
ENJAYMO infusionsvätska, lösning 50 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Potilas
Observera
▼Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje milliliter infusionsvätska, lösning innehåller 50 mg sutimlimab*.
En injektionsflaska innehåller 1 100 mg sutimlimab i 22 ml.
* Sutimlimab är en immunglobulin G4 (IgG4) monoklonal antikropp (mAb) framställd med rekombinant DNA‑teknik i ovarieceller från kinesisk hamster (CHO‑celler).
Hjälpämne med känd effekt
Varje milliliter infusionsvätska, lösning innehåller 3,5 mg natrium.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Infusionsvätska, lösning (infusion)
Kliniska uppgifter
Terapeutiska indikationer
Enjaymo är avsett för behandling av hemolytisk anemi hos vuxna patienter med köldagglutininsjukdom (CAD).
Villkor
Valmistetta saa antaa vain terveydenhoitoalan ammattilainen hyväksytyn käyttöaiheen hoitoon perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Enjaymo måste administreras av sjukvårdspersonal och övervakas av en läkare med erfarenhet av hantering av patienter med hematologiska sjukdomar.
Dosering
Patienterna ska vaccineras enligt de mest aktuella lokala rekommendationerna för patienter med bestående komplementbrist (se avsnitt Varningar och försiktighet).
Den rekommenderade dosen baseras på kroppsvikt. För patienter som väger 39 kg till mindre än 75 kg är den rekommenderade dosen 6 500 mg och för patienter som väger 75 kg eller mer är den rekommenderade dosen 7 500 mg. Enjaymo administreras intravenöst varje vecka under de första två veckorna och därefter varannan vecka. Enjaymo ska administreras vid de rekommenderade tidpunkterna enligt dosschemat eller inom två dagar före eller efter dessa tidpunkter (se avsnitt Varningar och försiktighet). Enjaymo är avsett för kontinuerlig användning som kronisk behandling, om inte utsättning av Enjaymo är kliniskt indicerad.
Glömd dos
Om en dos missas, ska den missade dosen administreras så snart som möjligt. Om det går mer än 17 dagar efter den sista dosen, ska behandlingen återupptas med administrering varje vecka under de första två veckorna och därefter varannan vecka.
Särskilda populationer
Äldre
Inga dosjusteringar är nödvändiga för patienter i åldern 65 år och äldre med köldagglutininsjukdom (se avsnitt Farmakodynamiska egenskaper och Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Inga dosjusteringar är nödvändiga för patienter med nedsatt leverfunktion.
Nedsatt njurfunktion
Inga dosjusteringar är nödvändiga för patienter med nedsatt njurfunktion.
Pediatrisk population
Det finns ingen relevant användning av Enjaymo för barn under 18 år vid behandling av köldagglutininsjukdom.
Administreringssätt
Enjaymo är endast avsett för intravenös infusion. Administrera det inte som en intravenös stöt- eller bolusdos. Anvisningar om beredning och administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Efter beredning ska Enjaymo infusionslösning administreras intravenöst med den infusionshastighet som anges i tabell 1.
Tabell 1 - Referenstabell för infusioner
| Kroppsviktsintervall | Dos (mg) | Antal injektionsflaskor som behövs | Volym (ml) | Maximal infusionshastighet |
| 39 kg eller mer till mindre än 75 kg | 6 500 | 6 | 130 | 130 ml/h |
| 75 kg eller mer | 7500 | 7 | 150 | 150 ml/h |
Patienter med hjärt-lungsjukdom kan få infusionen under 120 minuter.
Om en biverkning inträffar under administreringen av Enjaymo, kan infusionen saktas ner eller avbrytas enligt läkarens bedömning. Om överkänslighetsreaktioner uppstår, ska Enjaymo sättas ut och lämplig behandling påbörjas. Patienten ska övervakas i minst två timmar efter den första infusionen för tecken eller symtom på en infusionsrelaterad reaktion och/eller överkänslighetsreaktion. Patienten ska övervakas en timme efter de efterföljande infusionerna för tecken eller symtom på en infusionsrelaterad reaktion.
Infusion i hemmet
Infusioner i hemmet ska utföras av sjukvårdspersonal.
Beslutet att överväga infusion i hemmet ska baseras på patientens individuella kliniska egenskaper och individuella behov. Övergång av infusion från en klinisk enhet till administration i hemmet innebär att säkerställa att lämplig utrustning och sjukvårdspersonal finns tillgängliga enligt den behandlande läkarens anvisningar. Infusion av Enjaymo i hemmet kan övervägas för patienter som har tolererat sina infusioner väl på en klinisk enhet och inte har haft infusionsrelaterade reaktioner. Patientens underliggande samtidiga sjukdomar och förmåga att följa kraven för infusion i hemmet ska beaktas vid bedömning av patientens lämplighet för att få infusion i hemmet. Dessutom ska följande kriterier beaktas:
- Patienten får inte ha något pågående samtidigt tillstånd som enligt läkaren kan utsätta patienten för en högre risk vid infusion i hemmet än vid infusion på en klinik. En omfattande utvärdering ska göras innan infusion i hemmet påbörjas för att säkerställa att patienten är medicinskt stabil.
- Patienten ska ha fått infusion med Enjaymo utan problem på en klinik (på sjukhus eller inom öppenvård) åtminstone i tre månader under överinseende av läkare eller sjukvårdspersonal med erfarenhet av behandling av patienter med köldagglutininsjukdom.
- Patienten ska vara villig och ha förmågan att följa procedurerna för infusion i hemmet och den behandlande läkarens eller sjukvårdspersonalens rekommendationer.
