
VABYSMO injektioneste, liuos 120 mg/ml, injektioneste, liuos, esitäytetty ruisku 120 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Tämä potilasopas sisältää tärkeitä turvallisuutta koskevia tietoja, joista potilaan on oltava tietoinen ennen Vabysmo-hoidon aloittamista ja hoidon aikana.
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi ml injektionestettä, liuosta, sisältää 120 mg farisimabia.
Esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 21 mg farisimabia 0,175 ml:ssa liuosta. Tästä saadaan käyttöannokseksi 0,05 ml:n kerta-annos liuosta, joka sisältää 6 mg farisimabia.
Injektiopullo
Yksi injektiopullo sisältää 28,8 mg farisimabia 0,24 ml:ssa liuosta. Tästä saadaan käyttöannokseksi 0,05 ml:n kerta-annos liuosta, joka sisältää 6 mg farisimabia.
Farisimabi on nisäkässoluviljelmässä yhdistelmä-DNA-tekniikalla, kiinanhamsterin munasarjasoluissa (CHO), tuotettu humanisoitu vasta-aine.
Apuaineet, joiden vaikutus tunnetaan
0,05 ml liuosta sisältää 0,02 mg polysorbaattia ja 0,07 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Vabysmo on tarkoitettu aikuisille potilaille
- neovaskulaarisen (kostean) silmänpohjan ikärappeuman (nAMD) hoitoon
- diabeettisesta makulaturvotuksesta (DME) aiheutuneen näkökyvyn heikkenemisen hoitoon
- verkkokalvon laskimotukoksesta (verkkokalvon haaralaskimotukos (BRVO) tai verkkokalvon keskuslaskimotukos (CRVO)) johtuvan makulaturvotuksen aiheuttaman näkökyvyn heikkenemisen hoitoon.
Ehto
Valmistetta saa antaa lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus ja antotapa
Tämän lääkevalmisteen saa annostella lääkäri, jolla on kokemusta lasiaiseen annettavista injektioista.
Annostus
Kostea silmänpohjan ikärappeuma
Suositeltu annos on 6 mg (0,05 ml liuosta) injektiona lasiaiseen 4 viikon välein (kuukausittain) kolmen ensimmäisen annoksen ajan.
Tämän jälkeen tautiaktiivisuutta suositellaan arvioitavaksi näöntarkkuuden ja/tai anatomisen hoitovasteen perusteella 16 ja/tai 20 viikon kuluttua hoidon aloittamisesta, jotta hoitoa voidaan säädellä yksilöllisesti. Potilaille, joilla ei havaita tautiaktiivisuutta, tulee harkita farisimabin annostelua 16 viikon (4 kuukauden) välein. Potilaille, joilla havaitaan tautiaktiivisuutta, tulee harkita hoitoa 8 viikon (2 kuukauden) tai 12 viikon (3 kuukauden) välein. Jos potilaan näkökyky ja/tai anatominen hoitovaste muuttuu, pitää hoitoväliä muuttaa vastaavasti. Hoitoväliä pitää lyhentää, jos potilaan näkökyky ja/tai anatominen hoitovaste heikkenee (ks. kohta Farmakodynamiikka). Kahdeksan (8) viikon tai sitä lyhyemmällä annosteluvälillä annetusta hoidosta on rajallisesti turvallisuutta koskevaa tietoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoitokäyntien välinen seuranta perustuu lääkärin arvioon potilaan tilasta. Kuukausittainen seuranta injektioiden välillä ei ole välttämätöntä.
Diabeettisesta makulaturvotuksesta aiheutunut näkökyvyn heikkeneminen ja verkkokalvon laskimotukoksesta aiheutuva makulaturvotus (RVO)
Suositeltu annos on 6 mg (0,05 ml liuosta) injektiona lasiaiseen 4 viikon välein (kuukausittain); kolme tai sitä useampia peräkkäisiä kuukausittaisia injektioita voidaan tarvita.
Tämän jälkeen hoitoväliä pidennetään yksilöllisesti (treat-and-extend approach). Hoitovälin pidentäminen perustuu lääkärin arvioon potilaan näöntarkkuudesta ja/tai anatomisesta hoitovasteesta. Hoitoväliä voidaan pidentää enintään 4 viikkoa kerrallaan. Jos potilaan näkökyky ja/tai anatominen hoitovaste muuttuu, pitää hoitoväliä muuttaa vastaavasti. Hoitoväliä pitää lyhentää, jos potilaan näkökyky ja/tai anatominen hoitovaste heikkenee (ks. kohta Farmakodynamiikka). Alle 4 viikon välein ja yli 4 kuukauden välein annettavasta hoidosta ei ole tutkittua tietoa. Hoitokäyntien välinen seuranta perustuu lääkärin arvioon potilaan tilasta. Kuukausittainen seuranta injektioiden välillä ei ole välttämätöntä.
Hoidon kesto
Tämä lääkevalmiste on tarkoitettu pitkäaikaishoitoon. Jos näkökyky ja/tai anatomiavasteet osoittavat, että potilas ei hyödy hoidon jatkamisesta, hoito pitää lopettaa.
Annoksen viivästyminen tai jääminen väliin
Jos annos viivästyy tai jää väliin, potilaan tulee saapua lääkärin arvioitavaksi seuraavalle vapaalle vastaanottokäynnille, ja hoitoa tulee jatkaa harkinnan mukaan.
Erityispotilasryhmät
Iäkkäät
65-vuotiaiden ja sitä vanhempien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Turvallisuutta koskevia tietoja on rajallisesti ≥ 85-vuotiaista kosteaa silmänpohjan ikärappeumaa sairastavista potilaista ja potilaista, joilla on verkkokalvon laskimotukos (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Pediatrisille potilaille ei ole asiaankuuluvaa käyttöä tällä lääkevalmisteella kostean silmänpohjan ikärappeuman, diabeettisen makulaturvotuksen ja verkkokalvon laskimotukoksen hoidossa.
Antotapa
Vain injektiona silmän lasiaiseen. Yhtä esitäytettyä ruiskua tai injektiopulloa tulee käyttää vain yhden silmän hoitoon.
Vabysmo-valmiste on tarkastettava silmämääräisesti ennen antoa hiukkasten ja värimuutosten havaitsemiseksi. Jos näitä havaitaan, esitäytettyä ruiskua tai injektiopulloa ei pidä käyttää.
Lasiaisinjektio tulee antaa aseptisissa olosuhteissa käsittäen käsien kirurgisen desinfektion, sekä steriilin leikkausliinan ja steriilin luomenlevittimen (tai vastaavan) käytön. Potilaan aiemmat yliherkkyysreaktiot on selvitettävä tarkoin ennen intravitreaalista toimenpidettä (ks. kohta Haittavaikutukset). Ennen injektiota silmänympärysiho, silmäluomet ja silmän pinta desinfioidaan laajakirjoisella mikrobisidilla ja annetaan riittävä puudutus.
Esitäytetty ruisku
Esitäytetty ruisku sisältää ylimääräistä liuosta. Ylimääräinen tilavuus on poistettava ennen suositellun annoksen injisoimista. Esitäytetyn ruiskun koko tilavuuden injisointi voi johtaa yliannokseen.
Lääkevalmisteessa olevat ilmakuplat on poistettava painamalla mäntää hitaasti, kunnes kumitulpan alareuna on 0,05 ml:n annosmerkin kohdalla (ks. kohdat Yliannostus ja Käyttö- ja käsittelyohjeet).
Suodattimella varustettu injektioneula (mukana pakkauksessa) pistetään lasiaistilaan 3,5–4,0 mm limbuksesta posteriorisesti välttäen samalla horisontaalista meridiaania ja tähdätään silmämunan keskiosaan. Sen jälkeen annettava 0,05 ml:n injektiotilavuus injisoidaan hitaasti. Kovakalvon pistoskohtaa vaihdetaan seuraavien injektioiden yhteydessä.
Injektiopullo
Injektioneula (30G x ½”, ei mukana pakkauksessa) pistetään lasiaistilaan 3,5–4,0 mm limbuksesta posteriorisesti välttäen samalla horisontaalista meridiaania ja tähdätään silmämunan keskiosaan. Sen jälkeen annettava 0,05 ml:n injektiotilavuus injisoidaan hitaasti. Kovakalvon pistoskohtaa vaihdetaan seuraavien injektioiden yhteydessä.
Injektion jälkeinen seuranta
Injektion jälkeen käyttämättä jäävä lääkevalmiste tai jätemateriaali on hävitettävä paikallisten vaatimusten mukaisesti.
Potilasta on seurattava välittömästi intravitreaalisen injektion jälkeen silmänpaineen kohoamisen varalta. Asianmukaiseen seurantaan voi kuulua näköhermon pään perfuusion tarkistaminen tai tonometria. Steriilit parasenteesivälineet on oltava tarvittaessa saatavilla.
Intravitreaalisen injektion jälkeen potilaita tulee neuvoa ilmoittamaan viipymättä endoftalmiittiin viittaavista oireista (esim. näkökyvyn menetys, silmäkipu, silmän punoitus, valonarkuus, näön sumeneminen).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiiviset tai epäillyt silmän tai silmänympäryksen infektiot.
Aktiivinen silmänsisäinen tulehdus.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Lasiaiseen annettuun injektioon liittyvät reaktiot
Lasiaiseen annettuihin injektioihin, farisimabi-injektiot mukaan lukien, on liittynyt endoftalmiittia, silmänsisäistä tulehdusta, regmatogeenisia verkkokalvon irtaumia, verkkokalvon repeämiä sekä hoidosta johtuvaa traumaperäistä kaihia (ks. kohta Haittavaikutukset). Vabysmo-valmistetta annettaessa on aina noudatettava asianmukaista aseptista injektiotekniikkaa. Potilaita pitää kehottaa ilmoittamaan viipymättä kaikista endoftalmiittiin tai mistä tahansa edellä mainittuihin haittavaikutuksiin viittaavista oireista, kuten kivusta, näkökyvyn menetyksestä, valonarkuudesta, näön sumenemisesta, lasiaiskellujista tai punoituksesta, jotta asianmukainen hoito voidaan aloittaa nopeasti. Potilailla, joille injektioita annetaan tavanomaista tiheämmin, voi olla suurempi toimenpiteeseen liittyvien komplikaatioiden riski.
Silmänpaineen nousu
Ohimenevää silmänpaineen nousua on havaittu 60 minuutin kuluessa lasiaiseen annetusta injektiosta, farisimabi mukaan lukien (ks. kohta Haittavaikutukset). Erityinen varovaisuus on tarpeen potilailla, joilla on todettu huonossa hoitotasapainossa oleva glaukooma (Vabysmo-valmistetta ei saa injisoida, jos silmänpaine on ≥ 30 mmHg). Sekä silmänpainetta että näköhermon pään perfuusiota on aina seurattava ja hoidettava asianmukaisesti.
Systeemiset vaikutukset
Systeemisiä haittatapahtumia, mukaan lukien valtimotromboembolisia tapahtumia, on raportoitu lasiaiseen injektoitavan verisuonen endoteelin kasvutekijän (VEGF) estäjähoidon jälkeen. On olemassa teoreettinen riski, että nämä voivat liittyä VEGF:n estoon. Kliinisissä farisimabitutkimuksissa potilailla, joilla oli kostea silmänpohjan ikärappeuma, diabeettista makulaturvotusta ja verkkokalvon laskimotukos, havaittiin valtimotromboembolisia tapahtumia; niiden ilmaantuvuus oli vähäinen. Raportoidut tiedot ovat samankaltaisia kuin muissa kliinisissä tutkimuksissa, joissa on käytetty VEGF:n estäjiä. Farisimabihoidon turvallisuutta koskevat tiedot diabeettista makulaturvotusta sairastavista potilaista, joilla on korkea verenpaine (≥ 140/90 mmHg) ja verisuonitauti, sekä ≥ 85-vuotiaista potilaista, joilla on kostea silmänpohjan ikärappeuma tai verkkokalvon laskimotukos, ovat suppeita.
Immunogeenisuus
Farisimabi on terapeuttinen proteiini, joten siihen voi liittyä immunogeenisuutta (ks. kohta Haittavaikutukset). Potilaita pitää ohjeistaa kertomaan lääkärille silmänsisäisen tulehduksen oireista ja löydöksistä, kuten näkökyvyn menetyksestä, silmäkivusta, lisääntyneestä valonarkuudesta, lasiaiskellujista tai silmän punoituksen pahenemisesta, jotka voivat olla farisimabista aiheutuvaan yliherkkyyteen liittyviä kliinisiä oireita (ks. kohta Haittavaikutukset).
