
KERENDIA tabletti, kalvopäällysteinen 10 mg, 20 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Kerendia 10 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 10 mg finerenonia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 45 mg laktoosia (monohydraattina), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Kerendia 20 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 20 mg finerenonia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 40 mg laktoosia (monohydraattina), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Kerendia 40 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 40 mg finerenonia.
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 25 mg laktoosia (monohydraattina), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti)
Kliiniset tiedot
Käyttöaiheet
Kerendia on tarkoitettu tyypin 2 diabetekseen liittyvän kroonisen munuaistaudin (johon liittyy albuminuria) hoitoon aikuisille. Tutkimustulokset, jotka koskevat munuais- ja kardiovaskulaaritapahtumia, ks. kohta Farmakodynamiikka.
Kerendia on tarkoitettu oireisen kroonisen sydämen vajaatoiminnan hoitoon aikuisille, kun vasemman kammion ejektiofraktio (LVEF) on ≥ 40 %.
Annostus ja antotapa
Annostus
Tyypin 2 diabetekseen (T2D) liittyvä krooninen munuaistauti
Suositeltu tavoiteannos on 20 mg finerenonia kerran vuorokaudessa.
Suurin suositeltu annos on 20 mg finerenonia kerran vuorokaudessa.
Hoidon aloittaminen
Seerumin kaliumtaso ja arvioitu glomerulusten suodatusnopeus (eGFR) on mitattava, jotta voidaan määrittää, voiko finerenonihoidon aloittaa (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet) ja mikä on sopiva aloitusannos.
Seerumin kaliumtason seuraamisesta kerrotaan kohdassa "Hoidon jatkaminen".
Jos seerumin kaliumtaso on ≤ 4,8 mmol/l, finerenonihoito voidaan aloittaa.
Jos seerumin kaliumtaso on > 4,8–5,0 mmol/l, finerenonihoidon aloittamista voidaan harkita, mutta seerumin kaliumtasoa on seurattava ensimmäisen 4 viikon ajan potilaan ominaisuuksien ja seerumin kaliumtasojen mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos seerumin kaliumtaso on > 5,0 mmol/l, finerenonihoitoa ei pidä aloittaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Finerenonin suositeltu aloitusannos perustuu eGFR-arvoon, ks. taulukko 1.
Taulukko 1: Finerenonihoidon aloittaminen ja suositeltu annos
| eGFR (ml/min/1,73 m2) | Aloitusannos (kerran vuorokaudessa) |
| ≥ 60 | 20 mg |
| ≥ 25 – < 60 | 10 mg |
| < 25 | Hoitoa ei suositella |
Hoidon jatkaminen
Seerumin kaliumtaso ja eGFR on mitattava uudelleen 4 viikkoa finerenonihoidon aloittamisen tai uudelleenaloittamisen tai annoksen suurentamisen jälkeen (ks. taulukosta 2 lisätietoa finerenonihoidon jatkamisesta ja annoksen muuttamisesta).
Sen jälkeen seerumin kaliumtaso on mitattava uudelleen säännöllisesti ja tarpeen mukaan potilaan ominaisuuksien ja seerumin kaliumtasojen perusteella.
Katso lisätietoja kohdista Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Taulukko 2: Finerenonihoidon jatkaminen ja annoksen muuttaminen
| Nykyinen finerenoniannos (kerran vuorokaudessa) | |||
| 10 mg | 20 mg | ||
| Nykyinen seerumin kaliumtaso (mmol/l) | ≤ 4,8 | Nosta annos 20 mg:aan finerenonia kerran vuorokaudessa* | Jatka annoksella 20 mg kerran vuorokaudessa |
| > 4,8–5,5 | Jatka annoksella 10 mg kerran vuorokaudessa | Jatka annoksella 20 mg kerran vuorokaudessa | |
| > 5,5 | Keskeytä finerenonihoito. Harkitse hoidon aloittamista uudelleen annoksella 10 mg kerran vuorokaudessa, kun seerumin kaliumtaso ≤ 5,0 mmol/l. | Keskeytä finerenonihoito. Aloita hoito uudelleen annoksella 10 mg kerran vuorokaudessa, kun seerumin kaliumtaso ≤ 5,0 mmol/l. | |
* jatka annoksella 10 mg kerran vuorokaudessa, jos eGFR laskee > 30 % edelliseen mittausarvoon verrattuna
Sydämen vajaatoiminta ja vasemman kammion ejektiofraktio (LVEF) ≥ 40 %
Suositeltu tavoiteannos määräytyy finerenonihoidon aloittamisajankohtana vallitsevan munuaisten toiminnan (eGFR) perusteella (ks. taulukko 4):
- 40 mg kerran vuorokaudessa, jos eGFR on ≥ 60 ml/min/1,73 m2
- 20 mg kerran vuorokaudessa, jos eGFR on ≥ 25 – ˂ 60 ml/min/1,73 m2
Suurin suositeltu annos on 40 mg finerenonia kerran vuorokaudessa.
Hoidon aloittaminen
Jos seerumin kaliumtaso on ≤ 5,0 mmol/l, finerenonihoito voidaan aloittaa.
Seerumin kaliumtaso ja arvioitu glomerulusten suodatusnopeus (eGFR) on mitattava, jotta voidaan määrittää, voiko finerenonihoidon aloittaa (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet) ja mikä on sopiva aloitusannos.
Seerumin kaliumtason seuraamisesta kerrotaan kohdassa "Hoidon jatkaminen".
Finerenonin suositeltu aloitusannos perustuu eGFR-arvoon, ks. taulukko 3.
Taulukko 3: Finerenonihoidon aloittaminen ja suositeltu annos
| eGFR (ml/min/1,73 m2) | Aloitusannos (kerran vuorokaudessa) |
| ≥ 60 | 20 mg |
| ≥ 25 – < 60 | 10 mg |
| < 25 | Hoitoa ei suositella |
Hoidon jatkaminen
Seerumin kaliumtaso ja eGFR on mitattava uudelleen 4 viikkoa finerenonihoidon aloittamisen tai uudelleenaloittamisen tai annoksen muuttamisen jälkeen (ks. taulukosta 4 lisätietoa finerenonihoidon jatkamisesta ja annoksen muuttamisesta).
Sen jälkeen seerumin kaliumtaso ja eGFR on mitattava uudelleen säännöllisesti ja tarpeen mukaan potilaan ominaisuuksien ja seerumin kaliumtasojen perusteella.
Katso lisätietoja kohdista Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Taulukko 4: Finerenonihoidon jatkaminen ja annoksen muuttaminen
| Nykyinen finerenoniannos (kerran vuorokaudessa) | |||
10 mg | 20 mg | 40 mg | ||
Nykyinen seerumin kaliumtaso (mmol/l) | ˂ 5,0 | Nosta annos 20 mg:aan finerenonia kerran vuorokaudessa, jos eGFR ei ole pienentynyt > 30 % verrattuna edelliseen mittaukseen | Nosta annos 40 mg:aan finerenonia kerran vuorokaudessa, jos eGFR ei ole pienentynyt > 30 % verrattuna edelliseen mittaukseen Jatka annoksella 20 mg kerran vuorokaudessa, jos eGFR on < 60 ml/min/1,73 m2 hoidon aloittamishetkellä | Jatka annoksella 40 mg kerran vuorokaudessa Pienennä annos 20 mg:aan kerran vuorokaudessa, jos eGFR on pienentynyt > 30 % verrattuna edelliseen mittaukseen |
5,0 – ˂ 5,5 | Jatka annoksella 10 mg kerran vuorokaudessa | Jatka annoksella 20 mg kerran vuorokaudessa | Jatka annoksella 40 mg kerran vuorokaudessa Pienennä annos 20 mg:aan kerran vuorokaudessa, jos eGFR on pienentynyt > 30 % verrattuna edelliseen mittaukseen | |
5,5 – ˂ 6,0 | Keskeytä finerenonihoito. Aloita hoito uudelleen annoksella 10 mg kerran vuorokaudessa, kun seerumin kaliumtaso on ˂ 5,0 mmol/l. | Pienennä annos 10 mg:aan kerran vuorokaudessa | Pienennä annos 20 mg:aan kerran vuorokaudessa | |
≥ 6,0 | Keskeytä finerenonihoito. Aloita hoito uudelleen annoksella 10 mg kerran vuorokaudessa, kun seerumin kaliumtaso on ˂ 5,5 mmol/l, tai jos taso on toistuvasti ≥ 5,5 mmol/l, odota hoidon uudelleenaloituksessa, kunnes taso on ˂ 5,0 mmol/l. | |||
Mikäli eGFR pienenee ≥ 40 % verrattuna edelliseen mittaukseen, on harkittava finerenoniannoksen pienentämistä tai hoidon keskeyttämistä. Kun eGFR-tasot ovat vakautuneet, potilaan yksilöllisten ominaisuuksien mukaan voidaan harkita annoksen suurentamista tai hoidon aloittamista uudelleen.
Unohtunut annos
Potilaan on otettava unohtunut annos heti, kun hän huomaa sen unohtuneen, mutta vain saman päivän aikana.
Potilaan ei pidä ottaa kahta annosta unohtuneen annoksen korvaamiseksi.
Erityisryhmät
Iäkkäät (≥ 65-vuotiaat)
Iäkkäiden potilaiden annostusta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka), mutta munuaisten toiminnan säännöllistä seurantaa suositellaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaisten vajaatoiminta
Hoidon aloittaminen
Jos eGFR on < 25 ml/min/1,73 m2, finerenonihoitoa ei pidä aloittaa niukkojen kliinisten tietojen vuoksi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Hoidon jatkaminen
Jos eGFR on ≥ 15 ml/min/1,73 m2, finerenonihoitoa voidaan jatkaa niin, että annosta muutetaan seerumin kaliumarvoihin perustuen. eGFR on mitattava 4 viikkoa hoidon aloittamisen jälkeen, jotta voidaan määrittää, voidaanko aloitusannosta nostaa (ks. "Annostus", "Hoidon jatkaminen" ja taulukot 2 ja 4).
Finerenonihoito on lopetettava potilailla, joiden munuaistauti on edennyt loppuvaiheeseen (eGFR < 15 ml/min/1,73 m2), koska kliinisiä tietoja on vain vähän (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Potilaat, joilla on
-
vaikea maksan vajaatoiminta:
Finerenonihoitoa ei pidä aloittaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Tietoja ei ole saatavilla. -
keskivaikea maksan vajaatoiminta:
Aloitusannosta ei tarvitse muuttaa. Seerumin kaliumtasojen lisäseurantaa on harkittava ja seuranta mukautettava potilaan ominaisuuksiin (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). -
lievä maksan vajaatoiminta:
Aloitusannosta ei tarvitse muuttaa.
Samanaikainen muiden lääkevalmisteiden käyttö
Jos potilas käyttää finerenonia samanaikaisesti keskivahvojen tai heikkojen CYP3A4:n estäjien, kaliumlisien, trimetopriimin tai trimetopriimi/sulfametoksatsolin kanssa, on harkittava seerumin kaliumtason lisäseurantaa ja seurannan mukauttamista potilaan ominaisuuksien mukaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Finerenonihoitoa koskevat päätökset on tehtävä taulukon 2 ja 4 (ks. ”Annostus”, ”Hoidon jatkaminen”) ohjeiden mukaan.
Finerenonihoidon väliaikainen keskeyttäminen voi olla tarpeen, jos potilaan on otettava trimetopriimiä tai trimetopriimi/sulfametoksatsolia. Katso lisätietoja kohdista Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset.
Paino
Annosta ei tarvitse muuttaa painon perusteella (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Finerenonin turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Tabletit voi ottaa vesilasillisen kanssa aterian yhteydessä tai ilman ruokaa (ks. kohta Farmakokinetiikka).
