se avsnitt Varningar och försiktighet). Alla patienter som fick onasemnogen-abeparvovek hade anti‑AAV9‑titrar vid eller under ≤ 1:50 före behandling. Genomsnittliga ökningar från baslinjen av AAV9‑titrar observerades hos alla patienter vid alla tidpunkter utom en för antikroppstitrar mot peptiden AAV9, vilket speglar normalt svar på främmande viralt antigen. Vissa patienter fick AAV9‑titrar som överskred kvantifieringsnivån, men de flesta av dessa patienter hade inga potentiellt kliniskt signifikanta biverkningar. Således har inget samband mellan höga anti‑AAV9‑antikroppstitrar och potentialen för eventuella biverkningar eller effektparametrar fastställts.
I den kliniska studien AVXS‑101‑CL‑101 screenades 16 patienter för anti‑AAV9‑antikroppstitrar: 13 hade titrar under 1:50 och rekryterades till studien; tre patienter hade titrar över 1:50 och två av dessa testades på nytt efter att amning avbrutits och deras titrar uppmättes till under 1:50 och båda rekryterades till studien. Det saknas information om amning ska begränsas hos mödrar som kan vara seropositiva för anti‑AAV9‑antikroppar. Samtliga patienter hade AAV9‑antikroppstitrar under eller lika med 1:50 före behandling med onasemnogen-abeparvovek och uppvisade därefter en ökning av anti‑AAV9‑antikroppstitrar på minst 1:102 400 och upp till över 1:819 200.
Detektionen av antikroppsbildning är till stor del beroende av analysens sensitivitet och specificitet. Dessutom kan den observerade incidensen av antikroppspositivitet (inklusive neutraliserande antikroppar) i en analys påverkas av flera faktorer inklusive analysmetod, provhantering, tidpunkt för provtagning, samtidiga läkemedel och underliggande sjukdom.
Ingen patient behandlad med onasemnogen-abeparvovek uppvisade ett immunsvar mot transgenen.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Inga data från kliniska studier är tillgängliga avseende överdosering av onasemnogen-abeparvovek. Justering av prednisolondosen, noggrann klinisk observation och kontroll av laboratorieparametrar (inklusive klinisk kemi och hematologi) för systemiskt immunsvar rekommenderas (se avsnitt Varningar och försiktighet).
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Övriga medel för sjukdomar i rörelseapparaten, ATC-kod: M09AX09
Verkningsmekanism
Onasemnogen-abeparvovek är en genterapi utformad för att introducera en funktionell kopia av överlevnadsmotorneurongenen (SMN1) i transducerade celler för att behandla den monogena ursprungliga orsaken till sjukdomen. Genom att tillhandahålla en alternativ källa till uttryck av SMN‑protein i motorneuroner förväntas det främja överlevnad och funktion av transducerade motorneuroner.
Onasemnogen-abeparvovek är en icke‑replikerande rekombinant AAV‑vektor som använder AVV9‑kapsid för att ge en stabil, fullt funktionell human SMN‑transgen. Förmågan hos AAV9‑kapsiden att korsa blod‑hjärnbarriären och transducerade motorneuroner har påvisats. SMN1‑genen i onasemnogen-abeparvovek är utformad för att uppehålla sig som episomalt DNA i kärnan av transducerade celler och förväntas vara stabilt uttryckta under en längre tidsperiod i postmitotiska celler. AAV9‑viruset är inte känt för att orsaka sjukdom hos människa. Transgenen förs in i målcellerna som en självkomplementär dubbelsträngad molekyl. Uttryck av transgenen drivs av en konstituerande promotor (cytomegalovirusförstärkt β‑aktin‑hybrid från kyckling), som leder till kontinuerlig och ihållande uttryck av SMN‑protein. Bevis på verkningsmekanismen har stöd i icke‑kliniska studier och i humana biodistributionsdata.
Klinisk effekt och säkerhet
AVXS‑101‑CL‑303 fas 3‑studie på patienter med SMA typ 1
AVXS‑101‑CL‑303 (studie CL‑303) är en öppen enarmad studie i fas 3 med en engångsdos av intravenös onasemnogen-abeparvovek vid den terapeutiska dosen (1,1 × 1014 vg/kg). Tjugotvå patienter med SMA typ 1 och 2 kopior av SMN2 rekryterades. Innan behandling med onasemnogen-abeparvovek behövde ingen av de 22 patienterna icke-invasiv ventilatorbehandling (NIV), och alla patienter kunde uteslutande matas oralt (dvs. behövde inte icke-oral näring). Den genomsnittliga Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP‑INTEND)-poängen vid baslinjen var 32,0 (intervall 18 till 52). Den genomsnittliga åldern för de 22 patienterna vid tidpunkten för behandlingen var 3,7 månader (0,5 till 5,9 månader).
Av de 22 rekryterade patienterna överlevde 21 patienter utan permanent ventilation (dvs. händelsefri överlevnad) till ≥ 10,5 månaders ålder, 20 patienter överlevde till ≥ 14 månaders ålder (samprimärt effektmått) och 20 patienter överlevde händelsefria till 18 månaders ålder.