Molempien silmien hoito
Farisimabin turvallisuutta ja tehoa molempien silmien samanaikaiseen hoitoon ei ole tutkittu. Molempien silmien hoidosta voi aiheutua haittavaikutuksia molempiin silmiin ja/tai lisääntynyttä systeemistä altistusta, joka voi lisätä systeemisten haittavaikutusten riskiä. Kyseessä on farisimabiin liittyvä teoreettinen riski, kunnes molempien silmien hoidosta saadaan tietoja.
Samanaikainen käyttö muiden verisuonen endoteelin kasvutekijän (VEGF) estäjien kanssa
Farisimabin ja muiden verisuonen endoteelin kasvutekijän estäjien samanaikaisesta käytöstä samaan silmään ei ole tietoja saatavilla. Farisimabia ei pidä antaa samanaikaisesti muiden verisuonen endoteelin kasvutekijän estäjien (systeemisesti tai silmään annettavien) kanssa.
Muiden injektioneulojen käyttö esitäytetyn ruiskun kanssa
Esitäytetyn ruiskun kanssa tulee käyttää vain pakkauksen sisältämää suodattimella varustettua injektioneulaa. Muiden injektioneulojen käytöstä esitäytetyn ruiskun kanssa ei ole kliinisiä tietoja saatavissa.
Hoidon keskeyttäminen
Hoito pitää keskeyttää, jos
- potilaalla on regmatogeeninen verkkokalvon irtauma, 3. tai 4. asteen makulareikiä tai verkkokalvon repeämä. Hoitoa ei pidä aloittaa uudelleen ennen kuin verkkokalvon tila on hoidettu asianmukaisesti
- potilaan hoitoon liittyvä paras laseilla korjattu näöntarkkuus (BCVA) on laskenut ≥ 30 kirjainta verrattuna näöntarkkuuden edelliseen arviointiin. Hoitoa ei pidä jatkaa ennen seuraavaa sovittua hoitokertaa
- potilaan silmänpaine on ≥ 30 mmHg
- potilaalla on verkkokalvon alainen verenvuoto verkkokalvon keskikuopan alueella, tai jos verenvuodon laajuus on ≥ 50 % koko leesion alueesta
- potilaalle on edeltävien 28 päivän aikana tehty silmäleikkaus tai silmäleikkausta suunnitellaan seuraavien 28 päivän aikana. Hoitoa ei pidä jatkaa ennen seuraavaa sovittua hoitokertaa.
Verkkokalvon pigmenttiepiteelin repeämä
Verkkokalvon pigmenttiepiteelin repeämä on pigmenttiepiteelin irtauman komplikaatio kosteaa silmänpohjan ikärappeumaa sairastavilla potilailla. Kosteaan silmänpohjan ikärappeumaan annetun anti-VEGF-hoidon jälkeen verkkokalvon pigmenttiepiteelin repeämän kehittymiseen liittyviä riskitekijöitä ovat laaja-alainen ja/tai korkea pigmenttiepiteelin irtaumat. Farisimabihoitoa aloitettaessa on noudatettava varovaisuutta niiden potilaiden kohdalla, joilla on havaittu verkkokalvon pigmenttiepiteelin repeämälle altistavia riskitekijöitä. Verkkokalvon pigmenttiepiteelin repeämät ovat yleisiä niillä kosteaa silmänpohjan ikärappeumaa sairastavilla potilailla, joilla on pigmenttiepiteelin irtoama ja jotka saavat lasiaiseen annettavaa anti-VEGF-hoitoa, farisimabi mukaan lukien. Verkkokalvon pigmenttiepiteelin repeämän esiintyvyys oli farisimabiryhmässä (2,9 %) suurempi kuin afliberseptiryhmässä (1,5 %). Valtaosa tapahtumista ilmeni latausvaiheen aikana ja ne olivat lieviä tai keskivaikeita, eivätkä vaikuttaneet näkökykyyn.
Potilasryhmät, joista on rajallisesti tietoja
Hoitokokemusta on vain rajallisesti ≥ 85-vuotiaista potilaista, joilla on kostea silmänpohjan ikärappeuma tai verkkokalvon laskimotukos, sekä diabeettista makulaturvotusta sairastavista potilaista, joilla on tyypin I diabetes, HbA1c yli 10 %, korkean riskin proliferatiivinen diabeettinen retinopatia, korkea verenpaine (≥ 140/90 mmHg) ja verisuonitauti, ja joiden hoidon antoväli on pitkään lyhyempi kuin 8 viikkoa (Q8W). Hoitokokemusta on myös rajallisesti potilaista, joilla on kostea silmänpohjan ikärappeuma, diabeettista makulaturvotusta ja verkkokalvon laskimotukos ja joilla on aktiivisia systeemisiä infektioita. Pitkäaikaisesta hoidosta 8 viikon tai sitä lyhyemmällä antovälillä on rajallisesti tietoa, ja siihen voi liittyä suurentunut silmien ja systeemisten (myös vakavien) haittavaikutusten riski. Farisimabihoidosta ei myöskään ole kokemusta diabetespotilailla ja verkkokalvon laskimotukospotilailla, joilla on huonossa hoitotasapainossa oleva verenpainetauti, eikä myöskään verkkokalvon laskimotukospotilailla, joiden aiempi hoito on epäonnistunut. Lääkärin on otettava näiden potilasryhmien hoidossa huomioon yllämainittujen tietojen puuttuminen.
Natriumsisältö
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaattisisältö
Tämä lääkevalmiste sisältää 0,02 mg polysorbaattia per 0,05 ml:n annos. Potilaat, jotka ovat yliherkkiä polysorbaatille, eivät saa käyttää tätä lääkettä.
Koulutusmateriaali
Lääkettä määräävien lääkäreiden on tutustuttava potilasoppaaseen, jonka tarkoitus on varmistaa, että potilaat ovat tietoisia silmänsisäisen tulehduksen ja endoftalmiitin oireista ja löydöksistä. Lääkärin on annettava kyseinen opas potilaalle / potilasta hoitavalle henkilölle ja samalla kerrottava näistä tapahtumista.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Farisimabin biotransformaation ja eliminaation perusteella (ks. kohta Farmakokinetiikka) yhteisvaikutuksia ei oletettavasti esiinny. Farisimabia ei kuitenkaan pidä antaa samanaikaisesti muiden systeemisesti tai silmään annettavien anti-VEGF-lääkevalmisteiden kanssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, tulee käyttää tehokasta ehkäisyä hoidon aikana ja vähintään 3 kuukautta viimeisen lasiaiseen annetun farisimabi-injektion jälkeen.
Raskaus
Tietoja farisimabin käytöstä raskaana oleville naisille ei joko ole, tai tietoja on rajallisesti. Silmään annetusta farisimabista aiheutuva systeeminen altistus on vähäinen, mutta farisimabi on vaikutusmekanisminsa perusteella (verisuonten endoteelin kasvutekijän esto) katsottava mahdollisesti teratogeeniseksi ja alkio-sikiötoksiseksi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Farisimabia ei pidä käyttää raskauden aikana, ellei mahdollinen hyöty ole sikiölle aiheutuvaa mahdollista riskiä suurempi.
Imetys
Ei tiedetä erittyykö farisimabi ihmisen rintamaitoon. Rintaruokittavaan vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Vabysmo-valmistetta ei pidä käyttää imetyksen aikana. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko farisimabihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Kuusi kuukautta kestäneessä jaavanmakakeilla tehdyssä farisimabitutkimuksessa ei havaittu vaikutuksia lisääntymiselimiin tai hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vabysmo-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Lasiaiseen annetusta injektiosta ja siihen liittyvästä silmätutkimuksesta voi aiheutua tilapäisiä näköhäiriöitä. Potilaiden ei pidä ajaa moottoriajoneuvoa eikä käyttää koneita ennen kuin näkökyky on palautunut riittävästi.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia olivat kaihi (10 %), sidekalvon verenvuoto (7 %), lasiaisen irtauma (4 %), silmänpaineen nousu (4 %), lasiaiskellujat (4 %), silmäkipu (3 %) ja verkkokalvon pigmenttiepiteelin repeämä (3 %) (vain kostea silmänpohjan ikärappeuma).
Vakavimpia haittavaikutuksia olivat uveiitti (0,5 %), endoftalmiitti (0,4 %), vitreiitti (0,4 %), verkkokalvon repeämä (0,2 %), regmatogeeninen verkkokalvon irtauma (0,1 %) ja traumaperäinen kaihi (< 0,1 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa tai markkinoille tulon jälkeen raportoidut haittavaikutukset on lueteltu MedDRA-elinjärjestelmäluokituksen ja yleisyyden perusteella seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) tai tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutusten esiintyvyydet
| MedDRA-elinjärjestelmäluokka | Esiintyvyys |
| Silmät | |
| Kaihi | Yleinen |
| Sidekalvon verenvuoto | Yleinen |
| Lasiaisen irtauma | Yleinen |
| Kohonnut silmänpaine | Yleinen |
| Lasiaiskellujat | Yleinen |
| Verkkokalvon pigmenttiepiteelin repeämä (vain kostea silmänpohjan ikärappeuma) | Yleinen |
| Silmäkipu | Yleinen |
| Sarveiskalvon naarmu | Melko harvinainen |
| Silmä-ärsytys | Melko harvinainen |
| Lisääntynyt kyynelvuoto | Melko harvinainen |
| Sumentunut näkökyky | Melko harvinainen |
| Silmän kutina | Melko harvinainen |
| Epämukavuuden tunne silmässä | Melko harvinainen |
| Silmän hyperemia | Melko harvinainen |
| Iriitti | Melko harvinainen |
| Heikentynyt näöntarkkuus | Melko harvinainen |
| Uveiitti | Melko harvinainen |
| Endoftalmiitti | Melko harvinainen |
| Rikan tunne silmässä | Melko harvinainen |
| Lasiaisverenvuoto | Melko harvinainen |
| Vitreiitti | Melko harvinainen |
| Iridosykliitti | Melko harvinainen |
| Sidekalvon hyperemia | Melko harvinainen |
| Toimenpiteestä johtuva kipu | Melko harvinainen |
| Verkkokalvon repeämä | Melko harvinainen |
| Regmatogeeninen verkkokalvon irtauma | Melko harvinainen |
| Tilapäisesti heikentynyt näöntarkkuus | Harvinainen |
| Traumaperäinen kaihi | Harvinainen |
| Verkkokalvon vaskuliitti* | Tuntematon |
| Verkkokalvon okklusiivinen vaskuliitti* | Tuntematon |
Tähdellä (*) merkityt termit ovat haittavaikutuksia, jotka on tunnistettu markkinoilletulon jälkeisten spontaanien ilmoitusten perusteella. Näiden haittavaikutusten yleisyyttä ei voida aina luotettavasti arvioida, koska niiden raportointi on ollut vapaaehtoista ja populaation koko on tuntematon.
Valikoitujen haittavaikutusten kuvaus
Verkkokalvon vaskuliitti ja verkkokalvon okklusiivinen vaskuliitti
Verkkokalvon vaskuliittia ja/tai verkkokalvon okklusiivista vaskuliittia on raportoitu harvinaisina tapauksina spontaanisti markkinoille tulon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Verkkokalvon vaskuliittia ja verkkokalvon okklusiivista vaskuliittia on myös raportoitu lasiaiseen annettavaa hoitoa (IVT) saaneilla potilailla.
Lääkevalmisteluokkaan liittyvät haittavaikutukset
Verisuonen endoteelin kasvutekijän (VEGF) estäjien lasiaiseen annon jälkeen on olemassa valtimotromboembolisten tapahtumien, mukaan lukien aivohalvauksen ja sydäninfarktin teoreettinen riski. Kliinisissä farisimabitutkimuksissa potilailla, joilla oli kostea silmänpohjan ikärappeuma, diabeettista makulaturvotusta ja verkkokalvon laskimotukos, havaittiin vähäinen valtimotromboembolisten tapahtumien ilmaantuvuus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Farisimabilla ja vertailuvalmisteella hoitoa saaneiden ryhmien välillä ei havaittu missään käyttöaiheessa merkittävää eroa.
