
KAPRUVIA injektioneste, liuos 50 mikrog/ml
Huomioitavaa
▼Tähän lääkkeeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 1 ml:n injektiopullo sisältää 50 mikrogrammaa difelikefaliinia (asetaattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Kapruvia on tarkoitettu krooniseen munuaistautiin liittyvän keskivaikean tai vaikean kutinan hoitoon aikuisille hemodialyysipotilaille (ks. kohta Farmakodynamiikka).
Ehto
Valmisteen käyttö tulee rajoittaa ainoastaan hoitolaitoksessa annettavaan hemodialyysihoitoon. Se on tarkoitettu sellaisten terveydenhuollon ammattilaisten käyttöön, joilla on kokemusta difelikefaliinilla hoidettavien tilojen diagnosoinnista ja hoidosta.
Annostus ja antotapa
Kapruvia-valmisteen käyttö tulee rajoittaa ainoastaan hoitolaitoksessa annettavaan hemodialyysihoitoon.
Kapruvia on tarkoitettu sellaisten terveydenhuollon ammattilaisten käyttöön, joilla on kokemusta difelikefaliinilla hoidettavien tilojen diagnosoinnista ja hoidosta. Muut kutinan aiheuttajat kuin krooninen munuaistauti on suljettava pois ennen difelikefaliinihoidon aloittamista.
Annostus
Difelikefaliinia annetaan 3 kertaa viikossa laskimonsisäisenä bolusinjektiona dialyysikierron laskimolinjaan hemodialyysihoidon lopussa joko takaisinhuuhtelun aikana tai sen jälkeen.
Difelikefaliinin suositeltu annos on 0,5 mikrogrammaa/kuivapainokilo (eli dialyysin jälkeinen tavoitepaino). Injektiopullosta otettavan kokonaisannoksen tilavuus (ml) lasketaan seuraavasti: 0,01 × kuivapaino (kg) pyöristettynä lähimpään kymmenykseen (0,1 ml). Jos potilaan kuivapaino on 195 kg tai enemmän, suositeltu annos on 100 mikrogrammaa (2 ml). Injektiotilavuudet annetaan seuraavassa taulukossa:
Painoalue (kuivapaino kilogrammoina) | Injektiotilavuus1 (ml) | |
| 40–44 | 0,4 | |
| 45–54 | 0,5 | |
| 55–64 | 0,6 | |
| 65–74 | 0,7 | |
| 75–84 | 0,8 | |
| 85–94 | 0,9 | |
| 95–104 | 1,0 | |
| 105–114 | 1,1 | |
| 115–124 | 1,2 | |
| 125–134 | 1,3 | |
| 135–144 | 1,4 | |
| 145–154 | 1,5 | |
| 155–164 | 1,6 | |
| 165–174 | 1,7 | |
| 175–184 | 1,8 | |
| 185–194 | 1,9 | |
| ≥ 195 | 2,0 | |
1Jos tarvittava injektiotilavuus on yli 1 ml, saatetaan tarvita enemmän kuin yksi injektiopullo.
Difelikefaliinin kutinaa lievittävän vaikutuksen voidaan odottaa alkavan 2–3 viikon hoidon jälkeen.
Annosten jääminen väliin
Jos säännöllisen aikataulun mukainen hemodialyysihoito jää väliin, sama annos Kapruvia-valmistetta tulee antaa seuraavan hemodialyysihoidon yhteydessä.
Lisähoito
Jos viikon aikana annetaan 4. hemodialyysihoito, suositeltu annos Kapruvia-valmistetta tulee antaa hemodialyysin lopussa. Viikon aikana saa antaa enintään 4 annosta, vaikka hemodialyysihoitoja olisi viikon sisällä enemmän kuin 4. Neljäs Kapruvia-annos ei todennäköisesti aiheuta turvallisuuden vaarantavaa difelikefaliinin kertymistä, sillä hemodialyysi poistaa suurimman osan difelikefaliinista, jota elimistössä vielä on edellisen hoitokerran jäljiltä (ks. kohdat Yliannostus ja Farmakokinetiikka). Neljännen annoksen turvallisuutta ja tehoa ei kuitenkaan ole täysin varmistettu, sillä tietoa ei ole riittävästi.
Potilaat, joiden hemodialyysihoitoa ei ole suoritettu loppuun
Jos hemodialyysihoito kestää alle 1 tunnin, difelikefaliinia ei pidä antaa ennen seuraavaa hemodialyysikertaa.
Jopa 70 % hemodialyysipotilaille annetusta difelikefaliinista poistuu potilaan elimistöstä ennen seuraavaa hemodialyysikertaa (ks. kohdat Yliannostus ja Farmakokinetiikka). Seuraavan hemodialyysin ajankohtana plasmassa jäljellä oleva difelikefaliinipitoisuus pienenee noin 40–50 %:lla tunnin sisällä hemodialyysin aloittamisesta.