Tabletteja ei pidä ottaa greipin tai greippimehun kanssa (ks. kohta Yhteisvaikutukset).
Tablettien murskaaminen
Jos potilas ei kykene nielemään kokonaisia tabletteja, Kerendia-tabletit voidaan murskata ja sekoittaa veteen tai pehmeisiin ruokiin, kuten omenasoseeseen, juuri ennen niiden ottamista suun kautta (ks. kohta Farmakokinetiikka).
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Samanaikainen hoito vahvoilla CYP3A4:n estäjillä (ks. kohta Yhteisvaikutukset), esim.
- itrakonatsoli
- ketokonatsoli
- ritonaviiri
- nelfinaviiri
- kobisistaatti
- klaritromysiini
- telitromysiini
- nefatsodoni
- Addisonin tauti
Varoitukset ja käyttöön liittyvät varotoimet
Hyperkalemia
Finerenonihoitoa saaneilla potilailla on havaittu hyperkalemiaa (ks. kohta Haittavaikutukset).
Joillakin potilailla on suurempi hyperkalemian kehittymisen riski.
Riskitekijöitä ovat matala eGFR, korkea seerumin kaliumtaso ja aiemmat hyperkalemiaepisodit. Näitä potilaita on seurattava tiheämmin.
Jos seerumin kaliumtaso on > 5,0 mmol/l, finerenonihoitoa ei pidä aloittaa.
Seuranta
Seerumin kaliumtaso ja eGFR on mitattava uudelleen 4 viikkoa finerenonihoidon aloittamisen, uudelleenaloittamisen tai annoksen muutoksen jälkeen. Sen jälkeen seerumin kaliumtaso on arvioitava säännöllisesti ja tarpeen mukaan potilaan ominaisuuksien ja seerumin kaliumtasojen perusteella (ks. kohta Annostus ja antotapa).
Tyypin 2 diabetekseen liittyvä krooninen munuaistauti
Hoidon aloittaminen ja jatkaminen (ks. kohta Annostus ja antotapa)
Jos seerumin kaliumtaso on > 4,8–5,0 mmol/l, finerenonihoidon aloittamista voidaan harkita, kun seerumin kaliumtasoa seurataan ensimmäisen 4 viikon ajan potilaan ominaisuuksien ja seerumin kaliumtasojen mukaan.
Jos seerumin kaliumtaso on > 5,5 mmol/l, finerenonihoito pitää keskeyttää. Paikallisia ohjeita hyperkalemian hoidosta on noudatettava.
Kun seerumin kaliumtaso on ≤ 5,0 mmol/l, finerenonihoito voidaan aloittaa uudelleen annoksella 10 mg vuorokaudessa.
Sydämen vajaatoiminta ja vasemman kammion ejektiofraktio (LVEF) ≥ 40 %
Hoidon aloittaminen ja jatkaminen (ks. kohta Annostus ja antotapa)
Jos seerumin kaliumtaso on ≥6,0 mmol/l, finerenonihoito pitää keskeyttää. Paikallisia ohjeita hyperkalemian hoidosta on noudatettava. Kun seerumin kaliumtaso on < 5,5 mmol/l, finerenonihoito voidaan aloittaa uudelleen annoksella 10 mg kerran vuorokaudessa. Mikäli seerumin kaliumtaso on toistuvasti mittauksissa ≥ 5,5 mmol/l, finerenonihoidon voi aloittaa uudelleen vasta, kun seerumin kaliumtaso on < 5,0 mmol/l.
Samanaikainen muiden lääkevalmisteiden käyttö
Hyperkalemian riski voi kasvaa myös käytettäessä samanaikaisesti lääkevalmisteita, jotka suurentavat seerumin kaliumtasoja (ks. kohta Yhteisvaikutukset). Katso myös "Samanaikainen finerenonialtistukseen vaikuttavien aineiden käyttö".
Finerenonia ei pidä antaa samanaikaisesti seuraavien kanssa:
- kaliumia säästävät diureetit (esim. amiloridi, triamtereeni) ja
- muut mineralokortikoidireseptorin antagonistit (MRA), esim. eplerenoni, esakserenoni, spironolaktoni, kanrenoni.
Finerenonia on käytettävä varoen ja seerumin kaliumtasoja seurattava, jos sitä käytetään samanaikaisesti seuraavien kanssa:
- kaliumlisät tai kaliumia sisältävät suolan korvikkeet
- trimetopriimi tai trimetopriimi/sulfametoksatsoli. Finerenonihoidon väliaikainen keskeyttäminen voi olla tarpeen.
Munuaisten toiminnan heikkeneminen sydämen vajaatoimintapotilailla, joiden LVEF ≥ 40 %
Munuaisten toiminnan heikkenemistä on raportoitu enemmän sydämen vajaatoimintapotilailla, joiden LVEF ≥ 40 % ja joita hoidetaan finerenonilla (ks. kohta Haittavaikutukset). Säännöllistä munuaisten toiminnan seurantaa suositellaan hoidon aikana ja tarpeen mukaan potilaan ominaisuuksien mukaan. Munuaisten toiminnan heikkenemisen riski on suurempi iäkkäillä potilailla ja potilailla, joilla on munuaisten vajaatoimintaa (eGFR < 60 ml/min/1,73 m2), ja heitä on seurattava tiiviimmin (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Hyperkalemian riski kasvaa munuaisten toiminnan heikentyessä. Munuaisten toimintaa on seurattava tarpeen mukaan jatkuvasti tavanomaisten käytäntöjen mukaisesti (ks. kohta Annostus ja antotapa).
Hoidon aloittaminen
Jos eGFR on < 25 ml/min/1,73 m2, finerenonihoitoa ei pidä aloittaa niukkojen kliinisten tietojen vuoksi (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Hoidon jatkaminen
Kliinisiä tietoja on vain vähän, joten finerenonihoito on lopetettava potilailla, joiden munuaistauti on edennyt loppuvaiheeseen (eGFR < 15 ml/min/1,73 m2).
Maksan vajaatoiminta
Finerenonihoitoa ei pidä aloittaa potilaille, joilla on vaikea maksan vajaatoiminta (ks. kohta Annostus ja antotapa). Näitä potilaita ei ole tutkittu (ks. kohta Farmakokinetiikka), mutta finerenonialtistuksen odotetaan suurenevan merkittävästi.
Jos finerenonia käytetään potilaille, joilla on keskivaikea maksan vajaatoiminta, potilaiden tiheämpi seuranta saattaa olla tarpeen finerenonialtistuksen suurenemisen vuoksi. Seerumin kaliumtasojen lisäseurantaa on harkittava ja seuranta mukautettava potilaan ominaisuuksiin (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Potilaat, joiden New York Heart Association (NYHA) -luokka on IV
Kokemus finerenonihoidosta potilailla, joiden sydämen vajaatoiminta on NYHA-luokkaa IV, on niukkaa (ks. kohta Farmakodynamiikka).
Iäkkäät
Iäkkäillä esiintyy todennäköisemmin munuaisten vajaatoimintaa ja sellaisten lääkevalmisteiden käyttöä, jotka saattavat aiheuttaa munuaistoiminnan muutoksia. Siksi munuaisten toiminnan säännöllistä seurantaa suositellaan.
Samanaikainen finerenonialtistukseen vaikuttavien aineiden käyttö
Keskivahvat ja heikot CYP3A4:n estäjät
Jos keskivahvoja tai heikkoja CYP3A4:n estäjiä käytetään samanaikaisesti finerenonin kanssa, seerumin kaliumtasoa on seurattava (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Vahvat ja keskivahvat CYP3A4:n indusoijat
Finerenonia ei pidä käyttää samanaikaisesti vahvojen tai keskivahvojen CYP3A4:n indusoijien kanssa (ks. kohta Yhteisvaikutukset).
Greippi
Greippiä tai greippimehua ei pidä nauttia finerenonihoidon aikana (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Alkio-/sikiötoksisuus
Finerenonia ei pidä käyttää raskauden aikana, ellei äidille koituvaa hyötyä ja sikiöön kohdistuvaa vaaraa ole harkittu huolellisesti. Jos nainen tulee raskaaksi finerenonihoidon aikana, hänelle on kerrottava mahdollisista sikiöön kohdistuvista riskeistä.
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä finerenonihoidon aikana.
Naisia on neuvottava pidättäytymään imetyksestä finerenonihoidon aikana.
Katso lisätietoja kohdista Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta.
Tietoa apuaineista
Kerendia sisältää laktoosia
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi‑imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Kerendia sisältää natriumia
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Finerenoni eliminoituu lähes täysin sytokromi P450 (CYP) ‑välitteisen oksidatiivisen metabolian kautta (pääasiassa CYP3A4:n [90 %] ja vähäisessä määrin CYP2C8:n [10 %] välityksellä).
Samanaikainen käyttö vasta-aiheista
Vahvat CYP3A4:n estäjät
Kerendia-valmisteen samanaikainen käyttö itrakonatsolin, klaritromysiinin ja muiden vahvojen CYP3A4:n estäjien kanssa (esim. ketokonatsoli, ritonaviiri, nelfinaviiri, kobisistaatti, telitromysiini ja nefatsodoni) on vasta-aiheista (ks. kohta Vasta-aiheet), koska finerenonialtistuksen odotetaan kasvavan merkittävästi.
Samanaikaista käyttöä ei suositella
Vahvat ja keskivahvat CYP3A4:n indusoijat
Kerendia-valmistetta ei pidä käyttää samanaikaisesti rifampisiinin ja muiden vahvojen CYP3A4:n indusoijien (esim. karbamatsepiini, fenytoiini, fenobarbitaali, mäkikuisma) tai efavirentsin ja muiden keskivahvojen CYP3A4:n indusoijien kanssa. Näiden CYP3A4:n indusoijien odotetaan pienentävän finerenonin pitoisuutta plasmassa merkittävästi, mikä heikentää hoidon vaikutusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tietyt lääkevalmisteet, jotka nostavat seerumin kaliumtasoja
Kerendia-valmistetta ei pidä käyttää samanaikaisesti kaliumia säästävien diureettien (esim. amiloridi, triamtereeni) ja muiden MRA-valmisteiden kanssa (esim. eplerenoni, esakserenoni, spironolaktoni, kanrenoni). On odotettavissa, että nämä lääkevalmisteet suurentavat hyperkalemian riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Greippi
Finerenonihoidon aikana ei pidä nauttia greippiä tai greippimehua, koska sen odotetaan CYP3A4:n eston myötä nostavan finerenonin pitoisuuksia plasmassa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikainen käyttö varoen
Keskivahvat CYP3A4:n estäjät
Kliinisessä tutkimuksessa samanaikainen erytromysiinin (500 mg kolme kertaa vuorokaudessa) käyttö johti finerenonin AUC-arvon suurenemiseen 3,5‑kertaiseksi ja Cₘₐₓ-arvon suurenemiseen 1,9‑kertaiseksi. Toisessa kliinisessä tutkimuksessa verapamiili (240 mg:n säädellysti vapauttava tabletti kerran vuorokaudessa) johti finerenonin AUC-arvon suurenemiseen 2,7‑kertaiseksi ja Cₘₐₓ-arvon suurenemiseen 2,2‑kertaiseksi.
Seerumin kaliumtaso voi nousta, minkä vuoksi sen seuraaminen on suositeltavaa erityisesti silloin, kun finerenoni tai CYP3A4:n estäjä aloitetaan tai annosta muutetaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Heikot CYP3A4:n estäjät
Fysiologiaan perustuvat farmakokineettiset mallinnukset viittaavat siihen, että fluvoksamiini (100 mg kahdesti vuorokaudessa) suurentaa finerenonin AUC-arvoa (1,6‑kertaiseksi) ja Cₘₐₓ-arvoa (1,4‑kertaiseksi).