Immunogeenisuus
Farisimabihoitoa saaville potilaille saattaa kehittyä immuunivaste (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Enimmillään 112 viikon (kostea silmänpohjan ikärappeuma), 100 viikon (diabeettinen makulaturvotus) ja 72 viikon (verkkokalvon laskimotukos) ajan annetun farisimabihoidon jälkeen noin 13,8 %:lla kosteaa silmänpohjan ikärappeumaa sairastavista potilaista, 9,6 %:lla diabeettista makulaturvotusta sairastavista potilaista ja 14,4 %:lla verkkokalvon laskimotukospotilaista, jotka oli satunnaistettu farisimabihoitoon, havaittiin hoidon aikana ilmenneitä vasta-aineita farisimabia kohtaan. Anti-farisimabin vasta-aineiden kliininen merkitys turvallisuuden kannalta on toistaiseksi epäselvä. Silmänsisäisiä tulehduksia havaittiin 12 potilaalla 98:sta (12,2 %, kostea silmänpohjan ikärappeuma), 15 potilaalla 128:sta (11,7 %, diabeettinen makulaturvotus) ja 9 potilaalla 95:stä (9,5 %, verkkokalvon laskimotukos) anti-farisimabi vasta-ainepositiivisista potilaista. Anti-farisimabi vasta-ainenegatiivisilla potilailla silmänsisäisiä tulehduksia havaittiin 8 potilaalla 562:sta (1,4 %, kostea silmänpohjan ikärappeuma), 5 potilaalla 1124:stä (0,4 %, diabeettinen makulaturvotus) ja 10 potilaalla 543:sta (1,8 %, verkkokalvon laskimotukos). Vakavia silmän haittavaikutuksia ilmaantui 6 potilaalle 98:sta (6,1 %, kostea silmänpohjan ikärappeuma), 14 potilaalle 128:sta (10,9 %, diabeettinen makulaturvotus) ja 7 potilaalle 95:stä (7,4 %, verkkokalvon laskimotukos) anti-farisimabi vasta-ainepositiivisesta potilaasta. Anti-farisimabi vasta-ainenegatiivisilla potilailla vakavia silmän haittavaikutuksia ilmaantui 23 potilaalle 562:sta (4,1 %, kostea silmänpohjan ikärappeuma), 45 potilaalle 1124:stä (4,0 %, diabeettinen makulaturvotus) ja 34 potilaalle 543:sta (6,3 %, verkkokalvon laskimotukos). Anti-farisimabin vasta-aineet eivät vaikuttaneet kliiniseen tehoon tai systeemiseen farmakokinetiikkaan.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus suositeltua suuremmalla injektiotilavuudella voi aiheuttaa silmänpaineen nousua. Yliannostuksen yhteydessä on seurattava silmänpainetta. Asianmukainen hoito on aloitettava, jos hoitava lääkäri katsoo sen tarpeelliseksi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Silmätautien lääkkeet, uudissuonittumisen estoon käytettävät lääkkeet, ATC-koodi: S01LA09
Vaikutusmekanismi
Farisimabi on humanisoitu kaksoisspesifinen immunoglobuliini G1 (IgG1) vasta-aine, joka vaikuttaa estämällä kahta erillistä reittiä neutraloimalla sekä angiopoietiini-2:ta (ANG2) että verisuonen endoteelin kasvutekijä A:ta (VEGF-A).
ANG2 aiheuttaa verisuoniston epätasapainoa edistämällä endoteelin epävakautta, perisyyttikatoa ja patologista angiogeneesiä ja siten voimistamalla vuotoa verisuonistosta ja tulehdusta. Se myös herkistää verisuonet VEGF-A:n aktiivisuudelle, mikä lisää entisestään verisuoniston epävakautta. ANG2ja VEGF-A lisäävät synergistisesti verisuoniston läpäisevyyttä ja stimuloivat uudissuoniston muodostumista.
Farisimabi estää sekä ANG2:ta että VEGF-A:ta ja siten vähentää verisuoniston läpäisevyyttä ja tulehdusta, estää patologista verisuonten uudismuodostusta ja stabiloi verisuonistoa.
Farmakodynaamiset vaikutukset
Jäljempänä kuvatuissa kuudessa vaiheen III tutkimuksessa havaittiin 7. päivästä alkaen silmän vapaan ANG2:n ja vapaan VEGF-A:n pitoisuuden (mediaani) väheneminen lähtötilanteesta.
Kostea silmänpohjan ikärappeuma
TENAYA- ja LUCERNE-tutkimuksissa käytettiin objektiivisia, ennalta määritettyjä näkökyvyn ja anatomian kriteerejä sekä hoitavan lääkärin kliinistä arviota ohjaamaan hoitopäätöstä taudin aktiivisuuden arviointiajankohtina (viikko 20 ja viikko 24).
Keskimääräinen verkkokalvon keskikuopan (keskikentän) paksuuden (central subfield thickness, CST) alenema lähtötilanteesta ensisijaisen päätetapahtuman arviointikäynneille (keskiarvo viikoilta 40−48) oli verrannollinen farisimabi- ja afliberseptihoitoa saaneilla potilailla. TENAYA- ja LUCERNE-tutkimuksissa farisimabihoitoa enimmillään 16 viikon (enintään Q16W) välein saaneilla potilailla alenemat olivat -137 µm ja -137 µm. Afliberseptihoidolla vastaavat luvut olivat -129 µm ja -131 µm. CST:n keskimääräiset alenemat säilyivät toisen hoitovuoden ajan.
Viikolla 48 havaittiin kummassakin tutkimuksessa farisimabi- ja afliberseptihoidolla verrannolliset vaikutukset retinan sisäisen nesteen, subretinaalisen nesteen ja pigmenttiepiteelin irtaumien vähenemisessä. Nämä vaikutukset säilyivät toisen hoitovuoden ajan. Myös suonikalvon uudissuonittumisen pinta-alan ja vuotoalueen pieneneminen lähtötilanteesta olivat verrannolliset farisimabi- ja afliberseptihoitohaaroissa.
Diabeettinen makulaturvotus
YOSEMITE- ja RHINE-tutkimuksissa makulaturvotukseen liittyvät anatomiset parametrit olivat osa hoitopäätöksiä ohjaavia arvioita sairauden aktiivisuudesta.
Keskimääräinen verkkokalvon keskikuopan (keskikentän) paksuuden (central subfield thickness, CST) alenema lähtötilanteesta ensisijaisen päätetapahtuman arviointikäynneille (keskiarvo viikoilta 48−56) oli numeerisesti suurempaa kuin afliberseptihoidolla: YOSEMITE-tutkimuksessa CST pieneni -207 µm farisimabin Q8W-haarassa ja -197 µm joustavassa Q16W-farisimabihaarassa verrattuna -170 µm:iin afliberseptin Q8W-haarassa; RHINE-tutkimuksessa CST pieneni farisimabin Q8W-haarassa 196 µm ja joustavassa Q16W-farisimabihaarassa 188 µm verrattuna 170 µm:iin afliberseptin Q8W-haarassa. Verkkokalvon keskikuopan (keskikentän) paksuuden jatkuvaa vähenemistä havaittiin kahden vuoden ajan. Niiden potilaiden osuus, joilla ei ollut nesteen kertymistä verkkokalvon sisään eikä diabeettista makulaturvotusta (määriteltiin verkkokalvon keskikuopan paksuudeksi alle 325 µm), oli kummankin tutkimuksen kummassakin farisimabihaarassa kahden vuoden ajan suurempi kuin afliberseptihaarassa.
Verkkokalvon laskimotukos
Vaiheen III tutkimuksissa potilailla, joilla oli verkkokalvon haaralaskimotukos (BRVO; BALATON) ja verkkokalvon keskuslaskimotukos / verkkokalvon keskuslaskimon päähaaratukos (C/HRVO; COMINO), verkkokalvon keskikuopan (keskikentän) keskimääräisen paksuuden havaittiin vähentyneen farisimabin Q4W-haarassa lähtötilanteesta viikkoon 24, ja vähenemän havaittiin olleen verrannollista afliberseptin Q4W-haaraan nähden. BALATON-tutkimuksessa verkkokalvon keskikuopan (keskikentän) keskimääräinen paksuus väheni lähtötilanteesta viikkoon 24 farisimabin Q4W-haarassa 311,4 μm verrattuna 304,4 μm:iin afliberseptin Q4W-haarassa; COMINO-tutkimuksessa vastaavat luvut olivat farisimabin osalta 461,6 μm ja afliberseptin osalta 448,8 μm. Verkkokalvon keskikuopan (keskikentän) paksuuden vähenemä säilyi viikkoon 72 saakka, potilaiden siirryttyä farisimabin joustavaan annosteluun enintään Q16W-antovälillä.
Niiden potilaiden osuus, joilla ei ollut nesteen kertymistä verkkokalvon sisään, subretinaalista nestettä eikä makulaturvotusta (määriteltiin verkkokalvon keskikuopan paksuudeksi alle 325 µm), oli kummankin tutkimuksen farisimabin Q4W-haarassa ja afliberseptin Q4W-haarassa verrannollinen viikkoon 24 saakka. Nämä tulokset säilyivät viikkoon 72 saakka, potilaiden siirryttyä farisimabin joustavaan annosteluun enintään Q16W-antovälillä.
Kliininen teho ja turvallisuus
Kostea silmänpohjan ikärappeuma
Farisimabin turvallisuutta ja tehoa kosteaa silmänpohjan ikärappeumaa sairastaville potilaille arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, vertailuvalmisteella kontrolloidussa, kaksi vuotta kestäneessä hoitojen vähintään samanveroisuutta (non-inferiority) selvittäneessä monikeskustutkimuksessa (TENAYA ja LUCERNE). Tutkimuksiin otettiin mukaan yhteensä 1329 potilasta, joista 1135 potilasta (85 %) oli tutkimuksissa mukana 112 viikon ajan. Yhteensä 1326 potilasta sai vähintään yhden annoksen (664 sai farisimabiannoksen). Potilaiden ikä oli 50–99 vuotta, ja keskimääräinen ikä oli 75,9 vuotta (keskihajonta 8,6 vuotta).
Potilaat satunnaistettiin kummassakin tutkimuksessa suhteessa 1:1 toiseen kahdesta hoitohaarasta:
- farisimabi 6 mg enintään 16 viikon välein aluksi neljän kuukausittain annetun annoksen jälkeen (enintään Q16W)
- aflibersepti 2 mg 8 viikon välein aluksi kolmen kuukausittain annetun annoksen jälkeen (Q8W).
Neljä ensimmäistä annosta annettiin kuukauden välein (viikot 0, 4, 8 ja 12), jonka jälkeen potilaat satunnaistettiin farisimabihaaraan, jossa he saivat hoitoa 16 viikon välein (Q16W), 12 viikon välein (Q12W) tai 8 viikon välein (Q8W) tautiaktiivisuusarvion perusteella. Tautiaktiivisuusarvio tehtiin viikoilla 20 ja 24, ja se perustui objektiivisiin ennalta määriteltyihin kriteereihin parhaan laseilla korjatun näöntarkkuuden (BCVA, best corrected visual acuity) ja anatomian (CST) muutoksiin sekä hoitavan lääkärin kliiniseen arvioon makulaverenvuodon tai hoitoa vaativan silmänpohjan ikärappeuman ilmaantumisesta (vain viikolla 24). Potilaat saivat hoitoa viikkoon 60 saakka saman määritellyn antovälin mukaisesti ilman lisähoitoa. Farisimabihaaran potilaat siirtyivät viikosta 60 alkaen joustavaan annosteluun, jossa hoitoväliä voitiin pidentää enintään 4 viikkoa kerrallaan (Q16W-antoväliin asti) tai lyhentää enintään 8 viikkoa (Q8W-antoväliin asti). Päätös antovälin pidentämisestä tai lyhentämisestä perustui ennalta määritettyjen visuaalisten (BCVA) ja anatomisten (CST ja makulaverenvuoto) taudin aktiivisuuskriteereiden automatisoituun objektiiviseen arviointiin. Afliberseptihaaran potilaat saivat hoitoa Q8W-antovälillä koko tutkimusjakson ajan. Kummankin tutkimuksen kesto oli 112 viikkoa.
Tulokset
Kummassakin tutkimuksessa todettiin ensisijaista päätetapahtumaa koskeva teho, joksi määriteltiin viikkojen 40, 44 ja 48 käynneillä lähtötilanteesta todetun parhaan laseilla korjatun näöntarkkuuden (BCVA) muutoksen keskiarvo ETDRS-kirjaimina (Early Treatment Diabetic Retinopathy Study) pisteytettynä (taulukko 2 ja taulukko 3). Kummassakin tutkimuksessa farisimabihoitoa enintään 16 viikon välein saaneiden potilaiden parhaan laseilla korjatun näöntarkkuuden keskimääräinen muutos lähtötilanteessa yhden vuoden aikapisteessä oli vähintään samanveroinen (non-inferior) niihin potilaisiin nähden, jotka saivat afliberseptiä Q8W yhden vuoden aikapisteessä. Näöntarkkuuden paraneminen säilyi viikon 112 aikapisteeseen saakka. Parhaan laseilla korjatun näöntarkkuuden paraneminen lähtötilanteesta viikon 112 aikapisteessä esitetään kuvassa 1.