Maksan vajaatoimintapotilaat
Annosta ei tarvitse muuttaa potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Difelikefaliinia ei ole tutkittu vaikeaa maksan vajaatoimintaa (National Cancer Institute (NCI) Organ Dysfunction Working Group (ODWG) -työryhmän määritelmän mukaan) sairastavilla henkilöillä, eikä sitä siksi suositella käytettäväksi tässä potilasryhmässä.
Iäkkäät potilaat (≥ 65 vuoden ikäiset)
Annossuositukset iäkkäille potilaille ovat samat kuin aikuisille potilaille.
Pediatriset potilaat
Difelikefaliinin turvallisuutta ja tehoa 12‑17 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu.
Tällä hetkellä saatavilla olevia tietoja kuvataan kohdassa Farmakodynamiikka.
Difelikefaliinin turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu.
Tietoja alle 12-vuotiaista potilaista ei ole saatavilla.
Antotapa
Kapruvia-valmistetta ei pidä laimentaa eikä sekoittaa muiden lääkevalmisteiden kanssa.
Difelikefaliini poistuu dialyysikalvon läpi, joten se on annettava, kun verta ei enää kierrätetä dialysaattorin kautta. Difelikefaliinia annetaan 3 kertaa viikossa laskimonsisäisenä bolusinjektiona dialyysikierron laskimolinjaan hemodialyysihoidon lopussa joko takaisinhuuhtelun aikana tai sen jälkeen.
Jos annos annetaan takaisinhuuhtelun jälkeen, Kapruvia-injektion jälkeen tulee antaa takaisinhuuhteluna vähintään 10 ml 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä. Jos annos annetaan takaisinhuuhtelun aikana, letkun huuhteluun ei tarvitse käyttää ylimääräistä 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Hyperkalemia
Hyperkalemiaa esiintyy yleisesti kroonista munuaistautia sairastavilla, hemodialyysihoitoa saavilla potilailla. Lumekontrolloiduissa kliinisissä tutkimuksissa hyperkalemiaa raportoitiin haittavaikutuksena numeerisesti enemmän difelikefaliinihoitoa saaneilla potilailla (4,7 %; 20/424 potilasta) kuin lumelääkettä saaneilla potilailla (3,5 %; 15/424 potilasta). Syy-yhteyttä ei ole osoitettu. Kaliumpitoisuuden tiheää seurantaa suositellaan.
Sydämen vajaatoiminta ja eteisvärinä
Difelikefaliinia ei ole tutkittu potilailla, joilla on New York Heart Association -luokan IV sydämen vajaatoiminta. Kliinisissä avaintutkimuksissa difelikefaliinihoitoa saaneilla potilailla ja lumelääkettä saaneilla potilailla todetuissa sydämen vajaatoiminta- ja eteisvärinätapauksissa todettiin pientä numeerista epätasapainoa, erityisesti potilailla, joilla on ollut eteisvärinää ja jotka keskeyttivät eteisvärinän hoidon tai jättivät hoidon väliin. Syy-yhteyttä ei ole osoitettu.
Potilaat, joilla on veri-aivoesteen toimintahäiriö
Difelikefaliini on perifeerisesti vaikuttava kappa-opioidireseptorin agonisti, jonka pääsy keskushermostoon on rajallista. Veri-aivoesteen läpäisemättömyys on tärkeää, sillä se minimoi difelikefaliinin pääsyn keskushermostoon (ks. kohta Farmakodynamiikka). Jos potilaan veri-aivoesteen toiminta on häiriintynyt kliinisesti merkittävässä määrin (esim. primaarisen pahanlaatuisen aivokasvaimen, keskushermostometastaasien tai muiden tulehdustilojen, aktiivisen multippeliskleroosin tai pitkälle edenneen Alzheimerin taudin takia), voi olla olemassa riski, että difelikefaliinia pääsee keskushermostoon. Kapruvia-valmistetta tulee määrätä tällaisille potilaille varoen ja potilaan yksilöllisen hyöty-haittatasapainon huomioiden, ja mahdollisia keskushermostoon kohdistuvia vaikutuksia on tarkkailtava.
Heitehuimaus ja uneliaisuus
Difelikefaliinia saavilla potilailla on esiintynyt heitehuimausta ja uneliaisuutta, jotka saattavat hävitä ajan mittaan hoidon jatkuessa (ks. kohta Haittavaikutukset). Sedatiivisten antihistamiinien, opioidikipulääkkeiden tai muiden keskushermostoa lamaavien lääkevalmisteiden samanaikainen käyttö saattaa suurentaa näiden haittavaikutusten todennäköisyyttä, ja näitä lääkkeitä on käytettävä varoen difelikefaliinihoidon aikana (ks. kohta Yhteisvaikutukset).