- Sjukvårdspersonalen som ger infusionen i hemmet ska vara tillgänglig hela tiden under infusionen i hemmet och minst 1 timme efter infusionen.
Om patienten får biverkningar under infusionen i hemmet, ska infusionen avbrytas omedelbart, lämplig medicinsk behandling påbörjas (se avsnitt Varningar och försiktighet) och den behandlande läkaren informeras. I sådana fall ska den behandlande läkaren avgöra om efterföljande infusioner ska ges och om infusionerna ska ges på ett sjukhus eller inom öppenvård under övervakning.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Spårbarhet
För att underlätta spårbarhet av biologiska läkemedel ska läkemedlets namn och tillverkningssatsnummer dokumenteras.
Infektioner
Enjaymo riktas mot den klassiska vägen av komplementsystemet, specifik bindning till komplementproteinkomponent 1, s-subkomponenten (C1s) och förhindrar klyvningen av komplementproteinet C4. Även om lektinvägen och de alternativa vägarna förblir opåverkade kan patienterna ha ökad känslighet för allvarliga infektioner, särskilt infektioner orsakade av kapslade bakterier, till exempel Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae. Patienter ska vaccineras mot kapslade bakterier innan behandling med Enjaymo påbörjas, se ”Vaccinationer” nedan.
I kliniska studier avseende köldagglutininsjukdom har allvarliga infektioner, inklusive sepsis, rapporterats hos patienter som behandlats med Enjaymo (se avsnitt Biverkningar). Behandling med Enjaymo ska inte påbörjas hos patienter med aktiva, allvarliga infektioner. Patienter ska övervakas för tidiga tecken och symtom på infektioner och uppmanas att omedelbart uppsöka medicinsk vård om sådana symtom uppkommer.
Patienter med virushepatit och hiv uteslöts från de kliniska studierna. Patienter måste informera läkaren före och under behandlingen om de har diagnostiserats med hepatit B-infektion, hepatit C-infektion eller hivinfektion. Försiktighet ska iakttas vid behandling av patienter med hepatit B-infektion, hepatit C-infektion eller hivinfektion i anamnesen.
Vaccinationer
Patienterna ska vaccineras enligt aktuella lokala rekommendationer för patienter med bestående komplementbrist, inklusive meningokock- och streptokockvacciner. Patienterna ska återvaccineras i enlighet med lokala rekommendationer.
Patienter utan tidigare vaccination mot kapslade bakterier ska immuniseras minst 2 veckor före den första dosen av Enjaymo. Om akut behandling med Enjaymo är indicerad hos en ovaccinerad patient ska vaccin(erna) administreras så snart som möjligt. Fördelarna och riskerna med antibiotikaprofylax för förebyggande av infektioner hos patienter som behandlas med Enjaymo har inte fastställts.
Överkänslighetsreaktioner
Liksom med andra proteinprodukter kan administrering av Enjaymo orsaka överkänslighetsreaktioner, såsom anafylaxi. I kliniska studier observerades inga allvarliga överkänslighetsreaktioner med Enjaymo. Om överkänslighetsreaktioner uppstår, ska Enjaymo sättas ut och lämplig behandling påbörjas.
Infusionsrelaterade reaktioner
Administrering av Enjaymo kan orsaka infusionsrelaterade reaktioner under infusion eller omedelbart efter infusion (se avsnitt Biverkningar). Patienter ska övervakas med avseende på infusionsrelaterade reaktioner och infusionen ska avbrytas om en reaktion uppstår och lämplig behandling påbörjas.
Systemisk lupus erythematosus (SLE)
Individer med ärftlig klassisk komplementbrist har en högre risk för SLE. Patienter med SLE uteslöts från kliniska studier med Enjaymo. Patienter som behandlas med Enjaymo ska övervakas för tecken och symtom på SLE och utvärderas på lämpligt sätt. Enjaymo ska användas med försiktighet hos patienter med SLE eller tecken och symtom på SLE.
Övervakning av manifestationer av köldagglutininsjukdom efter utsättning av Enjaymo
Effekterna på hemolys minskar efter avslutad behandling. Patienterna ska därför övervakas med avseende på tecken och symtom på hemolys vid utsättande av behandling.
Natrium
Detta läkemedel innehåller 3,5 mg per ml eller 77 mg natrium per injektionsflaska, motsvarande 3,85 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna).
Interaktioner
Inga interaktionsstudier har utförts. Enjaymo orsakar sannolikt inte cytokrom P450-medierade läkemedelsinteraktioner, eftersom det är ett rekombinant humant protein. Interaktionen av sutimlimab med CYP-substrat har inte studerats. Sutimlimab minskar dock nivåerna av proinflammatoriska cytokiner hos patienter, liksom IL-6 som är känt att minska uttrycket av specifika CYP450-leverenzymer (CYP1A2, CYP2C9, CYP2C19 och CYP3A4). Därför ska försiktighet iakttas när behandling med sutimlimab påbörjas eller avslutas hos patienter som också får CYP450 3A4-, 1A2-, 2C9- eller C19-substrat, särskilt om de har ett smalt terapeutiskt index (såsom warfarin, karbamazepin, fenytoin och teofyllin), och doserna ska vid behov justeras.
Fertilitet, graviditet och amning
Graviditet
Det finns inga tillgängliga data från användning av sutimlimab hos gravida kvinnor. Djurstudier tyder inte på direkta eller indirekta reproduktionstoxikologiska effekter (se avsnitt Prekliniska säkerhetsuppgifter).