Potilaiden osuudet kunkin antovälin osalta TENAYA- ja LUCERNE-tutkimusten viikolla 112 olivat:
- Q16W 59 % (TENAYA) ja 67 % (LUCERNE)
- Q12W 15 % (TENAYA) ja 14 % (LUCERNE)
- Q8W 26 % (TENAYA) ja 19 % (LUCERNE)
Taulukko 2. Tehoa koskevat hoitotulokset TENAYA-tutkimuksen ensisijaisen päätetapahtuman käynneilläaja 2 vuoden aikapisteessäb
| Tehoa koskevat hoitotulokset | TENAYA | |||
| 1. vuosi | 2. vuosi | |||
Farisimabi enintään Q16W N = 334 | Aflibersepti Q8W N = 337 | Farisimabi enintään Q16W N = 334 | Aflibersepti Q8W N = 337 | |
| Keskimääräinen BCVAn muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (95 %:n luottamusväli) | 5,8 (4,6–7,1) | 5,1 (3,9–6,4) | 3,7 (2,1–5,4) | 3,3 (1,7–4,9) |
| Ero keskimääräisenä muutoksena, LS (95 %:n luottamusväli) | 0,7 (-1,1–2,5) | 0,4 (-1,9–2,8) | ||
| Niiden potilaiden osuus, joilla ≥ 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 20,0 % (15,6–24,4 %) | 15,7 % (11,9–19,6 %) | 22,5 % (17,8–27,2 %) | 16,9 % (12,7–21,1 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | 4,3 % (-1,6–10,1 %) | 5,6 % (-0,7–11,9 %) | ||
| Niiden potilaiden osuus, jotka välttivät ≥ 15 kirjaimen huononemisen lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 95,4 % (93,0–97,7 %) | 94,1 % (91,5–96,7 %) | 92,1 % (89,1–95,1 %) | 88,6 % (85,1–92,2 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | 1,3 % (-2,2–4,8 %) | 3,4 % (-1,2–8,1 %) | ||
aViikkojen 40, 44 ja 48 keskiarvo; bViikkojen 104, 108 ja 112 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Taulukko 3. Tehoa koskevat hoitotulokset LUCERNE-tutkimuksen ensisijaisen päätetapahtuman käynneilläa ja 2 vuoden aikapisteessäb
| Tehoa koskevat hoitotulokset | LUCERNE | |||
| 1. vuosi | 2. vuosi | |||
Farisimabi enintään Q16W N = 331 | Aflibersepti Q8W N = 327 | Farisimabi enintään Q16W N = 331 | Aflibersepti Q8W N = 327 | |
| Keskimääräinen BCVAn muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (95 %:n luottamusväli) | 6,6 (5,3–7,8) | 6,6 (5,3–7,8) | 5,0 (3,4–6,6) | 5,2 (3,6–6,8) |
| Ero keskimääräisenä muutoksena, LS (95 %:n luottamusväli) | 0,0 (-1,7–1,8) | -0,2 (-2,4–2,1) | ||
| Niiden potilaiden osuus, joilla ≥ 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 20,2 % (15,9–24,6 %) | 22,2 % (17,7–26,8 %) | 22,4 % (17,8–27,1 %) | 21,3 % (16,8–25,9 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | -2,0 % (-8,3–4,3 %) | 1,1 % (-5,4–7,6 %) | ||
| Niiden potilaiden osuus, jotka välttivät ≥ 15 kirjaimen huononemisen lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 95,8 % (93,6–98,0 %) | 97,3 % (95,5–99,1 %) | 92,9 % (90,1–95,8 %) | 93,2 % (90,2–96,2 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | -1,5 % (-4,4–1,3 %) | -0,2 % (-4,4–3,9 %) | ||
aViikkojen 40, 44 ja 48 keskiarvo; bViikkojen 104, 108 ja 112 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Kuva 1. Näöntarkkuuden keskimääräinen muutos lähtötilanteesta 2 vuoden aikapisteeseen (viikkoon 112) saakka; TENAYA- ja LUCERNE-tutkimusten yhdistetyt tiedot
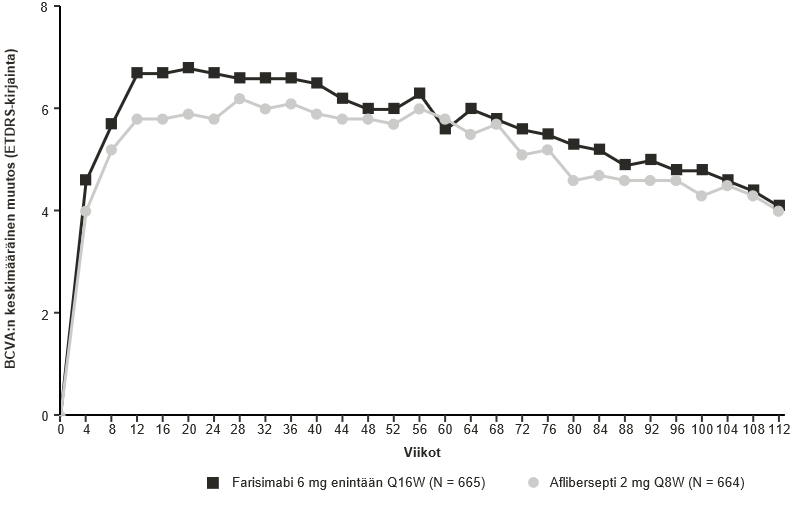
Sekä TENAYA- että LUCERNE-tutkimuksessa parhaan laseilla korjatun näöntarkkuuden ja verkkokalvon keskikuopan (keskikentän) paksuuden paraneminen lähtötilanteesta oli viikolla 60 verrannollista näissä kahdessa hoitohaarassa ja yhdenmukaista viikkoon 48 nähden.
Viikolla 60 sekä TENAYA- että LUCERNE-tutkimuksessa 46 %:lla potilaista hoidon antoväli oli Q16W. Näistä potilaista 69 % jatkoi kummassakin tutkimuksessa Q16W-antovälillä viikkoon 112 saakka eikä antoväliä lyhennetty.
Viikolla 60 TENAYA-tutkimuksessa 80 %:lla potilaista ja LUCERNE-tutkimuksessa 78 %:lla potilaista antoväli oli ≥ Q12W (Q16W tai Q12W). Näistä potilaista 67 % (TENAYA) ja 75 % (LUCERNE) jatkoi ≥ Q12W‑antovälillä viikkoon 112 saakka eikä antoväliä lyhennetty alle Q12W-antovälin.
Viikolla 60 sekä TENAYA- että LUCERNE-tutkimuksessa 33 %:lla potilaista antoväli oli Q12W. Näistä potilaista 3,2 % (TENAYA) ja 0 % (LUCERNE) jatkoi ≥ Q12W‑antovälillä viikkoon 112 saakka.
Viikolla 60 TENAYA-tutkimuksessa 20 %:lla ja LUCERNE-tutkimuksessa 22 %:lla antoväli oli Q8W. Näistä potilaista 34 % (TENAYA) ja 30 % (LUCERNE) jatkoi hoitoa Q8W-antovälillä viikkoon 112 saakka.
Tehon tulokset kummankin tutkimuksen kaikissa arvioitavissa olleissa alaryhmissä (esim. ikä, sukupuoli, etninen tausta, lähtötilanteen näöntarkkuus, leesiotyyppi, leesion koko) ja yhdistetyssä analyysissä olivat yhdenmukaisia koko potilasjoukkojen tulosten kanssa.
Tutkimuksissa farisimabi enintään Q16W-haarassa todettiin ennalta määritellyssä tehon päätetapahtumassa eli NEI VFQ-25 (National Eye Institute Visual Function Questionnaire) ‑yhdistelmäpisteiden keskimääräisessä muutoksessa lähtötilanteesta viikkoon 48 paranemista, joka oli verrannollista afliberseptiin Q8W nähden ja ylitti 4 pisteen raja-arvon. Näiden muutosten suuruusluokka vastaa parhaan laseilla korjatun näöntarkkuuden (BCVA) paranemista 15 kirjaimella.
Tutkittavassa silmässä viikkoon 112 mennessä havaittujen silmiin liittyvien haittavaikutusten ilmaantuvuus oli farisimabihaarassa 53,9 % ja afliberseptihaarassa 52,1 % ja silmiin liittymättömien haittavaikutusten ilmaantuvuus oli farisimabihaarassa 73,3 % ja afliberseptihaarassa 74,3 % (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Diabeettinen makulaturvotus
Farisimabin turvallisuutta ja tehoa diabeettista makulaturvotusta sairastavien potilaiden hoidossa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivisella vertailuvalmisteella kontrolloidussa, kaksi vuotta kestäneessä hoitojen vähintään samanveroisuutta (non-inferiority) selvittäneessä monikeskustutkimuksessa (YOSEMITE ja RHINE). Näihin kahteen tutkimukseen otettiin mukaan yhteensä 1 891 potilasta, joista 1 622 potilasta (86 %) oli tutkimuksissa mukana 100 viikon ajan. Yhteensä 1 887 potilasta sai vähintään yhden annoksen viikkoon 56 mennessä (1 262 sai farisimabihoitoa). Potilaiden ikä oli 24–91 vuotta, ja keskimääräinen ikä oli 62,2 vuotta (keskihajonta 9,9 vuotta). Koko potilasjoukossa oli mukana sekä verisuonen endoteelin kasvutekijän (VEGF) estäjillä aiemmin hoitamattomia potilaita (78 %) että potilaita, jotka olivat saaneet ennen tutkimukseen osallistumista hoitoa endoteelin kasvutekijän estäjällä (22 %). Potilaat satunnaistettiin kummassakin tutkimuksessa suhteessa 1:1:1 yhteen kolmesta hoitohaarasta:
- farisimabi 6 mg Q8W hoitoväli, aloitusannos neljän viikon välein 6 ensimmäistä annosta.
- farisimabi 6 mg joustavalla annostuksella enintään Q16W; aloitusannos neljän viikon välein 4 ensimmäistä annosta, ensimmäisten neljän kuukausittaisen annoksen jälkeen hoitoa annettiin 4, 8, 12 tai 16 viikon välein.
- aflibersepti 2 mg Q8W hoitoväli, aloitusannos neljän viikon välein 5 ensimmäistä annosta.
Hoitohaarassa, jossa käytettiin joustavaa Q16W-annostusta, antoväliä pidennettiin vakioidusti (standardized treat-and-extend approach). Antoväliä voitiin pidentää 4 viikkoa kerrallaan tai lyhentää 4 tai 8 viikkoa kerrallaan näkökyvyn ja/tai anatomisten hoitotulosten perusteella käyttämällä vain tutkimuslääkkeen antokäynneillä saatuja tietoja.
Tulokset
Kummassakin tutkimuksessa todettiin ensisijaista päätetapahtumaa koskeva teho, joksi määriteltiin 1 vuoden aikapisteessä (viikkojen 48, 52 ja 56 käyntien keskiarvo) lähtötilanteesta todetun parhaan laseilla korjatun näöntarkkuuden (BCVA) muutoksen keskiarvo ETDRS-kirjaimina pisteytettynä. Kummassakin tutkimuksessa farisimabihoitoa enintään Q16W saaneiden potilaiden parhaan laseilla korjatun näöntarkkuuden keskimääräinen muutos lähtötilanteesta oli 1 vuoden aikapisteessä vähintään samanveroinen (non-inferior) niihin potilaisiin nähden, jotka saivat afliberseptiä Q8W, ja näkökyvyssä todettu paraneminen säilyi kahden vuoden ajan.
Alkuvaiheessa annettujen neljän kuukausittaisen annoksen jälkeen farisimabihoitoa joustavalla enintään Q16W-annostuksella saaneen haaran potilaille voitiin antaa yhteensä vähintään 6 ja enintään 21 injektiota 96 viikon aikana. Viikolla 52 YOSEMITE-tutkimuksessa 74 % ja RHINE-tutkimuksessa 71 % potilaista, jotka saivat farisimabihoitoa joustavalla Q16W-annoksella, oli saavuttanut Q16W- tai Q12W-antovälin (Q16W 53 % YOSEMITE-tutkimuksessa ja 51 % RHINE-tutkimuksessa, Q12W 21 % YOSEMITE-tutkimuksessa ja 20 % RHINE-tutkimuksessa). Näistä potilaista 75 % (YOSEMITE) ja 84 % (RHINE) jatkoi ≥ Q12W‑annostuksella eikä antoväliä lyhennetty viikkoon 96 mennessä alle Q12W-antovälin; Q16W-antoväliä viikolla 52 käyttäneistä potilaista 70 % (YOSEMITE) ja 82 % (RHINE) jatkoi Q16W-antovälillä eikä antoväliä lyhennetty viikkoon 96 mennessä. Viikolla 96 kummassakin tutkimuksessa 78 % potilaista, jotka saivat farisimabihoitoa joustavalla enintään Q16W-annoksella, oli saavuttanut Q16W- tai Q12W-antovälin (Q16W 60 % YOSEMITE-tutkimuksessa ja 64 % RHINE-tutkimuksessa, Q12W 18 % YOSEMITE-tutkimuksessa ja 14 % RHINE-tutkimuksessa). YOSEMITE-tutkimuksessa 4 %:lla ja RHINE-tutkimuksessa 6 %:lla potilaista annostukseksi oli suurennettu Q8W ja he jatkoivat hoitoa antovälillä ≤ Q8W viikkoon 96 saakka; YOSEMITE-tutkimuksessa 3 % ja RHINE-tutkimuksessa 5 % sai hoitoa antovälillä vain Q4W viikkoon 96 saakka.