Lumelääkkeeseen verrattuna uneliaisuutta esiintyi yleisemmin vähintään 65‑vuotiailla (7,0 %) kuin alle 65‑vuotiailla (2,8 %) difelikefaliinia saaneilla tutkittavilla.
Mielentilan muutokset
Mielentilan muutoksia (mukaan lukien sekavuustila) voi esiintyä (ks. kohta Haittavaikutukset), mikä on otettava huomioon erityisesti ikääntyneiden riskiryhmässä.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia per injektiopullo eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Kliinisiä yhteisvaikutustutkimuksia ei ole tehty. Difelikefaliini ei estä eikä indusoi CYP450-entsyymejä eikä ole CYP450-entsyymien substraatti. Se ei ole myöskään glukuronisoivien entsyymien estäjä. Difelikefaliini ei ole ihmisen kuljettajaproteiinien substraatti eikä estäjä (ks. kohta Farmakokinetiikka). Näin ollen difelikefaliinin ja muiden lääkevalmisteiden väliset yhteisvaikutukset ovat epätodennäköisiä.
Lääkevalmisteiden, kuten sedatiivisten antihistamiinien, opioidikipulääkkeiden tai muiden keskushermostoa lamaavien lääkevalmisteiden (esim. klonidiini, ondansetroni, gabapentiini, pregabaliini, tsolpideemi, alpratsolaami, sertraliini, tratsodoni) samanaikainen anto voi suurentaa heitehuimauksen ja uneliaisuuden todennäköisyyttä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja difelikefaliinin käytöstä raskaana oleville naisille.
Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varmuuden vuoksi Kapruvia-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö difelikefaliini ihmisen rintamaitoon.
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Kapruvia-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Eläinkokeet ovat osoittaneet difelikefaliinin erittyvän rintamaitoon.
Hedelmällisyys
Difelikefaliinin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Rotilla tehdyissä difelikefaliinitutkimuksissa ei todettu hedelmällisyyteen kohdistuvia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kapruvia-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Difelikefaliinia saavilla potilailla on raportoitu uneliaisuutta ja/tai heitehuimausta (ks. kohta Haittavaikutukset).
Potilaita on varoitettava ajamasta autoa ja käyttämästä vaarallisia koneita, ennen kuin difelikefaliinin vaikutus hänen ajokykyynsä ja koneidenkäyttökykyynsä on tiedossa. Uneliaisuus ilmaantui ensimmäisten 3 hoitoviikon aikana ja yleensä väheni hoidon jatkuessa. Heitehuimaus ilmaantui ensimmäisten 9 hoitoviikon aikana ja oli yleensä ohimenevää.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Lumekontrolloiduissa ja kontrolloimattomissa vaiheen 3 kliinisissä tutkimuksissa noin 6,6 %:lla potilaista ilmeni vähintään yksi haittavaikutus difelikefaliinihoidon aikana. Yleisimpiä haittavaikutuksia olivat uneliaisuus (1,1 %), heitehuimaus (0,9 %), parestesia (mukaan lukien hypestesia, suun parestesia ja suun hypestesia) (1,1 %), päänsärky (0,6 %), pahoinvointi (0,7 %), oksentelu (0,7 %), ripuli (0,2 %) ja mielentilan muutokset (mukaan lukien sekavuustila) (0,3 %). Useimmat näistä tapahtumista olivat vaikeusasteeltaan lieviä tai keskivaikeita, niillä ei ollut haitallisia seurauksia, ja ne korjaantuivat hoidon jatkuessa. Mikään tapahtumista ei ollut vakava, ja hoidon lopettamiseen johtaneiden tapahtumien ilmaantuvuus oli ≤ 0,5 % kaikkien edellä mainittujen haittatapahtumien osalta.
Haittavaikutustaulukko
Lumekontrolloiduissa ja kontrolloimattomissa vaiheen 3 kliinisissä tutkimuksissa difelikefaliinihoitoa saaneilla potilailla (N = 1 306) todetut haittavaikutukset on lueteltu taulukossa 1 MedDRA-elinjärjestelmäluokan, suositeltujen termien ja yleisyyden mukaan.
Yleisyydet määritellään seuraavasti: yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100).
Taulukko 1: Hemodialyysipotilailla esiintyneet haittavaikutukset, joiden katsottiin liittyvän difelikefaliinihoitoon