Seerumin kaliumtaso voi nousta, minkä vuoksi sen seuraaminen on suositeltavaa erityisesti silloin, kun finerenoni tai CYP3A4:n estäjä aloitetaan tai annosta muutetaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Tietyt lääkevalmisteet, jotka nostavat seerumin kaliumtasoja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Kerendia-valmisteen samanaikaisen käytön kaliumlisien ja trimetopriimin tai trimetopriimi/sulfametoksatsolin kanssa odotetaan lisäävän hyperkalemian riskiä. Seerumin kaliumtasoja on seurattava.
Kerendia-hoidon väliaikainen keskeyttäminen trimetopriimi- tai trimetopriimi/sulfametoksatsolihoidon ajaksi voi olla tarpeen.
Verenpainelääkkeet
Hypotension riski kasvaa, jos samanaikaisesti käytetään useita muita verenpainelääkkeitä. Näillä potilailla suositellaan verenpaineen seurantaa.
40 mg:n finerenoniannoksen vaikutus CYP3A4- ja CYP2C8-substraatteihin
Kun annos on 40 mg kerran vuorokaudessa, finerenoni on CYP3A4-entsyymin heikko inhibiittori in vivo. Samanaikainen useiden 40 mg:n finerenoniannosten anto yhdessä CYP3A4-testisubstraatin, midatsolaamin, kanssa tuotti 1,31-kertaisen midatsolaamin keskimääräisen AUC-arvon suurenemisen ilman vaikutusta Cₘₐₓ-arvoon. Herkkien CYP3A4-substraattien, joilla on kapea terapeuttinen ikkuna, mahdollisesti suurentunut altistus on otettava huomioon käytettäessä samanaikaisesti 40 mg:n finerenoniannosta vuorokaudessa. Toistuvan annostelun tutkimuksessa kerran vuorokaudessa annostellulla 20 mg:n finerenoniannoksella 10 vuorokauden ajan ei ollut oleellista vaikutusta CYP3A4:n testisubstraatin, midatsolaamin, AUC-arvoon. Siten finerenonin kliinisesti relevantti CYP3A4:n esto tai induktio voidaan sulkea pois tällä annostasolla.
Kun annos on 40 mg kerran vuorokaudessa, finerenoni on CYP2C8-entsyymin heikko inhibiittori in vivo. Samanaikainen useiden 40 mg:n finerenoniannosten anto yhdessä CYP2C8-testisubstraatin, repaglinidin, kanssa tuotti 1,59-kertaisen repaglinidin keskimääräisen AUC-arvon suurenemisen sekä 1,30-kertaisen Cₘₐₓ-arvon suurenemisen. Herkkien CYP2C8-substraattien, joilla on kapea terapeuttinen ikkuna, mahdollisesti suurentunut altistus on otettava huomioon käytettäessä samanaikaisesti 40 mg:n finerenoniannosta vuorokaudessa.
Yksittäisellä 20 mg:n finerenoniannoksella ei ollut kliinisesti relevanttia vaikutusta CYP2C8:n testisubstraatin, repaglinidin, AUC- tai Cₘₐₓ-arvoon. Siten finerenoni ei inhiboi CYP2C8:a tällä annostasolla.
Raskaus ja imetys
Raskauden ehkäisy naisilla
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä finerenonihoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Ei ole olemassa tietoja finerenonin käytöstä raskaana oleville naisille.
Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Kerendia-valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa finerenonilla. Jos nainen tulee raskaaksi finerenonihoidon aikana, hänelle on kerrottava mahdollisista sikiöön kohdistuvista riskeistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö finerenoni/metaboliitit ihmisen rintamaitoon.
Olemassa olevat farmakokineettiset/toksikologiset tiedot koe-eläimistä ovat osoittaneet finerenonin ja sen metaboliittien erittyvän rintamaitoon. Tätä reittiä altistetuilla rottien poikasilla ilmeni haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
On päätettävä lopetetaanko rintaruokinta vai lopetetaanko Kerendia-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hedelmällisyys
Tietoa finerenonin vaikutuksesta ihmisen hedelmällisyyteen ei ole.
Eläinkokeissa on havaittu naaraiden hedelmällisyyden heikkenemistä ihmisen suurinta altistusta suuremmilla altistuksilla, mikä osoittaa kliinisen relevanssin olevan pieni (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kerendialla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Finerenonihoidon aikana useimmin ilmoitettu haittavaikutus oli hyperkalemia (12,6 %). Ks. "Valikoitujen haittavaikutusten kuvaus, Hyperkalemia" jäljempänä ja kohta Varoitukset ja käyttöön liittyvät varotoimet.
Haittavaikutustaulukko
Finerenonin turvallisuutta potilaille, joilla on krooninen munuaistauti (CKD) ja (T2D), arvioitiin kahdessa vaiheen III avaintutkimuksessa FIDELIO‑DKD (diabeteksen munuaistauti) ja FIGARO-DKD. FIDELIO-DKD-tutkimuksessa 2 818 potilasta sai finerenonia (10 mg tai 20 mg kerran vuorokaudessa) hoidon kestäessä keskimäärin 2,2 vuotta. FIGARO-DKD-tutkimuksessa 3 671 potilasta sai finerenonia (10 mg tai 20 mg kerran vuorokaudessa) hoidon kestäessä keskimäärin 2,9 vuotta.
Finerenonin turvallisuutta sydämen vajaatoimintapotilailla, joiden LVEF on ≥ 40 %, arvioitiin vaiheen III tutkimuksessa FINEARTS-HF. Tässä tutkimuksessa 2 993 potilasta sai finerenonia (10 mg, 20 mg tai 40 mg kerran vuorokaudessa) hoidon kestäessä keskimäärin 2,1 vuotta.
Havaitut haittavaikutukset on lueteltu taulukossa 5. Ne on luokiteltu MedDRA:n elinjärjestelmäluokkien sekä esiintymistiheyksien mukaan.
Haittavaikutukset on ryhmitelty niiden esiintymistiheyden mukaan laskevassa vakavuusjärjestyksessä.
Esiintymistiheydet ovat seuraavat:
hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 5: Haittavaikutukset
| Elinjärjestelmäluokka | Esiintymistiheys | CKD ja T2D | HF ja LVEF ≥ 40 % |
| Aineenvaihdunta ja ravitsemus | Hyvin yleinen | Hyperkalemia | – |
| Yleinen | Hyponatremia Hyperurikemia | Hyperkalemia Hyponatremia Hyperurikemia | |
| Verisuonisto | Yleinen | Hypotensio | Hypotensio |
| Ruoansulatuselimistö | Yleinen | – | Ripuli Ummetus |
| Iho ja ihonalainen kudos | Yleinen | Kutina | – |
| Munuaiset ja virtsatiet | Yleinen | – | Munuaisten vajaatoiminta Akuutti munuaisvaurio |
| Tutkimukset | Yleinen | Veren kreatiniiniarvon nousu / glomerulusten suodatusnopeuden pieneneminen | Veren kreatiniiniarvon nousu / glomerulusten suodatusnopeuden pieneneminen |
| Melko harvinainen | Hemoglobiiniarvon pienentyminen | – |
Valikoitujen haittavaikutusten kuvaus
Hyperkalemia
Ensimmäisen hoitokuukauden aikana seerumin keskimääräisen kaliumpitoisuuden havaittiin nousseen enintään 0,2 mmol/l lähtötilanteesta finerenoniryhmässä verrattuna lumelääkeryhmään, minkä jälkeen kaliumpitoisuus pysyi vakaana.
FIDELIO‑DKD- ja FIGARO-DKD-tutkimusten yhteisanalyysissä hyperkalemiatapauksia raportoitiin 14,0 %:lla finerenonia ja 6,9 %:lla lumelääkettä saaneista potilaista. Vakavia hyperkalemiatapauksia raportoitiin enemmän finerenonihoitoa saaneilla (1,1 %) kuin lumelääkettä saaneilla (0,2 %). Seerumin kaliumpitoisuuksia > 5,5 mmol/l raportoitiin 16,8 %:lla finerenonihoitoa saaneista ja 7,4 %:lla lumelääkettä saaneista potilaista ja pitoisuuksia > 6,0 mmol/l 3,3 %:lla finerenonihoitoa saaneista ja 1,3 %:lla lumelääkettä saaneista potilaista. Pysyvään hoidon lopetukseen johtanutta hyperkalemiaa ilmeni 1,7 %:lla finerenonihoitoa saaneista potilaista ja 0,6 %:lla lumelääkettä saaneista potilaista. Finerenoniryhmässä 0,9 % tutkittavista ja lumelääkeryhmässä 0,2 % tutkittavista joutui sairaalahoitoon hyperkalemian vuoksi.
FINEARTS-HF-tutkimuksessa hyperkalemiatapauksia raportoitiin 9,7 %:lla finerenonia ja 4,2 %:lla lumelääkettä saaneista potilaista. Pysyvään hoidon lopetukseen johtanutta hyperkalemiaa ilmeni 0,4 %:lla finerenonihoitoa saaneista potilaista ja 0,2 %:lla lumelääkettä saaneista potilaista. Finerenoniryhmässä 0,5 % tutkittavista ja lumelääkeryhmässä 0,2 % tutkittavista joutui sairaalahoitoon hyperkalemian vuoksi.
Kaikissa tutkimuksissa suurin osa hyperkalemiatapauksista oli lieviä tai keskivaikeita ja korjaantuvia finerenonihoitoa saaneilla potilailla.
Tarkat suositukset, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Munuaisten toiminnan heikkeneminen
FIDELIO-DKD- ja FIGARO-DKD-tutkimusten yhteisanalyysissä GFR:n pienentymistapauksia raportoitiin 5,4 %:lla finerenonihoitoa saaneista potilaista ja 4,2 %:lla lumelääkettä saaneista potilaista. Pysyvään hoidon lopetukseen johtanutta GFR:n pienentymistä ilmeni yhtä paljon finerenonihoitoa saaneilla potilailla ja lumelääkettä saaneilla potilailla (0,2 %). GFR:n pienentymisen vuoksi sairaalahoitoon joutuneiden osuus oli sama potilailla, jotka saivat finerenonia tai lumelääkettä (< 0,1 %).
Veren kreatiniiniarvojen nousun tapauksia raportoitiin 2,6 %:lla finerenonihoitoa saaneilla potilailla verrattuna lumelääkehoitoa saaneiden potilaiden 2,3 %:iin.
Suurin osa GFR:n pienentymistapauksista / veren kreatiniiniarvon nousun tapauksista oli lieviä tai keskivaikeita ja korjaantuvia finerenonihoitoa saaneilla potilailla. Finerenonihoitoa saaneilla potilailla ilmeni aluksi eGFR:n pienentymistä (keskimäärin 2 ml/min/1,73 m2), joka lieveni ajan myötä lumelääkkeeseen verrattuna. Tämä pienentyminen vaikutti häviävän hoidon jatkuessa.
FINEARTS-HF-tutkimuksessa munuaisten toiminnan heikkenemiseen liittyviä tapauksia raportoitiin useammin finerenonihoitoa saaneilla potilailla (17,7 %) kuin lumelääkehoitoa saaneilla (10,9 %). Raportoituja tapahtumia olivat munuaisten toiminnan heikentyminen (6,6 % vs. 3,9 %), GFR:n pienentyminen (5,2 % vs. 3,6 %), akuutti munuaisvaurio (3,7 % vs. 2,1 %), munuaisten vajaatoiminta (2,6 % vs. 1,6 %) ja veren kreatiniinitason nousu (1,2 % vs. 0,8 %).