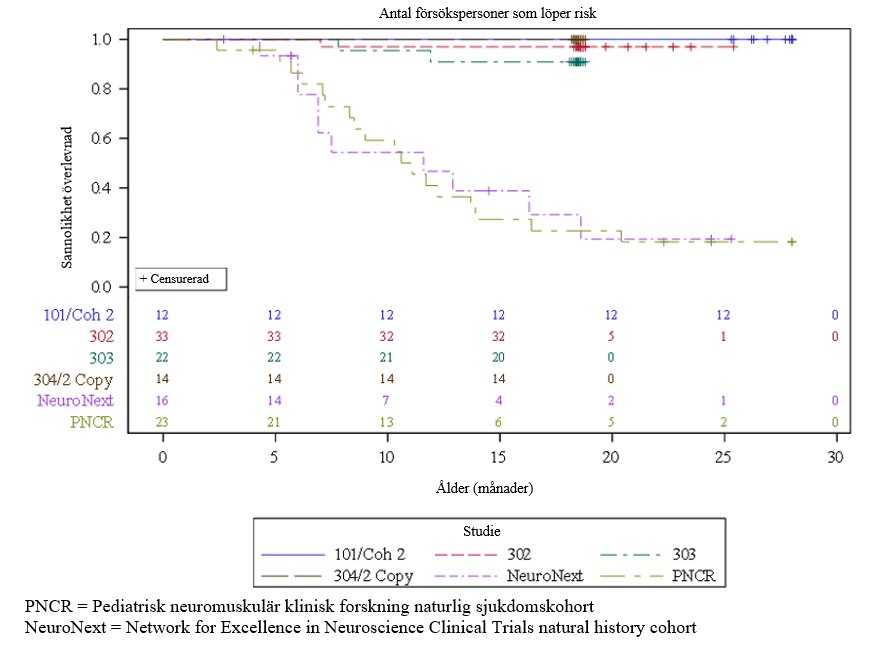
Tre patienter slutförde inte studien, av vilka 2 patienter hade en händelse (dödsfall eller permanent ventilation) vilket ledde till 90,9 % (95 % KI: 79,7 %, 100 %) händelsefri överlevnad (levande utan permanent ventilation) vid 14 månaders ålder, se figur 1.
Figur 1 Tid (månader) till dödsfall eller permanent ventilation poolad från studier av i.v. onasemnogen-abeparvovek (CL‑101, CL‑302, CL‑303, CL-304-kohort med 2 kopior)
För de 14 patienterna i studie CL‑303 som uppnådde milstolpen oberoende sittande under minst 30 sekunder vid varje besök under studien, var medianåldern när milstolpen först påvisades 12,6 månader (intervall: 9,2 till 18,6 månader). Tretton patienter (59,1 %) bekräftade milstolpen oberoende sittande under minst 30 sekunder vid 18‑månadersbesöket (co‑primärt effektmått, p<0,0001). En patient uppnådde milstolpen oberoende sittande i 30 sekunder vid 16 månaders ålder, men milstolpen bekräftades inte vid 18-månadersbesöket. Den videobekräftade utvecklingsmilstolpen för patienter i studie CL‑303 sammanfattas i tabell 4. Tre patienter uppnådde inte några motoriska milstolpar (13,6 %) och ytterligare 3 patienter (13,6 %) uppnådde huvudkontroll som den maximala motoriska milstolpen före det sista studiebesöket vid 18 månaders ålder.
Tabell 4 Mediantid till videodokumenterat uppnående av motoriska milstolpar studie CL-303
| Videodokumenterad milstolpe | Antal patienter som uppnådde milstolpe n/N (%) | Medianålder vid vilken ålder milstolpen uppnåddes (månader) | 95 % Konfidensintervall |
| Huvudkontroll | 17/20* (85,0) | 6,8 | (4,77; 7,57) |
| Rulla från rygg till sidor | 13/22 (59,1) | 11,5 | (7,77; 14,53) |
| Sitta utan stöd i 30 sekunder (Bayley) | 14/22 (63,6) | 12,5 | (10,17; 15,20) |
| Sitta utan stöd i minst 10 sekunder (WHO) | 14/22 (63,6) | 13,9 | (11,00; 16,17) |
* 2 patienter rapporterades ha huvudkontroll vid klinisk bedömning vid baslinjen.
En patient (4,5 %) kunde också gå med assistans vid 12,9 månader. Baserat på sjukdomens naturliga historik förväntas inte patienter som uppfyllde kriterierna för studiedeltagande att nå förmågan att sitta utan stöd. Dessutom var 18 av de 22 patienterna oberoende av ventilationsstöd vid 18 månaders ålder.