Humana IgG-antikroppar passerar placentabarriären och därför kan sutimlimab överföras från modern till det utvecklande fostret.
Som en försiktighetsåtgärd bör man undvika användning av sutimlimab under graviditet. Sutimlimab ska endast ges under graviditet om det är klart indicerat.
Amning
Det är känt att humana IgG-antikroppar utsöndras i bröstmjölk under de första dagarna efter födseln, men koncentrationen minskar till låga nivåer snart efter födseln. En risk för det ammade barnet kan därför inte uteslutas under denna korta period. Det är okänt om sutimlimab/metaboliter utsöndras i bröstmjölk. Ett beslut måste fattas om man ska avbryta amningen eller avbryta/avstå från behandling med sutimlimab efter att man tagit hänsyn till fördelen med amning för barnet och fördelen med behandling för kvinnan.
Fertilitet
Effekter av sutimlimab på manlig och kvinnlig fertilitet har inte studerats hos djur. I studier med upprepade doser av sutimlimab med exponering vid upp till cirka 4 gånger den rekommenderade dosen till människa observerades inga effekter på reproduktionsorganen hos cynomolgusapor.
Effekter på förmågan att framföra fordon och använda maskiner
Enjaymo har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
De vanligaste rapporterade biverkningarna med Enjaymo i de kliniska studierna CADENZA och CARDINAL var huvudvärk, hypertoni, urinvägsinfektion, övre luftvägsinfektion, nasofaryngit, illamående, buksmärta, infusionsrelaterade reaktioner och cyanos (rapporterad som akrocyanos).
Lista med biverkningar i tabellformat
Säkerhetsutvärderingen av Enjaymo hos patienter med köldagglutininsjukdom baserades främst på data från 66 patienter som deltog i den randomiserade placebokontrollerade fas 3-studien (CADENZA) och en öppen enarmad studie (CARDINAL).
I tabell 2 listas biverkningar som observerats i studierna CADENZA och CARDINAL uppdelade efter organsystem och frekvens enligt följande kategorier: mycket vanliga (≥ 1/10), vanliga (≥ 1/100, < 1/10), mindre vanliga (≥ 1/1 000, < 1/100), sällsynta (≥ 1/10 000, < 1/1 000), mycket sällsynta (< 1/10 000). Inom varje frekvensgrupp är biverkningar presenterade i fallande ordning efter allvarlighetsgrad.
Tabell 2 Lista med biverkningar i studierna CADENZA och CARDINAL
| MedDRA-organsystem | Mycket vanliga | Vanliga |
| Infektioner och infestationer | Urinvägsinfektion Cystit Övre luftvägsinfektionera Nasofaryngit b Gastroenterit Rinit | Nedre luftvägsinfektionerc Urosepsis Urinvägsinfektion orsakad av Escherichia coli Bakteriell urinvägsinfektion Bakteriell cystit Munherpes Herpes simplex-viremi Herpes zoster |
| Allmänna sjukdomar och/eller symtom vid administreringsstället | Pyrexif Köldkänslaf Infusionsrelaterade reaktionerf | |
| Centrala och perifera nervsystemet | Huvudvärk | Auraf Yrself* |
| Blodkärl | Hypertonid Cyanos (rapporterad som akrocyanos) Raynauds fenomen | Hypotonif* Stresskardiomyopatif |
| Magtarmkanalen | Buksmärtae Illamående | Diarréf Dyspepsif Aftösa sårf |
| Andningsvägar, bröstkorg och mediastinum | Obehag i bröstetf* | |
| Hud och subkutan vävnad | Klådaf* |
aÖvre luftvägsinfektioner: övre luftvägsinfektion, bronkit och virusinfektion i övre luftvägarna
bNasofaryngit: nasofaryngit, faryngit
cNedre luftvägsinfektion: Klebsiella pneumoniae, covid-19-pneumoni, nedre luftvägsinfektion, virusinfektion i luftvägarna, luftvägsinfektion, pneumoni
dHypertoni: hypertoni, förhöjt blodtryck, essentiell hypertoni, hypertensiv kris, vitrockshypertoni
eBuksmärta: buksmärta, smärta i nedre delen av buken, smärta i övre delen av buken, ömhet i buken
fInfusionsrelaterad reaktion: alla inträffade inom 24 timmar efter att infusionen med Enjaymo påbörjades. *Händelser som tyder på överkänslighetsreaktioner är inkluderade i tabellen.
Allvarliga infektioner
Av de 66 patienterna som deltog i studierna CADENZA och CARDINAL rapporterades allvarliga infektioner hos 10 (15,2 %) patienter. Allvarliga infektioner som listas i biverkningstabellen inkluderar luftvägsinfektion [Klebsiella pneumoniae (n = 1), luftvägsinfektion (n = 1), covid-19-pneumoni (n = 1)], urinvägsinfektion [urosepsis (n = 1), urinvägsinfektion (n = 1), bakteriell urinvägsinfektion (n = 1)], herpes zoster (n = 1). Sutimlimab avbröts hos en patient på grund av en allvarlig infektion med Klebsiella pneumoniae med dödlig utgång . Inga andra fall av infektioner med dödlig utgång rapporterades. Se avsnitt Varningar och försiktighet för information om vaccinationsrekommendationer för allvarliga infektioner och övervakning av tidiga tecken och symtom på infektioner.