| MedDRA-elinjärjestelmäluokka | Yleinen | Melko harvinainen |
| Psyykkiset häiriöt | Mielentilan muutokset1 | |
| Hermosto | Uneliaisuus, parestesia2 | Heitehuimaus, päänsärky |
| Ruoansulatuselimistö | Oksentelu, pahoinvointi, ripuli |
1 Mielentilan muutoksiin sisältyivät suositellut MedDRA-termit sekavuustila ja mielentilan muutokset.
2 Parestesiaan sisältyivät suositellut MedDRA-termit parestesia, hypestesia, suun parestesia ja suun hypestesia.
Valikoitujen haittavaikutusten kuvaus
Uneliaisuus
Uneliaisuutta raportoitiin hoidon aikana ilmenneenä haittavaikutuksena 2,2 %:lla difelikefaliinihoitoon satunnaistetuista tutkittavista. Valtaosa näistä tapahtumista oli vaikeusasteeltaan lieviä tai keskivaikeita. Uneliaisuus johti difelikefaliinihoidon lopettamiseen 0,3 %:lla potilaista. Uneliaisuutta raportoitiin vakavana haittatapahtumana < 0,1 %:lla difelikefaliinihoitoa saaneista tutkittavista. Uneliaisuuden ja difelikefaliinihoidon välillä raportoitiin syy-yhteys 1,1 %:lla potilaista. Uneliaisuus ilmaantui ensimmäisten 3 hoitoviikon aikana ja yleensä väheni hoidon jatkuessa.
Uneliaisuuden todennäköisyys saattaa suurentua, jos difelikefaliinia käytetään samanaikaisesti muiden lääkevalmisteiden kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Heitehuimaus
Heitehuimausta raportoitiin hoidon aikana ilmenneenä haittavaikutuksena 7,9 %:lla difelikefaliinihoitoon satunnaistetuista tutkittavista. Valtaosa näistä tapahtumista oli vaikeusasteeltaan lieviä tai keskivaikeita. Heitehuimaus johti difelikefaliinihoidon lopettamiseen 0,5 %:lla potilaista. Heitehuimausta raportoitiin vakavana haittatapahtumana 0,5 %:lla difelikefaliinihoitoa saaneista tutkittavista. Heitehuimauksen ja difelikefaliinihoidon välillä raportoitiin syy-yhteys 0,9 %:lla potilaista. Heitehuimaus ilmaantui ensimmäisten 9 hoitoviikon aikana ja oli yleensä ohimenevää.
Heitehuimauksen todennäköisyys saattaa suurentua, jos difelikefaliinia käytetään samanaikaisesti muiden lääkevalmisteiden kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Mielentilan muutokset
Mielentilan muutoksia (mukaan lukien sekavuustila) raportoitiin hoidon aikana ilmenneenä haittavaikutuksena 4,4 %:lla difelikefaliinihoitoon satunnaistetuista tutkittavista.
Suurin osa näistä tapahtumista oli vaikeusasteeltaan lieviä tai keskivaikeita. Mielentilan muutokset johtivat difelikefaliinihoidon lopettamiseen 0,2 %:lla potilaista.
Mielentilan muutoksia raportoitiin vakavana haittatapahtumana 2,2 %:lla difelikefaliinihoitoa saaneista tutkittavista. Mielentilan muutosten ja difelikefaliinihoidon välillä raportoitiin syy-yhteys 0,3 %:lla potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Hemodialyysipotilaille annettiin kliinisissä tutkimuksissa difelikefaliinia kerta-annoksina, jotka olivat enintään 12‑kertaisia kliiniseen annokseen 0,5 mikrogrammaa/kg nähden, ja toistuvina annoksina, jotka olivat enintään 5‑kertaisia kliiniseen annokseen 0,5 mikrogrammaa/kg nähden. Haittatapahtumien, kuten heitehuimauksen, uneliaisuuden, mielentilan muutosten, parestesian, väsymyksen, hypertension ja oksentelun, esiintyvyyden todettiin lisääntyvän annoksesta riippuvalla tavalla.
Yliannostustapauksessa tulee antaa asianmukaista hoitoa potilaan kliinisen tilan perusteella.
Neljä tuntia kestänyt hemodialyysi suurivirtauksisella dialysaattorilla poisti plasmasta tehokkaasti noin 70–80 % difelikefaliinista, eikä difelikefaliinia havaittu plasmassa enää toisen dialyysikierron lopussa (ks. kohta Farmakokinetiikka).
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut lääkevalmisteet, muut, ATC-koodi: V03AX04
Vaikutusmekanismi
Difelikefaliini on selektiivinen kappa-opioidireseptorin agonisti, joka penetroituu keskushermostoon huonosti.
Difelikefaliinin fysikaalis-kemialliset ominaisuudet (vesiliukoinen, synteettinen D‑aminohappopeptidi, jolla on suuri poolinen pinta-ala ja varaus fysiologisessa pH:ssa) minimoivat sen passiivisen diffuusion (läpäisevyyden) ja aktiivisen kuljetuksen solukalvojen läpi ja rajoittavat siten sen penetroitumista keskushermostoon.
Krooniseen munuaistautiin liittyvän kutinan patofysiologian arvellaan olevan monisyinen. Siihen liittyy systeemistä tulehdusta ja endogeenisen opioidijärjestelmän epätasapainoa (esim. myy-opioidireseptorien yli-ilmentymistä ja samanaikaista kappa-opioidireseptorien vaimennussäätelyä). Opioidireseptorien tiedetään vaikuttavan kutinasignaaleihin ja tulehdukseen, ja kappa-opioidireseptorien aktivaatio vähentää kutinaa ja saa aikaan immunomodulatorisia vaikutuksia.