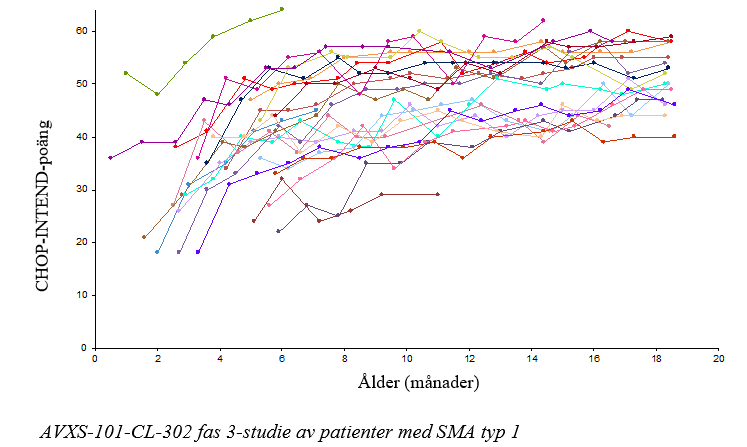
Förbättringar av motorisk funktion observerades också mätt med CHOP‑INTEND, se figur 2. Tjugoen patienter (95,5 %) uppnådde en CHOP‑INTEND-poäng ≥ 40, 14 patienter (63,6 %) uppnådde CHOP‑INTEND‑poäng ≥ 50 och 9 patienter (40,9 %) uppnådde en CHOP‑INTEND‑poäng ≥ 58. Patienter med obehandlad SMA typ 1 uppnår nästan aldrig en CHOP‑INTEND‑poäng ≥ 40. Uppnådd motorisk milstolpe observerades hos vissa patienter trots att de uppnådde platå i CHOP‑INTEND. Ingen tydlig korrelation observerades mellan CHOP‑INTEND‑poäng och uppnådd motorisk milstolpe.
Figur 2 CHOP-INTEND‑poäng motorisk funktionstudie CL‑303 (N=22)
AVXS-101-CL-302 (studie CL-302) är en fas 3, öppen, enarmad, studie med en engångsdos av intravenöst givet onasemnogen-abeparvovek vid terapeutisk dos (1,1 × 1014 vg/kg). Trettiotre patienter med SMA typ 1 och 2 kopior av SMN2 rekryterades. Innan behandling med onasemnogen-abeparvovek rapporterade 9 patienter (27,3 %) behov av ventilationsstöd och 9 patienter (27,3 %) rapporterade behov av matningsstöd. Den genomsnittliga CHOP-INTEND-poängen för de 33 patienterna vid studiestart var 27,9 (intervall, 14 till 55). Medelåldern för de 33 patienterna vid behandlingstillfället var 4,1 månader (intervall, 1,8 till 6,0 månader).
Av de 33 rekryterade patienterna (Efficacy Completers population, dvs. populationen som har följt hela studieprotokollet) doserades en patient (3 %) utanför protokollets åldersintervall och inkluderades därför inte i ITT-populationen (intention to treat). Av de 32 patienterna i ITT-populationen dog en patient (3 %) under studien på grund av sjukdomsprogression.
Av de 32 patienterna i ITT-populationen uppnådde 14 patienter (43,8 %) milstolpen att sitta utan stöd i minst 10 sekunder vid varje besök till och med 18-månadersbesöket (primärt effektmått). Medianåldern när denna milstolpe först uppnåddes var 15,9 månader (intervall, 7,7 till 18,6 månader). Trettioen patienter (96,9 %) i ITT-populationen överlevde utan permanent ventilation (dvs. händelsefri överlevnad) till ≥ 14 månaders ålder (sekundärt effektmått).
De ytterligare videobekräftade utvecklingsmilstolparna för patienter i populationen som har följt hela studieprotokollet i studie CL-302 vid varje besök till och med 18-månadersbesöket sammanfattas i tabell 5.
Tabell 5 Mediantid för att uppnå videodokumenterade motoriska milstolpar i studie CL-302 (Efficacy Completers population, dvs. populationen som har följt hela studieprotokollet)
| Videodokumenterad milstolpe | Antal patienter som uppnådde milstolpe n/N (%) | Medianålder vid vilken ålder milstolpen uppnåddes (månader) | 95 % Konfidensintervall |
| Huvudkontroll | 23/30* (76,7) | 8,0 | (5,8; 9,2) |
| Rulla från rygg till sidor | 19/33 (57,6) | 15,3 | (12,5; 17,4) |
| Sitta utan stöd i minst 30 sekunder | 16/33 (48,5) | 14,3 | (8,3; 18,3) |
* 3 patienter rapporterades ha huvudkontroll genom klinisk bedömning vid baslinjen.
En patient (3 %) uppnådde de motoriska milstolparna att krypa, stå med hjälp, stå ensam, gå med assistans och gå ensam, alla vid 18 månaders ålder.