Yksityiskohtaiset tulokset YOSEMITE- ja RHINE-tutkimusten analyyseistä luetellaan alla taulukossa 4, taulukossa 5 sekä kuvassa 2.
Taulukko 4. Tehoa koskevat hoitotulokset YOSEMITE-tutkimuksen 1 vuoden aikapisteessä ensisijaisen päätetapahtuman käynneilläa ja 2 vuoden aikapisteessäb
| Tehoa koskevat hoitotulokset | YOSEMITE | |||||
| 1 vuosi | 2 vuotta | |||||
Farisimabi Q8W N = 315 | Farisimabi joustava enintään Q16W N = 313 | Aflibersepti Q8W N = 312 | Farisimabi Q8W N = 315 | Farisimabi joustava enintään Q16W N = 313 | Aflibersepti Q8W N = 312 | |
| BCVAn keskimääräinen muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (1 vuoden 97,5 %:n luottamusväli ja 2 vuoden 95 %:n luottamusväli) | 10,7 (9,4–12,0) | 11,6 (10,3–12,9) | 10,9 (9,6–12,2) | 10,7 (9,4–12,1) | 10,7 (9,4–12,1) | 11,4 (10,0–12,7) |
| Ero keskimääräisenä muutoksena, LS (1 vuoden 97,5 %:n luottamusväli ja 2 vuoden 95 %:n luottamusväli) | -0,2 (-2,0–1,6) | 0,7 (-1,1–2,5) | -0,7 (-2,6–1,2) | -0,7 (-2,5–1,2) | ||
| Niiden potilaiden osuus, joilla BCVAn vähintään 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 1 ja 2 vuoden 95 %:n luottamusväli) | 29,2 % (23,9–34,5 %) | 35,5 % (30,1–40,9 %) | 31,8 % (26,6–37,0 %) | 37,2 % (31,4–42,9 %) | 38,2 % (32,8–43,7 %) | 37,4 % (31,7–43,0 %) |
| Ero CMH-painotuksessa, % (1 ja 2 vuoden 95 %:n luottamusväli) | -2,6 % (-10,0–4,9 %) | 3,5 % (-4,0–11,1 %) | -0,2 % (-8,2–7,8 %) | 0,2 % (-7,6–8,1 %) | ||
| Niiden potilaiden osuus, jotka välttivät BCVAn vähintään 15 kirjaimen huononemisen lähtötilanteesta (CMH-painotettu osuus, 1 ja 2 vuoden 95 %:n luottamusväli) | 98,1 % (96,5–99,7 %) | 98,6 % (97,2–100,0 %) | 98,9 % (97,6–100,0 %) | 97,6 % (95,7–99,5 %) | 97,8 % (96,1–99,5 %) | 98,0 % (96,2–99,7 %) |
| Ero CMH-painotuksessa, % (1 ja 2 vuoden 95 %:n luottamusväli) | -0,8 % (-2,8–1,3 %) | -0,3 % (-2,2–1,5 %) | -0,4 % (-2,9–2,2 %) | -0,2 % (-2,6–2,2 %) | ||
aViikkojen 48, 52 ja 56 keskiarvo; bViikkojen 92, 96 ja 100 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Huom.: Afliberseptihaaran CMH-painotus (%) esitetty farisimabihoidon 8 viikon välein ja afliberseptin vertailusta, mutta vastaava CMH-painotus (%) farisimabihoidon joustavina annoksina ja afliberseptin vertailusta on samankaltainen kuin edellä on esitetty.
Taulukko 5. Tehoa koskevat hoitotulokset RHINE-tutkimuksen 1 vuoden aikapisteessä ensisijaisen päätetapahtuman käynneilläa ja 2 vuoden aikapisteessäb
| Tehoa koskevat hoitotulokset | RHINE | ||||||
| 1 vuosi | 2 vuotta | ||||||
Farisimabi Q8W N = 317 | Farisimabi joustava enintään Q16W N = 319 | Aflibersepti Q8W N = 315 | Farisimabi Q8W N = 317 | Farisimabi joustava enintään Q16W N = 319 | Aflibersepti Q8W N = 315 | ||
| BCVAn keskimääräinen muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (1 vuoden 97,5 %:n luottamusväli ja 2 vuoden 95 %:n luottamusväli) | 11,8 (10,6–13,0) | 10,8 (9,6–11,9) | 10,3 (9,1–11,4) | 10,9 (9,5–12,3) | 10,1 (8,7–11,5) | 9,4 (7,9–10,8) | |
| Ero keskimääräisenä muutoksena, LS (1 vuoden 97,5 %:n luottamusväli ja 2 vuoden 95 %:n luottamusväli) | 1,5 (-0,1–3,2) | 0,5 (-1,1–2,1) | 1,5 (-0,5–3,6) | 0,7 (-1,3–2,7) | |||
| Niiden potilaiden osuus, joilla BCVAn vähintään 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 1 ja 2 vuoden 95 %:n luottamusväli) | 33,8 % (28,4–39,2 %) | 28,5 % (23,6–33,3 %) | 30,3 % (25,0–35,5 %) | 39,8 % (34,0–45,6 %) | 31,1 % (26,1–36,1 %) | 39,0 % (33,2–44,8 %) | |
| Ero CMH-painotuksessa, % (1 ja 2 vuoden 95 %:n luottamusväli) | 3,5 % (-4,0–11,1 %) | -2,0 % (-9,1–5,2 %) | 0,8 % (-7,4–9,0 %) | -8 % (‑15,7 – 0,3 %) | |||
| Niiden potilaiden osuus, jotka välttivät BCVAn vähintään 15 kirjaimen huononemisen lähtötilanteesta (CMH-painotettu osuus, 1 ja 2 vuoden 95 %:n luottamusväli) | 98,9 % (97,6–100,0 %) | 98,7 % (97,4–100,0 %) | 98,6 % (97,2–99,9 %) | 96,6 % (94,4–98,8 %) | 96,8 % (94,8–98,9 %) | 97,6 % (95,7–99,5 %) | |
| Ero CMH-painotuksessa, % (1 ja 2 vuoden 95 %:n luottamusväli) | 0,3 % (-1,6–2,1 %) | 0,0 % (-1,8–1,9 %) | -1,0 % (-3,9–1,9 %) | -0,7 % (-3,5–2,0 %) | |||
aViikkojen 48, 52 ja 56 keskiarvo; bViikkojen 92, 96 ja 100 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Huom.: Afliberseptihaaran CMH-painotus (%) esitetty farisimabihoidon 8 viikon välein ja afliberseptin vertailusta, mutta vastaava CMH-painotus (%) farisimabihoidon joustavina annoksina ja afliberseptin vertailusta on samankaltainen kuin edellä on esitetty.
Kuva 2. Näöntarkkuuden keskimääräinen muutos lähtötilanteesta vuoteen 2 (viikkoon 100); YOSEMITE- ja RHINE-tutkimusten yhdistetyt tiedot
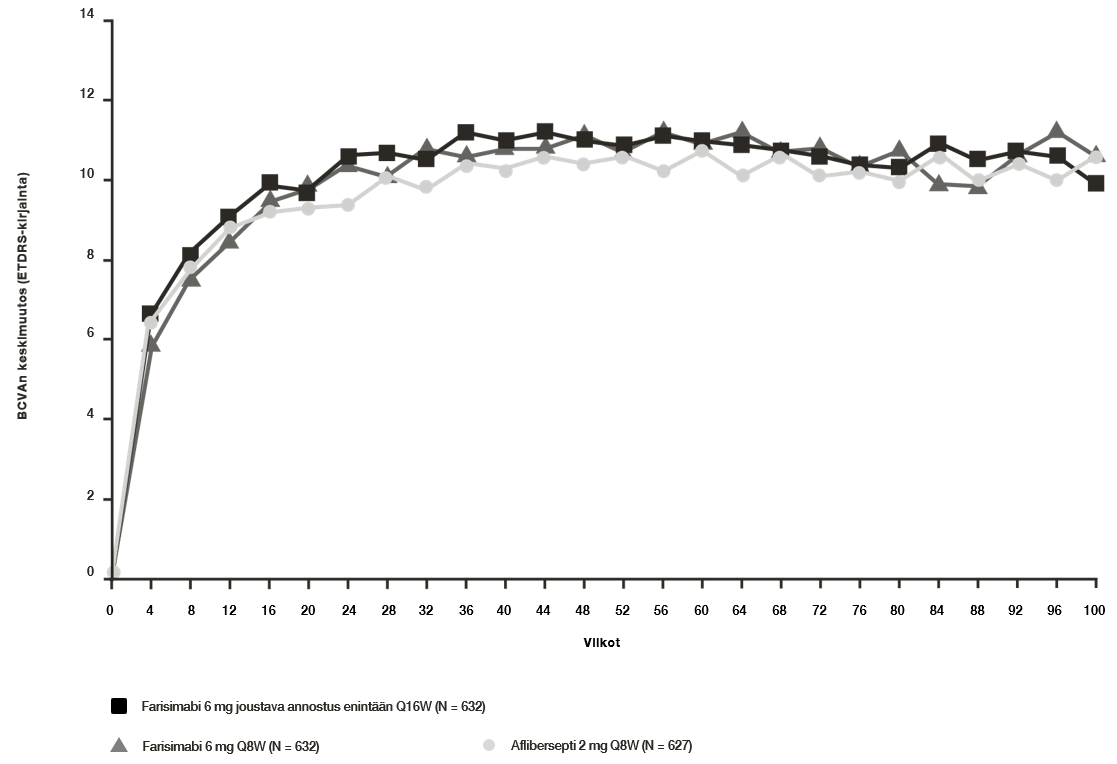
Kussakin tutkimuksessa tehon tulokset potilailla, jotka eivät olleet saaneet hoitoa verisuonen endoteelin kasvutekijän (VEGF) estäjillä ennen tutkimukseen osallistumista, ja kaikissa muissa arvioitavissa olleissa alaryhmissä (esim. iän, sukupuolen, etnisen taustan, lähtötilanteen HbA1c-pitoisuuden, lähtötilanteen näöntarkkuuden mukaan) olivat verrannolliset koko potilasjoukkojen tulosten kanssa.
Tutkimuksissa farisimabin Q8W- ja joustavalla Q16W-annosteluilla todettiin lähtötilanteesta viikkoon 52 keskimääräisen muutoksen ennalta määritellyssä tehon päätetapahtumassa paranemista NEI VFQ-25 -yhdistelmäpisteissä. Tulokset olivat verrattavissa Q8W-afliberseptihoitoon ja ylittivät 4 pisteen raja-arvon. Farisimabin Q8W- ja joustavalla Q16W-annosteluilla todettiin lähtötilanteesta viikkoon 52 kliinisesti merkityksellistä paranemista myös ennalta määritettyjen tehon päätetapahtumien muutoksessa; näitä olivat lähinäköä vaativien toimintojen, kaukonäköä vaativien toimintojen ja ajamista koskevat NEI VFQ-25 ‑pisteet, jotka olivat verrannolliset afliberseptin Q8W-antovälin kanssa. Näiden muutosten suuruusluokka vastaa BCVAn paranemista 15 kirjaimella. Samansuuruisella osalla farisimabihoitoa Q8W, farisimabihoitoa joustavalla enintään Q16W-annostelulla ja afliberseptin Q8W-annostelulla saaneista potilaista todettiin viikolla 52 ennalta määritellyssä tehon päätetapahtumassa eli NEI VFQ-25 -yhdistelmäpisteissä kliinisesti merkityksellinen ≥ 4 pisteen paraneminen lähtötilanteesta. Nämä tulokset säilyivät viikkoon 100 saakka.
Muu keskeinen tehon hoitotulos diabeettista makulaturvotusta koskeneissa tutkimuksissa oli ETDRS-DRSS-pisteiden (Early Treatment Diabetic Retinopathy Study Diabetic Retinopathy Severity Scale) muutos lähtötilanteesta viikkoon 52. YOSEMITE - ja RHINE-tutkimuksiin mukaan otetuista 1 891 potilaasta diabeettista retinopatiaa koskevien päätetapahtumien osalta arvioitavissa oli 708 (YOSEMITE) ja 720 potilasta (RHINE).
Lähtötilanteen ETDRS-DRSS-pisteet olivat 10–71.