Immunogenicitet
Immunogeniciteten av sutimlimab utvärderades hos patienter med köldagglutininsjukdom i studierna CARDINAL och CADENZA vid baslinjen, under behandlingsperioden och i slutet av behandlingen (vecka 26). Två av de 24 patienterna (8,3 %) som inkluderades i studien CARDINAL och som fick minst en dos av sutimlimab utvecklade behandlingsutlösta anti-läkemedelsantikroppar (ADA). I studien CADENZA utvecklade 6 av 42 patienter som behandlades med sutimlimab (14,3 %) behandlingsutlösta ADA. Dessa ADA var övergående med låg titer och förknippades inte med förändringar i den farmakokinetiska profilen, det kliniska svaret eller biverkningar.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till:
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
I händelse av överdosering rekommenderas omedelbart avbrytande av infusionen och noggrann övervakning.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Immunsuppressiva medel, komplementhämmare, ATC-kod: L04AJ04
Verkningsmekanism
Sutimlimab är en monoklonal antikropp av IgG, subtyp 4 (IgG4) som hämmar den klassiska vägen och specifikt binder till komplementproteinkomponenten 1, s-subkomponenten (C1s), ett serinproteas som klyver C4. Aktiviteten av lektinvägen och den alternativa vägen av komplementsystemet hämmas inte av sutimlimab. Hämningen av den klassiska vägen av komplementsystemet på C1s-nivå förhindrar depositionen av komplementopsoniner på ytan av röda blodkroppar, vilket leder till hämning av hemolys hos patienter med köldagglutininsjukdom, förhindrar bildningen av de proinflammatoriska anafylatoxinerna C3a och C5a och nedströms det terminala komplementkomplexet C5b-9.
Klinisk effekt och säkerhet
Större än 90 % hämning av den klassiska vägen observerades efter den första infusionen av Enjaymo och C4-nivåerna återställdes till normala nivåer (0,2 g/l) hos patienter med köldagglutininsjukdom inom en vecka efter den första dosen av Enjaymo.
Säkerheten och effekten av Enjaymo hos patienter med köldagglutininsjukdom utvärderades i en randomiserad, dubbelblind, placebokontrollerad fas 3-studie (CADENZA) hos 42 patienter (n = 22 med Enjaymo och n = 20 med placebo) och i en öppen, enarmad fas 3-studie (CARDINAL) hos 24 patienter under 26 veckor. Efter att de sex månaders behandlingsperioderna (del A) avslutades, fortsatte patienterna i båda studierna att få Enjaymo i en förlängningsfas (del B) som utvärderade den långvariga säkerheten och varaktigheten av svaret i ytterligare 12 månader (CADENZA) och 24 månader (CARDINAL) efter att den sista patienten gått ur del A. Båda studierna inkluderade en 9-veckors uppföljningsfas efter den sista dosen av Enjaymo. De viktigaste lämplighetskriterierna var hemoglobin (Hb) vid baslinjen ≤10 g/dl och aktiv hemolys med en bilirubinnivå över det normala referensområdet. Patienter med köldagglutininsyndrom (CAS) exkluderades. Patienter i studien CADENZA hade inte haft transfusion inom 6 månader, eller mer än en blodtransfusion under 12 månader före inskrivningen i studien, medan patienter som inkluderades i studien CARDINAL hade haft minst en dokumenterad blodtransfusion inom 6 månader före inskrivning i studien. Patienterna som vägde 39-< 75 kg fick 6 500 mg och patienter som vägde ≥ 75 kg fick 7 500 mg Enjaymo intravenöst under cirka 60 minuter dag 0, dag 7 och därefter var 14:e dag. De viktigaste baslinjekarakteristika för studiepopulationen sammanfattas i tabell 3 nedan.
Tabell 3 Baslinjekarakteristika för patienter inkluderade i de kliniska studierna
| Parameter | Statistik | CADENZA | CARDINAL | |
|---|---|---|---|---|
| Placebo N = 20 | Enjaymo N = 22 | Enjaymo N = 24 | ||
| Ålder | Genomsnittlig Min, max | 68,2 51, 83 | 65,3 46, 88 | 71,3 55, 85 |
Kön Man Kvinna |
n (%) |
4 (20,0) 16 (80,0) |
5 (22,7) 17 (77,3)
|
9 (37,5) 15 (62,5) |
| Kroppsvikt | Genomsnittlig, kg Min, max | 64,9 48, 95 | 66,8 39, 100 | 67,8 40, 112 |
| Hemoglobin | Genomsnittlig, g/dl | 9,33 | 9,15 | 8,59 |
| Bilirubin (total)* | µmol/l | 35,77 (1,75 X ULN) | 41,17 (2 X ULN) | 53,26 (2,6 × ULN†) |
Tidigare transfusion Inom de senaste 6 månaderna Inom de senaste 12 månaderna | Genomsnittligt antal transfusioner (intervall) |
0 0 |
0 0,14 (0, 1) |
3,2 (1, 19) 4,8 (1, 23) |
| FACIT†-trötthetsskala | Genomsnittlig
| 32,99 | 31,67 | 32,5 |
*N = 21 i CARDINAL; placebo N = 18 och Enjaymo N = 20 i CADENZA, för bilirubindata exklusive patienter med antingen positivt eller otillgängligt testresultat för Gilberts syndrom.
†ULN: Övre normalgräns, FACIT: Functional Assessment of Chronic Illness Therapy (FACIT-trötthet mäts på en skala från 0 (värst trötthet) till 52 (ingen trötthet)
Studien CADENZA
42 patienter randomiserades till att få Enjaymo (n = 22) eller placebo (n = 20) fram till vecka 25.