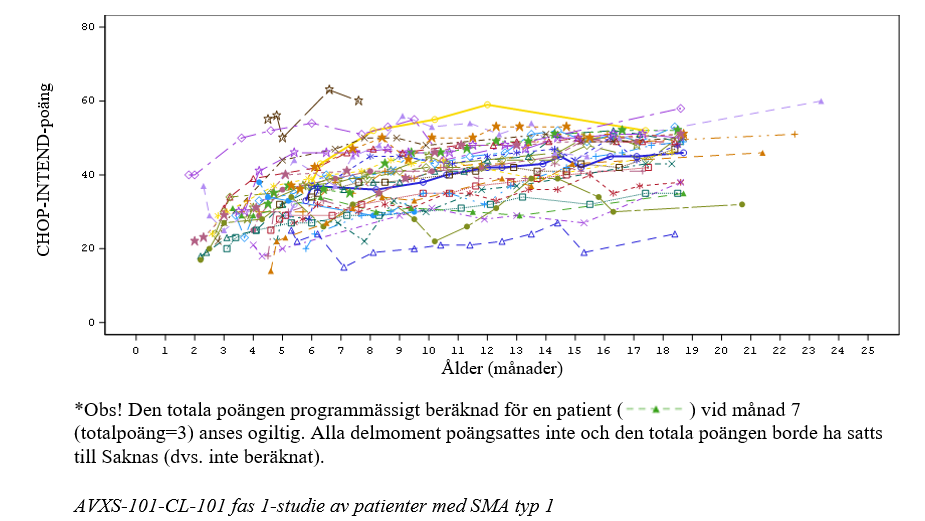
Av de 33 inkluderade patienterna uppnådde 24 patienter (72,7 %) en CHOP-INTEND-poäng ≥ 40, 14 patienter (42,4 %) uppnådde en CHOP-INTEND-poäng ≥ 50 och 3 patienter (9,1 %) uppnådde en CHOP-INTEND-poäng ≥ 58 (se figur 3). Patienter med obehandlad SMA typ 1 uppnår nästan aldrig en CHOP INTEND-poäng ≥ 40.
Figur 3 CHOP-INTEND-motorfunktionspoäng i studie CL-302 (Efficacy Completers population, dvs. populationen som har följt hela studieprotokollet; N=33)*
Resultaten som setts i studie CL-303 stöds av studie AVXS‑101‑CL‑101 (studie CL-101), en fas 1‑studie av patienter med SMA typ 1, i vilken onasemnogen-abeparvovek administrerades som en intravenös engångsinfusion till 12 patienter mellan 3,6 kg och 8,4 kg (0,9 till 7,9 månaders ålder). Vid 14 månaders ålder var alla patienter händelsefria, dvs. överlevde utan permanent ventilation, jämfört med 25 % i kohorten för det naturliga sjukdomsförloppet. I slutet av studien (24 månader efter dosering) var alla behandlade patienter händelsefria, jämfört med färre än 8 % i det naturliga sjukdomsförloppet, se figur 1.
Vid uppföljning 24 månader efter dosering kunde 10 av 12 patienter sitta utan stöd i ≥ 10 sekunder, 9 patienter kunde sitta utan stöd i ≥ 30 sekunder och 2 patienter kunde stå och gå utan assistans. En av 12 patienter uppnådde inte huvudkontroll som maximal motorisk milstolpe före 24 månaders ålder. Tio av 12 patienter från studie CL‑101 följs fortsatt i en långvarig studie (i upp till 6,6 år efter dosering) och alla 10 patienterna var vid liv och fria från permanent ventilation den 23 maj 2021. Alla patienter har antingen bibehållit tidigare uppnådda milstolpar eller nått nya milstolpar såsom sittande med stöd, stå med assistans och gå själva. Fem av de 10 patienterna fick samtidig behandling med nusinersen eller risdiplam vid någon tidpunkt under den långvariga studien. Underhåll av effekt och uppnådda milstolpar kan således inte tillskrivas endast onasemnogen-abeparvovek hos alla patienter. Milstolpen stå med assistans uppnåddes nyligen av 2 patienter som inte hade fått nusinersen eller risdiplam vid någon tidpunkt innan denna milstolpe uppnåddes.
AVXS-101-CL-304 fas 3‑studie på patienter med presymtomatisk SMA
Studie CL‑304 är en global, öppen, enarmad fas 3-studie med en engångsdos av intravenös administrering av onasemnogen-abeparvovek till presymtomatiska nyfödda patienter upp till 6 veckor gamla med 2 (kohort 1, n=4) eller 3 (kohort 2, n=15) kopior av SMN2.
Kohort 1
De 14 behandlade patienterna med 2 kopior av SMN2 följdes till 18 månaders ålder. Alla patienter överlevde händelsefritt till ≥ 14 månaders ålder utan permanent ventilation.
Alla 14 patienterna uppnådde oberoende sittande under minst 30 sekunder vid alla besök upp till besöket vid 18 månaders ålder (primärt effektmått), vid åldrar mellan 5,7 och 11,8 månader och 11 av de 14 patienterna uppnådde oberoende sittande vid eller före 279 dagars ålder, den 99:e percentilen för utveckling av denna milstolpe. Nio patienter uppnådde milstolpen gå själva (64,3 %). Alla 14 patienterna uppnådde en CHOP‑INTEND‑poäng ≥ 58 vid alla besök upp till besöket vid 18 månaders ålder. Inga patienter behövde något ventilationsstöd eller något matningsstöd under studien.
Kohort 2
De 15 behandlade patienterna med 3 kopior av SMN2 följdes till 24 månaders ålder. Alla patienter överlevde händelsefritt till 24 månaders ålder utan permanent ventilation.