Valtaosalla potilaista (noin 60 %:lla) oli lähtötilanteessa keskivaikea tai vaikea ei-proliferatiivinen diabeettinen retinopatia (DRSS 43/47/53).
Niiden potilaiden osuus, joilla todettiin ETDRS-DRSS-tutkimuksessa ≥ 2 askelen ja ≥ 3 askelen paranemista lähtötilanteesta viikolla 52 ja viikolla 96 esitetään jäljempänä taulukossa 6 ja taulukossa 7.
Taulukko 6. Niiden potilaiden osuus YOSEMITE-tutkimuksessa, joilla ETDRS-DRSS-tutkimuksessa ≥ 2 askeleen ja ≥ 3 askeleen parannus lähtötilanteesta viikolla 52 ja viikolla 96 (diabeettisen retinopatian suhteen arvioitavissa ollut potilasjoukko)
| YOSEMITE | ||||||
| 52 viikkoa | 96 viikkoa | |||||
Farisimabi Q8W n = 237 | Farisimabi joustava enintään Q16W n = 242 | Aflibersepti Q8W n = 229 | Farisimabi Q8W n = 220 | Farisimabi joustava enintään Q16W n = 234 | Aflibersepti Q8W n = 221 | |
Niiden potilaiden osuus, joilla ETDRS-DRSS-tutkimuksessa ≥ 2 askeleen parannus lähtötilanteesta (CMH-painotettu osuus) | 46,0 % | 42,5 % | 35,8 % | 51,4 % | 42,8 % | 42,2 % |
| Painotettu ero (1 vuoden 97,5 %:n luottamusväli, 2 vuoden 95 %:n luottamusväli) | 10,2 % (0,3–20,0 %) | 6,1 % (‑3,6–15,8 %) | 9,1 % (0,0–18,2 %) | 0,0 % (-8,9–8,9 %) | ||
| Niiden potilaiden osuus, joilla ETDRS-DRSS-tutkimuksessa ≥ 3 askeleen parannus lähtötilanteesta (CMH-painotettu osuus) | 16,8 % | 15,5 % | 14,7 % | 22,4 % | 14,6 % | 20,9 % |
| Painotettu ero (1 ja 2 vuoden 95 %:n luottamusväli) | 2,1 % (-4,3–8,6 %) | 0,6 % (-5,8–6,9 %) | 1,5 % (-6,0–9,0 %) | -6,7 % (-13,6–0,1 %) | ||
ETDRS-DRSS: Early Treatment Diabetic Retinopathy Study Diabetic Retinopathy Severity Scale
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Huom.: Afliberseptihaaran CMH-painotus (%) esitetty farisimabihoidon 8 viikon välein ja afliberseptin vertailusta, mutta vastaava CMH-painotus (%) farisimabihoidon joustavalla annostuksella ja afliberseptin vertailusta on samankaltainen kuin edellä on esitetty.
Taulukko 7. Niiden potilaiden osuus RHINE-tutkimuksessa, joilla ETDRS-DRSS-tutkimuksessa ≥ 2 askeleen ja ≥ 3 askeleen parannus lähtötilanteesta viikolla 52 ja viikolla 96 (diabeettisen retinopatian suhteen arvioitavissa ollut potilasjoukko)
| RHINE | ||||||
| 52 viikkoa | 96 viikkoa | |||||
Farisimabi Q8W n = 231 | Farisimabi joustava enintään Q16W n = 251 | Aflibersepti Q8W n = 238 | Farisimabi Q8W n = 214 | Farisimabi joustava enintään Q16W n = 228 | Aflibersepti Q8W n = 203 | |
Niiden potilaiden osuus, joilla ETDRS-DRSS-tutkimuksessa ≥ 2 askeleen parannus lähtötilanteesta (CMH-painotettu osuus) | 44,2 % | 43,7 % | 46,8 % | 53,5 % | 44,3 % | 43,8 % |
| Painotettu ero (1 vuoden 97,5 %:n luottamusväli, 2 vuoden 95 %:n luottamusväli) | -2,6 % (‑12,6–7,4 %) | -3,5 % (‑13,4–6,3 %) | 9,7 % (0,4–19,1 %) | 0,3 % (-8,9–9,5 %) | ||
| Niiden potilaiden osuus, joilla ETDRS-DRSS-tutkimuksessa ≥ 3 askeleen parannus lähtötilanteesta (CMH-painotettu osuus) | 16,7 % | 18,9 % | 19,4 % | 25,1 % | 19,3 % | 21,8 % |
| Painotettu ero (1 ja 2 vuoden 95 %:n luottamusväli) | -0,2 % (-5,8–5,3 %) | -1,1 % (-8,0–5,9 %) | 3,3 % (-4,6–11,3 %) | -2,7 % (-10,2–4,8 %) | ||
ETDRS-DRSS: Early Treatment Diabetic Retinopathy Study Diabetic Retinopathy Severity Scale
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Huom.: Afliberseptihaaran CMH-painotus (%) esitetty farisimabihoidon 8 viikon välein ja afliberseptin vertailusta, mutta vastaava CMH-painotus (%) farisimabihoidon joustavalla annostuksella ja afliberseptin vertailusta on samankaltainen kuin edellä on esitetty.
Kummassakin tutkimuksessa hoidon teho arvioitavissa olleissa alaryhmissä (esim. aiemman verisuonen endoteelin kasvutekijää (VEGF) estävän hoidon, iän, sukupuolen, etnisen taustan, lähtötilanteen HbA1c-pitoisuuden ja lähtötilanteen näöntarkkuuden mukaan) oli yleensä yhdenmukainen koko potilasjoukon tulosten kanssa.
Hoitovaikutus alaryhmissä oli erilainen diabeettisen retinopatian lähtötilanteen vaikeusasteen mukaan, ja DRSS-tutkimuksessa todettiin suurin ≥ 2 askeleen parannus niillä potilailla, joilla oli kohtalaisen vaikea-asteinen tai vaikea-asteinen ei-proliferatiivinen diabeettinen retinopatia, sillä kummankin tutkimuksen kaikissa hoitohaaroissa yhdenmukaisesti noin 90 %:lla potilaista todettiin tilan paranemista.
Tutkittavassa silmässä viikkoon 100 mennessä havaittujen silmiin liittyvien haittavaikutusten ilmaantuvuus oli farisimabi Q8W-haarassa 49,7 %, farisimabi enintään Q16W-haarassa 49,2 % ja aflibersepti Q8W-haarassa 45,4 % ja silmiin liittymättömien haittavaikutusten ilmaantuvuus oli farisimabi Q8W-haarassa 73,0 %, farisimabi enintään Q16W-haarassa 74,2 % ja aflibersepti Q8W-haarassa 75,7 % (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
RHONE-X-tutkimukseen otettiin mukaan 1 474 potilasta, jotka olivat aiemmin olleet joko YOSEMITE- tai RHINE-tutkimuksessa niiden päättymiseen saakka. RHONE-X oli 2 vuoden pituinen, monikeskustutkimuksena toteutettu pitkäkestoinen jatkotutkimus, jonka tarkoituksena oli arvioida yksilöllisellä hoitovälillä lasiaiseen annettavan 6 mg:n farisimabiannoksen pitkäaikaista turvallisuutta ja siedettävyyttä.
RHONE-X-tutkimuksessa havaittu farisimabin pitkäaikainen turvallisuusprofiili oli yhdenmukainen YOSEMITE- ja RHINE-tutkimusten havaintojen kanssa.
Verkkokalvon laskimotukos
Farisimabin turvallisuutta ja tehoa verkkokalvon haaralaskimotukoksen (BALATON) tai verkkokalvon keskuslaskimotukoksen (CRVO) / verkkokalvon keskuslaskimon päähaaratukoksen (HRVO) (COMINO) seurauksena kehittyneen makulaturvotuksen hoidossa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, 72 viikkoa kestäneessä monikeskustutkimuksessa. Aktiivisella vertailuvalmisteella kontrolloituja tietoja on saatavissa kuukauteen 6 saakka.
Näihin kahteen tutkimukseen otettiin mukaan yhteensä 1 282 potilasta (BALATON-tutkimukseen 553 potilasta ja COMINO-tutkimukseen 729 potilasta), ja 1 276 potilasta sai vähintään yhden annoksen viikkoon 24 mennessä (641 sai farisimabia). BALATON-tutkimuksessa potilaiden ikä oli 28–93 vuotta ja keskimääräinen ikä (keskihajonta) oli 64 (10,7) vuotta; COMINO-tutkimuksessa potilaiden ikä oli 22–100 vuotta ja keskimääräinen ikä (keskihajonta) oli 65 (13,2) vuotta.
BALATON-tutkimuksessa yhteensä 489 potilasta 553 satunnaistetusta potilaasta oli tutkimuksessa mukana viikon 72 aikapisteeseen saakka; 263 potilasta, jotka satunnaistettiin aluksi farisimabihoitoon (farisimabia aiemmin saaneet), ja 267 potilasta, jotka satunnaistettiin aluksi afliberseptihoitoon (afliberseptiä aiemmin saaneet), sai farisimabin joustavan annostelun jakson aikana vähintään yhden farisimabiannoksen.
COMINO-tutkimuksessa yhteensä 656 potilasta 729 satunnaistetusta potilaasta oli tutkimuksessa mukana 72 viikon aikapisteeseen saakka; 353 farisimabia aiemmin saanutta ja 342 afliberseptiä aiemmin saanutta potilasta sai farisimabin joustavan annostelun jakson aikana vähintään yhden farisimabiannoksen.
Potilaat satunnaistettiin kummassakin tutkimuksessa suhteessa 1:1 toiseen kahdesta tutkimushaarasta viikkoon 24 saakka
- farisimabi 6 mg Q4W hoitoväli; 6 peräkkäistä kuukausittaista annosta
- aflibersepti 2 mg Q4W hoitoväli; 6 peräkkäistä kuukausittaista annosta.
Alkuvaiheen 6 kuukausittaisen annoksen jälkeen 2 mg afliberseptiä aluksi saaneeseen haaraan satunnaistetut potilaat siirrettiin saamaan 6 mg farisimabia, jolloin heidän oli mahdollista saada 6 mg farisimabia joustavana annostuksena enintään Q16W-antovälillä, jolloin antoväliä voitiin pidentää 4 viikkoa kerrallaan tai lyhentää 4, 8 tai 12 viikolla näöntarkkuutta ja anatomista sairautta koskevien ennalta määriteltyjen aktiivisuuskriteerien automatisoidun objektiivisen arvioinnin perusteella.
Tulokset
Kummassakin tutkimuksessa todettiin ensisijaista päätetapahtumaa koskeva teho, joksi määriteltiin parhaassa laseilla korjatussa näöntarkkuudessa (BCVA) 24 viikon aikapisteessä todettu muutos lähtötilanteesta ETDRS-kirjaimina pisteytettynä. Kummassakin tutkimuksessa farisimabihoitoa Q4W saaneiden potilaiden parhaan laseilla korjatun näöntarkkuuden keskimääräinen muutos lähtötilanteesta oli vähintään samanveroinen (non-inferior) niihin potilaisiin nähden, jotka saivat afliberseptiä Q4W; tällainen näkökyvyn paraneminen säilyi viikkoon 72 saakka, potilaiden siirryttyä farisimabin joustavaan annosteluun enintään Q16W-antovälillä.
≥ Q12W-antovälin (Q16W tai Q12W) saavutti viikoilla 24–68 BALATON-tutkimuksessa 81,5 % farisimabia joustavalla annostuksella enintään Q16W-antovälillä saaneista potilaista ja COMINO-tutkimuksessa 74,0 % farisimabia joustavalla annostuksella enintään Q16W-antovälillä saaneista potilaista. Näistä potilaista 72,1 % (BALATON) ja 61,6 % (COMINO) sai vähintään yhden täyden Q12W-hoitosyklin ja jatkoi hoitoa viikkoon 68 saakka ≥ Q12W-antovälillä lyhentämättä antoväliä alle Q12W-antovälin; 1,2 % potilaista (BALATON) ja 2,5 % potilaista (COMINO) sai hoitoa viikkoon 68 saakka vain Q4W-antovälillä.
Tutkimuksissa farisimabi Q4W-haarassa todettiin ennalta määritellyssä tehon päätetapahtumassa eli NEI VFQ-25 ‑yhdistelmäpisteiden muutoksessa lähtötilanteesta viikkoon 24 paranemista, joka oli verrannollista afliberseptiin Q4W nähden. Farisimabi Q4W-haarassa todettiin ennalta määritellyssä tehon päätetapahtumassa eli NEI VFQ-25 ‑yhdistelmäpisteiden muutoksessa lähtötilanteesta viikkoon 24 myös lähinäköä vaativissa toiminnoissa ja kaukonäköä vaativissa toiminnoissa paranemista, joka oli verrannollista afliberseptiin Q4W nähden. Nämä tulokset säilyivät viikkoon 72 saakka, jolloin kaikki potilaat saivat farisimabia joustavalla annostuksella enintään Q16W-antovälillä.