Ääreishermoston tuntohermosoluissa ja immuunisoluissa tapahtuvan, difelikefaliinin aikaansaaman kappa-opioidireseptorien aktivaation katsotaan olevan mekanistisesti vastuussa lääkkeen kutinaa vähentävistä ja tulehdusta estävistä vaikutuksista.
Kliininen teho ja turvallisuus
Lumekontrolloidut tutkimukset
Kahdessa kliinisessä vaiheen 3 avaintutkimuksessa, joissa käytettiin samankaltaista kaksoissokkoutettua, satunnaistettua, lumekontrolloitua tutkimusasetelmaa (KALM‑1 ja KALM‑2), kroonista munuaistautia sairastaville hemodialyysipotilaille, joilla esiintyi keskivaikeaa tai vaikeaa kutinaa, annettiin joko lumelääkettä tai difelikefaliinia annoksena 0,5 mikrogrammaa/kg laskimoon 3 kertaa viikossa hemodialyysin jälkeen 12 viikon ajan. Annosten enimmäismäärä oli 4 annosta viikossa, mikäli potilas sai viikon aikana ylimääräisen dialyysihoidon. Molemmissa tutkimuksissa ensisijainen päätetapahtuma oli niiden potilaiden prosenttiosuus, joiden pahinta kutinaa mittaavan Worst Itching-Numerical Rating Scale (WI‑NRS) -arviointiasteikon pistearvo oli pienentynyt vähintään 3 pisteellä lähtötilanteesta viikolla 12. Molemmissa tutkimuksissa tärkeimpiä toissijaisia päätetapahtumia olivat niiden potilaiden prosenttiosuudet, joiden WI‑NRS-pisteet olivat parantuneet vähintään 4 pisteellä 12 viikon jälkeen, sekä kutinan vaikeusasteen ja kutinaan liittyvän elämänlaadun (QoL) muutokset Skindex‑10- ja 5‑D Itch -asteikoilla mitattuna. Mukana oli myös hoitovasteen saavuttaneiden analyysi, joka perustui potilaan yleisarvioon muutoksesta.
Avaintutkimuksiin otettiin yhteensä 851 potilasta, joilla oli keskivaikeaa tai vaikeaa kutinaa (lähtötilanteen WI-NRS > 4). Potilaiden keski-ikä oli 59 vuotta, 33,1 % potilaista oli vähintään 65‑vuotiaita ja 11,1 % vähintään 75‑vuotiaita, ja 60 % potilaista oli miehiä. Lähtötilanteen keskimääräiset WI-NRS-pisteet olivat 7,18 sekä difelikefaliini- että lumeryhmässä; lähtötilanteen mediaani WI-NRS-pisteet olivat 7,13 (vaihteluväli 4,2–10) difelikefaliini- ja 7,13 (vaihteluväli 4,1–10) lumeryhmässä. Muut taudin piirteet olivat lähtötilanteessa vertailukelpoiset difelikefaliini- ja lumeryhmissä: kroonisen munuaistaudin diagnosoinnista kulunut aika (8,22 vuotta vs. 8,54 vuotta), kutinan kesto (3,20 vuotta vs. 3,31 vuotta) ja kutinaa lievittävien lääkkeiden kuten antihistamiinien, kortikosteroidien, gabapentiinin tai pregabaliinin käyttö (37,5 % vs. 38 %). Tutkimuksissa difelikefaliini vähensi kutinan voimakkuutta merkitsevästi ja paransi kutinaan liittyvää elämänlaatua 12 viikon aikana taulukossa 2 kuvatulla tavalla.
Taulukko 2: Yhteenveto ensisijaisista ja tärkeimmistä toissijaisista tuloksista KALM‑1- ja KALM‑2-tutkimuksissa viikolla 12
| Päätetapahtuma viikon 12 loppuun mennessä | KALM‑1 (n = 378) | KALM‑2 (n = 473) | ||
difelikefaliini (n = 189) | Lumelääke (n = 189) | difelikefaliini (n = 237) | Lumelääke (n = 236) | |
| Ensisijainen päätetapahtuma | ||||
| WI‑NRS | ||||
| Potilaat, joiden pistearvo parani ≥ 3 pisteellä (%) | 51,0 % (p < 0,001) | 27,6 % | 54,0 % (p = 0,02) | 42,2 % |
| Toissijaiset päätetapahtumat | ||||
| WI‑NRS | ||||
| Potilaat, joiden pistearvo parani ≥ 4 pisteellä (%) | 38,9 % (p < 0,001) | 18,0 % | 41,2 % (p = 0,01) | 28,4 % |
| Skindex‑10 | ||||
Muutos lähtötilanteesta [kokonaispisteet] | -17,2 (p < 0,001) | -12,0 | -16,6 (p = 0,171) | -14,8 |
| 5‑D Itch | ||||
Muutos lähtötilanteesta [kokonaispisteet] | -5,0 (p < 0,001) | -3,7 | -4,9 Ei soveltuva1 | -3,8 |