Alla 15 patienterna kunde stå själva utan stöd under minst 3 sekunder (primärt effektmått), vid åldrar mellan 9,5 och 18,3 månader och 14 av de 15 patienterna uppnådde stående ensamma vid eller före 514 dagars ålder, den 99:e percentilen för utveckling av denna milstolpe. Fjorton patienter (93,3%) kunde gå minst fem steg själva. Alla 15 patienterna uppnådde ≥ 4 poäng på skalan Bayley-III Gross and Fine Motor Subtests inom 2 standardavvikelser från medelvärdet för ålder vid alla besök efter studiestart upp till 24 månaders ålder. Inga patienter behövde något ventilationsstöd eller något matningsstöd under studien.
COAV101A12306 fas 3‑studie på patienter med SMA som väger ≥ 8,5 kg till ≤ 21 kg
Studie COAV101A12306 är en avslutad, öppen, enarmad, multicenterstudie i fas 3 med en engångsdos av intravenös administrering av onasemnogen-abeparvovek vid den terapeutiska dosen (1,1 × 1014 vg/kg) till 24 pediatriska patienter med SMA som vägde ≥ 8,5 kg till ≤ 21 kg (medianvikt: 15,8 kg). Patienternas ålder varierade från cirka 1,5 till 9 år vid tidpunkten för administreringen. Patienterna hade 2 till 4 kopior av SMN2 (två [n=5], tre [n=18], fyra [n=1] kopior). Före behandling med onasemnogen-abeparvovek hade 19/24 patienter tidigare fått nusinersen under en mediantid på 2,1 år (intervall 0,17 till 4,81 år) och 2/24 patienter hade tidigare fått risdiplam under en mediantid på 0,48 år (intervall 0,11 till 0,85 år). Vid baslinjen hade patienterna en genomsnittlig Hammersmith Functional Motor Scale - Expanded (HFMSE)-poäng på 28,3 och en genomsnittlig Revised Upper Limb Module (RULM)-poäng på 22,0. Dessutom uppvisade samtliga patienter milstolparna för huvudkontroll och sittande med stöd, tjugoen kunde sitta utan stöd och sex uppvisade de högsta möjliga milstolparna att stå ensam och gå ensam.
Vid vecka 52 var den genomsnittliga förändringen från baslinjen av övergripande HFMSE-totalpoäng 3,7 (18/24 patienter). Den genomsnittliga ökningen av övergripande RULM-totalpoäng var 2,0 (17/24 patienter) vid vecka 52. Fyra patienter uppnådde nya utvecklingsmilstolpar. Milstolpar som observerats vid besöket vid studiestart bibehölls till vecka 52 för majoriteten av patienterna. Två patienter som inte uppvisade tidigare uppnådda utvecklingsmilstolpar visade förbättring av HFMSE-poängen från baslinjen till vecka 52.
Onasemnogen-abeparvovek har inte studerats på patienter med en biallelisk mutation i SMN1‑genen och bara en kopia av SMN2 i kliniska studier.
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för onasemnogen-abeparvovek för en eller flera grupper av den pediatriska populationen för spinal muskelatrofi för godkänd indikation (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Onasemnogen-abeparvovek vektorutsöndringsstudier, som bedömer den mängd vektor som eliminerats från kroppen via saliv, urin, feces och nasala sekret har utförts.
Onasemnogen-abeparvovek vektor‑DNA kunde detekteras i utsöndringsprover efter infusion. Utsöndring av onasemnogen-abeparvovek skedde primärt via feces. De högsta värdena för utsöndring hos de flesta patienter observerades inom 7 dagar efter dosering avseende avföring och inom 2 dagar efter dosering avseende saliv, urin och nasala sekret. Huvuddelen av vektorn eliminerades inom 30 dagar efter administrering av dos.
Biodistribution utvärderades hos 2 patienter som dog 5,7 månader respektive 1,7 månader efter infusion av onasemnogen-abeparvovek vid dosen 1,1 x 1014 vg/kg. Båda fallen visade att den högsta nivån av vektor‑DNA sågs i levern. Vektor‑DNA detekterades också i mjälte, hjärta, pankreas, inguinal lymfkörtel, skelettmuskulatur, perifera nerver, njurar, lungor, tarmar, könskörtlar, ryggmärg, hjärna och bräss. Immunfärgning för SMN‑protein visade generaliserad SMN‑expression i spinala motorneuroner, neuronala celler och gliaceller i hjärnan och i hjärta, lever, skelettmuskulatur samt annan utvärderad vävnad.
Prekliniska säkerhetsuppgifter
Efter intravenös administrering till neonatal mus distribuerades vektorn i stor omfattning och de högsta vektor-DNA-nivåerna detekterades generellt i hjärta, lever, lungor och skelettmuskulatur. Uttrycket av transgent mRNA visade liknande mönster. Efter intravenös administrering i juvenila icke-humana primater (NHP) distribuerades vektorn i stor omfattning med efterföljande uttryck av transgent mRNA, med de högsta koncentrationerna av vektor-DNA och transgent mRNA som tenderade att förekomma i lever, muskler och hjärta. Vektor-DNA och transgent mRNA i båda arterna detekterades i ryggmärg, hjärna och könskörtlar.