Effekten baserades på andelen patienter som uppfyllde de primära effektmåttskriterierna: en ökning från baslinjen i Hb-nivån ≥ 1,5 g/dl vid tidpunkten för utvärdering av behandlingen (medelvärde från vecka 23, 25 och 26), ingen blodtransfusion från vecka 5 fram till vecka 26 och ingen behandling för köldagglutininsjukdom utöver vad som var tillåtet enligt protokoll från vecka 5 till och med vecka 26. Patienter fick en blodtransfusion om de uppnådde följande gränsvärden för hemoglobin: Hb < 7 g/dl eller Hb < 9 g/dl med symtom. Förbjudna behandlingar inkluderade rituximab ensamt eller i kombination med cytotoxiska medel.
Effekten utvärderades ytterligare med följande två viktiga sekundära effektmått: baserat på effekten av Enjaymo på medelförändringen från baslinjen i Hb och FACIT-trötthetspoäng för bedömning av förändring i livskvaliteten. Ytterligare sekundära effektmått var: laboratorievärden för hemolys inklusive medelförändring från baslinjen i totalbilirubin. Understödjande effektdata som samlades in inkluderade användning av transfusion efter fem veckors behandling.
Effektresultaten beskrivs i tabell 4 och 5 nedan.
Tabell 4 Effektresultat hos patienter med köldagglutininsjukdom i studien CADENZA – del A
| Parameter | Statistik | Placebo N = 20 | Enjaymo N = 22 | Behandlingseffekt |
|---|---|---|---|---|
| Patient som svarar på behandlinga | % (95 % KI)
Oddskvot (95 % KI) p-värde
| 3 (15,0) (3,2, 37,9)
| 16 (72,7) (49,8, 89,3)
|
15,94 (2,88, 88,04) < 0,001 |
| Hemoglobin | Medelförändring från baslinjen (LS†-medelvärde), g/dl | 0,09 | 2,66 | 2,56 |
| 95 % KI av LS-medelvärde | (-0,5, 0,68) |
(2,09, 3,22) |
(1,75, 3,38) | |
| p-värde | < 0,001 | |||
| Genomsnittligt antal transfusioner (vecka 5 till vecka 26) | n (standardavvikelse) | 0,5 (1,1) | 0,05 (0,2) | Ej beräknat |
| FACIT†-trötthetsskala | Genomsnittlig | 33,66 | 43,15 | |
| Medelförändring från baslinjen (LS†-medelvärde) | 1,91 | 10,83 | 8,93 | |
| 95 % KI av LS-medelvärde | (-1,65, 5,46) | (7,45, 14,22) | (4, 13,85) | |
| p-värde | < 0,001 | |||
| Totalbilirubin* | Genomsnittlig, µmol/l | 33,95 | 12,12 | |
| Medelförändring från baslinjen | -1,83 | -22,13 | Ej beräknat | |
| Antal patienter med normaliserade värden (%) | 4 (22,2 %) | 15 (88,2) |
aEn patient som svarar på behandling definierades som en patient med en ökning från baslinjen i Hb-nivån ≥ 1,5 g/dl vid tidpunkten för utvärdering av behandlingen (medelvärde från vecka 23, 25 och 26), ingen blodtransfusion från vecka 5 fram till vecka 26 och ingen behandling för köldagglutininsjukdom utöver vad som var tillåtet enligt protokoll från vecka 5 fram till vecka 26.
*N = 18 för placebo och N = 17 för Enjaymo, för bilirubindata exklusive patienter med antingen positivt eller otillgängligt testresultat för Gilberts syndrom
†LS: Minsta kvadrat, FACIT: Functional Assessment of Chronic Illness Therapy, NC ej beräknat
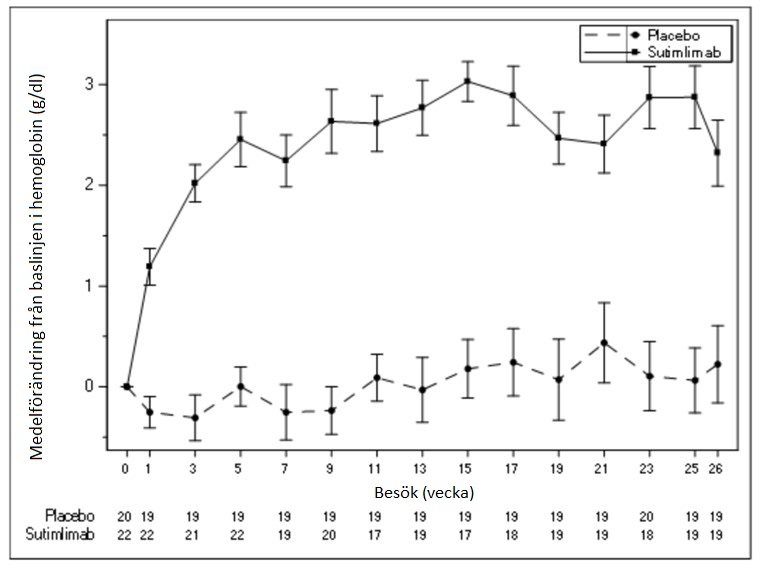
Genomsnittlig förändring från baslinjen i hemoglobin (Hb) visas i figur 1 nedan.