Taulukko 8: Tehoa koskevat hoitotulokset BALATON-tutkimuksen 24 viikon aikapisteessä ensisijaisen päätetapahtuman käynneillä ja tutkimuksen päättyessäa
| Tehoa koskevat hoitotulokset | BALATON | |||
| 24 viikkoa | 72 viikkoaa | |||
Farisimabi Q4W N = 276 | Aflibersepti Q4W N = 277 | Farisimabi Q4W-antovälistä farisimabin joustavaan annostukseen N = 276 | Aflibersepti Q4W-antovälistä farisimabin joustavaan annostukseen N = 277 | |
| BCVA:n keskimääräinen muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (95 %:n luottamusväli) | 16,9 (15,7–18,1) | 17,5 (16,3–18,6) | 18,1 (16,9–19,4) | 18,8 (17,5–20,0) |
| Ero keskimääräisenä muutoksena, LS (95 %:n luottamusväli) | ‑0,6 (‑2,2–1,1) | |||
| Niiden potilaiden osuus, joilla vähintään 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 56,1 % (50,4–61,9 %) | 60,4 % (54,7–66,0 %) | 61,5 % (56,0–67,0 %) | 65,8 % (60,3–71,2 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | ‑4,3 % (‑12,3–3,8 %) | |||
aViikkojen 64, 68, 72 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Taulukko 9: Tehoa koskevat hoitotulokset COMINO-tutkimuksen 24 viikon aikapisteessä ensisijaisen päätetapahtuman käynneillä ja tutkimuksen päättyessäa
| Tehoa koskevat hoitotulokset | COMINO | |||
| 24 viikkoa | 72 viikkoaa | |||
Farisimabi Q4W N = 366 | Aflibersepti Q4W N = 363 | Farisimabi Q4W-antovälistä farisimabin joustavaan annostukseen N = 366 | Aflibersepti Q4W-antovälistä farisimabin joustavaan annostukseen N = 363 | |
| BCVA:n keskimääräinen muutos lähtötilanteesta ETDRS-kirjaimin pisteytettynä (95 %:n luottamusväli) | 16,9 (15,4–18,3) | 17,3 (15,9–18,8) | 16,9 (15,2–18,6) | 17,1 (15,4–18,8) |
| Ero keskimääräisenä muutoksena, LS (95 %:n luottamusväli) | ‑0,4 (‑2,5–1,6) | |||
| Niiden potilaiden osuus, joilla vähintään 15 kirjaimen parannus lähtötilanteesta (CMH-painotettu osuus, 95 %:n luottamusväli) | 56,6 % (51,7–61,5 %) | 58,1 % (53,3–62,9 %) | 57,6 % (52,8–62,5 %) | 59,5 % (54,7–64,3 %) |
| Ero CMH-painotuksessa, % (95 %:n luottamusväli) | ‑1,5 % (‑8,4–5,3 %) | |||
aViikkojen 64, 68, 72 keskiarvo
BCVA: paras laseilla korjattu näöntarkkuus
ETDRS: Early Treatment Diabetic Retinopathy Study
LS: pienimmät neliösummat
CMH: Cochran–Mantel–Haenszelin menetelmä; tilastollinen testi, jolla muodostetaan arvio yhteydestä binaariseen hoitotulokseen ja jota käytetään luokkamuuttujien arviointiin.
Kuva 3: Näöntarkkuuden keskimääräinen muutos lähtötilanteesta viikkoon 72; BALATON-tutkimus
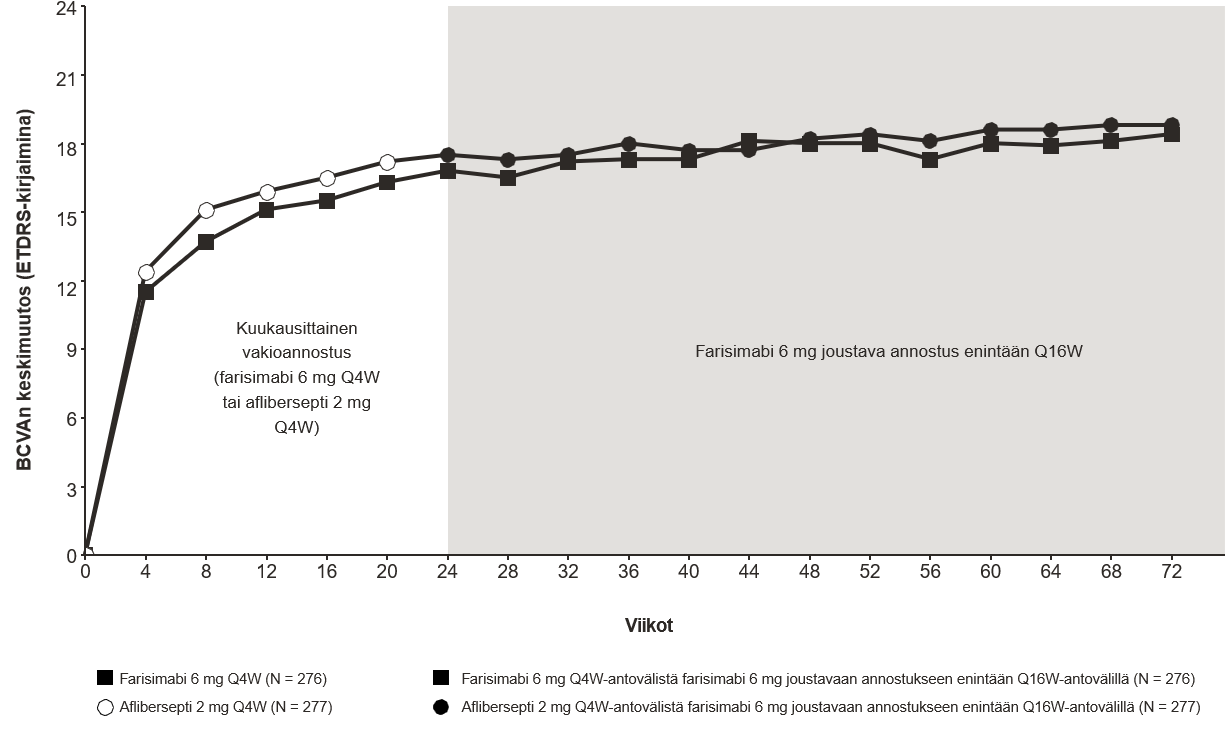
Farisimabihoito 6 mg:n joustavalla annostuksella enintään Q16W-antovälillä aloitettiin viikolla 24, mutta kaikki potilaat eivät saaneet farisimabia viikolla 24.
Kuva 4: Näöntarkkuuden keskimääräinen muutos lähtötilanteesta viikkoon 72; COMINO-tutkimus
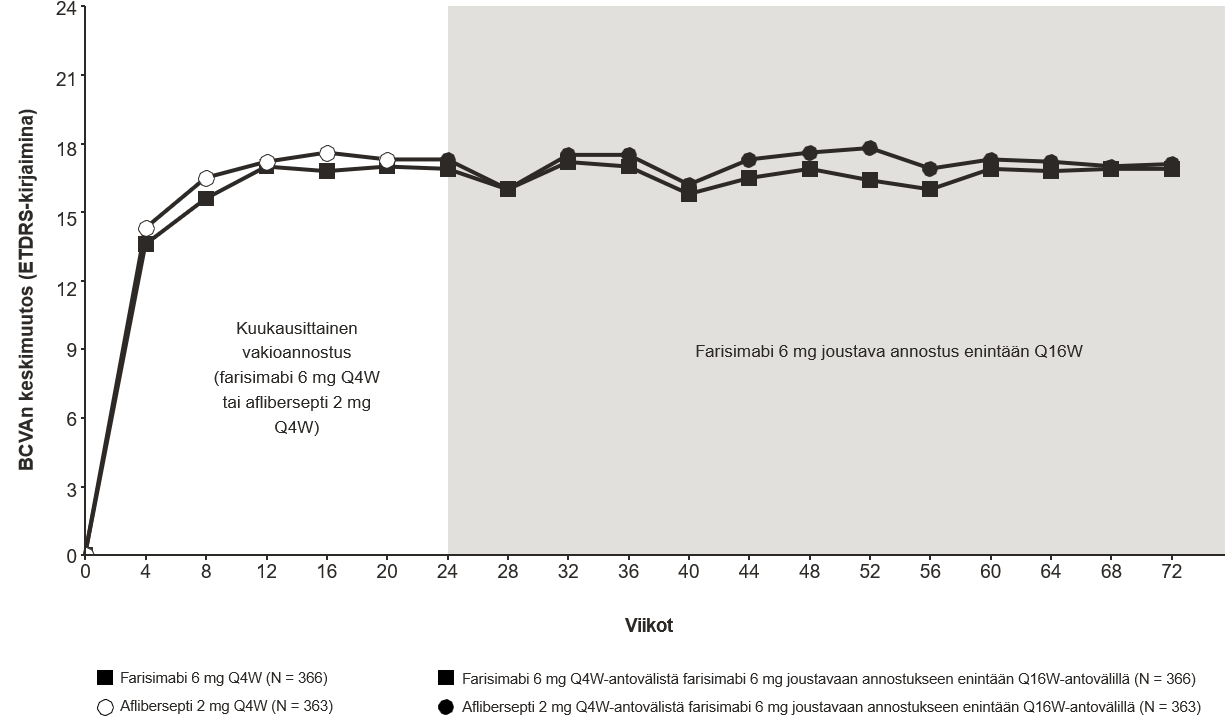
Farisimabihoito 6 mg:n joustavalla annostuksella enintään Q16W-antovälillä aloitettiin viikolla 24, mutta kaikki potilaat eivät saaneet farisimabia viikolla 24.
Viikkoon 24 mennessä silmiin liittyvien haittavaikutusten ilmaantuvuus tutkittavassa silmässä oli farisimabin Q4W-haarassa 20,1 % ja afliberseptin Q4W-haarassa 24,6 % ja silmiin liittymättömien haittavaikutusten ilmaantuvuus oli farisimabin Q4W-haarassa 32,9 % ja afliberseptin Q4W-haarassa 36,4 % (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset farisimabin käytöstä kostean silmänpohjan ikärappeuman, diabeettisen makulaturvotuksen ja verkkokalvon laskimotukoksen hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Farisimabi injektoidaan lasiaiseen, jotta silmään saadaan paikallinen vaikutus.
Imeytyminen ja jakautuminen
Populaatiofarmakokineettisen analyysin perusteella (kostea silmänpohjan ikärappeuma ja diabeettinen makulaturvotus mukaan lukien N = 2 246), vapaan (VEGF-A:han ja ANG2:een sitoutumattoman) farisimabin huippupitoisuus plasmassa (Cmax) saavutetaan arviolta noin 2 päivää annoksen jälkeen. Keskimääräiseksi (± keskihajonta) huippupitoisuudeksi (Cmax) plasmassa arvioidaan kosteaa silmänpohjan ikärappeumaa sairastavilla 0,23 (0,07) µg/ml ja diabeettista makulaturvotusta sairastavilla 0,22 (0,07) µg/ml. Toistettujen antokertojen jälkeen vapaan farisimabin pienimpien pitoisuuksien plasmassa ennustetaan olevan keskimäärin 0,002–0,003 µg/ml Q8W-annostelulla.
Farisimabin farmakokinetiikka annosvälillä 0,5–6 mg on suhteessa annokseen (Cmax- ja AUC-arvojen perusteella). Kuukausittaisessa annossa farisimabin ei havaittu kertyneen lasiaiseen eikä plasmaan.
Vapaan farisimabin huippupitoisuuksien plasmassa ennustetaan olevan noin 600 kertaa pienemmät kuin kammionesteessä ja 6000 kertaa pienemmät kuin lasiaisnesteessä. Sen vuoksi systeemiset farmakodynaamiset vaikutukset ovat epätodennäköisiä, mitä myös farisimabihoito kliinisissä tutkimuksissa tukee, sillä vapaan VEGF:n ja ANG2:n pitoisuuksissa plasmassa ei ollut merkittäviä muutoksia
Populaatiofarmakokineettinen analyysi on osoittanut, että ikä ja paino vaikuttavat farisimabin systeemiseen farmakokinetiikkaan tai sen farmakokinetiikkaan silmässä. Kumpaakaan vaikutusta ei katsottu kliinisesti merkittäväksi, joten annosta ei tarvitse muuttaa.
Biotransformaatio ja eliminaatio
Farisimabi on proteiini, joten sen metaboliaa ja eliminaatiota ei ole täysin selvitetty. Farisimabin oletetaan kataboloituvan lysosomeissa pieniksi peptideiksi ja aminohapoiksi, jotka saattavat erittyä munuaisten kautta samalla tavoin kuin endogeeninen IgG eliminoituu.