Yleisesti ottaen useimmat raportoidut tapahtumat, jotka liittyivät munuaisten toiminnan heikkenemiseen, olivat lieviä tai keskivaikeita ja korjaantuvia finerenonihoitoa saaneilla potilailla. Joissakin tapauksissa, joissa finerenonihoito lopetettiin pysyvästi, eGFR-arvo ei palannut lähtötasolle. Munuaisten toiminnan heikkenemiseen liittyvät tapahtumat, jotka johtivat pysyvään hoidon lopetukseen, olivat identtisiä finerenoni- ja lumelääkeryhmissä (0,3 %). Sairaalahoitoa edellyttäneitä munuaisten toiminnan heikkenemisen tapauksia raportoitiin useammin finerenonihoitoa saaneilla potilailla kuin lumelääkeryhmässä (2,0 % vs. 1,3 %). Akuutti munuaisvaurio oli useimmin raportoitu sairaalahoitoa edellyttävä tapahtuma (1,6 % finerenoniryhmässä vs. 0,8 % lumelääkeryhmässä).
Finerenonia saaneilla potilailla GFR pieneni aluksi, mutta se vaikutti korjaantuvan jatkuvan hoidon aikana. GFR:n keskimääräinen ero finerenoni- ja lumelääkeryhmän välillä oli noin 3‑4 ml/min/1,73 m2 kuukaudesta 1 kuukauteen 6. Kuukauden 6 jälkeen GFR:n pienentyminen oli hieman suurempaa lumelääkeryhmässä, ja keskimääräinen ero finerenoni- ja lumelääkeryhmän välillä oli noin 2‑3 ml/min/1,73 m2.
Hypotensio
Finerenonihoitoa saaneiden potilaiden keskimääräinen systolinen verenpaine laski 2–4 mmHg ja keskimääräinen diastolinen verenpaine laski 1–2 mmHg ensimmäisen kuukauden aikana ja pysyi vakaana sen jälkeen.
FIDELIO‑DKD- ja FIGARO-DKD-tutkimusten yhteisanalyysissä hypotensiotapauksia raportoitiin 4,7 %:lla finerenonihoitoa ja 3,0 %:lla lumelääkettä saaneista potilaista. Kolmen potilaan (< 0,1 %) finerenonihoito lopetettiin pysyvästi hypotension vuoksi. Hypotension takia sairaalahoitoon joutuneiden osuus oli sama finerenonihoitoa saaneilla potilailla ja lumelääkettä saaneilla potilailla (0,1 %).
FINEARTS‑HF-tutkimuksessa hypotensiotapauksia raportoitiin 7,6 %:lla finerenonihoitoa ja 4,7 %:lla lumelääkettä saaneista potilaista. Kolmen potilaan (0,1 %) finerenonihoito lopetettiin pysyvästi hypotension vuoksi. Finerenoniryhmässä 0,4 % tutkittavista ja lumelääkeryhmässä 0,3 % tutkittavista joutui sairaalahoitoon hypotension vuoksi.
Kaikissa tutkimuksissa suurin osa hypotensiotapauksista oli lieviä tai keskivaikeita ja ohimeneviä finerenonihoitoa saaneilla potilailla.
Hyperurikemia
FIDELIO‑DKD- ja FIGARO-DKD-tutkimusten yhteisanalyysissä hyperurikemiatapauksia raportoitiin 5,1 %:lla finerenonihoitoa ja 3,9 %:lla lumelääkettä saaneista potilaista. Seerumin keskimääräisen uraattipitoisuuden havaittiin nousseen 0,3 mg/dl lähtötilanteesta finerenoniryhmässä verrattuna lumelääkeryhmään kuukauteen 16 asti, mutta tämä lieveni ajan myötä. Finerenonihoitoa saaneilla potilailla kihtiä raportoitiin (3,1 %) verrattavissa yhtä paljon kuin lumelääkettä saaneilla potilailla (3,0 %).
FINEARTS-HF-tutkimuksessa finerenonihoitoa saaneilla potilailla hyperurikemiaa raportoitiin (2,7 %) verrattavissa samoja määriä kuin lumelääkettä saaneilla potilailla (2,4 %). Finerenonihoitoa saaneilla potilailla kihtiä raportoitiin (2,4 %) verrattavissa yhtä paljon kuin lumelääkettä saaneilla potilailla (2,8 %).
Missään tutkimuksessa hyperurikemiatapaukset eivät olleet vakavia eivätkä johtaneet hoidon pysyvään lopettamiseen finerenonia saaneilla potilailla.
Ruoansulatuselimistö
FINEARTS-HF-tutkimuksissa ripulia (5,7 % vs. 4,4 %) ja ummetusta (3,8 % vs. 2,7 %) raportoitiin useammin finerenoniryhmässä kuin lumelääkeryhmässä.
Hemoglobiiniarvon pienentyminen
FIDELIO-DKD- ja FIGARO-DKD-tutkimusten yhteisanalyysissä plasebokorjattu hemoglobiiniarvo oli pienentynyt finerenoniryhmässä keskimäärin 0,15 g/dl ja hematokriittiarvo 0,5 % hoidon kestettyä 4 kuukautta. Finerenonihoitoa saaneilla potilailla anemian raporointi (6,5 %) oli verrattavissa lumelääkettä saaneisiin potilaisiin (6,1 %). Vaikeita anemiatapauksia oli vähän sekä finerenonihoitoa saaneilla potilailla ja lumelääkettä saaneilla potilailla (0,5 %). Hemoglobiini- ja hematokriittiarvojen muutokset olivat ohimeneviä, ja arvot palautuivat 24–32 kuukauden kuluessa tasolle, joka oli verrattavissa lumelääkettä saaneiden potilaiden arvoihin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus ilmenee todennäköisimmin hyperkalemiana. Jos hyperkalemiaa esiintyy, siihen on annettava tavanomaista hoitoa.
Finerenoni ei todennäköisesti poistu tehokkaasti hemodialyysillä, koska sen plasmaproteiineihin sitoutumisen fraktio on noin 90 %.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: diureetit, aldosteroniantagonistit, ATC-koodi: C03DA05
Vaikutusmekanismi
Finerenoni on ei-steroidaalinen, selektiivinen antagonisti mineralokortikoidireseptorille (MR), joka aktivoituu aldosteronin ja kortisolin vaikutuksesta ja säätelee geenin transkriptiota. Se sitoutuu MR:ään muodostaen erityisen reseptoriligandikompleksin, joka estää tulehdusta ja fibroosia edistävien välittäjäaineiden ekspressioon osallistuvien transkriptiokoaktivaattorien sitoutumista.
Farmakodynaamiset vaikutukset
Satunnaistetuissa, kaksoissokkoutetuissa, lumelääkekontrolloiduissa vaiheen III monikeskustutkimuksissa FIDELIO-DKD ja FIGARO-DKD, joihin osallistui aikuisia CKD- ja T2D-potilaita, lumelääkekorjattu virtsan albumiinin ja kreatiniinin suhteen (UACR) suhteellinen pieneneminen oli finerenoniryhmään satunnaistetuilla potilailla 31 % ja 32 % neljän kuukauden kuluessa, ja UACR pysyi pienentyneenä molempien tutkimusten keston ajan.
Satunnaistettussa, kaksoissokkoutettussa, lumelääkekontrolloidussa, vaiheen III monikeskustutkimuksessa FINEARTS‑HF, johon osallistui aikuisia sydämen vajaatoimintapotilaita, joiden LVEF oli ≥ 40 %, lumelääkekorjattu UACR:n suhteellinen pieneneminen finerenoniryhmään satunnaistetuilla potilailla oli 30 % 6 kuukauden kohdalla, ja UACR pysyi pienentyneenä viimeiseen mittaukseen asti 2 vuoden kohdalla.
Satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa vaiheen IIb monikeskustutkimuksessa ARTS-DN, johon osallistui aikuisia CKD- ja T2D-potilaita, lumelääkekorjattu UACR:n suhteellinen pieneneminen 90 päivän kuluessa oli 25 % 10 mg finerenonia kerran vuorokaudessa saaneilla potilailla ja 38 % 20 mg finerenonia kerran vuorokaudessa saaneilla potilailla.
Sydämen elektrofysiologia
Erillisessä QT-tutkimuksessa, johon osallistui 57 tervettä vapaaehtoista, osoitettiin, että finerenonilla ei ole vaikutusta sydämen repolarisaatioon. Merkkejä finerenonin vaikutuksesta QT-/QTc-ajan pitenemiseen yksittäisten 20 mg:n (terapeuttinen) tai 80 mg:n (supraterapeuttinen) annosten jälkeen ei havaittu.
Kliininen teho ja turvallisuus
Tyypin 2 diabetekseen liittyvä krooninen munuaistauti
FIDELIO-DKD- ja FIGARO-DKD-tutkimuksissa tutkittiin finerenonin vaikutusta munuaisiin ja sydämeen ja verisuonistoon liittyviin päätetapahtumiin lumelääkkeeseen verrattuna aikuispotilailla, joilla oli CKD ja T2D.
Potilaiden edellytettiin saavan tavanomaista hoitoa, mukaan lukien suurin siedetty käyttöaiheen mukainen annos angiotensiinikonvertaasin estäjää (ACE:n estäjä) tai angiotensiinireseptorin salpaajaa (ARB).
Tutkimukseen ei otettu potilaita, joilla oli diagnosoitu sydämen vajaatoiminta, johon liittyi alentunut ejektiofraktio ja NYHA-luokka II–IV, koska heille luokan 1A-suosituksena on MRA-hoito.
Potilaat soveltuivat FIDELIO-DKD-tutkimukseen, mikäli heillä havaittiin seulontahetkellä todisteita pysyvästä albuminuriasta (> 30–5 000 mg/g), heidän eGFR-arvonsa oli 25–75 ml/min/1,73 m2 ja seerumin kaliumtasonsa ≤ 4,8 mmol/l.
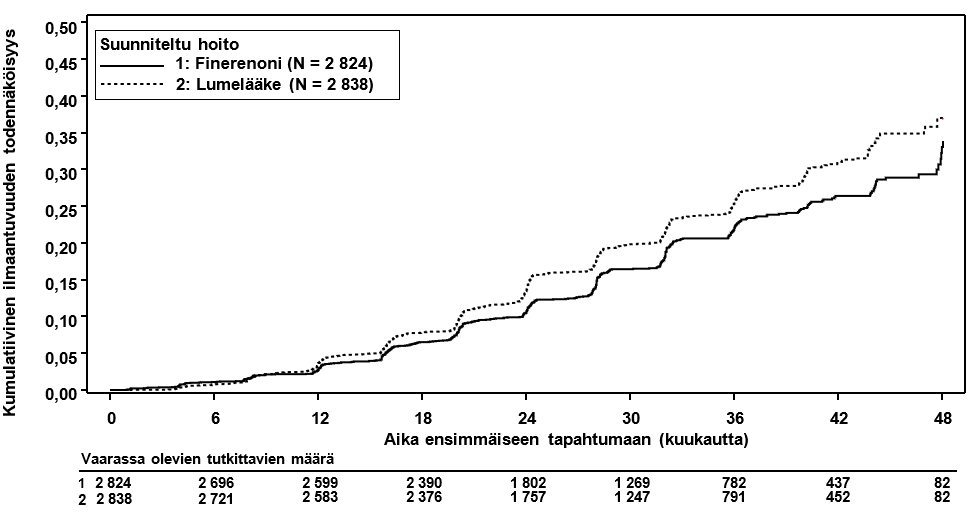
Ensisijainen päätetapahtuma oli seuraava yhdistelmä: aika ensimmäiseen munuaisen vajaatoiminnan esiintymiskertaan (määritelty kroonisena dialyysina tai munuaissiirteenä tai pysyvänä eGFR:n pienentymisenä tasolle < 15 ml/min/1,73 m2 vähintään 4 viikon ajan), pysyvään eGFR:n pienentymiseen vähintään 40 %:lla lähtötilanteeseen verrattuna vähintään 4 viikon ajan tai munuaiskuolemaan. Keskeinen toissijainen päätetapahtuma oli seuraava yhdistelmä: aika ensimmäiseen kardiovaskulaariseen kuolemaan, ei‑fataaliin sydäninfarktiin (MI), ei‑fataaliin aivohalvaukseen tai sairaalahoitoon sydämen vajaatoiminnan vuoksi.