1 Ei testattu hierarkkisen testausjärjestyksen perusteella.
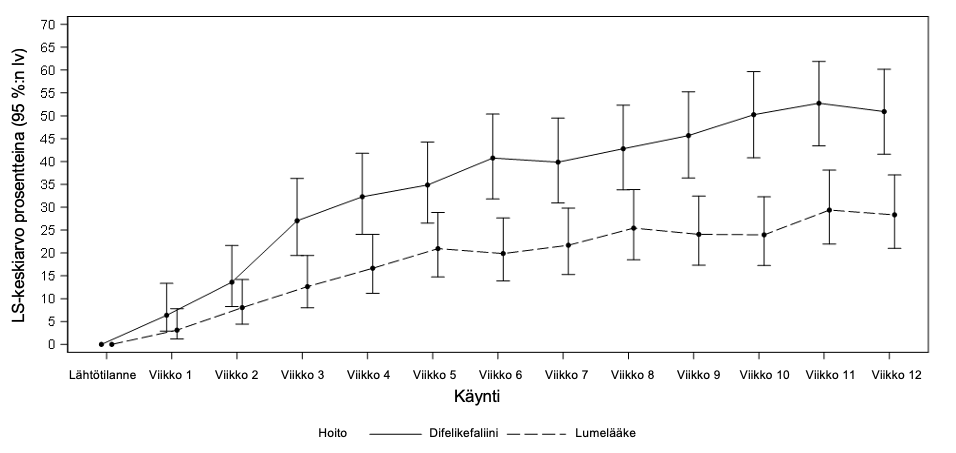
Kuvassa 1 esitetään tutkimusviikoittain niiden KALM‑1- ja KALM‑2-tutkimuksiin osallistuneiden potilaiden keskimääräiset prosenttiosuudet, joiden WI‑NRS-pistearvo parani ≥ 3 pisteellä lähtötilanteesta. Kerroinsuhteiden perusteella difelikefaliinia suosiva tilastollisesti merkitsevä paranema oli nähtävissä viikkoon 3 mennessä KALM‑1-tutkimuksessa ja viikkoon 2 mennessä KALM‑2‑tutkimuksessa, ja sama suuntaus jatkui molemmissa tutkimuksissa kaikilla seuraavilla viikoilla viikon 12 loppuun asti.
Kuva 1: Niiden potilaiden viikoittaiset prosenttiosuudet, joiden WI‑NRS-pisteet paranivat ≥ 3 pisteellä KALM‑1- ja KALM‑2-tutkimuksissa (ITT-populaatio)
KALM‑1
KALM‑2
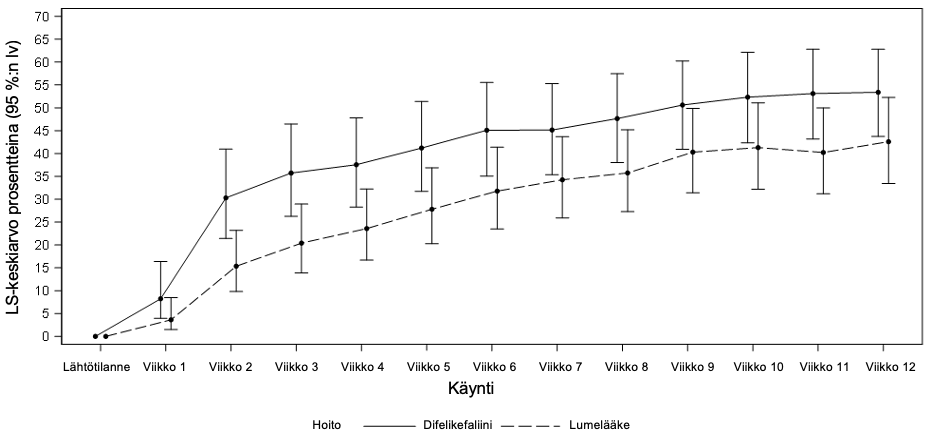
lv = luottamusväli; ITT = hoitoaikeen mukainen; LS = pienin neliösumma; WI-NRS = Worst Itching-Numerical Rating Scale -arviointiasteikko
Avoimet jatkotutkimukset
Difelikefaliinihoidon vaikutusta arvioitiin enintään 52 viikon ajalta 5‑D Itch -asteikolla KALM‑1- ja KALM‑2-tutkimusten yhdellä hoitoryhmällä toteutetuissa avoimissa jatkotutkimuksissa, joihin osallistui 712 potilasta.