Figur 1 studien CADENZA del A: Diagram över medelförändring från baslinjen i hemoglobin (g/dl) (+/- standardfel) per besök
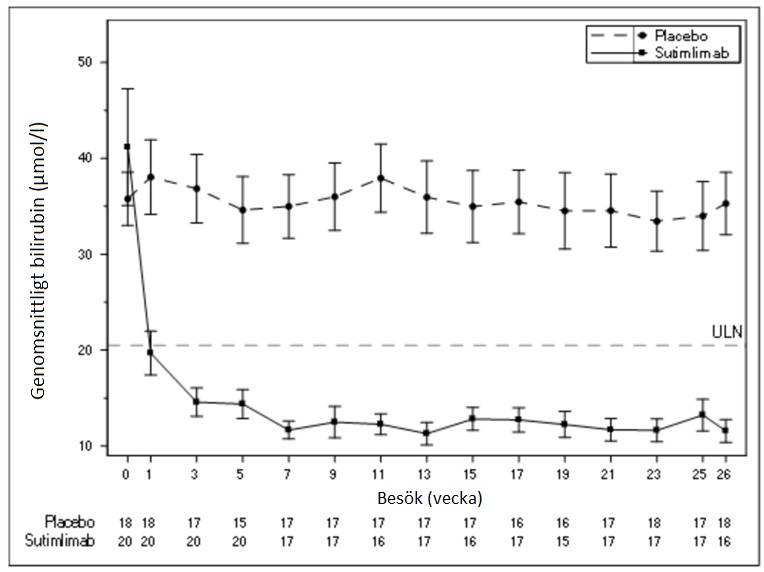
Genomsnittliga bilirubinnivåer per besök visas i figur 2 nedan.
Figur 2 - studien CADENZA del A: Diagram över genomsnittligt bilirubin (µmol/l) (+/- standardfel) per besök (exklusive försökspersoner med positivt eller okänt testresultat avseende Gilberts syndrom)
Hälsorelaterad livskvalitet
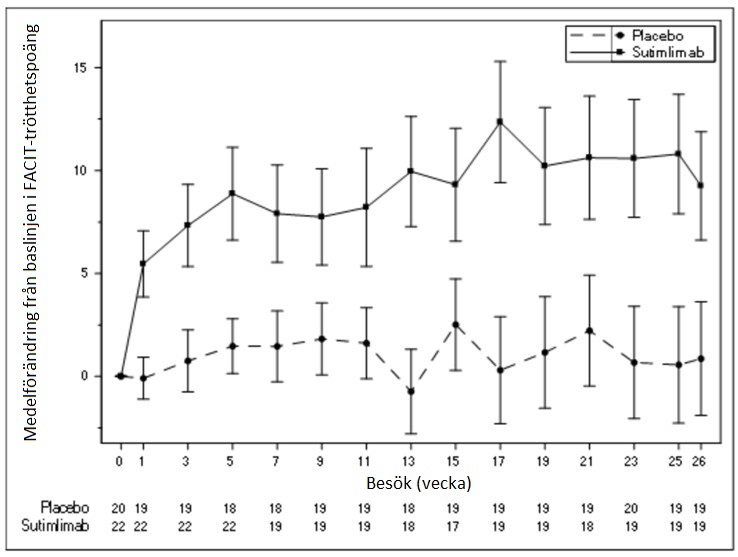
För del A visas ökningar i genomsnittliga FACIT-trötthetspoäng i figur 3 nedan.
Figur 3 – studien CADENZA del A: Diagram över medelförändring i FACIT-trötthetspoäng (standardfel) per besök – observerad – fullständigt analysset
I del B förblev de genomsnittliga hemoglobinnivåerna högre än 11 g/dl och en bestående normalisering av de genomsnittliga bilirubinnivåerna observerades, vilket tyder på en bestående minskning av hemolys. Ökningar i FACIT-trötthetspoängen som observerades i del A bibehölls.
Efter den sista dosen av Enjaymo i studien, observerades tecken och symtom på återkommande hemolys. De genomsnittliga hemoglobinnivåerna, nio veckor efter den sista dosen i del B, minskade med 2,41 g/dl (standardavvikelse: 2,21) och de genomsnittliga bilirubinnivåerna ökade med 21,80 µmol/l (standardavvikelse: 18,14) från de senaste värden som varit tillgängliga under behandlingen. De genomsnittliga FACIT-trötthetspoängen återställdes till nästan baslinjenivå, vid 31,29, och medelförändringen i standardavvikelse från baslinjen var ‑1,40 (11,48).
Studien CARDINAL
24 patienter fick Enjaymo fram till vecka 25.
Effekten baserades på andelen patienter som uppfyllde de primära effektmåttskriterierna: en ökning från baslinjen i Hb-nivån ≥ 2 g/dl eller ≥ 12 g/dl vid tidpunkten för utvärdering av behandlingen (medelvärde från vecka 23, 25 och 26), ingen blodtransfusion från vecka 5 fram till vecka 26 och ingen behandling för köldagglutininsjukdom utöver vad som var tillåtet enligt protokoll från vecka 5 fram till vecka 26. Patienter fick en blodtransfusion om de uppnådde följande gränsvärden för hemoglobin: Hb < 7 g/dl eller Hb < 9 g/dl med symtom. Förbjudna behandlingar inkluderade rituximab ensamt eller i kombination med cytotoxiska medel.
Effekten utvärderades ytterligare med följande sekundära effektmått: baserat på effekten av Enjaymo på Hb och laboratorievärden för hemolys inklusive medelförändring från baslinjen i totalbilirubin. Förändring i livskvalitet bedömdes enligt medelförändring från baslinjen i FACIT-trötthetspoäng som sekundärt effektmått. Understödjande effektdata som samlades in inkluderade användning av transfusion efter fem veckors behandling.
Tabell 5 visar effektresultat hos patienter med köldagglutininsjukdom i studien CARDINAL.