Kaikkiaan 5 662 potilasta satunnaistettiin saamaan joko finerenonia (N = 2 824) tai lumelääkettä (N = 2 838) ja analysoitiin. Seurannan mediaanikesto oli 2,6 vuotta. Finerenoni- tai lumelääkeannosta voitiin muuttaa 10 mg:n ja 20 mg:n vuorokausiannoksen välillä tutkimuksen aikana, pääasiassa seerumin kaliumpitoisuuden perusteella. Kuukauden 24 kohdalla finerenonilla hoidetuista tutkittavista 67 % käytti annosta 20 mg kerran vuorokaudessa, 31 % annosta 10 mg kerran vuorokaudessa ja 3 % oli keskeyttänyt hoidon.
Tutkimuksen päättymisen jälkeen elossaolostatus saatiin selville 99,7 %:sta potilaista. Tutkittavista 63 % oli valkoihoisia, 25 % aasialaisia ja 5 % tummaihoisia. Keskimääräinen ikä kirjautumishetkellä oli 66 vuotta, ja 70 % tutkittavista oli miehiä. Lähtötilanteessa keskimääräinen eGFR oli 44,4 ml/min/1,73 m2, ja 55 %:lla potilaista eGFR oli < 45 ml/min/1,73 m2, mediaani UACR oli 853 mg/g ja keskimääräinen HbA1c oli 7,7 %, 46 %:lla potilaista oli aiemmin todettu ateroskleroottinen sydän- ja verisuonitauti, 30 %:lla sepelvaltimotauti, 8 %:lla sydämen vajaatoiminta, ja potilaiden verenpaine oli keskimäärin 138/76 mmHg. T2D:n keskimääräinen kesto lähtötilanteessa oli 16,6 vuotta ja aiempaa diabeettista retinopatiaa raportoitiin 47 %:lla potilaista ja diabeettista neuropatiaa 26 %:lla potilaista. Lähtötilanteessa lähes kaikki potilaat saivat ACE:n estäjä- (34 %) tai ARB-hoitoa (66 %), ja 97 % potilaista käytti yhtä tai useampaa diabeteslääkettä (insuliini [64 %], biguanidit [44 %], glukagonin kaltaisen peptidi‑1:n [GLP‑1] reseptoriagonistit [7 %], natriumin- ja glukoosinkuljettajaproteiini 2:n [SGLT2] estäjät [5 %]). Yleisimmät muut lähtötilanteessa käytetyt lääkevalmisteet olivat statiinit (74 %) ja kalsiumkanavan salpaajat (63 %).
Tilastollisesti merkitsevä ero finerenonin eduksi osoitettiin ensisijaiseen yhdistettyyn päätetapahtumaan ja keskeiseen toissijaiseen yhdistettyyn päätetapahtumaan (ks. kuva 1 / taulukko 6 jäljempänä). Hoitovaikutus keskeisiin ensisijaisiin ja toissijaisiin päätetapahtumiin oli yleisesti ottaen yhdenmukainen eri alaryhmissä, mukaan lukien alue, eGFR, UACR, systolinen verenpaine (SBP) ja HbA1c lähtötilanteessa.
Potilaat soveltuivat FIGARO-DKD-tutkimukseen, mikäli heillä havaittiin seulontahetkellä todisteita pysyvästä albuminuriasta: heidän UACR-arvonsa oli ≥ 30 – < 300 mg/g ja eGFR-arvonsa 25‑90 ml/min/1,73 m2, tai heidän UACR-arvonsa oli ≥ 300 mg/g ja eGFR-arvonsa ≥ 60 ml/min/1,73 m2. Potilaiden seerumin kaliumtason edellytettiin olevan seulontahetkellä ≤ 4,8 mmol/l.
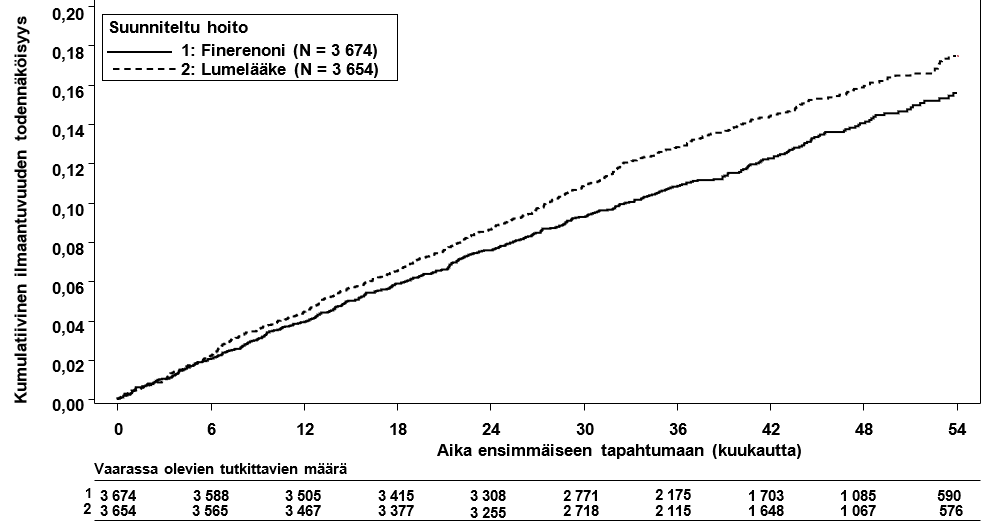
Ensisijainen päätetapahtuma oli seuraava yhdistelmä: aika ensimmäiseen kardiovaskulaariseen kuolemaan, ei‑fataaliin sydäninfarktiin (MI), ei‑fataaliin aivohalvaukseen tai sairaalahoitoon sydämen vajaatoiminnan vuoksi. Toissijainen päätetapahtuma oli seuraava yhdistelmä: aika ensimmäiseen munuaisen vajaatoiminnan esiintymiskertaan, pysyvään eGFR:n pienentymiseen vähintään 40 %:lla lähtötilanteeseen verrattuna vähintään 4 viikon ajan tai munuaiskuolemaan.
Kaikkiaan 7 328 potilasta satunnaistettiin saamaan joko finerenonia (N = 3 674) tai lumelääkettä (N = 3 654) ja analysoitiin. Seurannan mediaanikesto oli 3,4 vuotta. Finerenoni- tai lumelääkeannosta voitiin muuttaa 10 mg:n ja 20 mg:n vuorokausiannoksen välillä tutkimuksen aikana, pääasiassa seerumin kaliumpitoisuuden perusteella. Kuukauden 24 kohdalla finerenonilla hoidetuista tutkittavista 82 % käytti annosta 20 mg kerran vuorokaudessa, 15 % annosta 10 mg kerran vuorokaudessa ja 3 % oli keskeyttänyt hoidon. Tutkimuksen päättymisen jälkeen elossaolostatus saatiin selville 99,8 %:sta potilaista. Tutkittavista 72 % oli valkoihoisia, 20 % aasialaisia ja 4 % tummaihoisia. Keskimääräinen ikä kirjautumishetkellä oli 64 vuotta, ja 69 % tutkittavista oli miehiä. Lähtötilanteessa keskimääräinen eGFR oli 67,8 ml/min/1,73 m2, ja 62 %:lla potilaista eGFR oli ≥ 60 ml/min/1,73 m2, mediaani UACR oli 309 mg/g ja keskimääräinen HbA1c oli 7,7 %, 45 %:lla potilaista oli aiemmin todettu ateroskleroottinen sydän- ja verisuonitauti, 8 %:lla sydämen vajaatoiminta, ja potilaiden verenpaine oli keskimäärin 136/77 mmHg. T2D:n keskimääräinen kesto lähtötilanteessa oli 14,5 vuotta ja aiempaa diabeettista retinopatiaa raportoitiin 31 %:lla potilaista ja diabeettista neuropatiaa 28 %:lla potilaista. Lähtötilanteessa lähes kaikki potilaat saivat ACE:n estäjä- (43 %) tai ARB-hoitoa (57 %), ja 98 % potilaista käytti yhtä tai useampaa diabeteslääkettä (insuliini [54 %], biguanidit [69 %], GLP‑1-reseptoriagonistit [8 %], SGLT2-estäjät [8 %]). Yleisimmät muut lähtötilanteessa käytetyt lääkevalmisteet olivat statiinit (71 %).
Tilastollisesti merkitsevä ero finerenonin eduksi osoitettiin ensisijaiseen yhdistettyyn kardiovaskulaariseen päätetapahtumaan (ks. kuva 2 / taulukko 7 jäljempänä). Hoitovaikutus ensisijaiseen päätetapahtumaan oli yhdenmukainen eri alaryhmissä, mukaan lukien alue, eGFR, UACR, systolinen verenpaine ja HbA1c lähtötilanteessa.
Toissijaisessa yhdistetyssä päätetapahtumassa (munuaisten vajaatoiminta, pysyvä eGFR:n pienentyminen vähintään 40 %:lla tai munuaiskuolema) ilmaantuvuuden havaittiin olevan pienempi finerenoniryhmässä verrattuna lumelääkeryhmään, mutta tämä ero ei ollut tilastollisesti merkitsevä (ks. taulukko 7 jäljempänä). Hoitovaikutus toissijaisessa munuaisia koskevassa yhdistetyssä päätetapahtumassa oli yhdenmukainen kaikissa eGFR:n alaryhmissä lähtötilanteessa, mutta potilaiden alaryhmässä, joiden UACR oli < 300 mg/g, HR oli 1,16 (95 % CI 0,91; 1,47) ja potilaiden alaryhmällä, joiden UACR oli ≥ 300 mg/g, HR oli 0,74 (95 % CI 0,61; 0,89).
Etukäteen määritellyt toissijaiset tapahtumaan kuluvan ajan (time-to-event) päätetapahtumat sisältyvät taulukkoon 7.