Potilailla, jotka siirtyivät lumelääkkeestä difelikefaliiniin kaksoissokkoutetun vaiheen lopussa, 5‑D Itch -pisteissä todettiin paranemista 4 viikkoa kestäneen hoidon jälkeen. Lähtötilanteen jälkeen tapahtuneen muutoksen pienimmän neliösumman keskiarvo (keskivirhe) oli verrattavissa potilaisiin, jotka olivat saaneet difelikefaliinia tutkimuksen alusta asti: -6,0 (0,22) vs. -5,7 (0,23). 5‑D Itch -pisteiden paranema säilyi molemmissa hoitoryhmissä koko 52 viikkoa kestäneen hoidon ajan.
Pediatriset potilaat
Kaikkiaan 8 hemodialyysihoitoa saavaa nuorta (12-17 -vuotiasta) otettiin mukaan avoimeen, yhden haaran tutkimukseen, jossa arvioitiin laskimonsisäisen difelikefaliinin kerta-annoksen farmakokinetiikkaa. On osoitettu, että difelikefaliinin kehon kuivapainoon perustuva 0,5 μg/kg kerta-annos antaa vertailukelpoisen altistuksen hemodialyysihoidossa oleville nuorille ja aikuisille. Nuorille laskimoon annetun difelikefaliinin kuivapainoon perustuvan 0,5 μg/kg kerta-annoksen turvallisuusprofiili vastasi difelikefaliinin tunnettua turvallisuusprofiilia aikuisilla.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset difelikefaliinin käytöstä krooniseen munuaistautiin liittyvän kutinan hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Vaikeaa munuaisten vajaatoimintaa sairastavilla hemodialyysipotilailla difelikefaliinin kokonaispuhdistuma vähenee terveisiin henkilöihin verrattuna, ja plasman lääkeainepitoisuudet pienenevät hitaasti, kunnes dialyysi poistaa lääkkeen. Koska difelikefaliinista poistuu dialyysissä 70‑80 %, difelikefaliini annetaan näille potilaille kunkin hemodialyysikerran jälkeen. Saatavilla olevat tiedot yksilöiden välisestä vaihtelusta hemodialyysipotilailla, jotka saavat difelikefaliinia annoksena 0,5 mikrogrammaa/kg, viittaavat siihen, että AUC-arvo voi vaihdella yli 30 %.
Jakautuminen
Difelikefaliinin sitoutuminen plasman proteiineihin on vähäistä tai kohtalaista (24–32 %), eikä munuaisten vajaatoiminta vaikuta siihen. Keskimääräinen jakautumistilavuus vakaassa tilassa oli terveillä tutkittavilla 145–189 ml/kg ja keskivaikeasta tai vaikeasta kutinasta kärsivillä hemodialyysipotilailla 214–301 ml/kg. Difelikefaliinin penetroituminen keskushermostoon on vähäistä (alle määritysrajan) fysikaalis-kemiallisten tietojen, in vitro -tietojen ja eläimistä saatujen tietojen perusteella.
Eliminaatio
Terveillä henkilöillä difelikefaliini poistuu elimistöstä pääasiassa munuaisteitse, sillä noin 81 % annoksesta erittyy virtsaan ja 11 % ulosteeseen. Sekä terveillä vapaaehtoisilla että hemodialyysipotilailla suurin osa virtsaan ja ulosteeseen erittyneestä annoksesta oli muuttumattomassa muodossa olevaa difelikefaliinia ja vain pieni määrä oletettuja metaboliitteja, joista yhdenkään osuus ei ollut yli 2,5 %. Keskimääräinen kokonaispuhdistuma oli 54–71 ml/h/kg ja keskimääräiset puoliintumisajat 2–3 tuntia. Sitä vastoin hemodialyysipotilailla eliminaatio tapahtui pääasiassa ulosteen kautta, sillä keskimäärin noin 59 % annoksesta erittyi ulosteeseen, 19 % hemodialysaattiin ja noin 11 % virtsaan. Verrattuna tutkittaviin, joiden munuaiset toimivat normaalisti, keskimääräinen kokonaispuhdistuma pieneni ja puoliintumisajat suurenivat noin 10‑kertaisesti, ja niiden vaihteluvälit olivat 5,3–7,5 ml/h/kg ja vastaavasti 23–31 tuntia.
Yhteisvaikutukset muiden lääkevalmisteiden kanssa
Difelikefaliini ei ole CYP1A2:n, CYP2C8:n, CYP2C9:n, CYP2C19:n, CYP2D6:n tai CYP3A4:n substraatti eikä CYP1A2:n, CYP2B6:n, CYP2C8:n, CYP2C9:n, CYP2C19:n, CYP2D6:n tai CYP3A4/5:n estäjä, eikä se indusoi ihmisen CYP1A2:ta, CYP2B6:ta tai CYP3A:ta lainkaan tai juuri lainkaan. Se ei ole myöskään glukuronidaatioentsyymien (UGT1A3, UGT1A9 tai UGT2B7) estäjä.