I pivotala toxikologiska studier på mus under 3 månader var de huvudsakliga målorganen för toxicitet identifierade som hjärta och lever. Resultat som hade samband med onasemnogen-abeparvovek i hjärtats ventriklar bestod av dosrelaterad inflammation, ödem och fibros. I hjärtats förmak observerades inflammation, trombos, myokardiell degeneration/nekros och fibroplasi. NoAEL (No Adverse Effect Level) identifierades inte för onasemnogen-abeparvovek i musstudier eftersom ventrikulär myokardinflammation/ödem/fibros och atriell inflammation observerades vid den lägsta dosen (1,5 × 1014 vg/kg). Denna dos anses vara den maximala tolererade dosen och motsvarar cirka 1,4 gånger den rekommenderade kliniska dosen. Den mortalitet på majoriteten av möss som hade samband med onasemnogen-abeparvovek var associerad med atriell trombos och observerades vid 2,4 × 1014 vg/kg. Orsaken till mortaliteten hos resten av djuren kunde inte fastställas, även om mikroskopisk degeneration/regeneration hittades i hjärtana på dessa djur.
Leverfynd från möss bestod av hepatocellulär hypertrofi, aktivering av Kupfferceller och spridd hepatocellulär nekros. I långtidstoxicitetsstudier med intravenös och intratekal (ej godkänt för användning) administrering av onasemnogen-abeparvovek hos juvenila icke-humana primater (NHP), visade leverfynd, inklusive individuell cellnekros (single cell necrosis) av hepatocyter och oval cellhyperplasi, partiell (IV) eller fullständig (IT) reversibilitet.
I en 6-månaders toxikologisk studie utförd på juvenila icke‑humana primater (NHP) ledde administrering av en engångsdos av onasemnogen-abeparvovek vid den kliniskt rekommenderade intravenösa dosen, med eller utan kortikoidsteroidbehandling, till akut, minimal till lätt mononukleär cellinflammation och neuronal degeneration i de dorsala rotganglierna (DRG) och trigeminusganglierna (TG), samt axonal degeneration och/eller glios i ryggmärgen. Efter 6 månader resulterade dessa icke-progressiva fynd i full upplösning i TG och partiell upplösning (minskad incidens och/eller svårighetsgrad) i DRG och ryggmärgen. Efter intratekal (ej godkänd för användning) administrering av onasemnogen-abeparvovek noterades dessa akuta, icke-progressiva fynd med minimal till måttlig svårighetsgrad hos juvenila icke-humana primater (NHP) med partiell till full upplösning efter 12 månader. Dessa fynd hos icke-humana primater (NHP) hade inga korrelativa kliniska observationer, därför är den kliniska relevansen hos människor inte känd.
Studier av gentoxicitet, karcinogenicitet och reproduktionstoxicitet har inte utförts med onasemnogen-abeparvovek.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Trometamin
Magnesiumklorid
Natriumklorid
Poloxamer 188
Saltsyra (för pH‑justering)
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
2 år
Efter upptining
När läkemedlet har tinat upp ska det inte frysas igen och kan förvaras i kylskåp vid 2 °C till 8 °C i originalkartongen under 14 dagar.
När dosvolymen dragits upp i sprutan måste den administreras inom 8 timmar. Kasta sprutan som innehåller vektorn om den inte har administrerats inom tidsramen på 8 timmar.
Särskilda förvaringsanvisningar
Förvaras och transporteras i djupfryst tillstånd (≤ −60 °C).
Förvaras i kylskåp (2 °C - 8 °C) omedelbart vid mottagande.
Förvaras i originalkartongen.
Förvaringsanvisningar för läkemedlet efter tining finns i avsnitt Hållbarhet
Datum för mottagandet ska anges på originalkartongen innan produkten förvaras i kylskåpet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
ZOLGENSMA infuusioneste, liuos
2 x 10exp13 vektorigenomia/ml (L:ei) 2x5,5 ml+10x8,3 ml (-), 5,5 ml+10x8,3 ml (-), 2x5,5 ml+11x8,3 ml (-), 5,5 ml+11x8,3 ml (-), 2x5,5 ml+12x8,3 ml (-), 5,5 ml+12x8,3 ml (-), 5,5 ml+13x8,3 ml (-), 2x5,5 ml+2x8,3 ml (-), 5,5 ml+2x8,3 ml (-), 2x5,5 ml+3x8,3 ml (-), 5,5 ml+3x8,3 ml (-), 2x5,5 ml+4x8,3 ml (-), 5,5 ml+4x8,3 ml (-), 2x5,5 ml+5x8,3 ml (-), 5,5 ml+5x8,3 ml (-), 2x5,5 ml+6x8,3 ml (-), 5,5 ml+6x8,3 ml (-), 2x5,5 ml+7x8,3 ml (-), 5,5 ml+7x8,3 ml (-), 2x5,5 ml+8,3 ml (-), 2x5,5 ml+8x8,3 ml (-), 5,5 ml+8x8,3 ml (-), 2x5,5 ml+9x8,3 ml (-), 5,5 ml+9x8,3 ml (-), 2 x 8,3 ml (-), 3 x 8,3 ml (-), 4 x 8,3 ml (-), 5 x 8,3 ml (-), 6 x 8,3 ml (-), 7 x 8,3 ml (-), 8 x 8,3 ml (-), 9 x 8,3 ml (-), 10 x 8,3 ml (-), 11 x 8,3 ml (-), 12 x 8,3 ml (-), 13 x 8,3 ml (-), 14 x 8,3 ml (-)
PF-selosteen tieto
Onasemnogen-abeparvovek levereras i en injektionsflaska (10 ml polymer Crystal Zenith) med propp (20 mm klorobutylgummi) och försegling (aluminium, snäpp) med ett färgat lock (plast), i två olika fyllnadsvolymer, antingen 5,5 ml eller 8,3 ml.