Tabell 5 Effektresultat hos patienter med köldagglutininsjukdom i studien CARDINAL – del A
| Parameter | Statistik | ENJAYMO N = 24 |
|---|---|---|
| Patient som svarar på behandlinga | n (%) | 13 (54) |
| Hemoglobin | Medelförändring från baslinjen (LS†-medelvärde), g/dl 95 % KI av LS-medelvärde | 2,60 (0,74, 4,46) |
| Genomsnittligt antal transfusioner (vecka 5 till vecka 26) | n | 0,9 |
| Totalbilirubin* | Genomsnittlig, µmol/l Medelförändring från baslinjen (LS†-medelvärde) Antal patienter med normaliserade värden (%) | 15,48 (0,76 × ULN†) -38,18 13 (54,2) |
| FACIT†-trötthetsskala | Genomsnittlig Medelförändring från baslinjen (LS†-medelvärde) 95 % KI av LS-medelvärde | 44,26 10,85 (8,0, 13,7) |
a En patient som svarar på behandling definierades som en patient med en ökning från baslinjen i Hb-nivån ≥ 2 g/dl eller ≥ 12 g/dl vid tidpunkten för utvärdering av behandlingen (medelvärde från vecka 23, 25 och 26), ingen blodtransfusion från vecka 5 fram till vecka 26 och ingen behandling för köldagglutininsjukdom utöver vad som var tillåtet enligt protokoll från vecka 5 fram till vecka 26.
*N = 21 för bilirubindata, exklusive patienter med Gilberts syndrom
†LS: Minsta kvadrat, ULN: Övre normalgräns, FACIT: Functional Assessment of Chronic Illness Therapy
I del B bibehölls de genomsnittliga hemoglobinnivåerna högre än 11 g/dl och en bestående normalisering av de genomsnittliga bilirubinnivåerna observerades, vilket tyder på en bestående minskning av hemolys.
Efter den sista dosen av Enjaymo i studien, observerades tecken och symtom på återkommande hemolys. De genomsnittliga hemoglobinnivåerna, nio veckor efter den sista dosen i del B, minskade med 2,28 g/dl (standardavvikelse: 1,80) och de genomsnittliga bilirubinnivåerna ökade med 24,27 µmol/l (standardavvikelse: 13,51) från de senaste värden som varit tillgängliga under behandlingen. De genomsnittliga FACIT-trötthetspoängen återställdes till nära baslinjenivån, och medelförändringen i standardavvikelse från värden vid baslinjen före behandlingen var 1,05 (8,15).
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Enjaymo för alla grupper av den pediatriska populationen för behandling av primär köldagglutininsjukdom (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Äldre
Majoriteten av patienterna (43/66, 65 %) som inkluderades i de kliniska studierna med Enjaymo för köldagglutininsjukdom var 65 år eller äldre. Enligt rapporterad klinisk erfarenhet har inga skillnader i svaren mellan patienter över 65 år och yngre patienter identifierats.
Farmakokinetiska egenskaper
Farmakokinetiken av sutimlimab utvärderades hos 24 patienter (CARDINAL) och 42 patienter (CADENZA), som inkluderade 51 patienter som behandlades med 6 500 mg och 15 patienter som behandlades med 7 500 mg enligt rekommenderad dosering. Den totala exponeringen vid steady state för föreslaget dosschema visas i tabell 6.
Tabell 6 Genomsnittliga (standardavvikelse) exponeringsparametrar vid steady state
CARDINAL och CADENZA | Dos (mg) | Cmin (µg/ml)* | AUCSS (µg·h/ml)* |
Genomsnittlig |
7500 (n = 15) |
1 107 (661) |
|
*Förkortningar: AUCss = arean under kurvan mellan 2 på varandra följande doser efter att steady state uppnåtts; Cmin = dalkoncentration vid steady state definierad som 1 timme före nästa dos
Steady state uppnåddes vid vecka 7 efter påbörjad behandling med sutimlimab, med en ackumuleringskvot på mindre än 2.
Distribution
Distributionsvolymen vid steady state i centrala och perifera kompartment var ungefär 5,8 l hos patienter med köldagglutininsjukdom.
Metabolism
Sutimlimab är ett protein. Det är allmänt känt att antikroppar metaboliseras genom nedbrytning till små peptider och enskilda aminosyror.
Eliminering
Halveringstiden för sutimlimab är beroende av plasmakoncentrationen. Den terminala elimineringshalveringstiden för sutimlimab vid steady state baserad på total clearance (linjär och icke-linjär) är 16 dagar.
Linjäritet/icke-linjäritet
Efter engångsdoser visade clearance av sutimlimab en kraftig initial minskning vid doser under 30 mg/kg (~ 2 g) och blev dosoberoende mellan 60 och 100 mg/kg sutimlimab.
Särskilda populationer
Inga kliniskt signifikanta skillnader observerades i sutimlimabs farmakokinetik baserat på kön, ålder, nedsatt leverfunktion eller nedsatt njurfunktion. Exponeringsnivåerna (Cmax, Cmin och AUC) vid steady state beräknades baserat på 6 500 mg (< 75 kg) och 7 500 mg (> = 75 kg) som gavs dag 0, dag 7 och därefter var 14:e dag. Den farmakokinetiska populationsanalysen visade liknande exponeringsparametrar mellan könen med 101 manliga och 95 kvinnliga försökspersoner.
Den farmakokinetiska populationsanalysen visade liknande exponeringsparametrar avseende försökspersonens ras (94 vita, 10 svarta, 42 asiater).