Difelikefaliini ei ole myöskään BCRP:n, BSEP:n, LAT1:n, MATE1:n, MATE2-K:n, MRP2:n, OAT1:n, OAT3:n, OATP1A2:n, OATP1B1:n, OATP1B3:n, OCT1:n, OCT2:n, OCT3:n, P‑glykoproteiinin, PEPT1:n tai PEPT2:n estäjä eikä ASBT:n, BCRP:n, BSEP:n, LAT1:n, MATE1:n, MATE2-K:n, MRP2:n, OAT1:n, OAT2:n, OAT3:n, OATP1A2:n, OATP1B1:n, OATP1B3:n, OATP2B1:n, OCT1:n, OCT2:n, OCT3:n, OCTN1:n, OCTN2:n, OSTαβ:n, P‑glykoproteiinin, PEPT1:n tai PEPT2:n substraatti.
Lineaarisuus/ei-lineaarisuus
Terveillä tutkittavilla difelikefaliinin farmakokinetiikan osoitettiin olevan lineaarista ja suhteessa annokseen (testatut annosalueet olivat kerta-annostutkimuksissa 1–40 mikrogrammaa/kg ja toistuvan altistuksen tutkimuksissa 1–20 mikrogrammaa/kg). Suhteellisuus annokseen vakaassa tilassa vahvistettiin myös kroonista munuaistautia sairastavilla hemodialyysipotilailla, jotka saivat toistuvasti 0,5–2,5 mikrogrammaa/kg 3 kertaa viikossa 1 viikon ajan. Eräässä toisessa tutkimuksessa suhteellisuus annokseen todettiin kuitenkin annoksilla 0,5 ja 1 mikrogrammaa/kg mutta ei annoksella 1,5 mikrogrammaa/kg. Plasman pienimmät lääkeainepitoisuudet saavuttivat vakaan tilan toiseen annokseen mennessä, ja annoksella 0,5 mikrogrammaa/kg keskimääräinen kertymissuhde oli yhdessä tutkimuksessa 1,144 AUC0–48h-arvon perusteella ja toisessa tutkimuksessa 1,33 AUC0–44h-arvon perusteella, mikä osoittaa, että kertymisparametrit voivat vaihdella yli 30 %.
Ominaisuudet tutkittavien tai potilaiden erityisryhmissä
Saatavilla olevan näytön perusteella ei ole viitteitä siitä, että iän, sukupuolen, etnisen alkuperän tai lievän tai keskivaikean maksan vajaatoiminnan kaltaisilla tekijöillä olisi mitään vaikutusta difelikefaliinin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Lisääntymistoksisuus
Rotilla ei todettu vaikutuksia urosten tai naaraiden hedelmällisyyteen, varhaisvaiheen alkionkehitykseen eikä prenataaliseen tai postnataaliseen kehitykseen altistuksella, joka oli jopa 2 000‑kertainen ihmisen AUC-arvoon verrattuna. Myöskään kaniineilla ei todettu prenataaliseen kehitykseen kohdistuvia vaikutuksia, vaikka ihmisen AUC-arvoon nähden 30‑kertainen altistus aiheutti emoille huomattavia haittavaikutuksia.
Rotilla difelikefaliini läpäisee istukan.
Väärinkäytön ja riippuvuuden mahdollisuus
Rotilla tehtyjen väärinkäytön ja riippuvuuden mahdollisuutta selvittäneiden tutkimusten mukaan difelikefaliiniin ei todennäköisesti liity fyysisen riippuvuuden tai väärinkäytön riskiä.
Farmaseuttiset tiedot
Apuaineet
Etikkahappo (pH:n säätöön)
Natriumasetaattitrihydraatti (pH:n säätöön)
Natriumkloridi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KAPRUVIA injektioneste, liuos
50 mikrog/ml (L:ei) 12 x 1 ml (647,04 €)
PF-selosteen tieto
Kapruvia toimitetaan kertakäyttöisissä 2 ml:n lasisissa (tyypin I lasista valmistetuissa) injektiopulloissa, joissa on bromobutyylikumitulppa, alumiinisinetti ja sininen irti napsautettava muovikorkki.
Pakkauskoot: 3 ja 12 injektiopulloa, jotka sisältävät 1 ml injektionestettä.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön liuos, jossa ei ole hiukkasia (pH 4,5).
Käyttö- ja käsittelyohjeet
Vain kertakäyttöön.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KAPRUVIA injektioneste, liuos
50 mikrog/ml 12 x 1 ml
- Ei korvausta.
ATC-koodi
V03AX04
Valmisteyhteenvedon muuttamispäivämäärä
01.05.2026
Yhteystiedot
Gustav III:s Boulevard 46
SE-169 73 Solna
Sverige
+46 8 558 066 00
www.viforpharma.se
Info.nordic@viforpharma.com