Dosen onasemnogen-abeparvovek och exakt antal injektionsflaskor som krävs för varje patient beräknas baserat på patientens vikt (se avsnitt Dosering och administreringssätt och Tabell 6 nedan).
Tabell 6 Kartong-/kittyper
| Patientvikt (kg) | 5,5 ml injektionsflaskaa | 8,3 ml injektionsflaskab | Totalt antal injektionsflaskor per kartong |
| 2,6‑3,0 | 0 | 2 | 2 |
| 3,1‑3,5 | 2 | 1 | 3 |
| 3,6‑4,0 | 1 | 2 | 3 |
| 4,1‑4,5 | 0 | 3 | 3 |
| 4,6‑5,0 | 2 | 2 | 4 |
| 5,1‑5,5 | 1 | 3 | 4 |
| 5,6‑6,0 | 0 | 4 | 4 |
| 6,1‑6,5 | 2 | 3 | 5 |
| 6,6‑7,0 | 1 | 4 | 5 |
| 7,1‑7,5 | 0 | 5 | 5 |
| 7,6‑8,0 | 2 | 4 | 6 |
| 8,1‑8,5 | 1 | 5 | 6 |
| 8,6‑9,0 | 0 | 6 | 6 |
| 9,1‑9,5 | 2 | 5 | 7 |
| 9,6‑10,0 | 1 | 6 | 7 |
| 10,1‑10,5 | 0 | 7 | 7 |
| 10,6‑11,0 | 2 | 6 | 8 |
| 11,1‑11,5 | 1 | 7 | 8 |
| 11,6‑12,0 | 0 | 8 | 8 |
| 12,1‑12,5 | 2 | 7 | 9 |
| 12,6‑13,0 | 1 | 8 | 9 |
| 13,1‑13,5 | 0 | 9 | 9 |
| 13,6–14,0 | 2 | 8 | 10 |
| 14,1–14,5 | 1 | 9 | 10 |
| 14,6–15,0 | 0 | 10 | 10 |
| 15,1–15,5 | 2 | 9 | 11 |
| 15,6–16,0 | 1 | 10 | 11 |
| 16,1–16,5 | 0 | 11 | 11 |
| 16,6–17,0 | 2 | 10 | 12 |
| 17,1–17,5 | 1 | 11 | 12 |
| 17,6–18,0 | 0 | 12 | 12 |
| 18,1–18,5 | 2 | 11 | 13 |
| 18,6–19,0 | 1 | 12 | 13 |
| 19,1–19,5 | 0 | 13 | 13 |
| 19,6–20,0 | 2 | 12 | 14 |
| 20,1–20,5 | 1 | 13 | 14 |
| 20,6–21,0 | 0 | 14 | 14 |
a Injektionsflaskans nominella koncentration är 2 × 1013 vg/ml och innehåller en extraherbar volym på minst 5,5 ml.
b Injektionsflaskans nominella koncentration är 2 × 1013 vg/ml och innehåller en extraherbar volym på minst 8,3 ml.
Läkemedlets utseende:
En klar till lätt ogenomskinlig, färglös till lätt vitaktig lösning.
Särskilda anvisningar för destruktion och övrig hantering
Mottagande och upptining av injektionsflaskor
- Injektionsflaskor ska transporteras i djupfryst tillstånd (≤ −60 °C). Vid mottagande ska injektionsflaskor omedelbart placeras i kylskåp vid 2 °C till 8 °C och förvaras i originalkartongen. Behandling med onasemnogen-abeparvovek ska initieras inom 14 dagar från mottagande av injektionsflaskor.
- Injektionsflaskor måste tinas före användning. Använd inte onasemnogen-abeparvovek om det inte har tinat.
- För förpackningar innehållande upp till 9 injektionsflaskor tinar läkemedlet efter cirka 12 timmar i kylskåp. För förpackningar innehållande upp till 14 injektionsflaskor tinar läkemedlet efter cirka 16 timmar i kylskåp. Alternativt, och för omedelbar användning, kan tining ske i rumstemperatur.
- För förpackningar innehållande upp till 9 injektionsflaskor sker tining från fruset tillstånd efter cirka 4 timmar vid rumstemperatur (20 °C till 25 °C). För förpackningar innehållande upp till 14 injektionsflaskor sker tining från fruset tillstånd efter cirka 6 timmar vid rumstemperatur (20 °C till 25 °C).