Taulukko 6: Ensisijaisten ja toissijaisten tapahtumaan kuluvan ajan (time‑to‑event) ‑päätetapahtumien (ja niiden yksittäisten osien) analyysi vaiheen III tutkimuksessa FIDELIO‑DKD
| Kerendia* (N = 2 824) | Lumelääke (N = 2 838) | Hoitovaikutus | |||
|---|---|---|---|---|---|---|
| N (%) | Tapahtumia/ | N (%) | Tapahtumia/ | HR (95 %:n CI) | |
| Ensisijainen munuaisia koskevat yhdistetty päätetapahtuma ja sen osat | ||||||
Yhdistetty munuaisten vajaatoiminta, pysyvä eGFR:n pienentyminen ≥ 40 % tai munuaiskuolema | 498 (17,6)
| 7,53
| 600 (21,1)
| 9,09
| 0,82 (0,73; 0,92) p = 0,0009 | |
Munuaisten vajaatoiminta
| 205 (7,3)
| 2,96
| 235 (8,3)
| 3,39
| 0,86 (0,72; 1,05)
| |
Pysyvä eGFR:n pienentyminen ≥ 40 %
| 473 (16,7)
| 7,15
| 577 (20,3)
| 8,74
| 0,81 (0,72; 0,91)
| |
Munuaiskuolema
| 2 (< 0,1)
| –
| 2 (< 0,1)
| –
| –
| |
| Keskeinen toissijainen yhdistetty kardiovaskulaarinen päätetapahtuma ja sen osat | ||||||
Yhdistetty kardiovaskulaarikuolema, ei‑fataali MI, ei‑fataali aivohalvaus tai sairaalahoito sydämen vajaatoiminnan vuoksi | 366 (13,0)
| 5,11
| 420 (14,8)
| 5,93
| 0,86 (0,75; 0,99) p = 0,0344 | |
Kardiovaskulaarikuolema
| 128 (4,5)
| 1,70
| 150 (5,3)
| 1,99
| 0,86 (0,68; 1,09)
| |
Ei‑fataali MI
| 70 (2,5)
| 0,94
| 87 (3,1)
| 1,18
| 0,80 (0,58; 1,09)
| |
Ei‑fataali aivohalvaus
| 90 (3,2)
| 1,22
| 87 (3,1)
| 1,18
| 1,03 (0,77; 1,38)
| |
Sairaalahoito sydämen vajaatoiminnan vuoksi
| 138 (4,9)
| 1,88
| 162 (5,7)
| 2,22
| 0,85 (0,68; 1,07)
| |
| Toissijaiset tehoa koskevat päätetapahtumat | ||||||
Kuolema mistä tahansa syystä
| 219 (7,8)
| 2,90
| 244 (8,6)
| 3,24
| 0,90 (0,75; 1,08) **
| |
Sairaalahoito mistä tahansa syystä
| 1 259 (44,6)
| 22,59
| 1 321 (46,5)
| 23,91
| 0,95 (0,88; 1,02) **
| |
Yhdistetty munuaisten vajaatoiminta, pysyvä eGFR:n pienentyminen ≥ 57 % tai munuaiskuolema | 248 (8,8)
| 3,60
| 326 (11,5)
| 4,74
| 0,75 (0,65; 0,90) **
| |
*Hoito annoksella 10 tai 20 mg kerran vuorokaudessa suurimpien siedettyjen käyttöaiheen mukaisten ACE:n estäjä- tai ARB-annosten lisäksi.
** p = ei tilastollisesti merkittävä, kun monivertailu otetaan huomioon
CI: luottamusväli
HR: riskisuhde
pv: potilasvuosia
Kuva 1: Ensimmäiseen munuaisten vajaatoiminnan esiintymiseen, pysyvään eGFR:n pienentymiseen ≥ 40 % lähtötilanteesta tai munuaiskuolemaan kuluva aika FIDELIO‑DKD-tutkimuksessa
Taulukko 7: Ensisijaisten ja toissijaisten tapahtumaan kuluvan ajan (time‑to‑event) päätetapahtumien (ja niiden yksittäisten osien) analyysi vaiheen III tutkimuksessa FIGARO-DKD
| Kerendia* (N = 3 674) | Lumelääke (N = 3 654) | Hoitovaikutus | |||
|---|---|---|---|---|---|---|
| N (%) | Tapahtumia/ | N (%) | Tapahtumia/ | HR (95 %:n CI) | |
| Ensisijainen yhdistetty kardiovaskulaarinen päätetapahtuma ja sen osat | ||||||
Yhdistetty kardiovaskulaarikuolema, ei‑fataali MI, ei‑fataali aivohalvaus tai sairaalahoito sydämen vajaatoiminnan vuoksi | 457 (12,4)
| 3,88
| 518 (14,2)
| 4,46
| 0,87 (0,76; 0,98) p = 0,0254 | |
Kardiovaskulaarikuolema
| 193 (5,3)
| 1,56
| 214 (5,9)
| 1,75
| 0,89 (0,73; 1,08)
| |
Ei‑fataali MI
| 103 (2,8)
| 0,85
| 101 (2,8)
| 0,84
| 1,00 (0,76; 1,32)
| |
Ei‑fataali aivohalvaus
| 108 (2,9)
| 0,89
| 111 (3,0)
| 0,93
| 0,97 (0,74; 1,26)
| |
Sairaalahoito sydämen vajaatoiminnan vuoksi
| 117 (3,2) | 0,97 | 163 (4,5) | 1,36 | 0,71 (0,56; 0,90) | |
| Toissijainen munuaisia koskeva yhdistetty päätetapahtuma ja sen osat | ||||||
Yhdistetty munuaisten vajaatoiminta, pysyvä eGFR:n pienentyminen ≥ 40 % tai munuaiskuolema | 350 (9,5)
| 3,17
| 395 (10,8)
| 3,59
| 0,87 (0,75; 1,01) p = 0,0635 ** | |
Munuaisten vajaatoiminta
| 46 (1,3)
| 0,40
| 62 (1,7)
| 0,55
| 0,72 (0,49; 1,05)
| |
Pysyvä eGFR:n pienentyminen ≥ 40 %
| 338 (9,2)
| 3,06
| 385 (10,5)
| 3,50
| 0,86 (0,74; < 1,00)
| |
Munuaiskuolema
| 0
| –
| 2 (< 0,1)
| –
| –
| |
| Toissijaiset tehoa koskevat päätetapahtumat | ||||||
Kuolema mistä tahansa syystä
| 332 (9,0)
| 2,69
| 370 (10,1)
| 3,03
| 0,89 (0,77; 1,03) **
| |
Sairaalahoito mistä tahansa syystä
| 1 569 (42,7)
| 16,94
| 1 599 (43,8)
| 17,54
| 0,97 (0,90; 1,04) **
| |
Yhdistetty munuaisten vajaatoiminta, pysyvä eGFR:n pienentyminen ≥ 57 % tai munuaiskuolema | 108 (2,9)
| 0,95
| 139 (3,8)
| 1,23
| 0,77 (0,60; 0,99) **
| |
* Hoito annoksella 10 tai 20 mg kerran vuorokaudessa suurimpien siedettyjen käyttöaiheen mukaisten ACE:n estäjä- tai ARB-annosten lisäksi.
** p = ei tilastollisesti merkittävä, kun monivertailu otetaan huomioon
CI: luottamusväli
HR: riskisuhde
pv: potilasvuosia
Kuva 2: Ensimmäiseen kardiovaskulaariseen kuolemaan, ei‑fataaliin sydäninfarktiin (MI), ei‑fataaliin aivohalvaukseen tai sairaalahoitoon sydämen vajaatoiminnan vuoksi kuluva aika FIGARO-DKD-tutkimuksessa
Sydämen vajaatoiminta potilailla, joiden vasemman kammion ejektiofraktio (LVEF) on ≥ 40 %
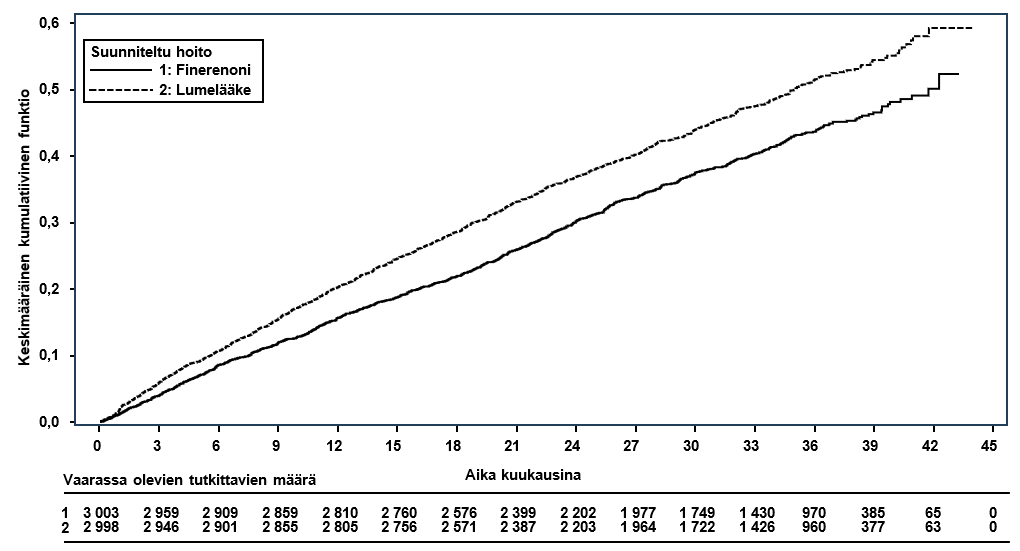
FINEARTS‑HF-tutkimuksessa tutkittiin finerenonin vaikutusta kardiovaskulaarisiin päätetapahtumiin lumelääkkeeseen verrattuna aikuisilla sydämen vajaatoimintapotilailla.
Potilaat kelpuutettiin tutkimukseen, jos heillä oli sydämen vajaatoiminnan diagnoosi NYHA-luokassa II–IV, he olivat avohoidossa tai sairaalahoidossa ensisijaisesti sydämen vajaatoiminnan vuoksi ja jos heidän dokumentoitu LVEF oli ≥ 40 %. Lisäksi potilailla oli eGFR ≥ 25 ml/min/1,73 m2 ja seerumin kaliumtaso ≤ 5,0 mmol/l seulonta- ja satunnaistushetkellä ja he saivat vakiintunutta lääkehoitoa sairauteensa, mukaan lukien hoitoa diureeteilla.
Ensisijainen päätetapahtuma oli seuraavien päätetapahtumien yhdistelmä: kardiovaskulaarinen kuolema ja sydämen vajaatoimintatapahtumien (ensimmäisten ja uusiutuvien) kokonaismäärä, joka koostui tapahtumista, joissa potilas joutui sairaalahoitoon sydämen vajaatoiminnan vuoksi ja kiireellisistä sydämen vajaatoimintaan liittyvistä sairaalakäynneistä. Toissijaisten päätetapahtumien, joihin kuului sydämen vajaatoimintatapahtumien (ensimmäisten ja uusiutuvien) kokonaismäärä sekä muutos lähtötilanteesta kuukausiin 6, 9 ja 12 Kansas City Cardiomyopathy Questionnaire (KCCQ) -kyselyn oireiden kokonaispisteissä /TSS (kysely kvantifioi sydämen vajaatoiminnan oireiden tiheyttä ja vaikeutta) analyysissä käytettiin monitestausmenetelmää.
Tutkimuksessa analysoitiin 6 001 potilasta, jotka oli satunnaisesti allokoitu saamaan joko finerenonia (N = 3 003) tai lumelääkettä (N = 2 998). Seurannan mediaanikesto oli 2,7 vuotta. Tutkimukseen osallistui 3 247 (54 %) potilasta, joilla oli ollut sydämen vajaatoimintatapahtuma edellisten 3 kuukauden aikana, mukaan lukien 1 219 (20 %) potilasta, jotka satunnaistettiin sairaalahoidon aikana tai 7 vuorokauden kuluessa kotiutuksesta.
Munuaisparametrien mukaisesti potilaat saivat joko 10 mg:n, 20 mg:n tai 40 mg:n finerenoniannoksen tai lumelääkettä kerran vuorokaudessa tutkimuksen ajan. Kuukauden 24 kohdalla finerenonilla hoidetuista tutkittavista 35 % käytti annosta 40 mg kerran vuorokaudessa, 32 % annosta 20 mg kerran vuorokaudessa. 12 % annosta 10 mg kerran vuorokaudessa ja 1 % oli keskeyttänyt hoidon. Kaikkina aikoina hoidon aikana 80 % potilaista saavutti tavoiteannoksensa.