Farisimabin pitoisuus-aikaprofiili plasmassa pieneni rinnan lasiaisen ja kammionesteen pitoisuus-aikaprofiilin kanssa. Farisimabin laskennallisen puoliintumisajan silmässä ja näennäisen systeemisen puoliintumisajan keskiarvo on noin 7,5 vuorokautta.
Farmakokineettinen analyysi potilaista, joilla on neovaskulaarinen (kostea) silmänpohjan ikärappeuma, diabeettista makulaturvotusta tai verkkokalvon laskimotukos (N = 2 977), on osoittanut, että farisimabin farmakokinetiikka on verrannollinen potilailla, joilla on neovaskulaarinen (kostea) silmänpohjan ikärappeuma, diabeettista makulaturvotusta tai verkkokalvon laskimotukos.
Erityispotilasryhmät
Iäkkäät
Kuudessa vaiheen III kliinisessä tutkimuksessa noin 58 % (1 496/2 571) potilaista, jotka satunnaistettiin saamaan farisimabihoitoa, oli ≥ 65-vuotiaita. Populaatiofarmakokineettinen analyysi on osoittanut, että ikä vaikuttaa farisimabin farmakokinetiikkaan silmässä. Vaikutuksella ei katsota olevan kliinistä merkitystä. 65-vuotiaiden ja vanhempien potilaiden annosta ei tarvitse muuttaa (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Farisimabilla ei ole tehty spesifisiä tutkimuksia munuaisten vajaatoimintaa sairastavilla potilailla. Farmakokineettinen analyysi kaikkiin kliinisiin tutkimuksiin osallistuneista potilaista, joista 63 %:lla oli munuaisten vajaatoimintaa (lievä 38 %, keskivaikea 23 % ja vaikea 2 %), ei tuonut esiin eroja farisimabin systeemisessä farmakokinetiikassa, kun farisimabi annettiin lasiaiseen. Munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavilla potilailla ei ole tehty spesifisiä farisimabitutkimuksia. Tämän potilasryhmän osalta ei tarvitse huomioida erityisiä seikkoja, koska metabolia tapahtuu proteolyysin välityksellä eikä ole riippuvainen maksan toiminnasta. Maksan vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Annostus ja antotapa).
Muut potilasryhmät
Etninen tausta ei vaikuta farisimabin systeemiseen farmakokinetiikkaan. Sukupuolella ei osoitettu kliinisesti oleellista vaikutusta farisimabin systeemiseen farmakokinetiikkaan. Annosta ei tarvitse muuttaa.
Prekliiniset tiedot turvallisuudesta
Farisimabin karsinogeenisuuden tai mutageenisuuden selvittämiseksi ei ole tehty tutkimuksia.
Tiineille jaavanmakakeille laskimoon annetuista farisimabi-injektioista aiheutui seerumiin yli 500-kertainen altistus (Cmax) verrattuna ihmisen maksimialtistukseen, eikä siitä aiheutunut kehitystoksisuutta eikä teratogeenisuutta; se ei myöskään vaikuttanut istukan painoon tai rakenteeseen, vaikka farisimabi pitää farmakologisen vaikutuksensa perusteella katsoa mahdollisesti teratogeeniseksi ja alkio-/sikiötoksiseksi.
Silmään annetusta farisimabista aiheutuva systeeminen altistus on hyvin vähäinen.
Farmaseuttiset tiedot
Apuaineet
L-histidiini
Etikkahappo 30 % (pH:n säätöön) (E 260)
L-metioniini
Polysorbaatti 20 (E 432)
Natriumkloridi
D-sakkaroosi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Esitäytetty ruisku: 2 vuotta
Injektiopullo: 30 kuukautta
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Pidä esitäytetty ruisku avaamattomassa repäisypakkauksessa alkuperäisessä kartonkikotelossa. Herkkä valolle.
Avaamatonta esitäytettyä ruiskua tai injektiopulloa voidaan säilyttää ennen käyttöä huoneenlämmössä (20–25 °C) alkuperäisessä kartonkikotelossa enintään 24 tuntia.
Varmista, että injektiot annetaan heti annoksen valmistelun jälkeen.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VABYSMO injektioneste, liuos
120 mg/ml (L:ei) 1 kpl (siirtoneula, 0,24 ml) (748,01 €)
VABYSMO injektioneste, liuos, esitäytetty ruisku
120 mg/ml (L:ei) 1 kpl (suodattimella varustettu injektionneula, 0,175 ml) (748,01 €)
PF-selosteen tieto
Esitäytetty ruisku
Injektioneste, liuos, esitäytetyssä ruiskussa, jossa on (tyypin I) lasinen runko-osa ja siinä annosmerkki, butyylikumitulppa ja peukaloinnin ilmaiseva suljin (joka koostuu kärjen jäykästä korkista, kärjen butyylikumikorkista ja Luer lock ‑liittimestä). Esitäytetyssä ruiskussa on mäntä ja pitkä sormituki. Yksi esitäytetty ruisku sisältää 21 mg farisimabia 0,175 ml:ssa liuosta.
Pakkauskoko: yksi erittäin ohut steriili, suodattimella varustettu injektioneula (30G x ½”, 0,30 mm x 12,7 mm, 5 µm) yhteispakkauksessa yhden esitäytetyn ruiskun kanssa.
Kärjen kumikorkki, kumitulppa, lasinen runko-osa ja suodattimella varustettu injektioneula ovat kosketuksissa lääkevalmisteen kanssa.
Injektiopullo
0,24 ml steriiliä liuosta lasisessa injektiopullossa, jossa on päällystetty kumitulppa ja joka on sinetöity alumiinikorkilla ja keltaisella irti napsautettavalla (flip-off) muovilevyllä.
Pakkauskoko: yksi injektiopullo ja yksi tylppä suodattimella varustettu siirtoneula (18G x 1½”, 1,2 mm x 40 mm, 5 µm).
Valmisteen kuvaus:
Kirkas tai opalisoiva, väritön tai ruskehtavankeltainen liuos, jonka pH on 5,5 ja osmolaliteetti on 270−370 mosm/kg.
Käyttö- ja käsittelyohjeet
Ei saa ravistaa.
Vabysmo pitää jääkaapista otettaessa ja ennen antoa tarkistaa silmämääräisesti. Jos havaitaan hiukkasia tai samentumaa, Vabysmo-valmistetta ei saa käyttää.
Esitäytetty ruisku
Esitäytetty ruisku on kertakäyttöinen ja tarkoitettu vain yhden silmän hoitoon. Steriili esitäytetty ruisku tulee avata vain aseptisissa olosuhteissa. Liuos pitää ennen antoa tarkistaa silmämääräisesti. Jos havaitaan hiukkasia tai samentumaa, esitäytettyä ruiskua ei saa käyttää.
Esitäytetty ruisku sisältää farisimabia enemmän kuin suositellun 6 mg:n annoksen (vastaa 0,05 ml:aa). Yksi esitäytetty ruisku sisältää 21 mg farisimabia 0,175 ml:ssa liuosta. Ylimääräinen tilavuus on poistettava ennen injektiota.
Älä käytä, jos pakkaus, esitäytetty ruisku ja/tai suodattimella varustettu injektioneula ovat vaurioituneet tai niiden viimeinen käyttöpäivämäärä on ohitettu. Yksityiskohtaiset käyttöohjeet ovat pakkausselosteessa.
Injektiopullo
Injektiopullo sisältää enemmän kuin suositellun 6 mg:n annoksen. Injektiopullon koko täyttötilavuutta (0,24 ml) ei ole tarkoitus käyttää. Ylimääräinen tilavuus on poistettava ennen injektiota. Injektiopullon koko tilavuuden injisointi johtaa yliannokseen. Injisoitava annos pitää asettaa 0,05 ml:n annosmerkin kohdalle eli 6 mg:aan farisimabia.
Injektiopullon sisältö ja suodattimella varustettu siirtoneula ovat steriilejä ja kertakäyttöisiä. Älä käytä, jos pakkaus, injektiopullo ja/tai suodattimella varustettu siirtoneula ovat vaurioituneet tai niiden viimeinen käyttöpäivämäärä on ohitettu. Yksityiskohtaiset käyttöohjeet ovat pakkausselosteessa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Esitäytetyn ruiskun käyttöohjeet:
| Noudata ruiskun poistamisessa repäisypakkauksestaan (vaihe 1) ja kaikissa seuraavissa vaiheissa aseptista tekniikkaa. | |
| Huom. Annos on asetettava 0,05 ml:n annosmerkin kohdalle. | |
| Avaa repäisypakkaus ja poista ruiskun korkki | |
| 1 | Vedä ruiskun repäisypakkauksen kansi irti ja poista esitäytetty ruisku repäisypakkauksesta aseptisesti. |
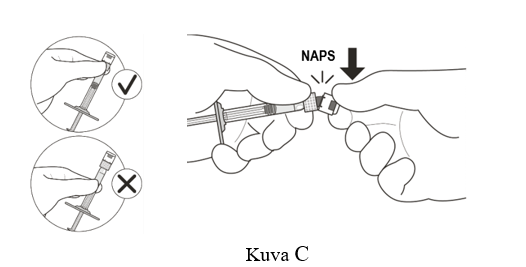
| 2 | Pitele ruiskua valkoisesta kaulusosasta ja napsauta ruiskun korkki irti (ks. kuva C). |
| Älä kierrä korkkia irti. | |
 | |
| Kiinnitä suodattimella varustettu injektioneula | |
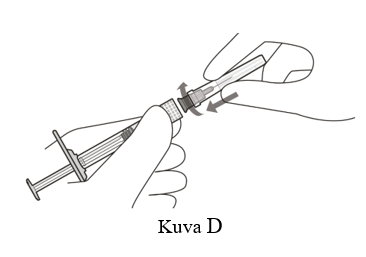
| 3 | Poista suodattimella varustettu injektioneula pakkauksestaan aseptisesti. |
| 4 | Kiinnitä suodattimella varustettu injektioneula aseptisesti ja tukevasti kiinni Luer lock ‑ruiskuun (ks. kuva D). |
 | Käytä antoon vain pakkaukseen sisältyvää suodattimella varustettua injektioneulaa |
| 5 | Irrota neulansuojus vetämällä se varovasti suoraan irti. |
| Poista ilmakuplat | |
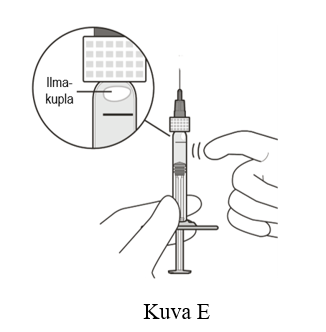
| 6 | Pitele ruiskua suodattimella varustettu injektioneula ylöspäin osoittaen ja tarkista, onko ruiskussa ilmakuplia. |
| 7 | Jos näet ilmakuplia, naputtele ruiskua varovasti sormella, kunnes kuplat nousevat yläosaan (ks. kuva E). |
 | |
| Säädä lääkevalmisteannos ja poista ilma | |
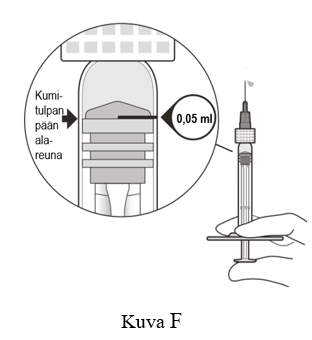
| 8 | Pitele ruiskua silmiesi tasolla ja paina mäntää hitaasti, kunnes kumitulpan pään alareuna on 0,05 ml:n annosmerkin kohdalla (ks. kuva F). Näin ilma ja ylimääräinen tilavuus poistuvat ruiskusta ja jäljelle jää 0,05 ml:n annos. |
| Varmista, että injektio annetaan heti annoksen valmistelun jälkeen. | |
 | |
| Injektion antaminen | |
| 9 | Injektio on annettava aseptisissa olosuhteissa. Injisoi hitaasti, kunnes kumitulppa on painettu ruiskun pohjaan ja 0,05 ml:n tilavuus on siten annettu. Älä laita neulansuojusta takaisin suodattimella varustetun injektioneulan päälle tai irrota neulaa ruiskusta. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti. |
Korvattavuus
VABYSMO injektioneste, liuos
120 mg/ml 1 kpl
VABYSMO injektioneste, liuos, esitäytetty ruisku
120 mg/ml 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Aflibersepti ja farisimabi: Aikuisten neovaskulaarisen (kostean) silmänpohjan ikärappeuman (AMD) hoito (3094).
ATC-koodi
S01LA09
Valmisteyhteenvedon muuttamispäivämäärä
08.05.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com