Den farmakokinetiska populationsanalysen visade att kroppsvikt och etnicitet (japansk vs. icke-japansk) påverkade farmakokinetiken för sutimlimab. Lägre exponering observerades hos patienter med högre kroppsvikt. Baserat på en jämförelse mellan studier var AUC0-168 för sutimlimab efter 30 till 100 mg/kg upp till 38 % högre hos japanska försökspersoner än hos icke-japanska försökspersoner.
Farmakokinetiskt (Farmakokinetiska)/farmakodynamiskt (farmakodynamiska) förhållande(n)
Sutimlimabkoncentration över 100 µg/ml resulterade i maximal hämning av den klassiska vägen. Det föreslagna dosschemat resulterade i adekvat sutimlimabexponering vid steady state för att ge kliniskt relevanta effekter på nivåerna av Hb, bilirubin och totalt C4.
Prekliniska säkerhetsuppgifter
En utvidgad pre‑ och postnatal utvecklingsstudie (ePPND) på cynomolgusapor visade ingen evidens på skadliga effekter på utvecklingen vid intravenös administrering av sutimlimab under organogenesen fram till förlossningen, vid exponering 2–3 gånger högre än AUC hos människa vid den maximala rekommenderade dosen. I studier med upprepade doser av sutimlimab med exponering vid upp till cirka 4 gånger den rekommenderade dosen till människa observerades inga effekter på reproduktionsorganen hos cynomolgusapor.
Inga djurstudier har utförts för att utvärdera sutimlimabs karcinogena potential.
Icke-kliniska data visade inte några särskilda risker för människa baserat icke-kliniska studier på cynomolgusapor.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Polysorbat 80 (E 433)
Natriumklorid
Dibasiskt natriumfosfat (E 339)
Monobasiskt natriumfosfat (E 339)
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
Oöppnad injektionsflaska:
4 år
Förvaring av läkemedel efter öppnandet:
Kemisk och fysikalisk hållbarhet vid användning har visats för 16 timmar vid 18 ºC–25 ºC eller för 72 timmar vid 2 ºC–8 ºC. Ur ett mikrobiologiskt perspektiv, ska produkten användas omedelbart.
Om den inte används omedelbart, är lagringstiden och lagringsförhållanden före användning användarens ansvar och är normalt inte längre än 24 timmar vid 2 °C–8 °C eller 8 timmar vid rumstemperatur, om inte öppnande av injektionsflaskan och poolning i infusionspåsen har skett under kontrollerade och validerade aseptiska förhållanden.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C–8 °C).
Förvaras i originalkartongen. Ljuskänsligt.
Får ej frysas.
Förvaringsanvisningar för läkemedlet efter första öppnande av injektionsflaskan finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
ENJAYMO infuusioneste, liuos
50 mg/ml (L:ei) 1 kpl (22 ml) (1466,33 €)
PF-selosteen tieto
22 ml lösning i injektionsflaska (typ I glas) med en propp (butylgummi), försegling (aluminium) och ett snäpplock
En förpackning innehåller 1 eller 6 injektionsflaskor.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Opaliserande, färglös till svagt gulaktig lösning, näst intill fri från partiklar, med ett pH på cirka 6,1 och osmolalitet på 268–312 mOsm/kg.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Enjaymo tillhandahålls som en lösning i en engångsinjektionsflaska och ska beredas av sjukvårdspersonal med aseptisk teknik.
Beredning
- Ta ut Enjaymo ur kylskåpet. För att minska skumbildning, skaka inte.
- Inspektera injektionsflaskorna visuellt med avseende på partiklar och missfärgning före administrering. Lösningen är en opaliserande och färglös till svagt gul lösning. Administrera inte om lösningen är missfärgad eller innehåller främmande partiklar.
- Dra upp den beräknade volymen från lämpligt antal injektionsflaskor baserat på den rekommenderade dosen (se tabell 1) och tillsätt i en tom infusionspåse. Kassera oanvänd lösning som finns kvar i injektionsflaskan.
- Den beredda lösningen ska administreras omedelbart. Förvaringsanvisningar för läkemedlet finns i avsnitt Hållbarhet.
Administrering
- Låt infusionslösningen anta rumstemperatur (18 °C–25 °C) före administrering. Se tabell 1 för infusionshastighet (avsnitt Dosering och administreringssätt). Infusionen ska administreras under 1–2 timmar beroende på patientens kroppsvikt. Infusionen ska administreras genom ett 0,22 mikrometers filter med polyetersulfonmembran. Infusionsvärmare kan användas, överskrid inte en temperatur på 40 °C.
- Infusionskatetern och -slangen ska beredas med doseringslösningen omedelbart före infusionen och sköljas omedelbart efter avslutad infusion med tillräcklig mängd (ungefär 20 ml) 9 mg/ml (0,9 %) natriumklorid injektionsvätska, lösning.
- Inga inkompabiliteter har observerats mellan Enjaymo infusionslösning och infusionspåsar av Di-(2-etylhexyl)ftalat (DEHP) mjukgjord polyvinylklorid (PVC), etylvinylacetat (EVA) och polyolefin (PO); administreringsset av DEHP-mjukgjord PVC, DEHP-fri polypropylen (PP) och polyetylen (PE) och adaptrar för injektionsflaskor av polykarbonat (PC) och akrylnitrilbutadienstyren (ABS).
Ersättning
ENJAYMO infuusioneste, liuos
50 mg/ml 1 kpl
- Ei korvausta.
Atc-kod
L04AJ04
Datum för översyn av produktresumén
01.08.2025
Yhteystiedot
Berzelius väg 8
171 65 Solna
Sverige
+46 8 545 802 30
www.recordati.com