- Före uppdragning av dosvolymen i sprutan ska det tinade läkemedlet snurras varsamt. Skaka INTE.
- Använd inte detta läkemedel om du noterar partiklar eller missfärgning när den frysta produkten har tinat upp och före administrering.
- Läkemedlet ska inte frysas igen efter att det har tinat upp.
- Onasemnogen-abeparvovek ska ges så snart som möjligt efter att det har tinat upp. När dosvolymen dragits upp i sprutan måste den administreras inom 8 timmar. Kasta sprutan som innehåller vektorn om den inte har administrerad inom tidsramen på 8 timmar.
Administrering av onasemnogen-abeparvovek till patienten
Administrera onasemnogen-abeparvovek genom att dra upp hela dosvolymen i sprutan. Ta bort all luft i sprutan före intravenös infusion via en venkateter införd i en perifer extremitetsven på patienten.
Försiktighetsåtgärder för hantering, kassering och oavsiktlig exponering av läkemedlet
Detta läkemedel innehåller genetiskt modifierade organismer. Lämpliga försiktighetsåtgärder för hantering, kassering och oavsiktlig exponering av onasemnogen-abeparvovek ska beaktas:
- Sprutan med onasemnogen-abeparvovek ska hanteras aseptiskt under sterila förhållanden.
- Personlig skyddsutrustning (inklusive handskar, skyddsglasögon, laboratorierock och ärmskydd) ska användas vid hantering eller administrering av onasemnogen-abeparvovek. Personal med skär- eller skrapmärken på huden ska inte arbeta med onasemnogen-abeparvovek.
- Allt spill av onasemnogen-abeparvovek måste torkas upp med absorberande gasväv och spillområdet måste desinficeras med en blekningslösning följt av sprittorkar. Allt material som använts för upptorkning måste läggas i dubbla påsar och kasseras i enlighet med lokala riktlinjer för hantering av biologiskt avfall.
- Ej använt läkemedel och avfall ska kasseras enligt gällande lokala riktlinjer för hantering av biologiskt avfall.
- Allt material som har kommit i kontakt med onasemnogen-abeparvovek (t.ex. injektionsflaska, allt material som använts för injektion, inklusive sterila dukar och nålar) måste kasseras i enlighet med lokala riktlinjer för hantering av biologiskt avfall.
- Oavsiktlig exponering för onasemnogen-abeparvovek måste undvikas. I händelse av exponering av huden måste det drabbade området rengöras noggrant med tvål och vatten under minst 15 minuter. I händelse av exponering av ögon måste det drabbade området sköljas noggrant med vatten under minst 15 minuter.
Utsöndring
Tillfällig utsöndring av onasemnogen-abeparvovek kan uppkomma, främst genom kroppsavfall. Vårdgivare och patientens familj ska få råd om att använda följande anvisningar för korrekt hantering av patientens kroppsvätskor och avföring:
- God handhygien (användning av skyddshandskar och noggrann handtvätt efteråt med tvål och rinnande varmt vatten eller ett alkoholbaserat handdesinfektionsmedel) krävs vid direktkontakt med patientens kroppsvätskor och avföring under minst 1 månad efter behandling med onasemnogen-abeparvovek.
- Engångsblöjor ska förseglas i dubbla plastpåsar och kan kastas i hushållssoporna.
Ersättning
ZOLGENSMA infuusioneste, liuos
2 x 10exp13 vektorigenomia/ml 2x5,5 ml+10x8,3 ml, 5,5 ml+10x8,3 ml, 2x5,5 ml+11x8,3 ml, 5,5 ml+11x8,3 ml, 2x5,5 ml+12x8,3 ml, 5,5 ml+12x8,3 ml, 5,5 ml+13x8,3 ml, 2x5,5 ml+2x8,3 ml, 5,5 ml+2x8,3 ml, 2x5,5 ml+3x8,3 ml, 5,5 ml+3x8,3 ml, 2x5,5 ml+4x8,3 ml, 5,5 ml+4x8,3 ml, 2x5,5 ml+5x8,3 ml, 5,5 ml+5x8,3 ml, 2x5,5 ml+6x8,3 ml, 5,5 ml+6x8,3 ml, 2x5,5 ml+7x8,3 ml, 5,5 ml+7x8,3 ml, 2x5,5 ml+8,3 ml, 2x5,5 ml+8x8,3 ml, 5,5 ml+8x8,3 ml, 2x5,5 ml+9x8,3 ml, 5,5 ml+9x8,3 ml, 2 x 8,3 ml, 3 x 8,3 ml, 4 x 8,3 ml, 5 x 8,3 ml, 6 x 8,3 ml, 7 x 8,3 ml, 8 x 8,3 ml, 9 x 8,3 ml, 10 x 8,3 ml, 11 x 8,3 ml, 12 x 8,3 ml, 13 x 8,3 ml, 14 x 8,3 ml
- Ei korvausta.
Atc-kod
M09AX09
Datum för översyn av produktresumén
12.06.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com