Tutkimuksen päättymisen jälkeen elossaolostatus saatiin selville 99,7 %:sta potilaita. Tutkittavista 79 % oli valkoihoisia, 17 % aasialaisia ja 1,5 % tummaihoisia. Keskimääräinen ikä kirjautumishetkellä oli 72 vuotta, ja 46 % tutkittavista oli naisia. Lähtötilanteessa keskimääräinen LVEF oli 53 %, 64 %:lla potilaista LVEF oli ≥ 50 % ja 69 % oli NYHA-luokassa II, 30 % luokassa III ja 1 % luokassa IV. Keskimääräinen verenpaine oli 129/75 mmHg, ja painoindeksi 30 kg/m2. B-tyypin natriureettisen N-terminaalisen propeptidin (NT-pro-BNP) mediaani oli 1 041 pg/ml, keskimääräinen eGFR oli 62 ml/min/1,73 m2, 48 %:lla potilaista oli eGFR ˂ 60 ml/min/1,73 m2 ja mediaani UACR oli 18 mg/g. Eteisvärinää esiintyi 38 %:lla potilaista, ja 41 %:lla oli diabetes mellitus. Suurin osa potilaista sai loop-diureetteja (87 %), ACEi- tai ARB-hoitoa (79 %) tai angiotensiinireseptorin neprilysiini-inhibiittoria (9 %), ja 14 % sai SGLT2-inhibiittoria.
Tilastollisesti merkitsevä ero finerenonin eduksi osoitettiin ensisijaisen yhdistetyn päätetapahtuman osalta (ks. taulukko 8 jäljempänä). Vaikutus havaittiin varhain, ja se säilyi koko tutkimuksen ajan (ks. kuva 3 jäljempänä). Tilastollisesti merkitsevä ero finerenonin eduksi osoitettiin myös toissijaisissa päätetapahtumissa, sydämen vajaatoimintatapahtumien kokonaismäärän kohdalla. Myös ennalta määritetyt toissijaiset tehon päätetapahtumat on sisällytetty taulukkoon 8 jäljempänä. Ensisijaisten ja keskeisten toissijaisten päätetapahtumien hoitovaikutus oli yhdenmukainen kaikissa ennalta määritetyissä alaryhmissä, mukaan lukien sukupuoli, LVEF, NYHA-luokka, eGFR, aika edellisestä sydämen vajaatoimintatapahtumasta, SGLT2-inhibiittorihoito ja diabetes mellitus -tila.
Taulukko 8: Ensisijaisten ja toissijaisten päätetapahtumien (ja tapahtuma-aikapäätetapahtumien osalta niiden yksittäisten osien) analyysi vaiheen III tutkimuksessa FINEARTS-HF
| Kerendia* (N = 3 003) | Lumelääke (N = 2 998) | Hoitovaikutus | |||
|---|---|---|---|---|---|---|
| [Tapahtumat yhteensä] N (%) | Tapah-tumia/ | [Tapahtumat yhteensä] N (%) | Tapah-tumia/ | (95 %:n CI) | |
| Ensisijainen yhdistetty kardiovaskulaarinen päätetapahtuma ja sen osat | ||||||
Kardiovaskulaarisen kuoleman ja sydämen vajaatoimintatapahtumien kokonaismäärän yhdistelmä | [1 083] 624 (20,8)
| 14,88
| [1 283] 719 (24,0)
| 17,70
| RR 0,84 (0,74; 0,95) p = 0,0072
| |
Sydämen vajaatoimintatapahtumat yhteensä**
| [842] 479 (16,0)
| 11,57
| [1 024] 573 (19,1)
| 14,12
| RR 0,82 (0,71; 0,94) p = 0,0062
| |
Kardiovaskulaarikuolema
| 242 (8,1)
| 3,33
| 260 (8,7)
| 3,59
| HR 0,93 (0,78; 1,11)
| |
| Toissijaiset tehoa koskevat päätetapahtumat | ||||||
KCCQ-TSS-pisteiden muutos lähtötilanteesta
| LSM 7,99
| -
| LSM 6,43
| -
| LSM-ero 1,56 (0,79; 2,34) p ˂ 0,0001
| |
NYHA-luokan parannus
| 557 (18,6) N = 3 002 | -
| 553 (18,4)
| -
| OR 1,01 (0,88; 1,15) p = 0,9295†
| |
Munuaisten yhdistelmäpäätetapahtuma | 75 (2,5)
| 1,16
| 55 (1,8)
| 0,85
| HR 1,33 (0,94; 1,89) p†† | |
Pysyvä eGFR:n pienentyminen ≥ 50 %
| 68 (2,3)
| 1,05
| 51 (1,7)
| 0,79
| -
| |
Pysyvä eGFR:n pienentyminen arvoon ˂ 15 ml/min/1,73 m2
| 5 (0,2)
| 0,08
| 2 (˂ 0,1)
| 0,03
| -
| |
Dialyysin aloitus
| 2 (˂ 0,1)
| 0,03
| 2 (˂ 0,1)
| 0,03
| - | |
Munuaisen siirto
| 0 (0,0)
| 0,00
| 0 (0,0)
| -
| -
| |
Kuolema mistä tahansa syystä
| 491 (16,4)
| 6,71
| 522 (17,4)
| 7,17
| HR 0,93 (0,83; 1,06)
| |
Lyhenteet: CI: luottamusväli; HR: riskitiheyssuhde; LSM: pienimmän neliösumman keskiarvo; OR: vetosuhde; RR: riskisuhde; pv: potilasvuotta
* Hoitoa annettiin 10 mg, 20 mg tai 40 mg kerran vuorokaudessa vakiintuneen lääkehoidon (kuten diureettihoidon) lisäksi
** Sydämen vajaatoimintapahtumien (ensimmäisten ja uusiutuvien) kokonaismäärä oli myös keskeinen toissijainen päätetapahtuma
† Ei merkitsevä (testaus lopetettu)
†† Päätetapahtumaa ei testattu virallisesti (edeltävä testauksen päätetapahtuma ei ollut merkitsevä)
Kuva 3: Kardiovaskulaarisen kuoleman ja sydämen vajaatoimintatapahtumien (ensimmäisten ja uusiutuvien) kokonaismäärä (ensisijainen ja yhdistetty päätetapahtuma) FINEARTS-HF-tutkimuksessa
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Kerendia-valmisteen käytöstä kroonisen munuaistaudin ja kroonisen sydämen vajaatoiminnan hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Finerenoni imeytyy lähes kokonaan oraalisen annostelun jälkeen. Imeytyminen on nopeaa, ja huippupitoisuudet plasmassa (Cₘₐₓ) saavutetaan 0,5–1,25 tunnissa, kun tabletti otetaan paastotilassa. Finerenonin absoluuttinen biologinen hyötyosuus on 43,5 % suolen seinämässä ja maksassa tapahtuvan ensikierron metabolian vuoksi. Finerenoni on effluksitransportteri P‑glykoproteiinin substraatti in vitro, mitä ei kuitenkaan pidetä olennaisena sen imeytymisessä in vivo finerenonin suuren permeabiliteetin vuoksi.
Ruoan vaikutus
Finerenonin ottaminen runsaasti rasvaa ja kaloreita sisältävän ruoan kanssa suurensi finerenonin AUC-arvoa enintään 21 %, pienensi Cₘₐₓ-arvoa 19–23 % ja pidensi Cₘₐₓ:n saavuttamiseen kuluvaa aikaa enintään 2,5 tuntiin. Koska tällä ei katsota olevan kliinistä merkitystä, finerenoni voidaan ottaa joko aterian yhteydessä tai ilman ruokaa.
Jakautuminen
Finerenonin jakautumistilavuus vakaassa tilassa (Vₛₛ) on 52,6 l. Finerenonin sitoutuminen ihmisen plasmaproteiiniin in vitro on 91,7 %, ja sitoutuminen tapahtuu pääasiassa seerumin albumiiniin.
Biotransformaatio
Noin 90 % finerenonin metaboliasta on CYP3A4-välitteistä ja 10 % CYP2C8-välitteistä. Plasmasta löytyi neljä merkittävää metaboliittia. Kaikki metaboliitit ovat farmakologisesti inaktiivisia.
Eliminaatio
Finerenonin eliminaatio plasmasta on nopeaa eliminaation puoliintumisajan (t½) ollessa noin 2–3 tuntia. Finerenonin systeeminen puhdistuma verestä on noin 25 l/h. Noin 80 % annetusta annoksesta erittyi virtsaan ja noin 20 % ulosteisiin. Eritys tapahtui lähes täysin metaboliitteina, kun taas muuttumattoman finerenonin erittyminen on vähäpätöinen reitti (< 1 % annoksesta virtsaan glomerulussuodatuksen kautta, < 0,2 % ulosteisiin).
Lineaarisuus
Finerenonin farmakokinetiikka on lineaarista tutkitulla annosalueella 1,25–80 mg, kun valmiste annetaan kerta-annoksena tabletteina.
Erityisryhmät
Iäkkäät
Finerenonia FIDELIO‑DKD-tutkimuksessa saaneista kaikkiaan 2 818 potilaasta 58 % oli vähintään 65-vuotiaita ja 15 % oli vähintään 75-vuotiaita. Finerenonia FIGARO‑DKD-tutkimuksessa saaneista kaikkiaan 3 671 potilaasta 53 % oli vähintään 65-vuotiaita ja 14 % oli vähintään 75‑vuotiaita. Finerenonia FINEARTS-HF-tutkimuksessa saaneista kaikkiaan 2 993 potilaasta 79 % oli vähintään 65-vuotiaita ja 43 % oli vähintään 75-vuotiaita.
Vaiheen I tutkimuksessa (N = 48) iäkkäillä terveillä osallistujilla (≥ 65-vuotiaat) ilmeni suurempia finerenonin pitoisuuksia plasmassa kuin nuoremmilla terveillä osallistujilla (≤ 45-vuotiaat), keskimääräisen AUC-arvon ollessa iäkkäillä 34 % suurempi ja Cₘₐₓ-arvon 51 % suurempi (ks. kohta Annostus ja antotapa). Populaatiofarmakokineettisessä analyysissä ikää ei tunnistettu finerenonin AUC:n tai Cₘₐₓ:n kovariaatiksi.
Munuaisten vajaatoiminta
Lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma [CLCR] 60–< 90 ml/min) ei vaikuttanut finerenonin AUC- tai Cₘₐₓ-arvoon.
Verrattuna potilaisiin, joilla on normaali munuaistoiminta (CLCR ≥ 90 ml/min), keskivaikean (CLCR 30–< 60 ml/min) tai vaikean (CLCR < 30 ml/min) munuaisten vajaatoiminnan vaikutus finerenonin AUC-arvoon oli samanlainen, AUC suureni 34–36 %. Keskivaikealla tai vaikealla munuaisten vajaatoiminnalla ei ollut vaikutusta Cₘₐₓ-arvoon (ks. kohta Annostus ja antotapa).
Suuren plasmaproteiineihin sitoutumisen vuoksi finerenonin ei odoteta olevan dialysoitavissa.
Maksan vajaatoiminta
Finerenonialtistus ei muuttunut kirroosipotilailla, joilla oli lievä maksan vajaatoiminta (ks. kohta Annostus ja antotapa).
Kirroosipotilailla, joilla oli keskivaikea maksan vajaatoiminta, finerenonin kokonais-AUC-arvo kasvoi 38 % ja sitoutumattoman finerenonin AUC-arvo 55 %. Cₘₐₓ-arvossa ei havaittu eroa suhteessa terveisiin verrokkeihin (ks. kohta Annostus ja antotapa).
Ei ole tietoja potilaista, joilla on vaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Paino
Populaatiofarmakokineettisessä analyysissä paino tunnistettiin finerenonin Cₘₐₓ:n ja AUC-arvon kovariaatiksi. Alle 57 kg painavien tutkittavien Cₘₐₓ- arvon arvioitiin olevan keskimäärin 52 % suurempi ja AUC-arvon arvioitiin olevan keskimäärin 30 % suurempi verrattuna 57–122 kg painaviin tutkittaviin. Yli 122 kg painavien tutkittavien Cₘₐₓ- arvon arvioitiin olevan keskimäärin 32 % pienempi ja AUC-arvon arvioitiin olevan keskimäärin 20 % pienempi verrattuna 57–122 kg painaviin tutkittaviin. Annoksen muuttaminen painon perusteella ei ole tarpeellista (ks. kohta Annostus ja antotapa).
Farmakokineettiset/farmakodynaamiset suhteet
Pitoisuus-vaikutussuhdetta UACR:ään ajan suhteen luonnehti enimmäistehon malli, joka osoitti saturaatiota suurilla altistuksilla. Mallin ennustama aika, jossa saavutetaan täysi (99 %) vakaan tilan lääkevaikutus UACR:ään, oli 138 päivää. Farmakokineettinen puoliintumisaika oli 2–3 tuntia ja vakaa tila saavutettiin 2 vuorokauden kuluttua, mikä viittaa epäsuoriin ja viivästyneisiin vaikutuksiin farmakodynaamisissa vasteissa.
Kliiniset tutkimukset, joissa ei ollut relevantteja lääkkeiden välisiä yhteisvaikutuksia
Samanaikainen gemfibrotsiilin (600 mg kahdesti vuorokaudessa) eli vahvan CYP2C8:n estäjän käyttö suurensi finerenonin AUC-arvon 1,1‑kertaiseksi ja Cₘₐₓ-arvon 1,2‑kertaiseksi. Tällä ei katsota olevan kliinistä merkitystä.
Edeltävä ja samanaikainen hoito protonipumpun estäjällä omepratsolilla (40 mg kerran vuorokaudessa) ei vaikuttanut finerenonin keskimääräiseen AUC- tai Cₘₐₓ-arvoon.
Samanaikaisella antasidin (alumiinihydroksidi ja magnesiumhydroksidi (70 mval)) käytöllä ei ollut vaikutusta finerenonin keskimääräiseen AUC-arvoon ja se pienensi sen keskimääräistä Cₘₐₓ-arvoa 19 %. Tällä ei katsota olevan kliinistä merkitystä.
Keskinäisen farmakokineettisen yhteisvaikutuksen puute osoitettiin finerenonin ja CYP2C9:n substraatin varfariinin sekä finerenonin ja P‑gp -substraatin digoksiinin välillä.
Useilla suun kautta kerran vuorokaudessa otetuilla 40 mg:n finerenoniannoksilla ei ollut kliinisesti oleellista vaikutusta rintasyövän resistenssiproteiinin (BCRP) ja orgaanisten anionien transportteripolypeptidien (OATP) substraatin rosuvastatiinin AUC-arvoon ja Cₘₐₓ-arvoon.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, yhden annoksen toksisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta, fototoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Toistuvan annon toksisuus
Koirilla havaittiin eturauhasen painon ja koon pienenemistä noin 4–60-kertaisella AUCsitoutumaton‑arvolla ihmisten annokseen verrattuna. Annoksella, jolla löydöksiä ei havaita, jää turvallisuusmarginaaliksi noin 1-2.
Karsinogeeninen potentiaali
2 vuotta kestäneissä karsinogeenisuustutkimuksissa finerenonilla ei osoitettu karsinogeenista potentiaalia uros- tai naarasrotilla tai naarashiirillä. Uroshiirillä finerenoni aiheutti Leydigin solujen adenooman lisääntymistä annoksilla, jotka olivat 10–26‑kertaisia suhteessa ihmisen AUCsitoutumaton-arvoon. Annokset, jotka vastasivat 7-kertaista altistusta suhteessa 40 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna sekä annokset, jotka vastasivat 17‑kertaista altistusta suhteessa 20 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna eivät aiheuttaneet kasvaimia. Jyrsijöillä on tunnettu alttius kehittää näitä kasvaimia, minkä lisäksi supraterapeuttisiin annoksiin liittyvien farmakologisten mekanismien ja riittävien turvallisuusrajojen perusteella voidaan todeta, ettei Leydigin solujen kasvainten lisääntymisellä uroshiirissä ole kliinistä relevanssia.
Kehitystoksisuus
Rotilla tehdyssä alkion ja sikiön toksisuustutkimuksessa finerenoni aiheutti istukan painon pienenemistä ja sikiötoksisuuden merkkejä, mukaan lukien sikiön pienempää painoa ja myöhästynyttä luutumista, emolle toksisella annoksella 10 mg/kg/vrk, joka vastasi vähintään 7-kertaista altistusta suhteessa ihmisen altistukseen AUCsitoutumaton-arvolla mitattuna. Annoksella 30 mg/kg/vrk sisäelin- ja luustomuutosten (lievä turvotus, lyhentynyt napanuora, hieman suurentunut aukile) ilmaantuvuus suureni ja yhdellä sikiöllä todettiin monimutkaisia epämuodostumia, mukaan lukien harvinainen epämuodostuma (kaksi aortankaarta) käytettäessä noin 10-kertaista altistusta suhteessa 40 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna ja noin 25-kertaista altistusta suhteessa 20 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna. Löydöksistä vapaiden annosten (pieni annos rotilla, suuri annos kaniineilla) perusteella turvallisuusraja vastaa 4–13‑kertaista AUCsitoutumaton-arvoa. Siten rottatutkimusten löydökset eivät viittaa sikiöön kohdistuvien haittojen lisääntymiseen.
Kun rotat altistettiin tiineyden ja imetyksen aikana pre- ja postnataalista kehitystä koskevassa toksisuustutkimuksessa, poikasten kuolleisuutta ja muita haittavaikutuksia (pienempi poikasten paino, viivästynyt korvien avautuminen) havaittiin altistuksella, joka vastasi noin kaksinkertaista 40 mg annoksella odotettua altistusta ihmisellä AUCsitoutumaton-arvolla mitattuna tai nelinkertaista 20 mg annoksella odotettua ihmisen altistusta AUCsitoutumaton-arvolla mitattuna. Lisäksi poikasten liikkumisaktiivisuus oli hieman kohonnut, mutta niillä ei havaittu muita neurobehavioraalisia muutoksia altistuksella, joka vastasi minimissään noin kaksinkertaista odotettua AUCsitoutumaton-arvolla mitattua altistusta ihmisellä annoksen ollessa 40 mg tai noin nelinkertaista odotettua AUCsitoutumaton-arvolla mitattua altistusta ihmisellä annoksen ollessa 20 mg. Löydöksistä vapaiden annosten perusteella turvallisuusraja vastaa suunnilleen AUCsitoutumaton-arvoa 2 annoksella 20 mg ja on terapeuttisella vaihteluvälillä 40 mg:n annoksella. Poikasten lisääntynyt liikkumisaktiivisuus voi viitata mahdolliseen riskiin sikiölle. Lisäksi poikaslöydösten perusteella vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea.
Naaraiden hedelmällisyys
Finerenoni heikensi naaraiden hedelmällisyyttä (keltarauhasten ja kiinnittymiskohtien lukumäärän väheneminen) sekä aiheutti merkkejä varhaisesta alkiotoksisuudesta (lisääntynyt implantaation jälkeinen kuolleisuus ja elinkykyisten sikiöiden pienempi lukumäärä) annoksella, joka vastasi noin 9-kertaista altistusta suhteessa 40 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna ja noin 21-kertaista altistusta suhteessa 20 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna. Lisäksi havaittiin munasarjojen painon pienentymistä annoksilla, jotka vastasivat noin 7-kertaista altistusta suhteessa 40 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna ja 17-kertaista altistusta suhteessa 20 mg annoksella saavutettuun altistukseen ihmisellä AUCsitoutumaton-arvolla mitattuna. Vaikutuksia naaraiden hedelmällisyyteen ja varhaiseen alkionkehitykseen ei havaittu annoksilla, jotka vastasivat noin 4-kertaista altistusta ihmisellä AUCsitoutumaton-arvolla mitattuna käytettäessä 40 mg:n annosta ja 10-kertaista altistusta ihmisellä AUCsitoutumaton-arvolla mitattuna käytettäessä 20 mg:n annosta. Näin ollen naarasrotista tehdyillä löydöksillä ei ole kliinistä relevanssia (ks. kohta Raskaus ja imetys).
Ympäristöön kohdistuvien riskien arviointi
Ympäristöön kohdistuvien riskien arviointitutkimukset ovat osoittaneet, että finerenoni saattaa aiheuttaa riskin pinta- ja pohjavedelle (ks. kohta Käyttö- ja käsittelyohjeet).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Selluloosa, mikrokiteinen (E 460)
Kroskarmelloosinatrium
Hypromelloosi 2910 (E 464)
Laktoosimonohydraatti
Magnesiumstearaatti (E 470b)
Natriumlauryylisulfaatti (E 487)
Tabletin päällys
Hypromelloosi 2910 (E 464)
Titaanidioksidi (E 171)
Talkki (E 553b)
Kerendia 10 mg kalvopäällysteiset tabletit
Punainen rautaoksidi (E 172)
Kerendia 20 mg kalvopäällysteiset tabletit
Keltainen rautaoksidi (E 172)
Kerendia 40 mg kalvopäällysteiset tabletit
Punainen rautaoksidi (E 172)
Keltainen rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KERENDIA tabletti, kalvopäällysteinen
10 mg (L:kyllä) 28 fol (64,30 €), 98 fol (213,70 €)
20 mg (L:kyllä) 28 fol (64,30 €), 98 fol (213,70 €)
PF-selosteen tieto
Läpinäkyvät PVC/PVDC/alumiini-läpipainopakkaukset, joihin on merkitty viikonpäivät ja joissa on 14 kalvopäällysteistä tablettia. Pakkauskoko 14, 28 tai 98 kalvopäällysteistä tablettia.
Perforoidut, läpinäkyvät yksittäispakatut PVC/PVDC/alumiini-läpipainopakkaukset, joissa on 10 x 1 kalvopäällysteistä tablettia. Pakkaus sisältää 100 × 1 kalvopäällysteistä tablettia.
Valkoinen läpinäkymätön HDPE-purkki, jossa valkoinen läpinäkymätön polypropeenista valmistettu turvakierrekorkki ja tiiviste. Pakkaus sisältää 100 kalvopäällysteistä tablettia (vain Kerendia 10 mg:n ja 20 mg:n kalvopäällysteiset tabletit).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kerendia 10 mg kalvopäällysteiset tabletit
Vaaleanpunainen, soikea, pitkänomainen kalvopäällysteinen tabletti, jonka pituus on 10 mm ja leveys 5 mm ja jonka toisella puolella on merkintä "10" ja toisella puolella merkintä "FI".
Kerendia 20 mg kalvopäällysteiset tabletit
Vaaleankeltainen, soikea, pitkänomainen kalvopäällysteinen tabletti, jonka pituus on 10 mm ja leveys 5 mm ja jonka toisella puolella on merkintä "20" ja toisella puolella merkintä "FI".
Kerendia 40 mg kalvopäällysteiset tabletit
Harmaanoranssi, soikea, pitkänomainen kalvopäällysteinen tabletti, jonka pituus on 11 mm ja leveys 5 mm ja jonka toisella puolella on merkintä "40" ja toisella puolella merkintä "FI".
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste voi aiheuttaa ympäristöön kohdistuvan riskin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KERENDIA tabletti, kalvopäällysteinen
10 mg 28 fol, 98 fol
20 mg 28 fol, 98 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Finerenoni: Aikuisten tyypin 2 diabetekseen liittyvän kroonisen munuaistaudin hoito erityisin edellytyksin (3071).
ATC-koodi
C03DA05
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2026
Yhteystiedot
 BAYER OY
BAYER OY Tuulikuja 2, PL 73
02151 Espoo
020 785 21
www.bayer.fi